 Patrick D. Lyden1*
Patrick D. Lyden1* Kent E. Pryor2Jennifer Minigh3Thomas P. Davis4
Kent E. Pryor2Jennifer Minigh3Thomas P. Davis4 John H. Griffin5Howard Levy6
John H. Griffin5Howard Levy6 Berislav V. Zlokovic1
Berislav V. Zlokovic1- 1Department of Physiology and Neuroscience, Keck School of Medicine, Zilkha Neurogenetic Institute, University of Southern California, Los Angeles, CA, United States
- 2ZZ Biotech LLC, Houston, TX, United States
- 3inSeption Group LLC, Lansdale, PA, United States
- 4Department of Medical Pharmacology, University of Arizona College of Medicine, Tucson, AZ, United States
- 5Department of Molecular Medicine, The Scripps Research Institute, La Jolla, CA, United States
- 6Howard Levy Consulting LLC, Hopewell, NJ, United States
Ischemic stroke is the most widespread cause of disability and a leading cause of death in developed countries. To date, the most potent approved treatment for acute stroke is recanalization therapy with thrombolytic drugs such as tissue plasminogen activator (rt-PA or tPA) or endovascular mechanical thrombectomy. Although tPA and thrombectomy are widely available in the United States, it is currently estimated that only 10–20% of stroke patients get tPA treatment, in part due to restrictive selection criteria. Recently, however, tPA and thrombectomy selection criteria have loosened, potentially allowing more patients to qualify. The relatively low rate of treatment may also reflect the perceived risk of brain hemorrhage following treatment with tPA. In translational research and a single patient study, protease activated receptor 1 (PAR-1) targeted therapies given along with thrombolysis and thrombectomy appear to reduce hemorrhagic transformation after recanalization. Such adjuncts may likely enhance the availability of recanalization and encourage more physicians to use the recently expanded selection criteria for applying recanalization therapies. This narrative review discusses stroke therapies, the role of hemorrhagic transformation in producing poor outcomes, and presents the data suggesting that PAR-1 acting agents show promise for decreasing hemorrhagic transformation and improving outcomes.
Introduction
Each year about 795,000 people in the United States experience a stroke (1). Of all types of stroke, 87% are ischemic (i.e., caused by an interruption of blood supply), 10% are intracerebral hemorrhage strokes (i.e., caused by a ruptured blood vessel), and 3% are subarachnoid hemorrhage strokes (bleeding into the outermost layer of the brain) (2). Ischemic stroke is the most widespread cause of disability and a leading cause of death in developed countries (3).
The most potent treatment for stroke is recanalization, that is, treatment with intravenous thrombolytics, mechanical revascularization (removal of the clot) known as intra-arterial thrombectomy (IAT), or both. Not all patients respond fully to recanalization; therefore, adjunctive cytoprotective treatments are needed and many development efforts are ongoing to overcome the long history of failed neuroprotection trials (likely due to lack of recanalization documentation). Agents acting on the protease activated receptor 1 (PAR-1) exhibit pleiotropic actions on neurons, glia, and cerebral vascular cells, including cytoprotection and anti-inflammation (4). In the RHAPSODY trial, the PAR-1 acting drug 3K3A-APC appeared to reduce hemorrhagic transformation (5).
In 2015, several successful trials proved the efficacy of IAT for acute ischemic stroke with large vessel occlusions (6–10). Then, it was shown that multimodal imaging permits clinicians to select patients for IAT with great success. Recently, the feasibility of combining IAT with a putative cytoprotectant has been shown in 2 trials. The RHAPSODY trial was the first to include IAT in the clinical trial of a cytoprotectant (5). A larger recent trial allowed IAT use, but only in patients with evidence of good collateral flow (11). These results confirm that recanalization may powerfully influence the effect of putative cytoprotectants.
Stroke continues to be a major public health concern despite significant previous research that has produced treatment approaches addressing acute reperfusion and revascularization (12–14), neuronal protection (12), and regeneration of damaged brain tissue (15, 16). All these tactics were based on scientific principles and preclinical data, yet no candidate cytoprotective therapy has successfully entered clinical practice (15). It is now clear that single-action, single-target agents fail to treat stroke because ischemia produces a combination of pathologic pathways proceeding in parallel that damage neural tissue (17).
This narrative review presents a discussion of stroke therapies, the role of hemorrhagic transformation in producing poor outcomes, and presents the data suggesting that PAR-1 acting agents show promise for decreasing hemorrhagic transformation and improving outcomes.
Stroke Therapy
Recanalization therapy with thrombolytic drugs such as recombinant tissue plasminogen activator (rt-PA or tPA) is the most common treatment for acute stroke. tPA is approved for intravenous administration within 3 h of onset of acute ischemic stroke in the United States and for up to 4.5 h following the stroke in Europe (1, 18). Thrombolytic therapy with intravenous tPA beyond 4.5 h in select subjects with diffusion/fluid attenuated inversion recovery mismatch on magnetic resonance imaging (MRI) is also recommended, but less frequently possible (19). The most widely feared adverse effect of tPA is symptomatic intracranial hemorrhage (SICH; 3–6%); other risks include systemic bleeding, myocardial rupture (when used to treat acute myocardial infarction), and, in rare cases, anaphylaxis, or angioedema (20). Although tPA is widely available in the United States, only 10–20% of stroke patients receive such treatment (21, 22), primarily because patients may present with mild deficits, are beyond 4.5 h after onset, have conditions or concomitant medications that increase bleeding risk, or for other reasons.
Another effective (however, less frequently used) recanalization therapy for stroke is mechanical thrombectomy (7–9, 23). Use of mechanical thrombectomy (with or without tPA) is considered standard-of-care treatment in patients with documented large vessel occlusion, defined as thromboembolic blockage of the distal internal carotid artery, the M1 or proximal M2 portions of the middle cerebral artery, or the proximal anterior cerebral artery. As shown in Table 1, several well-controlled randomized clinical trials showed benefit following combination therapy of thrombectomy and tPA. In some of these trials, however, patients benefited who were ineligible for tPA and were treated with thrombectomy alone. With careful imaging selection, recanalization with thrombolysis or thrombectomy may be successful as late as 16 or 24 h after last known well time (12, 25, 26).
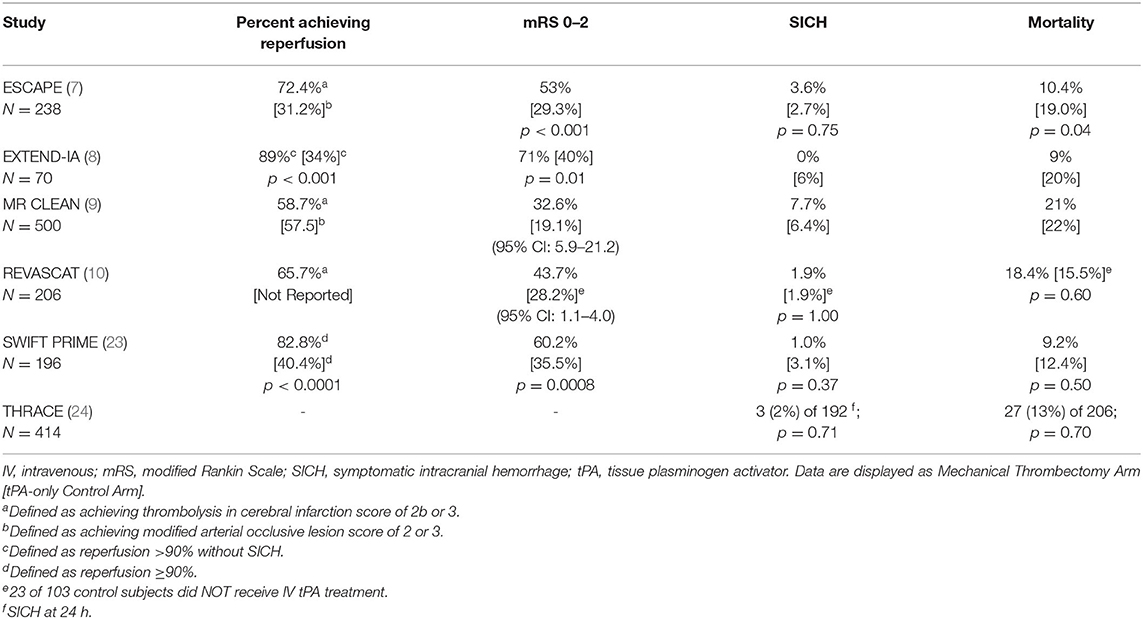
Table 1. Summary of mechanical thrombectomy study outcomes.
Although recent trials suggest that recanalization therapy for stroke offers great promise, there remains a very large unmet need to reduce stroke-related deficit and ensure improved outcomes. First, not all patients treated with thrombolysis or thrombectomy recover full function. Second, the risk of hemorrhage after recanalization therapies dissuades some practitioners from using them. Thus, adjuvant cytoprotectants are needed to complement recanalization therapies in such patients, or to provide improved outcomes in patients unable to receive thrombolytic or thrombectomy therapy. In past clinical trials that did not include mechanical thrombectomy as a treatment option, it is likely that many patients failed to reperfuse; the candidate adjuvant cytoprotectants may therefore have appeared less likely to benefit the patients. In modern clinical stroke trial design, candidate adjuvant therapy is studied in concert with recanalization. In patients with large vessel occlusion, more than 80% receiving mechanical thrombectomy do recanalize. In patients without documented large vessel occlusion, thrombolytic therapy alone is generally sufficient to reperfuse the microvasculature.
Hemorrhagic Transformation
Hemorrhagic transformation (HT) is a consequence of ischemic blood–brain barrier breakdown that occurs mainly within 2 weeks of ischemic stroke (27). Following an acute stroke, the cerebral vasculature is damaged, which increases the risk for HT.
The presentation of HT includes minor petechial bleeding (hemorrhagic infarct) and large mass-producing hemorrhages (parenchymal hematoma). Intracranial hemorrhages are classified by both imaging characteristics and the presentation of clinical worsening.
Radiologic classification uses the location and extent of hemorrhage to distinguish among hemorrhage subtypes (see Table 2).
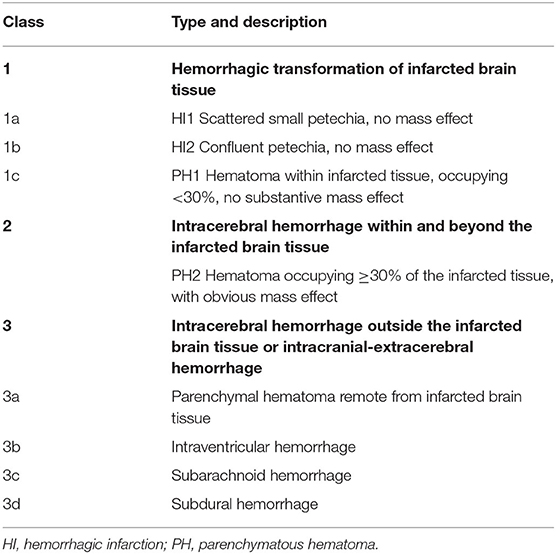
Table 2. Anatomic descriptions of intracranial hemorrhages according to the heidelberg bleeding classification (28).
Symptomatic vs. Asymptomatic Hemorrhagic Transformation
In addition to radiologic classification, intracranial hemorrhages are labeled asymptomatic, or symptomatic based on an accompaniment of observable neurologic decline.
The term SICH was first used by Levy et al. (29). The National Institute of Neurological Disorders and Stroke (NINDS) trials, defined SICH as “any hemorrhagic transformation temporally related to any worsening in neurologic condition (30).”Over the next 2 decades, this definition was recognized as over-inclusive. Other groups such as the Safe Implementation of Thrombolysis in Stroke-Monitoring Study (SITS-MOST) investigators (31), the European Cooperative Acute Stroke Study (ECASS) II and III investigators (18, 32), and the International Stroke Trial-3 (IST-3) investigators (33) have sought more comprehensive definitions of SICH. Widely used definitions are the SITS-MOST and ECASS II:
• SITS-MOST definition of SICH: Local or remote parenchymatous hematoma (PH)-2 with a worsening (i.e., increase of ≥4) on the National Institutes of Health Stroke Scale (NIHSS) score.
• ECASS II definition of SICH: Any intracranial hemorrhage with a clinical worsening (indicated by clinical deterioration or adverse events) or causing a worsening (i.e., increase of ≥4) in NIHSS score.
Asymptomatic intracranial hemorrhage (AICH) does not have a rigorous definition like SICH. In general, it is described as an imaging-documented brain bleed without a concomitant marked deterioration in the patient's neurologic state observable using a neurological rating scale. Thus, the descriptor “asymptomatic” is a misnomer, as the patient very well may exhibit subtle findings, or more robust findings were they to be examined weeks or months later. In many studies, AICH classification is generally not included so that when meta-analyses are performed, those patients with AICH can only be identified as those patients not having SICH. To complicate matters, there are only a handful of studies that specifically enroll subjects with AICH.
Symptomatic Hemorrhage—Clearly a Detriment
Regardless of how SICH is defined, it is consistently associated with worse clinical outcomes (34, 35). Hao et al. (34) reported that patients with and without SICH differed significantly using the modified Rankin score (mRS) scores (odds ratio: 1.45; 95% confidence interval [CI]: 1.10–1.81), 90-day mortality (higher in patients with SICH [65.3%] vs. without [18.8%]; p < 0.001); furthermore, favorable neurological outcome (defined as mRS 0–2) at 90 days was proportionally lower in patients with SICH (8.9%) than without (51.2%) (p < 0.001).
Asymptomatic Hemorrhage—Likely a Detriment as Well
Whether AICH fosters a negative prognosis remains controversial. Some studies confirmed that AICH has a negative effect on functional outcome. Although there is little clinical trial information regarding possible adverse effects of AICH, the limited available evidence indicates it may not be harmless.
In a study by Kent et al. (36), patients with AICH tended toward worse outcomes, even after adjusting for other prognostic variables (odds ratio: 0.69); however, this trend did not reach statistical significance. The investigators cautioned against concluding that AICH are clinically innocuous based on a lack of statistical effect.
Dzialowski et al. (37) used data obtained from the Canadian Alteplase for Stroke Effectiveness Study to investigate the association between HT type and functional outcome. The authors concluded that the likelihood of a poor outcome following thrombolysis was associated with the extent of hemorrhage. The proportion of patients with a good outcome was 41% with no HT, 30% with HI-1, 17% with HI-2, 15% with PH-1, and 7% with PH-2 (p < 0.0001). Although HI-1 was not a predictor of outcome, other types of bleeds were after adjusting for covariates: HI-2 (odds ratio: 0.38; 95% CI: 0.17–0.83), PH-1 (odds ratio: 0.32; 95% CI: 0.12–0.80), and PH-2 (odds ratio: 0.14; 95% CI: 0.04–0.48), thereby suggesting that HI grades of hemorrhagic transformation may not be benign.
Park et al. (38) who sought to determine the impact of asymptomatic hemorrhage transformation on the 3-month outcome, found the odds of a worse outcome were increased by a factor of 2 in patients with AICH compared with those without after acute ischemic stroke. The crude and adjusted odds ratios of AICH for an increment of mRS score at 3 months were 2.94 (95% CI: 2.05–4.24) and 1.90 (95% CI: 1.27–2.82), respectively.
Lei et al. (39) examined whether AICH affects risk of stroke recurrence and a long-term poor outcome. Both SICH and AICH post acute ischemic stroke impacted long-term clinical outcomes. Moreover, patients with SICH or AICH suffered a lower survival rate than did patients without HT in the 1st year following stroke (p < 0.001). The investigators suggested that AICH should not be considered clinically innocuous.
In acute ischemic stroke patients undergoing thrombectomy, AICH appeared to be associated with high mortality and worse functional outcomes (40). Specifically, AICH appeared to result in lower odds of functional independence (61.9% of patients without AICH and 35.9% with AICH achieved functional independence at the 3-month follow-up; adjusted p = 0.117) and higher odds of deaths (35.9% of patients with AICH vs. 11.1% without AICH died; adjusted p = 0.015).
Hao et al. (41) reported that in an Asian population, patients with AICH after endovascular treatment had a lower ratio of excellent outcome (odds ratio: 0.53; 95% CI: 0.33–0.84; p = 0.007) compared with patients without ICH. According to the researchers: “Considering the relatively higher incidence (33.5%) and negative impacts on functional outcomes in this study, AICH after endovascular treatment may not be innocuous.”
In a recent study, Li et al. (42) evaluated the prevalence of previous chronic cerebral hemorrhage, especially asymptomatic cases, and the associated factors in patients who experienced an acute ischemic stroke. Overall, 9.4% of patients were determined to have had a previous chronic cerebral hemorrhage, with almost half of these being asymptomatic, indicating that previous chronic cerebral hemorrhage is not uncommon in acute ischemic stroke patients. Furthermore, there were no differences in the clinical characteristics of symptomatic vs. asymptomatic previous chronic cerebral hemorrhage, which complicates the detection of asymptomatic hemorrhage, and according to the authors, could increase the risk of re-bleeding.
While AICH may not be associated with acute observable neurologic deterioration, its presence may undermine long-term neurological functions. As the red blood cells in the microbleeds break down over the following days to weeks, neural toxic effects can emerge including heme-induced cerebral inflammation, neuronal apoptosis, and demyelination (43, 44).
Although many studies report the rate of AICH to be ~10% (30, 45, 46), other studies indicate the rate may be as high as 30–40% when using CT scan (24, 34, 47). AICH occurs at a sufficient frequency such that hemorrhages initially presenting as asymptomatic can eventually result in substantial complications, cause an increase in hospital length of stay, lead to poorer long-term outcomes, and incur higher healthcare costs (48); thus, any bleeding, asymptomatic or not, is a concern following stroke.
APC and APC Analogs
A promising approach for stroke therapy is based on recently discovered biological properties of APC, which is an endogenous plasma protease with multiple properties including antithrombotic action, cytoprotective propensity, and anti-inflammatory activity in the brain and spinal cord (4). Based on known cellular and molecular mechanisms, an APC approach showed promise in experiments consistent with Stroke Therapy Academic Industry Roundtable and other guidelines (49). Wildtype APC shows potent anticoagulant effects along with cytoprotection and reduced inflammation, and anticoagulants often carry an increased risk for serious bleeding. Therefore, protein engineering of APC was undertaken to reduce bleeding risk.
Signaling-selective APC analogs were engineered to retain normal cell-signaling activity (50–53) but to have greatly diminished anticoagulant activity (<10%) (54), thereby reducing in vivo risk for bleeding compared with wildtype APC (55–57).
An engineered form of APC, called 3K3A-APC reflecting lysine to alanine substitutions at positions 191, 192, and 193, offers advantages over wildtype APC (4, 53, 58, 59). 3K3A-APC is a 405-residue APC variant engineered to maximize neuroprotective and cytoprotective activities and minimize anticoagulant activity. It was developed by altering factor Va binding exosites (reducing anticoagulation) on APC without modifying the exosites that recognize and bind to the G-protein coupled receptors, protease-activated receptor 1 (PAR-1), and PAR-3. This mutant retains the cytoprotective cell-signaling effects of native (wildtype) APC but has >90% less of the wildtype anticoagulant effects (54). Glycosylation of recombinant 3K3A-APC differs from wildtype APC because it is expressed in Chinese hamster ovary cells.
Anticoagulants do not improve outcome following stroke (60–62). Thus, the residual anticoagulant activity of 3K3A-APC is not responsible for the benefits seen in animal models (63).
Rationale for APC and APC Analogs in Treating Stroke
APC is an endogenous serine protease with systemic anticoagulant activity as well as cell-signaling actions that convey endothelial stabilizing, anti-inflammatory, and anti-apoptotic activities, and that promote neurogenesis (59, 64–67). APC is normally generated in vivo from zymogen protein C through activation by thrombin on the surface of endothelial cells. This activation requires 2 membrane receptors: the thrombomodulin receptor (which binds thrombin) and the endothelial protein C receptor (which binds protein C). The multiple properties of APC should combine in reversing the effects of an ischemic stroke and in protecting ischemic brain tissue from further damage.
The anticoagulant activity of APC is independent of its direct cellular effects and is mediated by irreversible proteolytic degradation of factors Va and VIIIa with contributions by other cofactors. Its cytoprotective cell-signaling activities require multiple cell-surface receptors and, in most cases, proteolytic activation of PAR-1 (Figure 1) (51, 59, 65–68).
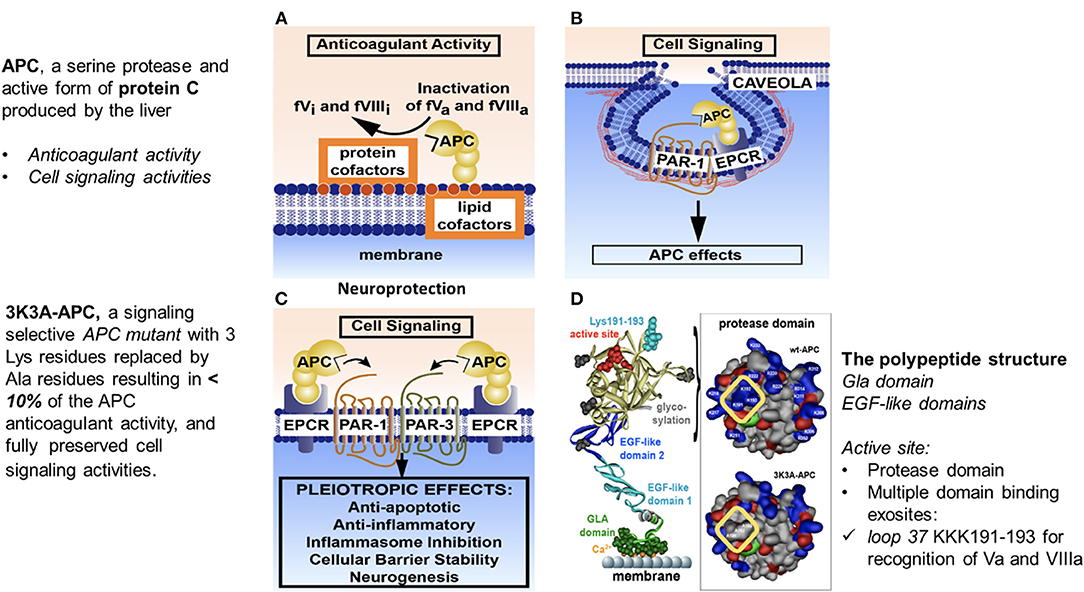
Figure 1. Anticoagulant and cell-signaling pathways of APC and the structure of signaling-selective 3K3A-APC. APC, activated protein C; BBB, blood–brain barrier; EGF, endothelial growth factor; EPCR, endothelial protein C receptor; GLA, gamma-carboxyglutamic acid; PAR, protease-activated receptor. Reprinted from blood, vol. 132(2), Griffin et al. (67) activated protein C, protease activated receptor 1 and neuroprotection; 159–169, 2018, with permission from the American Society of Hematology. (A) Anticoagulant activity of APC involves the proteolytic inactivation of factors Va and VIIIa on membrane surfaces containing phospholipids that are derived from cells, platelets, lipoproteins, or cellular microparticles. The irreversible inactivation of factors Va and VIIIa to yield inactive factors Vi and VIIIi by APC is accelerated by a variety of lipid and protein cofactors (e.g., glucosyl ceramide, protein S, etc). (B) Beneficial direct effects of APC on cells require the EPCR and PAR-1. One distinction between pro-inflammatory thrombin signaling and cytoprotective APC signaling is the localization of APC signaling in the caveolin-1–rich microdomains (caveolae). (C) Neuroprotective mechanisms for APC effects on cells may also involve other receptors including PAR-3. APC-initiated signaling effects on cells can include anti-apoptotic activities, anti-inflammatory activities, inhibition of the inflammasome, stabilization of endothelial barrier functions, including the BBB, and neurogenesis. (D) The polypeptide structure of APC comprises an N-terminal GLA domain (green) that binds to negatively charged lipids and EPCR, 2 EGF-like domains (light blue and dark blue), and the protease domain containing the active site triad of serine, histidine, and aspartic acid residues (red). Four glycosylation sites are indicated by gray-shaded moieties. Substrate selectivity of this protease is determined by interactions between the targeted substrates and the active site and also by multiple unique binding exosites on APC that vary for different substrates. The protease domain space–filled model (see insert in D) highlights in the yellow box 3 positively charged lysine (K) residues within the so-called 37 loop (KKK 191–193), which is an exosite for APC's recognition of factors Va and VIIIa. Mutation of these 3 lysine residues to alanine (3K3A-APC) reduces APC's anticoagulant activity by >90% but does not affect its interactions with the cytoprotective substrates, PAR-1, PAR-3, or its other known cell-signaling receptors. Thus, 3K3A-APC is very “signaling-selective”.
The cellular signaling by APC gives rise to cytoprotective alterations in gene expression profiles resulting in multiple cytoprotective actions due to anti-inflammatory and anti-apoptotic activities, as well as a reduction of endothelial barrier disruption (Figures 1, 2) (70–74). APC crosses the blood brain barrier via an active transport mechanism (75).
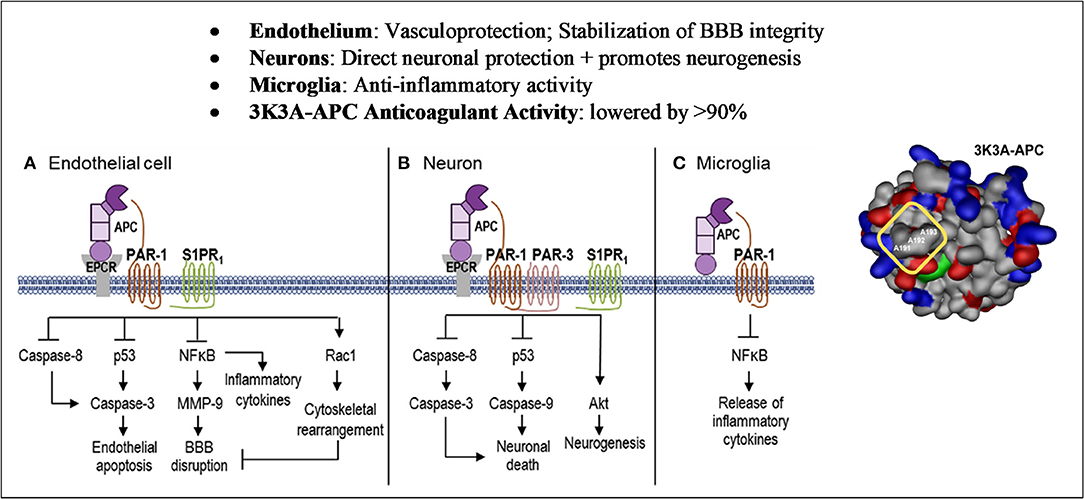
Figure 2. Cell-Specific APC protective signaling pathways. Akt, protein kinase B; APC, activated protein C; BBB, blood–brain barrier; EPCR, endothelial protein C receptor; MMP, matrix metallopeptidase; NFkB, nuclear factor kappa-light-chain-enhancer of activated B cells; PAR, protease-activated receptor; Rac1, Ras-related C3 botulinum toxin substrate 1; S1PR1, sphingosine 1-phosphate receptor 1. Reprinted from neuropharmacology, vol. 134, Amar et al. (69) can adjunctive therapies augment the efficacy of endovascular thrombolysis? A potential role for activated protein C, 293–301, 2018, with permission from Elsevier. 3D structure reprinted from blood, vol. 132(2), Griffin et al. (67) activated protein C, protease activated receptor 1 and neuroprotection; 159–169, 2018, with permission from the American Society of Hematology. (A) In endothelial cells, APC helps to seal the BBB and is vasculoprotective. APC/EPCR activates PAR-1 and inhibits caspase-8 activation of caspase-3, thereby limiting the extrinsic apoptotic pathway in endothelium. APC/EPCR-dependent PAR-1 activation suppresses the pro-apoptotic p53 transcription factor inhibiting caspase-3 activation blocking the intrinsic apoptotic pathway. Also, APC suppresses the NFkB-dependent transcriptional activation of MMP-9, thereby blocking degradation of the BBB basement membrane. Furthermore, APC blocks the expression of pro-inflammatory cytokines, limiting inflammation by controlling NFkB nuclear translocation. APC's cytoprotective effects on endothelial cells require EPCR and PAR-1 to cross-activate S1PR1. Cross-activation of S1PR1 activates Rac1, leading to stabilization of the BBB cytoskeleton, thereby supporting the integrity of the BBB. (B) In neurons, APC/EPCR is cytoprotective via PAR-1 and PAR-3, which inhibits caspase-8 upstream of caspase-3 and thereby limits the extrinsic apoptotic pathway. Also, an APC-PAR-1-PAR-3 pathway blocks p53 activation in injured neurons, thereby blocking the caspase-9-dependent intrinsic apoptotic pathway. Furthermore, APC promotes neurogenesis via a PAR-1-PAR-3-S1PR1-Akt pathway. (C) APC's inhibition of NFkB-dependent transcriptional expression of different pro-inflammatory cytokines suppresses microglial activation. Suppression of NLRP3 inflammasome development by APC is another activity but is not shown in this figure.
For 3K3A-APC cytoprotective actions in murine preclinical ischemic stroke studies, not only is PAR-1 required but also the arginine 46 residue in PAR-1. The requirement for arginine 46 strongly supports the concept that APC cytoprotection requires “biased” signaling initiated by the G-protein coupled receptor PAR-1 (50, 68). Activation of PAR-1 by APC occurs after proteolysis of the PAR-1 extracellular N-terminal domain at arginine 46, producing a tethered ligand peptide that begins at asparagine 47 causing APC biased, β-arrestin-2-dependent cytoprotective signaling (Figure 3) (50, 68, 76–79). In contrast, activation of PAR-1 by thrombin involves cleavage at arginine 16, which generates thrombin-receptor activated peptide, a tethered ligand peptide that begins at residue 42, initiating cytotoxic effects via G-protein-dependent signaling and causing human platelet activation, pro-inflammatory changes, endothelial vascular leakage and CNS toxicity (67, 68, 80). PAR-1-tethered ligand peptides beginning at asparagine 47, but not those beginning at amino acid 42, exert cytoprotective effects (Figure 3) (68). Similarly, APC activates human PAR-3 by non-canonical cleavage at arginine 41, whereas thrombin cleaves PAR-3 at lysine 38 (81). PAR-3-tethered ligand peptides beginning at amino acid 42, but not those beginning at amino acid 39, exert cytoprotective effects (82), suggesting that human PAR-3 cleavage at arginine 41 by APC causes cytoprotection, whereas PAR-3 cleavage at lysine 38 initiates thrombin-like cytotoxic pro-inflammatory effects (82).
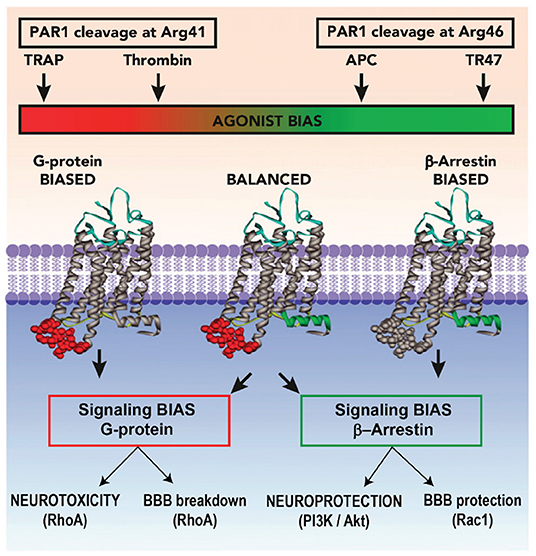
Figure 3. Biased Agonism of PAR-1 by APC. Akt, protein kinase B; APC, activated protein C; BBB, blood–brain barrier; PAR, protease-activated receptor; P13K, phosphoinositide 3-kinase; Rac, Ras-related C3 botulinum toxin substrate; RhoA, ras homolog gene family member A; TRAP, thrombin-receptor activated peptide. Reprinted from Blood, vol. 120(26), Mosnier et al. (68) biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46; 5237–5246, 2012, with permission from the American Society of Hematology. Activation of PAR-1 by APC and its cytoprotective analogs involves cleavage of PAR-1 N-terminal domain at Arg46, which reveals a tethered ligand peptide that begins at Asn47 causing APC's biased, β-arrestin-2-dependent cytoprotective signaling. Activation of PAR-1 by thrombin involves cleavage at Arg41, which generates a tethered ligand that begins at Thr42, initiating cytotoxic effects via G-protein-dependent signaling causing human platelet activation, inflammatory changes, vascular leakage, and CNS toxicity.
Studies With the APC Analog 3K3A-APC
Signaling-selective APC analogs, such as 3K3A-APC, were engineered to retain normal cell-signaling activity (50–53) but to have greatly diminished anticoagulant activity (54), thereby reducing in vivo risk for bleeding compared with wildtype APC (55–57). In in vitro assays, 3K3A-APC retains the cytoprotective activity of recombinant wildtype APC but has <10% of its anticoagulant activity (e.g., see Table 3) (54).

Table 3. Cytoprotective and anticoagulant activity of recombinant wildtype APC vs. 3K3A-APC.
The effects of 3K3A-APC on the fibrinolytic activity of tPA has also been studied in vitro; no statistically significant effects were noted when rt-PA was applied to induce clot lysis in the presence of either wildtype APC or 3K3A-APC (83).
3K3A-APC has beneficial effects in rodent models of stroke (50, 55–57, 72, 84–89), brain trauma (90, 91), amyotrophic lateral sclerosis (92–94), multiple sclerosis (95) and Alzheimer's disease (96), as well as ischemic injury of heart, kidney or liver, organ transplant, total body radiation, diabetes, sepsis, and wound healing (59, 67). In the CNS, PAR-1 and PAR-3 are both necessary for neuronal protection by APC (84, 85, 92), PAR-1 and endothelial protein C receptor for vasculoprotection and stabilization of the blood–brain barrier (4, 67, 69, 71–73, 84, 85, 97, 98), and PAR-1 for suppression of microglia activation and anti-inflammatory activity.(4, 66, 91, 94) The extensive preclinical studies of the cytoprotective actions of APC and 3K3A-APC have been summarized in several reviews (Figure 2) (4, 59, 65–67, 69, 99).
In studies with human progenitor and fetal neural cells, 3K3A-APC promoted neurogenesis in vitro (52) as well as in vivo using a mouse middle cerebral artery occlusion (MCAO) stroke model (57).
3K3A-APC acts synergistically with tPA in both mouse and rat stroke models (55). tPA alone or in combination with 3K3A-APC, was administered 4 h after MCAO, followed by 3K3A-APC for 3–4 consecutive days afterward. In this delayed treatment paradigm, tPA alone had no beneficial effects on infarct volume, or behavior (neurological score, foot-fault, forelimb asymmetry, adhesive removal) compared with controls. In contrast, the combination of tPA plus 3K3A-APC as compared with control significantly reduced infarct volume at 24 h (65% reduction) and at 7 days (63% reduction) following MCAO in mice and at 7 days (52% reduction) after embolic stroke in rats (p < 0.05). Furthermore, the combination significantly improved behavioral outcomes and eliminated tPA-related intracerebral microhemorrhages (p < 0.01–0.05).
These positive effects of 3K3A-APC extend to elderly animals and animals with comorbidities such as might be seen in the target patient population of this study. 3K3A-APC alone or with tPA was given 4 h after transient MCAO in aged female mice and 4 h after embolic stroke in spontaneously hypertensive rats (56). 3K3A-APC was then administered from 3 to 7 days afterward. Assessments included neurological scores, foot-fault, forelimb asymmetry, and adhesive removal. In both models, tPA alone given 4 h after stroke had no effect on infarct volume or behavior. Treatment with 3K3A-APC alone or 3K3A-APC in combination with tPA reduced the infarct volume determined at 7 days by 62–66% (MCAO in aged mice) and 50–53%, (embolic stroke in spontaneously hypertensive rats), as well as improved behavior (p < 0.05) and significantly reduced tPA-induced intracerebral microhemorrhages (Figure 4).
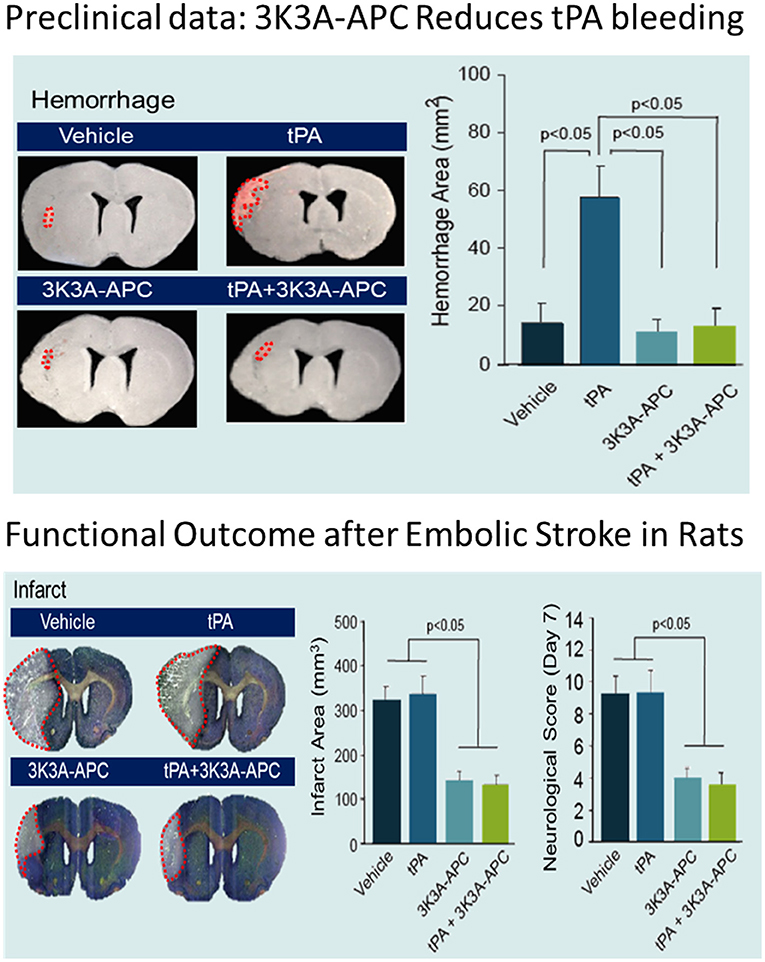
Figure 4. Effects of 3K3A-APC and 3K3A-APC combined with tPA on hemorrhage (upper panel) and neuropathological (hematoxylin and eosin staining; infarct volume) and neurological (neurological score) outcomes (lower panel) in young male spontaneously hypertensive rats within 7 days after embolic stroke. APC, activated protein C; SD, standard deviation; tPA, tissue plasminogen activator. Reprinted from Stroke, vol. 44(12), Wang et al. (56) activated protein C analog protects from ischemic stroke and extends the therapeutic window of tissue-type plasminogen activator in aged female mice and hypertensive rats, 3529–3536, 2013, with permission from Wolters Kluwer Health, Inc. 3K3A-APC and tPA were administered 4 h after embolic stroke. 3K3A-APC was administered for 3 consecutive days afterward. Mean + SD, N = 8–9 rats per group.
Overall, in preclinical studies, 3K3A-APC appears to have a reduced risk for bleeding and provides at least equivalent if not greater cytoprotection compared with recombinant wildtype APC in mouse models of stroke. When 3K3A-APC is combined with tPA, infarct volumes are reduced and intracerebral microhemorrhages are greatly reduced and/or eliminated. At the same time, behavioral outcomes in both mouse and rat models of stroke are improved. Furthermore, 3K3A-APC expands the tPA therapeutic window, supporting further development of tPA and 3K3A-APC combination therapy (69, 99). Of note, the transient suture model of stroke that has been used in several of these studies closely mimics the clinical procedure of thrombectomy as recently reviewed (69, 99). This implies the combination of thrombectomy and 3K3A-APC for focal ischemic stroke in humans may be efficacious.
Currently, 3K3A-APC is in clinical development for stroke therapy and other indications (5).
Need for a Stroke Therapy With Decreased Bleeding
As previously mentioned, bleeding is a risk following stroke therapies. In a randomized, controlled trial in patients who had had an arterial occlusion, the control group received standard care alone (including the use of tPA) and the thrombectomy group received mechanical thrombectomy in addition to standard care (100). AICH was more common in the thrombectomy group (51%) compared with the control group (25%).
Although mechanical thrombectomy is associated with greater bleeding and despite tPA being widely available in the United States, it is currently estimated that only 10–20% of stroke patients get tPA treatment (21, 22). According to von Kummer in 2002 (101): “The risk of brain hemorrhage is the main argument of the European authorities not to approve rt-PA, and the fear of hurting patients with rt-PA explains some of its limited use in North America. The common argument is, ‘Treatment with rt-PA may have some beneficial effect, but that is traded off by a considerable risk of symptomatic hemorrhage.’”
Fear of thrombolytic-related hemorrhage influences physicians away from treating stroke (102). To quantify the effect on physicians' prescribing behavior from fear that tPA will cause intracerebral bleeding, a biopharmaceutical company obtained focus group and survey data from a private polling service for its developmental therapy for stroke, 3K3A-APC. The market research firm interviewed 34 key opinion leaders and high-volume practitioners who were practicing stroke specialists, split evenly between the United States and Europe. The majority of interviewees were neurologists who routinely treat stroke patients at a comprehensive stroke center. Each interviewee received an honorarium for his or her time. The interviewees remained anonymous to the company and to each other. They were interviewed one-on-one over the telephone in November, 2014, using a formatted target product profile and standardized questionnaire. The interviews were digitally recorded for subsequent data capture and aggregation. See Table 4 and Figure 5.

Table 4. Market research survey data from stroke specialists about stroke therapies.
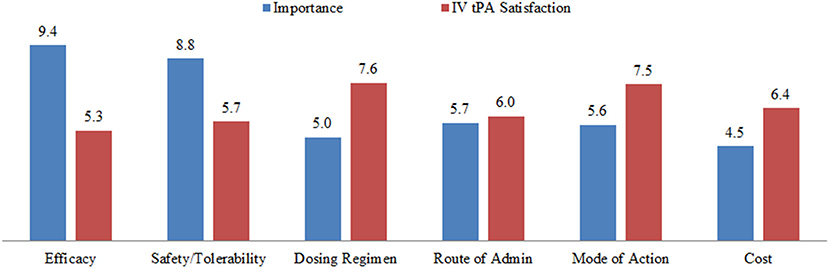
Figure 5. Unmet medical needs concerning physician satisfaction with tPA therapy in the United States and Europe. IV, intravenous; tPA, tissue plasminogen activator. The terms in the figure were provided to the physician interviewees without further definitions, and interviewees were asked to rank each term on a 10-point scale. For importance: 0 = unimportant and 10 = essential. For satisfaction: 0 = fully unsatisfactory and 10 = entirely satisfactory. The numbers are mean responses of the 32 interviewees.
The interviews provided insights into physicians' perceptions of the relative importance of several aspects of tPA treatment, compared with their perceived satisfaction with the therapy (Figure 5). The data revealed that physicians perceive the “efficacy” of tPA to be very important (9.4/10) but not fully satisfactory (5.3/10). In contrast, the cost of the treatment was rated less important (4.5/10). Of most relevance to the present discussion, the “safety” of the drug was perceived to be very important, and rated an 8.8/10, but physicians are not satisfied with the current safety profile, giving a rating of 5.7/10. These data suggest that physicians perceive there to be a critical need for improving the safety of tPA for acute ischemic stroke.
Approximately 20% of interviewed physicians' patients eligible for intravenous (IV) tPA were not being prescribed IV tPA (Table 4) because of patient, family, or physician assessment that bleeding risk outweighed benefit (Table 4). However, the percentage of eligible patients not administered tPA was much higher in the emergency room setting (46%) relative to stroke centers (16%) (Table 4).
Physician responses suggest ~90% of patients currently being prescribed IV tPA would also be prescribed 3K3A-APC (Table 4); according to the interviewers, many physicians stated it would be a requirement in their opinion to prescribe 3K3A-APC to those patients who received IV tPA.
Conclusion
Hemorrhagic transformation after ischemic stroke—even if labeled “asymptomatic”—may lead to long term disability and cognitive impairment. Fear of hemorrhagic transformation leads some physicians to hesitate to use indicated recanalization therapies. The PAR-1 acting agent, 3K3A-APC, reduces hemorrhagic transformation, and in animal models appears to improve long term outcomes after ischemic stroke. Agents that reduce hemorrhagic transformation may lead to wider acceptance of recanalization therapies and improved long-term outcome for ischemic stroke patients.
Author Contributions
All authors contributed to the creation and revision of the manuscript, as well as read and approved the submitted version.
Funding
This work was supported by U24 NS113452 and R01 NS075930 (to PL), R01 HL142975 and R01 HL133728 (to JG), and R01 NS090904 and NS117827 (to BZ).
Conflict of Interest
KP is employed by ZZ Biotech. JG, HL, JM, and BZ are paid consultants to ZZ Biotech. PL has received research funding from NIH to conduct trials of 3K3A-APC; received royalties from sales of the book Thrombolytic Therapy for Acute Ischemic Stroke, 3rd Edition; and received fees for occasional expert witness testimony. The Scripps Research Institute has intellectual property related to the topic of this review.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, et al. Heart disease and stroke statistics-2010 update: a report from the American Heart Association. Circulation. (2010) 121:e46–215. doi: 10.1161/CIRCULATIONAHA.109.192667
2. Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, et al. Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation. (2017) 135:e146–603. doi: 10.1161/CIR.0000000000000491
3. Thom T, Haase N, Rosamond W, Howard VJ, Rumsfeld J, Manolio T, et al. Heart disease and stroke statistics-2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. (2006) 113:e85–151. doi: 10.1161/CIRCULATIONAHA.105.171600
4. Zlokovic BV, Griffin JH. Cytoprotective protein C pathways and implications for stroke and neurological disorders. Trends Neurosci. (2011) 34:198–209. doi: 10.1016/j.tins.2011.01.005
5. Lyden P, Pryor KE, Coffey CS, Cudkowicz M, Conwit R, Jadhav A, et al. Final results of the RHAPSODY trial: a multi-center, phase 2 trial using a continual reassessment method to determine the safety and tolerability of 3K3A-APC, a recombinant variant of human activated protein C, in combination with tissue plasminogen activator, mechanical thrombectomy or both in moderate to severe acute ischemic stroke. Ann Neurol. (2019) 85:125–36. doi: 10.1002/ana.25383
6. Saver JL, Goyal M, Bonafe A, Diener HC, Levy EI, Pereira VM, et al. Stent-retriever thrombectomy after intravenous t-PA vs. t-PA alone in stroke. N Engl J Med. (2015) 372:2285–95. doi: 10.1056/NEJMoa1415061
7. Goyal M, Demchuk AM, Menon BK, Eesa M, Rempel JL, Thornton J, et al. Randomized assessment of rapid endovascular treatment of ischemic stroke. N Engl J Med. (2015) 372:1019–30. doi: 10.1056/NEJMoa1414905
8. Campbell BC, Mitchell PJ, Kleinig TJ, Dewey HM, Churilov L, Yassi N, et al. Endovascular therapy for ischemic stroke with perfusion-imaging selection. N Engl J Med. (2015) 372:1009–18. doi: 10.1056/NEJMoa1414792
9. Berkhemer OA, Fransen PS, Beumer D, van den Berg LA, Lingsma HF, Yoo AJ, et al. A randomized trial of intraarterial treatment for acute ischemic stroke. N Engl J Med. (2015) 372:11–20. doi: 10.1056/NEJMoa1411587
10. Jovin TG, Chamorro A, Cobo E, de Miquel MA, Molina CA, Rovira A, et al. Thrombectomy within 8 hours after symptom onset in ischemic stroke. N Engl J Med. (2015) 372:2296–306. doi: 10.1056/NEJMoa1503780
11. Hill MD, Goyal M, Menon BK, Nogueira RG, McTaggart RA, Demchuk AM, et al. Efficacy and safety of nerinetide for the treatment of acute ischaemic stroke (ESCAPE-NA1): a multicentre, double-blind, randomised controlled trial. Lancet. (2020) 395:878–87. doi: 10.1016/S0140-6736(20)30258-0
12. Albers GW, Marks MP, Kemp S, Christensen S, Tsai JP, Ortega-Gutierrez S, et al. Thrombectomy for stroke at 6 to 16 hours with selection by perfusion imaging. N Engl J Med. (2018) 378:708–18. doi: 10.1056/NEJMoa1713973
13. Mullen MT, Pisapia JM, Tilwa S, Messe SR, Stein SC. Systematic review of outcome after ischemic stroke due to anterior circulation occlusion treated with intravenous, intra-arterial, or combined intravenous+intra-arterial thrombolysis. Stroke. (2012) 43:2350–5. doi: 10.1161/STROKEAHA.111.639211
14. Ellis JA, Youngerman BE, Higashida RT, Altschul D, Meyers PM. Endovascular treatment strategies for acute ischemic stroke. Int J Stroke. (2011) 6:511–22. doi: 10.1111/j.1747-4949.2011.00670.x
15. Tymianski M. Can molecular and cellular neuroprotection be translated into therapies for patients? Yes, but not the way we tried it before. Stroke. (2010) 41:S87–90. doi: 10.1161/STROKEAHA.110.595496
16. Xiong Y, Mahmood A, Chopp M. Angiogenesis, neurogenesis and brain recovery of function following injury. Curr Opin Investig Drugs. (2010) 11:298–308.
17. Lapchak PA. Emerging therapies: pleiotropic multi-target drugs to treat stroke victims. Transl Stroke Res. (2011) 2:129–35. doi: 10.1007/s12975-011-0074-4
18. Hacke W, Kaste M, Bluhmki E, Brozman M, Dávalos A, Guidetti D, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. (2008) 359:1317–29. doi: 10.1056/NEJMoa0804656
19. Thomalla G, Simonsen CZ, Boutitie F, Andersen G, Berthezene Y, Cheng B, et al. MRI-guided thrombolysis for stroke with unknown time of onset. N Engl J Med. (2018) 379:611–22. doi: 10.1056/NEJMoa1804355
20. National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. N Engl J Med. (1995) 333:1581–7. doi: 10.1056/NEJM199512143332401
21. Grotta JC, Burgin WS, El-Mitwalli A, Long M, Campbell M, Morgenstern LB, et al. Intravenous tissue-type plasminogen activator therapy for ischemic stroke: houston experience 1996 to 2000. Arch Neurol. (2001) 58:2009–13. doi: 10.1001/archneur.58.12.2009
22. Stemer A, Lyden P. Evolution of the thrombolytic treatment window for acute ischemic stroke. Curr Neurol Neurosci Rep. (2010) 10:29–33. doi: 10.1007/s11910-009-0076-8
23. Saver JL, Goyal M, Bonafe A, Diener HC, Levy EI, Pereira VM, et al. Solitaire™ with the Intention for thrombectomy as primary endovascular treatment for acute ischemic stroke (SWIFT PRIME) trial: protocol for a randomized, controlled, multicenter study comparing the Solitaire revascularization device with IV tPA with IV tPA alone in acute ischemic stroke. Int J Stroke. (2015) 10:439–48. doi: 10.1111/ijs.12459
24. Bracard S, Ducrocq X, Mas JL, Soudant M, Oppenheim C, Moulin T, et al. Mechanical thrombectomy after intravenous alteplase vs. alteplase alone after stroke (THRACE): a randomised controlled trial. Lancet Neurol. (2016) 15:1138–47. doi: 10.1016/S1474-4422(16)30177-6
25. Dankbaar JW, Bienfait HP, van den Berg C, Bennink E, Horsch AD, van Seeters T, et al. Wake-up stroke vs. stroke with known onset time: clinical and multimodality CT imaging characteristics. Cerebrovasc Dis. (2018) 45:236–44. doi: 10.1159/000489566
26. Nogueira RG, Jadhav AP, Haussen DC, Bonafe A, Budzik RF, Bhuva P, et al. Thrombectomy 6 to 24 hours after stroke with a mismatch between deficit and infarct. N Engl J Med. (2018) 378:11–21. NEJMoa1706442. doi: 10.1056/NEJMoa1706442
27. del Zoppo GJ, von Kummer R, Hamann GF. Ischaemic damage of brain microvessels: inherent risks for thrombolytic treatment in stroke. J Neurol Neurosurg Psychiatry. (1998) 65:1–9. doi: 10.1136/jnnp.65.1.1
28. von Kummer R, Broderick JP, Campbell BC, Demchuk A, Goyal M, Hill MD, et al. The Heidelberg bleeding classification: classification of bleeding events after ischemic stroke and reperfusion therapy. Stroke. (2015) 46:2981–6. doi: 10.1161/STROKEAHA.115.010049
29. Levy DE, Brott TG, Haley EC Jr, Marler JR, Sheppard GL, Barsan W, et al. Factors related to intracranial hematoma formation in patients receiving tissue-type plasminogen activator for acute ischemic stroke. Stroke. (1994) 25:291–7. doi: 10.1161/01.STR.25.2.291
30. Trouillas P, von Kummer R. Classification and pathogenesis of cerebral hemorrhages after thrombolysis in ischemic stroke. Stroke. (2006) 37: 556–61. doi: 10.1161/01.STR.0000196942.84707.71
31. Wahlgren N, Ahmed N, Dávalos A, Ford GA, Grond M, Hacke W, et al. Thrombolysis with alteplase for acute ischaemic stroke in the safe implementation of thrombolysis in stroke-monitoring study (SITS-MOST): an observational study. Lancet. (2007) 369:275–82. doi: 10.1016/S0140-6736(07)60149-4
32. Hacke W, Kaste M, Fieschi C, von Kummer R, Davalos A, Meier D, et al. Randomised double-blind placebo-controlled trial of thrombolytic therapy with intravenous alteplase in acute ischaemic stroke (ECASS II). Second European-Australasian Acute Stroke Study Investigators. Lancet. (1998) 352:1245–51. doi: 10.1016/S0140-6736(98)08020-9
33. Emberson J, Lees KR, Lyden P, Blackwell L, Albers G, Bluhmki E, et al. Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: a meta-analysis of individual patient data from randomised trials. Lancet. (2014) 384:1929–35. doi: 10.1016/S0140-6736(14)60584-5
34. Hao Y, Yang D, Wang H, Zi W, Zhang M, Geng Y, et al. Predictors for symptomatic intracranial hemorrhage after endovascular treatment of acute ischemic stroke. Stroke. (2017) 48:1203–09. doi: 10.1161/STROKEAHA.116.016368
35. Powers WJ, Rabinstein AA, Ackerson T, Adeoye OM, Bambakidis NC, Becker K, et al. 2018 Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. (2018) 49:e46–110. doi: 10.1161/STR.0000000000000158
36. Kent DM, Hinchey J, Price LL, Levine SR, Selker HP. In acute ischemic stroke, are asymptomatic intracranial hemorrhages clinically innocuous? Stroke. (2004) 35:1141–6. doi: 10.1161/01.STR.0000125712.02090.6e
37. Dzialowski I, Pexman JH, Barber PA, Demchuk AM, Buchan AM, Hill MD, et al. Asymptomatic hemorrhage after thrombolysis may not be benign: prognosis by hemorrhage type in the Canadian Alteplase for Stroke Effectiveness Study registry. Stroke. (2007) 38:75–9. doi: 10.1161/01.STR.0000251644.76546.62
38. Park JH, Ko Y, Kim W-J, Jang MS, Yang MH, Han M-K, et al. Is asymptomatic hemorrhagic transformation really innocuous? Neurology. (2012) 78:421–6. doi: 10.1212/WNL.0b013e318245d22c
39. Lei C, Wu B, Liu M, Chen Y. Asymptomatic hemorrhagic transformation after acute ischemic stroke: is it clinically innocuous? J Stroke Cerebrovasc Dis. (2014) 23:2767–72. doi: 10.1016/j.jstrokecerebrovasdis.2014.06.024
40. Jiang F, Zhao W, Wu C, Zhang Z, Li C, Che R, et al. Asymptomatic intracerebral hemorrhage may worsen clinical outcomes in acute ischemic stroke patients undergoing thrombectomy. J Stroke Cerebrovasc Dis. (2019) 28:1752–8. doi: 10.1016/j.jstrokecerebrovasdis.2019.02.006
41. Hao Y, Liu W, Wang H, Zi W, Yang D, Wang W, et al. Prognosis of asymptomatic intracranial hemorrhage after endovascular treatment. J Neurointerv Surg. (2019) 11:123–6. doi: 10.1136/neurintsurg-2018-013848
42. Li GF, Wu YL, Wang S, Shi YH, Zhao R, Liu FD, et al. Previous chronic symptomatic and asymptomatic cerebral hemorrhage in patients with acute ischemic stroke. Neuroradiology. (2019) 61:103–7. doi: 10.1007/s00234-018-2141-y
43. Wang X, Lo EH. Triggers and mediators of hemorrhagic transformation in cerebral ischemia. Mol Neurobiol. (2003) 28:229–44. doi: 10.1385/MN:28:3:229
44. Gáll T, Balla G, Balla J. Heme, heme oxygenase, and endoplasmic reticulum stress-a new insight into the pathophysiology of vascular diseases. Int J Mol Sci. (2019) 20:3675. doi: 10.3390/ijms20153675
45. Kernan WN, Ovbiagele B, Black HR, Bravata DM, Chimowitz MI, Ezekowitz MD, et al. Guidelines for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. (2014) 45:2160–236. doi: 10.1161/STR.0000000000000024
46. Tan S, Wang D, Liu M, Zhang S, Wu B, Liu B. Frequency and predictors of spontaneous hemorrhagic transformation in ischemic stroke and its association with prognosis. J Neurol. (2014) 261:905–12. doi: 10.1007/s00415-014-7297-8
47. Zhao W, Che R, Shang S, Wu C, Li C, Wu L, et al. Low-dose tirofiban improves functional outcome in acute ischemic stroke patients treated with endovascular thrombectomy. Stroke. (2017) 48:3289–94. doi: 10.1161/STROKEAHA.117.019193
48. Boccuzzi SJ, Martin J, Stephenson J, Kreilick C, Fernandes J, Beaulieu J, et al. Retrospective study of total healthcare costs associated with chronic nonvalvular atrial fibrillation and the occurrence of a first transient ischemic attack, stroke, or major bleed. Curr Med Res Opin. (2009) 25:2853–64. doi: 10.1185/03007990903196422
49. O'Collins VE, Macleod MR, Cox SF, Van Raay L, Aleksoska E, Donnan GA, et al. Preclinical drug evaluation for combination therapy in acute stroke using systematic review, meta-analysis, and subsequent experimental testing. J Cereb Blood Flow Metab. (2011) 31:962–75. doi: 10.1038/jcbfm.2010.184
50. Sinha RK, Wang Y, Zhao Z, Xu X, Burnier L, Gupta N, et al. PAR1 biased signaling is required for activated protein C in vivo benefits in sepsis and stroke. Blood. (2018) 131:1163–71. doi: 10.1182/blood-2017-10-810895
51. Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. (2007) 109:3161–72. doi: 10.1182/blood-2006-09-003004
52. Guo H, Zhao Z, Yang Q, Wang M, Bell RD, Wang S, et al. An activated protein C analog stimulates neuronal production by human neural progenitor cells via a PAR1-PAR3-S1PR1-Akt pathway. J Neurosci. (2013) 33:6181–90. doi: 10.1523/JNEUROSCI.4491-12.2013
53. Mosnier LO, Zlokovic BV, Griffin JH. Cytoprotective-selective activated protein C therapy for ischaemic stroke. Thromb Haemost. (2014) 112:883–92. doi: 10.1160/th14-05-0448
54. Mosnier LO, Gale AJ, Yegneswaran S, Griffin JH. Activated protein C variants with normal cytoprotective but reduced anticoagulant activity. Blood. (2004) 104:1740–4. doi: 10.1182/blood-2004-01-0110
55. Wang Y, Zhang Z, Chow N, Davis TP, Griffin JH, Chopp M, et al. An activated protein C analog with reduced anticoagulant activity extends the therapeutic window of tissue plasminogen activator for ischemic stroke in rodents. Stroke. (2012) 43:2444–9. doi: 10.1161/STROKEAHA.112.658997
56. Wang Y, Zhao Z, Chow N, Rajput PS, Griffin JH, Lyden PD, et al. An activated protein C analog protects from ischemic stroke and extends the therapeutic window of tissue-type plasminogen activator in aged female mice and hypertensive rats. Stroke. (2013) 44:3529–36. doi: 10.1161/STROKEAHA.113.003350
57. Wang Y, Zhao Z, Rege SV, Wang M, Si G, Zhou Y, et al. 3K3A-activated protein C stimulates postischemic neuronal repair by human neural stem cells in mice. Nat Med. (2016) 22:1050–5. doi: 10.1038/nm.4154
58. Williams PD, Zlokovic BV, Griffin JH, Pryor KE, Davis TP. Preclinical safety and pharmacokinetic profile of 3K3A-APC, a novel, modified activated protein C for stroke. Curr Pharm Des. (2012) 18:4215–22. doi: 10.2174/138161212802430413
59. Griffin JH, Zlokovic BV, Mosnier LO. Activated protein C: biased for translation. Blood. (2015) 125:2898–907. doi: 10.1182/blood-2015-02-355974
60. Whiteley WN, Adams HP Jr, Bath PM, Berge E, Sandset PM, Dennis M, et al. Targeted use of heparin, heparinoids, or low-molecular-weight heparin to improve outcome after acute ischaemic stroke: an individual patient data meta-analysis of randomised controlled trials. Lancet Neurol. (2013) 12:539–45. doi: 10.1016/S1474-4422(13)70079-6
61. Bath PM, Lindenstrom E, Boysen G, De Deyn P, Friis P, Leys D, et al. Tinzaparin in acute ischaemic stroke (TAIST): a randomised aspirin-controlled trial. Lancet. (2001) 358:702–10. doi: 10.1016/S0140-6736(01)05837-8
62. Lyden PD, Zivin JA, Soll M, Sitzer M, Rothrock JF, Alksne J. Intracerebral hemorrhage after experimental embolic infarction: anticoagulation. Arch Neurol. (1987) 44:848–50. doi: 10.1001/archneur.1987.00520200052018
63. Rajput PS, Lamb JA, Fernández JA, Bai J, Pereira BR, Lei I, et al. Neuroprotection and vasculoprotection using genetically targeted protease-ligands. Brain Res. (2019) 1715:13–20. doi: 10.1016/j.brainres.2019.03.010
64. Mosnier LO, Yang XV, Griffin JH. Activated protein C mutant with minimal anticoagulant activity, normal cytoprotective activity, and preservation of thrombin activable fibrinolysis inhibitor-dependent cytoprotective functions. J Biol Chem. (2007) 282:33022–33. doi: 10.1074/jbc.M705824200
65. Griffin JH, Fernández JA, Lyden PD, Zlokovic BV. Activated protein C promotes neuroprotection: mechanisms and translation to the clinic. Thromb Res. (2016) 141(Suppl. 2):S62–4. doi: 10.1016/S0049-3848(16)30368-1
66. Griffin JH, Mosnier LO, Fernández JA, Zlokovic BV. 2016 scientific sessions Sol Sherry distinguished lecturer in thrombosis: thrombotic stroke: neuroprotective therapy by recombinant-activated protein C. Arterioscler Thromb Vasc Biol. (2016) 36:2143–51. doi: 10.1161/ATVBAHA.116.308038
67. Griffin JH, Zlokovic BV, Mosnier LO. Activated protein C, protease activated receptor 1 and neuroprotection. Blood. (2018) 132:159–69. doi: 10.1182/blood-2018-02-769026
68. Mosnier LO, Sinha RK, Burnier L, Bouwens EA, Griffin JH. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood. (2012) 120:5237–46. doi: 10.1182/blood-2012-08-452169
69. Amar AP, Sagare AP, Zhao Z, Wang Y, Nelson AR, Griffin JH, et al. Can adjunctive therapies augment the efficacy of endovascular thrombolysis? A potential role for activated protein C. Neuropharmacology. (2018) 134(Pt. B):293–301. doi: 10.1016/j.neuropharm.2017.09.021
70. Joyce DE, Gelbert L, Ciaccia A, DeHoff B, Grinnell BW. Gene expression profile of antithrombotic protein c defines new mechanisms modulating inflammation and apoptosis. J Biol Chem. (2001) 276:11199–203. doi: 10.1074/jbc.C100017200
71. Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. (2002) 296:1880–2. doi: 10.1126/science.1071699
72. Cheng T, Liu D, Griffin JH, Fernández JA, Castellino F, Rosen ED, et al. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med. (2003) 9:338–42. doi: 10.1038/nm826
73. Dömötör E, Benzakour O, Griffin JH, Yule D, Fukudome K, Zlokovic BV. Activated protein C alters cytosolic calcium flux in human brain endothelium via binding to endothelial protein C receptor and activation of protease activated receptor-1. Blood. (2003) 101:4797–801. doi: 10.1182/blood-2002-12-3680
74. Mosnier LO, Griffin JH. Inhibition of staurosporine-induced apoptosis of endothelial cells by activated protein C requires protease-activated receptor-1 and endothelial cell protein C receptor. Biochem J. (2003) 373(Pt.1):65–70. doi: 10.1042/bj20030341
75. Deane R, LaRue B, Sagare AP, Castellino FJ, Zhong Z, Zlokovic BV. Endothelial protein C receptor-assisted transport of activated protein C across the mouse blood-brain barrier. J Cereb Blood Flow Metab. (2009) 29:25–33. doi: 10.1038/jcbfm.2008.117
76. Schuepbach RA, Madon J, Ender M, Galli P, Riewald M. Protease-activated receptor-1 cleaved at R46 mediates cytoprotective effects. J Thromb Haemost. (2012) 10:1675–84. doi: 10.1111/j.1538-7836.2012.04825.x
77. Soh UJ, Trejo J. Activated protein C promotes protease-activated receptor-1 cytoprotective signaling through β-arrestin and dishevelled-2 scaffolds. Proc Natl Acad Sci U S A. (2011) 108:E1372–80. doi: 10.1073/pnas.1112482108
78. Kiseleva EV, Sidorova MV, Gorbacheva LR, Strukova SM. Peptide-agonist of protease-activated receptor (PAR 1), similar to activated protein C, promotes proliferation in keratinocytes and wound healing of epithelial layer. Biomed Khim. (2014) 60:702–6. doi: 10.18097/PBMC20146006702
79. de Oliveira AS, de Almeida VH, Gomes FG, Rezaie AR, Monteiro RQ. TR47, a PAR1-based peptide, inhibits melanoma cell migration in vitro and metastasis in vivo. Biochem Biophys Res Commun. (2018) 495:1300–4. doi: 10.1016/j.bbrc.2017.11.174
80. Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. (2000) 407:258–64. doi: 10.1038/35025229
81. Stavenuiter F, Mosnier LO. Noncanonical PAR3 activation by factor Xa identifies a novel pathway for Tie2 activation and stabilization of vascular integrity. Blood. (2014) 124:3480–9. doi: 10.1182/blood-2014-06-582775
82. Burnier L, Mosnier LO. Novel mechanisms for activated protein C cytoprotective activities involving noncanonical activation of protease-activated receptor 3. Blood. (2013) 122:807–16. doi: 10.1182/blood-2013-03-488957
83. Mosnier LO, Fernández JA, Davis TP, Zlokovic BV, Griffin JH. Influence of the 3K3A-activated protein C variant on the plasma clot lysis activity of t-PA and of t-PA on the variant's anticoagulant activity. J Thromb Haemost. (2013) 11:2059–2062. doi: 10.1111/jth.12400
84. Guo H, Wang Y, Singh I, Liu D, Fernández JA, Griffin JH, et al. Species-dependent neuroprotection by activated protein C mutants with reduced anticoagulant activity. J Neurochem. (2009) 109:116–24. doi: 10.1111/j.1471-4159.2009.05921.x
85. Guo H, Singh I, Wang Y, Deane R, Barrett T, Fernández JA, et al. Neuroprotective activities of activated protein C mutant with reduced anticoagulant activity. Eur J Neurosci. (2009) 29:1119–30. doi: 10.1111/j.1460-9568.2009.06664.x
86. Liu D, Cheng T, Guo H, Fernández JA, Griffin JH, Song X, et al. Tissue plasminogen activator neurovascular toxicity is controlled by activated protein C. Nat Med. (2004) 10:1379–83. doi: 10.1038/nm1122
87. Zlokovic BV, Zhang C, Liu D, Fernandez J, Griffin JH, Chopp M. Functional recovery after embolic stroke in rodents by activated protein C. Ann Neurol. (2005) 58:474–7. doi: 10.1002/ana.20602
88. Thiyagarajan M, Fernández JA, Lane SM, Griffin JH, Zlokovic BV. Activated protein C promotes neovascularization and neurogenesis in postischemic brain via protease-activated receptor 1. J Neurosci. (2008) 28:12788–97. doi: 10.1523/JNEUROSCI.3485-08.2008
89. Wang Y, Thiyagarajan M, Chow N, Singh I, Guo H, Davis TP, et al. Differential neuroprotection and risk for bleeding from activated protein C with varying degrees of anticoagulant activity. Stroke. (2009) 40:1864–9. doi: 10.1161/STROKEAHA.108.536680
90. Walker CT, Marky AH, Petraglia AL, Ali T, Chow N, Zlokovic BV. Activated protein C analog with reduced anticoagulant activity improves functional recovery and reduces bleeding risk following controlled cortical impact. Brain Res. (2010) 1347:125–31. doi: 10.1016/j.brainres.2010.05.075
91. Petraglia AL, Marky AH, Walker C, Thiyagarajan M, Zlokovic BV. Activated protein C is neuroprotective and mediates new blood vessel formation and neurogenesis after controlled cortical impact. Neurosurgery. (2010) 66:165–71; discussion 171–2. doi: 10.1227/01.NEU.0000363148.49779.68
92. Shi Y, Hung S-T, Rocha G, Lin S, Linares GR, Staats KA, et al. Identification and therapeutic rescue of autophagosome and glutamate receptor defects in C9ORF72 and sporadic ALS neurons. JCI Insight. (2019) 5:127736. doi: 10.1172/jci.insight.127736
93. Zhong Z, Ilieva H, Hallagan L, Bell R, Singh I, Paquette N, et al. Activated protein C therapy slows ALS-like disease in mice by transcriptionally inhibiting SOD1 in motor neurons and microglia cells. J Clin Invest. (2009) 119:3437–49. doi: 10.1172/JCI38476
94. Winkler EA, Sengillo JD, Sagare AP, Zhao Z, Ma Q, Zuniga E, et al. Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc Natl Acad Sci U S A. (2014) 111:E1035–42. doi: 10.1073/pnas.1401595111
95. Han MH, Hwang SI, Roy DB, Lundgren DH, Price JV, Ousman SS, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. (2008) 451:1076–81. doi: 10.1038/nature06559
96. Lazic D, Sagare AP, Nikolakopoulou AM, Griffin JH, Vassar R, Zlokovic BV. 3K3A-activated protein C blocks amyloidogenic BACE1 pathway and improves functional outcome in mice. J Exp Med. (2019) 216:279–93. doi: 10.1084/jem.20181035
97. Finigan JH, Dudek SM, Singleton PA, Chiang ET, Jacobson JR, Camp SM, et al. Activated protein C mediates novel lung endothelial barrier enhancement: role of sphingosine 1-phosphate receptor transactivation. J Biol Chem. (2005) 280:17286–93. doi: 10.1074/jbc.M412427200
98. Uchiba M, Okajima K, Oike Y, Ito Y, Fukudome K, Isobe H, et al. Activated protein C induces endothelial cell proliferation by mitogen-activated protein kinase activation in vitro and angiogenesis in vivo. Circ Res. (2004) 95:34–41. doi: 10.1161/01.RES.0000133680.87668.FA
99. Amar AP, Griffin JH, Zlokovic BV. Combined neurothrombectomy or thrombolysis with adjunctive delivery of 3K3A-activated protein C in acute ischemic stroke. Front Cell Neurosci. (2015) 9:344. doi: 10.3389/fncel.2015.00344
100. Martins SO, Mont'Alverne F, Rebello LC, Abud DG, Silva GS, Lima FO, et al. Thrombectomy for stroke in the public health care system of Brazil. N Engl J Med. (2020) 382:2316–26. NEJMoa2000120. doi: 10.1056/NEJMoa2000120
101. von Kummer R. Brain hemorrhage after thrombolysis: good or bad? Stroke. (2002) 33:1446–7. doi: 10.1161/01.STR.0000016923.99605.75
Keywords: hemorrhagic transformation, ischemic stroke, tissue plasminogen activator, intracranial hemorrhage, activated protein C, stroke therapy, thrombectomy, bleeding
Citation: Lyden PD, Pryor KE, Minigh J, Davis TP, Griffin JH, Levy H and Zlokovic BV (2021) Stroke Treatment With PAR-1 Agents to Decrease Hemorrhagic Transformation. Front. Neurol. 12:593582. doi: 10.3389/fneur.2021.593582
Received: 11 August 2020; Accepted: 08 February 2021;
Published: 15 March 2021.
Edited by:
Thiruma Valavan Arumugam, La Trobe University, AustraliaReviewed by:
Asaf Honig, University of British Columbia, CanadaDavid Howells, University of Tasmania, Australia
Copyright © 2021 Lyden, Pryor, Minigh, Davis, Griffin, Levy and Zlokovic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrick D. Lyden, cGx5ZGVuQHVzYy5lZHU=