 Marie J. Pietrowski1†
Marie J. Pietrowski1† Amr Ahmed Gabr1,2†
Amr Ahmed Gabr1,2† Stanislav Kozlov1†
Stanislav Kozlov1† David Blum3,4
David Blum3,4 Annett Halle1,5*‡
Annett Halle1,5*‡ Kevin Carvalho3,4‡
Kevin Carvalho3,4‡- 1Microglia and Neuroinflammation Laboratory, German Center for Neurodegenerative Diseases (DZNE), Bonn, Germany
- 2Department of Physiology, Faculty of Veterinary Medicine, Cairo University, Giza, Egypt
- 3University of Lille, Inserm, CHU Lille, U1172 LilNCog - Lille Neuroscience and Cognition, Lille, France
- 4Alzheimer and Tauopathies, Labex DISTALZ, Lille, France
- 5Institute of Neuropathology, University of Bonn, Bonn, Germany
Purinergic signaling regulates neuronal and glial cell functions in the healthy CNS. In neurodegenerative diseases, purinergic signaling becomes dysregulated and can affect disease-associated phenotypes of glial cells. In this review, we discuss how cell-specific expression patterns of purinergic signaling components change in neurodegeneration and how dysregulated glial purinergic signaling and crosstalk may contribute to disease pathophysiology, thus bearing promising potential for the development of new therapeutical options for neurodegenerative diseases.
Introduction
Neurodegenerative diseases are characterized by a progressive loss of structure and function of the CNS. An estimated 50 million patients worldwide are currently affected by neurodegenerative diseases and it has been projected that this number will rise to 131 million patients by 2050 (1). Many of these diseases are idiopathic/polygenic proteinopathies, which are characterized by accumulation and/or aggregation of proteins such as TAU, amyloid-β or α-synuclein (α-SYN) in the CNS and are multifactorial, with both genetic and environmental risk factors. The most common ones are the sporadic forms of Alzheimer's disease (AD; TAU/amyloid-β (Aβ) aggregates), Parkinson's disease (PD; α-SYN aggregates), amyotrophic lateral sclerosis (ALS; TAR DNA binding protein 43 (TDP-43)/FUS RNA binding protein (FUS) aggregates), Lewy body dementia (α-SYN aggregates) and frontotemporal lobar degeneration (FTLD; TAU/TDP-43/FUS aggregates) (2).
Despite decades of research, no causal treatment is available for any of these diseases. One promising approach to help overcome the lack of therapeutical options is to shift the focus from a linear neuron-centered view on how neurodegenerative diseases develop to a broader and integrative view in which interactions between all cell types in the brain are considered. This paradigm shift has led to an increased interest in the role of glial cells in neurodegeneration.
Indeed, there is strong evidence that changes in glial cells, microglia, astrocytes and oligodendrocytes, are causally involved in neurodegenerative diseases. Around half of the known genetic risk factors in sporadic AD are glial genes related to immune function such as TREM2, CD33 or CR1 (3, 4). Furthermore, recent data have highlighted phenotypic changes of glial cells over the disease course with the discovery of disease-associated microglial (DAM) and disease-associated astrocytic markers (DAA) (5, 6). Accordingly, new therapeutic strategies targeting glial signaling pathways are currently being tested in pre-clinical intervention studies, for example, treatments modulating the NLRP3 inflammasome (7), astrocytic nuclear factor kappa-β (8) and TREM2-mediated signaling (9), among others.
The targeting of purinergic signaling—i.e., signaling pathways mediated by extracellular nucleotides and nucleosides such as ATP or adenosine—belongs to these novel emerging therapeutic strategies (10). All CNS cell types, including glial cells, express purinergic receptors and purinergic signaling influences key CNS functions [for review see Agostinho et al. (11)] such as synaptic transmission, proliferation, maturation and neuroinflammation (12) that are altered in neurodegeneration. Thus, in the present manuscript, we discuss the current knowledge on purinergic signaling in glial cells and its potential relevance in neurodegenerative disease. We provide a comprehensive overview and cell-specific expression tables based on available transcriptomic data of purinergic genes in glial cells in neurodegeneration and link this data with data from functional studies. Finally, we discuss glial purinergic signaling as a potential target for future therapeutic intervention.
The Components of Purinergic Signaling
Purines are a family of small molecules involved in DNA/RNA structure and key cellular processes such as cell metabolism, intracellular signaling and extracellular signaling. Geoffrey Burnstock coined the term of “purinergic signaling” for the latter in 1972 (13), in which he referred to cell signaling pathways that are activated by engagement of nucleosides and nucleotides with specific cell receptors. ATP, the main energy storage of the cell, can be hydrolyzed in the extracellular space through specific enzymes called ectonucleotidases into ADP, AMP and adenosine. These include ectonucleoside triphosphate diphosphohydrolases (E-NTPDases), ectonucleotide pyrophosphatase/ phosphodiesterases (ENPPs), alkaline phosphatases and ecto-5′-nucleotidase (NT5E/CD73) (14). Adenosine can also be produced through the S-adenosyl-L-homocysteine pathways and degraded by adenosine deaminase and adenosine kinase. Furthermore, ATP and its derivatives can be transported between cell compartments and released extracellularly through different transporters like the equilibrative nucleoside transporters (ENTs) or concentrative nucleoside transporters (CNTs), but also through channels such as Connexin-43 (CX43), Connexin-32 (CX32) or Pannexin-1 (PANX1), secretion involving vesicular nucleotide transporter (SLC17A9/VNUT) or ultimately by membrane rupture during cellular injury, allowing them to trigger purinergic receptor signaling (15, 16).
Purinergic receptors are subclassified into two large families: P1 receptors that are activated by nucleosides, and P2 receptors that are activated by nucleotides. P2 receptors are further subdivided into ionotropic P2X receptors and metabotropic P2Y receptors (17, 18). So-called P0 receptors have recently been defined and constitute adenine receptors (19, 20). The P1 receptor family consists of four receptors (adenosine A1, A2A, A2B and A3 receptor), the P2X family of seven receptors (P2X1-P2X7) and the P2Y family of eight receptors (P2Y1, P2Y2, P2Y4, P2Y6, P2Y11-P2Y14).
Glial Purinergic Signaling and Crosstalk Under Physiological Conditions
Purinergic signaling controls important physiological processes in the healthy CNS, such as synaptic transmission, cell proliferation and innate immune response (11, 12). ATP and adenosine are produced and released upon neuronal or glial activation and initiate various cellular pathways corresponding to the activation of P2X, P2Y and adenosine receptors. Neurons, microglia, astrocytes and oligodendrocytes express a unique repertoire of purinergic receptors (Figure 1, left), which due to receptor-specific intracellular downstream signaling cascades lead to specific responses in each CNS cell type. Many previous studies have contributed to our understanding of the purinergic signaling repertoires in microglia (21–24), astrocytes (25–27) and oligodendrocytes (28, 29). However, a full overview of the expression patterns of all components of purinergic signaling in the main mouse and human CNS cells has only become available through the seminal work of the laboratory of Ben Barres. The group created an unbiased RNA sequencing database of purified specific CNS cells from human and mouse cortex (30–32). Using this database, we here assembled expression data of purinergic genes from neurons, microglia, astrocytes and oligodendrocytes. With the caveat of missing validation on the protein level, possible cell impurities, region-specific differences or age-dependent modifications, this makes it possible to compare expression patterns of purinergic genes in these CNS cell types in human and mouse (Figure 2).
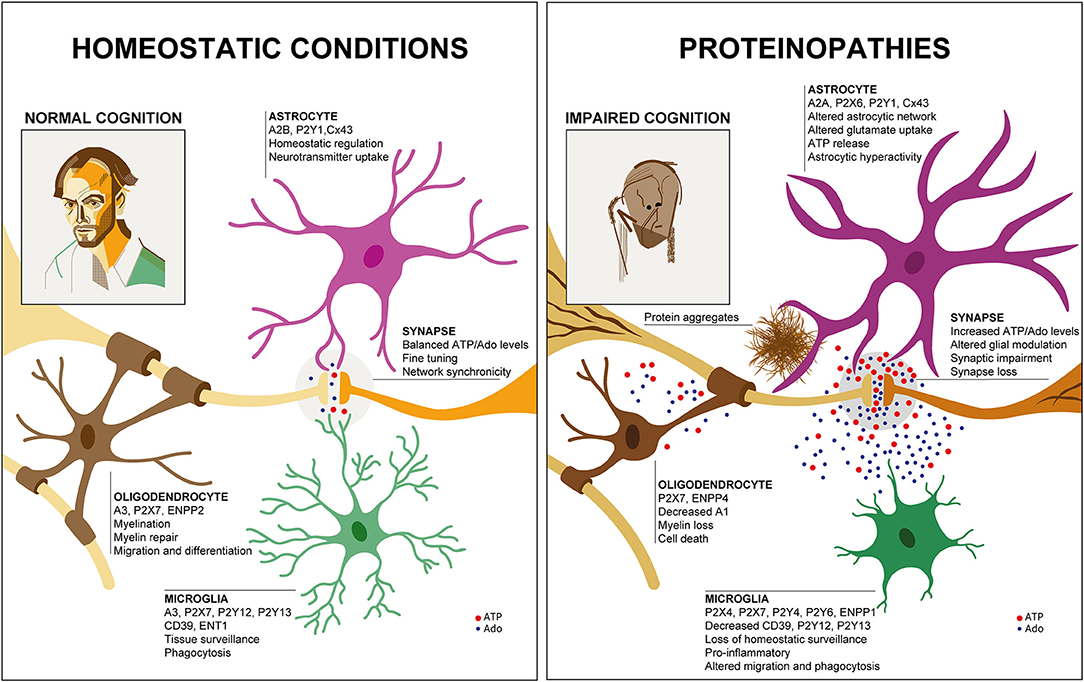
Figure 1. Glial purinergic signaling under homeostatic conditions and in proteinopathies. Under healthy conditions (Left), cell-specific glial purinergic signaling contributes to fine-tuning of synapse function and thus, normal cognitive abilities. In proteinopathies (Right), accumulation of protein aggregates leads to glial phenotype changes that are associated with altered expression of purinergic signaling components and altered ATP and adenosine (Ado) levels. This impacts synaptic function and ultimately results in synapse loss, contributing to impaired cognition. Two self-portraits are shown to illustrate the cognitive status in health and disease, similar to the famous work of William Utermohlen, who continued to create self-portraits after being diagnosed with AD.
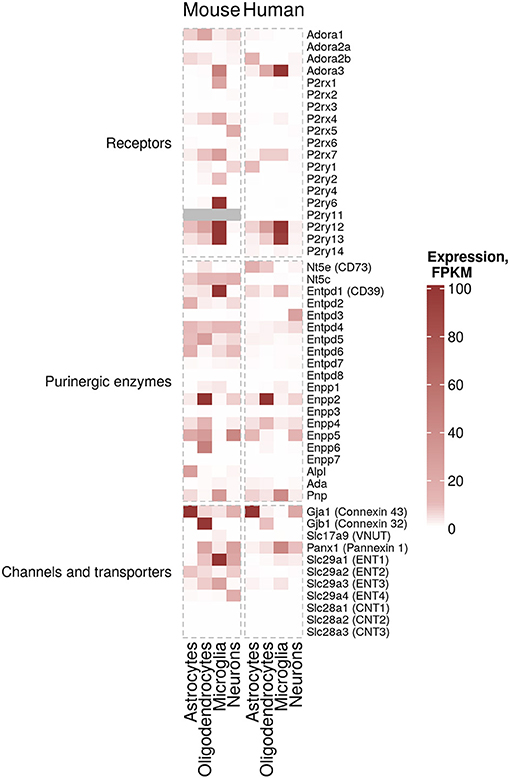
Figure 2. Cell-specific expression of purinergic genes. Estimated expression levels FPKM (Fragments Per Kilobase Million) of genes involved in purinergic signaling pathways in microglia, myelinating oligodendrocytes, mature astrocytes and neurons was obtained from existing data sets from Zhang et al. (30) (mouse data) and Zhang et al. (31) (human data) (30, 31). FPKM values of 100 and above are shown in the same color for visualization purposes.
According to this data, microglia are the cells with the highest expression and largest number of expressed purinergic receptors (in particular ADORA3, P2RY12, P2RY13, and at lower levels P2RX7 and P2RY14 genes) and ectonucleotidases (in particular ENTPD1 gene also known as CD39) in the human cortex. There are significant differences in murine microglial gene expression of purinergic components compared to human. For example, Adora1, P2rx4 and Entpd2 show higher expression levels in mouse. Additionally, genes encoding ENTs (ENT1, 2 and 3, encoded by Slc29a1, Slc29a2 and Slc29a3, correspondingly) are expressed at higher levels in mouse microglia, suggesting that nucleoside transport processes may differ between humans and mice. Similarly, cell-specific expression of some ectonucleotidases, including Entpd1 in microglia are considerably different in human and mouse (Figure 2).
Human astrocytes, despite displaying–in some cases considerably–lower expression values than microglia, also express a variety of purinergic receptors (ADORA2B, P2RY1, P2RY12) and ectonucleotidases (ENTPD1, ENPP5 and NT5E also known as CD73). Similar patterns can be found in mouse astrocytes (Figure 2).
Lastly, human oligodendrocytes express ADORA3, P2RX7, P2RY12, P2RY13, and NT5E. Interestingly, ectonucleotide pyrophosphatases/phosphodiesterases (ENPPs, namely Enpp2, Enpp4, Enpp5, and Enpp6) in oligodendrocytes show a higher expression in mouse (Figure 2).
Although some of the data largely overlaps with previous findings in rodent and human CNS tissue, this cell type-specific gene expression table allows to better appreciate the considerable inter-species similarities–and differences–between human and mouse. It also shows the large spectrum of purinergic gene expression patterns in the different CNS cell types.
How is this cell type-specific purinergic signaling repertoire linked to glial cell function? Although some aspects have remained ill-defined, especially regarding the role of purinergic enzymes and transporters, a lot is known about how purinergic receptor signaling shapes glial cell function in the healthy brain [Figure 1 left; for a recent detailed review see Agostinho et al. (11)].
For example, different P1 and P2 receptors such as A3, P2Y12, P2Y13, P2X4 control the motility of the ramified and dynamic cell processes of microglia, the tissue-resident immune cells of the brain (33–36). This motility of cell processes is important for surveillance and chemotactic cell process movement toward localized brain damage. Furthermore, phagocytosis and release of inflammatory molecules, both major effector functions of microglia, are modulated by purinergic signaling pathways, involving P2X4, P2X7, and P2Y6 (37–40). In addition, there is evidence that microglial contribution to synaptic pruning and synaptic function is at least partly controlled by purinergic mechanisms (41). It has also been shown that innate immune responses of microglia are positively modulated by ATP through P2X7 receptors (42) and negatively by adenosine through A2A receptors (43).
Astrocytes are involved in a wide variety of functions in the CNS, such as metabolic support, synaptic function, neuronal and synaptic maturation and blood brain barrier permeability (44–47). The relevance of astrocytic P2 receptor signaling in some of these cell functions has been implicated in a number of previous studies. More specifically, P2Y1 mediates calcium signaling in astrocytes, which is critical for modulating synapse function and blood-brain barrier maintenance (48). Neurotransmitter recapture is also modulated by astrocytic A2A receptor-mediated signaling, allowing for a reduced uptake of glutamate and greater uptake of GABA, reinforcing synaptic activation (49, 50).
Oligodendrocytes synthesize myelin sheaths, which insulate CNS axons, enabling a rapid action potential propagation. This process occurs life-long as oligodendrocytes perform myelin remodeling. Additionally, oligodendrocytes contribute to the metabolic support of axons through a wide variety of transporters (51). Oligodendrocyte progenitor cells (OPC, also called NG2 cells as they express the NG2 chondroitin sulfate proteoglycan) can differentiate into new mature oligodendrocytes. As for microglia and astrocytes, there is evidence that purinergic signaling controls a number of cell functions in oligodendrocytes. Adenosine and ATP/ADP have been shown to contribute to proliferation, migration and maturation of oligodendrocytes through P1 (A1 and A2A) and P2 receptors (P2X7 and P2Y1).
Apart from its importance in shaping cell functions of particular glial cells, purinergic signaling plays a key role in neuro-glial interactions (11). Neurons and glial cells reside in spatial proximity to each other, which is particularly important at the synaptic site. This proximity enables an efficient inter-cellular crosstalk through purinergic signaling, based on ATP/adenosine release, ectonucleotidase activity and receptor stimulation, affecting the neighboring cells in an autocrine and paracrine manner.
For example, ATP and adenosine released by synaptic stimulation trigger calcium wave signaling in the astrocytic syncytium through P2Y receptors, which in response decrease the activation of neighboring synapses through adenosine A1 receptor, a phenomenon called “heterosynaptic depression” (52, 53). Moreover, neuronal ATP release at the synapse recruits microglial cell processes, allowing cleavage of ATP into adenosine through microglial CD39 and ubiquitous CD73, leading to dampening of synapse activity upon A1 receptor stimulation (41).
Furthermore, ectonucleotidases such as CD73 are required for adenosine A2A receptor potentiation of synapses, and such mechanism could be similar in a glial context (54). For instance, ENTPD1 (CD39) and NT5E (CD73) are expressed in all CNS cell types in the human CNS, albeit in case of CD73 at very low and in case of CD39 at high levels in microglia compared to the other cell types (31). This suggests that local degradation of ATP and ADP and production of adenosine may also occur in a cell-autonomous manner.
Glial Purinergic Signaling and Crosstalk in Neurodegeneration
Since purinergic signaling is a multi-cellular, dynamic and complex signaling system, some of its aspects in neurodegenerative diseases have been difficult to evaluate and will require more specific quantitative tools in the future.
For instance, one major question that has remained unresolved due to the lack of specific tools with sufficient temporal and spatial resolution, is how the spatio-temporal kinetics of ligand availability and purinergic receptor activation is regulated in neurodegeneration (55). However, it has recently been shown that purinergic metabolites are strongly modified in AD. Adenosine was shown to be the most affected purine, increasing in temporal and parietal cortices of AD brains (56). Similarly, increased levels of adenosine are detectable in the CSF of ALS patients (57).
Another question that has not been sufficiently addressed until recently has been whether and how expression of purinergic signaling components is altered in glial cells in neurodegeneration. To get a better understanding of disease-associated changes, we took advantage of the increasing number of unbiased transcriptomic studies on human and murine glial cells in neurodegeneration. These studies did not have the primary goal to investigate differential purinergic gene expression in glial cells. However, they now constitute valuable resources to detect potential patterns of purinergic transcriptomic response in microglia, astrocytes and oligodendrocytes in neurodegeneration. To assemble the available data in a comprehensive manner, we searched for studies from transgenic AD, PD, tauopathy and ALS mouse models and human AD, PD, tauopathy and ALS patients, in which transcriptomic analyses on single cells or nuclei and bulk-sorted glial populations were performed. Among the available studies that mainly covered AD and ALS, we selected the data sets, in which at least one component of purinergic signaling was significantly dysregulated. We thus included 18 data sets from 14 studies on microglia (5, 58–68), nine data sets from eight studies on astrocytes (61, 68–74) and 4 data sets from three studies on oligodendrocytes (61, 68, 72) and used log2 fold change expression values between diseased and control samples to visualize shifts of gene expression involved in purinergic signaling pathways for each cell type (Figures 3–5).
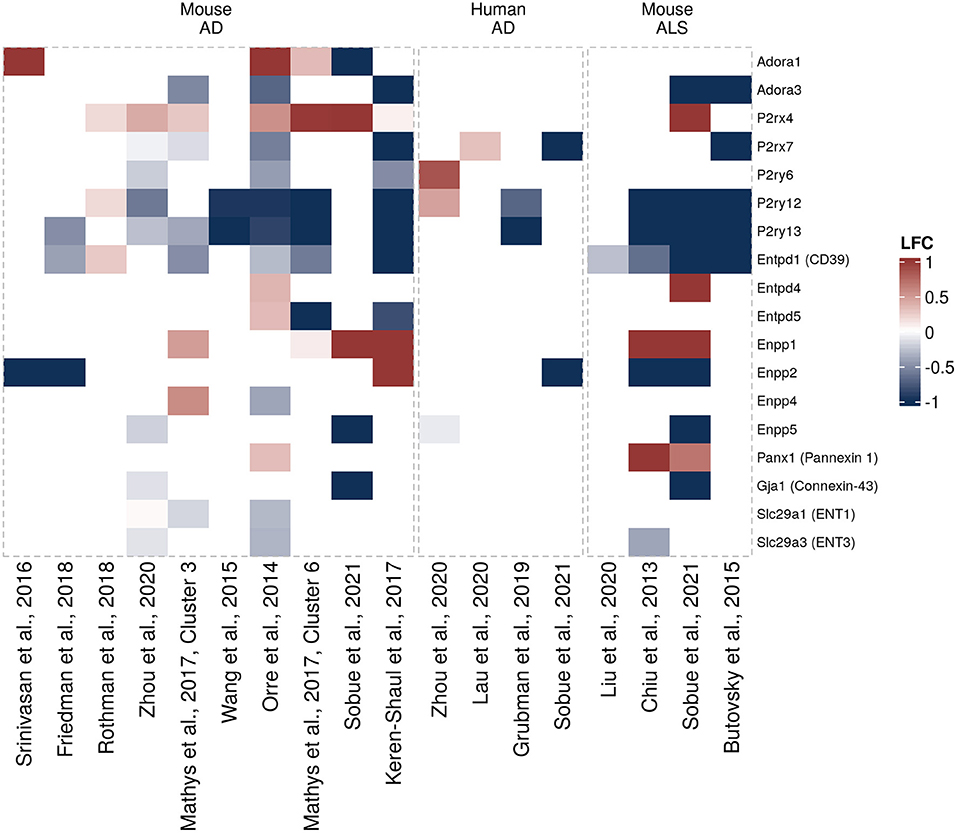
Figure 3. Differentially expressed purinergic genes in microglia in neurodegeneration. Heatmap shows Log2 fold changes in expression of genes involved in purinergic signaling pathways between disease (AD or ALS, or relevant mouse models) and control samples. LFC values above 1 and below −1 are shown in the same color intensity for visualization purposes. Studies were selected according to the following criteria: transcriptomic analysis was performed on single cells or nuclei and allows disease/control comparisons for selected cell types, or on bulk-sorted glial populations; differential expression analysis results were provided by authors as supplementary material and were accessible. In total, 18 data sets from 14 studies were included for microglia [Orre et al. (69) (mouse, AD model, bulk), Chiu et al. (63) (mouse, ALS model, bulk), Wang et al. (64) (mouse, AD model, bulk), Srinivasan et al. (65) (mouse, AD model, bulk), Friedman et al. (66) (mouse, AD model, bulk), Rothman et al. (58) (mouse, AD model, bulk), Mathys et al. (72) (mouse, AD model, single-cell, two disease-associated clusters), Keren-Shaul et al., (5) (mouse, AD model, single-cell, DAM cluster), Zhou et al. (68) (two data sets, mouse AD model and human AD patients, single-nuclei), Liu et al. (59) (mouse, ALS model, single-cell), Lau et al. (61) (human, ALS, single-nuclei), Grubman et al. (74) (human, AD, single-nuclei), Butovsky et al. (75) (mouse, ALS model, bulk), Sobue et al. (60) (three data sets, mouse AD and ALS models, and human AD patients, bulk)] (5, 58–68).
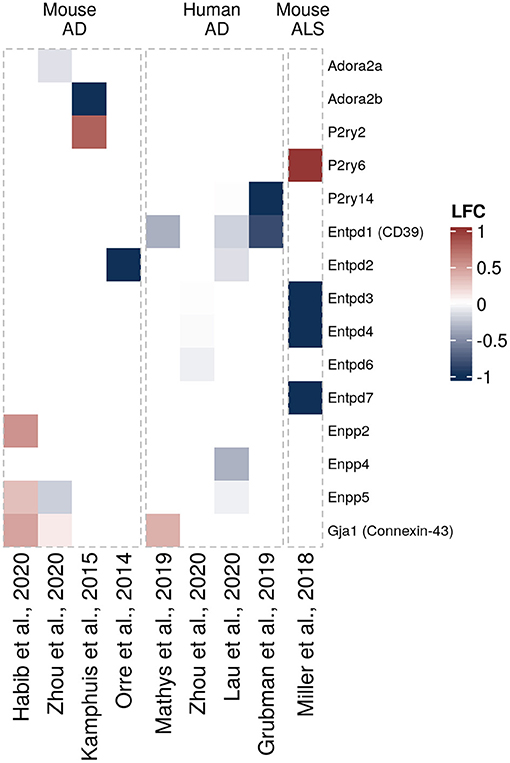
Figure 4. Differentially expressed purinergic genes in astrocytes in neurodegeneration. Heatmap shows Log2 fold changes in expression of genes involved in purinergic signaling pathways between disease (AD or ALS, or relevant mouse models) and control samples. LFC values above 1 and below −1 are shown in the same color intensity for visualization purposes. Studies were selected according to the following criteria: transcriptomic analysis was performed on single cells or nuclei and allows disease/control comparisons for selected cell types, or on bulk-sorted glial populations; differential expression analysis results were provided by authors as supplementary material and were accessible. In total, nine data sets from eight studies were included for astrocytes [Orre et al. (69) (mouse, AD model, bulk), Kamphuis et al. (70) (mouse, AD model, bulk), Miller et al. (71) (mouse, ALS model, bulk), Mathys et al. (72) (human, AD, single-nuclei), Zhou et al. (68) (two data sets, mouse AD model and human AD patients, single-nuclei), Habib et al. (73) (mouse, AD model, single-nuclei), Lau et al. (61) (human, ALS, single-nuclei), Grubman et al. (74) (human, AD, single-nuclei)] (61, 68–74).
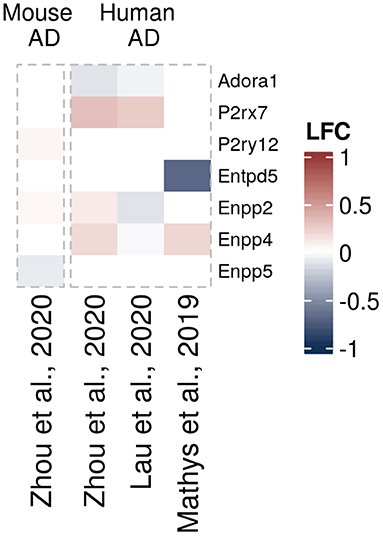
Figure 5. Differentially expressed purinergic genes in oligodendrocytes in neurodegeneration. Heatmap shows Log2 fold changes in expression of genes involved in purinergic signaling pathways between disease (AD or relevant mouse models) and control samples. LFC values above 1 and below −1 are shown in the same color intensity for visualization purposes. Studies were selected according to the following criteria: transcriptomic analysis was performed on single cells or nuclei and allows disease/control comparisons for selected cell types, or on bulk-sorted glial populations; differential expression analysis results were provided by authors as supplementary material and were accessible. In total, four data sets from three studies were included for oligodendrocytes [Mathys et al. (72) (human, AD, single-nuclei), Zhou et al. (68) (two datasets, mouse AD model and human AD patients, single-nuclei), Lau et al. (61) (human, ALS, single-nuclei)] (61, 68, 72).
Using this data and functional studies on glial purinergic signaling, we will discuss in the following sections what is known about purinergic signaling in microglia, astrocytes and oligodendrocytes in neurodegeneration.
It should be noted, however, that glial populations in neurodegeneration are spatially and temporally heterogeneous, depending on disease stage and proximity to specific pathological hallmarks. This heterogeneity is well-described for microglial populations in AD, in which “early” and “late” subsets of disease-associated microglia occur on both gene and protein expression levels (5, 76, 77). Moreover, single-cell transcriptomic studies have shown divergent but co-existent microglial subsets with different directions of gene expression changes (67, 76, 78). Therefore, it is likely that functionally and spatially distinct glial subsets co-exist within disease-associated reactive populations of glial cells [disease-associated microglia (DAM, (5)), disease-associated astrocytes (DAA, (73)) and pathology-associated populations of oligodendrocytes (67, 79)]. This may explain some of the discrepancies between purinergic gene expression changes and evidence from functional studies.
Microglia
Microglia are centrally involved in the pathophysiology of neurodegenerative diseases. Microglia not only drive inflammation in neurodegenerative diseases, but, as the main phagocytes in the CNS, are important for clearing protein aggregates, debris and apoptotic cells (80–86). Furthermore, the elaborate cellular morphology and process motility that enable efficient tissue surveillance are altered in neurodegeneration and this capability is therefore strongly affected (87–90). Some of these changes in microglial cellular functions may be caused by alterations in microglial purinergic signaling upon neurodegeneration (Figure 1, right).
When interrogating the microglia purinergic transcriptomic data from AD patients and AD mouse models we summarized here, a microglial response pattern emerges, in which Adora1 and P2rx4 are upregulated and Adora3, P2rx7, P2ry6, P2ry12, and P2ry13 downregulated (Figure 3). There are also changes in ectonucleotidase expression, particularly in Enpp1 (upregulated) and Entpd1 (downregulated) that are consistent between several data sets in mice. However, the majority of differentially expressed genes is only detected as differentially expressed in a few studies. Some inconsistencies in expression changes of some genes including upregulation of P2RY6 and P2RY12 in Zhou et al. (68), or upregulation of P2ry12 and Entpd1 in Rothman et al. (58), may be explained by differences in the experimental approaches (58, 68). A number of ALS-related changes in gene expression are similar to gene expression patterns from AD studies, including P2ry12 and P2ry13 (91) that are downregulated both in murine AD and ALS mouse models. Transcriptomic data sets for glial cells in Parkinson's disease are still largely missing, both from mouse models and human brain tissue. However, there was no clear association between PD risk genes and microglia or astrocyte populations in a recent study, in which single nuclei from substantia nigra and cortex were extracted and analyzed, suggesting that microglial purinergic alterations may play a less prominent role in PD than in AD (92).
In contrast to these transcriptomic data, numerous experimental studies have shown that microglial P2X7 protein levels are elevated in human AD brains, in multiple rodent amyloidosis models (93–98), in the PD rat model 6-OHDA as well as in the spinal cord of ALS patients and SOD1 mutant mice that serve as ALS animal models (99–101).
These findings are functionally important since enhanced activity of P2X7 drives cellular inflammation in several neurodegenerative pathologies. On a molecular level, Aβ-induced ATP release activates P2X7, which in turn results in ROS production (98, 102–104) as well as activation of the inflammasome and subsequent release of the cytokine IL-1β (82, 105–108). Similarly, SOD1G93A and TDP-43Q331K in ALS and α-synuclein in PD, contribute to oxidative stress and inflammation via microglial P2X7 activation (83, 109–111). On a functional level, it was demonstrated that inhibition of P2X7 decreases migration of microglia in vitro, whereas phagocytosis is enhanced in vivo in the J20 AD model (93) making it an interesting target. The functional role of P2X7 in the pathogenesis of ALS is less clear than in AD. On the one hand, SOD1 mutant microglia show reduced ATP degradation and enhanced ATP sensitivity in vitro (100). On the other hand, the onset of clinical symptoms seemed to be accelerated and disease progression exacerbated (112) in P2X7-deficient SOD1 mutant mice. This indicates a complex scenario, which has implications for potential treatment strategies, as the time point of P2RX7 counteraction seems to play a key role. Microglia seem to play a dual role in ALS, acting beneficially at early stages of the disease, while exacerbating disease pathology at later stages (113). Taken together, P2RX7 activation and subsequent inflammatory response represent a common purinergic dysregulation in proteinopathies such as AD, PD and ALS. However, further research needs to be conducted, especially in the fields of ALS and PD research.
As mentioned earlier, microglial morphology, motility and chemotaxis are regulated by the interplay of multiple different purinergic receptors including P1 and P2 receptors. The adenosine A1 and A3 receptors both play a role in microglial migration (33, 114). A1 is involved in morphological changes of microglia (115) and A3 contributes to the regulation of process extension (33). Consequently, the transcriptomic changes of Adora1 (A1) and Adora3 (A3) that can be detected in the transcriptomic studies in neurodegeneration (Figure 3) may contribute to alterations in microglial morphology, although functional studies are currently missing. In contrast to Adora1 and Adora3, expression of Adora2a (encoding A2A) is barely detectable under normal conditions (Figure 2). Nevertheless, A2A has been shown to mediate microglia process retraction (116), to induce Ptgs2 (Cyclooxygenase 2) expression (117) and proliferation of microglia (118). Interestingly, A2A was found to be increased in microglia in AD when using immunolabelling (119). Consequently, this upregulation could contribute to increased microglia cell number and decreased morphology around Aβ plaques. However, microglia-specific studies investigating the effects of P1 receptors in neurodegeneration are limited and further investigations will be required.
P2Y12 and P2Y13, encoded by P2ry12 and P2ry13, are the main P2 receptors that regulate microglial surveillance motility and chemotaxis. More specifically, P2Y13 regulates microglial morphology and surveillance (35), whereas P2Y12 mediates directed motility in microglia, process extension and microglia migration toward stimuli including ATP release (34, 41, 88). P2ry12 and P2ry13 are strongly downregulated in the transcriptomic studies summarized here (Figure 3), a finding that has been confirmed in mouse and human tissue at the RNA and protein levels (74, 75, 91, 120). Given the functional role of these two receptors, a strong decrease of P2Y13 may contribute to a loss of microglial surveillance function in neurodegeneration that normally supports tissue integrity and downregulation of P2Y12 may affect microglial directed migration, including migration to the plaque site in AD. Furthermore, P2Y12 is important to balance hyperexcitability of neurons, most likely through the involvement of P2Y12 in cell process extension toward the synaptic site (41) and P2Y12 deletion exacerbates experimentally induced epileptic events in mice (121). It is thus tempting to speculate that a reduction of P2Y12 in microglia in AD may contribute to the increased occurrence of epileptic events in AD patients (122).
In addition, brain-derived neurotrophic factor (BDNF) serves, among other functions, as a signal to regulate neuronal activity, since BDNF-mediated disruption of chloride gradient and following disturbance of chloride-dependent GABAergic inhibitory function was reported (123–125). Exocytosis of BDNF is mediated by ATP-evoked P2X4 activation (123, 126–130) and regulated by A2A (118). Decrease of inhibitory signals favors increased neuronal activity and hyperexcitability, eventually causing epileptic seizures (131–134). Both of the mentioned purinergic receptors, P2X4 and A2A, that regulate BDNF release are upregulated in AD [Figure 3, (119)]. The upregulation and following release of BDNF may therefore also contribute to hyperexcitability and epileptic events in neurodegeneration.
Besides, P2X4 is involved in the chemotactic response of microglia, as pharmacological inhibition and downregulation of P2X4 lead to impaired chemotaxis in vitro (36). The transcriptomic studies we summarize here provide evidence that P2rx4 is upregulated in different murine AD models (Figure 3), implicating that it could play a functional role in altering microglial chemotaxis in AD.
In summary, a number of purinergic receptors that regulate microglial morphology, motility and migration are dysregulated in neurodegeneration and may thus constitute pathways that contribute to impairment of these microglial motility functions. It will be important to validate this assumption in functional studies in the future.
Another important microglial cell function that is affected in neurodegeneration is the phagocytic capacity (87), which among many other mechanisms, is regulated by the UDP-sensitive purinergic receptor P2Y6. Interestingly, it has been shown that the ion channel function of P2X4 is inhibited by P2Y6 upregulation. This suggests an interesting mechanism, in which P2RY6 would be key to switch from migratory to phagocytic behavior of microglia (135). Because P2ry6 is downregulated in several murine AD models (Figure 3), this could mean that UDP-sensitive phagocytosis may be decreased in neurodegeneration. This could also be relevant for a process called “phagoptosis,” i.e., the uptake of live neurons by microglia involving P2Y6 (136). Inhibition of P2Y6 prevents neuronal loss induced by low levels of Aβ, suggesting that microglial phagocytosis could be responsible for neuronal loss (136). However, the absence of P2Y6-dependent inhibition of ex vivo phagocytosis in 5xFAD mice indicates that in AD, purinergic receptors other than P2Y6 may regulate microglial phagocytosis (137). Unlike in AD models, P2RY6 was upregulated in a PD model and in human PD brains, implying an opposite functional impact of P2Y6 in PD (101, 138).
In addition to P2Y6, there is also evidence that P2Y2 contributes to phagocytosis functions in microglia. Specifically, Aβ-induced microglial ATP release enhances phagocytosis of Aβ via P2Y2 activation in cultured cells (103, 139). In vivo data confirmed that heterozygous deletion of P2Y2 enhances β-amyloid plaque burden, indicating that downregulation of P2Y2 could contribute to AD pathology (140, 141). Apart from phagocytosis, pinocytosis induced by ATP or UTP is another mechanism that contributes to Aβ uptake by microglia. This process can be accelerated by yet another purinergic receptor, namely P2Y4 (142).
Altogether, since purinergic signaling regulates phagocytosis, a key function of microglia allowing clearance of pathologic protein aggregates, dysregulation of these purinergic pathways is likely to contribute to neurodegenerative disease pathology.
In light of the importance of purinergic signaling for the regulation of many microglia functions, the balance of extracellular purinergic ligands such as ATP, ADP, AMP and adenosine gains in significance. The concentration of extracellular purinergic elements is regulated by purinergic enzymes and transporters. Ectonucleotidases, namely CD39 and CD73, through ATP degradation and adenosine production, modulate microglial migration, microglia morphology and process elongation (114, 143, 144) and reduce phagocytosis (145). It is thus conceivable that downregulation and therefore loss of CD39 function, as observed in the spinal cord of ALS subjects (75) and in transcriptomic data from SOD1 mutant mice and murine amyloidosis models (Figure 3), could have detrimental consequences. Additionally, Enpp1 in turn is upregulated, while microglial ENT1 and ENT3 are downregulated (Figure 3). This indicates that the regulation of extracellular ligands by ectonucleotidases and by cross-membrane transport may become disturbed in neurodegeneration, although the exact impact on microglia themselves and other CNS cells will require further investigation.
Astrocytes
Astrocytes become reactive upon neurodegeneration and display morphological, functional, and molecular changes. It has recently been re-confirmed in a consensus statement that reactive astrocytes should not be subclassified into binary categories but defined at multiple levels using morphological, molecular and functional parameters and considering temporal and spatial aspects (6). Interestingly, the presence of reactive astrocytes correlates with the cognitive status in AD (146, 147). This may be due to the fact that astrocytes are involved in a wide variety of functions in the CNS and during neurodegeneration lose some of these properties (Figure 1, right), such as their ability to maintain the integrity of the blood brain barrier, contribute to gliotransmitter release and glutamate uptake. This loss of homeostatic defensive function, which may correlate with astroglial atrophy or “asthenia,” has been attributed to contribute to the propagation of cognitive decline in AD (148). At the same time, astrocytes gain potentially toxic functions and release pro-inflammatory molecules.
Based on the transcriptomic data we assembled here (Figure 2), it appears clear that mouse and human astrocytes show distinct expression patterns. P2ry2, Enpp2, Enpp5, and Gja1 (encoding CX43) are upregulated in AD mouse models, although not consistently across the different studies, while only Gja1 was found upregulated in human astrocytes in AD (Figure 4). Additionally, Adora2a, Adora2b, P2y14, Entpd1 (encoding CD39), Entpd2, and Enpp4 are downregulated in AD models or human AD. Furthermore, different sets of purinergic genes were found upregulated (P2ry6) or downregulated (Entpd3, Entpd4, Entpd7) in an ALS mouse model.
Functionally, the P2Y1 receptor signaling pathway is one of the major purinergic signaling pathways that may contribute to altered astrocyte properties in neurodegeneration, especially in AD. P2Y1 mediates calcium waves in astrocytes, which are linked to gliotransmitter release and synchronization of astrocytic syncytium, which is critical for synaptic plasticity (149–151). Unlike in the transcriptomic studies we summarized here, previous studies showed that astrocytic P2Y1 receptor expression is elevated in AD mouse models and human AD brain, especially in close vicinity to amyloid plaques (151, 152). This spatially restricted expression pattern may explain some of the discrepancies to the transcriptome data. P2Y1 activation has been linked to cognitive decline by enhancing astrocyte hyperactivity (153) and mediating astroglial network defects (151). Acute pharmacological inhibition of P2Y1 allows normalization of such defects (151), highlighting the therapeutical potential of targeting P2Y1 in AD. In particular, P2Y1 inhibitor injected through chronic ICV infusion reduced dystrophic neurite burden, improved astroglial function and long-term potentiation in an AD animal model (152).
Another study that shows discrepancies to the transcriptomic analyses shown here, found increased levels of CD39L1 (encoded by ENTPD2) at Braak stage III-IV in AD (154). Since CD39L1 modulates ATP metabolism and is particularly expressed in astrocytes (25), increased CD39L expression would reduce local ATP levels and thus dampen P2Y1-mediated astrocytic hyperactivity in CNS tissue with AD pathology, while its reduced expression, as suggested by the transcriptomic analyses, would have the opposite effect. Further studies are required to resolve these potential effects.
In agreement with the upregulation of Gja1/Connexin-43 (CX43) in astrocytes found in the transcriptomic analyses from AD mouse models and human AD brains we summarized here (Figure 4), Gja1 was detected among the astrocytic genes dysregulated in the proximity of amyloid plaques (155). CX43 contributes to ATP release (156). These effects seem to be mediated by amyloid pathology, as amyloid exposure triggers increased expression of CX43, both in vitro and in AD models (157–159). Deletion of astroglial CX43 in an AD mouse model was able to reduce astrocyte reactivity, ATP release, plaque-associated neuronal damage and improved synaptic function (160). In both, an in vitro and a mouse model of PD, astroglial CX43 was increased following exposure of rotenone, a neurotoxic substance (161, 162). Interestingly, CX43 activity is downstream of P2X7 and contributes to ATP release from astrocytes (163). Using a CX43 inhibitor, reduced α-synuclein deposits and attenuated neuroinflammation in a PD rat model were observed (164), highlighting the therapeutic potential of CX43 in several neurodegenerative diseases. CX43 and PANX1 are downregulated in an ALS mouse model (SOD1 mutant) at pre-symptomatic stage, while being upregulated at symptomatic stage (165), which again suggests that disease-dependent activation stages should be taken into account when designing therapeutic intervention.
Finally, despite weak expression in astrocytes under physiological conditions, adenosine A2A receptor was shown to be increased in astrocytes in both human AD brains (166) and amyloid models (49, 166, 167), although this finding is not supported by the transcriptomic data assembled here (Figure 4). Astrocytic A2A receptor is known for its regulation of glutamate and GABA uptake (49, 50) but also its in vitro effect on astrocyte gene regulation (168). Blocking A2A-mediated signaling was sufficient to mitigate memory defects in animal models (166).
Further investigations will be needed in the future to better understand whether A2A is clearly dysregulated in astrocytes or astrocytic subpopulations and whether astrocyte-specific A2A directly impacts astrocyte function in neurodegeneration.
Oligodendrocytes
In neurodegenerative diseases, oligodendrocytes become progressively damaged from various causes, such as neuroinflammation, direct effects from protein aggregates or oxidative stress, leading to myelin loss, disruption of energy transfer to neurons, and ultimately cell death (169). Regarding oligodendrocytes in neurodegenerative diseases, we could only find few transcriptomic data sets with significant differentially expressed purinergic genes. Of those, P2rx7 and Enpp4 genes appear to be upregulated in AD mouse models, and Adora1 downregulated (61, 68, 72) (Figure 5).
All members of the oligodendrocyte lineage are very sensitive to adenosine and ATP released by neurons or other glial cells as they are able to migrate, differentiate and proliferate upon activation of A1, A2A and P2Y1 receptors through calcium signaling (29, 170–172). In AD, impaired repair of myelin is observed, which was postulated to contribute to disease initiation (173). During neurodegenerative conditions, increased ATP release induces activation of P2X7 in oligodendrocytes, which could serve as an early sensor of neuronal damage, promoting OPC migration in vivo (174). Despite several studies pointing out P2X7 implication in different demyelinating context as well as oligodendrocyte death, no experimental data was found in proteinopathies (175–178).
Also, the involvement of oligodendrocyte-specific connexins such as connexin-29 (CX29 or Gjc3), connexin-32 (CX32 or Gjb1) or connexin-47 (CX47 or Gjc2) in a manner similar to CX43 in astrocytes cannot be ruled out (179). A study showed a downregulation of oligodendrocytic CX47 in an AD model, contrasting with the increase of CX43 observed in astrocytes (180). The authors stipulate that such modification could favor astrocyte-astrocyte connection at the expense of astrocyte-oligodendrocyte communication, contributing to oligodendrocyte function impairment. Additionally, CX32 was suggested to be increased in PD and correlated with increased alpha-synuclein uptake (181). Thus, consequences of ATP release from oligodendrocyte connexins and pannexins in neurodegeneration remain to be investigated.
Overall, proliferation and maturation of oligodendrocyte lineage are strongly influenced by purinergic signaling. As such, an imbalanced purinergic signaling upon neurodegeneration could contribute to myelin loss. Noteworthy, most studies on oligodendrocytic purinergic signaling have been conducted on multiple sclerosis and other inflammatory diseases [for review see Welsh and Kucenas (182)]. The lack of data on the present topic should be addressed in the future as P2X7 was found significantly upregulated in oligodendrocytes in AD (61), which could imply a deleterious role in disease progression.
Purinergic Signaling Crosstalk in Neurodegeneration
Given the importance of purinergic signaling for inter-glial and neuro-glial communication, alterations in cell-specific glial purinergic signaling in neurodegeneration will inevitably lead to disruptions in purinergic inter-cellular crosstalk (Figure 1, right).
For example, although adenosine has been reported to be the main purine metabolite that is elevated in neurodegeneration, at least in AD (56), it cannot be ruled out that opposite changes may take place in a more restricted spatial or temporal manner. For instance, CD39, a critical enzyme for the extracellular hydrolysis of ATP, is downregulated in microglia in AD (Figure 3) (41), especially in the vicinity of amyloid-beta plaques. This could have two immediate consequences: firstly, a local lack of adenosine production, which would reduce adenosine receptor activation, and secondly, high local levels of ATP, which could trigger microglial P2X7 receptor. In line with this, global CD39 deletion was shown to increase ATP and decrease adenosine in the CSF (183). Furthermore, global and microglia-specific Entpd1/CD39 deletion is associated with neuronal hyperactivity and increased vulnerability to epileptic events, mediated by a decreased activation of the inhibitory neuronal adenosine A1 receptor (41, 183). The reduced expression of astrocytic ENTPD1/CD39 in human AD cases (Figure 4), could further amplify these events. Additionally, a local ATP increase would trigger other P2 receptors such as astrocytic P2Y1, which would further impair neuronal synchronicity through astrocytic hyperactivation (151, 152). Adding further complexity, the ectonucleotidase ENPP1 is increased in microglia in AD (Figure 3). Whether this can–at least partially–compensate for decreased CD39-mediated ATPase activity and whether this affects astrocytic and oligodendrocyte functions has not been investigated. Hopefully, future studies will be able to answer these questions.
Furthermore, there is evidence that neuro-glial communication is affected by upregulation of glial purinergic receptors in neurodegeneration. For instance, microglial upregulated P2X7 (184–186) leads to an exacerbation of a pro-inflammatory microglial phenotype and increased release of proinflammatory molecules, which disrupt synaptic communication (187–189). Pro-inflammatory molecules like chemokines or reactive oxygen species also affect oligodendrocytes and astrocytes, causing oligodendrocyte cell death (190–192) and astrocytic reactivity (6), leading to further cell and network impairment. P2X7 is also upregulated in oligodendrocytes and has been implicated in oligodendrocytic cell death, which would critically alter inter-neuron communication and neuronal metabolic support. Furthermore, increased astrocytic P2Y1 in neurodegeneration, which is associated with astrocyte hyperexcitability, network synchronicity loss and increased ATP/glutamate release (152, 193) could in turn reinforce P2X7 activation in microglia and oligodendrocytes. Some studies have also reported increased A2A receptor expression in astrocytes, which has been shown to reduce glutamate intake and increase GABA intake (49, 50), thus favoring an excitotoxicity state, which is detrimental for synaptic function and reinforces reactive phenotypes of microglia and astrocytes.
In summary, increased ligand availability (ATP and adenosine) together with a distinct set of activated glial purinergic signaling pathways (P2X7, P2Y1, A2A) and loss of homeostatic neuronal and glial purinergic signaling pathways (A1, P2Y12, P2Y13) during neurodegeneration may alter the balance of purinergic signaling homeostasis. This in turn could lead to more cellular damage (excitotoxicity, neuroinflammation, myelin loss), loss of homeostatic functions (reduced energy delivery, reduced trophic factors, impaired synchronicity between networks) and promote protein aggregation (reduced phagocytosis of protein aggregates), thus ultimately favoring the progression of disease pathology in neurodegenerative diseases.
Therapeutical Perspectives
Recent advances have highlighted the beneficial effect of targeting purinergic signaling in neurodegenerative diseases, notably with the authorization of istradefylline, a selective antagonist of A2A receptors, in the co-treatment of Parkinson's disease in the USA in 2019 (194), following the approval in Japan in 2013. These first steps show that targeting purinergic signaling can be safe for use in neurodegenerative diseases and help to slow disease progression. Additionally, several drugs targeting purinergic signaling have gone into clinical trial in order to treat inflammatory diseases like rheumatoid arthritis, that are beyond the scope of this article [for review see Antonioli et al. (195)]. Noteworthy, proteinopathies are often associated with chronic low-grade neuroinflammation, reinforcing the potential benefits of using drugs targeting purinergic signaling (196).
P2X7 is one of the most evident targets of glial purinergic signaling in neurodegeneration, being upregulated in both microglia and oligodendrocytes at the protein levels (61). It was postulated that P2X7 is also expressed in neurons, but this was disproved by unbiased studies and the use of specific tools (197). Indeed, activation of P2X7 requires high level of ATP, which is found in neurodegeneration (198). It has been hypothesized that P2X7 acts as an early sensor, which represents a prerequisite for glial response to insults. However, chronic activation of P2X7 results in cell death, contributing to disease progression. Blocking P2X7 mitigates amyloid burden in AD models and improves synaptic plasticity, integrity and memory (94, 108, 199). Additionally, increased motor neuron survival and decreased microgliosis and inflammatory markers were shown after P2X7 inhibition at late pre-onset in SOD1 mutant mice (110). Furthermore, using P2X7- or P2Y6-selective antagonists, BBG or MRS2578 respectively, in an animal model of PD, neuroprotection and a reduced microglia reactive phenotype were observed (101, 200).
Adenosine receptors also represent suitable targets for glial modulation. Administration of the A2A receptor antagonist preladenant partly decreased ex vivo hyperactive motility around Aβ plaques in the 5xFAD amyloidosis model (89). In addition, preladenant restored microglial process extension toward tissue damage in the MPTP-induced PD model (201). Several studies have shown increased astrocytic A2A in pathological context such as AD, which suggests an abnormal function (166, 167). Additionally, A2A has also been studied as a therapeutical target in neuroinflammatory conditions involving myelin loss and T cell activation (202). Altogether, these results suggest that adenosine receptors like A2A could be targeted therapeutically to improve the disease-associated phenotype of glial cells in proteinopathies.
Apart from purinergic receptors, several studies have highlighted elevated astrocytic CX43 in neurodegenerative diseases, leading to increased ATP release. Together with other nucleotide transporter such as pannexin-1, they could be targets of choice in order to decrease ATP release and aberrant purinergic signaling in neurodegeneration (203).
Conclusion
As outlined here, purinergic signaling in neurodegeneration is not only altered in neurons, but in all CNS cell types, including glial cells. This highlights the potential to target purinergic signaling in a multi-cellular fashion. However, to develop this as a valuable strategy in the future, many functional aspects of purinergic signaling in glial cells need to be further elucidated. In particular, purinergic signaling in astrocytes and oligodendrocytes have remained ill-defined, as well as purinergic signaling pathways in proteinopathies involving FUS or TDP-43 aggregates. Furthermore, novel tools are needed that help to better define the cell-specific and the spatio-temporal aspect of purinergic signaling in neurodegeneration. These challenges will need to be faced in the future to better understand this fascinating system, as within it potentially lies the hopes of tomorrow's treatments.
Author Contributions
MP, AG, SK, AH, and KC wrote the manuscript. MP, AG, SK, DB, AH, and KC reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
AG was supported by a DAAD fellowship of the German Egyptian Research Long-Term Scholarship Program (GERLS). DB was supported by grants from ANR (ADORASTrAU and METABOTAU), CoEN 5008, and Programs d'Investissements d'Avenir LabEx (excellence laboratory) DISTALZ (Development of Innovative Strategies for a Transdisciplinary approach to ALZheimer's disease). The lab of DB is also supported by Fondation pour la Recherche Médicale, France Alzheimer/Fondation de France, FHU VasCog research network (Lille, France), Fondation Vaincre Alzheimer, Fondation Plan Alzheimer as well as Inserm, CNRS, Université Lille, Lille Métropole Communauté Urbaine, DN2M. AH was supported by the German Center of Neurodegenerative Diseases (DZNE), by Helmholtz Grant Funding (ExNet-0008-Phase2-3) and CoEN 5008.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to BooBoo Tannenbaum for her artistic support with Figure 1.
References
1. Arvanitakis Z, Shah RC, Bennett DA. Diagnosis and management of dementia: review. JAMA. (2019) 322:1589–99. doi: 10.1001/jama.2019.4782
2. Kovacs GG. Molecular pathology of neurodegenerative diseases: principles and practice. J Clin Pathol. (2019) 72:725–35. doi: 10.1136/jclinpath-2019-205952
3. Karch CM, Goate AM. Alzheimer's disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. (2015) 77:43–51. doi: 10.1016/j.biopsych.2014.05.006
4. Heneka MT, Carson MJ, Khoury JEl, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. (2015) 14:388–405. doi: 10.1016/S1474-4422(15)70016-5
5. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer's disease. Cell. (2017) 169:1276–90.e17. doi: 10.1016/j.cell.2017.05.018
6. Escartin C, Galea E, Lakatos A, O'Callaghan JP, Petzold GC, Serrano-Pozo A, et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. (2021) 24:312–25. doi: 10.1038/s41593-020-00783-4
7. Zahid A, Li B, Kombe AJK, Jin T, Tao J. Pharmacological inhibitors of the nlrp3 inflammasome [Internet]. Front Immunol. (2019) 10:2538. doi: 10.3389/fimmu.2019.02538
8. Lindsay A, Hickman D, Srinivasan M. A nuclear factor-kappa B inhibiting peptide suppresses innate immune receptors and gliosis in a transgenic mouse model of Alzheimer's disease. Biomed Pharmacother. (2021) 138:111405. doi: 10.1016/j.biopha.2021.111405
9. Lewcock JW, Schlepckow K, Di Paolo G, Tahirovic S, Monroe KM, Haass C. Emerging microglia biology defines novel therapeutic approaches for Alzheimer's disease. Neuron. Cell Press. (2020) 108:801–21. doi: 10.1016/j.neuron.2020.09.029
10. Calzaferri F, Ruiz-Ruiz C, de Diego AMG, de Pascual R, Méndez-López I, Cano-Abad MF, et al. The purinergic P2X7 receptor as a potential drug target to combat neuroinflammation in neurodegenerative diseases. Med Res Rev. (2020) 40:2427–65. doi: 10.1002/med.21710
11. Agostinho P, Madeira D, Dias L, Simões AP, Cunha RA, Canas PM. Purinergic signaling orchestrating neuron-glia communication. Pharmacol Res. (2020) 162:105253. doi: 10.1016/j.phrs.2020.105253
12. Burnstock G. Introduction to purinergic signalling in the brain. In: Advances in Experimental Medicine and Biology. Dordrecht: Springer. (2020). p. 1–12.
14. Zimmermann H, Zebisch M, Sträter N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. (2012) 8:437–502. doi: 10.1007/s11302-012-9309-4
15. Young JD. The SLC28 (CNT) and SLC29 (ENT) nucleoside transporter families: a 30-year collaborative odyssey. Biochem Soc Trans. (2016) 44:869–76. doi: 10.1042/BST20160038
16. Burnstock G, Verkhratsky A, Burnstock G, Verkhratsky A. Mechanisms of ATP release and inactivation. In: Purinergic Signalling and the Nervous System. Berlin: Springer (2012). p. 79–118.
17. Burnstock G. Purinergic receptors. J Theor Biol. (1976) 62:491–503. doi: 10.1016/0022-5193(76)90133-8
18. Burnstock G. A basis for distinguishing two types of purinergic receptor. Cell Membr Recept Drugs Horm A Multidiscip Approach. (1978) 107–18.
19. Knospe M, Müller CE, Rosa P, Abdelrahman A, von Kügelgen I, Thimm D, et al. The rat adenine receptor: pharmacological characterization and mutagenesis studies to investigate its putative ligand binding site. Purinergic Signal. (2013) 9:367–81. doi: 10.1007/s11302-013-9355-6
20. Thimm D, Knospe M, Abdelrahman A, Moutinho M, Alsdorf BBA, von Kügelgen I, et al. Characterization of new G protein-coupled adenine receptors in mouse and hamster. Purinergic Signal. (2013) 9:415–26. doi: 10.1007/s11302-013-9360-9
21. Crain JM, Nikodemova M, Watters JJ. Expression of P2 nucleotide receptors varies with age and sex in murine brain microglia. J Neuroinflammation. (2009) 6:24. doi: 10.1186/1742-2094-6-24
22. Lai AY, Dibal CD, Armitage GA, Winship IR, Todd KG. Distinct activation profiles in microglia of different ages: a systematic study in isolated embryonic to aged microglial cultures. Neuroscience. (2013) 254:185–95. doi: 10.1016/j.neuroscience.2013.09.010
23. Bianco F, Pravettoni E, Colombo A, Schenk U, Möller T, Matteoli M, et al. Astrocyte-derived ATP induces vesicle shedding and IL-1β release from microglia. J Immunol. (2005) 174:7268–77. doi: 10.4049/jimmunol.174.11.7268
24. Lim HM, Heo W, Han JW, Lee MG, Kim JY. NPP1 is responsible for potent extracellular ATP hydrolysis as NTPDase1 in primary cultured murine microglia. Purinergic Signal. (2018) 14:157–66. doi: 10.1007/s11302-018-9601-z
25. Wink MR, Braganhol E, Tamajusuku ASK, Lenz G, Zerbini LF, Libermann TA, et al. Nucleoside triphosphate diphosphohydrolase-2 (NTPDase2/CD39L1) is the dominant ectonucleotidase expressed by rat astrocytes. Neuroscience. (2006) 138:421–32. doi: 10.1016/j.neuroscience.2005.11.039
26. Fam SR, Gallagher CJ, Salter MW. P2Y1 purinoceptor-mediated Ca2+ signaling and Ca2+ wave propagation in dorsal spinal cord astrocytes. J Neurosci. (2000) 20:2800–8. doi: 10.1523/JNEUROSCI.20-08-02800.2000
27. Hashioka S, Wang YF, Little JP, Choi HB, Klegeris A, McGeer PL, et al. Purinergic responses of calcium-dependent signaling pathways in cultured adult human astrocytes. BMC Neurosci. (2014) 15:18. doi: 10.1186/1471-2202-15-18
28. Agresti C, Meomartini ME, Amadio S, Ambrosini E, Volonté C, Aloisi F, et al. ATP regulates oligodendrocyte progenitor migration, proliferation, and differentiation: involvement of metabotropic P2 receptors. In: Brain Research Reviews. San Diego, CA: Elsevier (2005). p. 157–65.
29. Othman T, Yan H, Rivkees SA. Oligodendrocytes express functional A1 adenosine receptors that stimulate cellular migration. Glia. (2003) 44:166–72. doi: 10.1002/glia.10281
30. Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. (2014) 34:11929–47. doi: 10.1523/JNEUROSCI.1860-14.2014
31. Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. (2016) 89:37–53. doi: 10.1016/j.neuron.2015.11.013
32. Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, Okamoto J, et al. Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron. (2019) 101:207–23.e10. doi: 10.1016/j.neuron.2018.12.006
33. Ohsawa K, Sanagi T, Nakamura Y, Suzuki E, Inoue K, Kohsaka S. Adenosine A3 receptor is involved in ADP-induced microglial process extension and migration. J Neurochem. (2012) 121:217–27. doi: 10.1111/j.1471-4159.2012.07693.x
34. Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. (2006) 9:1512–9. doi: 10.1038/nn1805
35. Kyrargyri V, Madry C, Rifat A, Arancibia-Carcamo IL, Jones SP, Chan VTT, et al. P2Y 13 receptors regulate microglial morphology, surveillance, and resting levels of interleukin 1β release. Glia. (2020) 68:328–44. doi: 10.1002/glia.23719
36. Ohsawa K, Irino Y, Nakamura Y, Akazawa C, Inoue K, Kohsaka S. Involvement of P2X 4 and P2Y 12 receptors in ATP-induced microglial chemotaxis. Glia. (2007) 55:604–16. doi: 10.1002/glia.20489
37. Shieh CH, Heinrich A, Serchov T, van Calker D, Biber K. P2X7-dependent, but differentially regulated release of IL-6, CCL2, and TNF-α in cultured mouse microglia. Glia. (2014) 62:592–607. doi: 10.1002/glia.22628
38. Hung SC, Choi CH, Said-Sadier N, Johnson L, Atanasova KR, Sellami H, et al. P2X4 Assembles with P2X7 and Pannexin-1 in Gingival Epithelial Cells and Modulates ATP-induced Reactive Oxygen Species Production and Inflammasome Activation. PLoS ONE. (2013) 8:70210. doi: 10.1371/journal.pone.0070210
39. Koizumi S, Shigemoto-Mogami Y, Nasu-Tada K, Shinozaki Y, Ohsawa K, Tsuda M, et al. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature. (2007) 446:1091–5. doi: 10.1038/nature05704
40. Cox MA, Gomes B, Palmer K, Du K, Wiekowski M, Wilburn B, et al. The pyrimidinergic P2Y6 receptor mediates a novel release of proinflammatory cytokines and chemokines in monocytic cells stimulated with UDP. Biochem Biophys Res Commun. (2005) 330:467–73. doi: 10.1016/j.bbrc.2005.03.004
41. Badimon A, Strasburger HJ, Ayata P, Chen X, Nair A, Ikegami A, et al. Negative feedback control of neuronal activity by microglia. Nature. (2020) 586:417–23. doi: 10.1038/s41586-020-2777-8
42. Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 Receptor in Infection and Inflammation. Immunity. (2017) 47:15–31. doi: 10.1016/j.immuni.2017.06.020
43. Sitkovsky MV, Lukashev D, Apasov S, Kojima H, Koshiba M, Caldwell C, et al. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu Rev Immunol. (2004) 22:657–82. doi: 10.1146/annurev.immunol.22.012703.104731
44. Wang DD, Bordey A. The astrocyte odyssey. Prog Neurobiol. (2008) 86:342–67. doi: 10.1016/j.pneurobio.2008.09.015
45. Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on Astrocyte-neuron metabolic cooperation. Cell Metabolism. (2011) 14:724–38. doi: 10.1016/j.cmet.2011.08.016
46. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. (2010) 119:7–35. doi: 10.1007/s00401-009-0619-8
47. Verkhratsky A, Nedergaard M. Physiology of astroglia. Physiol Rev. (2018) 98:239–389. doi: 10.1152/physrev.00042.2016
48. Bazargani N, Attwell D. Astrocyte calcium signaling: the third wave. Nat Neurosci. (2016) 19:182–9. doi: 10.1038/nn.4201
49. Matos M, Augusto E, MacHado NJ, Dos Santos-Rodrigues A, Cunha RA, Agostinho P. Astrocytic adenosine A2A receptors control the amyloid-β peptide-induced decrease of glutamate uptake. J Alzheimer's Dis. (2012) 31:555–67. doi: 10.3233/JAD-2012-120469
50. Cristóvão-Ferreira S, Navarro G, Brugarolas M, Pérez-Capote K, Vaz SH, Fattorini G, et al. A1R-A2AR heteromers coupled to Gs and Gi/0 proteins modulate GABA transport into astrocytes. Purinergic Signal. (2013) 9:433–49. doi: 10.1007/s11302-013-9364-5
51. Philips T, Rothstein JD. Oligodendroglia: metabolic supporters of neurons. J Clin Invest. (2017) 127:3271–80. doi: 10.1172/JCI90610
52. Chen J, Tan Z, Zeng L, Zhang X, He Y, Gao W, et al. Heterosynaptic long-term depression mediated by ATP released from astrocytes. Glia. (2013) 61:178–91. doi: 10.1002/glia.22425
53. Cunha RA. Different cellular sources and different roles of adenosine: A1 receptor-mediated inhibition through astrocytic-driven volume transmission and synapse-restricted A2A receptor-mediated facilitation of plasticity. Neurochem Int. (2008) 52:65–72. doi: 10.1016/j.neuint.2007.06.026
54. Gonçalves FQ, Lopes JP, Silva HB, Lemos C, Silva AC, Gonçalves N, et al. Synaptic and memory dysfunction in a β-amyloid model of early Alzheimer's disease depends on increased formation of ATP-derived extracellular adenosine. Neurobiol Dis. (2019) 132:104570. doi: 10.1016/j.nbd.2019.104570
55. Cunha RA. How does adenosine control neuronal dysfunction and neurodegeneration? J Neurochem. (2016) 139:1019–55. doi: 10.1111/jnc.13724
56. Alonso-Andrés P, Albasanz JL, Ferrer I, Martín M. Purine-related metabolites and their converting enzymes are altered in frontal, parietal and temporal cortex at early stages of Alzheimer's disease pathology. Brain Pathol. (2018) 28:933–46. doi: 10.1111/bpa.12592
57. Ng S kah, Higashimori H, Tolman M, Yang Y.Suppression of adenosine 2a receptor (A2aR)-mediated adenosine signaling improves disease phenotypes in a mouse model of amyotrophic lateral sclerosis. Exp Neurol. (2015) 267:115–22. doi: 10.1016/j.expneurol.2015.03.004
58. Rothman SM, Tanis KQ, Gandhi P, Malkov V, Marcus J, Pearson M, et al. Human Alzheimer's disease gene expression signatures and immune profile in APP mouse models: a discrete transcriptomic view of Aβ plaque pathology. J Neuroinflammation. (2018) 15:256. doi: 10.1186/s12974-018-1265-7
59. Liu W, Venugopal S, Majid S, Ahn IS, Diamante G, Hong J. Single-cell RNA-seq analysis of the brainstem of mutant SOD1 mice reveals perturbed cell types and pathways of amyotrophic lateral sclerosis. Neurobiol Dis. (2020) 141:104877. doi: 10.1016/j.nbd.2020.104877
60. Sobue A, Komine O, Hara Y, Endo F, Mizoguchi H, Watanabe S, et al. Microglial gene signature reveals loss of homeostatic microglia associated with neurodegeneration of Alzheimer's disease. Acta Neuropathol Commun. (2021) 9:1. doi: 10.1186/s40478-020-01099-x
61. Lau SF, Cao H, Fu AKY, Ip NY. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer's disease. Proc Natl Acad Sci USA. (2020) 117:25800–9. doi: 10.1073/pnas.2008762117
62. Orre M, Kamphuis W, Osborn LM, Melief J, Kooijman L, Huitinga I, et al. Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol Aging. (2014) 35:1–14. doi: 10.1016/j.neurobiolaging.2013.07.008
63. Chiu IM, Morimoto ETA, Goodarzi H, Liao JT, O'Keeffe S, Phatnani HP, et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. (2013) 4:385–401. doi: 10.1016/j.celrep.2013.06.018
64. Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell. (2015) 160:1061–71. doi: 10.1016/j.cell.2015.01.049
65. Srinivasan K, Friedman BA, Larson JL, Lauffer BE, Goldstein LD, Appling LL, et al. Untangling the brain's neuroinflammatory and neurodegenerative transcriptional responses. Nat Commun. (2016) 7:11295. doi: 10.1038/ncomms11295
66. Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, et al. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer's disease not evident in mouse models. Cell Rep. (2018) 22:832–47. doi: 10.1016/j.celrep.2017.12.066
67. Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M, et al. Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep. (2017) 21:366–80. doi: 10.1016/j.celrep.2017.09.039
68. Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer's disease. Nat Med. (2020) 26:131–42. doi: 10.1038/s41591-019-0695-9
69. Orre M, Kamphuis W, Osborn LM, Jansen AHP, Kooijman L, Bossers K, et al. Isolation of glia from Alzheimer's mice reveals inflammation anddysfunction. Neurobiol Aging. (2014) 35:2746–60. doi: 10.1016/j.neurobiolaging.2014.06.004
70. Kamphuis W, Kooijman L, Orre M, Stassen O, Pekny M, Hol EM. GFAP and vimentin deficiency alters gene expression in astrocytes and microglia in wild-type mice and changes the transcriptional response of reactive glia in mouse model for Alzheimer's disease. Glia. (2015) 63:1036–56. doi: 10.1002/glia.22800
71. Miller SJ, Glatzer JC, Hsieh Y, chun Rothstein JD. Cortical astroglia undergo transcriptomic dysregulation in the G93A SOD1 ALS mouse model. J Neurogenet. (2018) 32:322–35. doi: 10.1080/01677063.2018.1513508
72. Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, et al. Single-cell transcriptomic analysis of Alzheimer's disease. Nature. (2019) 570:332–7. doi: 10.1038/s41586-019-1195-2
73. Habib N, Mccabe C, Medina S, Varshavsky M, Kitsberg D, Dvir-szternfeld R, et al. Disease-associated astrocytes in Alzheimer ' s disease and aging. Nat Neurosci. (2020) 23:701–6. doi: 10.1038/s41593-020-0624-8
74. Grubman A, Chew G, Ouyang JF, Sun G, Choo XY, McLean C, et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer's disease reveals cell-type-specific gene expression regulation. Nat Neurosci. (2019) 22:2087–97. doi: 10.1038/s41593-019-0539-4
75. Butovsky O, Jedrychowski MP, Cialic R, Krasemann S, Murugaiyan G, Fanek Z, et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann Neurol. (2015) 77:75–99. doi: 10.1002/ana.24304
76. Lee S-H, Meilandt WJ, Xie L, Gandham VD, Ngu H, Barck KH, et al. Trem2 restrains the enhancement of tau accumulation and neurodegeneration by β-amyloid pathology. Neuron. (2021) 9:1283–1301.e6. doi: 10.1016/j.neuron.2021.02.010
77. Monasor LS, Müller SA, Colombo AV, Tanrioever G, König J, Roth S, et al. Fibrillar aβ triggers microglial proteome alterations and dysfunction in alzheimer mouse models. Elife. (2020) 9:1–33. doi: 10.7554/eLife.54083
78. Sala Frigerio C, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, et al. The major risk factors for Alzheimer's disease: age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep. (2019) 27:1293–306.e6. doi: 10.1016/j.celrep.2019.03.099
79. Li W, Silva HB, Real J, Wang YM, Rial D, Li P, et al. Inactivation of adenosine A2A receptors reverses working memory deficits at early stages of Huntington's disease models. Neurobiol Dis. (2015) 79:70–80. doi: 10.1016/j.nbd.2015.03.030
80. Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, et al. Decreased clearance of CNS amyloid-β in Alzheimer's disease. Science. (2010) 330:1774. doi: 10.1126/science.1197623
81. Hickman SE, Allison EK, Khoury JEl. Neurobiology of disease microglial dysfunction and defective-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. (2008) 28:8354–60. doi: 10.1523/JNEUROSCI.0616-08.2008
82. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat Immunol. (2008) 9:857–65. doi: 10.1038/ni.1636
83. Deora V, Lee JD, Albornoz EA, McAlary L, Jagaraj CJ, Robertson AAB, et al. The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia. (2020) 68:407–21. doi: 10.1002/glia.23728
84. Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. (2018) 21:1359–69. doi: 10.1038/s41593-018-0242-x
85. Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. (2013) 4:1562. doi: 10.1038/ncomms2534
86. Muth C, Hartmann A, Sepulveda-Falla D, Glatzel M, Krasemann S. Phagocytosis of apoptotic cells is specifically upregulated in apoe4 expressing microglia in vitro. Front Cell Neurosci. (2019) 13:181. doi: 10.3389/fncel.2019.00181
87. Krabbe G, Halle A, Matyash V, Rinnenthal JL, Eom GD, Bernhardt U, et al. Functional impairment of microglia coincides with beta-amyloid deposition in mice with alzheimer-like pathology. PLoS ONE. (2013) 8:e60921. doi: 10.1371/journal.pone.0060921
88. Walker DG, Tang TM, Mendsaikhan A, Tooyama I, Serrano GE, Sue LI, et al. Patterns of expression of purinergic receptor P2RY12, a putative marker for non-activated microglia, in aged and alzheimer's disease brains. Int J Mol Sci. (2020) 21:678. doi: 10.3390/ijms21020678
89. Gyoneva S, Swanger SA, Zhang J, Weinshenker D, Traynelis SF. Altered motility of plaque-associated microglia in a model of Alzheimer's disease. Neuroscience. (2016) 330:410–20. doi: 10.1016/j.neuroscience.2016.05.061
90. Plescher M, Seifert G, Hansen JN, Bedner P, Steinhäuser C, Halle A. Plaque-dependent morphological and electrophysiological heterogeneity of microglia in an Alzheimer's disease mouse model. Glia. (2018) 66:1464–80. doi: 10.1002/glia.23318
91. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. (2017) 47:566–81.e9. doi: 10.1016/j.immuni.2017.08.008
92. Agarwal D, Sandor C, Volpato V, Caffrey TM, Monzón-Sandoval J, Bowden R, et al. A single-cell atlas of the human substantia nigra reveals cell-specific pathways associated with neurological disorders. Nat Commun. (2020) 11:4183. doi: 10.1038/s41467-020-17876-0
93. Martínez-Frailes C, Di Lauro C, Bianchi C, De Diego-García L, Sebastián-Serrano Á, Boscá L, et al. Amyloid peptide induced neuroinflammation increases the P2X7 receptor expression in microglial cells, impacting on its functionality. Front Cell Neurosci. (2019) 13:1–15. doi: 10.3389/fncel.2019.00143
94. Martin E, Amar M, Dalle C, Youssef I, Boucher C, Le Duigou C, et al. New role of P2X7 receptor in an Alzheimer's disease mouse model. Mol Psychiatry. (2019) 24:108–25. doi: 10.1038/s41380-018-0108-3
95. Qin J, Zhang X, Wang Z, Li J, Zhang Z, Gao L, et al. Presenilin 2 deficiency facilitates Aβ-induced neuroinflammation and injury by upregulating P2X7 expression. Sci China Life Sci. (2017) 60:189–201. doi: 10.1007/s11427-016-0347-4
96. Lee HG, Won SM, Gwag BJ, Lee YB. Microglial P2X7 receptor expression is accompanied by neuronal damage in the cerebral cortex of the APPswe/PS1dE9 mouse model of alzheimer's disease. Exp Mol Med. (2011) 43:7–14. doi: 10.3858/emm.2011.43.1.001
97. McLarnon JG, Ryu JK, Walker DG, Choi HB. Upregulated expression of purinergic P2X7 receptor in Alzheimer disease and amyloid-β peptide-treated microglia and in peptide-injected rat hippocampus. J Neuropathol Exp Neurol. (2006) 65:1090–7. doi: 10.1097/01.jnen.0000240470.97295.d3
98. Parvathenani LK, Tertyshnikova S, Greco CR, Roberts SB, Robertson B, Posmantur R. P2X7 mediates superoxide production in primary microglia and is up-regulated in a transgenic mouse model of Alzheimer's disease. J Biol Chem. (2003) 278:13309–17. doi: 10.1074/jbc.M209478200
99. Yiangou Y, Facer P, Durrenberger P, Chessell IP, Naylor A, Bountra C, et al. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. (2006) 6:1–14. doi: 10.1186/1471-2377-6-12
100. D'Ambrosi N, Finocchi P, Apolloni S, Cozzolino M, Ferri A, Padovano V, et al. The proinflammatory action of microglial P2 receptors is enhanced in SOD1 models for amyotrophic lateral sclerosis. J Immunol. (2009) 183:4648–56. doi: 10.4049/jimmunol.0901212
101. Oliveira-Giacomelli ÁM. Albino C, de Souza HDN, Corrêa-Velloso J, de Jesus Santos AP, Baranova J, et al. P2Y6 and P2X7 receptor antagonism exerts neuroprotective/ neuroregenerative effects in an animal model of Parkinson's disease. Front Cell Neurosci. (2019) 13:476. doi: 10.3389/fncel.2019.00476
102. Soo YK, Ju HM, Hwan GL, Seung UK, Yong BL. ATP released from β-amyloid-stimulated microglia induces reactive oxygen species production in an autocrine fashion. Exp Mol Med. (2007) 39:820–7. doi: 10.1038/emm.2007.89
103. Kim HJ, Ajit D, Peterson TS, Wang Y, Camden JM, Wood WG, et al. Nucleotides released from Aβ 1-42-treated microglial cells increase cell migration and Aβ 1-42 uptake through P2Y 2 receptor activation. J Neurochem. (2012) 121:228–38. doi: 10.1111/j.1471-4159.2012.07700.x
104. Narcisse L, Scemes E, Zhao Y, Lee SC, Brosnan CF. The cytokine IL-1β transiently enhances P2X7 receptor expression and function in human astrocytes. Glia. (2005) 49:245–58. doi: 10.1002/glia.20110
105. Chiozzi P, Sarti AC, Sanz JM, Giuliani AL, Adinolfi E, Vultaggio-Poma V, et al. Amyloid β-dependent mitochondrial toxicity in mouse microglia requires P2X7 receptor expression and is prevented by nimodipine. Sci Rep. (2019) 9:1–15. doi: 10.1038/s41598-019-42931-2
106. Rampe D, Wang L, Ringheim GE. P2X7 receptor modulation of β-amyloid- and LPS-induced cytokine secretion from human macrophages and microglia. J Neuroimmunol. (2004) 147:56–61. doi: 10.1016/j.jneuroim.2003.10.014
107. Sanz JM, Chiozzi P, Ferrari D, Colaianna M, Idzko M, Falzoni S, et al. Activation of microglia by amyloid β requires P2X 7 receptor expression. J Immunol. (2009) 182:4378–85. doi: 10.4049/jimmunol.0803612
108. Diaz-Hernandez JI, Gomez-Villafuertes R, León-Otegui M, Hontecillas-Prieto L, del Puerto A, Trejo JL, et al. In vivo P2X7 inhibition reduces amyloid plaques in Alzheimer's disease through GSK3β and secretases. Neurobiol Aging. (2012) 33:1816–28. doi: 10.1016/j.neurobiolaging.2011.09.040
109. Apolloni S, Parisi C, Pesaresi MG, Rossi S, Carrì MT, Cozzolino M, et al. The NADPH oxidase pathway is dysregulated by the P2X 7 receptor in the SOD1-G93A microglia model of amyotrophic lateral sclerosis. J Immunol. (2013) 190:5187–95. doi: 10.4049/jimmunol.1203262
110. Apolloni S, Amadio S, Parisi C, Matteucci A, Potenza RL, Armida M, et al. Spinal cord pathology is ameliorated by P2X7 antagonism in a SOD1-mutant mouse model of amyotrophic lateral sclerosis. DMM Dis Model Mech. (2014) 7:1101–9. doi: 10.1242/dmm.017038
111. Jiang T, Hoekstra J, Heng X, Kang W, Ding J, Liu J, et al. P2X7 receptor is critical in α-synuclein-mediated microglial NADPH oxidase activation. Neurobiol Aging. (2015) 36:2304–18. doi: 10.1016/j.neurobiolaging.2015.03.015
112. Apolloni S, Amadio S, Montilli C, Volonté C, D'ambrosi N. Ablation of p2X7 receptor exacerbates gliosis and motoneuron death in the SOD1-G93A mouse model of amyotrophic lateral sclerosis. Hum Mol Genet. (2013) 22:4102–16. doi: 10.1093/hmg/ddt259
113. Volonté C, Apolloni S, Parisi C, Amadio S. Purinergic contribution to amyotrophic lateral sclerosis. Neuropharmacology. (2016) 104:180–93. doi: 10.1016/j.neuropharm.2015.10.026
114. Färber K, Markworth S, Pannasch U, Nolte C, Prinz V, Kronenberg G, et al. The ectonucleotidase cd39/ENTPDase1 modulates purinergic-mediated microglial migration. Glia. (2008) 56:331–41. doi: 10.1002/glia.20606
115. Luongo L, Guida F, Imperatore R, Napolitano F, Gatta L, Cristino L, et al. The A1 adenosine receptor as a new player in microglia physiology. Glia. (2014) 62:122–32. doi: 10.1002/glia.22592
116. Orr AG, Orr AL, Li X-J, Gross RE, Traynelis SF, Neurosci N. Adenosine A 2A receptor mediates microglial process retraction. Nat Neurosci. (2009) 12:872–8. doi: 10.1038/nn.2341
117. Fiebich BL, Biber K, Lieb K, Van Calker D, Berger M, Bauer J, et al. Cyclooxygenase-2 expression in rat microglia is induced by adenosine A2a-receptors. Glia. (1996) 18:152–80. doi: 10.1002/(SICI)1098-1136(199610)18:2 <152::AID-GLIA7>3.0.CO;2-2
118. Gomes C, Ferreira R, George J, Sanches R, Rodrigues DI, Gonçalves N, et al. Activation of microglial cells triggers a release of brain-derived neurotrophic factor (BDNF) inducing their proliferation in an adenosine A 2A receptor-dependent manner: a 2A receptor blockade prevents BDNF release and proliferation of microglia. J Neuroinflammation. (2013) 10:16. doi: 10.1186/1742-2094-10-16
119. Angulo E, Casadó V, Mallol J, Canela EI, Viñals F, Ferrer I, et al. A1 adenosine receptors accumulate in neurodegenerative structures in Alzheimer disease and mediate both amyloid precursor protein processing and tau phosphorylation and translocation. Brain Pathol. (2003) 13:440–51. doi: 10.1111/j.1750-3639.2003.tb00475.x
120. Mildner A, Huang H, Radke J, Stenzel W, Priller J. P2Y12 receptor is expressed on human microglia under physiological conditions throughout development and is sensitive to neuroinflammatory diseases. Glia. (2017) 65:375–87. doi: 10.1002/glia.23097
121. Eyo UB, Peng J, Swiatkowski P, Mukherjee A, Bispo A, Wu LJ. Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus. J Neurosci. (2014) 34:10528–40. doi: 10.1523/JNEUROSCI.0416-14.2014
122. Vossel KA, Tartaglia MC, Nygaard HB, Zeman AZ, Miller BL. Epileptic activity in Alzheimer's disease: causes and clinical relevance. Lancet Neurol. (2017) 16:311–22. doi: 10.1016/S1474-4422(17)30044-3
123. Ferrini F, Trang T, Mattioli TAM, Laffray S, Del'Guidice T, Lorenzo LE, et al. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl-homeostasis. Nat Neurosci. (2013) 16:183–92. doi: 10.1038/nn.3295
124. Coull JAM, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. (2005) 438:1017–21. doi: 10.1038/nature04223
125. Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, et al. BDNF-induced TrkB activation down-regulates the K-Cl cotransporter KCC2 and impairs neuronal Cl extrusion. J Cell Biol. (2002) 159:747–52. doi: 10.1083/jcb.200209011
126. Ulmann L, Hatcher JP, Hughes JP, Chaumont S, Green PJ, Conquet F, et al. Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J Neurosci. (2008) 28:11263–8. doi: 10.1523/JNEUROSCI.2308-08.2008
127. Trang T, Beggs S, Wan X, Salter MW. P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J Neurosci. (2009) 29:3518–28. doi: 10.1523/JNEUROSCI.5714-08.2009
128. Jin Xhong, Wang LN, Zuo JL, Yang JP, Liu Slan. P2X4 receptor in the dorsal horn partially contributes to brain-derived neurotrophic factor oversecretion and toll-like receptor-4 receptor activation associated with bone cancer pain. J Neurosci Res. (2014) 92:1690–702. doi: 10.1002/jnr.23443
129. Zabala A, Vazquez-Villoldo N, Rissiek B, Gejo J, Martin A, Palomino A, et al. P2X4 receptor controls microglia activation and favors remyelination in autoimmune encephalitis. EMBO Mol Med. (2018) 10:e8743. doi: 10.15252/emmm.201708743
130. Long T, He W, Pan Q, Zhang S, Zhang D, Qin G, et al. Microglia P2X4R-BDNF signalling contributes to central sensitization in a recurrent nitroglycerin-induced chronic migraine model. J Headache Pain. (2020) 21:4. doi: 10.1186/s10194-019-1070-4
131. Buchin A, Chizhov A, Huberfeld G, Miles R, Boris X, Gutkin S. Neurobiology of disease reduced efficacy of the KCC2 cotransporter promotes epileptic oscillations in a subiculum network model. J Neurosci. (2016) 36:11619–33. doi: 10.1523/JNEUROSCI.4228-15.2016
132. Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, et al. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci. (2007) 27:9866–73. doi: 10.1523/JNEUROSCI.2761-07.2007
133. Ferrini F, De Koninck Y. Microglia control neuronal network excitability via BDNF signalling. Neural Plast. (2013) 2013:11. doi: 10.1155/2013/429815
134. Binder DK, Routbort MJ, McNamara JO. Immunohistochemical evidence of seizure-induced activation of trk receptors in the mossy fiber pathway of adult rat hippocampus. J Neurosci. (1999) 19:4616–26. doi: 10.1523/JNEUROSCI.19-11-04616.1999
135. Bernier L-P, Ase AR, Boué-Grabot É, Séguéla P. Inhibition of P2X4 function by P2Y6 UDP receptors in microglia. Glia. (2013) 61:2038–49. doi: 10.1002/glia.22574
136. Neher JJ, Neniskyte U, Hornik T, Brown GC. Inhibition of UDP/P2Y6 purinergic signaling prevents phagocytosis of viable neurons by activated microglia in vitro and in vivo. Glia. (2014) 62:1463–75. doi: 10.1002/glia.22693
137. Wendt S, Maricos M, Vana N, Meyer N, Guneykaya D, Semtner M, et al. Changes in phagocytosis and potassium channel activity in microglia of 5xFAD mice indicate alterations in purinergic signaling in a mouse model of Alzheimer's disease. Neurobiol Aging. (2017) 58:41–53. doi: 10.1016/j.neurobiolaging.2017.05.027
138. Yang X, Lou Y, Liu G, Wang X, Qian Y, Ding J, et al. Microglia P2Y6 receptor is related to Parkinson's disease through neuroinflammatory process. J Neuroinflammation. (2017) 14:1–12. doi: 10.1186/s12974-017-0795-8
139. Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. (2009) 461:282–6. doi: 10.1038/nature08296
140. Ajit D, Woods LT, Camden JM, Thebeau CN, El- FG, Glen WG, et al. Loss of P2Y2 nucleotide receptors enhances early pathology in the TgCRND8 mouse model of Alzheimer's disease. Mol Neurobiol. (2015) 49:1031–42. doi: 10.1007/s12035-013-8577-5
141. Lai MKP, Tan MGK, Kirvell S, Hobbs C, Lee J, Esiri MM, et al. Selective loss of P2Y2 nucleotide receptor immunoreactivity is associated with Alzheimer's disease neuropathology. J Neural Transm. (2008) 115:1165–72. doi: 10.1007/s00702-008-0067-y
142. Li Hq, Chen C, Dou Y, Wu Hj, Liu Yj, Lou HF, et al. P2Y4 Receptor-mediated pinocytosis contributes to amyloid beta-induced self-uptake by microglia. Mol Cell Biol. (2013) 33:4282–93. doi: 10.1128/MCB.00544-13
143. Braun N, Sévigny J, Robson SC, Enjyoji K, Guckelberger O, Hammer K, et al. Assignment of ecto-nucleoside triphosphate diphosphohydrolase-1/cd39 expression to microglia and vasculature of the brain. Eur J Neurosci. (2000) 12:4357–66. doi: 10.1046/j.1460-9568.2000.01342.x
144. Matyash M, Zabiegalov O, Wendt S, Matyash V, Kettenmann H. The adenosine generating enzymes CD39/CD73 control microglial processes ramification in the mouse brain. PLoS ONE. (2017) 12:e0175012. doi: 10.1371/journal.pone.0175012
145. Bulavina L, Szulzewsky F, Rocha A, Krabbe G, Robson SC, Matyash V, et al. NTPDase1 activity attenuates microglial phagocytosis. Purinergic Signal. (2013) 9:199–205. doi: 10.1007/s11302-012-9339-y
146. Osborn LM, Kamphuis W, Wadman WJ, Hol EM. Astrogliosis: an integral player in the pathogenesis of Alzheimer's disease. Prog Neurobiol. (2016) 144:121–41. doi: 10.1016/j.pneurobio.2016.01.001
147. Kashon ML, Ross GW, Callaghan JPO, Miller DB, Petrovitch H, Burchfiel CM, et al. Associations of cortical astrogliosis with cognitive performance and dementia status. J Alzheimers Dis. (2004) 6:595–604. doi: 10.3233/JAD-2004-6604
148. Verkhratsky A, Augusto-Oliveira M, Pivoriunas A, Popov A, Brazhe A, Semyanov A. Astroglial asthenia and loss of function, rather than reactivity, contribute to the ageing of the brain. Pflugers Archiv Eur J Physio. (2020) 473:753–74. doi: 10.1007/s00424-020-02465-3
149. Zolkipli-Cunningham Z, Naviaux JC, Nakayama T, Hirsch CM, Monk JM, Li K, et al. Metabolic and behavioral features of acute hyperpurinergia and the maternal immune activation mouse model of autism spectrum disorder. PLoS ONE. (2021) 16:e0248771. doi: 10.1371/journal.pone.0248771
150. Chisari M, Scuderi A, Ciranna L, Volsi GL, Licata F, Sortino MA. Purinergic P2Y1 Receptors control rapid expression of plasma membrane processes in hippocampal astrocytes. Mol Neurobiol. (2017) 54:4081–93. doi: 10.1007/s12035-016-9955-6
151. Delekate A, Füchtemeier M, Schumacher T, Ulbrich C, Foddis M, Petzold GC.Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer's disease mouse model. Nat Commun. (2014) 5:5422. doi: 10.1038/ncomms6422
152. Reichenbach N, Delekate A, Breithausen B, Keppler K, Poll S, Schulte T, et al. P2Y1 receptor blockade normalizes network dysfunction and cognition in an Alzheimer's disease model. J Exp Med. (2018) 215:1649–63. doi: 10.1084/jem.20171487
153. Kuboyama K, Harada H, Tozaki-Saitoh H, Tsuda M, Ushijima K, Inoue K.Astrocytic P2Y1 receptor is involved in the regulation of cytokine/chemokine transcription and cerebral damage in a rat model of cerebral ischemia. J Cereb Blood Flow Metab. (2011). 31:1930–41. doi: 10.1038/jcbfm.2011.49
154. Ansoleaga B, Jové M, Schlüter A, Garcia-Esparcia P, Moreno J, Pujol A, et al. Deregulation of purine metabolism in Alzheimer's disease. Neurobiol Aging. (2015) 36:68–80. doi: 10.1016/j.neurobiolaging.2014.08.004
155. Chen WT, Lu A, Craessaerts K, Pavie B, Sala Frigerio C, Corthout N, et al. Spatial transcriptomics and in situ sequencing to study Alzheimer's disease. Cell. (2020) 182:976–91.e19. doi: 10.1016/j.cell.2020.06.038
156. Kang J, Kang N, Lovatt D, Torres A, Zhao Z, Lin J, et al. Connexin 43 hemichannels are permeable to ATP. J Neurosci. (2008) 28:4702–11. doi: 10.1523/JNEUROSCI.5048-07.2008
157. He JT, Ll XY, Yang L, Zhao X.Astroglial connexins and cognition: memory formation or deterioration? Biosci Reports. (2020) 40:BSR20193510. doi: 10.1042/BSR20193510
158. Koulakoff A, Mei X, Orellana JA, Sáez JC, Giaume C. Glial connexin expression and function in the context of Alzheimer's disease. Biochim Biophys Acta. (2012) 1818:2048–57. doi: 10.1016/j.bbamem.2011.10.001
159. Orellana JA, Shoji KF, Abudara V, Ezan P, Amigou E, Sáez PJ, et al. Amyloid β-induced death in neurons involves glial and neuronal hemichannels. J Neurosci. (2011) 31:4962–77. doi: 10.1523/JNEUROSCI.6417-10.2011
160. Yi C, Mei X, Ezan P, Mato S, Matias I, Giaume C, et al. Astroglial connexin43 contributes to neuronal suffering in a mouse model of Alzheimer's disease. Cell Death Differ. (2016) 23:1691–701. doi: 10.1038/cdd.2016.63
161. Freitas-Andrade M, Naus CC.Astrocytes in neuroprotection and neurodegeneration: The role of connexin43 and pannexin1. Neuroscience. (2016) 323:207–21. doi: 10.1016/j.neuroscience.2015.04.035
162. Xie HY, Cui Y, Deng F, Feng JC. Connexin: a potential novel target for protecting the central nervous system? Neural Regen Res. (2015) 10:659–66. doi: 10.4103/1673-5374.155444
163. Garré JM, Retamal MA, Cassina P, Barbeito L, Bukauskas FF, Sáez JC, et al. FGF-1 induces ATP release from spinal astrocytes in culture and opens pannexin and connexin hemichannels. Proc Natl Acad Sci USA. (2010) 107:22659–64. doi: 10.1073/pnas.1013793107
164. Thakur P, Nehru B. Inhibition of neuroinflammation and mitochondrial dysfunctions by carbenoxolone in the rotenone model of Parkinson's disease. Mol Neurobiol. (2014) 51:209–19. doi: 10.1007/s12035-014-8769-7
165. Cunha C, Santos C, Gomes C, Fernandes A, Correia AM, Sebastião AM, et al. Downregulated Glia Interplay and Increased miRNA-155 as Promising Markers to Track ALS at an Early Stage. Mol Neurobiol. (2018) 55:4207–24. doi: 10.1007/s12035-017-0631-2
166. Orr AG, Hsiao EC, Wang MM, Ho K, Kim DH, Guo W, et al. Astrocytic adenosine receptor A2A and Gs -coupled signaling regulate memory. Nat Neurosci. (2015) 18:423–34. doi: 10.1038/nn.3930
167. Orr AG, Lo I, Schumacher H, Ho K, Gill M, Guo W, et al. Istradefylline reduces memory deficits in aging mice with amyloid pathology. Neurobiol Dis. (2018) 110:29–36. doi: 10.1016/j.nbd.2017.10.014
168. Paiva I, Carvalho K, Santos P, Cellai L, Pavlou MAS, Jain G, et al. A2AR-induced transcriptional deregulation in astrocytes: an in vitro study. Glia. (2019) 67:2329–42. doi: 10.1002/glia.23688
169. Ferrer I. Oligodendrogliopathy in neurodegenerative diseases with abnormal protein aggregates: the forgotten partner. Progress Neurobiol. (2018) 169:24–54. doi: 10.1016/j.pneurobio.2018.07.004
170. Stevens B, Porta S, Haak LL, Gallo V, Fields RD. Adenosine: a neuron-glial transmitter promoting myelination in the CNS in response to action potentials. Neuron. (2002) 36:855–68. doi: 10.1016/S0896-6273(02)01067-X
171. Agresti C, Meomartini ME, Amadio S, Ambrosini E, Serafini B, Franchini L, et al. Metabotropic P2 receptor activation regulates oligodendrocyte progenitor migration and development. Glia. (2005) 50:132–44. doi: 10.1002/glia.20160
172. Kastritsis CH, McCarthy KD. Oligodendroglial lineage cells express neuroligand receptors. Glia. (1993) 8:106–13. doi: 10.1002/glia.440080206
173. Mitew S, Kirkcaldie MTK, Vickers JC, Dickson TC. Focal demyelination in Alzheimer's disease and transgenic mouse models. Acta Neuropathol. (2010) 119:567–77. doi: 10.1007/s00401-010-0657-2
174. Feng JF, Gao XF, Pu Yyan, Burnstock G, Xiang Z, He C. P2X7 receptors and Fyn kinase mediate ATP-induced oligodendrocyte progenitor cell migration. Purinergic Signal. (2015) 11:361–9. doi: 10.1007/s11302-015-9458-3
175. Skaper SD, Debetto P, Giusti P. The P2X 7 purinergic receptor: from physiology to neurological disorders. FASEB J. (2010) 24:337–45. doi: 10.1096/fj.09-138883
176. Matute C. Glutamate and ATP signalling in white matter pathology. J Anat. (2011) 219:53–64. doi: 10.1111/j.1469-7580.2010.01339.x
177. Matute C, Torre I, Pérez-Cerdá F, Pérez-Samartín A, Alberdi E, Etxebarria E, et al. P2X7 receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J Neurosci. (2007) 27:9525–33. doi: 10.1523/JNEUROSCI.0579-07.2007
178. Wang LY, Cai WQ, Chen PH, Deng QY, Zhao CM. Downregulation of P2X7 receptor expression in rat oligodendrocyte precursor cells after hypoxia ischemia. Glia. (2009) 57:307–19. doi: 10.1002/glia.20758
179. Vejar S, Oyarzún JE, Retamal MA, Ortiz FC, Orellana JA. Connexin and pannexin-based channels in oligodendrocytes: implications in brain health and disease. Front Cell Neurosci. (2019) 13:3. doi: 10.3389/fncel.2019.00003
180. Angeli S, Kousiappa I, Stavrou M, Sargiannidou I, Georgiou E, Papacostas SS, et al. Altered Expression of Glial Gap Junction Proteins Cx43, Cx30, and Cx47 in the 5XFAD Model of Alzheimer's Disease. Front Neurosci. (2020) 14:582934. doi: 10.3389/fnins.2020.582934
181. Reyes JF, Sackmann C, Hoffmann A, Svenningsson P, Winkler J, Ingelsson M, et al. Binding of α-synuclein oligomers to Cx32 facilitates protein uptake and transfer in neurons and oligodendrocytes. Acta Neuropathol. (2019) 138:23–47. doi: 10.1007/s00401-019-02007-x
182. Welsh TG, Kucenas S. Purinergic signaling in oligodendrocyte development and function. J Neurochem. (2018) 145:6–18. doi: 10.1111/jnc.14315
183. Lanser AJ, Rezende RM, Rubino S, Lorello PJ, Donnelly DJ, Xu H, et al. Disruption of the ATP/adenosine balance in CD39–/– mice is associated with handling-induced seizures. Immunology. (2017) 152:589–601. doi: 10.1111/imm.12798
184. Choi HB, Ryu JK, Kim SU, McLarnon JG. Modulation of the purinergic P2X7 receptor attenuates lipopolysaccharide-mediated microglial activation and neuronal damage in inflamed brain. J Neurosci. (2007) 27:4957–68. doi: 10.1523/JNEUROSCI.5417-06.2007
185. Fan X, Ma W, Zhang Y, Zhang L. P2X7 Receptor (P2X7R) of microglia mediates neuroinflammation by regulating (NOD)-like receptor protein 3 (NLRP3) inflammasome-dependent inflammation after spinal cord injury. Med Sci Monit. (2020) 26:e925491. doi: 10.12659/MSM.925491
186. Zhou J, Zhang X, Zhou Y, Wu B, Tan ZY. Up-regulation of P2X7 receptors contributes to spinal microglial activation and the development of pain induced by BmK-I. Neurosci Bull. (2019) 35:624–36. doi: 10.1007/s12264-019-00345-0
187. Bernardino L, Balosso S, Ravizza T, Marchi N, Ku G, Randle JC, et al. Inflammatory events in hippocampal slice cultures prime neuronal susceptibility to excitotoxic injury: a crucial role of P2X7 receptor-mediated IL-1β release. J Neurochem. (2008) 106:271–80. doi: 10.1111/j.1471-4159.2008.05387.x
188. Roy ER, Wang B, Wan YW, Chiu G, Cole A, Yin Z, et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest. (2020) 130:1912–30. doi: 10.1172/JCI133737
189. Yang J, Wise L, Fukuchi KI. TLR4 Cross-talk with NLRP3 inflammasome and complement signaling pathways in Alzheimer's disease. Fronti Immunol. (2020) 11:724. doi: 10.3389/fimmu.2020.00724
190. Buntinx M, Gielen E, Van Hummelen P, Raus J, Ameloot M, Steels P, et al. Cytokine-induced cell death in human oligodendroglial cell lines. II: Alterations in gene expression induced by interferon-γ and tumor necrosis factor-α. J Neurosci Res. (2004) 76:846–61. doi: 10.1002/jnr.20117
191. Li Y, Zhang R, Hou X, Zhang Y, Ding F, Li F, et al. Microglia activation triggers oligodendrocyte precursor cells apoptosis via HSP60. Mol Med Rep. (2017) 16:603–8. doi: 10.3892/mmr.2017.6673
192. Yeo YA, Martínez Gómez JM, Croxford JL, Gasser S, Ling EA, Schwarz H. CD137 ligand activated microglia induces oligodendrocyte apoptosis via reactive oxygen species. J Neuroinflammation. (2012) 9:173. doi: 10.1186/1742-2094-9-173
193. Shen W, Nikolic L, Meunier C, Pfrieger F, Audinat E. An autocrine purinergic signaling controls astrocyte-induced neuronal excitation. Sci Rep. (2017) 7:1–13. doi: 10.1038/s41598-017-11793-x
194. Ogawa Y, Furusawa E, Saitoh T, Sugimoto H, Omori T, Shimizu S, et al. Inhibition of astrocytic adenosine receptor A2A attenuates microglial activation in a mouse model of Sandhoff disease. Neurobiol Dis. (2018) 118:142–54. doi: 10.1016/j.nbd.2018.07.014
195. Antonioli L, Blandizzi C, Pacher P, Haskó G. The purinergic system as a pharmacological target for the treatment of immune-mediated inflammatory diseases. Pharmacol Rev. (2019) 71:345–82. doi: 10.1124/pr.117.014878
196. Thawkar BS, Kaur G. Inhibitors of NF-κB and P2X7/NLRP3/Caspase 1 pathway in microglia: Novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer's disease. J Neuroimmunol. (2019) 326:62–74. doi: 10.1016/j.jneuroim.2018.11.010
197. Kaczmarek-Hajek K, Zhang J, Kopp R, Grosche A, Rissiek B, Saul A, et al. Re-evaluation of neuronal P2X7 expression using novel mouse models and a P2X7-specific nanobody. Elife. (2018) 7:e36217. doi: 10.7554/eLife.36217
198. Xu J, Chalimoniuk M, Shu Y, Simonyi A, Sun AY, Gonzalez FA, et al. Prostaglandin E2 production in astrocytes: regulation by cytokines, extracellular ATP, and oxidative agents. Prostaglandins Leukot Essent Fat Acids. (2003) 69:437–48. doi: 10.1016/j.plefa.2003.08.016
199. Chen X, Hu J, Jiang L, Xu S, Zheng B, Wang C, et al. Brilliant Blue G improves cognition in an animal model of Alzheimer's disease and inhibits amyloid-β-induced loss of filopodia and dendrite spines in hippocampal neurons. Neuroscience. (2014) 279:94–101. doi: 10.1016/j.neuroscience.2014.08.036
200. Carmo MRS, Menezes APF, Nunes ACL, Pliássova A, Rolo AP, Palmeira CM, et al. The P2X7 receptor antagonist Brilliant Blue G attenuates contralateral rotations in a rat model of Parkinsonism through a combined control of synaptotoxicity, neurotoxicity and gliosis. Neuropharmacology. (2014) 81:142–52. doi: 10.1016/j.neuropharm.2014.01.045
201. Gyoneva S, Shapiro L, Lazo C, Garnier-Amblard E, Smith Y, Miller GW, et al. Adenosine A2A receptor antagonism reverses inflammation-induced impairment of microglial process extension in a model of Parkinson's disease. Neurobiol Dis. (2014) 67:191–202. doi: 10.1016/j.nbd.2014.03.004
202. Ingwersen J, Wingerath B, Graf J, Lepka K, Hofrichter M, Schröter F, et al. Dual roles of the adenosine A2a receptor in autoimmune neuroinflammation. J Neuroinflammation. (2016) 13:1–11. doi: 10.1186/s12974-016-0512-z
Keywords: Alzheimer, neurodegeneration, ATP, adenosine, oligodendrocyte, microglia, astrocyte, purine
Citation: Pietrowski MJ, Gabr AA, Kozlov S, Blum D, Halle A and Carvalho K (2021) Glial Purinergic Signaling in Neurodegeneration. Front. Neurol. 12:654850. doi: 10.3389/fneur.2021.654850
Received: 17 January 2021; Accepted: 16 April 2021;
Published: 14 May 2021.
Edited by:
Andras Lakatos, University of Cambridge, United KingdomReviewed by:
Alexei Verkhratsky, The University of Manchester, United KingdomAude Panatier, Centre National de la Recherche Scientifique (CNRS), France
Copyright © 2021 Pietrowski, Gabr, Kozlov, Blum, Halle and Carvalho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annett Halle, QW5uZXR0LkhhbGxlQGR6bmUuZGU=
†These authors share first authorship
‡These authors share last authorship