 Rosie Bell
Rosie Bell Michele Vendruscolo
Michele Vendruscolo- Centre for Misfolding Disease, Department of Chemistry, University of Cambridge, Cambridge, United Kingdom
Parkinson's disease is characterised by the presence in brain tissue of aberrant inclusions known as Lewy bodies and Lewy neurites, which are deposits composed by α-synuclein and a variety of other cellular components, including in particular lipid membranes. The dysregulation of the balance between lipid homeostasis and α-synuclein homeostasis is therefore likely to be closely involved in the onset and progression of Parkinson's disease and related synucleinopathies. As our understanding of this balance is increasing, we describe recent advances in the characterisation of the role of post-translational modifications in modulating the interactions of α-synuclein with lipid membranes. We then discuss the impact of these advances on the development of novel diagnostic and therapeutic tools for synucleinopathies.
Introduction
Physiological Functions of α-Synuclein That Involve the Binding to Lipid Membranes
α-Synuclein is a 140-residue protein encoded by the SCNA gene and comprised of three domains, an N-terminal amphipathic region (residues 1-60), a central aggregation-prone region (residues 61-95) known as the non-Aβ component (NAC), and a negatively charged C-terminal domain (residues 96-140) (1). In the cellular environment, α-synuclein can populate different states, including a disordered monomer in solution (2–4), and a partially structured α-helical state at the surface of lipid membranes (5, 6). Many of the synaptic functions that have been proposed for α-synuclein involve its association with cell membranes, including regulation of synaptic plasticity, dopamine levels, and synaptic vesicle trafficking (Figure 1) (7, 9).
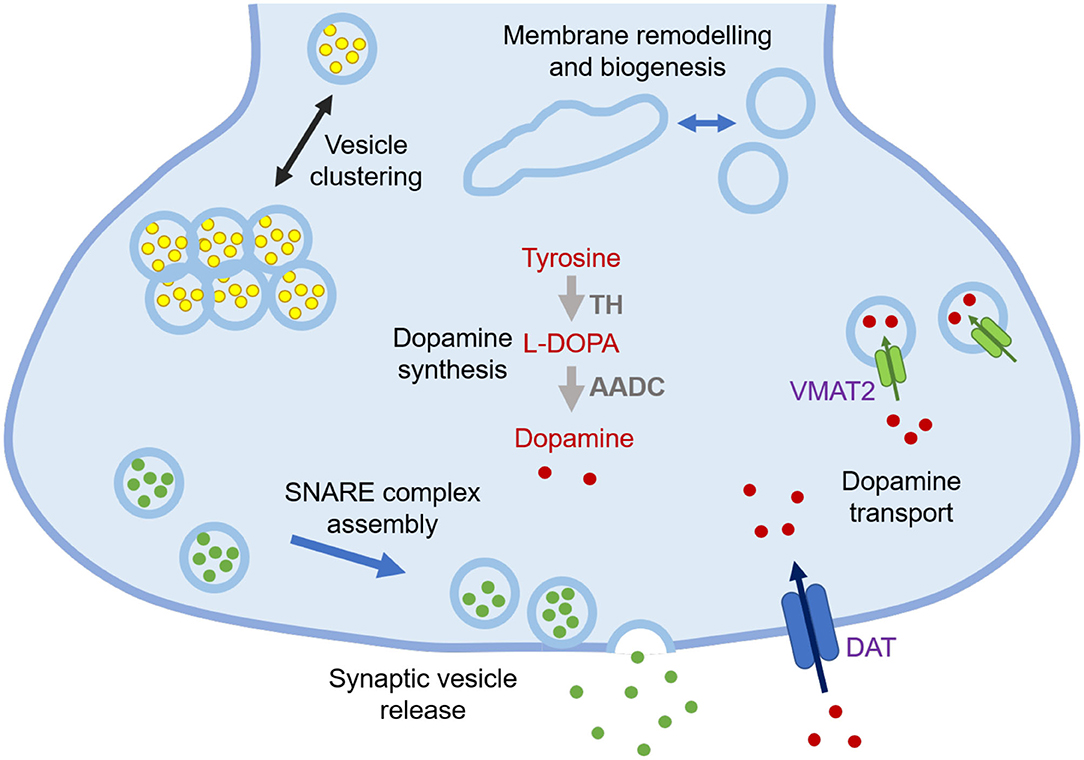
Figure 1. Schematic illustration of currently known functions of α-synuclein at synapses. The functions of α-synuclein are still to be fully elucidated, but at synapses they may include lipid-related functions, such as regulation of synaptic vesicle pools via sorting and clustering, remodelling of lipid membranes, in particular tubulation and biogenesis, and promotion of SNARE complex assembly and synaptic vesicle release, as well as non-lipid related functions, including modulation of dopamine synthesis via tyrosine hydroxylase (TH) that converts tyrosine into L-3,4-dihydroxyphenylalanine (L-DOPA) and regulation of dopamine transport through dopamine transporter (DAT) and vesicular monoamine transporter (VMAT2) (7, 8).
The regulation of synaptic vesicles by α-synuclein may include the coordination of their availability for neurotransmitter release by binding, ordering, localizing, and sequestering them (10). The N-terminal amphipathic domain is crucial for the interaction of α-synuclein with lipid membranes. Upon interacting with highly curved lipid membranes, the N-terminal and NAC regions can form an antiparallel broken α-helical conformation, with the two α-helices comprising residues 1-37 and 45-94, respectively (11). In the presence of lipid membranes with lower curvature, the two α-helices can merge into a continuous one (12). A more recent study has shown that upon binding to small unilamellar lipid vesicles (SUVs) the first 25 residues of α-synuclein tend to be anchored to the vesicle surfaces, while the central region (residues 26-98) can transiently bind other vesicles in a “double anchor” mechanism, with the C-terminal residues remaining disordered (5, 13).
Cell membranes exhibit complex and heterogeneous compositions, with the prime constituents being phospholipids (14), which are molecules composed of a glycerol head group esterified by two fatty acid chains with varying degrees of unsaturation (14, 15). The most abundant phospholipids in the outer leaflets of cell membranes are phosphatidic acid (PA), phosphatidylcholine (PC), phosphatidylethanolamine (PE), sphingomyelin (SM), and cholesterol (14), while in the inner leaflets phosphatidylinositol (PI), and phosphatidylserine (PS) are most abundant (10). The composition of phospholipids in lipid membranes alters their interactions with proteins (16), and in particular the acidic non-bulky head groups of phospholipids tend to attract the positively charged N-terminus of α-synuclein (17).
α-Synuclein in Parkinson's Disease
Parkinson's disease is the most prevalent neurodegenerative movement disorder, effecting over 6 million individuals worldwide (18, 19). This disease is characterised by a loss of dopaminergic neurons in the substantia nigra and by the presence of Lewy body and Lewy neurites, which are deposits containing α-synuclein (20) as well as a variety of other cellular components (21, 22). Mutations within the SNCA gene (A53T/E, A30P, E46K, H50Q, and G51D) have linked α-synuclein to familial Parkinson's disease and related synucleinopathies (23–28). In addition, SNCA duplication and triplication were also found to promote the early onset form of the disease (29–31).
These considerations firmly link α-synuclein to Parkinson's disease (8, 32–34), in particular through its interactions with lipid membranes (15, 35). This protein thus represents a primary therapeutic target for this disease and related synucleinopathies (36–41).
Aggregation of α-Synuclein
α-Synuclein is an amyloidogenic protein that can forms characteristic cross-β amyloid fibrils (42). The aggregation process of α-synuclein can be followed in vitro as a function of time by measuring the fluorescence of amyloid-binding dyes, and appears a sigmoidal curve with lag, exponential and plateau phases. This macroscopic behaviour is the result of a complex interplay between different microscopic processes, which include primary nucleation, which is usually promoted by the presence of lipid membranes, fibril elongation, and secondary nucleation whereby the surfaces of existing fibrils catalyse the formation of new seeds (Figure 2) (45, 48). Since aggregates of α-synuclein are toxic and enhance the formation of further aggregates, blocking this process is a promising therapeutic route (39). Amyloid aggregation is toxic in multiple ways, including in particular through the formation of misfolded oligomers, which can disrupt mitochondrial and synaptic functions, and induce endoplasmic reticulum (ER) stress and membrane damage (49–52). In addition, larger α-synuclein deposits can sequester key cellular proteins and with consequential loss of function (21, 22, 53), and travel from cell to cell (54).
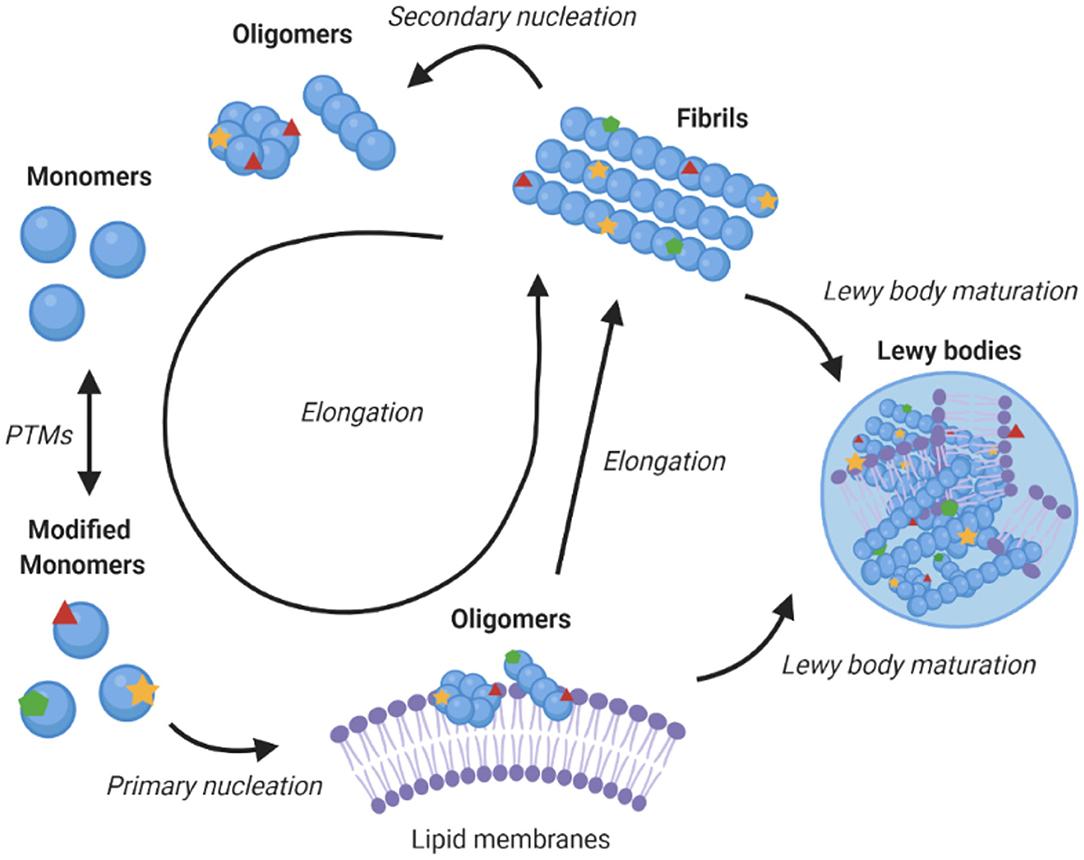
Figure 2. Role of lipid membranes in α-synuclein aggregation and its links with Parkinson's disease. The figure shows a schematic illustration of a kinetic network model for the aggregation of α-synuclein into amyloid fibrils. Initially, α-synuclein monomers combine to form oligomers, in a heterogeneous nucleation process promoted by lipid membranes (43). These oligomers can then either redissolve or mature and elongate into highly structured amyloid fibrils (44). Once fibrils have been formed, secondary processes, including fragmentation and secondary nucleation, accelerate the aggregation process (45). Downstream to fibril formation, the mechanism of Lewy body formation and maturation remains to be established, although it is likely to involve interaction of different α-synuclein species with lipid membranes (21, 22, 46). The post-translational modifications of α-synuclein can affect essentially all the steps in the aggregation process, including by modulating the binding of α-synuclein to lipid membranes, which may increase the local concentration of α-synuclein and facilitate the initial nucleation events, compared to the slow nucleation rate of free monomers in solution (47).
α-Synuclein does not readily aggregate spontaneously (55, 56) and this process is affected by the environmental conditions (57–59). The binding to lipid membranes offers a possible interface for primary nucleation events to initiate the toxic cascade of α-synuclein aggregation (47, 55). For example, aggregation has been shown to be accelerated by exosomes (60–62).
Intersection of α-Synuclein Homeostasis and Lipid Homeostasis
Although Parkinson's disease is often described a proteinopathy, emerging evidence suggests that it could be considered as a lipidopathy, or most likely as a combination of the two (63). Multiple links between lipid homeostasis and genes associated with Parkinson's disease have been reported (64). An analysis of three genome-wide association studies (GWAS) revealed four major processes relevant to Parkinson's disease—oxidative stress response, endosomal-lysosomal functions, ER stress response, and immune response, with lipids and lipoproteins being key to all four processes (65). A link has also been uncovered between Parkinson's disease and PLA2G6 (PARK14), the gene encoding phospholipase A2 group VI (PLA2G6). This enzyme catalyses glycerophospholipid and phospholipid hydrolysis to produce free fatty acid and lysophospholipids (66). Mutations in the GBA gene, encoding β-glucocerebrosidase (GBA), are a common risk factor for Parkinson's disease for both homozygous and heterozygous carriers. GBA catalyses the breakdown of glucocerebroside into glucose and ceramide, and a loss of function leads to the accumulation of glycosphingolipids (67, 68), influencing the sensitivity of neurons to α-synuclein aggregation and spreading (69).
In knock-out mice models, loss of α-synuclein function is not particularly detrimental, but SCNA triplication or α-synuclein mutations that affect its expression have been linked with Parkinson's disease (8). Although a gain of toxic function by α-synuclein was initially thought to involve primarily the processes of amyloid aggregation, recent studies have shown an increasing role for lipid homeostasis in α-synuclein induced toxicity (Figure 3). In this perspective, all the known familial mutations of α-synuclein occur within its lipid membrane binding region. A study in yeast found that α-synuclein toxicity was not dependent on fibril formation, and that mutations that lead to defective lipid membrane binding were less toxic, suggesting toxicity was caused by interaction with cell membranes (71). Another study observed α-synuclein toxicity correlated with accumulation of lipid vesicles in yeast, which induced ER to Golgi trafficking defects, suggesting that the reliance of neurons on lipid vesicle transport may be the reason they are so vulnerable to α-synuclein toxicity (72).
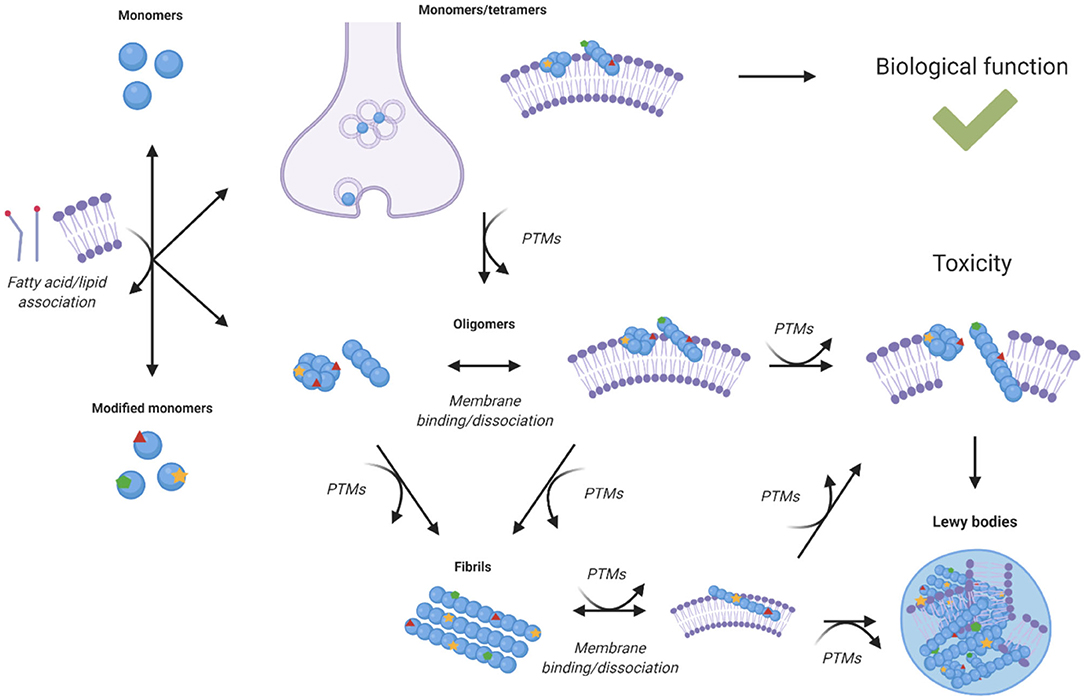
Figure 3. Effects of post-translational modifications on the relationship between α-synuclein homeostasis and lipid homeostasis. Lipid binding is associated with the cellular functions of α-synuclein, but it is also with its toxicity upon aggregation. Different species of α-synuclein can interact with cell membranes, lipid vesicles, and free fatty acids. The interactions of aggregated species of α-synuclein with lipid membranes can lead to altered structure, composition, function, and integrity of the membranes themselves (73–75, 77, 84–88, 90, 91). The binding of α-synuclein to lipids and lipid membranes can also impact its aggregation, toxicity, expression, and localisation. Post-translational modifications can alter the interaction of α-synuclein species with lipid species, modulate the binding, and dissociation of α-synuclein with lipid membranes and influence the aggregation propensity of α-synuclein (15, 16, 47, 55, 80–83, 92, 94).
The existence of a soluble conformation of α-synuclein with α-helical secondary structure was recently reported and suggested to originate from transient interaction with lipid vesicles and may be the species that initiates aggregation (73). In the familial mutant E46K, one of the KTKEGV motifs important for lipid membrane binding is disrupted, and so this mutation has been utilised to study the effects of α-synuclein membrane binding. Two further mutations of E to K within other KTKEGV motifs have been made in E46K α-synuclein to create a binding-deficient α-synuclein form termed 3K (74, 75). In neurons, 3K α-synuclein led to increased inclusion formation (74) and in mice to a Parkinson's disease-like phenotype with inclusions containing α-synuclein and lipid vesicles (75).
Recently, it was observed that α-synuclein overexpression in yeast, rodent neurons and induced pluripotent stem cells (IPSCs) led to changes in cellular lipid profiles by increasing the formation of mono-unsaturated fatty acids (MUFAs). Surplus MUFAs, specifically oleic acid, subsequently induced enhanced α-synuclein toxicity by altering the equilibrium of membrane bound to soluble α-synuclein. These findings revealed a new therapeutic target for ameliorating Parkinson's disease, as stearoyl-CoA-desaturase inhibition could reduce biosynthesis of MUFAs and lead to reduced α-synuclein cytotoxicity (63, 76). α-Synuclein oligomerisation was found to be regulated by poly-unsaturated FA (PUFA) levels in neural cells, and deletion of the SNCA gene in mice led to a decrease in docosahexaenoic acid (DHA) and α-linolenic acid (ALA) (77). Free fatty acids, specifically DHA and ALA, have been shown to bind to α-synuclein and increase its aggregation at low ratios, but to reduce it at high ratios. α-Synuclein binding to DHA and ALA was protective against oxidative damage induced by fatty acids (15, 78–80). Altered levels of PE, PI, PS, and PC have been observed in Parkinson's disease patients (63). Under the oxidative stress conditions seen in Parkinson's disease, the balance between the cholesterol derivatives 24-hydroxysterol and 27-hydroxysterol can become disrupted (15, 81–83). These results suggest that the levels, modifications and toxicity of α-synuclein are intricately linked with lipids in vivo.
α-Synuclein has been shown to bind to mitochondrial membranes and stimulate mitochondrial tubulation, fragmentation and dysfunction (84–86). Mitochondrial dysfunction caused by α-synuclein leads to an increase in reactive oxygen species which may in turn lead to increased toxicity of α-synuclein (86). More generally, α-synuclein can remodel lipid membranes, for example POPG vesicles were remodelled into tubules (87), POPC/POPA bilayers were thinned (88), POPG vesicles remodelled into nanoparticles (89), sphingomyelin was remodelled into nanodiscs (90), and arachidonic acid was released from liposomes (91), upon interaction with α-synuclein.
Modulation of the Interactions Between α-Synuclein and Lipid Membranes
The interaction of α-synuclein with lipid membranes is modulated by the properties of the phospholipid head groups. For example, 27-hydroxycholesterol, a product of cholesterol oxidation, induces expression and accumulation of α-synuclein in human dopaminergic neurons via transcription (82), dioleoyl-PA induces aggregation of α-synuclein (92), docosahexaenoic acid (DHA) can induce oligomerisation (80), and cholesterol modulates the binding of α-synuclein to vesicles (16). The lipid composition of cell membranes can affect the way in which α-synuclein interacts with the membranes themselves, as demonstrated by the findings that cardiolipin can promote the pore-forming activity of α-synuclein oligomers (93). The familial mutants of α-synuclein have been shown to have different membrane interaction properties (94), so small chemical changes can have an impact on how α-synuclein binds and responds to lipid membranes.
Post-Translational Modifications of α-Synuclein
α-Synuclein is subject to multiple post-translational modifications (Figure 4) including acetylation, phosphorylation, ubiquitination, SUMOylation, nitration, truncation, and glycation (49, 95–97). Much work has been undertaken to understand how these post-translational modifications affect the aggregation of α-synuclein (98). By contrast, the impact of post-translational modifications on the affinity of α-synuclein for biological membranes requires further study.
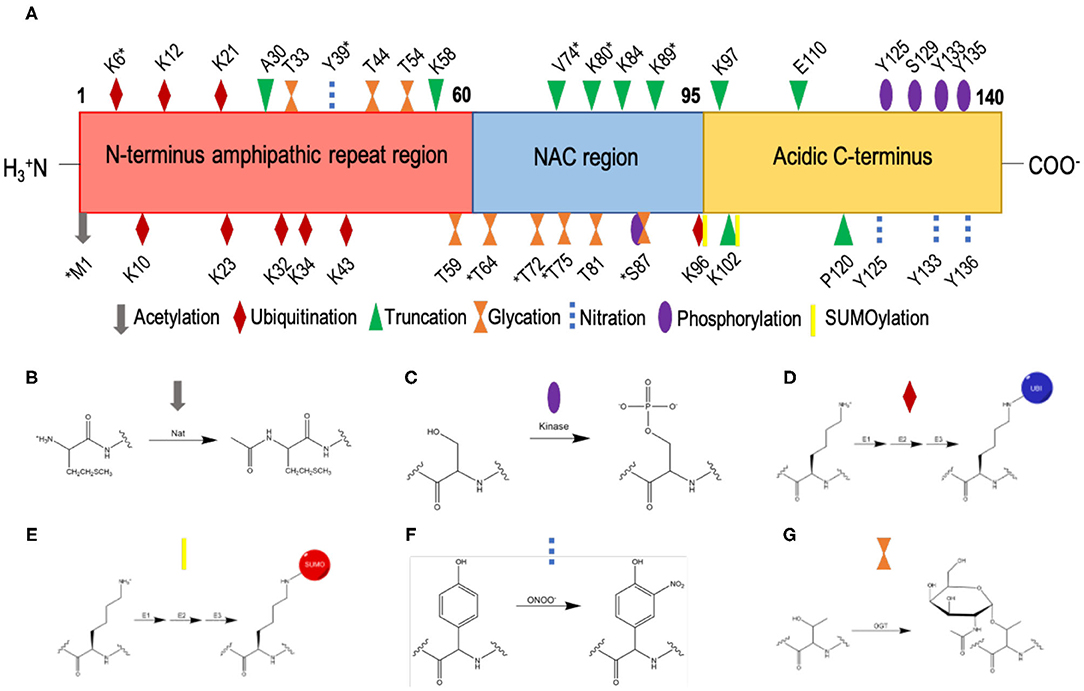
Figure 4. List of currently known post-translational modifications of α-synuclein. (A) The amino acid sequence of α-synuclein comprises an N-terminal amphipathic region (residues 1-60), a non-Aβ component (NAC) region (residues 61-95), and an acidic C-terminal region (residue 96-140). Post-translation modification sites are labelled; *indicates post-translation modifications within low-solubility areas using CamSol (70). (B) N-terminal acetylation, which is carried out by N-acetyltransferases (Nat). (C) Phosphorylation, which occurs by the esterification by a phosphate group of the side-chain hydroxyl moiety of serine or threonine residues; shown here is the phosphorylation of a serine residue. (D) Ubiquitination, where ubiquitin is added to lysine residues via an isopeptide bond, catalysed by sequential action of 3 enzymes, E1 (ubiquitin-activating enzyme), E2 (ubiquitin-conjugating enzyme) and E3 (ubiquitin ligase). (E) SUMOylation, where SUMO is added to lysine residues via a thioester bond, catalysed by sequential action of 3 enzymes, E1 (SUMO-activating enzyme), E2 (SUMO-conjugating enzyme), and E3 (SUMO ligase). (F) Nitration, where tyrosine residues are nitrated by peroxynitrite; the phenolic R group is converted to 3-nitro-tyrosine, by the addition of a nitro (NO2) group onto the ortho position of the ring by peroxynitrite. (G) O-GlcNAcylation, where the enzyme O-GlcNAc transferase (OGT) catalyses the addition of N-acetylglucosamine (GlcNAc) to the hydroxyl group (O-linked) of the side-chains of serine and threonine residues; shown here is the O-GlcNAcylation of a threonine residue.
N-Terminal Acetylation
N-terminal acetylation is a common co-translational and post-translational modification, which affects about 70–90% of the proteins in eukaryote proteomes (99, 100). In humans, there are 7 types of N-terminal acetyltransferases (NatA-F and NatH), which are enzymes that catalyse the covalent attachment of an acetyl group (CH3CO) to the free α-amino group () of the N-terminal residue of a protein, depending on its N-terminal dipeptide (101). This post-translational modification impacts a wide range of properties of proteins, including their stability, folding, interactions and subcellular localisation, and has been implicated in a wide range of human disorders, including cancer and developmental diseases (102).
It has been estimated that over 80% of α-synuclein molecules are constitutively N-terminal acetylated (103), the large majority of which by the N-acetyltransferase B (NatB) complex (104) (Figures 4A,B). The addition of the acetyl group to the amine group of the N-terminus results in the loss of a positive charge in an area of poor solubility (105). Given that phospholipid headgroups tend to be negatively charged, modifications in this region impact the binding behaviour of α-synuclein to cell membranes. In a recent mass spectrometric analysis, Lewy bodies have been found to be enriched in N-terminal acetylated α-synuclein (106).
In addition, as N-terminal acetylation induces more α-helical structure in monomeric α-synuclein in solution (107), the entropic barrier from intrinsically disordered free α-synuclein to a membrane bound α-helical α-synuclein may be lowered. Indeed, N-terminal acetylation has been shown to generally enhance the affinity of α-synuclein for lipid membrane binding (Table 1). Recently, it was shown by nuclear magnetic resonance (NMR) spectroscopy that N-terminal acetylation increases the binding affinity but does not alter not structure of the bound form α-synuclein to synaptic vesicle mimics (DOPE:DOPS:DOPC SUVs, 5:3:2 w/w) (108). Similarly, an enhanced binding affinity to synaptic-like vesicles has been reported, when α-synuclein is N-terminally acetylated (111, 112). N-terminal acetylation was observed to not only effect the interaction of the N-terminus of α-synuclein with phospholipid vesicles, but also to impact the entire protein (112). However, no significant difference was observed in the membrane binding affinity in acetylated and non-acetylated α-synuclein with synaptic vesicle mimics (POPC/POPS/Chol large unilamellar vesicles, LUVs) (113).
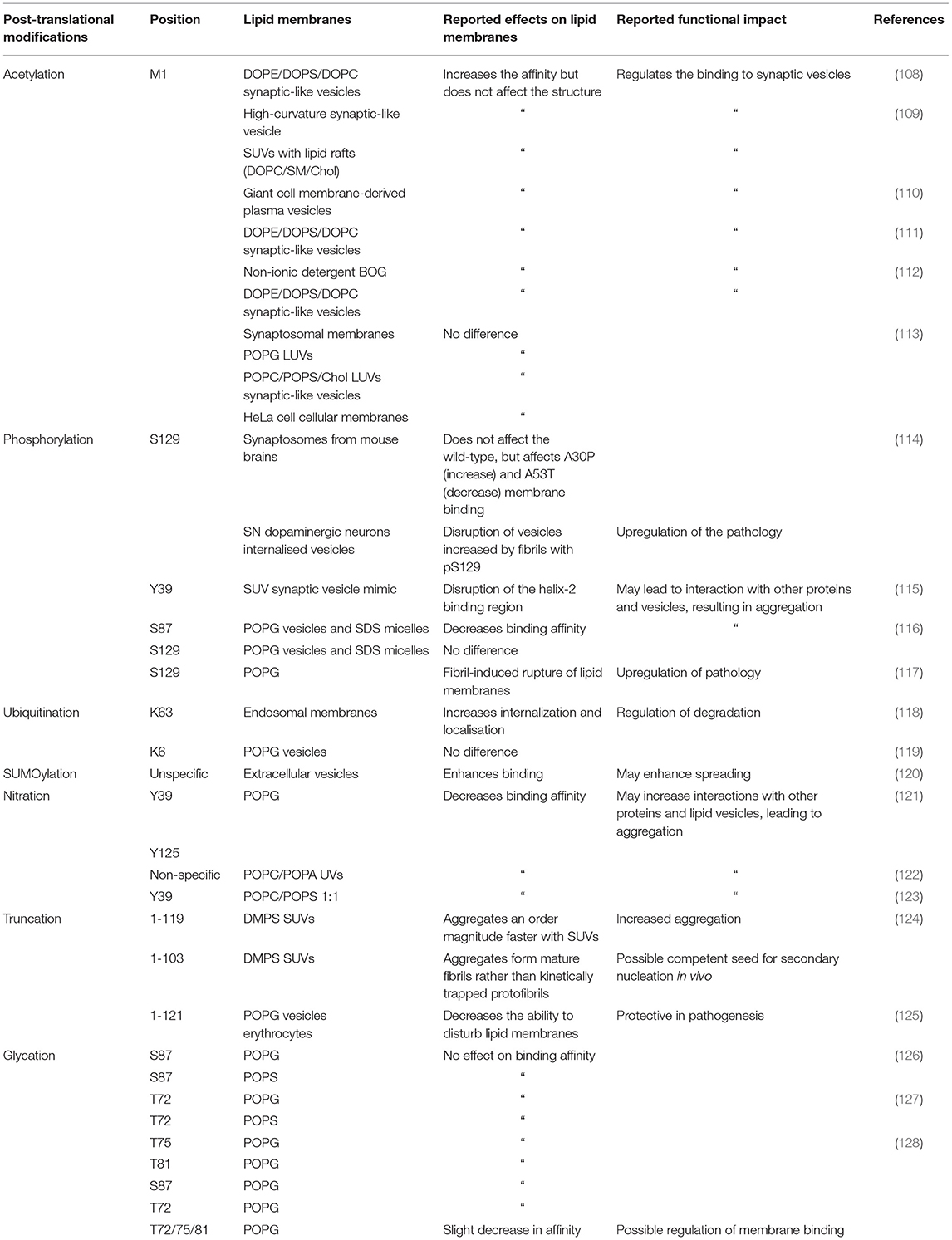
Table 1. Summary of the known post-translational modifications of α-synuclein, and of their effects on its binding to lipid membranes.
A study of LUVs comprised of POPG and mouse synaptosomes found no significant effects of N-terminal acetylation on α-synuclein membrane binding (113). Binding to SDS micelles was also examined, but again no significant effect was detected, although this modification induced a new conformation of α-synuclein when bound to β-octyl-glucoside (BOG) vesicles (111). It was also found that acetylation of α-synuclein enhances binding to phospholipid vesicles that have similar size and high curvature to synaptic vesicles (15:0, 12:0 lyso-, and 16:0 lyso-PC SUVs). A decrease in binding affinity to DPPC vesicles containing cholesterol was also observed when α-synuclein is acetylated, and enhanced binding to lipid raft SUVs with high curvature (DOPC/SM/Chol) (109). An investigation of the effects of N-terminal acetylation on α-synuclein in yeast revealed that N-terminal acetylation was required for proper plasma membrane targeting (129). N-terminal acetylation has been shown to increase the binding affinity of N-terminus of α-synuclein to giant cell membrane-derived plasma vesicles (110).
Phosphorylation
Phosphorylation is the most common post-translational modification of eukaryotic proteins, which acts as a molecular switch to regulate protein interactions (130, 131). This process is carried out by kinases, which are a large class of enzymes (132) and reversed by phosphatases (133). The phosphorylation of proteins is an esterification reaction that involves the attachment of a phosphoryl group to the hydroxyl group of the side-chains of specific amino acids, most commonly serine, threonine and tyrosine, and in some cases arginine, lysine, aspartic acid, glutamic acid and cysteine (130, 134). The dysregulation of protein phosphorylation is associated with a wide range of human diseases, and kinases are a major target for pharmacological intervention, in particular for cancer (135, 136).
α-Synuclein can be most commonly phosphorylated at serine and tyrosine residues (Figures 4A,C) and it is typically phosphorylated at S129 and S87 in Lewy bodies (103). The impact of phosphorylation can be studied by co-expressing kinases to phosphorylate α-synuclein, or by using phosphomimetics such as mutations to aspartic acid or glutamic acid. Alternatively, since kinases may lead to unspecific phosphorylation, semi-synthetic strategies (137), or post-translational chemical mutagenesis (138) can enable site-specific phosphorylation.
Phosphorylation at S129 (pS129) is closely linked to Parkinson's disease (139–141) and increases to about 90% levels in Lewy bodies from the 5% levels observed in healthy brains (142). However, it still remains to be established whether phosphorylation at S129 precedes Lewy body formation or follows it as a downstream event, whether it inhibits or promotes α-synuclein aggregation, and whether it enhances or reduces α-synuclein binding to lipid membranes. In vitro studies have offered conflicting conclusions about the effect of pS129 on α-synuclein aggregation (114, 143). By co-expressing polo-like kinase 2 (PLK2) with α-synuclein to phosphorylate S129, an increased rate pS129-α-synuclein aggregation was observed, but synaptosome membrane binding was not affected (114). However, the mutants A30P and A53T were affected, as A30P-α-synuclein binding increased and A53T-α-synuclein binding was lowered by the presence of pS129. The internalisation of pS129-α-synuclein fibrils was not significantly different with respect to unmodified α-synuclein in dopaminergic neurons in the ventral tegmental area but was increased in those in the substantia nigra. Disruption of lipid membranes was also increased by S129 phosphorylation of A30P-α-synuclein (114). By synthetic phosphorylation at S129 it was found that this modification induced a different strain and increased the toxicity of fibrils, as pS129-α-synuclein fibrils more readily ruptured POPG vesicles (117). Monomeric pS129-α-synuclein bound to POPG vesicles exhibited less α-helical secondary structure, suggesting that phosphorylation may impede lipid membrane binding (117). Aggregates of pS129-α-synuclein were found to co-localise with mitochondria, impairing their function (144).
A study of the influence of phosphorylation of Y39 on synaptic vesicle mimics (DOPS/DOPC/DOPE SUVs) revealed that pY39 disrupted binding but had no effect when binding to SDS micelles. It was also found the phosphomimetic Y39E perturbed peripheral lipid membrane localisation compared to wild-type α-synuclein in yeast. The effects of pY39 were similar to that of the familial mutation G51D (115). An analysis of the effects of phosphorylation at S87 showed that pS87 enhanced conformational flexibility, reduced aggregation, and decreased α-synuclein binding to lipid membranes (SDS micelles and POPG vesicles) (116).
Phosphorylation at different residues may impact in distinct manners the interaction of N-terminus of α-synuclein with phospholipid membranes. Since residues S87 and Y39 lie within the α-helical domain when α-synuclein binds to membranes (5, 11) and in an areas of low solubility (Figure 4A), they may have more of an effect on the binding affinity of the N-terminus of α-synuclein and the resulting aggregation of the protein. It is still unclear when phosphorylation occurs in the aggregation process, but it has been suggested to happen during or post aggregation, rather than before (116, 145–147). The disruption of phosphorylation is linked with Parkinson's disease, with the mutation of PARK6 encoding an protein kinase PINK1 being the second most common cause of autosomal recessive familial Parkinson's disease (148).
Ubiquitination
Ubiquitination is a reversible post-translational modification that is essential for the maintenance of protein homeostasis. Ubiquitin is a 76-residue protein that is added via an isopeptide bond to lysine residues to its target as a marker for degradation or to alter the targets interactions (149) (Figure 4D). The process is mediated by three enzymes E1 (ubiquitin-activating enzyme), E2 (ubiquitin-conjugating enzyme), and E3 (ubiquitin ligase) and ubiquitin can be removed by deubiquitinating enzymes (150). Poly-ubiquitin chains can be built up on a protein via sequential addition of ubiquitin to one of the seven lysine residues within the previous ubiquitin molecule (151). The specificity of the ubiquitin-lysine linked chains influences the effect that ubiquitination has on the target, for example K48-linked ubiquitin chains trigger degradation of the target, whereas K63-linked ubiquitin chains regulate the formation of complexes (149). The ubiquitination system is known to be deregulated in diseases such as cancer, immune disorders, muscle-wasting disorders, diabetes, and neurodegeneration (152).
Lewy bodies have shown immunoreactivity to anti-ubiquitin antibodies and contain ubiquitin-α-synuclein (103, 153). α-Synuclein has eight lysine residues that can be ubiquitinated (Figure 4A), and ubiquitin has multiple internal lysine residues from which poly-ubiquitin chains can form. The combinations of ubiquitin-α-synuclein sites and linkages are vast, and so there is much more to learn about the effects of different ubiquitination on α-synuclein, especially on the interactions of N-terminus of α-synuclein with phospholipid membranes.
Multiple groups have found that ubiquitination of α-synuclein affects the aggregation. Tetra- ubiquitinated K-48 linked chains at K12 of α-synuclein was shown to increase aggregation propensity of α-synuclein, but the aggregates formed were amorphous, and did not convert into mature fibrils (154). The effects on mono-ubiquintation at multiple lysine residues in α-synuclein were investigated by mutating K to C to allow the addition of ubiquitin via a disulphide-mediated reaction (155). This study revealed that the ubiquitinated lysine residue was capable of modulating the aggregation of α-synuclein. K10C-ubiquitin and K23C-ubiquitin reduced the rate of α-synuclein fibril formation, and that K6C-ubiquitin, K12C-ubiquitin and K21C-ubiquitin had a moderate inhibitory effect and K32C-ubiquitin, K34C-ubiquitin, K43C-ubiquitin, and K96C-ubiquitin had strong inhibitory effects (155).
In a recent study, mono-ubiquitin was synthetically added to α-synuclein via a BTA linkage, to allow ubiquitination to remain in reducing environments (156). This approach revealed that ubiquitination at K6, K23, K43, and K96 had no effect on monomeric α-synuclein change to secondary structure via CD, but all reduced amyloid aggregation. Furthermore, the ubiquitin-α-synuclein aggregates were less toxic to SH-SY5Y cells (156). In a related study, lysine residues of α-synuclein were semi-synthetically modified via disulphide directed mono-ubiquitination. This approach showed that K6-ubiquitin and K23-ubiquitin both inhibit fibril formation, but do not alter the structure of fibrils. However, K96-ubiquitin inhibits aggregation and also alters structure of aggregates formed (157).
The effect of ubiquitin on the interaction of α-synuclein with phospholipid membranes revealed the mono-ubiquitination at position K6 along the sequence of α-synuclein inhibited aggregation of monomeric α-synuclein, but did not affect the secondary structure when bound to POPG vesicles (119). However, it was also found that K63 poly-ubiquitinated α-synuclein was preferentially internalised and translocated to endosomes (118). It is worth noting that lysine residues within the KTKEGV regions are thought to be essential in the mechanism of α-synuclein binding to anionic membranes, and so ubiquitination of these residues may prevent membrane binding of monomers, oligomers, and fibrils (158).
SUMOylation
Related to ubiquitination, the addition of small ubiquitin-like modifier (SUMO) proteins is a reversible, covalent, post-translational modification of lysine residues (159). SUMOylation acts to regulate the function, localization, and interactions of the modified proteins. There are several SUMO proteins within the SUMO family, which is highly conserved across eukaryotes, with the most abundant SUMOylation found in mammalian cells being SUMO-1 addition (160). Similarly to ubiquitination, SUMOylation is carried out by the sequential action of three enzymes, E1, E2, and E3, while de-SUMOylation is catalysed by SUMO-specific proteases (150, 160). This post-translational modification is carried out through the conjugation of the ε-amino group of a lysine residue within the target protein via a thioester bond to a C-terminal glycine in a SUMO protein (Figure 4E) (161). SUMOylation of lysine residues occurs at consensus sequences consisting of Ψ-K-x-D/E, where Ψ is a branched aliphatic amino acid, x is any amino acid, and K is the modified lysine (162). Protein SUMOylation has been linked to a variety of diseases including cancer, heart disease, and neurodegeneration (160, 163, 164).
SUMOylation of α-synuclein is closely linked to Parkinson's disease (165). SUMO proteins have been detected in Lewy bodies as colocalised with α-synuclein (166). In α-synuclein there are two major sites for SUMOylation (Figure 4A), K96 and K102 (167), although seven more lysine residues have been reported to be SUMOylated (168). It was also shown that α-synuclein aggregation was stimulated by SUMOylation upon proteasomal inhibition. SUMO proteins have been observed to co-localise with lysosomes and may recruit them to aggregated α-synuclein (169), or target α-synuclein to the autophagy pathway (170). α-Synuclein mixed with SUMO-α-synuclein was found to be less prone to aggregation compared to wild-type α-synuclein, even if a small fraction of α-synuclein monomers had been SUMOylated, leading to the suggestion that a role of SUMOylation is to increase the solubility of α-synuclein (168). Similarly, synthetically SUMOylated α-synuclein at K96 and K102 was observed to reduce amyloid aggregation in a site-specific manner (156, 171). The amorphous aggregates of K102-SUMO α-synuclein were also found to be significantly less toxic to SH-SY5Y cells than the amyloid aggregates of wild-type α-synuclein (156).
The observation that the release of α-synuclein within extracellular vesicles is SUMO-dependent led to the suggestion that SUMOylation acts as a regulator of α-synuclein sorting into the extracellular vesicle pathway which may facilitate the spreading of α-synuclein pathology (120). The affinity of SUMO for SUVs was shown to vary depending on the phospholipid makeup, with SUMO having a high affinity for the phospholipids PI (3)P, PI (3–5)P3, and POPS, but a lower one for uncharged POPC. SUMOylated-α-synuclein was also found to be enriched in the mouse neuroblastoma N2A cell membrane pellets compared to that of unmodified α-synuclein, suggesting that SUMOylation promotes membrane binding (120). Three familial variants of α-synuclein (A30P, A53T, and E46K) were found to be more susceptible to PIAS2 (an E3 ligase) mediated SUMOylation in vitro and in SH-SY5Y cells, and this was associated with an increase in aggregation and inclusion formation. It was also observed that SUMOylation of α-synuclein decreased the ubiquitination both in vitro and in vivo (172).
SUMOylation is an essential, intricate and dynamic process, the complexities of which are still not fully understood. Although little is known about how SUMOylation affects the binding and function of α-synuclein at phospholipid membranes, it is clear SUMOylation has an impact on α-synuclein aggregation. To fully understand the role of SUMOylation in Parkinson's disease, it will be important to investigate how the membrane binding of α-synuclein is affected as this is a possible site for aggregation and key to the function of α-synuclein.
Nitration
Nitration is an irreversible post-translational modification that occurs on tyrosine residues, in particular in the presence of nitric oxide radicals (173). In this process, the phenol group of tyrosine is converted to 3-nitro-tyrosine in a reaction mediated by reactive nitrogen species, such as peroxynitrite (ONOO−, Figure 4F), an oxidant formed by the reaction of the nitric oxide and superoxide radicals (.NO and ) (174). These radicals are normally rapidly removed by superoxide dismutases (SODs), but can form spontaneously peroxynitrite via a diffusion-limited reaction (175, 176). Nitration is often highly selective and depends on the structure and environment surrounding the tyrosine residues (177). This post-translational modification has been associated with over 50 diseases, including cancer, cardiovascular disorders, and neurodegeneration (178) and can affect proteins structure, function, and other post-translational modifications (179).
α-Synuclein has four tyrosine residues (Y39, Y125, Y133, and Y136), which can be nitrated to form nitrated-α-synuclein (Figure 4A) (180). Nitrated α-synuclein is enriched in Lewy bodies compared to control brains (181, 182). Nitrated tyrosine residues are reactive and can form dityrosine bonds via cross-linking, which can induce oligomerisation of α-synuclein (183). Nitrated α-synuclein has been shown to induce cytotoxicity in SH-SY5Y cells (184), and in the substantia nigra of rats (185). It was also found that Y39 is preferentially nitrated (nY39) in a cell model of Parkinson's disease overexpressing monoamine oxidase B (MOA-B), an enzyme involved in dopamine metabolism and known to create reactive oxygen species, and that this effect can be abrogated using selegiline, an inhibitor of MOA-B approved for the treatment of Parkinson's disease (186). Furthermore, using the Y39F mutant, it was shown that nY39 is key to dityrosine cross-linking and the subsequent induction of oligomer formation in α-synuclein (121). Therefore, assessing the effects of nitration could help develop a better understanding of the pathogenesis of Parkinson's disease.
To study the effects of nitration on α-synuclein one can expose α-synuclein to nitrating substances such as peroxynitrite or tetranitromethane (122, 187, 188), leading to unspecific nitration. A study of the effects of non-specific nitration revealed that in the presence of phospholipid vesicles (POPC/POPA SUVs), nitrated-α-synuclein bound with lower affinity than wild-type α-synuclein. It was also observed that nitration induced a change in secondary structure, an increase in disorder in solution, and a decreased propensity to adopt α-helical structures in presence of phospholipid vesicles. The effects of non-specific nitration were found to be largely due to Y39, as the mutation Y39F ameliorated them (122).
As an alternative non-specific nitration, one can use semi-synthetic or mutational approaches to individually modify one tyrosine residue to examine site-specific effects (121, 123). The selective nitration at Y39 was obtained by mutating the other three tyrosine residues in α-synuclein to phenylalanine residues (123). It was thus demonstrated that nY39 decreased the binding affinity of α-synuclein to negatively charged lipid vesicles, due to the electrostatic repulsion of the partial negative charge of the nitro group. With the mutant Y39F, it was also shown that although the C-terminus is not directly involved in binding phospholipid vesicles, but that nitration of Y125, Y133, and Y136 disrupted the binding affinity of α-synuclein. The disruption of binding by C-terminal nitration was attributed to nitration leading to a compaction of the C-terminus that may disrupt the long range contacts and allosterically regulates binding of α-synuclein to phospholipids (123). Further, by nitrating residues Y39 or Y125, a significant reduction in the α-helix formation upon α-synuclein binding to POPG vesicles was observed, while nitration at Y39 or Y125 lead to the formation of fibrils with distinct morphology compared to that of wild-type α-synuclein. It was also found binding to liposome-containing phospholipids protected α-synuclein against nitration (189).
Proteolytic Cleavage and C-Terminal Truncations
Protein truncations can result from a variety of causes, including genetic variations, post-translational modifications, or incomplete degradation. Truncations can alter the structure and interactions of proteins, and cause loss or gain of function (190, 191). Abnormal proteolytic processing is a common feature of proteins that drive neurodegenerative diseases, which may be induced by the release of proteases from their cellular compartments under conditions of stress (192). Notably, the aberrant proteolytic processing of amyloid precursor protein (APP) is linked to Alzheimer's disease by producing an excess of the aggregation prone 42-residue form of the amyloid-β peptide (193). Protein truncations are also prevalent in Pick's disease, Huntington's disease, spinocerebellar ataxia, amyotrophic lateral sclerosis and Parkinson's disease (192).
Irreversible truncation of α-synuclein at the N and C termini is common, with over 15% of α-synuclein in Lewy bodies being truncated (106, 194–196). Truncation is thought to occur due to incomplete digestion of α-synuclein by a number of enzymes, including calpain, plasmin, neurosin, 20S proteasome, cathepsin D, caspase 1, matrix metallo proteinase-1, and asparagine endopeptidase (Figure 4A), and may be promoted by dysfunction of protein homeostasis machinery such as the lysosomal chaperone-mediated autophagy (194, 197–205). The N-terminal of α-synuclein is essential for binding to phospholipid membranes, and so truncation at N-terminal sites may reduce or inhibit cell membrane binding. Truncation can stimulate aggregation and toxicity in vitro (198, 206–208) and in vivo (197, 209–211) and increase prion-like spreading (197, 208).
The impact of C-terminal truncations (α-synuclein 1-119 and 1-103) on the aggregation of α-synuclein in the presences of phospholipid vesicles was recently investigated (124). Similar secondary structures were observed for both truncated variants and wild-type α-synuclein. α-Synuclein 1-119 aggregated in a comparable way, but an order of magnitude faster than the wild-type form. However, the α-synuclein 1-103 variant aggregated following a distinct mechanism forming morphologically different aggregates that resembled mature fibrils compared to the protofibrils produced by the aggregation of wild-type α-synuclein in the presence of DMPS SUVs (124). An analysis of the biophysical properties of α-synuclein 1-121 revealed that in the proximity of POPG vesicles both the secondary structure and aggregation of full-length or truncated α-synuclein were similar. With both POPG vesicles and erythrocytes, truncated α-synuclein exhibited a decreased ability to distort the phospholipid membranes. Conversely, α-synuclein 1-121 was found to have higher toxicity compared to full-length α-synuclein, possibly following the activation of apoptosis signalling pathways and upregulation of phosphorylation at S129 of α-synuclein (125).
Truncation can occur at multiple sites along the sequence of α-synuclein (Figure 4A) (196) but the effects many of these modifications are not known in detail. In all cases, C-terminal truncation of α-synuclein reduces the solubility of α-synuclein in solution and affects its membrane binding properties. Further research, however, will be required to fully understand how truncations affect the behaviour of α-synuclein in the cellular milieu. A distinct species of α-synuclein, termed p-asyn*, truncated at both N- and C-termini and phosphorylated, was detected in Lewy bodies in mice models of Parkinson's disease and primary neurons exposed to α-synuclein fibrils (212). It was found that p-asyn* preferentially associated with mitochondria and ER to induce toxicity (212).
O-GlcNAcylation
O-GlcNAcylation is a reversible enzymatic post-translational modification in which N-acetylglucosamine (GlcNAc), an amide derivative of glucose, is transferred from uridine diphosphate GlcNAc (UDP-GlcNAc) to the hydroxyl group of a serine or threonine side-chain in a protein (Figure 4G) (213). Thousands of proteins in the nucleus, cytoplasm and mitochondria have been identified to be targets of O-GlcNAcylation, which modulates their functions, interactions, and maintenance. The addition and removal of GlcNAc is catalysed by two enzymes, O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA), respectively (214). The levels of O-GlcNAcylation depend on environmental stimuli, as well as on the levels of cellular glucose entering the hexosamine biosynthetic pathway to produce UDP-GlcNAc (215). O-GlcNAcylation is an important modification in the brain, and has been observed to modulate synaptic signalling, memory formation, and neuron growth (216) and is also key in cellular response to stress (217). The dysregulation of O-GlcNAcylation has been linked to metabolic diseases, cancer, cardiovascular diseases, and neurodegeneration (213).
At least nine residues of α-synuclein have been reported to be O-GlcNAcylated in vivo (128, 218), five of which are in the NAC region (Figure 4A). Semi-synthetic O-GlcNAcylation of α-synuclein at various positions inhibited aggregation, reduced toxicity of aggregates to both SH-SY5Y cells and rat primary cortical neurons (127). A study of the impact of S87 O-GlcNAcylation revealed a 5-fold reduction in fibril formation, and the formation of shorter fibrils (126). Single O-GlcNAcylation at T72, S87, T75 or T81, and of triple O-GlcNAcylation at T72, T75, and T81, reported site-specific effects (128). All mono-glycations had limited effects on the binding affinity of α-synuclein to lipid vesicles, whereas the triple-glycation decreased the helicity of α-synuclein upon membrane binding. All glycations inhibited seeded aggregation, with T81 and the triple-glycation having the most profound effects. Fibrils of both T75 and triple-glycations added to primary cultured mouse neurons with monomeric α-synuclein, were less toxic that wild-type α-synuclein (128). T72 and T75 are both within an area of low solubility in α-synuclein (Figure 4A) and so glycation at that site may increase the solubility of α-synuclein. These studies also showed that this modification has essentially no effect on phospholipid vesicle binding (126, 128). O-GlcNAcylation also impacted phosphorylation of α-synuclein, inhibiting S129 phosphorylation by CK1, PLK3, and GRK5 but increasing S87 phosphorylation by CK1 (127).
Overall, although O-GlcNAcylation does not affect α-synuclein binding to certain phospholipid membranes (128), other mixtures of phospholipids should be analysed, such as synaptic vesicle mimics, to determine if this effect would remain the same in vivo.
Post-translational Modifications of Proteins Interacting With α-Synuclein
The behaviour of α-synuclein can also be affected by post-translational modifications of other proteins interacting with it. For example, palmitoylation, which is the post-translational addition of the fatty acid palmitate to cysteine residues, mediates the interactions of SNARE proteins with lipid membranes. Thus, although α-synuclein is not known to be palmitoylated, its interactions with lipid membranes can be affected indirectly by this post-translational modification (219).
Post-Translational Modifications in the Diagnostics of Parkinson's Disease
Parkinson's disease has been traditionally diagnosed at the clinical level by the presence of motor symptoms, including bradykinesia, rigidity and resting tremor, and a range of non-motor symptoms, including constipation, anosmia, depression, and sleep disorder (220, 221). A definitive diagnosis is performed at the neuropathological level through the post-mortem detection of Lewy bodies (220). Without a well-established aetiology of the disease, however, its diagnosis remains challenging, as other movement disorders exhibit similar symptoms, such as multiple system atrophy and progressive supranuclear palsy (222, 223). Given this situation, it is crucial to establish accurate diagnostic methods at the molecular level, and indeed the search for biomarkers for Parkinson's disease has been a highly researched topic over the past several years (224).
Neuroimaging techniques offer a promising avenue of Parkinson's disease biomarker research (225), in particular through the use of positron emission tomography (PET) (226), although further developments are still required (227). In parallel efforts, many groups have focused on biomarkers present in biofluids, including cerebrospinal fluid (CSF), blood and saliva, or biopsies (228). The total levels of α-synuclein and of α-synuclein aggregate species have been investigated as potential biomarkers (229), and many more have been proposed, including through genetics, transcriptomics, and proteomics (227, 230, 231). In this review, we focus in particular on the opportunities offered by the detection and quantification of post-translationally modified forms of α-synuclein for the development of effective diagnostic tools (232).
Phosphorylation
As phosphorylation of α-synuclein is highly prevalent in Lewy bodies, the extent of α-synuclein phosphorylation have been studied as a potential biomarker for Parkinson's disease. The levels of pS129-α-synuclein in CSF have been to be significantly increased in Parkinson's patients compared to controls (233–237). pS129-α-Synuclein levels in CSF have been correlated with disease progression as measured by clinical assessment through the Unified Parkinson's Disease Rating Scale (UPDRS) (233) and disease duration (238). pS129-α-Synuclein levels in blood plasma (236), olfactory mucosa (239, 240), salivary glands (241, 242) colonic biopsies (243), and skin biopsies (232, 244–246) have also been investigated.
Nitration
Because of the links between α-synuclein nitration and Parkinson's disease, this modification has been investigated for the development of diagnostic tools for this condition. In a recent study, nitrated α-synuclein in salivary glands was detected in Parkinson's disease patients but not observed in healthy controls (247). Another study found the abundance of nitrated α-synuclein peripheral blood mononuclear cells to be significantly higher in Parkinson's disease patients than in controls. The levels of nitrated α-synuclein were also correlated with the those of reactive oxygen species, although no correlation was found between disease severity or duration (248). Nitrated α-synuclein levels were detected in colonic tissue and found to increase during ageing, and a loss of neurons was correlated with accumulation of both α-synuclein and nitrated α-synuclein (249).
Other Post-translational Modifications
Further post-translational modifications of α-synuclein have received less attention, however some studies show promise in using post-translational modifications as biomarkers for Parkinson's disease. The abundance of post-translationally modified α-synuclein was studied in α-synuclein enriched erythrocytes extracts (250). Phosphorylated Y125, nitrated Y39, and lysine-glycated α-synuclein levels were found to be increased in Parkinson's disease, while SUMOylated α-synuclein levels were reduced. Combining all the post-translational modifications led to a predictive score of Parkinson's disease with increased sensitivity and specificity. Furthermore, each post-translational modification alone, and combined correlated with Parkinson's disease severity (UPDRS scores) and all but SUMOylation correlated with duration of disease (250).
The plasma levels of ubiquitin C-terminal hydrolase L1 (UCHL1) were significantly higher in late Parkinson's disease patients compared to healthy controls and the amount of UCHL1 correlated to disease severity. UCHL1 is associated with increased ubiquitin levels and stability in neurons (251). The abundance of truncated α-synuclein in platelets was also studied, but not found to be significantly different between Parkinson's disease patients and controls (252).
Therapeutic Targeting of Post-Translational Modifications of α-Synuclein
Currently available treatments for Parkinson's disease are aimed at managing symptoms, but they do not stop the progression of the disease (253–256). Therapeutic interventions targeting α-synuclein aggregation and interactions offer promising opportunities for developing disease-modifying drugs (257). Since post-translational modifications modify how α-synuclein aggregates and interacts with lipid membranes, they offer promising opportunities for the treatment of Parkinson's disease and related synucleinopathies.
Phosphorylation
The modulation of phosphorylation of α-synuclein is an important therapeutic target, in particular through the pharmacological modulation of kinases and phosphatases (258–260). Inhibition of the α-synuclein kinase c-Abl by nilotinib, an FDA-approved cancer treatment, enhanced clearance of α-synuclein in mice, protected neurons from α-synuclein toxicity and improved motor behaviour in a mouse model of Parkinson's disease (261). By increasing methylation of phosphoprotein phosphatase 2A (PP2A), the activity of PP2A was enhanced leading to decreased α-synuclein phosphorylation at S129, which decreased α-synuclein aggregation and toxicity in mice (262).
Leucine-rich repeat kinase 2 (LRRK2) has emerged from GWAS as one of the most important risk loci for Parkinson's disease (263, 264). LRRK2 is protein kinase that regulates secretory and endocytic vesicle trafficking by phosphorylating a group of RAB proteins (265). LRRK2 is associated with both the familial and sporadic forms of Parkinson's disease, with G2019S being the most common mutation, which increases its kinase activity (266). LRRK2 has been observed to co-localise with α-synuclein in nigral Lewy bodies (267, 268), and two studies reported an increase in phosphorylated α-synuclein inclusions in mice carrying the A53T-α-synuclein and G2019S-LRRK2 mutations (269, 270). Further studies in rodents detected higher levels of pS129-α-synuclein in the striatum in G2019S-LRRK2 compared to wild-type LRRK2 carriers (271, 272). However, other studies concluded that LRKK2 is unlikely to directly phosphorylate α-synuclein (263). Other protein kinases have also been linked to Parkinson's disease, including a cyclin G-associated kinase (GAK) encoded by the GAK gene, which was identified by GWAS (273). Since GAK has been found to interact with LRRK2 (274, 275), it may impact its activity. The gene Rab29, which encodes Ras-related protein Rab-7L1, has also been linked with risk of Parkinson's disease (273). Rab-7L1 stimulates LRKK2 kinase activity (276) and so may also be involved in regulating LRKK2-induced phosphorylation.
Ubiquitination
As ubiquitination is a process closely involved in the pathology of Parkinson's disease, numerous enzymes in the ubiquitin pathway have been targeted as potential targets for therapies. Mutations in the Parkin gene, encoding an E3 ligase, can lead to familial Parkinson's disease (277, 278). Parkin co-expression with α-synuclein ameliorated α-synuclein toxicity and neuronal loss (279). Co-expression of parkin ameliorated toxicity induced by α-synuclein over expression in substantia nigra neurons of rodents (279). More recently, antibodies were developed to target and inhibit the ubiquitin E3 ligase seven in absentia homologue 1 (SIAH-1). Treatment of cells with the antibodies decreased expression and aggregation of α-synuclein and improved cell viability (280). Knock-down of USP13, a ubiquitin specific protease, in a mouse model mitigated α-synuclein-induced toxicity, as USP13 regulates parkin ubiquitination and therefore indirectly regulates α-synuclein ubiquitination (281).
Furthermore, PARK6, is also linked to familial Parkinson's disease (282, 283), and encodes PINK1 (PTEN-induced putative kinase protein 1), which has been shown to phosphorylate and stimulate the E3 ligase activity of parkin (284). The Park5 and Park15 genes have also been linked to Parkinson's disease through their involvement in the ubiquitination process. Park5 encodes a de-ubiquitinating enzyme UCHL1 (285). Protective effects of a UCHL1 variant were observed in a mouse model of Parkinson's disease, indicating this protein may also serve as a target for Parkinson's therapies (286). Park15 encodes F-box only protein 7 (FBXO7), which functions as an adaptor for an E3 ubiquitin ligase complex (the SKP1/cullin-1/F-box protein), which enables the E3 complex to recognise and ubiquitinate its substrates (287). Loss of function mutations of Park15 have been identified as causative mutations in familial Parkinson's disease (288), and have been found to colocalise with α-synuclein in Lewy bodies (289). A polymorphism of Park15 was found to be a protective factor against Parkinson's disease and so targeting FBXO7 may also be relevant for therapeutics (290).
SUMOylation
SUMOylation may play a role in the intracellular targeting, cellular levels, membrane binding, propagation and aggregation of α-synuclein, and so also could be targeted in the search for a Parkinson's disease therapeutic (165). A recent study examined the effects of α-synuclein SUMOylation, finding that overexpression of a SUMO-conjugase enzyme increased α-synuclein SUMOylation and reduced the toxicity in Parkinson's disease models (291). This finding indicates that increasing SUMOylation of α-synuclein or preventing SUMO removal may be viable targets for Parkinson's disease therapeutics.
Proteolytic Cleavage and C-Terminal Truncations
Targeting C-terminal truncations could be a viable strategy in Parkinson's disease therapeutic research, as this α-synuclein modification has been observed to be present in Lewy bodies, to accelerate aggregation in vitro and in vivo, and to enhance prion-like spreading in Parkinson's disease models (124, 125, 208, 292). Antibodies targeting the C-terminus of α-synuclein prevented C-terminal truncation improved Parkinson's pathology and motor symptoms in a mouse model, and reduced propagation of α-synuclein pathology in a cell system (293). Reducing the C-terminal truncation by the pharmacological inhibition of caspase-1, which cleaves α-synuclein at D121, was shown to mitigated neurodegeneration in a transgenic model of multiple system atrophy (294).
O-GlcNAcylation
O-GlcNAcylation of α-synuclein may offer novel opportunities for the treatment of Parkinson's disease. By building on the observation that O-GlcNAcylation can inhibit α-synuclein aggregation and ameliorate its associated toxicity (128), O-GlcNAcylated α-synuclein peptides from the NAC region were developed and shown to reduce α-synuclein aggregation (295). It has also been proposed that the pharmacological inhibition of O-GlcNAcase can increase the O-GlcNAcylation levels of α-synuclein, resulting in a lower aggregation propensity and in a reduced cellular intake of α-synuclein aggregates (296). O-GlcNAcylation has also been reported to inhibit calpain-mediated C-terminal α-synuclein truncations, which as discussed above increase the aggregation propensity of this protein. Similarly, O-GlcNAcylation competes with phosphorylation in targeting hydroxyl groups on serine and threonine side-chains (127), thus protecting α-synuclein from the increase in aggregation propensity caused by phosphorylation. In a recent study, the pharmacological inhibition of O-GlcNAcase was shown to reduce the accumulation of pS129 α-synuclein in the substantia nigra in a mouse model of Parkinson's disease (297).
Conclusions
α-Synuclein has been associated with Parkinson's disease over two decades ago (20). This discovery, however, has not yet led to the development of effective diagnostic tools and disease-modifying treatments for this disease. Such a slow progress can be attributed at least in part to the complexity of the structure, function, and interactions of α-synuclein with other cellular constituents.
In particular, the interaction of α-synuclein with cell membranes is important to determine both the function and the dysfunction of this protein. While α-synuclein does not readily spontaneously, lipid membranes provide a surface for the initial nucleation events (15). Following this observation, compounds have been identified that reduce the aggregation of α-synuclein by displacing it from lipid membranes, and reducing oligomer formation in membranes in vitro, in primary neuronal cells and in mice models of Parkinson's disease (298, 299).
In this review, we have discussed the role of post-translational modifications of α-synuclein in altering the behaviour of this protein in the presence of lipid membranes (Table 1) (137). N-terminal acetylation has been shown to regulate the binding of α-synuclein to phospholipid membranes, particularly to those of synaptic vesicles (16, 107). Phosphorylation and nitration appear to aggravate the pathology of α-synuclein by decreasing lipid membrane interactions, SUMOylation may be involved in the cell-to-cell spreading of α-synuclein aggregates by enhancing binding to extracellular vesicles, and α-synuclein truncations could promote aggregation through either primary or secondary events (Table 1).
Furthermore, post-translational modifications of α-synuclein can affect and modulate each other. For example, O-GlcNAcylation at S87 was found to regulate phosphorylation of S129 and S87 (126). S87 can be glycated or phosphorylated, K96 can be SUMOylated or ubiquitinated, and K102 can be SUMOylated or the site of truncation. Post-translational modifications might also upregulate other modifications, for example most of the ubiquitinated α-synuclein in Lewy bodies was found to be phosphorylated at S129 (103).
In conclusion, we have described how the investigation of the effects of post-translational modifications on the interaction of α-synuclein with lipid membranes is increasing our understanding the molecular origins of Parkinson's disease, and contributing to the identification of novel targets for therapeutic (253–256) and diagnostic (300, 301) interventions.
Author Contributions
RB and MV reviewed the literature and wrote the article. Both authors contributed to the article and approved the submitted version.
Funding
We acknowledge funding from the Centre for Misfolding Diseases and from the Wellcome Trust (203249/Z/16/Z).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AA, Arachidonic acid; ALA, α-lipoic acid; α-syn, α-synuclein; BOG, β-octyl-glucoside; CD, circular dichroism; Chol, cholesterol; DHA, docosahexaenoic acid 22:6(n-3); DMPS, 1, 2-dimyristoyl-sn-glycero-3-phospho-L-serine; DAT, dopamine transporter; DOPC, 1, 2-dioleoyl-sn-glycero-3-phosphocholine; DOPE, 1, 2-dioleoyl-sn-glycero-3-phosphoethanolamine; DOPS, 1, 2-dioleoyl-sn-glycero-3-phospho-L-serine; GMPV, giant cell-membrane derived plasma vesicle; LUV, large unilamellar vesicle; L-DOPA, L-3, 4-dihydroxyphenylalanine; MUFA, mono-unsaturated fatty acid; GlcNAc, N-acetylglucosamine; NMR, nuclear magnetic resonance; OGT, O-GlcNAc transferase; OA, oleic acid; PL, phospholipids; PA, phosphatidic acid; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PI, phosphatidylinositol; PS, phosphatidylserine; POPA, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate; POPC, 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine; POPG, 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol); POPS, 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine; PUFA, poly-unsaturated fatty acid; PTM, post-translational modification; SUMO, small ubiquitin-like modifier; SUV, small unilamellar vesicle; SDS, sodium dodecyl sulphate; SM, sphingomyelin; SN, substantia nigra; TH, tyrosine hydroxylase; Ub, ubiquitin; VMAT2, vesicular monoamine transporter; WT, wild-type.
References
1. Breydo L, Wu JW, Uversky VN. α-Synuclein misfolding and Parkinson's disease. Biochim Biophys Acta - Mol Basis Dis. (2012) 1822:261–85. doi: 10.1016/j.bbadis.2011.10.002
2. Dedmon MM, Lindorff-Larsen K, Christodoulou J, Vendruscolo M, Dobson CM. Mapping long-range interactions in α-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J Am Chem Soc. (2005) 127:476–7. doi: 10.1021/ja044834j
3. Theillet FX, Binolfi A, Bekei B, Martorana A, Rose HM, Stuiver M, et al. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature. (2016) 530:45–50. doi: 10.1038/nature16531
4. Waudby CA, Camilloni C, Fitzpatrick AWP, Cabrita LD, Dobson CM, Vendruscolo M, et al. In-Cell NMR characterization of the secondary structure populations of a disordered conformation of α-synuclein within E. coli cells. PLoS ONE. (2013) 8:e72286. doi: 10.1371/journal.pone.0072286
5. Fusco G, De Simone A, Gopinath T, Vostrikov V, Vendruscolo M, Dobson CM, et al. Direct observation of the three regions in α-synuclein that determine its membrane-bound behaviour. Nat Commun. (2014) 5:3827. doi: 10.1038/ncomms4827
6. Ulmer TS, Bax A. Comparison of structure and dynamics of micelle-bound human α-synuclein and Parkinson disease variants. J Biol Chem. (2005) 280:43179–87. doi: 10.1074/jbc.M507624200
7. Burré J, Sharma M, Südhof TCTC. Cell biology and pathophysiology of α-synuclein. Cold Spring Harb Perspect Med. (2018) 8:a024091. doi: 10.1101/cshperspect.a024091
8. Sulzer D, Edwards RH. The physiological role of α-synuclein and its relationship to Parkinson's disease. J Neurochem. (2019) 150:475–86. doi: 10.1111/jnc.14810
9. Bendor JT, Logan TP, Edwards RH. The function of α-synuclein. Neuron. (2013) 79:1044–66. doi: 10.1016/j.neuron.2013.09.004
10. Alza NP, Iglesias González PA, Conde MA, Uranga RM, Salvador GA. Lipids at the crossroad of α-synuclein function and dysfunction: biological and pathological implications. Front Cell Neurosci. (2019) 13:175. doi: 10.3389/fncel.2019.00175
11. Chandra S, Chen X, Rizo J, Jahn R, Südhof TC. A broken α-helix in folded α-synuclein. J Biol Chem. (2003) 278:15313–8. doi: 10.1074/jbc.M213128200
12. Georgieva ER, Ramlall TF, Borbat PP, Freed JH, Eliezer D. The lipid-binding domain of wild type and mutant α-synuclein: compactness and interconversion between the broken and extended helix forms. J Biol Chem. (2010) 285:28261–74. doi: 10.1074/jbc.M110.157214
13. Fusco G, De Simone A, Arosio P, Vendruscolo M, Veglia G, Dobson CM. Structural ensembles of membrane-bound α-synuclein reveal the molecular determinants of synaptic vesicle affinity. Sci Rep. (2016) 6:27125. doi: 10.1038/srep27125
14. Van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. (2008) 9:112–24. doi: 10.1038/nrm2330
15. Galvagnion C. The role of lipids interacting with α-synuclein in the pathogenesis of Parkinson's disease. J Parkinsons Dis. (2017) 7:433–50. doi: 10.3233/JPD-171103
16. Man WK, De Simone A, Barritt JD, Vendruscolo M, Dobson CM, Fusco G. A role of cholesterol in modulating the binding of α-synuclein to synaptic-like vesicles. Front Neurosci. (2020) 14:18. doi: 10.3389/fnins.2020.00018
17. Rhoades E, Ramlall TF, Webb WW, Eliezer D. Quantification of α-synuclein binding to lipid vesicles using fluorescence correlation spectroscopy. Biophys J. (2006) 90:4692–700. doi: 10.1529/biophysj.105.079251
18. Ray Dorsey E, Elbaz A, Nichols E, Abd-Allah F, Abdelalim A, Adsuar JC, et al. Global, regional, and national burden of Parkinson's disease, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. (2018) 17:939–53. doi: 10.1016/S1474-4422(18)30295-3
19. Balestrino R, Schapira AHV. Parkinson disease. Eur J Neurol. (2020) 27:27–42. doi: 10.1111/ene.14108
20. Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc Natl Acad Sci USA. (1998) 95:6469–73. doi: 10.1073/pnas.95.11.6469
21. Shahmoradian SH, Lewis AJ, Genoud C, Hench J, Moors TE, Navarro PP, et al. Lewy pathology in Parkinson's disease consists of crowded organelles and lipid membranes. Nat Neurosci. (2019) 22:1099–109. doi: 10.1038/s41593-019-0423-2
22. Mahul-Mellier AL, Burtscher J, Maharjan N, Weerens L, Croisier M, Kuttler F, et al. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc Natl Acad Sci USA. (2020) 117:4971–82. doi: 10.1073/pnas.1913904117
23. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science. (1997) 276:2045–7. doi: 10.1126/science.276.5321.2045
24. Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, et al. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's disease. Nat Genet. (1998) 18:106–8. doi: 10.1038/ng0298-106
25. Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The new mutation, E46K, of α-Synuclein causes Parkinson and lewy body dementia. Ann Neurol. (2004) 55:164–73. doi: 10.1002/ana.10795
26. Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, et al. A novel α-synuclein missense mutation in Parkinson disease. Neurology. (2013) 80:1062–4. doi: 10.1212/WNL.0b013e31828727ba
27. Lesage S, Anheim M, Letournel F, Bousset L, Honoré A, Rozas N, et al. G51D α-synuclein mutation causes a novel Parkinsonian-pyramidal syndrome. Ann Neurol. (2013) 73:459–71. doi: 10.1002/ana.23894
28. Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, et al. A novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson's disease-type pathology. Neurobiol Aging. (2014) 35:2180.e1–e5. doi: 10.1016/j.neurobiolaging.2014.03.024
29. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. α-synuclein locus triplication causes Parkinson's disease. Science. (2003) 302:841. doi: 10.1126/science.1090278
30. Chartier-Harlin M-C, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, et al. α-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. (2004) 364:1167–9. doi: 10.1016/S0140-6736(04)17103-1
31. Ferese R, Modugno N, Campopiano R, Santilli M, Zampatti S, Giardina E, et al. Four copies of SNCA responsible for autosomal dominant Parkinson's disease in two italian siblings. Parkinsons Dis. (2015) 2015:546462. doi: 10.1155/2015/546462
32. Kim WS, Kagedal K, Halliday GM. Alpha-synuclein biology in Lewy body diseases. Alzheimer's Res Ther. (2014) 6:73. doi: 10.1186/s13195-014-0073-2
33. Del Tredici K, Braak H. Sporadic Parkinson's disease: development and distribution of α-synuclein pathology. Neuropathol Appl Neurobiol. (2016) 42:33–50. doi: 10.1111/nan.12298
34. Henderson MX, Trojanowski JQ, Lee VMY. α-Synuclein pathology in Parkinson's disease and related α-synucleinopathies. Neurosci Lett. (2019) 709:134316. doi: 10.1016/j.neulet.2019.134316
35. Xicoy H, Wieringa B, Martens GJM. The role of lipids in Parkinson's disease. Cells. (2019) 8:27. doi: 10.3390/cells8010027
36. Brundin P, Dave KD, Kordower JH. Therapeutic approaches to target alpha-synuclein pathology. Exp Neurol. (2017) 298:225–35. doi: 10.1016/j.expneurol.2017.10.003
37. Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. (2013) 14:38–48. doi: 10.1038/nrn3406
38. Dehay B, Bourdenx M, Gorry P, Przedborski S, Vila M, Hunot S, et al. Targeting α-synuclein for treatment of Parkinson's disease: mechanistic and therapeutic considerations. Lancet Neurol. (2015) 14:855–66. doi: 10.1016/S1474-4422(15)00006-X
39. Weihofen A, Liu YT, Arndt JW, Huy C, Quan C, Smith BA, et al. Development of an aggregate-selective, human-derived α-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson's disease models. Neurobiol Dis. (2019) 124:276–88. doi: 10.1016/j.nbd.2018.10.016
40. Merchant KM, Cedarbaum JM, Brundin P, Dave KD, Eberling J, Espay AJ, et al. A proposed roadmap for Parkinson's disease proof of concept clinical trials investigating compounds targeting alpha-synuclein. J Parkinsons Dis. (2019) 9:31–61. doi: 10.3233/JPD-181471
41. Staats R, Michaels TCT, Flagmeier P, Chia S, Horne RI, Habchi J, et al. Screening of small molecules using the inhibition of oligomer formation in α-synuclein aggregation as a selection parameter. Commun Chem. (2020) 3:1–9. doi: 10.1038/s42004-020-00412-y
42. Guerrero-Ferreira R, Kovacik L, Ni D, Stahlberg H. New insights on the structure of alpha-synuclein fibrils using cryo-electron microscopy. Curr Opin Neurobiol. (2020) 61:89–95. doi: 10.1016/j.conb.2020.01.014
43. Buell AK, Galvagnion C, Gaspar R, Sparr E, Vendruscolo M, Knowles TPJ, et al. Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc Natl Acad Sci USA. (2014) 111:7671–6. doi: 10.1073/pnas.1315346111
44. Cremades N, Cohen SIA, Deas E, Abramov AY, Chen AY, Orte A, et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell. (2012) 149:1048–59. doi: 10.1016/j.cell.2012.03.037
45. Gaspar R, Meisl G, Buell AK, Young L, Kaminski CF, Knowles TPJ, et al. Secondary nucleation of monomers on fibril surface dominates α-synuclein aggregation and provides autocatalytic amyloid amplification. Q Rev Biophys. (2017) 50:e6. doi: 10.1017/S0033583516000172
46. Hardenberg M, Sinnige T, Casford S, Dada S, Poudel C, Robinson L, et al. Observation of an α-synuclein liquid droplet state and its maturation into Lewy body-like assemblies. J Mol Cell Biol. (2020). doi: 10.1101/2020.06.08.140798. [Epub ahead of print].
47. Galvagnion C, Brown JWP, Ouberai MM, Flagmeier P, Vendruscolo M, Buell AK, et al. Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of α-synuclein. Proc Natl Acad Sci USA. (2016) 113:7065–70. doi: 10.1073/pnas.1601899113
48. Knowles TPJ, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol. (2014) 15:384–96. doi: 10.1038/nrm3810
49. Zhang J, Li X, Li JD. The roles of post-translational modifications on α-synuclein in the pathogenesis of Parkinson's diseases. Front Neurosci. (2019) 13:381. doi: 10.3389/fnins.2019.00381
50. Gonzalez-Garcia M, Fusco G, De Simone A. Membrane interactions and toxicity by misfolded protein oligomers. Front Cell Dev Biol. (2021) 9:642623. doi: 10.3389/fcell.2021.642623
51. Musteikyte G, Jayaram AK, Xu CK, Vendruscolo M, Krainer G, Knowles TPJ. Interactions of α-synuclein oligomers with lipid membranes. Biochim Biophys Acta - Biomembr. (2021) 1863:183536. doi: 10.1016/j.bbamem.2020.183536
52. Kulenkampff K, Wolf Perez AM, Sormanni P, Habchi J, Vendruscolo M. Quantifying misfolded protein oligomers as drug targets and biomarkers in Alzheimer and Parkinson diseases. Nat Rev Chem. (2021) 5:277–94. doi: 10.1038/s41570-021-00254-9
53. Olzscha H, Schermann SM, Woerner AC, Pinkert S, Hecht MH, Tartaglia GG, et al. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell. (2011) 144:67–78. doi: 10.1016/j.cell.2010.11.050
54. Uemura N, Uemura MT, Luk KC, Lee VMY, Trojanowski JQ. Cell-to-cell transmission of Tau and α-Synuclein. Trends Mol Med. (2020) 26:936–52. doi: 10.1016/j.molmed.2020.03.012
55. Galvagnion C, Buell AK, Meisl G, Michaels TCT, Vendruscolo M, Knowles TPJ, et al. Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat Chem Biol. (2015) 11:229–34. doi: 10.1038/nchembio.1750
56. Campioni S, Carret G, Jordens S, Nicoud L, Mezzenga R, Riek R. The presence of an air-water interface affects formation and elongation of α-synuclein fibrils. J Am Chem Soc. (2014) 136:2866–75. doi: 10.1021/ja412105t
57. Peng C, Gathagan RJ, Covell DJ, Medellin C, Stieber A, Robinson JL, et al. Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature. (2018) 557:558–63. doi: 10.1038/s41586-018-0104-4
58. Stephens AD, Zacharopoulou M, Kaminski Schierle GS. The cellular environment affects monomeric α-synuclein structure. Trends Biochem Sci. (2019) 44:453–66. doi: 10.1016/j.tibs.2018.11.005
59. Puentes L, Lengyel-Zhand Z, Lee JY, Hsieh C-J, Schneider M, Edwards K, et al. Poly (ADP-ribose) induces α-synuclein aggregation in neuronal-like cells and interacts with phosphorylated α-synuclein in post mortem PD samples. bioRxiv [preprint]. (2020) 2020.04.08.032250. doi: 10.1101/2020.04.08.032250
60. Marie G, Dunning CJ, Gaspar R, Grey C, Brundin P, Sparr E, et al. Acceleration of α-synuclein aggregation by exosomes. J Biol Chem. (2015) 290:2969–82. doi: 10.1074/jbc.M114.585703
61. Lööv C, Scherzer CR, Hyman BT, Breakefield XO, Ingelsson M. α-synuclein in extracellular vesicles: functional implications and diagnostic opportunities. Cell Mol Neurobiol. (2016) 36:437–48. doi: 10.1007/s10571-015-0317-0
62. Thompson AG, Gray E, Heman-Ackah SM, Mäger I, Talbot K, El Andaloussi S, et al. Extracellular vesicles in neurodegenerative disease-pathogenesis to biomarkers. Nat Rev Neurol. (2016) 12:346–57. doi: 10.1038/nrneurol.2016.68
63. Fanning S, Selkoe D, Dettmer U. Parkinson's disease: proteinopathy or lipidopathy? NPJ Park Dis. (2020) 6:3. doi: 10.1038/s41531-019-0103-7
64. Stok R, Ashkenazi A. Lipids as the key to understanding α-synuclein behaviour in Parkinson disease. Nat Rev Mol Cell Biol. (2020) 21:357–8. doi: 10.1038/s41580-020-0235-y
65. Klemann CJHM, Martens GJM, Sharma M, Martens MB, Isacson O, Gasser T, et al. Integrated molecular landscape of Parkinson's disease. npj Park Dis. (2017) 3:155–9. doi: 10.1038/s41531-017-0015-3
66. Morgan NV, Westaway SK, Morton JEV, Gregory A, Gissen P, Sonek S, et al. PLA2G6, encoding a phospholipase A 2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet. (2006) 38:752–4. doi: 10.1038/ng1826
67. Thaler A, Gurevich T, Bar Shira A, Gana Weisz M, Ash E, Shiner T, et al. A “dose” effect of mutations in the GBA gene on Parkinson's disease phenotype. Park Relat Disord. (2017) 36:47–51. doi: 10.1016/j.parkreldis.2016.12.014
68. Gan-Or Z, Amshalom I, Kilarski LL, Bar-Shira A, Gana-Weisz M, Mirelman A. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology. (2015) 84:880–7. doi: 10.1212/WNL.0000000000001315
69. Henderson MX, Sedor S, McGeary I, Cornblath EJ, Peng C, Riddle DM, et al. Glucocerebrosidase activity modulates neuronal susceptibility to pathological α-synuclein insult. Neuron. (2020) 105:822–36.e7. doi: 10.1016/j.neuron.2019.12.004
70. Sormanni P, Aprile FA, Vendruscolo M. The CamSol method of rational design of protein mutants with enhanced solubility. J Mol Biol. (2015) 427:478–90. doi: 10.1016/j.jmb.2014.09.026
71. Volles MJ, Lansbury PT. Relationships between the sequence of α-synuclein and its membrane affinity, fibrillization propensity, and yeast toxicity. J Mol Biol. (2007) 366:1510–22. doi: 10.1016/j.jmb.2006.12.044
72. Gitler AD, Bevis BJ, Shorter J, Strathearn KE, Hamamichi S, Su LJ, et al. The Parkinson's disease protein α-synuclein disrupts cellular Rab homeostasis. Proc Natl Acad Sci USA. (2008) 105:145–50. doi: 10.1073/pnas.0710685105
73. Rovere M, Sanderson JB, Fonseca-Ornelas L, Patel DS, Bartels T. Refolding of helical soluble α-synuclein through transient interaction with lipid interfaces. FEBS Lett. (2018) 592:1464–72. doi: 10.1002/1873-3468.13047
74. Dettmer U, Newman AJ, Soldner F, Luth ES, Kim NC, Von Saucken VE, et al. Parkinson-causing α-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat Commun. (2015) 6:8008. doi: 10.1038/ncomms9008
75. Nuber S, Rajsombath M, Minakaki G, Winkler J, Müller CP, Ericsson M, et al. Abrogating native α-Synuclein tetramers in mice causes a L-DOPA-responsive motor syndrome closely resembling Parkinson's disease. Neuron. (2018) 100:75–90.e5. doi: 10.1016/j.neuron.2018.09.014
76. Fanning S, Haque A, Imberdis T, Baru V, Barrasa MI, Nuber S, et al. Lipidomic analysis of α-Synuclein neurotoxicity identifies stearoyl CoA desaturase as a target for Parkinson treatment. Mol Cell. (2019) 73:1001–14.e8. doi: 10.1016/j.molcel.2018.11.028
77. Sharon R, Bar-Joseph I, Mirick GE, Serhan CN, Selkoe DJ. Altered fatty acid composition of dopaminergic neurons expressing α-Synuclein and human brains with α-Synucleinopathies. J Biol Chem. (2003) 278:49874–81. doi: 10.1074/jbc.M309127200
78. De Franceschi G, Fecchio C, Sharon R, Schapira AHV, Proukakis C, Bellotti V, De Laureto PP. α-Synuclein structural features inhibit harmful polyunsaturated fatty acid oxidation, suggesting roles in neuroprotection. J Biol Chem. (2017) 292:6927–37. doi: 10.1074/jbc.M116.765149
79. De Franceschi G, Frare E, Pivato M, Relini A, Penco A, Greggio E, et al. Structural and morphological characterization of aggregated species of α-synuclein induced by docosahexaenoic acid. J Biol Chem. (2011) 286:22262–74. doi: 10.1074/jbc.M110.202937
80. Broersen K, Van Den Brink D, Fraser G, Goedert M, Davletov B. α-synuclein adopts an α-helical conformation in the presence of polyunsaturated fatty acids to hinder micelle formation. Biochemistry. (2006) 45:15610–6. doi: 10.1021/bi061743l
81. Rantham Prabhakara JP, Feist G, Thomasson S, Thompson A, Schommer E, Ghribi O. Differential effects of 24-hydroxycholesterol and 27-hydroxycholesterol on tyrosine hydroxylase and α-synuclein in human neuroblastoma SH-SY5Y cells. J Neurochem. (2008) 107:1722–9. doi: 10.1111/j.1471-4159.2008.05736.x
82. Marwarha G, Rhen T, Schommer T, Ghribi O. The oxysterol 27-hydroxycholesterol regulates α-synuclein and tyrosine hydroxylase expression levels in human neuroblastoma cells through modulation of liver X receptors and estrogen receptors-relevance to Parkinson's disease. J Neurochem. (2011) 119:1119–36. doi: 10.1111/j.1471-4159.2011.07497.x
83. Cheng D, Kim WS, Garner B. Regulation of α-synuclein expression by liver X receptor ligands in vitro. Neuroreport. (2008) 19:1685–9. doi: 10.1097/WNR.0b013e32831578b2
84. Nakamura K, Nemani VM, Azarbal F, Skibinski G, Levy JM, Egami K, et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein α-synuclein. J Biol Chem. (2011) 286:20710–26. doi: 10.1074/jbc.M110.213538
85. Varkey J, Isas JM, Mizuno N, Jensen MB, Bhatia VK, Jao CC, et al. Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J Biol Chem. (2010) 285:32486–93. doi: 10.1074/jbc.M110.139576
86. Parihar MS, Parihar A, Fujita M, Hashimoto M, Ghafourifar P. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell Mol Life Sci. (2008) 65:1272–84. doi: 10.1007/s00018-008-7589-1
87. Jiang Z, Flynn JD, Teague WE, Gawrisch K, Lee JC. Stimulation of α-synuclein amyloid formation by phosphatidylglycerol micellar tubules. Biochim Biophys Acta - Biomembr. (2018) 1860:1840–7. doi: 10.1016/j.bbamem.2018.02.025
88. Pfefferkorn CM, Heinrich F, Sodt AJ, Maltsev AS, Pastor RW, Lee JC. Depth of α-synuclein in a bilayer determined by fluorescence, neutron reflectometry, and computation. Biophys J. (2012) 102:613–21. doi: 10.1016/j.bpj.2011.12.051
89. Mizuno N, Varkey J, Kegulian NC, Hegde BG, Cheng N, Langen R, et al. Remodeling of lipid vesicles into cylindrical micelles by α-synuclein in an extended α-helical conformation. J Biol Chem. (2012) 287:29301–11. doi: 10.1074/jbc.M112.365817
90. Eichmann C, Kumari P, Riek R. High-density lipoprotein-like particle formation of Synuclein variants. FEBS Lett. (2017) 591:304–11. doi: 10.1002/1873-3468.12543
91. Broersen K, Ruiperez V, Davletov B. Structural and aggregation properties of alpha-synuclein linked to phospholipase A2 action. Protein Pept Lett. (2018) 25:368–78. doi: 10.2174/0929866525666180326120052
92. Mizuno S, Sasai H, Kume A, Takahashi D, Satoh M, Kado S, et al. Dioleoyl-phosphatidic acid selectively binds to α-synuclein and strongly induces its aggregation. FEBS Lett. (2017) 591:784–91. doi: 10.1002/1873-3468.12592
93. Ghio S, Camilleri A, Caruana M, Ruf VC, Schmidt F, Leonov A, et al. Cardiolipin promotes pore-forming activity of alpha-synuclein oligomers in mitochondrial membranes. ACS Chem Neurosci. (2019) 10:3815–29. doi: 10.1021/acschemneuro.9b00320
94. Snead D, Eliezer D. Alpha-synuclein function and dysfunction on cellular membranes. Exp Neurobiol. (2014) 23:292. doi: 10.5607/en.2014.23.4.292
95. Barrett PJ, Timothy Greenamyre J. Post-translational modification of α-synuclein in Parkinson's disease. Brain Res. (2015) 1628:247–53. doi: 10.1016/j.brainres.2015.06.002
96. Pajarillo E, Rizor A, Lee J, Aschner M, Lee E. The role of posttranslational modifications of α-synuclein and LRRK2 in Parkinson's disease: potential contributions of environmental factors. Biochim Biophys Acta - Mol Basis Dis. (2019) 1865:1992–2000. doi: 10.1016/j.bbadis.2018.11.017
97. Oueslati A, Fournier M, Lashuel HA. Role of post-translational modifications in modulating the structure, function and toxicity of α-synuclein. Implications for Parkinson's disease pathogenesis and therapies. In: Björklund A, Cenci M, editors. Progress in Brain Research. Lund: Elsevier B.V. p. 115–45. doi: 10.1016/S0079-6123(10)83007-9
98. Stephens AD, Zacharopoulou M, Moons R, Fusco G, Seetaloo N, Chiki A, et al. Extent of N-terminus exposure by altered long-range interactions of monomeric alpha-synuclein determines its aggregation propensity. Nat Commun. (2020) 11:740241. doi: 10.1101/740241
99. Arnesen T, Van Damme P, Polevoda B, Helsens K, Evjenth R, Colaert N, et al. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc Natl Acad Sci USA. (2009) 106:8157–62. doi: 10.1073/pnas.0901931106
100. Starheim KK, Gevaert K, Arnesen T. Protein N-terminal acetyltransferases: when the start matters. Trends Biochem Sci. (2012) 37:152–61. doi: 10.1016/j.tibs.2012.02.003
101. Aksnes H, Ree R, Arnesen T. Co-translational, post-translational, and non-catalytic roles of N-terminal acetyltransferases. Mol Cell. (2019) 73:1097–114. doi: 10.1016/j.molcel.2019.02.007
102. Ree R, Varland S, Arnesen T. Spotlight on protein N-terminal acetylation. Exp Mol Med. (2018) 50:1–13. doi: 10.1038/s12276-018-0116-z
103. Anderson JP, Walker DE, Goldstein JM, De Laat R, Banducci K, Caccavello RJ, et al. Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic lewy body disease. J Biol Chem. (2006) 281:29739–52. doi: 10.1074/jbc.M600933200
104. Vinueza-Gavilanes R, Íñigo-Marco I, Larrea L, Lasa M, Carte B, Santamaría E, Fernández-Irigoyen J, et al. N-terminal acetylation mutants affect alpha-synuclein stability, protein levels and neuronal toxicity. Neurobiol Dis. (2020) 137:104781. doi: 10.1016/j.nbd.2020.104781
105. Aksnes H, Drazic A, Marie M, Arnesen T. First things first: vital protein marks by N-terminal acetyltransferases. Trends Biochem Sci. (2016) 41:746–60. doi: 10.1016/j.tibs.2016.07.005
106. Bhattacharjee P, Öhrfelt A, Lashley T, Blennow K, Brinkmalm A, Zetterberg H. Mass spectrometric analysis of lewy body-enriched α-synuclein in Parkinson's disease. J Proteome Res. (2019) 18:2109–20. doi: 10.1021/acs.jproteome.8b00982
107. Kang L, Moriarty GM, Woods LA, Ashcroft AE, Radford SE, Baum J. N-terminal acetylation of α-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci. (2012) 21:911–7. doi: 10.1002/pro.2088
108. Runfola M, De Simone A, Vendruscolo M, Dobson CM, Fusco G. The N-terminal acetylation of α-synuclein changes the affinity for lipid membranes but not the structural properties of the bound state. Sci Rep. (2020) 10:204. doi: 10.1038/s41598-019-57023-4
109. O'Leary EI, Jiang Z, Strub MP, Lee JC. Effects of phosphatidylcholine membrane fluidity on the conformation and aggregation of N-terminally acetylated α-Synuclein. J Biol Chem. (2018) 293:11195–205. doi: 10.1074/jbc.RA118.002780
110. Birol M, Wojcik SP, Miranker AD, Rhoades E. Identification of N-linked glycans as specific mediators of neuronal uptake of acetylated α-Synuclein. PLoS Biol. (2019) 17:e3000318. doi: 10.1371/journal.pbio.3000318
111. Dikiy I, Eliezer D. N-terminal acetylation stabilizes N-terminal helicity in lipid- and micelle-bound α-Synuclein and increases its affinity for physiological membranes. J Biol Chem. (2014) 289:3652–65. doi: 10.1074/jbc.M113.512459
112. Maltsev AS, Ying J, Bax A. Impact of N-terminal acetylation of α-synuclein on its random coil and lipid binding properties. Biochemistry. (2012) 51:5004–13. doi: 10.1021/bi300642h
113. Fauvet B, Fares MB, Samuel F, Dikiy I, Tandon A, Eliezer D, et al. Characterization of semisynthetic and naturally N α- acetylated α-synuclein in vitro and in intact cells: implications for aggregation and cellular properties of α-synuclein. J Biol Chem. (2012) 287:28243–62. doi: 10.1074/jbc.M112.383711
114. Samuel F, Flavin WP, Iqbal S, Pacelli C, Renganathan SDS, Trudeau LE, et al. Effects of serine 129 phosphorylation on α-synuclein aggregation, membrane association, and internalization. J Biol Chem. (2016) 291:4374–85. doi: 10.1074/jbc.M115.705095
115. Dikiy I, Fauvet B, Jovičić A, Mahul-Mellier AL, Desobry C, El-Turk F, et al. Semisynthetic and in vitro phosphorylation of alpha-synuclein at Y39 promotes functional partly helical membrane-bound states resembling those induced by PD mutations. ACS Chem Biol. (2016) 11:2428–37. doi: 10.1021/acschembio.6b00539
116. Paleologou KE, Oueslati A, Shakked G, Rospigliosi CC, Kim HY, Lamberto GR, et al. Phosphorylation at S87 is enhanced in synucleinopathies, inhibits α-synuclein oligomerization, and influences synuclein-membrane interactions. J Neurosci. (2010) 30:3184–98. doi: 10.1523/JNEUROSCI.5922-09.2010
117. Ma MR, Hu ZW, Zhao YF, Chen YX, Li YM. Phosphorylation induces distinct alpha-synuclein strain formation. Sci Rep. (2016) 6:eaau8645. doi: 10.1038/srep37130
118. Sugeno N, Hasegawa T, Tanaka N, Fukuda M, Wakabayashi K, Oshima R, et al. Lys-63-linked ubiquitination by E3 ubiquitin ligase Nedd4-1 facilitates endosomal sequestration of internalized α-Synuclein. J Biol Chem. (2014) 289:18137–51. doi: 10.1074/jbc.M113.529461
119. Hejjaoui M, Haj-Yahya M, Kumar KSA, Brik A, Lashuel HA. Towards elucidation of the role of ubiquitination in the pathogenesis of parkinson's disease with semisynthetic ubiquitinated α-synuclein. Angew Chemie - Int Ed. (2011) 50:405–9. doi: 10.1002/anie.201005546
120. Kunadt M, Eckermann K, Stuendl A, Gong J, Russo B, Strauss K, et al. Extracellular vesicle sorting of α-Synuclein is regulated by sumoylation. Acta Neuropathol. (2015) 129:695–713. doi: 10.1007/s00401-015-1408-1
121. Burai R, Ait-Bouziad N, Chiki A, Lashuel HA. Elucidating the role of site-specific nitration of α-synuclein in the pathogenesis of Parkinson's disease via protein semisynthesis and mutagenesis. J Am Chem Soc. (2015) 137:5041–52. doi: 10.1021/ja5131726
122. Hodara R, Norris EH, Giasson BI, Mishizen-Eberz AJ, Lynch DR, Lee VMY, et al. Functional consequences of α-synuclein tyrosine nitration: Diminished binding to lipid vesicles and increased fibril formation. J Biol Chem. (2004) 279:47746–53. doi: 10.1074/jbc.M408906200
123. Sevcsik E, Trexler AJ, Dunn JM, Rhoades E. Allostery in a disordered protein: oxidative modifications to α-synuclein act distally to regulate membrane binding. J Am Chem Soc. (2011) 133:7152–8. doi: 10.1021/ja2009554
124. van der Wateren IM, Knowles TPJ, Buell AK, Dobson CM, Galvagnion C. C-terminal truncation of α-synuclein promotes amyloid fibril amplification at physiological pH. Chem Sci. (2018) 9:5506–16. doi: 10.1039/C8SC01109E
125. Ma L, Yang C, Zhang X, Li Y, Wang S, Zheng L, et al. C-terminal truncation exacerbates the aggregation and cytotoxicity of α-Synuclein: A vicious cycle in Parkinson's disease. Biochim Biophys Acta - Mol Basis Dis. (2018) 1864:3714–25. doi: 10.1016/j.bbadis.2018.10.003
126. Lewis YE, Galesic A, Levine PM, De Leon CA, Lamiri N, Brennan CK, et al. O-GlcNAcylation of α-Synuclein at Serine 87 reduces aggregation without affecting membrane binding. ACS Chem Biol. (2017) 12:1020–7. doi: 10.1021/acschembio.7b00113
127. Marotta NP, Lin YH, Lewis YE, Ambroso MR, Zaro BW, Roth MT, et al. O-GlcNAc modification blocks the aggregation and toxicity of the protein α-synuclein associated with Parkinson's disease. Nat Chem. (2015) 7:913–20. doi: 10.1038/nchem.2361
128. Levine PM, Galesic A, Balana AT, Mahul-Mellier AL, Navarro MX, De Leon CA, et al. α-Synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson's disease. Proc Natl Acad Sci USA. (2019) 116:1511–9. doi: 10.1073/pnas.1808845116
129. Zabrocki P, Bastiaens I, Delay C, Bammens T, Ghillebert R, Pellens K, et al. Phosphorylation, lipid raft interaction and traffic of α-synuclein in a yeast model for Parkinson. Biochim Biophys Acta - Mol Cell Res. (2008) 1783:1767–80. doi: 10.1016/j.bbamcr.2008.06.010
130. Cohen P. The origins of protein phosphorylation. Nat Cell Biol. (2002) 4:E127–30. doi: 10.1038/ncb0502-e127
131. Ochoa D, Jarnuczak AF, Viéitez C, Gehre M, Soucheray M, Mateus A, et al. The functional landscape of the human phosphoproteome. Nat Biotechnol. (2020) 38:365–73. doi: 10.1038/s41587-019-0344-3
132. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. (2002) 298:1912–34. doi: 10.1126/science.1075762
133. Cohen P. The structure and regulation of protein phosphatases. Annu Rev Biochem. (1989) 58:453–508. doi: 10.1146/annurev.bi.58.070189.002321
134. Needham EJ, Parker BL, Burykin T, James DE, Humphrey SJ. Illuminating the dark phosphoproteome. Sci Signal. (2019) 12 eaau8645. doi: 10.1126/scisignal.aau8645
135. Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. (2005) 353:172–87. doi: 10.1056/NEJMra044389
136. Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. (2009) 9:28–39. doi: 10.1038/nrc2559
137. Chen H, Zhao YF, Chen YX, Li YM. Exploring the roles of post-translational modifications in the pathogenesis of parkinson's disease using synthetic and semisynthetic modified α-synuclein. ACS Chem Neurosci. (2019) 10:910–21. doi: 10.1021/acschemneuro.8b00447
138. Lindstedt PR, Taylor RJ, Bernardes GJL, Vendruscolo M. Facile installation of post-translational modifications on the tau protein via chemical mutagenesis. ACS Chem Neurosci. (2021) 12:557–61. doi: 10.1021/acschemneuro.0c00761
139. Oueslati A. Implication of alpha-synuclein phosphorylation at S129 in synucleinopathies: what have we learned in the last decade? J Parkinsons Dis. (2016) 6:39–51. doi: 10.3233/JPD-160779
140. Walker DG, Lue LF, Adler CH, Shill HA, Caviness JN, Sabbagh MN, et al. Changes in properties of serine 129 phosphorylated α-synuclein with progression of Lewy-type histopathology in human brains. Exp Neurol. (2013) 240:190–204. doi: 10.1016/j.expneurol.2012.11.020
141. Perfeito R, Lázaro DF, Outeiro TF, Rego AC. Linking alpha-synuclein phosphorylation to reactive oxygen species formation and mitochondrial dysfunction in SH-SY5Y cells. Mol Cell Neurosci. (2014) 62:51–9. doi: 10.1016/j.mcn.2014.08.002
142. Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, et al. α-synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. (2002) 4:160–4. doi: 10.1038/ncb748
143. Paleologou KE, Schmid AW, Rospigliosi CC, Kim HY, Lamberto GR, Fredenburg RA, et al. Phosphorylation at Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of α-synuclein. J Biol Chem. (2008) 283:16895–905. doi: 10.1074/jbc.M800747200
144. Wang X, Becker K, Levine N, Zhang M, Lieberman AP, Moore DJ, et al. Pathogenic alpha-synuclein aggregates preferentially bind to mitochondria and affect cellular respiration. Acta Neuropathol Commun. (2019) 7:41. doi: 10.1186/s40478-019-0696-4
145. Mbefo MK, Paleologou KE, Boucharaba A, Oueslati A, Schell H, Fournier M, et al. Phosphorylation of synucleins by members of the polo-like kinase family. J Biol Chem. (2010) 285:2807–22. doi: 10.1074/jbc.M109.081950
146. Waxman EA, Giasson BI. Characterization of kinases involved in the phosphorylation of aggregated α-synuclein. J Neurosci Res. (2011) 89:231–47. doi: 10.1002/jnr.22537
147. Waxman EA, Giasson BI. Specificity and regulation of casein kinase-mediated phosphorylation of α-synuclein. J Neuropathol Exp Neurol. (2008) 67:402–16. doi: 10.1097/NEN.0b013e3186fc995
148. Klein C, Westenberger A. Genetics of Parkinson's disease. Cold Spring Harb Perspect Med. (2012) 2:a008888. doi: 10.1101/cshperspect.a008888
149. Yau R, Rape M. The increasing complexity of the ubiquitin code. Nat Cell Biol. (2016) 18:579–86. doi: 10.1038/ncb3358
150. Komander D. The emerging complexity of protein ubiquitination. Biochem Soc Trans. (2009) 37:937–53. doi: 10.1042/BST0370937
151. Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. (2012) 81:203–29. doi: 10.1146/annurev-biochem-060310-170328
152. Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med. (2014) 20:1242–53. doi: 10.1038/nm.3739
153. Kuzuhara S, Mori H, Izumiyama N, Yoshimura M, Ihara Y. Lewy bodies are ubiquitinated - a light and electron microscopic immunocytochemical study. Acta Neuropathol. (1988) 75:345–53. doi: 10.1007/BF00687787
154. Haj-Yahya M, Fauvet B, Herman-Bachinsky Y, Hejjaoui M, Bavikar SN, Karthikeyan SV, et al. Synthetic polyubiquitinated -Synuclein reveals important insights into the roles of the ubiquitin chain in regulating its pathophysiology. Proc Natl Acad Sci USA. (2013) 110:17726–31. doi: 10.1073/pnas.1315654110
155. Meier F, Abeywardana T, Dhall A, Marotta NP, Varkey J, Langen R, et al. Semisynthetic, site-specific ubiquitin modification of α-synuclein reveals differential effects on aggregation. J Am Chem Soc. (2012) 134:5468–71. doi: 10.1021/ja300094r
156. Lewis YE, Abeywardana T, Lin YH, Galesic A, Pratt MR. Synthesis of a Bis-thio-acetone (BTA) analogue of the lysine isopeptide bond and its application to investigate the effects of ubiquitination and SUMOylation on α-synuclein aggregation and toxicity. ACS Chem Biol. (2016) 11:931–42. doi: 10.1021/acschembio.5b01042
157. Moon SP, Balana AT, Galesic A, Rakshit A, Pratt MR. Ubiquitination can change the structure of the α-Synuclein amyloid fiber in a site selective fashion. J Org Chem. (2020) 85:1548–55. doi: 10.1021/acs.joc.9b02641
158. Iyer A, Claessens MMAE. Disruptive membrane interactions of alpha-synuclein aggregates. Biochim Biophys Acta - Proteins Proteomics. (2019) 1867:468–82. doi: 10.1016/j.bbapap.2018.10.006
159. Melchior F, Schergaut M, Pichler A. SUMO: ligases, isopeptidases and nuclear pores. Trends Biochem Sci. (2003) 28:612–8. doi: 10.1016/j.tibs.2003.09.002
160. Yang Y, He Y, Wang X, Liang Z, He G, Zhang P, et al. Protein SUMOylation modification and its associations with disease. Open Biol. (2017) 7:170167. doi: 10.1098/rsob.170167
161. Mukhopadhyay D, Dasso M. Modification in reverse: the SUMO proteases. Trends Biochem Sci. (2007) 32:286–95. doi: 10.1016/j.tibs.2007.05.002
162. Flotho A, Melchior F. Sumoylation: a regulatory protein modification in health and disease. Annu Rev Biochem. (2013) 82:357–85. doi: 10.1146/annurev-biochem-061909-093311
163. Seeler JS, Dejean A. SUMO and the robustness of cancer. Nat Rev Cancer. (2017) 17:184–97. doi: 10.1038/nrc.2016.143
164. Da Silva-Ferrada E, Ribeiro-Rodrigues TM, Rodríguez MS, Girão H. Proteostasis and SUMO in the heart. Int J Biochem Cell Biol. (2016) 79:443–50. doi: 10.1016/j.biocel.2016.09.015
165. Savyon M, Engelender S. SUMOylation in α-Synuclein homeostasis and pathology. Front Aging Neurosci. (2020) 12:167. doi: 10.3389/fnagi.2020.00167
166. Kim YM, Jang WH, Quezado MM, Oh Y, Chung KC, Junn E, et al. Proteasome inhibition induces α-synuclein SUMOylation and aggregate formation. J Neurol Sci. (2011) 307:157–61. doi: 10.1016/j.jns.2011.04.015
167. Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol. (2007) 8:947–56. doi: 10.1038/nrm2293
168. Krumova P, Meulmeester E, Garrido M, Tirard M, Hsiao HH, Bossis G, et al. Sumoylation inhibits α-synuclein aggregation and toxicity. J Cell Biol. (2011) 194:49–60. doi: 10.1083/jcb.201010117
169. Wong MB, Goodwin J, Norazit A, Meedeniya ACB, Richter-Landsberg C, Gai WP, et al. SUMO-1 is associated with a subset of lysosomes in glial protein aggregate diseases. Neurotox Res. (2013) 23:1–21. doi: 10.1007/s12640-012-9358-z
170. Popova B, Kleinknecht A, Braus GH. Posttranslational modifications and clearing of α-synuclein aggregates in yeast. Biomolecules. (2015) 5:617–34. doi: 10.3390/biom5020617
171. Abeywardana T, Pratt M. Extent of inhibition of α-synuclein aggregation in vitro by SUMOylation is conjugation site- and SUMO isoform-selective. Biochemistry. (2015) 54:959–61. doi: 10.1021/bi501512m
172. Rott R, Szargel R, Shani V, Hamza H, Savyon M, Elghani FA, et al. SUMOylation and ubiquitination reciprocally regulate α-synuclein degradation and pathological aggregation. Proc Natl Acad Sci USA. (2017) 114:13176–81. doi: 10.1073/pnas.1704351114
173. Mata-Pérez C, Begara-Morales JC, Chaki M, Sánchez-Calvo B, Valderrama R, Padilla MN, et al. Protein tyrosine nitration during development and abiotic stress response in plants. Front Plant Sci. (2016) 7:1699. doi: 10.3389/fpls.2016.01699
174. Souza JM, Peluffo G, Radi R. Protein tyrosine nitration-Functional alteration or just a biomarker? Free Radic Biol Med. (2008) 45:357–66. doi: 10.1016/j.freeradbiomed.2008.04.010
175. Huie RE, Padmaja S. The reaction of no with superoxide. Free Radic Res. (1993) 18:195–9. doi: 10.3109/10715769309145868
176. Bartesaghi S, Radi R. Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol. (2018) 14:618–25. doi: 10.1016/j.redox.2017.09.009
177. Bayden AS, Yakovlev VA, Graves PR, Mikkelsen RB, Kellogg GE. Factors influencing protein tyrosine nitration-structure-based predictive models. Free Radic Biol Med. (2011) 50:749–62. doi: 10.1016/j.freeradbiomed.2010.12.016
178. Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. (2007) 87:315–424. doi: 10.1152/physrev.00029.2006
179. Yeo WS, Kim YJ, Kabir MH, Kang JW, Kim KP. Mass spectrometric analysis of protein tyrosine nitration in aging and neurodegenerative diseases. Mass Spectrom Rev. (2015) 34:166–83. doi: 10.1002/mas.21429
180. He Y, Yu Z, Chen S. Alpha-synuclein nitration and its implications in Parkinson's disease. ACS Chem Neurosci. (2018) 10:777–82. doi: 10.1021/acschemneuro.8b00288
181. Giasson BI, Duda JE, Murray IVJ, Chen Q, Souza JM, Hurtig HI, et al. Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science. (2000) 290:985–9. doi: 10.1126/science.290.5493.985
182. Good PF, Hsu A, Werner P, Perl DP, Warren Olanow C. Protein nitration in Parkinson's disease. J Neuropathol Exp Neurol. (1998) 57:338–42. doi: 10.1097/00005072-199804000-00006
183. Souza JM, Giasson BI, Chen Q, Lee VMY, Ischiropoulos H. Dityrosine cross-linking promotes formation of stable α-synuclein polymers: implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J Biol Chem. (2000) 275:18344–49. doi: 10.1074/jbc.M000206200
184. Liu Y, Qiang M, Wei Y, He R. A novel molecular mechanism for nitrated α-synuclein-induced cell death. J Mol Cell Biol. (2011) 3:239–49. doi: 10.1093/jmcb/mjr011
185. Yu Z, Xu X, Xiang Z, Zhou J, Zhang Z, Hu C, et al. Nitrated α-synuclein induces the loss of dopaminergic neurons in the substantia nigra of rats. PLoS ONE. (2010) 5:e9956. doi: 10.1371/journal.pone.0009956
186. Danielson SR, Held JM, Schilling B, Oo M, Gibson BW, Andersen JK. Preferentially increased nitration of α-synuclein at tyrosine-39 in a cellular oxidative model of Parkinson's disease. Anal Chem. (2009) 81:7823–8. doi: 10.1021/ac901176t
187. Uversky VN, Yamin G, Munishkina LA, Karymov MA, Millett IS, Doniach S. Effects of nitration on the structure and aggregation of α-synuclein. Mol Brain Res. (2005) 134:84–102. doi: 10.1016/j.molbrainres.2004.11.014
188. Yamin G, Uversky VN, Fink AL. Nitration inhibits fibrillation of human α-synuclein in vitro by formation of soluble oligomers. FEBS Lett. (2003) 542:147–52. doi: 10.1016/S0014-5793(03)00367-3
189. Trostchansky A, Lind S, Hodara R, Oe T, Blair IA, Ischiropoulos H, et al. Interaction with phospholipids modulates α-synuclein nitration and lipid-protein adduct formation. Biochem J. (2006) 393:343–9. doi: 10.1042/BJ20051277
190. Zilka N, Kovacech B, Barath P, Kontsekova E, Novák M. The self-perpetuating tau truncation circle. In: Biochemical Society Transactions. London: Portland Press. p. 681–6. doi: 10.1042/BST20120015
191. Yao Y, Tsirka SE. Truncation of monocyte chemoattractant protein 1 by plasmin promotes blood-brain barrier disruption. J Cell Sci. (2011) 124:1486–95. doi: 10.1242/jcs.082834
192. Jadhav S, Zilka N, Novak M. Protein truncation as a common denominator of human neurodegenerative foldopathies. Mol Neurobiol. (2013) 48:516–32. doi: 10.1007/s12035-013-8440-8
193. Chow VW, Mattson MP, Wong PC, Gleichmann M. An overview of APP processing enzymes and products. Neuromol Med. (2010) 12:1–2. doi: 10.1007/s12017-009-8104-z
194. Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VMY, et al. Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol. (1998) 152:879–84.
195. Kellie JF, Higgs RE, Ryder JW, Major A, Beach TG, Adler CH, et al. Quantitative measurement of intact alpha-synuclein proteoforms from post-mortem control and parkinson's disease brain tissue by intact protein mass spectrometry. Sci Rep. (2014) 4:5797. doi: 10.1038/srep05797
196. Sorrentino ZA, Giasson BI. The emerging role of α-synuclein truncation in aggregation and disease. J Biol Chem. (2020) 295:10224–44. doi: 10.1074/jbc.REV120.011743
197. Terada M, Suzuki G, Nonaka T, Kametani F, Tamaoka A, Hasegawa M. The effect of truncation on prion-like properties of -synuclein. J Biol Chem. (2018) 293:13910–20. doi: 10.1074/jbc.RA118.001862
198. Liu CW, Giasson BI, Lewis KA, Lee VM, DeMartino GN, Thomas PJ. A precipitating role for truncated α-synuclein and the proteasome in α-synuclein aggregation: implications for pathogenesis of parkinson disease. J Biol Chem. (2005) 280:22670–8. doi: 10.1074/jbc.M501508200
199. Iwata A, Maruyama M, Akagi T, Hashikawa T, Kanazawa I, Tsuji S, et al. Alpha-synuclein degradation by serine protease neurosin: implication for pathogenesis of synucleinopathies. Hum Mol Genet. (2003) 12:2625–35. doi: 10.1093/hmg/ddg283
200. Wang W, Nguyen LTT, Burlak C, Chegini F, Guo F, Chataway T, et al. Caspase-1 causes truncation and aggregation of the Parkinson's disease-associated protein α-synuclein. Proc Natl Acad Sci USA. (2016) 113:9587–92. doi: 10.1073/pnas.1610099113
201. Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. Fiber diffraction of synthetic α-synuclein filaments shows amyloid-like cross-β conformation. Proc Natl Acad Sci USA. (2000) 97:4897–902. doi: 10.1073/pnas.97.9.4897
202. Sung JY, Park SM, Lee CH, Um JW, Lee HJ, Kim J, et al. Proteolytic cleavage of extracellular secreted α-synuclein via matrix metalloproteinases. J Biol Chem. (2005) 280:25216–24. doi: 10.1074/jbc.M503341200
203. Kim KS, Choi YR, Park JY, Lee JH, Kim DK, Lee SJ, et al. Proteolytic cleavage of extracellular α-synuclein by plasmin: implications for Parkinson disease. J Biol Chem. (2012) 287:24862–72. doi: 10.1074/jbc.M112.348128
204. Sevlever D, Jiang P, Yen SHC. Cathepsin D is the main lysosomal enzyme involved in the degradation of α-synuclein and generation of its carboxy-terminally truncated species. Biochemistry. (2008) 47:9678–87. doi: 10.1021/bi800699v
205. Zhang Z, Kang SS, Liu X, Ahn EH, Zhang Z, He L, et al. Asparagine endopeptidase cleaves α-synuclein and mediates pathologic activities in Parkinson's disease. Nat Struct Mol Biol. (2017) 24:632–42. doi: 10.1038/nsmb.3433
206. Murray IVJ, Giasson BI, Quinn SM, Koppaka V, Axelsen PH, Ischiropoulos H, et al. Role of α-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry. (2003) 42:8530–40. doi: 10.1021/bi027363r
207. Levitan K, Chereau D, Cohen SIA, Knowles TPJ, Dobson CM, Fink AL, et al. Conserved C-terminal charge exerts a profound influence on the aggregation rate of α-synuclein. J Mol Biol. (2011) 411:329–33. doi: 10.1016/j.jmb.2011.05.046
208. Sorrentino ZA, Vijayaraghavan N, Gorion KM, Riffe CJ, Strang KH, Caldwell J, et al. Physiological C-terminal truncation of -synuclein potentiates the prion-like formation of pathological inclusions. J Biol Chem. (2018) 293:18914–32. doi: 10.1074/jbc.RA118.005603
209. Perez RG, Waymire JC, Lin E, Liu JJ, Guo F, Zigmond MJ. A role for α-synuclein in the regulation of dopamine biosynthesis. J Neurosci. (2002) 22:3090–9. doi: 10.1523/JNEUROSCI.22-08-03090.2002
210. Michell AW, Tofaris GK, Gossage H, Tyers P, Spillantini MG, Barker RA. The effect of truncated human α-synuclein (1-120) on dopaminergic cells in a transgenic mouse model of Parkinson's disease. Cell Transplant. (2007) 16:461–74. doi: 10.3727/000000007783464911
211. Ulusoy A, Febbraro F, Jensen PH, Kirik D, Romero-Ramos M. Co-expression of C-terminal truncated alpha-synuclein enhances full-length alpha-synuclein-induced pathology. Eur J Neurosci. (2010) 32:409–22. doi: 10.1111/j.1460-9568.2010.07284.x
212. Grassi D, Howard S, Zhou M, Diaz-Perez N, Urban NT, Guerrero-Given D, et al. Identification of a highly neurotoxic α-synuclein species inducing mitochondrial damage and mitophagy in Parkinson's disease. Proc Natl Acad Sci USA. (2018) 115:E2634–43. doi: 10.1073/pnas.1713849115
213. Nie H, Yi W. O-GlcNAcylation, a sweet link to the pathology of diseases. J Zhejiang Univ Sci B. (2019) 20:437–48. doi: 10.1631/jzus.B1900150
214. Hart GW, Housley MP, Slawson C. Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. (2007) 446:1017–22. doi: 10.1038/nature05815
215. Harwood KR, Hanover JA. Nutrient-driven O-GlcNAc cycling -think globally but act locally. J Cell Sci. (2014) 127:1857–67. doi: 10.1242/jcs.113233
216. Vaidyanathan K, Durning S, Wells L. Functional O-GlcNAc modifications: implications in molecular regulation and pathophysiology. Crit Rev Biochem Mol Biol. (2014) 49:140–63. doi: 10.3109/10409238.2014.884535
217. Groves JA, Lee A, Yildirir G, Zachara NE. Dynamic O-GlcNAcylation and its roles in the cellular stress response and homeostasis. Cell Stress Chaperones. (2013) 18:535–58. doi: 10.1007/s12192-013-0426-y
218. Ryan P, Xu M, Davey AK, Danon JJ, Mellick GD, Kassiou M, et al. O-GlcNAc modification protects against protein misfolding and aggregation in neurodegenerative disease. ACS Chem Neurosci. (2019) 10:2209–21. doi: 10.1021/acschemneuro.9b00143
219. Ho GP, Imberdis T, Ramalingam N, Imberdis T, Wilkie EC, Imberdis T, Dettmer U, Selkoe DJ. Upregulation of Cellular Palmitoylation Mitigates α-Synuclein Accumulation and Neurotoxicity. Mov Disord. (2021) 36:348–59. doi: 10.1002/mds.28346
220. Tarakad A, Jankovic J. Diagnosis and management of Parkinson's disease. Semin Neurol. (2017) 37:118–26. doi: 10.1055/s-0037-1601888
221. Noyce AJ, R'Bibo L, Peress L, Bestwick JP, Adams-Carr KL, Mencacci NE, et al. PREDICT-PD: an online approach to prospectively identify risk indicators of Parkinson's disease. Mov Disord. (2017) 32:219–26. doi: 10.1002/mds.26898
222. Williams DR, Litvan I. Parkinsonian syndromes. Contin Lifelong Learn Neurol. (2013) 19:1189–212. doi: 10.1212/01.CON.0000436152.24038.e0
223. Reichmann H. Premotor diagnosis of Parkinson's disease. Neurosci Bull. (2017) 33:526–34. doi: 10.1007/s12264-017-0159-5
224. Marek K, Chowdhury S, Siderowf A, Lasch S, Coffey CS, Caspell-Garcia C, et al. The Parkinson's progression markers initiative (PPMI) – establishing a PD biomarker cohort. Ann Clin Transl Neurol. (2018) 5:1460–77. doi: 10.1002/acn3.644
225. Heller J, Brcina N, Dogan I, Holtbernd F, Romanzetti S, Schulz JB, et al. Brain imaging findings in idiopathic REM sleep behavior disorder (RBD) – a systematic review on potential biomarkers for neurodegeneration. Sleep Med Rev. (2017) 34:23–33. doi: 10.1016/j.smrv.2016.06.006
226. MJFF. Alpha-Synuclein Imaging Prize | Parkinson's Disease. Alpha-Synuclein Imaging Prize (2020). Available online at: https://www.michaeljfox.org/news/alpha-synuclein-imaging-prize (accessed December 10, 2020).
227. Miller DB, O'Callaghan JP. Biomarkers of Parkinson's disease: present and future. Metabolism. (2015) 64:S40–6. doi: 10.1016/j.metabol.2014.10.030
228. Lotankar S, Prabhavalkar KS, Bhatt LK. Biomarkers for Parkinson's disease: recent advancement. Neurosci Bull. (2017) 33:585–97. doi: 10.1007/s12264-017-0183-5
229. Sanderson JB, De S, Jiang H, Rovere M, Jin M, Zaccagnini L, et al. Analysis of α-synuclein species enriched from cerebral cortex of humans with sporadic dementia with Lewy bodies. Brain Commun. (2020) 2:fcaa010. doi: 10.1093/braincomms/fcaa010
230. Dos Santos MCT, Bell R, Da Costa AN. Recent developments in circulating biomarkers in Parkinson's disease: the potential use of miRNAs in a clinical setting. Bioanalysis. (2016) 8:2497–518. doi: 10.4155/bio-2016-0166
231. Mollenhauer B, Zhang J. Biochemical premotor biomarkers for Parkinson's disease. Mov Disord. (2012) 27:644–50. doi: 10.1002/mds.24956
232. Fayyad M, Salim S, Majbour N, Erskine D, Stoops E, Mollenhauer B, et al. Parkinson's disease biomarkers based on α-synuclein. J Neurochem. (2019) 150:626–36. doi: 10.1111/jnc.14809
233. Stewart T, Sossi V, Aasly JO, Wszolek ZK, Uitti RJ, Hasegawa K, et al. Phosphorylated α-synuclein in Parkinson's disease: correlation depends on disease severity. Acta Neuropathol Commun. (2015) 3:7. doi: 10.1186/s40478-015-0185-3
234. Wang Y, Shi M, Chung KA, Zabetian CP, Leverenz JB, Berg D, et al. Phosphorylated α-synuclein in Parkinson's disease. Sci Transl Med. (2012) 4:121ra20. doi: 10.1126/scitranslmed.3002566
235. Hansson O, Hall S, Öhrfelt A, Zetterberg H, Blennow K, Minthon L, Nägga K, et al. Levels of cerebrospinal fluid α-synuclein oligomers are increased in Parkinson's disease with dementia and dementia with Lewy bodies compared to Alzheimer's disease. Alzheimer's Res Ther. (2014) 6:25. doi: 10.1186/alzrt255
236. Foulds PG, Mitchell JD, Parker A, Turner R, Green G, Diggle P, et al. Phosphorylated α-synuclein can be detected in blood plasma and is potentially a useful biomarker for Parkinson's disease. FASEB J. (2011) 25:4127–37. doi: 10.1096/fj.10-179192
237. Majbour NK, Vaikath NN, Van Dijk KD, Ardah MT, Varghese S, Vesterager LB, et al. Oligomeric and phosphorylated alpha-synuclein as potential CSF biomarkers for Parkinson's disease. Mol Neurodegener. (2016) 11:7. doi: 10.1186/s13024-016-0072-9
238. Majbour NK, Vaikath NN, Eusebi P, Chiasserini D, Ardah M, Varghese S, et al. Longitudinal changes in CSF alpha-synuclein species reflect Parkinson's disease progression. Mov Disord. (2016) 31:1535–42. doi: 10.1002/mds.26754
239. Beach TG, White CL, Hladik CL, Sabbagh MN, Connor DJ, Shill HA, et al. Olfactory bulb α-synucleinopathy has high specificity and sensitivity for Lewy body disorders. Acta Neuropathol. (2009) 117:169–74. doi: 10.1007/s00401-008-0450-7
240. Saito Y, Shioya A, Sano T, Sumikura H, Murata M, Murayama S. Lewy body pathology involves the olfactory cells in Parkinson's disease and related disorders. Mov Disord. (2016) 31:135–8. doi: 10.1002/mds.26463
241. Beach TG, Adler CH, Dugger BN, Serrano G, Hidalgo J, Henry-Watson J, et al. Submandibular gland biopsy for the diagnosis of parkinson disease. J Neuropathol Exp Neurol. (2013) 72:130–6. doi: 10.1097/NEN.0b013e3182805c72
242. Campo F, Carletti R, Fusconi M, Pellicano C, Pontieri FE, Di Gioia CR, et al. Alpha-synuclein in salivary gland as biomarker for Parkinson's disease. Rev Neurosci. (2019) 30:455–62. doi: 10.1515/revneuro-2018-0064
243. Lebouvier T, Neunlist M, des Varannes SB, Coron E, Drouard A, N'Guyen JM, et al. Colonic biopsies to assess the neuropathology of parkinson's disease and its relationship with symptoms. PLoS ONE. (2010) 5:e12728. doi: 10.1371/journal.pone.0012728
244. Donadio V, Incensi A, Leta V, Giannoccaro MP, Scaglione C, Martinelli P, et al. Skin nerve a-synuclein deposits A biomarker for idiopathic Parkinson disease. Neurology. (2014) 82:1362–9. doi: 10.1212/WNL.0000000000000316
245. Donadio V, Incensi A, El-Agnaf O, Rizzo G, Vaikath N, Del Sorbo F, et al. Skin α-synuclein deposits differ in clinical variants of synucleinopathy: an in vivo study. Sci Rep. (2018) 8:14246. doi: 10.1038/s41598-018-32588-8
246. Doppler K, Ebert S, Üçeyler N, Trenkwalder C, Ebentheuer J, Volkmann J, Sommer C. Cutaneous neuropathy in Parkinson's disease: a window into brain pathology. Acta Neuropathol. (2014) 128:99–109. doi: 10.1007/s00401-014-1284-0
247. Ma LY, Gao yan L, Li X, Ma HZ, Feng T. Nitrated alpha-synuclein in minor salivary gland biopsies in Parkinson's disease. Neurosci Lett. (2019) 704:45–9. doi: 10.1016/j.neulet.2019.03.054
248. Prigione A, Piazza F, Brighina L, Begni B, Galbussera A, DiFrancesco JC, et al. Alpha-synuclein nitration and autophagy response are induced in peripheral blood cells from patients with Parkinson disease. Neurosci Lett. (2010) 477:6–10. doi: 10.1016/j.neulet.2010.04.022
249. Xuan Q, Zhang YX, Liu DG, Chan P, Xu SL, Cui YQ. Post-translational modifications of α-synuclein contribute to neurodegeneration in the colon of elderly individuals. Mol Med Rep. (2016) 13:5077–83. doi: 10.3892/mmr.2016.5166
250. Miranda HV, Cássio R, Correia-Guedes L, Gomes MA, Chegão A, Miranda E, et al. Posttranslational modifications of blood-derived alpha-synuclein as biochemical markers for Parkinson's disease. Sci Rep. (2017) 7:13713. doi: 10.1038/s41598-017-14175-5
251. Ng ASL, Tan YJ, Lu Z, Ng EY, Ng SYE, Chia NSY, et al. Plasma ubiquitin C-terminal hydrolase L1 levels reflect disease stage and motor severity in Parkinson's disease. Aging. (2020) 12:1488–95. doi: 10.18632/aging.102695
252. Michell AW, Luheshi LM, Barker RA. Skin and platelet α-synuclein as peripheral biomarkers of Parkinson's disease. Neurosci Lett. (2005) 381:294–8. doi: 10.1016/j.neulet.2005.02.030
253. Cacabelos R. Parkinson's disease: from pathogenesis to pharmacogenomics. Int J Mol Sci. (2017) 18:551. doi: 10.3390/ijms18030551
254. McFarthing K, Buff S, Rafaloff G, Dominey T, Wyse RK, Stott SRW. Parkinson's disease drug therapies in the clinical trial pipeline: 2020. J Parkinsons Dis. (2020) 10:757–74. doi: 10.3233/JPD-202128
255. Charvin D, Medori R, Hauser RA, Rascol O. Therapeutic strategies for Parkinson disease: beyond dopaminergic drugs. Nat Rev Drug Discov. (2018) 17:804–22. doi: 10.1038/nrd.2018.136
256. Kingwell K. Zeroing in on neurodegenerative α-synuclein. Nat Rev Drug Discov. (2017) 16:371–3. doi: 10.1038/nrd.2017.95
257. Kreiser RP, Wright AK, Block NR, Hollows JE, Nguyen LT, Leforte K, et al. Therapeutic strategies to reduce the toxicity of misfolded protein oligomers. Int J Mol Sci. (2020) 21:1–33. doi: 10.3390/ijms21228651
258. Braithwaite SP, Voronkov M, Stock JB, Mouradian MM. Targeting phosphatases as the next generation of disease modifying therapeutics for Parkinson's disease. Neurochem Int. (2012) 61:899–906. doi: 10.1016/j.neuint.2012.01.031
259. Braithwaite SP, Stock JB, Mouradian MM. α-Synuclein phosphorylation as a therapeutic target in Parkinson's disease. Rev Neurosci. (2012) 23:191–8. doi: 10.1515/revneuro-2011-0067
260. Guttuso T, Andrzejewski KL, Lichter DG, Andersen JK. Targeting kinases in Parkinson's disease: a mechanism shared by LRRK2, neurotrophins, exenatide, urate, nilotinib and lithium. J Neurol Sci. (2019) 402:121–30. doi: 10.1016/j.jns.2019.05.016
261. Hebron ML, Lonskaya I, Moussa CEH. Nilotinib reverses loss of dopamine neurons and improvesmotorbehavior via autophagic degradation of α-synuclein in parkinson's disease models. Hum Mol Genet. (2013) 22:3315–28. doi: 10.1093/hmg/ddt192
262. Lee KW, Chen W, Junn E, Im JY, Grosso H, Sonsalla PK, et al. Enhanced phosphatase activity attenuates α-synucleinopathy in a mouse model. J Neurosci. (2011) 31:6963–71. doi: 10.1523/JNEUROSCI.6513-10.2011
263. Cresto N, Gardier C, Gubinelli F, Gaillard MC, Liot G, West AB, et al. The unlikely partnership between LRRK2 and α-synuclein in Parkinson's disease. Eur J Neurosci. (2019) 49:339–63. doi: 10.1111/ejn.14182
264. Whiffin N, Armean IM, Kleinman A, Marshall JL, Minikel EV, Goodrich JK, et al. The effect of LRRK2 loss-of-function variants in humans. Nat Med. (2020) 26:869. doi: 10.1038/s41591-020-0893-5
265. Abeliovich A, Gitler AD. Defects in trafficking bridge Parkinson's disease pathology and genetics. Nature. (2016) 539:207–16. doi: 10.1038/nature20414
266. Henry AG, Aghamohammadzadeh S, Samaroo H, Chen Y, Mou K, Needle E, et al. Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum Mol Genet. (2015) 24:6013–28. doi: 10.1093/hmg/ddv314
267. Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. (2004) 44:601–7. doi: 10.1016/j.neuron.2004.11.005
268. Higashi S, Biskup S, West AB, Trinkaus D, Dawson VL, Faull RLM, et al. Localization of Parkinson's disease-associated LRRK2 in normal and pathological human brain. Brain Res. (2007) 1155:208–19. doi: 10.1016/j.brainres.2007.04.034
269. Ramonet D, Daher JPL, Lin BM, Stafa K, Kim J, Banerjee R, et al. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S Mutant LRRK2. PLoS ONE. (2011) 6:e18568. doi: 10.1371/journal.pone.0018568
270. Daher JPL, Pletnikova O, Biskup S, Musso A, Gellhaar S, Galter D, et al. Neurodegenerative phenotypes in an A53T α-synuclein transgenic mouse model are independent of LRRK2. Hum Mol Genet. (2012) 21:2420–31. doi: 10.1093/hmg/dds057
271. Longo F, Mercatelli D, Novello S, Arcuri L, Brugnoli A, Vincenzi F, et al. Age-dependent dopamine transporter dysfunction and Serine129 phospho-α-synuclein overload in G2019S LRRK2 mice. Acta Neuropathol Commun. (2017) 5:22. doi: 10.1186/s40478-017-0426-8
272. Volpicelli-Daley LA, Abdelmotilib H, Liu Z, Stoyka L, Daher JPL, Milnerwood AJ, et al. G2019s-LRRK2 expression augments α-synuclein sequestration into inclusions in neurons. J Neurosci. (2016) 36:7415–27. doi: 10.1523/JNEUROSCI.3642-15.2016
273. Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet Neurol. (2019) 18:1091–102. doi: 10.1016/S1474-4422(19)30320-5
274. Beilina A, Rudenko IN, Kaganovich A, Civiero L, Chau H, Kalia SK, et al. Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc Natl Acad Sci USA. (2014) 111:2626–31. doi: 10.1073/pnas.1318306111
275. Tomkins JE, Dihanich S, Beilina A, Ferrari R, Ilacqua N, Cookson MR, et al. Comparative protein interaction network analysis identifies shared and distinct functions for the human ROCO proteins. Proteomics. (2018) 18:e1700444. doi: 10.1002/pmic.201870081
276. Purlyte E, Dhekne HS, Sarhan AR, Gomez R, Lis P, Wightman M, et al. Rab29 activation of the Parkinson's disease-associated LRRK2 kinase. EMBO J. (2018) 37:1–8. doi: 10.15252/embj.201798099
277. Kumar D, Ambasta RK, Kumar P. Ubiquitin biology in neurodegenerative disorders: from impairment to therapeutic strategies. Ageing Res Rev. (2020) 61:101078. doi: 10.1016/j.arr.2020.101078
278. Ferreira M, Massano J. An updated review of Parkinson's disease genetics and clinicopathological correlations. Acta Neurol Scand. (2017) 135:273–84. doi: 10.1111/ane.12616
279. Yamada M, Mizuno Y, Mochizuki H. Parkin gene therapy for α-synucleinopathy: a rat model of Parkinson's disease. Hum Gene Ther. (2005) 16:262–70. doi: 10.1089/hum.2005.16.262
280. Cai ZL, Xu J, Xue SR, Liu YY, Zhang YJ, Zhang XZ, et al. The E3 ubiquitin ligase seven in absentia homolog 1 may be a potential new therapeutic target for Parkinson's disease. Neural Regen Res. (2015) 10:1286–91. doi: 10.4103/1673-5374.162763
281. Liu X, Hebron M, Shi W, Lonskaya I, Moussa CE-H. Ubiquitin specific protease-13 independently regulates parkin ubiquitination and alpha-synuclein clearance in alpha-synucleinopathies. Hum Mol Genet. (2019) 28:548–60. doi: 10.1093/hmg/ddy365
282. Valente EM, Bentivoglio AR, Dixon PH, Ferraris A, Lalongo T, Frontali M, et al. Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am J Hum Genet. (2001) 68:895–900. doi: 10.1086/319522
283. Valente EM, Abou-Sleiman PM, Caputo V, Muqit MMK, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. (2004) 304:1158–60. doi: 10.1126/science.1096284
284. Seirafi M, Kozlov G, Gehring K. Parkin structure and function. FEBS J. (2015) 282:2076–88. doi: 10.1111/febs.13249
285. Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, et al. The ubiquitin pathway in Parkinson's disease [6]. Nature. (1998) 395:451–2. doi: 10.1038/26652
286. Xilouri M, Kyratzi E, Pitychoutis PM, Papadopoulou-Daifoti Z, Perier C, Vila M, et al. Selective neuroprotective effects of the S18Y polymorphic variant of UCH-L1 in the dopaminergic system. Hum Mol Genet. (2012) 21:874–89. doi: 10.1093/hmg/ddr521
287. Deng H, Wang P, Jankovic J. The genetics of Parkinson disease. Ageing Res Rev. (2018) 42:72–85. doi: 10.1016/j.arr.2017.12.007
288. Fonzo AD, Dekker MCJ, Montagna P, Baruzzi A, Yonova EH, Guedes LC, et al. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian- pyramidal syndrome. Neurology. (2009) 72:240–5. doi: 10.1212/01.wnl.0000338144.10967.2b
289. Zhao T, Severijnen LA, Van Der Weiden M, Zheng PP, Oostra BA, Hukema RK, et al. FBXO7 Immunoreactivity in α-synuclein-containing inclusions in parkinson disease and multiple system atrophy. J Neuropathol Exp Neurol. (2013) 72:482–8. doi: 10.1097/NEN.0b013e318293c586
290. Chen CM, Chen IG, Huang YC, Juan HF, Chen YL, Chen YC, et al. FBXO7 Y52C polymorphism as a potential protective factor in Parkinson's disease. PLoS ONE. (2014) 9:e101392. doi: 10.1371/journal.pone.0101392
291. Verma DK, Ghosh A, Ruggiero L, Cartier E, Janezic E, Williams D, et al. The SUMO conjugase Ubc9 protects Dopaminergic cells from cytotoxicity and enhances the stability of α-synuclein in Parkinson's disease models. eneuro. (2020) 7:ENEURO.0134-20.2020. doi: 10.1523/ENEURO.0134-20.2020
292. Games D, Seubert P, Rockenstein E, Patrick C, Trejo M, Ubhi K, et al. Axonopathy in an α-synuclein transgenic model of Lewy body disease is associated with extensive accumulation of c-terminal-truncated α-synuclein. Am J Pathol. (2013) 182:940–53. doi: 10.1016/j.ajpath.2012.11.018
293. Games D, Valera E, Spencer B, Rockenstein E, Mante M, Adame A, et al. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson's disease-like models. J Neurosci. (2014) 34:9441–54. doi: 10.1523/JNEUROSCI.5314-13.2014
294. Bassil F, Fernagut PO, Bezard E, Pruvost A, Leste-Lasserre T, Hoang QQ, et al. Reducing C-terminal truncation mitigates synucleinopathy and neurodegeneration in a transgenic model of multiple system atrophy. Proc Natl Acad Sci USA. (2016) 113:9593–8. doi: 10.1073/pnas.1609291113
295. Ryan P, Xu ming M, Davey AK, Kassiou M, Mellick GD, Rudrawar S. O-GlcNAcylation of truncated NAC segment alters peptide-dependent effects on α-synuclein aggregation. Bioorg Chem. (2020) 94:103389. doi: 10.1016/j.bioorg.2019.103389
296. Tavassoly O, Yue J, Vocadlo DJ. Pharmacological inhibition and knockdown of O-GlcNAcase reduces cellular internalization of α-synuclein preformed fibrils. FEBS J. (2020) 288:452–70. doi: 10.1111/febs.15349
297. Lee BE, Kim HY, Kim H-J, Jeong H, Kim B-G, Lee H-E, et al. O-GlcNAcylation regulates dopamine neuron function, survival and degeneration in Parkinson disease. Brain. (2020) 143:3699–716. doi: 10.1093/brain/awaa320
298. Wrasidlo W, Tsigelny IF, Price DL, Dutta G, Rockenstein E, Schwarz TC, et al. A de novo compound targeting α-synuclein improves deficits in models of Parkinson's disease. Brain. (2016) 139:3217–36. doi: 10.1093/brain/aww238
299. Perni M, Galvagnion C, Maltsev A, Meisl G, Müller MBD, Challa PK, et al. A natural product inhibits the initiation of α-synuclein aggregation and suppresses its toxicity. Proc Natl Acad Sci USA. (2017) 114:E1009–17. doi: 10.1073/pnas.1610586114
300. Dos Santos MCT, Scheller D, Schulte C, Mesa IR, Colman P, Bujac SR, et al. Evaluation of cerebrospinal fluid proteins as potential biomarkers for early stage Parkinson's disease diagnosis. PLoS ONE. (2018) 13:e0206536. doi: 10.1371/journal.pone.0206536
Keywords: Parkinson's disease, protein aggregation, lipid membranes, post-translational modifications, lipid homeostasis, protein homeostais
Citation: Bell R and Vendruscolo M (2021) Modulation of the Interactions Between α-Synuclein and Lipid Membranes by Post-translational Modifications. Front. Neurol. 12:661117. doi: 10.3389/fneur.2021.661117
Received: 30 January 2021; Accepted: 18 May 2021;
Published: 15 July 2021.
Edited by:
Franziska Richter, University of Veterinary Medicine Hannover, GermanyReviewed by:
Zhong Pei, Sun Yat-sen University, ChinaFrancisco José Pan-Montojo, Ludwig Maximilian University of Munich, Germany
Copyright © 2021 Bell and Vendruscolo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michele Vendruscolo, bXYyNDVAY2FtLmFjLnVr