 Elisabetta Indelicato
Elisabetta Indelicato Sylvia Boesch*
Sylvia Boesch*- Center for Rare Movement Disorders Innsbruck, Department of Neurology, Medical University of Innsbruck, Innsbruck, Austria
Ion channel dysfunction is a key pathological substrate of episodic neurological disorders. A classical gene associated to paroxysmal movement disorders is CACNA1A, which codes for the pore-forming subunit of the neuronal calcium channel P/Q. Non-polyglutamine CACNA1A variants underlie familial hemiplegic ataxia type 1 (FHM1) and episodic ataxia type 2 (EA2). Classical paroxysmal manifestations of FHM1 are migraine attacks preceded by motor aura consisting of hemiparesis, aphasia, and disturbances of consciousness until coma. Patients with EA2 suffer of recurrent episodes of vertigo, unbalance, diplopia, and vomiting. Beyond these typical presentations, several reports highlighted manifold clinical features associated with P/Q channelopathies, from chronic progressive cerebellar ataxia to epilepsy and psychiatric disturbances. These manifestations may often outlast the burden of classical episodic symptoms leading to pitfalls in the diagnostic work-up. Lately, the spreading of next generation sequencing techniques linked de novo CACNA1A variants to an even broader phenotypic spectrum including early developmental delay, autism spectrum disorders, epileptic encephalopathy, and early onset paroxysmal dystonia. The age-dependency represents a striking new aspect of these phenotypes und highlights a pivotal role for P/Q channels in the development of the central nervous system in a defined time window. While several reviews addressed the clinical presentation and treatment of FHM1 and EA2, an overview of the newly described age-dependent manifestations is lacking. In this Mini-Review we present a clinical update, delineate genotype-phenotype correlations as well as summarize evidence on the pathophysiological mechanisms underlying the expanded phenotype associated with CACNA1A variants.
Introduction
The gene CACNA1A encodes the α1A pore-forming subunit of the neuronal calcium channel P/Q (1). The first association of CACNA1A with human diseases dates back to 1996, as Ophoff et al. (2) described its mutations in two allelic episodic neurological disorders, familial hemiplegic migraine type 1 (FHM1) and episodic ataxia type 2 (EA2). FHM1 presents with episodes of migraine with motor symptoms during the aura (3), while EA2 features attacks of paroxysmal gait disturbances, dysarthria and diplopia (3, 4). Already in 1997, a third disease, the spinocerebellar ataxia type 6 (SCA6), was mapped to CACNA1A locus (5). Differently from paroxysmal CACNA1A disorders, SCA6 is a late-onset disease manifesting as progressive isolated cerebellar ataxia (6). Far from being the first anomaly in the paradigm, our knowledge on CACNA1A disorders continued expanding over the past 25 years, revealing an unexpected variability both from a genotypic and phenotypic point of view. Established manifestations of CACNA1A variants comprehend neuropsychiatric disorders (7, 8), paroxysmal dystonia (9–11), epilepsy (12, 13), as well as complex phenotypes characterized by a various combination of early developmental delay and epileptic encephalopathy (14–16). Nowadays, an emerging hypothesis highlights an age-dependency of CACNA1A phenotypes (see Figure 1).
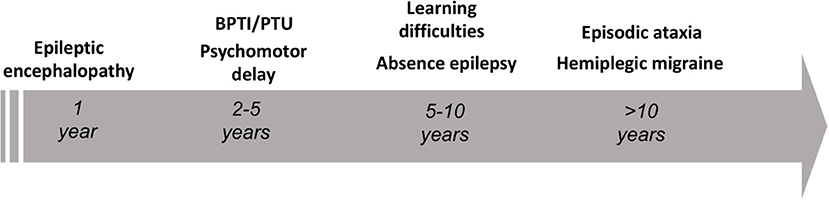
Figure 1. Schematic representation of the age-dependent phenotypes encountered in the setting of pathogenic CACNA1A variants. BPTI, benign paroxysmal torticollis of the infancy; PTU, paroxysmal tonic upward gaze.
The present review is an update on the expanded disease spectrum associated to CACNA1A variants. After a brief introduction on molecular genetics of P/Q channels, we review the related literature, delineate genotype-phenotype correlations and highlight translational aspects. Our aim is to offer a guide for the clinician as to when to apply a CACNA1A testing beyond the typical FHM1/EA2 phenotype.
Methods
Pertinent literature was retrieved through a Medline search using the following mesh terms in various combinations: CACNA1A, P/Q calcium channels, epilepsy, developmental delay, mental retardation/intellectual disability, epileptic encephalopathies, dystonia, and animal models. Furthermore, we considered relevant references from selected papers and consulted publicly accessible databases (ClinVar, Genecards, Uniprot).
P/Q Calcium Channels: Pathophysiology and Molecular Genetics
The P/Q calcium channels are voltage-gated channels consisting of a pore-forming α-subunit connected with further regulatory subunits (α2δ dimer and one or more β subunits) (17). The α-subunit determines the ion-specificity as well as the kinetic properties of the channels (18). Figure 2 offers a schematic representation of the unfolded α1A-subunit coded by CACNA1A. The P/Q channels are ubiquitous at central synapses and particularly abundant in cerebellar granules and Purkinje cells (18). From the presynaptic terminals P/Q channels orchestrate neurotransmitter release (19). Notably, calcium currents are not only involved in the electrochemical coupling during neurotransmission. Indeed, calcium acts also as a second messenger in the cells (20), being involved in the development of synaptic plasticity (21–23) and further regulatory processes in the central nervous system (24–26).
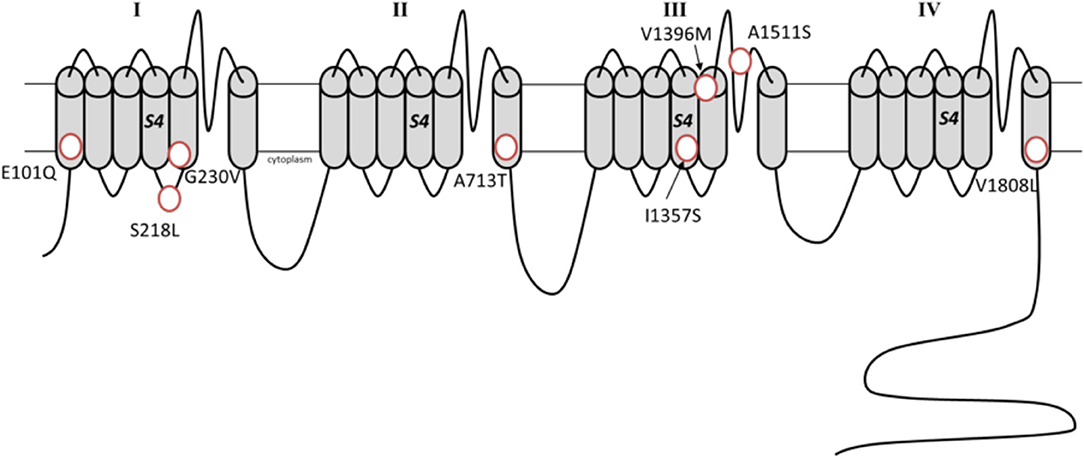
Figure 2. Schematic representation of the α1A subunit with the de novo missense variants associated with developmental epileptic encephalopathies (Transcript variant 2, NM_023035.2).
The CACNA1A gene is located on chromosome 19. It contains 47 exons, many of which undergo a canonical alternative splicing that results in myriad isoforms with different functional properties and regional expression patterns (27). CACNA1A scores among the 2% most intolerant genes of the human genome (28). Particularly the transmembrane region shows paucity of variations (29). The spectrum of disease associated CACNA1A variants spans from point mutations (missense and nonsense) to short indels and larger deletions (30–32). Pathogenic variants can also affect intronic sequences leading to translational frameshift (33). Several disease-associated missense variants lead to substitution of positively charged arginine in the segment S4 (29, 34–36). An updated graphical overview of the known mutations in CACNA1A can be found in (37) and (38). Irrespective of the mutation type, CACNA1A diseases shown an autosomal dominant pattern of inheritance. Concerning genotype-phenotype correlations, FHM1 is generally associated with missense gain-of-function mutations (39), while EA2 usually results from variants with loss-of-function effect, including truncating mutations and small deletions (40). Although sharing some paroxysmal symptoms at the beginning of disease, SCA6 is not directly affecting the channel kinetics as its allelic disorders. SCA6 is caused by an expanded CAG repeat coding for a polyglutamine tract (41). Recent research revealed that CACNA1A contains an “internal ribosome entry site,” which initiates the translation of a second peptide, referred to as α1ACT. This second product consists of a C-terminal fragment containing the expanded polyglutamine tract causing SCA6 (42). The peptide α1ACT is not involved in channel formation, but acts as a transcription factor and regulates the expression of key genes involved in synaptic formation, neurogenesis, and cell adhesion (42). The functionality of α1ACT in a specific time-window drives the maturation of the Purkinje cells in the early development (43).
The Expanded Phenotype of CACNA1A Variants
Classical Manifestation: Familial Hemiplegic Migraine Type 1 and Episodic Ataxia Type 2
Clinical Presentation and Genotype-Phenotype Correlation
FHM1 is a monogenic form of migraine characterized by the occurrence of motor deficits during the aura (44). The classical motor aura in FHM1 consists of a transient hemiparesis, but a variety of other neurological symptoms can be present, such as dysphasia, sensory loss, visual disturbance and vertigo (3). These deficits often outlast the associated headache and are extended for hours to days (3). The frequency of attacks is very variable and may range from 1 to 2 in an entire lifespan to several per month (39, 45). FHM1 is usually underlain by missense CACNA1A mutation with gain of function effect on calcium currents (2). The onset is usually in the first or second decade (3).
EA2 is the most common form of episodic ataxia (2). EA2 manifests with attacks of ataxia with nausea and vomiting. Attacks typically last minutes to days and can be associated with diplopia, dysarthria, tinnitus, dystonia, hemiplegia, and headache, also migraine headaches (4, 46). The frequency of attacks is very variable. Attacks can range from once or twice a year to several per day (47). Stress, caffeine, alcohol, exertion, fever, heat, and phenytoin can trigger attacks (48). The typical onset is in early adolescence or childhood (47). Truncating CACNA1A mutations with loss of function effect are the typical genotypic finding in EA2.
In FHM1 and EA2 symptoms may often overlap (13, 40, 49). Even if several patients present with isolated paroxysmal symptoms, the majority will eventually develop mild to moderate chronic cerebellar signs in the course of the disease (4, 39).
Developmental Delay
Clinical Presentation
Episodes of hemiplegic migraine or paroxysmal ataxia typically arise in the first two decades of life. Though, a focused clinical history may reveal earlier symptoms in a substantial proportion of patients. Indeed, the parents of older children and adolescents with EA2 and FHM1 can often recall some delay in the acquisition of the early milestones, for instance delayed walking or speaking (7, 45). Learning deficits as well as behavioral abnormalities consistent with attention-deficit hyperactivity disorder or autistic spectrum disorders can dominate the clinical picture in schoolchildren (4, 14, 34, 35, 50, 51). Although natural history data are lacking, the available literature and our clinical experience suggests that a compensation of apparently marked deficits can take place in the disease course. Adult patients who suffered from early delay or required special education can still conduct a normal working and social life (45, 52). Reports of rapid cognitive deterioration starting in adulthood are exceptional (53). In the past decade, the increasing availability of next generation sequencing (NGS) techniques added de novo missense CACNA1A mutations to the genetic background of intellectual disability. Indeed, several studies applying NGS discovered pathogenic CACNA1A variants in children with severe psychomotor delay born from healthy parents (54–56). Intellectual disability was the prominent symptom in these cases. Although motor examination can reveal a marked hypotonia (54, 55), clear signs of ataxia may become apparent only later in small children. In several cases, intellectual disability occurs along with therapy-resistant epilepsy, leading to severe clinical pictures in the spectrum of developmental epileptic encephalopathies (see next paragraphs).
Pathophysiology
The pathophysiological basis of intellectual disability due to pathogenic CACNA1A variants is largely unexplored. In a single animal study, the role of P/Q channels in circuitry underlying cognition and memory was investigated by means of a conditional murine model with selective knock-out of CACNA1A in the forebrain. The loss of P/Q currents in the neocortex and hippocampus led to marked deficits in memory, spatial learning and in increased exploratory behavior in the mouse (57). As mentioned above, both P/Q calcium currents and the transcription factor α1ACT have an established role in the early maturation of the cerebellum. Perturbations in the developing cerebellum underlid by P/Q channel and α1ACT dysfunction may contribute to occurrence of neuropsychiatric disorders early in life by altering the cerebellar tuning on cognitive cortical networks (58). This hypothesis is in line with the notion of a “cerebellar cognitive-affective syndrome” (59), which can arise independently from an ataxic motor phenotype in the setting of cerebellar disorders of various etiologies.
In the last decade, a syndrome with severe developmental delay and dysmorphic features has been described in association with microdeletion of chromosome 19 (19p13.13 or 19p13.2) containing also the CACNA1A gene (60–62). These findings corroborate on one hand the association between CACNA1A and neurodevelopmental disorders, on the other underlie the likely relevant contribute of multiple genetic factors—along with single gene variants—as background of such severe phenotypes.
Epilepsy in Classical Paroxysmal CACNA1A Disorders
Clinical Presentation and Genotype-Phenotype Correlation
An association between epilepsy and CACNA1A disorders has been postulated early (12) and then corroborated by several independent reports describing co-occurrence of epileptic features in CACNA1A pedigrees [reviewed in (63)]. Epileptic features display some genotype-specificity. Indeed, seizures and epileptiform activity in EEG are a recurrent finding in EA2 (12, 14, 35, 38, 40, 47, 64–67). Epileptic features are observed in childhood and adolescence, often before onset of episodic ataxia (31, 38, 40, 64, 68). Absences and 3 Hz Spike-Wave discharges are typical findings (13, 14, 38, 65, 67). Conversely, in pedigrees with FHM1 seizures mostly occur concomitantly to hemiplegic attacks (69–74) and independent epileptic manifestations are unusual (51, 75–77).
Pathophysiology
Beyond the clinical field, several lines of evidence support the association between a loss-of-function of P/Q channels and generalized epilepsy. Notably, mutant mice bearing loss-of-function CACNA1A variants represent established models of absence epilepsy (78). In these models (Tottering, Leaner, Rocker) absence-like seizures go along with an ataxic motor phenotype (79–82). Mice with CACNA1A null mutation develop a similar phenotype with absence epilepsy and progressive neurological deficits before dying in the age of 3–4 weeks (83).
From a pathophysiological point of view, the EEG changes driving absence depend on an abnormal synchronization of thalamocortical circuitry (80, 84). Several studies showed that a loss-of-function of P/Q channels results in increased T-type current in the thalamocortical network (78, 79). T-type calcium current are directly involved in the generation 3 Hz Spike-Wave rhythmicity and overexpression of T-type currents in mice is sufficient to induce absence epilepsy (85). Interestingly, a study in a conditional murine model showed that absence epilepsy occurs also when P/Q loss in limited to Purkinje cells (86). This notion suggests a relevant regulatory role for early cerebellar inputs in thalamocortical circuitry. Indeed, a reduced firing in Purkinje cells with knock-out of P/Q currents decreases inhibition on deep cerebellar nuclei and in turn leads to increasing output of cerebellar projections to the thalamus.
Epileptic Encephalopathy
Clinical Presentation and Genotype-Phenotype Correlation
Latest clinical studies in early onset epileptic encephalopathies suggest that the mechanisms subserving epileptogenesis in the setting of pathogenic CACNA1A variants is likely more complex than anticipated. Epileptic encephalopathies are devastating neurological disorders characterized by early onset of multiple seizure types, accompanied by psychomotor regression and a variety of focal neurological signs including ataxia and cerebral palsy (87). The severe and frequent epileptic activity is believed to be the pivotal trigger for further psychomotor deterioration. Originally, epileptic encephalopathies were considered to result mostly from a symptomatic etiology, such as perinatal hypoxic-ischemic insults. The discovery of mutations in the sodium channel gene SCN1A in the Dravet syndrome in 2001 paved the way to the exploration of their genetic background (88). Since then, cumulative research highlighted how de novo mutations in channel genes are a recurrent cause of epileptic encephalopathies and recently revealed a role also for CACNA1A variants (15). Indeed, CACNA1A variants have been found in the setting of severe epileptic syndromes including Ohtahara syndrome, epilepsy of infancy with migrating focal seizures and Lennox-Gastaut syndrome.
A phenotype compatible with epileptic encephalopathy has first been described in association with deletions and truncating mutations in CACNA1A (14). Afterwards, the application of NGS techniques in large collectives detected de novo missense CACNA1A variants in this setting (15, 89–92). The observed phenotype in the affected individuals reflected the typical course of an epileptic encephalopathy with the beginning of multiple seizure types in the first days of life as well as various degrees of intellectual disability. The pathophysiological mechanisms linking such a set of functionally heterogeneous genotypes with this devastating clinical picture are unclear (93). Detected variants include both newly described and well-known ones, such as the S218L, which is typically associated with a severe form of FHM1 (70). Despite current evidence is limited to few studies, some mutations have been already recurrently observed [e.g., A713T (15, 94, 95) and E101Q (15, 91)].
Pathophysiology
A recent study investigated the functional effect of the variants associated with epileptic encephalopathies (96). For example, the variants G239V and I1357S resulted in altered protein trafficking with accumulation in intracellular inclusions in HEK293 cells, a finding compatible with a dominant negative effect. The variants A713T and V1396M, showed a gain-of-function effect, which appeared more profound compared to that of classical gain-of-function mutations seen in FHM1 (96). Notably, the variant S218L, which underlies both a severe FHM1 form and epileptic encephalopathy, has also a well-established more deleterious effect on current activation and inactivation (97, 98). These notions suggest that the absolute degree of channel dysfunction, more than the specific effect on currents kinetics, drives severe phenotypes. The missense mutations associated with developmental epileptic encephalopathies are situated in transmembrane segments, except for the variant S218L which localizes in a cytosolic region (see Figure 2). The transmembrane located variants likely affect the voltage-sensor or pore domains, while it remains speculative if the intracellular located S218L variant may additionally alter the binding with intracellular protein/intracellular signaling.
Early Onset Dystonia
Clinical Presentation
Dystonia can occur in a number of primary cerebellar disorders including the autosomal-dominant spinocerebellar ataxias and several autosomal recessive ataxias. CACNA1A disorders may also feature dystonic symptoms, with some typical age-specific patterns. Several reports highlighted the occurrence of a periodic movement disorders such the benign paroxysmal torticollis of the infancy (BPTI) (9–11, 45, 99) and the paroxysmal tonic upward gaze (PTU) (10, 100, 101) in infants from CACNA1A pedigrees. Both conditions are rare and their occurrence is typically limited to the infancy. BPTI consists of transient episodes of cervical dystonia, which may be accompanied by vomiting, ataxia, pallor. The attacks usually remit spontaneously within 3–5 years of age (102). Infants with BPTI often have a positive family history for migraine. BPTI can later evolve in migraine or further migraine-related disorders and has been included in the third edition of the International Classification of Headache Disorders (44). Its “benign” course is an object of controversy, since a number of cases show a developmental delay (102). Notably, a recent study screening a larger group of infants with BPTI but without further neurological symptoms did not find CACNA1A variants (103). In general, a suspect for an underlying pathogenic CACNA1A variant is strong when BPTI arises in infants with positive family history for FHM/EA or presence of delayed milestones. PTU consists of episodes of sustained conjugate upward deviation of the eyes, with neck flexion and sometimes impaired coordination, lasting from minutes to hours (104). PTU was originally also believed to be a benign phenomenon, resolving without sequelae. Later reviews demonstrated that it may be associated to structural brain pathologies and predate a severe neurological phenotype with chronic ataxia, epilepsy and cognitive impairment (105, 106). Very recently, a multicentric study on 47 infants with CACNA1A variants revealed a high frequency of paroxysmal dystonia, especially of PTU (found in 47% of cases, while torticollis was evident in 19%) (107). In adult patients with EA2, dystonia, mostly cervical, can occur during paroxysmal manifestations (46, 108) or persist as chronic symptoms later in the disease course (49, 109–111). Pathophysiological background of paroxysmal dyskinesias has been investigated in tottering and rocker mice bearing loss-of-function CACNA1A mutations (112).
Discussion
The advent of NGS techniques revolutionized our understanding of a relevant number of neurological diseases. Especially in the field of neurodevelopmental disorders, the identification of de novo pathogenic variants in ion channel genes opened a new window of opportunities for unraveling their pathophysiological mechanisms. Within these studies, CACNA1A variants also emerged as a relevant novel genetic etiology.
When to Choose a Genetic Testing for CACNA1A
CACNA1A testing belongs to the standard assessment of hemiplegic migraine or episodic ataxia, especially in the setting of a positive family history or when chronic cerebellar signs are present (113).
Based on the current literature, CACNA1A sequencing should be considered also in the settings of early epilepsy and/or developmental delay when chronic cerebellar signs or a positive family history for FHM/EA are present. Concerning the chronic cerebellar signs, an isolated downbeat nystagmus may be a decisive clue (14). Similarly, a clinical picture with early onset paroxysmal dystonia, such as BPTI, plus cerebellar signs supports the application of CACNA1A sequencing, even in the presence of a negative family history. Furthermore, current evidence from the field of epileptic encephalopathies supports the application of diagnostic panels covering also CACNA1A in this setting. A flow chart in Figure 3 summarizes the present recommendations.
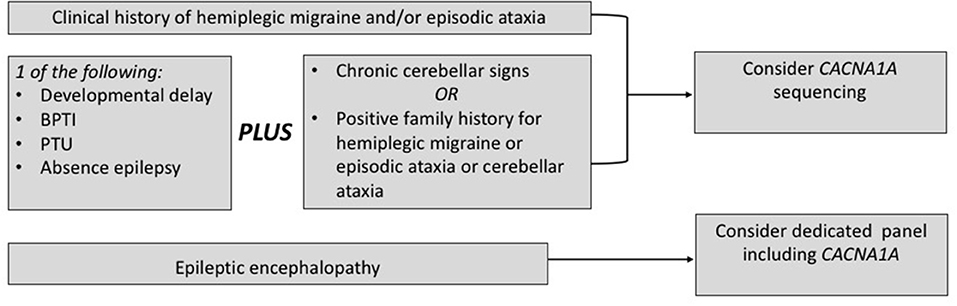
Figure 3. When to choose a genetic testing for CACNA1A. BPTI, benign paroxysmal torticollis of the infancy; PTU, paroxysmal tonic upward gaze.
Therapeutic Implications
The detection of a causal CACNA1A variant in the setting of a paroxysmal disorder bears relevant therapeutic consequences. Indeed, the classical paroxysmal manifestations such as hemiplegic migraine and episodic ataxia usually display a good to excellent response to the interval therapy with acetazolamide (40, 114). The exact mechanism underlying this therapeutic effect is unclear. Acetazolamide is a carbonic anhydrase inhibitor and induces a pH shift in liquor toward more acidic values. Changes in extracellular pH influence membrane conductance and may induce a stabilization of P/Q channel permeability (40). Second line compounds in non-responsive cases are 4-aminopyridine and flunarizine (114, 115). Generally, these therapies are not expected to improve chronic neurological symptoms, although their stabilization upon beginning of medical treatment can be occasionally reported (46, 116). A further exception is represented by improvement of down-beat nystagmus under 4-aminopyridine treatment (115).
Therapy-resistant seizures markedly contribute to developmental involution in epileptic encephalopathies and thus the timely initiation of an effective antiepileptic treatment represents a crucial issue. Since seizures are episodic manifestations sensu strictu, the question raises whether they may also improve upon acetazolamide as other paroxysmal symptoms. Acetazolamide has a known anticonvulsive effect, which also depends on the induction of an acidotic state (117). Because of its tolerance developing properties and side effects (electrolytes derangements, nephrolithiasis), acetazolamide is currently used mostly as add-on therapy in refractory epilepsies or in special situations, e.g., catamenial epilepsy (118). Interestingly, the established anticonvulsive drug topiramate also exhibits carbonic anhydrase inhibition properties and it has occasionally been applied with success in the treatment of FHM1/EA2 (45). In the largest reported series of CACNA1A-related epileptic encephalopathy, four out of six cases were upon treatment with acetazolamide or topiramate but information about the treatment response was not provided (15).
A detailed presentation of the treatment of manifestations associated with CACNA1A mutations is beyond the scope of this mini-review and for that we refer to the several reviews on the topic (40, 114).
Open Questions for Future Research
Despite the expanding knowledge on molecular genetics and clinical presentation of CACNA1A variants, the lack of prospective data from large collectives limits our understanding of this channelopathy. For example, it remains to be elucidated if CACNA1A-related neurodevelopmental disorders represent separate entities or rather an age-dependent stage within an evolving phenotype. The responsivity of early developmental/epileptic features to the classical interval therapy remains also an open question for clinicians. Future clinical studies with a prospective design may clarify these issues. A further puzzling issue remains the coexistence of marked intrafamilial variability as well as phenotypic overlap across different mutation types. These issues limit the definition of a clear-cut genotype-phenotype correlation for the clinician. On the bench side, a deeper understanding of implications of CACNA1A dysfunction on cerebrocerebellar circuitries and local plasticity phenomena at the cellular membrane may help to conceive common therapeutic strategies targeting symptoms across different phenotypes (37).
Author Contributions
EI and SB both contributed to conceive, write, and review the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Prof. Jörg Striessnig for the discussion and critical revision of the paper. The authors of this publication are members of the European Reference Network for Rare Neurological Diseases - Project ID No 739510.
References
1. Catterall WA. Voltage-gated sodium channels at 60: structure, function and pathophysiology. J Physiol. (2012) 590:2577–89. doi: 10.1113/jphysiol.2011.224204
2. Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. (1996) 87:543–52. doi: 10.1016/S0092-8674(00)81373-2
3. Russell MB, Ducros A. Sporadic and familial hemiplegic migraine: pathophysiological mechanisms, clinical characteristics, diagnosis, and management. Lancet Neurol. (2011) 10:457–70. doi: 10.1016/S1474-4422(11)70048-5
4. Jen J, Kim GW, Baloh RW. Clinical spectrum of episodic ataxia type 2. Neurology. (2004) 62:17–22. doi: 10.1212/01.WNL.0000101675.61074.50
5. Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet. (1997) 15:62–9. doi: 10.1038/ng0197-62
6. Geschwind DH, Perlman S, Figueroa KP, Karrim J, Baloh RW, Pulst SM. Spinocerebellar ataxia type 6. Frequency of the mutation and genotype-phenotype correlations. Neurology. (1997) 49:1247–51. doi: 10.1212/WNL.49.5.1247
7. Indelicato E, Nachbauer W, Karner E, Eigentler A, Wagner M, Unterberger I, et al. The neuropsychiatric phenotype in CACNA1A mutations: a retrospective single center study and review of the literature. Eur J Neurol. (2019) 26:66–e7. doi: 10.1111/ene.13765
8. Humbertclaude V, Riant F, Krams B, Zimmermann V, Nagot N, Annequin D, et al. Cognitive impairment in children with CACNA1A mutations. Dev Med Child Neurol. (2020) 62:330–7. doi: 10.1111/dmcn.14261
9. Giffin NJ, Benton S, Goadsby PJ. Benign paroxysmal torticollis of infancy: four new cases and linkage to CACNA1A mutation. Dev Med Child Neurol. (2002) 44:490–3. doi: 10.1111/j.1469-8749.2002.tb00311.x
10. Roubertie A, Echenne B, Leydet J, Soete S, Krams B, Rivier F, et al. Benign paroxysmal tonic upgaze, benign paroxysmal torticollis, episodic ataxia and CACNA1A mutation in a family. J Neurol. (2008) 255:1600–2. doi: 10.1007/s00415-008-0982-8
11. Vila-Pueyo M, Gene GG, Flotats-Bastardes M, Elorza X, Sintas C, Valverde MA, et al. A loss-of-function CACNA1A mutation causing benign paroxysmal torticollis of infancy. Eur J Paediatr Neurol. (2014) 18:430–3. doi: 10.1016/j.ejpn.2013.12.011
12. Jouvenceau A, Eunson LH, Spauschus A, Ramesh V, Zuberi SM, Kullmann DM, et al. Human epilepsy associated with dysfunction of the brain P/Q-type calcium channel. Lancet. (2001) 358:801–7. doi: 10.1016/S0140-6736(01)05971-2
13. Rajakulendran S, Graves TD, Labrum RW, Kotzadimitriou D, Eunson L, Davis MB, et al. Genetic and functional characterisation of the P/Q calcium channel in episodic ataxia with epilepsy. J Physiol. (2010) 588(Pt 11):1905–13. doi: 10.1113/jphysiol.2009.186437
14. Damaj L, Lupien-Meilleur A, Lortie A, Riou E, Ospina LH, Gagnon L, et al. CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. Eur J Hum Genet. (2015) 23:1505–12. doi: 10.1038/ejhg.2015.21
15. Epi KC. De novo mutations in SLC1A2 and CACNA1A are important causes of epileptic encephalopathies. Am J Hum Genet. (2016) 99:287–98. doi: 10.1016/j.ajhg.2016.06.003
16. Guerin AA, Feigenbaum A, Donner EJ, Yoon G. Stepwise developmental regression associated with novel CACNA1A mutation. Pediatr Neurol. (2008) 39:363–4. doi: 10.1016/j.pediatrneurol.2008.07.030
17. Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. (2011) 3:a003947. doi: 10.1101/cshperspect.a003947
18. Rajakulendran S, Kaski D, Hanna MG. Neuronal P/Q-type calcium channel dysfunction in inherited disorders of the CNS. Nat Rev Neurol. (2012) 8:86–96. doi: 10.1038/nrneurol.2011.228
19. Mintz IM, Sabatini BL, Regehr WG. Calcium control of transmitter release at a cerebellar synapse. Neuron. (1995) 15:675–88. doi: 10.1016/0896-6273(95)90155-8
21. Ito M. Cerebellar circuitry as a neuronal machine. Prog Neurobiol. (2006) 78:272–303. doi: 10.1016/j.pneurobio.2006.02.006
22. Higley MJ, Sabatini BL. Calcium signaling in dendrites and spines: practical and functional considerations. Neuron. (2008) 59:902–13. doi: 10.1016/j.neuron.2008.08.020
23. Adams PJ, Rungta RL, Garcia E, van den Maagdenberg AM, MacVicar BA, Snutch TP. Contribution of calcium-dependent facilitation to synaptic plasticity revealed by migraine mutations in the P/Q-type calcium channel. Proc Natl Acad Sci U S A. (2010) 107:18694–9. doi: 10.1073/pnas.1009500107
24. D'Adamo MC, Liantonio A, Conte E, Pessia M, Imbrici P. Ion channels involvement in neurodevelopmental disorders. Neuroscience. (2020) 440:337–59. doi: 10.1016/j.neuroscience.2020.05.032
25. Xu JH, Tang FR. Voltage-dependent calcium channels, calcium binding proteins, and their interaction in the pathological process of epilepsy. Int J Mol Sci. (2018) 19:2735. doi: 10.3390/ijms19092735
26. Nanou E, Catterall WA. Calcium channels, synaptic plasticity, and neuropsychiatric disease. Neuron. (2018) 98:466–81. doi: 10.1016/j.neuron.2018.03.017
27. Lipscombe D, Allen SE, Toro CP. Control of neuronal voltage-gated calcium ion channels from RNA to protein. Trends Neurosci. (2013) 36:598–609. doi: 10.1016/j.tins.2013.06.008
28. Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. (2013) 9:e1003709. doi: 10.1371/journal.pgen.1003709
29. Luo X, Rosenfeld JA, Yamamoto S, Harel T, Zuo Z, Hall M, et al. Clinically severe CACNA1A alleles affect synaptic function and neurodegeneration differentially. PLoS Genet. (2017) 13:e1006905. doi: 10.1371/journal.pgen.1006905
30. Sintas C, Carreno O, Fernandez-Castillo N, Corominas R, Vila-Pueyo M, Toma C, et al. Mutation spectrum in the CACNA1A gene in 49 patients with episodic ataxia. Sci Rep. (2017) 7:2514. doi: 10.1038/s41598-017-02554-x
31. Labrum RW, Rajakulendran S, Graves TD, Eunson LH, Bevan R, Sweeney MG, et al. Large scale calcium channel gene rearrangements in episodic ataxia and hemiplegic migraine: implications for diagnostic testing. J Med Genet. (2009) 46:786–91. doi: 10.1136/jmg.2009.067967
32. Riant F, Mourtada R, Saugier-Veber P, Tournier-Lasserve E. Large CACNA1A deletion in a family with episodic ataxia type 2. Arch Neurol. (2008) 65:817–20. doi: 10.1001/archneur.65.6.817
33. Eunson LH, Graves TD, Hanna MG. New calcium channel mutations predict aberrant RNA splicing in episodic ataxia. Neurology. (2005) 65:308–10. doi: 10.1212/01.wnl.0000169020.82223.dd
34. Blumkin L, Michelson M, Leshinsky-Silver E, Kivity S, Lev D, Lerman-Sagie T. Congenital ataxia, mental retardation, and dyskinesia associated with a novel CACNA1A mutation. J Child Neurol. (2010) 25:892–7. doi: 10.1177/0883073809351316
35. Tantsis EM, Gill D, Griffiths L, Gupta S, Lawson J, Maksemous N, et al. Eye movement disorders are an early manifestation of CACNA1A mutations in children. Dev Med Child Neurol. (2016) 58:639–44. doi: 10.1111/dmcn.13033
36. Sanchez-Albisua I, Schoning M, Jurkat-Rott K, Lerche H. Possible effect of corticoids on hemiplegic attacks in severe hemiplegic migraine. Pediatr Neurol. (2013) 49:286–8. doi: 10.1016/j.pediatrneurol.2013.04.011
37. Jaudon F, Baldassari S, Musante I, Thalhammer A, Zara F, Cingolani LA. Targeting alternative splicing as a potential therapy for episodic ataxia type 2. Biomedicines. (2020) 8:332. doi: 10.3390/biomedicines8090332
38. Stendel C, D'Adamo MC, Wiessner M, Dusl M, Cenciarini M, Belia S, et al. Association of a novel splice site mutation in P/Q-type calcium channels with childhood epilepsy and late-onset slowly progressive non-episodic cerebellar ataxia. Int J Mol Sci. (2020) 21:3810. doi: 10.3390/ijms21113810
39. Ducros A, Denier C, Joutel A, Cecillon M, Lescoat C, Vahedi K, et al. The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. N Engl J Med. (2001) 345:17–24. doi: 10.1056/NEJM200107053450103
40. Strupp M, Zwergal A, Brandt T. Episodic ataxia type 2. Neurotherapeutics. (2007) 4:267–73. doi: 10.1016/j.nurt.2007.01.014
41. Klockgether T. The clinical diagnosis of autosomal dominant spinocerebellar ataxias. Cerebellum. (2008) 7:101–5. doi: 10.1007/s12311-008-0023-2
42. Du X, Wang J, Zhu H, Rinaldo L, Lamar KM, Palmenberg AC, et al. Second cistron in CACNA1A gene encodes a transcription factor mediating cerebellar development and SCA6. Cell. (2013) 154:118–33. doi: 10.1016/j.cell.2013.05.059
43. Du X, Wei C, Hejazi Pastor DP, Rao ER, Li Y, Grasselli G, et al. α1ACT is essential for survival and early cerebellar programming in a critical neonatal window. Neuron. (2019) 102:770–85.e7. doi: 10.1016/j.neuron.2019.02.036
44. Headache Classification Committee of the International Headache Society. The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia. (2013) 33:629–808. doi: 10.1177/0333102413485658
45. Indelicato E, Nachbauer W, Eigentler A, Donnemiller E, Wagner M, Unterberger I, et al. Ten years of follow-up in a large family with familial hemiplegic migraine type 1: clinical course and implications for treatment. Cephalalgia. (2017) 38:1167–76. doi: 10.1177/0333102417715229
46. Nachbauer W, Nocker M, Karner E, Stankovic I, Unterberger I, Eigentler A, et al. Episodic ataxia type 2: phenotype characteristics of a novel CACNA1A mutation and review of the literature. J Neurol. (2014) 261:983–91. doi: 10.1007/s00415-014-7310-2
47. Mantuano E, Romano S, Veneziano L, Gellera C, Castellotti B, Caimi S, et al. Identification of novel and recurrent CACNA1A gene mutations in fifteen patients with episodic ataxia type 2. J Neurol Sci. (2010) 291:30–6. doi: 10.1016/j.jns.2010.01.010
48. Subramony SH, Schott K, Raike RS, Callahan J, Langford LR, Christova PS, et al. Novel CACNA1A mutation causes febrile episodic ataxia with interictal cerebellar deficits. Ann Neurol. (2003) 54:725–31. doi: 10.1002/ana.10756
49. Kinder S, Ossig C, Wienecke M, Beyer A, von der Hagen M, Storch A, et al. Novel frameshift mutation in the CACNA1A gene causing a mixed phenotype of episodic ataxia and familiar hemiplegic migraine. Eur J Paediatr Neurol. (2015) 19:72–4. doi: 10.1016/j.ejpn.2014.10.005
50. Bertholon P, Chabrier S, Riant F, Tournier-Lasserve E, Peyron R. Episodic ataxia type 2: unusual aspects in clinical and genetic presentation. Special emphasis in childhood. J Neurol Neurosurg Psychiatry. (2009) 80:1289–92. doi: 10.1136/jnnp.2008.159103
51. Riant F, Ducros A, Ploton C, Barbance C, Depienne C, Tournier-Lasserve E. De novo mutations in ATP1A2 and CACNA1A are frequent in early-onset sporadic hemiplegic migraine. Neurology. (2010) 75:967–72. doi: 10.1212/WNL.0b013e3181f25e8f
52. Wada T, Kobayashi N, Takahashi Y, Aoki T, Watanabe T, Saitoh S. Wide clinical variability in a family with a CACNA1A T666m mutation: hemiplegic migraine, coma, and progressive ataxia. Pediatr Neurol. (2002) 26:47–50. doi: 10.1016/S0887-8994(01)00371-X
53. Freilinger T, Ackl N, Ebert A, Schmidt C, Rautenstrauss B, Dichgans M, et al. A novel mutation in CACNA1A associated with hemiplegic migraine, cerebellar dysfunction and late-onset cognitive decline. J Neurol Sci. (2011) 300:160–3. doi: 10.1016/j.jns.2010.09.032
54. Ohba C, Osaka H, Iai M, Yamashita S, Suzuki Y, Aida N, et al. Diagnostic utility of whole exome sequencing in patients showing cerebellar and/or vermis atrophy in childhood. Neurogenetics. (2013) 14:225–32. doi: 10.1007/s10048-013-0375-8
55. Weyhrauch DL, Ye D, Boczek NJ, Tester DJ, Gavrilova RH, Patterson MC, et al. Whole exome sequencing and heterologous cellular electrophysiology studies elucidate a novel Loss-of-Function mutation in the CACNA1A-encoded neuronal P/Q-type calcium channel in a child with congenital hypotonia and developmental delay. Pediatr Neurol. (2016) 55:46–51. doi: 10.1016/j.pediatrneurol.2015.10.014
56. Yamamoto T, Imaizumi T, Yamamoto-Shimojima K, Lu Y, Yanagishita T, Shimada S, et al. Genomic backgrounds of Japanese patients with undiagnosed neurodevelopmental disorders. Brain Dev. (2019) 41:776–82. doi: 10.1016/j.braindev.2019.05.007
57. Mallmann RT, Elgueta C, Sleman F, Castonguay J, Wilmes T, van den Maagdenberg A, et al. Ablation of Ca(V)2.1 voltage-gated Ca(2)(+) channels in mouse forebrain generates multiple cognitive impairments. PLoS One. (2013) 8:e78598. doi: 10.1371/journal.pone.0078598
58. Sokolov AA, Miall RC, Ivry RB. The cerebellum: adaptive prediction for movement and cognition. Trends Cogn Sci. (2017) 21:313–32. doi: 10.1016/j.tics.2017.02.005
59. Schmahmann JD. The cerebellum and cognition. Neurosci Lett. (2019) 688:62–75. doi: 10.1016/j.neulet.2018.07.005
60. Auvin S, Holder-Espinasse M, Lamblin MD, Andrieux J. Array-CGH detection of a de novo 0.7-Mb deletion in 19p13.13 including CACNA1A associated with mental retardation and epilepsy with infantile spasms. Epilepsia. (2009) 50:2501–3. doi: 10.1111/j.1528-1167.2009.02189.x
61. Nimmakayalu M, Horton VK, Darbro B, Patil SR, Alsayouf H, Keppler-Noreuil K, et al. Apparent germline mosaicism for a novel 19p13.13 deletion disrupting NFIX and CACNA1A. Am J Med Genet A. (2013) 161A:1105–9. doi: 10.1002/ajmg.a.35790
62. Kuroda Y, Mizuno Y, Mimaki M, Oka A, Sato Y, Ogawa S, et al. Two patients with 19p13.2 deletion (Malan syndrome) involving NFIX and CACNA1A with overgrowth, developmental delay, and epilepsy. Clin Dysmorphol. (2017) 26:224–7. doi: 10.1097/MCD.0000000000000185
63. Indelicato E, Unterberger I, Nachbauer W, Eigentler A, Amprosi M, Zeiner F, et al. The electrophysiological footprint of CACNA1A disorders. J Neurol. (2021). doi: 10.1007/s00415-021-10415-x. [Epub ahead of print].
64. Choi JH, Seo JD, Choi YR, Kim MJ, Shin JH, Kim JS, et al. Exercise-induced downbeat nystagmus in a Korean family with a nonsense mutation in CACNA1A. Neurol Sci. (2015) 36:1393–6. doi: 10.1007/s10072-015-2157-6
65. Imbrici P, Eunson LH, Graves TD, Bhatia KP, Wadia NH, Kullmann DM, et al. Late-onset episodic ataxia type 2 due to an in-frame insertion in CACNA1A. Neurology. (2005) 65:944–6. doi: 10.1212/01.wnl.0000176069.64200.28
66. Imbrici P, Jaffe SL, Eunson LH, Davies NP, Herd C, Robertson R, et al. Dysfunction of the brain calcium channel CaV2.1 in absence epilepsy and episodic ataxia. Brain. (2004) 127(Pt 12):2682–92. doi: 10.1093/brain/awh301
67. Jung J, Testard H, Tournier-Lasserve E, Riant F, Vallet AE, Berroir S, et al. Phenotypic variability of episodic ataxia type 2 mutations: a family study. Eur Neurol. (2010) 64:114–6. doi: 10.1159/000315145
68. Chan YC, Burgunder JM, Wilder-Smith E, Chew SE, Lam-Mok-Sing KM, Sharma V, et al. Electroencephalographic changes and seizures in familial hemiplegic migraine patients with the CACNA1A gene S218L mutation. J Clin Neurosci. (2008) 15:891–4. doi: 10.1016/j.jocn.2007.01.013
69. Beauvais K, Cave-Riant F, De Barace C, Tardieu M, Tournier-Lasserve E, Furby A. New CACNA1A gene mutation in a case of familial hemiplegic migraine with status epilepticus. Eur Neurol. (2004) 52:58–61. doi: 10.1159/000079546
70. Debiais S, Hommet C, Bonnaud I, Barthez MA, Rimbaux S, Riant F, et al. The FHM1 mutation S218L: a severe clinical phenotype? A case report and review of the literature. Cephalalgia. (2009) 29:1337–9. doi: 10.1111/j.1468-2982.2009.01884.x
71. Kors EE, Terwindt GM, Vermeulen FL, Fitzsimons RB, Jardine PE, Heywood P, et al. Delayed cerebral edema and fatal coma after minor head trauma: role of the CACNA1A calcium channel subunit gene and relationship with familial hemiplegic migraine. Ann Neurol. (2001) 49:753–60. doi: 10.1002/ana.1031
72. Malpas TJ, Riant F, Tournier-Lasserve E, Vahedi K, Neville BG. Sporadic hemiplegic migraine and delayed cerebral oedema after minor head trauma: a novel de novo CACNA1A gene mutation. Dev Med Child Neurol. (2010) 52:103–4. doi: 10.1111/j.1469-8749.2009.03493.x
73. Stam AH, Luijckx GJ, Poll-The BT, Ginjaar IB, Frants RR, Haan J. Early seizures and cerebral oedema after trivial head trauma associated with the CACNA1A S218L mutation. J Neurol Neurosurg Psychiatry. (2009) 80:1125–9. doi: 10.1136/jnnp.2009.177279
74. Vahedi K, Denier C, Ducros A, Bousson V, Levy C, Chabriat H, et al. CACNA1A gene de novo mutation causing hemiplegic migraine, coma, and cerebellar atrophy. Neurology. (2000) 55:1040–2. doi: 10.1212/WNL.55.7.1040
75. Alonso I, Barros J, Tuna A, Coelho J, Sequeiros J, Silveira I, et al. Phenotypes of spinocerebellar ataxia type 6 and familial hemiplegic migraine caused by a unique CACNA1A missense mutation in patients from a large family. Arch Neurol. (2003) 60:610–4. doi: 10.1001/archneur.60.4.610
76. Romaniello R, Zucca C, Tonelli A, Bonato S, Baschirotto C, Zanotta N, et al. A wide spectrum of clinical, neurophysiological and neuroradiological abnormalities in a family with a novel CACNA1A mutation. J Neurol Neurosurg Psychiatry. (2010) 81:840–3. doi: 10.1136/jnnp.2008.163402
77. Stam AH, Vanmolkot KR, Kremer HP, Gartner J, Brown J, Leshinsky-Silver E, et al. CACNA1A R1347Q: a frequent recurrent mutation in hemiplegic migraine. Clin Genet. (2008) 74:481–5. doi: 10.1111/j.1399-0004.2008.00996.x
78. Miao QL, Herlitze S, Mark MD, Noebels JL. Adult loss of Cacna1a in mice recapitulates childhood absence epilepsy by distinct thalamic bursting mechanisms. Brain. (2020) 143:161–74. doi: 10.1093/brain/awz365
79. Llinas RR, Choi S, Urbano FJ, Shin HS. Gamma-band deficiency and abnormal thalamocortical activity in P/Q-type channel mutant mice. Proc Natl Acad Sci U S A. (2007) 104:17819–24. doi: 10.1073/pnas.0707945104
80. Maheshwari A, Noebels JL. Monogenic models of absence epilepsy: windows into the complex balance between inhibition and excitation in thalamocortical microcircuits. Prog Brain Res. (2014) 213:223–52. doi: 10.1016/B978-0-444-63326-2.00012-0
81. Mori Y, Wakamori M, Oda S, Fletcher CF, Sekiguchi N, Mori E, et al. Reduced voltage sensitivity of activation of P/Q-type Ca2+ channels is associated with the ataxic mouse mutation rolling Nagoya (tg(rol)). J Neurosci. (2000) 20:5654–62. doi: 10.1523/JNEUROSCI.20-15-05654.2000
82. Noebels JL, Sidman RL. Inherited epilepsy: spike-wave and focal motor seizures in the mutant mouse tottering. Science. (1979) 204:1334–6. doi: 10.1126/science.572084
83. Jun K, Piedras-Renteria ES, Smith SM, Wheeler DB, Lee SB, Lee TG, et al. Ablation of P/Q-type Ca(2+) channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha(1A)-subunit. Proc Natl Acad Sci U S A. (1999) 96:15245–50. doi: 10.1073/pnas.96.26.15245
84. Bomben VC, Aiba I, Qian J, Mark MD, Herlitze S, Noebels JL. Isolated P/Q calcium channel deletion in layer VI corticothalamic neurons generates absence epilepsy. J Neurosci. (2016) 36:405–18. doi: 10.1523/JNEUROSCI.2555-15.2016
85. Ernst WL, Zhang Y, Yoo JW, Ernst SJ, Noebels JL. Genetic enhancement of thalamocortical network activity by elevating alpha 1g-mediated low-voltage-activated calcium current induces pure absence epilepsy. J Neurosci. (2009) 29:1615–25. doi: 10.1523/JNEUROSCI.2081-08.2009
86. Mark MD, Maejima T, Kuckelsberg D, Yoo JW, Hyde RA, Shah V, et al. Delayed postnatal loss of P/Q-type calcium channels recapitulates the absence epilepsy, dyskinesia, and ataxia phenotypes of genomic Cacna1a mutations. J Neurosci. (2011) 31:4311–26. doi: 10.1523/JNEUROSCI.5342-10.2011
87. McTague A, Howell KB, Cross JH, Kurian MA, Scheffer IE. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. (2016) 15:304–16. doi: 10.1016/S1474-4422(15)00250-1
88. Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. (2001) 68:1327–32. doi: 10.1086/320609
89. Epi KC, Epilepsy Phenome/Genome P, Allen AS, Berkovic SF, Cossette P, Delanty N, et al. De novo mutations in epileptic encephalopathies. Nature. (2013) 501:217–21. doi: 10.1038/nature12439
90. Epperson MV, Haws ME, Standridge SM, Gilbert DL. An atypical rett syndrome phenotype due to a novel missense mutation in CACNA1A. J Child Neurol. (2018) 33:286–9. doi: 10.1177/0883073818754987
91. Mitta N, Menon RN, McTague A, Radhakrishnan A, Sundaram S, Cherian A, et al. Genotype-phenotype correlates of infantile-onset developmental & epileptic encephalopathy syndromes in South India: a single centre experience. Epilepsy Res. (2020) 166:106398. doi: 10.1016/j.eplepsyres.2020.106398
92. Hayashida T, Saito Y, Ishii A, Yamada H, Itakura A, Minato T, et al. CACNA1A-related early-onset encephalopathy with myoclonic epilepsy: a case report. Brain Dev. (2018) 40:130–3. doi: 10.1016/j.braindev.2017.08.006
93. Striessnig J. Voltage-gated Ca2+-channel de novo missense mutations: gain or loss of function - implications for potential therapies. Front Synaptic Neurosci. (2020). doi: 10.3389/fnsyn.2021.634760
94. Hamdan FF, Myers CT, Cossette P, Lemay P, Spiegelman D, Laporte AD, et al. High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. Am J Hum Genet. (2017) 101:664–85. doi: 10.1016/j.ajhg.2017.09.008
95. Balck A, Hanssen H, Hellenbroich Y, Lohmann K, Munchau A. Adult-onset ataxia or developmental disorder with seizures: two sides of missense changes in CACNA1A. J Neurol. (2017) 264:1520–2. doi: 10.1007/s00415-017-8494-z
96. Jiang X, Raju PK, D'Avanzo N, Lachance M, Pepin J, Dubeau F, et al. Both gain-of-function and loss-of-function de novo CACNA1A mutations cause severe developmental epileptic encephalopathies in the spectrum of Lennox-Gastaut syndrome. Epilepsia. (2019) 60:1881–94. doi: 10.1111/epi.16316
97. Loonen ICM, Jansen NA, Cain SM, Schenke M, Voskuyl RA, Yung AC, et al. Brainstem spreading depolarization and cortical dynamics during fatal seizures in Cacna1a S218L mice. Brain. (2019) 142:412–25. doi: 10.1093/brain/awy325
98. van den Maagdenberg AM, Pizzorusso T, Kaja S, Terpolilli N, Shapovalova M, Hoebeek FE, et al. High cortical spreading depression susceptibility and migraine-associated symptoms in Ca(v)2.1 S218L mice. Ann Neurol. (2010) 67:85–98. doi: 10.1002/ana.21815
99. Cuenca-Leon E, Corominas R, Fernandez-Castillo N, Volpini V, Del Toro M, Roig M, et al. Genetic analysis of 27 Spanish patients with hemiplegic migraine, basilar-type migraine and childhood periodic syndromes. Cephalalgia. (2008) 28:1039–47. doi: 10.1111/j.1468-2982.2008.01645.x
100. Quade A, Thiel A, Kurth I, Holtgrewe M, Elbracht M, Beule D, et al. Paroxysmal tonic upgaze: A heterogeneous clinical condition responsive to carbonic anhydrase inhibition. Eur J Paediatr Neurol. (2020) 25:181–6. doi: 10.1016/j.ejpn.2019.11.002
101. Blumkin L, Leshinsky-Silver E, Michelson M, Zerem A, Kivity S, Lev D, et al. Paroxysmal tonic upward gaze as a presentation of de-novo mutations in CACNA1A. Eur J Paediatr Neurol. (2015) 19:292–7. doi: 10.1016/j.ejpn.2014.12.018
102. Rosman NP, Douglass LM, Sharif UM, Paolini J. The neurology of benign paroxysmal torticollis of infancy: report of 10 new cases and review of the literature. J Child Neurol. (2009) 24:155–60. doi: 10.1177/0883073808322338
103. Shin M, Douglass LM, Milunsky JM, Rosman NP. The genetics of benign paroxysmal torticollis of infancy: is there an association with mutations in the CACNA1A gene? J Child Neurol. (2016) 31:1057–61. doi: 10.1177/0883073816636226
104. Ouvrier RA, Billson F. Benign paroxysmal tonic upgaze of childhood. J Child Neurol. (1988) 3:177–80. doi: 10.1177/088307388800300305
105. Hayman M, Harvey AS, Hopkins IJ, Kornberg AJ, Coleman LT, Shield LK. Paroxysmal tonic upgaze: a reappraisal of outcome. Ann Neurol. (1998) 43:514–20. doi: 10.1002/ana.410430416
106. Luat AF, Asano E, Chugani HT. Paroxysmal tonic upgaze of childhood with co-existent absence epilepsy. Epileptic Disord. (2007) 9:332–6. doi: 10.1684/epd.2007.0119
107. Gur-Hartman T, Berkowitz O, Yosovich K, Roubertie A, Zanni G, et al. Clinical phenotypes of infantile onset CACNA1A-related disorder. Eur J Neurol Paedriatr Neurol. (2020) S1090-3798(20)30198-7. doi: 10.1016/j.ejpn.2020.10.004. [Epub ahead of print].
108. Hu Y, Jiang H, Wang Q, Xie Z, Pan S. Identification of a novel nonsense mutation p.Tyr1957Ter of CACNA1A in a Chinese family with episodic ataxia 2. PLoS One. (2013) 8:e56362. doi: 10.1371/journal.pone.0056362
109. Cuenca-Leon E, Banchs I, Serra SA, Latorre P, Fernandez-Castillo N, Corominas R, et al. Late-onset episodic ataxia type 2 associated with a novel loss-of-function mutation in the CACNA1A gene. J Neurol Sci. (2009) 280:10–4. doi: 10.1016/j.jns.2009.01.005
110. Fuerte-Hortigon A, Perez-Noguera R, Dotor Garcia-Soto J, Royo Boronat I, Alvarez de Andres S, Garcia-Moreno JM. Novel CACNA1A variant may cause cervical dystonia and cerebellar ataxia syndrome. J Neurol Sci. (2020) 415:116909. doi: 10.1016/j.jns.2020.116909
111. Spacey SD, Materek LA, Szczygielski BI, Bird TD. Two novel CACNA1A gene mutations associated with episodic ataxia type 2 and interictal dystonia. Arch Neurol. (2005) 62:314–6. doi: 10.1001/archneur.62.2.314
112. Hess EJ, Jinnah HA. Dystonia: Animal Models. EncyclMov Disord. (2017) 393–7. doi: 10.1016/B978-0-12-374105-9.00095-2
113. Pelzer N, Haan J, Stam AH, Vijfhuizen LS, Koelewijn SC, Smagge A, et al. Clinical spectrum of hemiplegic migraine and chances of finding a pathogenic mutation. Neurology. (2018) 90:e575–e82. doi: 10.1212/WNL.0000000000004966
114. Pelzer N, Stam AH, Haan J, Ferrari MD, Terwindt GM. Familial and sporadic hemiplegic migraine: diagnosis and treatment. Curr Treat Options Neurol. (2013) 15:13–27. doi: 10.1007/s11940-012-0208-3
115. Strupp M, Kalla R, Dichgans M, Freilinger T, Glasauer S, Brandt T. Treatment of episodic ataxia type 2 with the potassium channel blocker 4-aminopyridine. Neurology. (2004) 62:1623–5. doi: 10.1212/01.WNL.0000125691.74109.53
116. Yabe I, Sasaki H, Takeichi N, Takei A, Hamada T, Fukushima K, et al. Positional vertigo and macroscopic downbeat positioning nystagmus in spinocerebellar ataxia type 6 (SCA6). J Neurol. (2003) 250:440–3. doi: 10.1007/s00415-003-1020-5
117. Ansell B, Clarke E. Acetazolamide in treatment of epilepsy. Br Med J. (1956) 1:650–4. doi: 10.1136/bmj.1.4968.650
Keywords: CACNA1A, calcium channels, developmental delay, epileptic encephalopathy, dystonia, next generation sequencing, de novo mutation, psychiatric manifestations
Citation: Indelicato E and Boesch S (2021) From Genotype to Phenotype: Expanding the Clinical Spectrum of CACNA1A Variants in the Era of Next Generation Sequencing. Front. Neurol. 12:639994. doi: 10.3389/fneur.2021.639994
Received: 10 December 2020; Accepted: 08 February 2021;
Published: 02 March 2021.
Edited by:
Kishore Raj Kumar, Garvan Institute of Medical Research, AustraliaReviewed by:
Jose M. Fernandez-Fernandez, Pompeu Fabra University, SpainArushi Gahlot Saini, Post Graduate Institute of Medical Education and Research (PGIMER), India
Copyright © 2021 Indelicato and Boesch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sylvia Boesch, U3lsdmlhLmJvZXNjaEBpLW1lZC5hYy5hdA==