 Davide Gobbo
Davide Gobbo Anja Scheller
Anja Scheller Frank Kirchhoff
Frank Kirchhoff- Molecular Physiology, Center for Integrative Physiology and Molecular Medicine (CIPMM), University of Saarland, Homburg, Germany
The electrographic hallmark of childhood absence epilepsy (CAE) and other idiopathic forms of epilepsy are 2.5–4 Hz spike and wave discharges (SWDs) originating from abnormal electrical oscillations of the cortico-thalamo-cortical network. SWDs are generally associated with sudden and brief non-convulsive epileptic events mostly generating impairment of consciousness and correlating with attention and learning as well as cognitive deficits. To date, SWDs are known to arise from locally restricted imbalances of excitation and inhibition in the deep layers of the primary somatosensory cortex. SWDs propagate to the mostly GABAergic nucleus reticularis thalami (NRT) and the somatosensory thalamic nuclei that project back to the cortex, leading to the typical generalized spike and wave oscillations. Given their shared anatomical basis, SWDs have been originally considered the pathological transition of 11–16 Hz bursts of neural oscillatory activity (the so-called sleep spindles) occurring during Non-Rapid Eye Movement (NREM) sleep, but more recent research revealed fundamental functional differences between sleep spindles and SWDs, suggesting the latter could be more closely related to the slow (<1 Hz) oscillations alternating active (Up) and silent (Down) cortical activity and concomitantly occurring during NREM. Indeed, several lines of evidence support the fact that SWDs impair sleep architecture as well as sleep/wake cycles and sleep pressure, which, in turn, affect seizure circadian frequency and distribution. Given the accumulating evidence on the role of astroglia in the field of epilepsy in the modulation of excitation and inhibition in the brain as well as on the development of aberrant synchronous network activity, we aim at pointing at putative contributions of astrocytes to the physiology of slow-wave sleep and to the pathology of SWDs. Particularly, we will address the astroglial functions known to be involved in the control of network excitability and synchronicity and so far mainly addressed in the context of convulsive seizures, namely (i) interstitial fluid homeostasis, (ii) K+ clearance and neurotransmitter uptake from the extracellular space and the synaptic cleft, (iii) gap junction mechanical and functional coupling as well as hemichannel function, (iv) gliotransmission, (v) astroglial Ca2+ signaling and downstream effectors, (vi) reactive astrogliosis and cytokine release.
Introduction
Epilepsy is a highly heterogeneous neurological condition characterized by enduring predisposition to unpredictable pathological discharge of rhythmic activity in the brain networks, which is commonly referred as seizure activity (1). In virtue of the severity and nature of the pathological alteration (abnormal, excessive, or excessively synchronous activation) as well as the cellular and anatomical composition of the affected brain networks, seizures can cause changes in the level of consciousness, behavior, memory, and emotional status. Although the etiology of epileptiform activity is still unknown in half of the cases, understanding the pathological alteration at the basis of the epileptic phenotype may not only be of fundamental therapeutical importance but also provide further insights into the functioning of the affected neural networks in the physiology of the healthy brain. The identification of the molecular and cellular mechanisms underlying physiological oscillations is critical for a full comprehension of their relationship to the respective pathological activity. In this regard, an exceptional case of study is the cortico-thalamo-cortical network, physiologically engaged during sleep and pathologically altered in the context of non-motor (absence) seizures (2).
Absence seizures are transient non-convulsive generalized epileptic events and are also referred as petit mal seizures (2, 3). Phenotypically, absence seizures are coupled with sudden and brief impairment of consciousness and lack of responsiveness to external stimuli as well as variable secondary clinical symptoms (e.g., automatisms, atonic, and tonic muscular components etc.) (4, 5). Absence seizures are the sole clinical symptom of childhood absence epilepsy (CAE) but are also associated with several other idiopathic generalized epilepsies (4, 6–11). Although CAE has up to 70% remission rate (7, 12), the gold standard monotherapy, based on ethosuximide and valproic acid, is still ineffective in 30% of the cases (13). Moreover, clinical conditions displaying absence seizures are often associated with severe neuropsychiatric comorbid conditions such as impaired attention, learning, memory and cognition, which are often left unaltered or even worsened by common antiepileptic drugs (14–17).
Although absence seizures display inter- and intraindividual variability (17, 18), they exhibit generalized bilateral 2.5–4 Hz spike and wave discharges (SWDs) with no aura or post-ictal depression (Figures 1A,B) (4, 27–29). It is widely accepted that the sharp spike and the slow wave component of SWDs are functionally coupled and correspond to a state of neuronal excitation and silence in the cortico-thalamo-cortical network, respectively (30). Blood oxygenation level-dependent (BOLD) functional magnetic resonance imaging (fMRI) studies in humans consistently showed cortical network engagement in correspondence of and even preceding the appearance of SWDs in electrographic traces as well as an increased interictal synchrony in the sensorimotor cortex (Figure 1C) (22, 23, 31–36). Most advancements on the understanding of the cellular and synaptic mechanisms underlying SWDs derive from the extensive use of genetic animal models, particularly the genetic absence epilepsy rats from Strasbourg (GAERS) and Wistar-Albino-Glaxo rats from Rijswijk (WAG/Rij) (20, 37–42) as well as many monogenic mouse mutants (43–45). Although sharing most electrographic and behavioral hallmarks of absence seizures, animal models are characterized by higher SWD frequencies (5–11 Hz) (Figure 1B). Ex vivo multi-site local field potential studies identified the peri-oral primary somatosensory cortex as initiation site of absence seizures in WAG/Rij (46) and GAERS rats (47–50). Notably, this has been proven wrong for the acute pharmacological γ-hydroxybutyric acid (GHB) model (51–54) in mice, where the prefrontal cortex was suggested as the initiation site of SWDs (55). With this in mind and considering the many areas contributing to the cortical pre-ictal BOLD changes of absence seizures, one can probably not identify a unique canonical focal onset or initiation site for absence seizures. Instead, the denomination cortical initiation network has been recently proposed (17), thereby settling the long-standing controversy about the SWDs initiation site (56–59). However, the existence of a cortical initiation network does not imply that manipulation of the sole thalamic components of the cortico-thalamo-cortical network is not sufficient to induce SWDs, as it is indeed the case (60–62), or that the wide thalamo-cortical innervation is not crucial for SWDs generalization, as suggested by the existence of subclinical SWDs restricted to the cortical network (48). In particular, the thalamic posterior nucleus plays a crucial role in the generalization of SWDs (61, 63–66). Till recently, ex vivo studies performed in different mammalian models identified the hyperexcitability and T-type Ca2+ channel-mediated burst activity of glutamatergic thalamo-cortical neurons and GABAergic neurons from the thalamic reticular nucleus (or nucleus reticularis thalami, NRT) as the rhythmogenic cortico-thalamo-cortical network mechanism of SWDs (Figures 1D,E) (24, 41, 67–71). Nevertheless, recent in vivo studies performed in rodents showed that only a small fraction of thalamo-cortical and cortico-thalamic neurons are synchronously active at each SWD cycle and the cellular composition of this neuronal subpopulation changes between subsequent cycles, thus excluding the existence of distinctive neuronal subpopulations (Figures 1F,G) (25, 26). This explains why, with SWD progression, the activities of the cortico-thalamic and thalamo-cortical neurons undergo a phase-shift in time (46) since different neuronal subpopulations participate in this excitatory feedback-loop with slightly different kinetics. Moreover, this progressive phase-shift between different subpopulations active at the same time accounts for the overlapping average electrical activity in the cortico-thalamic, thalamo-cortical, and NRT neurons within any SWD cycle. Moreover, although interictal T-type Ca2+ channel burst activity in the thalamo-cortical neurons increases right before SWD onset, overall in vivo ictal thalamic activity decreases and only cortical and NRT T-type channels are essentials for SWDs (25). Interestingly, all NRT neurons fire within each SWD cycle, even though a fraction of those neurons fires relatively asynchronous tonic spikes rather than T-type Ca2+ channel-mediated bursts in phase with the SWDs (Figures 1E,F) (25). The enhanced tonic inhibition of thalamo-cortical neurons as well as the increased thalamic GABA level are key aspects of absence seizures (25, 72–78). Moreover, the fact that SWDs can be induced by the impairment of the cortico-thalamic glutamate release due to deletion of P/Q-type Ca2+ channels in the projecting cortical neurons from layer VI could suggest that a balance shift toward GABAergic inhibition more than an absolute increase of GABA levels is the key mechanism of SWD generalization (79). Additionally, the decreased glutamate release could lead to reduced activity of cortical interneurons, thus contributing to cortical hyperexcitability.
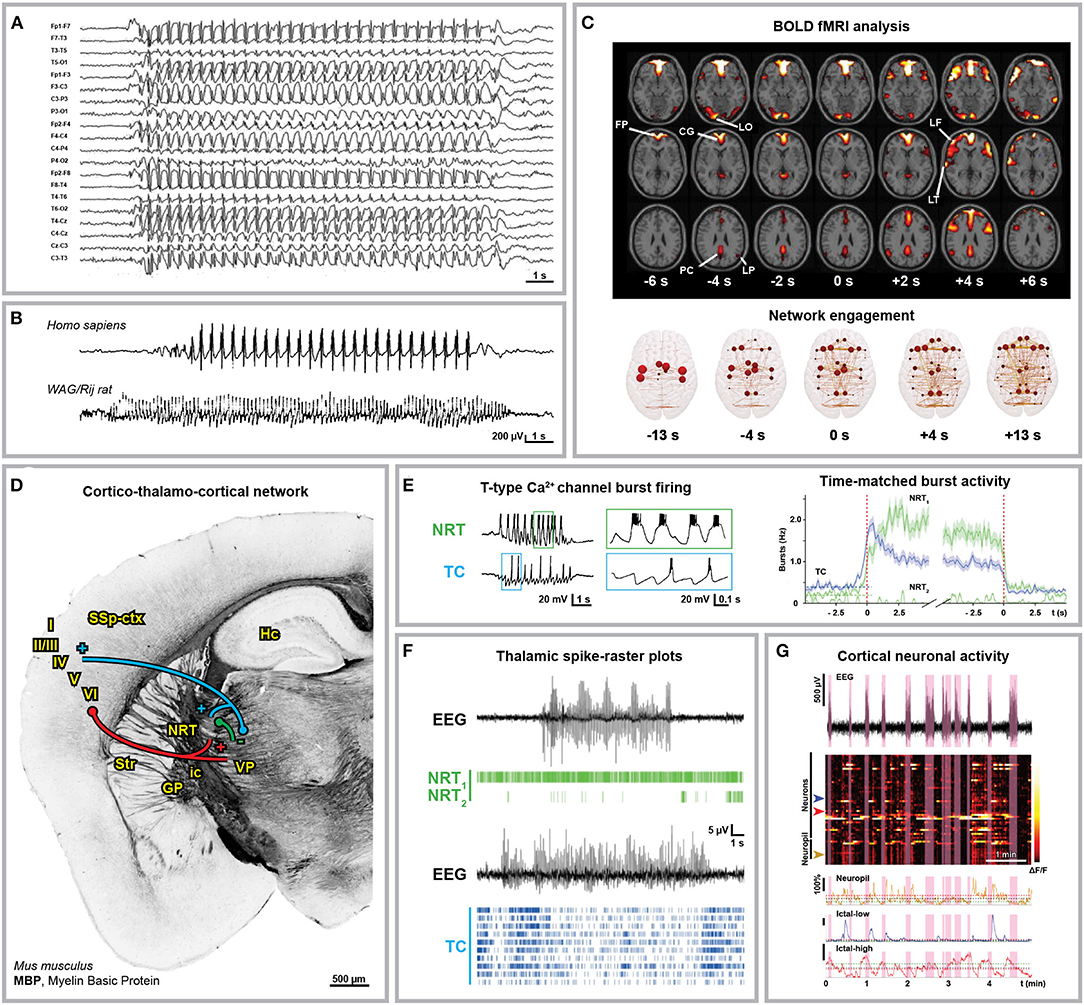
Figure 1. Anatomical and electrophysiological characterization of absence seizures. (A) Human electroencephalographical recording (EEG) displaying typical 3 Hz spike-and-wave discharges (SWDs). (B) 3 Hz SWDs associated with Childhood Absence Epilepsy (top trace, 8-year-old boy) and 8 Hz SWDs recorded in an adult WAG/Rij rat (bottom trace). (C) Blood Oxygenation Level Dependent (BOLD) functional Magnetic Resonance Imaging (fMRI) changes associated with absence seizures before (FP, frontal polar; CG, cingulate; LO, lateral occipital; PC, precuneus; LP, lateral parietal cortex) and after (LF, lateral frontal; LT, lateral temporal cortex) seizure onset (top) and brain network engagement analysis around SWDs events (bottom). (D) Anatomical organization of the cortico-thalamo-cortical network in C57BL/6N mice. Myelin Basic Protein (MBP) immunostaining indicates the axonal fiber tracts connecting the network (own data; mouse monoclonal MBP antibody, BioLegend, AB_2564741, Cat. No. 808401, 1:500). Cortico-thalamic excitatory neurons from cortical layers V and VI (red) project to both NRT and thalamus; thalamo-cortical excitatory neurons (blue) project back to cortical layer IV and NRT; NRT GABAergic neurons (green) inhibit the thalamic nuclei. I-VI, cortical layers; GP, globus pallidus; Hc, hippocampus; ic, internal capsule; NRT, nucleus reticularis thalami; SSp-ctx, primary somatosensory cortex; Str, striatum; VP, ventral posterior thalamic nuclei. (E) Glutamatergic thalamo-cortical neurons (TC) as well as GABAergic NRT neurons display T-type Ca2+ channel-mediated burst firing during SWDs (left, ex vivo recording from ferret thalamic slices). (F) Spike-time raster plots of two representative NRT neurons (NRT1, top trace; NRT2, bottom trace) and 10 TC neurons with time-matched EEG in GAERS rats. The overall TC activity decreases during SWDs and only a small portion of TC neurons fire synchronously. (G) 2-Photon laser scanning microscopy of neuronal cortical Ca2+ activity in stargazer mice during absence seizures. Heatmap of neuronal Ca2+ activity shows that only a subpopulation of neurons displays ictal synchronous firing. Modified from (A), (19); (B), (20, 21); (C), (22, 23); (E), (24, 25); (F), (25); (G), (26).
Astrocytes Contribute to Network Priming and Synchronization as Well as Swd Induction, Propagation, and Termination
After more than three decades of accumulating evidence, nowadays it is widely established that astroglia constitute a ubiquitous non-neuronal communication system in the brain involved in virtually every physiological and pathological scenario of the central nervous system (80–82). Not only do they support synapses from a mechanical, metabolical as well as functional point of view, but they also participate in synaptic transmission and plasticity, neural network excitability and balance between excitation and inhibition (E/I) as active information integrators and processors (83–86). The contribution of the astroglial network to the pathophysiology of epilepsy encompasses a plethora of different molecular mechanisms which currently represent one of the most fruitful research topics in neuroscience (87–96). Pathological priming mechanisms of the astroglial network ultimately involve either E/I imbalance or enhanced network synchronization (or both simultaneously). Alternatively, astrocytes may influence spatial and temporal propagation of seizures, thus playing a key role in the phenotypical outcome of seizures and their severity and therefore representing a promising target for the development of new non-neurocentric drugs. Most of the recent evidence focuses on astroglial contribution to convulsive epileptic activity. Nevertheless, we discuss in the following the putative involvement of astrocytes in network priming as well as seizure induction and propagation which could have a role in pathological epileptic scenarios including SWDs, as well. We focus on the evidence that links to observations coming from the clinics as well as genetic and pharmacological models of SWDs, aiming to point at specific topics which may be worth further research in the field of SWDs.
The Astroglial Network Controls Extracellular Space Homeostasis Through K+, Water and Solute Clearance
By means of their close juxtaposition to synapses, their expression of an extraordinary assortment of membrane transporters and receptors as well as their physical and functional coupling through gap junctions (GJs), astroglial networks provide a perfect spatial buffering for neural activity (Figure 2) (97–99). Astrocytes are key regulators of the extracellular K+ concentration. Their high K+ permeability mediated by inwardly-rectifying Kir and two-pore-domain K2P channels, Na+/K+ pumps and Na+/K+/Cl− transporters associated with their extensive GJ coupling enables them to uptake and redistribute excessive extracellular K+ resulting from neuronal firing (100–103). Astroglial K+ and glutamate uptake is altered in cultured cortical astrocytes after Kir4.1 channel downregulation (104) as well as in astroglial-specific Kir4.1 knock-out mice (105, 106). Gain-of-function (107) as well as loss-of-function mutations (108, 109) in the human KCNJ10 gene encoding Kir4.1 have been linked to forms of childhood epilepsies associated with ataxia and cognitive impairment, but not to CAE. Notably, artificially increasing extracellular K+ concentration ex vivo is associated with propagating epileptiform discharges induced by focal optogenetic activation of parvalbumin-expressing interneurons (110). In vivo though, K+ clearance impairment induced by blocking GJ was not sufficient to induce neocortical seizures (111). Interestingly, valproic acid (but not ethosuximide) induces Kir4.1 overexpression in the cortex of healthy rats (112). Nevertheless, further research is required to address the actual contribution of Kir4.1 overexpression in the anti-absence effects of valproic acid, as well as the putative role of astroglial Kir4.1 itself in the development and propagation of SWDs. The combined use of cell-specific conditional knock-out of these channels and pharmacological models of SWDs could shine new light on the topic.
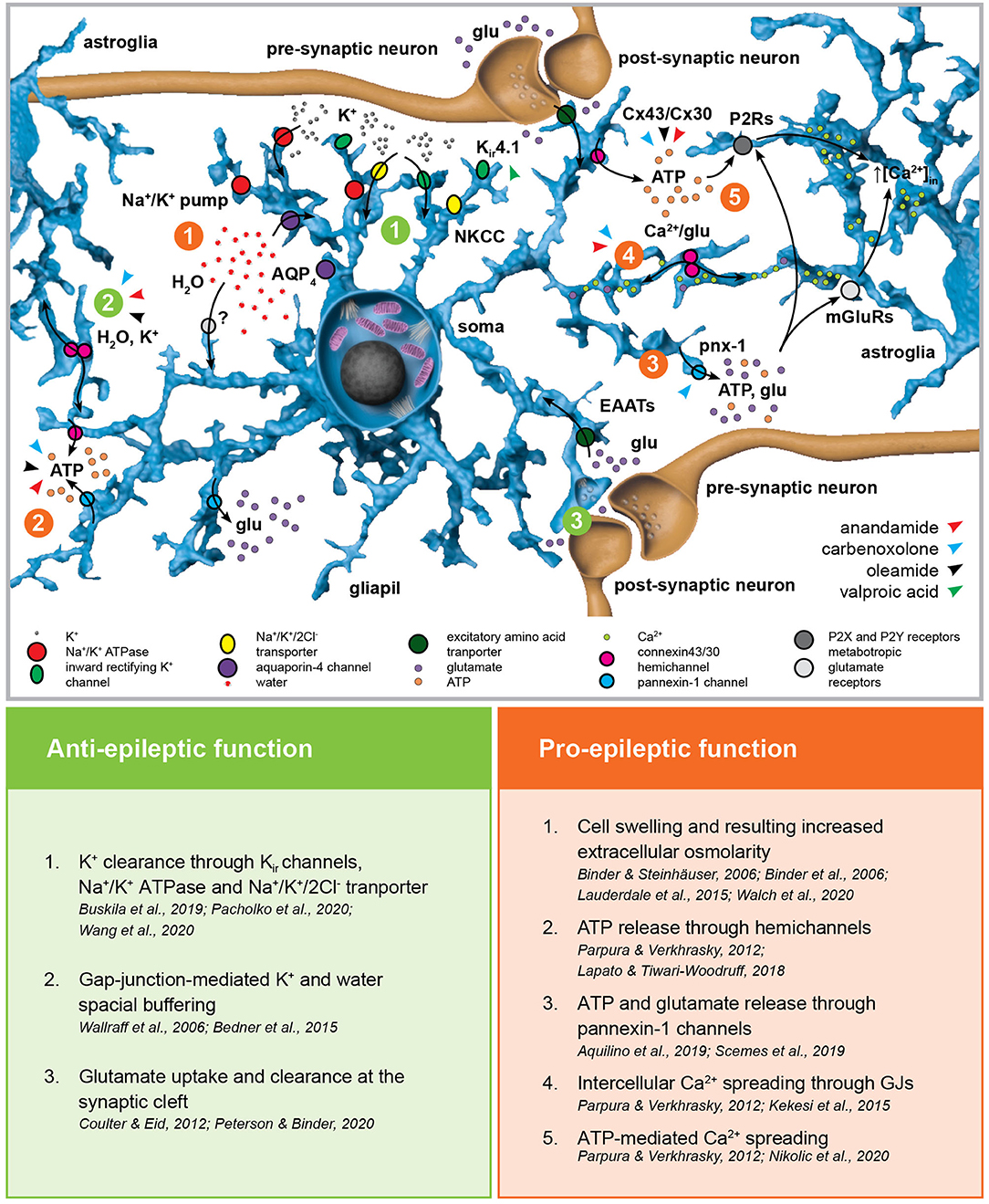
Figure 2. Astroglial homeostatic control of the extracellular space has opposite effects on epileptogenesis. The astroglial network is responsible for extracellular K+ uptake by means of inward rectifying K+ channels (Kir), Na+/K+ pump and Na+/K+/2Cl− transporter (NKCC). K+ clearance is coupled with water uptake through the water channel aquaporin-4 (AQP4) and possibly via yet unknown additional pathways. The excitatory amino acid transporters EAAT1 and EAAT2 are responsible for glutamate uptake. Astroglial connexins Cx43 and Cx30 enable gap-junction (GJ) coupling responsible for spatial ionic and metabolic buffering. Connexin hemichannels as well as pannexin-1 channels (Panx1) mediate glutamate and ATP release in the extracellular space possibly activating astroglial metabotropic glutamate receptors (mGluRs) and purinergic P2X and P2Y receptors (P2Rs), respectively. This, in term, induces intracellular Ca2+ increases in the neighboring astroglia. The figure summarizes the pro- and anti-epileptic roles of the mechanisms described above and points to the putative targets of valproic acid and the GJ blockers carbenoxolone, anandamide, and oleamide in this scenario.
K+ uptake is associated with cellular swelling due to Na+/K+-pump dependent water influx (113). Although the exact molecular mechanisms are still under debate, water fluxes across the astrocytic membrane are associated with K+ homeostasis and they influence local interstitial osmolarity as well as seizure generation and progression (114–120). Given the accumulating evidence against the predominant contribution of the astroglial water channel aquaporin-4 (AQP4) in water homeostasis (113, 121), the use of AQP4 conditional knock-out as a model of disrupted water homeostasis has been recently challenged. Nevertheless, water homeostasis impairment and the resulting volume and osmolarity dysregulation should affect neural network excitability. Indeed, a recent structural MRI study on CAE showed significant gray matter volume abnormalities in both frontotemporal cortical region and posterior thalami compared to controls (122, 123).
GJ coupling, particularly mediated by connexins Cx30 and Cx43, provides the astroglial network with a high level of intercellular structural, metabolic and functional connectivity, enabling the exchange of ions and small molecules (124–131). In the context of epilepsy, connexins mediate ATP release (into the extracellular space through hemichannels), the spreading of intercellular Ca2+ waves (132) and are fundamental in the spatial buffering required for K+ and water homeostasis as well as glutamate clearance (133, 134). With respect to absence epilepsy, most advancement in unraveling the role of GJs has been obtained employing GJ blockers in well-established genetic animal models (135). The broad-spectrum GJ blocker carbenoxolone (CBX) decreased both amplitude and duration of 4-aminopyridine-induced seizure-like events (SLEs) in thalamocortical slices obtained from mice with spontaneous SWDs (136, 137) as well as the duration of SWDs seen in GAERS rats in vivo after systemic application (138). Interestingly, in vivo injection of CBX in the NRT of rats with atypical absence seizures and spontaneous SWDs decreased the duration of SWDs (139), whereas no alteration of SWD phenotype was observed if CBX was injected in the posterior thalami of WAG/Rij rats and the lethargic mouse genetic model of absence epilepsy (140). Recently, intraperitoneal injection of CBX was associated with absence seizures worsening in WAG/Rij rats (141), hinting at non-obvious and non-trivial differences across the absence epilepsy models. The endocannabinoids anandamide (N-arachidonoylethanolamine, ANA) and oleamide (cis-9,10-octadecenoamide, OLE) are specific Cx43 blockers (142, 143). Intracerebroventricular injection of ANA decreased in a dose-dependent manner the recurrence and duration of SWDs, although its mechanism of action likely involves type-1 cannabinoid (CB1) receptor activation (144) or even direct inhibition of T-type Ca2+ channels (145). Interestingly, although specific studies addressing the impact of OLE in absence epilepsy are still required, OLE has a sleep-inducing effect and enhances GABAA receptor-mediated responses, thus possibly affecting the physiological, temporal-spatial pattern of cortico-thalamo-cortical oscillations (146, 147). CBX as well as ANA and OLE block both GJ activity and connexin hemichannels regulating water and solute (notably ATP) exchanges between the intra- and extracellular space, thus challenging the attribution of any observed phenotype to the sole GJ coupling (131, 148). Moreover, regional differences in connexin isoform expression may be at the basis of different contributions of GJ and hemichannel inhibition in different neural networks, and thus the net phenotypical outcome of the pharmacological manipulation (149). CBX is also known to block pannexin-1, which bears significant topological and pharmacological similarities with the connexins and forms single-membrane channels which have been linked to network hyperexcitability and hypersynchronization by mediating both ATP and glutamate release (150, 151). The use of antibodies or small peptides targeting specific amino acid sequences of different connexins (152–155) could shed new light into the differential contribution of GJ coupling and hemichannel function as well as into the role of different connexin isoforms and pannexin-1 channels in the generation and propagation of SWDs. Finally, ANA, but not OLE, can block Ca2+ wave propagation in astrocytes, which has to be taken into consideration in the interpretation of the results (142, 143).
In summary, astroglial networks contribute to the imbalance of neural excitation/inhibition through K+ and neurotransmitter (glutamate but also GABA) clearance under physiological conditions, thus counteracting network priming through aberrant shifts in the E/I balance possibly leading to network synchronization. Astrocytes rely on their extensive GJ coupling enabling effective spatial ionic, osmotic, and functional buffering. GJ hemichannels as well as pannexin-1 channels may be responsible for augmented synchronous activity through ATP and glutamate release and following Ca2 spreading throughout the astroglial network. So far, we are still missing evidence for linking the astroglial fine-tuning of the extracellular ion and transmitter homeostasis to SWDs. However, as it is the case for other kinds of epileptiform activity, their role in regulating such network excitability is very likely.
Astrocytes Are Actively Involved in Network Dynamics and E/I Balance Through Neurometabolic Coupling, Neurotransmission Modulation and Gliotransmission
Astrocytes do not only contribute to neural excitability and functioning by responding to neurotransmitter release and modification of extracellular ionic composition, they are also actively involved in neurotransmitter uptake and release, thus having a direct control of E/I balance (Figure 3) (86, 156–158). One of the key features of absence epilepsy are altered GABA levels (72, 159) and GABAergic tonic and phasic inhibition in the cortico-thalamo-cortical network (25, 73). In both GAERS rats and stargazer mice, astroglial GABA transporter GAT-1 malfunction leads to increased GABA levels in the thalamus resulting in altered tonic inhibition of GABAA receptors on the thalamo-cortical neurons (72, 74, 75, 77). Notably, a number of human mutations in SLC6A1 encoding GAT-1 leads to reduced GABA transport activity, and some of the mutations are associated with CAE or clinical conditions associated with absence seizures (160–164). Moreover, GABA released by astrocytes was proven to activate GABAA receptors on the membrane of thalamocortical neurons in rodents (165) and blocking astroglial GATs increased extrasynaptic GABAA receptor-mediated tonic inhibition (166). On the other hand, thalamic astrocytes express GABAA receptors themselves (167), whose specific role has not been fully resolved yet. Neuronal presynaptic GABAB receptor expression and function is impaired in the neocortex of WAG/Rij rats, possibly contributing to network hyperexcitability (168, 169). There is plenty of evidence that GABAB receptors contribute to network priming in absence seizures facilitating thalamo-cortical burst firing, as supported by the exacerbation of SWDs after baclofen or GHB treatment (170–176). Interestingly, the activation of extrasynaptic GABAB receptors require GABA spillover resulting from an intense GABAergic stimulation, which is in accordance with a predominant role of astrocytic GAT-1 in regulating SWDs, given its expression in close proximity of neuronal synapses compared to a more distal location of GAT-3 (175). A further level of complexity is given by the fact that astrocytes themselves express GABAB receptors and their activation leads to downstream Ca2+ signaling and possibly gliotransmission as shown in the thalamus upon local ex vivo baclofen and GHB application (177).

Figure 3. Regional specific imbalance of E/I at the basis of absence seizures. The main components of the cortico-thalamo-cortical network (SSp-ctx, primary somatosensory cortex; NRT, nucleus reticularis thalami; thalamic nuclei) display regional specific shifts toward either excitation or inhibition associated with absence seizures. The figure summarizes the pro- (+) and anti- (-) epileptic effects of specific alterations of the regional E/I balance in the pathological phenotype of absence seizures. A1R, adenosine receptor type 1; EAAT1/2, excitatory amino acid transporters 1/2; CB1, cannabinoid receptor type 1; GABA, γ-aminobutyric acid; GABABR, metabotropic GABAB receptor; GAT-1, GABA transporter 1; mGluR1/5, metabotropic glutamate receptors 1/5; TC, thalamo-cortical; TCA, tricarboxylic acid cycle; Y2R, neuropeptide Y receptor type 2.
As expected, the injection of the GAT inhibitor tiagabine in the thalamus enhances SWDs (178), while its injection in the somatosensory cortex suppresses SWDs, as does the injection of positive allosteric modulators of glutamate metabotropic receptors mGluR1 and mGluR5 in both somatosensory cortex and thalamus (178). A line of experimental evidence suggests that possibly all metabotropic glutamate receptors, including the mGluR2/3 and mGluR5 expressed on the astroglial membrane, are involved in SWDs through modulation of NMDA receptors and GABA uptake (178–182). Indeed, a subpopulation of astrocytes in the thalamus expresses mGluR5 and respond to cortico-thalamic glutamatergic afferents via intracellular Ca2+ oscillations (183). Therefore, it is very likely that astrocytes contribute to SWD phenotype by processing glutamate signaling. Astroglial glutamate transporters EAAT1 (GLutamate ASpartate Transporter, GLAST) and EAAT2 (GLutamate Transporter-1, GLT-1) (184, 185) as well as astroglial glutamine-glutamate-GABA cycle impairment (186, 187) have already been associated with the development of various forms of epileptic activities. GAERS rats display decreased protein expression of both astroglial GLT-1 and GLAST proteins before the development of absence seizures (188). Notably, GLAST is overexpressed at the mRNA level, possibly due to a compensatory mechanism of gene transcription (189). Moreover, excessive neuronal firing is known to induce astroglial swelling and subsequent glutamate release (190). This may add a further level of complexity in the already complex temporal firing dynamics of the thalamo-cortical neurons and NRT neurons both ictally and at interictal-to-ictal transitions (25). Although not yet proven in the context of SWDs, astrocytes possess the extraordinary capability of converting intensive glutamatergic neuronal activity into tonic inhibition, by coupling the glutamate/Na+ symport with the glutamine and GABA/Na+ symport (191). Notably, the only ATP expenditure associated with this process relies on the replenishment of the intracellular GABA storage since the driving force of the glutamine and GABA release is the re-establishment of the physiological Na+ homeostasis altered by the glutamate/Na+ symport. Finally, a comprehensive study of metabolic alterations in GAERS rats provides further insight into the cortical and thalamic astroglial contribution to the pathology of SWDs. Most strikingly, cortical astroglial metabolism and glutamine-glutamate-GABA cycle are enhanced in GAERS rats, leading to increased glutamate and glutamine levels and decreased GABA labeling (192). Interestingly, the expression of astroglial glutamate dehydrogenase is increased, in the cortex before the development of absence seizures and in the thalamus before and after the development of absence seizures, thus possibly leading to a decreased glutamate availability and a shift to the thalamic GABAergic inhibition fundamental for the generalization of SWDs (193). In line with this hypothesis, the intraperitoneal injection of branched-chain amino acids and α-ketoisocaproate pushing the chemical equilibrium toward the synthesis of glutamine led to decreased thalamic glutamate levels and the worsening of absence seizures (194). Moreover, a gain-of-function mutation of the glutamate dehydrogenase gene leading to aberrant glutamate availability and hyperammonemia has been associated with myoclonic absence epilepsy (195). Although further research in the field of absence epilepsy is still required, this evidence supports the role of astrocytic metabolism and glutamine-glutamate-GABA cycle in providing adequate energy supply and network homeostasis required for epileptic activity generation and propagation (94, 196).
In situ hybridization and Western blot analysis showed reduced levels of CB1 receptor mRNA and protein in the NRT and of the CB1 receptor in the thalamus of WAG/Rij rats at the protein level, thereby suggesting an impaired depolarization-induced CB1-mediated suppression of inhibition (197). Indeed, acute systemic injection of the synthetic CB1 receptor agonist WIN55,212-2 resulted in a transient reduction in SWDs frequency, however surprisingly followed by an increase in SWD duration in subchronic treatment (144, 197–199). Since the beneficial effects of the endocannabinoid ANA, previously described, last longer than the transient reduction in SWD frequency induced by the synthetic CB1 agonist and since ANA does indeed shorten SWDs, its mechanism of action is likely not only dependent on CB1 activation but a more complex molecular process (144).
The release of ATP through connexin and pannexin-1 hemichannels and the resulting spread of Ca2+ waves largely contribute to the astrocyte-mediated purinergic signaling in epilepsy (200). However, the net impact on the neural network is often context-dependent and may include the conversion of ATP into adenosine. Adenosine levels depend on extracellular ectonucleotidases as well as on the astroglial adenosine kinase (ADK) and its contribution encompasses antiepileptic A1 receptor-mediated as well as proepileptic A2 and A3 receptor-mediated effects (200–203). Once again, most research results have been derived from the analysis of convulsive seizures. Nevertheless, there is a number of evidence suggesting that purinergic signaling is altered in SWDs, too. To which extent this is related to astroglial contribution is still elusive. With respect to SWDs, GAERS rats show lower expression of A1 receptors in the NRT (204) and WAG/Rij rats are characterized by altered expression of A2A receptors in the somatosensory cortex, NRT and thalamus (205). Absence epileptic activity in WAG/Rij rats increases after activation of A2A receptors directly by the specific synthetic agonist 2-[4-(-2-carboxyethyl)-phenylamino]-5′-N-ethylcarboxamido-adenosine (CGS21680) (205) or indirectly after intraperitoneal injection of guanosine (206) as well as of adenosine (207). Conversely, acute caffeine administration, which is a mixed non-specific A1 and A2A receptor antagonist, reduced both amplitude and duration of SWDs in GAERS rats (208). However, the administration of the specific A1 antagonist 1,3.dipropyl-8-cyclopentylxanthine (DPCPX) in WAG/Rij rats had a proepileptic effect on SWDs (209). Notably, a duplication in the chromosomal region containing the gene coding for the extracellular catabolic enzyme adenosine deaminase was associated with a case of early-onset absence epilepsy, possibly leading to an impairment in adenosine homeostasis (210, 211).
The neuropeptide Y (NPY) released by thalamic neurons promotes phase-specific long-term depression of neuronal excitability in the NRT as well as in the thalamus itself and thus possibly contributing to thalamocortical synchronization and the altered dynamics of T-type Ca2+ channel-mediated bursting activity in the thalamic nuclei (212). Interestingly, valproic acid treatment increases thalamic levels of NPY mRNA in GAERS rats (213). Moreover, NPY intracerebroventricular injection as well as focal administration of NPY in the somatosensory cortex of GAERS rats had a strong antiepileptic effect mediated by the NPY receptor Y2 (214–216). This was confirmed by the analysis of specific NPY receptor knock-out mice (217, 218) and injection of the specific Y2 receptor agonist Ac[Leu (28, 31)] NPY24-36 and the specific Y2 receptor antagonist BIIE0246 in GAERS rats (215). Notably, viral overexpression of NPY as well as the mRNA of its receptor Y2, both in thalamus and somatosensory cortex of GAERS rats, reduced the number of seizures and the time spent in seizure activity (219). Since astrocytes produce (220) and release (221) NPY and express NPY receptors, including Y2 receptor (222, 223), one can imagine that astrocytes may play a role in NPY signaling in the pathophysiology of cortico-thalamo-cortical networks.
Alterations of astroglial neurometabolic coupling and contribution to the glutamine-glutamate-GABA cycle may be at the basis of SWDs, possibly through enhanced metabolism and glutamate presentation to cortical neurons. Moreover, astroglial control of extracellular neurotransmitter level, based on the expression of glutamate and GABA transporters (EAATs and GATs, respectively) and receptors (both metabotropic and ionotropic) and direct and indirect release of glutamate and GABA, plays a fundamental role in maintaining the E/I balance in the cortex, thalamus and NRT. Astroglial ATP release and subsequent adenosine production seem to have context-dependent effects on neural excitability, but generally in line with observations derived from convulsive seizures pointing at an antiepileptic and proepileptic role of A1 and A2 receptors, respectively. Shifts in the E/I toward inhibition in the thalamus (possibly through altered endocannabinoid signaling) and toward excitation in the NRT and cortex have a pro-epileptic effect on SWDs. Unexpected net outcomes of pharmacological or genetic manipulation may be due to differential impact on different key nodes of the cortico-thalamo-cortical network and/or to astrocytic ability to both preserve and reverse the sign of the input signal.
The Classic Chicken and Egg Situation. Which Comes First: Astroglial Ca2+ Or Seizures?
Intracellular Ca2+ oscillations are one of the most studied indicators of astroglial activity and information coding mechanism at the core of the astroglial signaling cascade resulting, among others, in gliotransmission (86). In the context of convulsive epilepsy, excitotoxic spilling of glutamate, GABA and ATP resulting from excessive network activity as well as dying cells induce perturbation in astroglial Ca2+ signals (224, 225). Conversely, spontaneous as well as induced Ca2+ oscillations lead to gliotransmission thus influencing neuronal synchrony and E/I balance (226–234). Notably, astroglial Ca2+ elevations precede temporally neuronal engagement and their attenuation results in reduction of the epileptic activity in an in vivo model of temporal lobe epilepsy (TLE) (235). Moreover, astroglial Ca2+ activity is associated with spreading depolarization-mediated seizure termination (236). However, current research is far from understanding astroglial Ca2+ contribution to seizure generation, propagation, severity, and termination both in mechanistic and logical (sufficiency and/or necessity) terms. In particular, research on the contribution of astroglial Ca2+ signaling in seizure phenotype has not yet provided causative links to the SWD pathophysiology. Nevertheless, in the following paragraph we include some observations that encourage further research on the topic.
Thalamic astroglial networks display multi-cellular Ca2+ oscillations in absence of neuronal input and induce glutamate release and NMDA-receptor mediated long lasting inward currents in thalamocortical neurons as studied in acute brain slice preparations (226, 237). Thalamic astrocytes segregate into two groups: a first group with mGluR5-dependent and no voltage-dependent Ca2+ oscillations in response to cortico-thalamic activation, and a second group with no mGluR5- but voltage-dependent Ca2+ responses (183). Moreover, thalamic astroglial Ca2+ responses were recorded after acute ex vivo application of the weak GABAB receptor agonist GHB (177), thus suggesting a putative role of astrocytes in the regulation of GABAergic signaling in the thalamus and possible in the phenomenology of SWDs. Notably, sustained GABAB receptor activation led to a decrease in glutamate release from astrocytes (177). In addition, Ca2+ signaling and GABA seem to be connected since artificial inhibition of Ca2+ oscillation in striatal astrocytes leads to GAT3 functional upregulation and increased GABA uptake (238). Further evidence suggesting an integrative role of thalamic astrocytes in cortico-thalamic interactions comes from the observation that astroglial glutamate- and NMDA receptor- mediated slow inward currents (SICs) in the thalamo-cortical neurons are largely resistant to afferent cortico-thalamic inputs in their emergence but not in their frequency upon sustained input (239, 240). Moreover, cortico-thalamic glutamatergic input induced disinhibition of thalamo-cortical neurons through astroglial mGluR2 activation, Ca2+-dependent glutamate release and inhibition of presynaptic GABAergic projections from the NRT (241). In the NRT astrocytes also enhance GABAA receptor signaling (242). Astrocyte-induced glutamate-mediated SICs of thalamo-cortical neurons seem to be dependent on extracellular glutamate levels, since exogenous exposure to the glutamate-mimetic D-aspartate increased the frequency of SICs (243). Although it is still unclear if abnormal or hypersynchronous astroglial Ca2+ signals could promote epileptiform network activity by itself, this evidence further supports an astroglial contribution to the propagation and self-sustain of seizure-like activity (244, 245).
The role of astrocytic Ca2+ signaling in epilepsy, and particularly in SWD-displaying epilepsies, is far from being understood. Yet, association studies on CAE and other idiopathic epileptic forms displaying SWDs as well as the evaluation of the genetic etiology of rodent absence epilepsy models point to a plethora of genes involved in voltage-gated Ca2+ channel signaling and G protein-coupled receptor signaling that is worth further assessment (7).
Astroglia display spontaneous Ca2+ oscillations responsible for gliotransmission and homeostatic control of the E/I balance as well as network synchronicity. Moreover, astrocytes respond to physiological network activity and pathological neurotransmitter spilling and release from dying cells by Ca2+ elevations, typically further contributing to network priming, seizure initiation and progression. Conversely, Ca2+ signaling-induced gliotransmitter release and modulation of astroglial neurotransmitter receptors and transporters may underlie putative (or potential) anti-epileptic roles of Ca2+ signaling. Notably, astroglial Ca2+ signaling may also contribute to seizure suppression. To which extent this applies to SWDs is still unclear.
Reactive Astrogliosis and the Astrocyte-Derived Inflammatory Response May Contribute to the Pathology of SWDs
Astroglial proliferation and morphological, biochemical, and functional changes associated with epilepsy as well as with other neurodegenerative diseases are commonly referred to as reactive astrogliosis (246–248). The term is misleading since it implies that the pathological phenotype of astrocytes results from the epileptiform activity and oversees the possible causative role of astrocyte modifications in its genesis (249–251). In GAERS rats, cortical as well as thalamic astrocytes display enhanced expression of the glial fibrillary acidic protein (GFAP) even before the onset of absence seizures (193). Similarly, increased levels of GFAP expression can be found in adult WAG/Rij rats, though to a lesser extent than in GAERS rats (252). Astonishingly, the number of glial cells in the somatosensory cortex is significantly decreased (253). This suggests that biochemical and functional changes may contribute to a greater extent to the pathology of absence seizures than morphological alteration or that the latter involves qualitative astroglial reorganization, e.g., overlap of the astroglial processes, astroglial domain reorganization, structural and quantitative alteration of synaptic contacts or blood-brain barrier dysfunction. Notably, valproic acid diminishes the overlap of astroglial processes observed in correspondence of epileptic foci in several pathological models of convulsive seizures (254). Nevertheless, it is not clear if the same is happening in the pharmacodynamics of valproic acid in the context of SWDs. The same is true for the alterations of the blood-brain barrier (BBB) which have been associated with many pathological scenarios, including epilepsy (255), but whose role in SWDs has not been extensively addressed yet.
Pathological stimulation of astrocytes during convulsive epileptiform activity leads to astrocytic upregulation and release of proinflammatory cytokines, with IL-1β, Il-6, and TNFα as the most prominent ones. These factors, in turn, can induce astroglial dysfunction leading to, among others, increased glutamate release, decreased glutamate uptake, down-regulation of Kir4.1, AQP4, connexins, and glutamine synthetase as well as upregulation of adenosine kinase (256–258). IL-1β is induced in reactive astroglia in the somatosensory cortex (and not in other regions of the cortex) in adult GAERS rats with mature SWDs and interestingly also in some young GAERS in association with immature forms of SWDs (259). Furthermore, inhibition of IL-1β biosynthesis in adult GAERS reduced both the number as well as the duration of SWDs. Conversely, IL-1β intraperitoneal administration in WAG/Rij rats induced a significant increase in SWDs and worsened the proepileptic effects of the GABA reuptake inhibitor tiagabine (260). TNFα administration also aggravates SWDs but with kinetics incompatible with a direct effect and therefore possibly through de novo production of IL-1β itself. Moreover, before the onset of SWDs, young WAG/Rij rats showed increased TNFα blood levels, which gradually decreased with age and returned to physiological levels in adult rats displaying mature SWDs, thus possibly suggesting a neuroprotective role of TNFα (260). The precise mechanism of TNFα action in this scenario is not clear, although it is known that TNFα reduces astroglial glutamate uptake and decreases neuronal GABAA receptor expression (261, 262). Notably, IL-1β-, TNFα-, and IL-6-inducing lipopolysaccharide (LPS) injection in WAG/Rij also promoted SWDs and the increase in the latter was prevented by blocking the inflammatory response with indomethacin (263, 264) as well as blockers of the mTOR pathway (265, 266). Similarly, LPS effects on SWDs were later confirmed in GAERS rats (267). Although IL-1β is believed to increase the levels of glutamate, co-administration of LPS and the NMDA receptor antagonist D-(-)-2-Amino-5-phosphonopentanoic acid (AP-5) did not counteract LPS effects as expected, but conversely prolonged them (264). Recently, it has been reported that IL-6 receptor (IL-6R) blockage via tocilizumab (a humanized monoclonal antibody against IL-6R) reduces SWDs in WAG/Rij rats and inhibits their LPS-induced worsening (268). In line with that, human CAE is known to be associated with detectable levels of IL-6 and IL-8 in the cerebrospinal fluid (269) and treatment with valproic acid reduces IL-6 serum levels in children with tonic-clonic generalized seizures (270).
Several lines of evidence support a role for a direct contribution of pro-inflammatory cytokines in the genesis and worsening of SWDs. Notably, astroglial alterations and cytokine release precede SWD onset, although it cannot be excluded that these cell responses may be due to subclinical epileptiform activities or genetic predispositions. IL-1β, IL-6 and TNFα may contribute to the pathology of SWDs, possibly through impaired K+ clearance, glutamine-glutamate-GABA cycle, adenosine metabolism, gliotransmission, and neurotransmitter reuptake. Other morphological alterations, such as astroglial overlap, connectivity, and synaptic coverage, may play a role as well.
Absence Seizures and Nrem Sleep: Two Sides of the Same Coin?
The cortico-thalamo-cortical network processes behaviourally relevant internal and external information and determines vigilance states as well as neuronal network oscillation during sleep (Figure 4A), thus playing a fundamental role in both physiology and pathology (25, 276–281). Several lines of evidence suggest that epilepsy and sleep are strongly related (282). Notably, various forms of epilepsy display different incidences across the 24 h sleep/wake cycle and among different sleep stages, possibly due to specific seizure susceptibility dependent on brain excitability and network engagement (283–285).
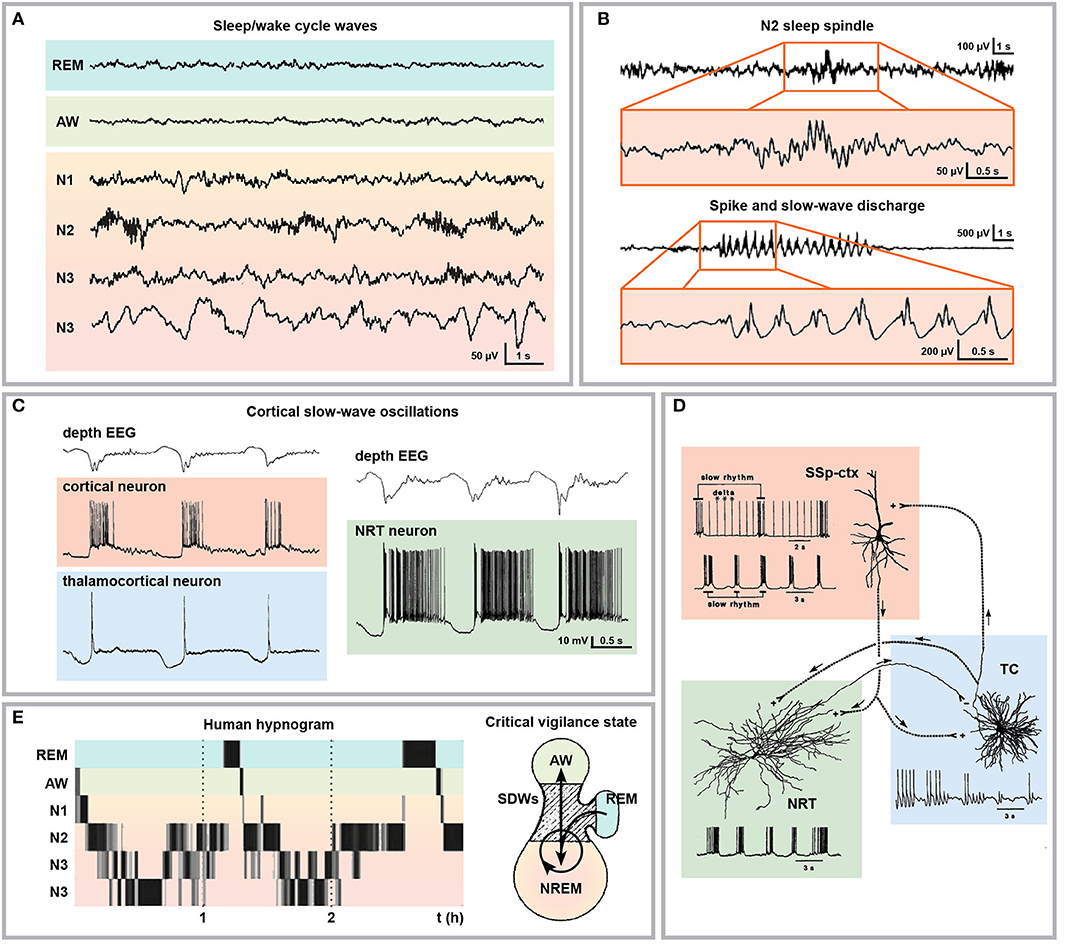
Figure 4. Electrophysiological and cellular bases of sleep and SWD relationship. (A) Representative human electroencephalographical wave recordings during wakefulness (AW), REM sleep and different NREM sleep stages (N1, passive wakefulness or light sleep; N2, light slow-wave sleep; N3, deep slow-wave sleep). (B) Sleep spindle typically occurring during stage N2 of NREM sleep with respective magnification and comparison with SWDs. (C) Depth cortical EEG recording displaying cortical slow wave oscillations (upper traces) and time-matched intracellular recordings from cortical, thalamocortical, and NRT neurons with typical burst firing activity. (D) Schematic representation of the cellular and electrical components of cortico-thalamo-cortical oscillations. (E) Human hypnogram displaying two typical sleep cycles characterized by the succession of the NREM sleep stages followed by one episode of REM sleep (left) and schematic representation of the critical vigilance level (hatched area) promoting SWD occurrence during transitions between NREM and wakefulness, between NREM stages and from (but not to) REM sleep (right). Modified from (A), (271); (B), (272), (C), (273); (D), (274); (E), (271, 275).
Till recently, SWDs were considered the pathological transformation of sleep spindles (also known also thalamocortical spindles) occurring during stage II NREM sleep (Figure 4B) (272, 286, 287). This concept was mainly supported by studies on the temporal coincidence of sleep spindles and SWDs (288, 289) and on the progressive transformation of sleep spindles into SWDs observed after intramuscular injection of penicillin in cats (24, 290). Indeed, to some extent both sleep spindles and SWDs share some anatomical, cellular and molecular mechanisms (291) and they are functionally correlated (292, 293). However, the identification of SWDs as pathological transitions from sleep spindles has been recently challenged (294–296), in favor of a predominant role of cortical slow (<1 Hz) oscillations alternating active (Up) and silent (Down) cortical activity and concomitantly occurring during NREM sleep (Figures 4C,D) (273, 274, 297–301). SWDs largely arise in a specific critical vigilance window in correspondence with passive wakefulness, transitions to NREM slow-wave sleep as well as during transitions between internal substages of NREM sleep (stage I to III; N1: light sleep or passive wakefulness, N2: light slow-wave sleep and N3: deep slow-wave sleep, respectively) (Figure 4E). Moreover, SWDs are disrupted by arousing stimuli and do not transition to REM sleep directly (38, 275, 302–308), thus suggesting that absence seizures prefer low and shifting-vigilance periods during superficial slow-wave NREM sleep (282). With respect to the incidence of seizures across the 24 h cycle, the distribution of generalized SWDs is still under debate. Seizures originating in the frontal lobe (as absence seizures are currently believed to be) are more frequent at night and in sleep (309, 310). Conversely though, dialeptic and atonic seizures occur more often during daytime (310). Generalized pediatric seizures, including absence seizures, were reported to occur predominantly during wakefulness (311, 312) but were restricted to NREM sleep stages I and II when occurring during the night and were almost absent during REM sleep (313, 314). Moreover, a study on idiopathic generalized epilepsies including CAE and other SWD-displaying epilepsies showed that interictal epileptic discharges are more frequent during NREM sleep and occur mainly at sleep onset (315). In WAG/Rij rats, SWDs are most frequent in the beginning of the dark phase and are at their minimum frequency at the onset of the light phase (316, 317). If rats are artificially kept in dim light (thus disrupting the 12:12 light-dark cycle), SWDs still display 24 h cyclicity, proving its endogenous rhythmicity, but the cycle is desynchronized with respect to the rhythm of the general motor activity, thus suggesting that the mechanism governing SWDs and sleep/wake cycles are different (317, 318). Interestingly, after an artificial shifting in the light-dark cycle, SWDs resynchronized at the same speed of light slow-wave speed in comparison with both REM and deep slow-wave sleep (319), pointing at the existence of a common circadian mechanism governing SWDs and light slow-wave sleep. Taken together, it seems that conditions associated with highly desynchronized (active wakefulness and REM sleep) and highly synchronized (deep slow-wave sleep) cortical activity tend to inhibit SWDs. In line with this hypothesis, the anti-absence molecule uridine (320) impacts sleep architecture by fragmenting sleep, thus increasing the frequency of NREM-REM transitions and by inducing preferentially REM sleep (321).
With respect to the putative interdependency of SWDs and NREM sleep waves, it was reported that sleep deprivation has a proepileptic effect on both humans (322–325) and rodents (304, 305, 326, 327). On the other side, epilepsy is associated with sleep alterations, including sleep fragmentation, day-time drowsiness and difficulties in sleep initiation (328). To date, the field still lacks a systematic clinical study on the effect of absence epilepsy on sleep. Nevertheless, it was shown in WAG/Rij rats that SWDs disrupt NREM sleep and sleep architecture (329). Moreover, epilepsy-induced sleep alterations depend on the timing of the epileptiform activity. In a time-controlled kindling epileptic model in rats, seizure induction at the transition from light to dark (zeitgeber time (ZT) 0) and from dark to light (ZT13) altered both NREM and REM duration without affecting sleep/wake cycles and the sole seizure induction at ZT13 induced increased levels of IL-1 and increased NREM sleep specifically (330). Interestingly, both IL-1β and TNFα increase the amount of NREM sleep (331), which could contribute to the increase of SWD number after LPS injection (263, 264) by an increased state of passive awareness and slow-wave sleep. Indeed, in silico meta-analysis of differentially expressed proteins from the fronto-parietal cortex and thalamus of LPS-treated WAG/Rij rats supports this scenario, given the overrepresentation of proteins associated with sleep regulation (332). Moreover, the pathological activation of the mTOR pathway involved in LPS-induced increase in SWDs (265, 266) is responsible for the upregulation of the core clock gene product aryl hydrocarbon receptor nuclear translocator-like protein 1 (ARNTL), also known as brain and muscle ARNT-Like 1 (BMAL1), as observed in a model of tuberous sclerosis complex, a neurological disorder displaying epileptic activity (333). BMAL1 not only is a key component of both circadian and sleep/wake cycles (334) as well as susceptibility to seizures and epilepsy (335), but it is also at the basis of cell-autonomous circadian clock of astrocytes (336, 337).
SWD occurrence varies during the sleep/wake cycle with respect to both sleep and vigilance states. Notably, SWDs peak in correspondence of low and shifting-vigilance periods during superficial slow-wave NREM sleep and are underrepresented during active wakefulness and REM sleep. Resynchronization studies after shifting in the light-dark cycle suggest a common circadian mechanism governing SWDs and NREM sleep. Moreover, even though the nature and the causative link between the two are far from being clearly understood, SWDs and NREM sleep are similarly and consistently altered by a number of pathological and pharmacological alterations.
Both Sleep Architecture and Sleep/Wake Cycle Are Shaped by Astroglial Activity
Given the fact that both SWDs and NREM recruit the cortico-thalamo-cortical network, further insights into the role of astrocytes in the pathophysiology of SWDs may derive from evidence in their contribution to sleep (particularly sleep architecture and sleep/wake cycle) (Figure 5) as well as the circadian cycle. Astroglial impact on circadian clock mechanisms generated in deep structures may contribute to epilepsy in non-intuitive ways (335). In the hypothalamic primary timekeeping center, the suprachiasmatic nucleus (SCN), astrocyte-derived glutamate inhibits neuronal firing through presynaptic NMDA receptors specifically during the night (338). Moreover, astrocytes release adenosine in a CB1 receptor- and intracellular Ca2+ signaling-dependent manner and induce the disinhibition of SCN neurons (339). WAG/Rij rats are also characterized by astrogliosis and impaired GABAergic transmission in the thalamic intergeniculate leaflet, which coordinates inputs from the retina and outputs to the SCN (340). The research on the role of astrocytes on timekeeping is still at its early days, but several lines of evidence support astroglial contribution to sleep, namely the modulation of sleep homeostasis, sleep pressure, vigilance states and sleep-dependent cognitive function, brain energetics and network metabolic supply, network excitability and sleep-associated waste clearance (341, 342).
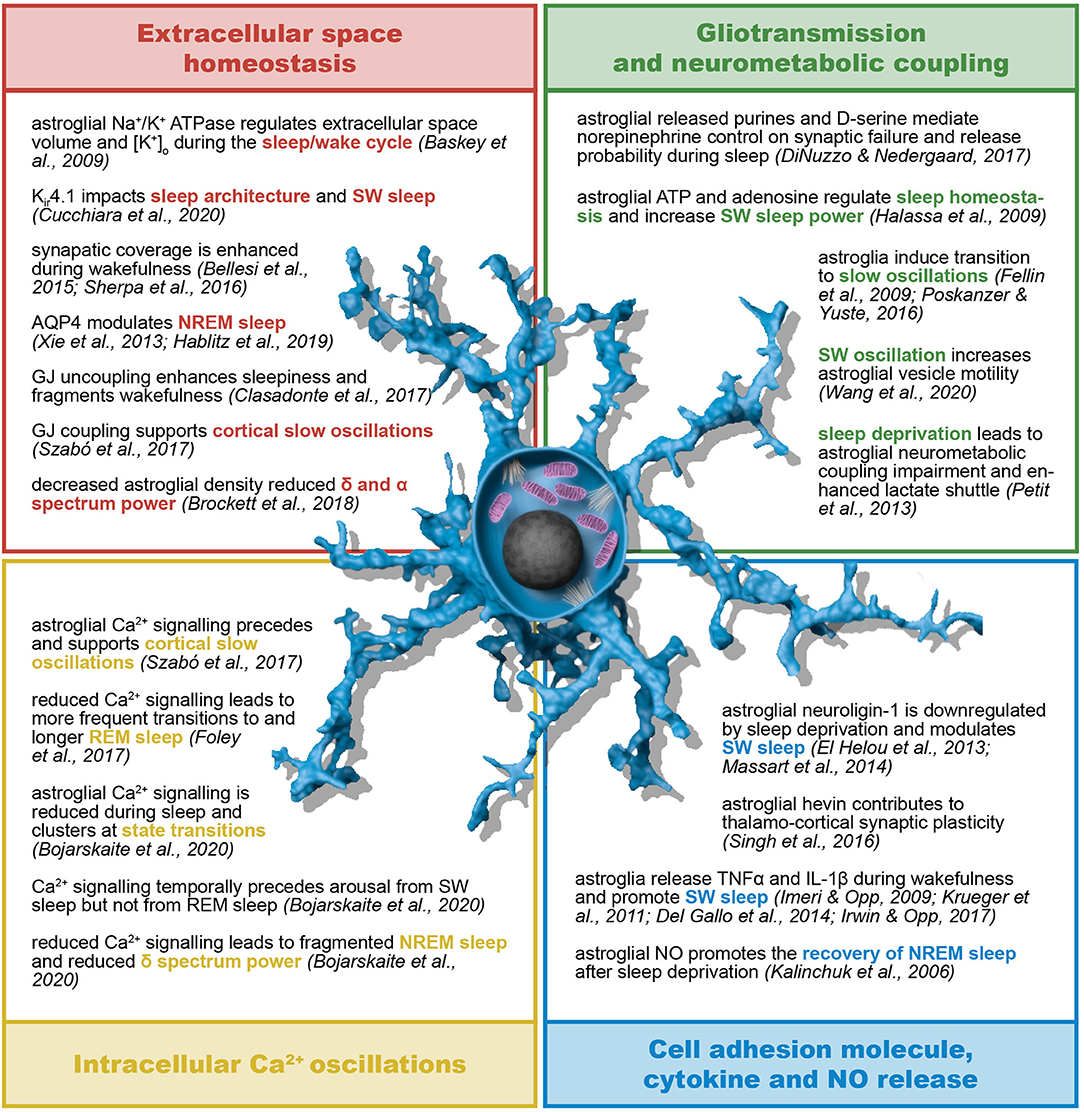
Figure 5. Astroglial role in sleep/wake cycle and sleep architecture. Astrocytes contribute to sleep homeostasis in terms of both sleep/wake cycle regulation and sleep architecture and dynamics. The astroglial regulation of sleep relies on their control of extracellular K+ concentration, interstitial fluid exchanges and spatial buffering through gap junctions. Moreover, astroglia support and influence neural activity by means of their neurometabolic coupling to neurons, intracellular Ca2+ oscillations, gliotransmission, and release of, among other, cell adhesion molecules, cytokines and nitric oxide. AQP4, aquaporin-4; ATP, adenosine triphosphate; GJ, gap junction; IL-1β, interleukin-1β; Kir, inward rectifying K+ channels; NO, nitric oxide; (N)REM, (non-) rapid eye movement; SW, slow wave; TNFα, tumor necrosis factor α.
The sleep/wake cycle is associated with changes in interstitial fluid and of the ionic composition with increased extracellular space and decreased interstitial K+ concentration during sleep (343–345), a process that involves norepinephrine-mediated inhibition of the astroglial Na+/K+ pump during wakefulness (346). This process is responsible for widespread neuronal hyperpolarization and decreased firing rate particularly during NREM sleep (347, 348). In parallel, it was recently reported that children with an autism-associated epilepsy phenotype carrying a gain-of-function mutation in the Kir4.1 coding KCHJ10 gene display abnormal slow-wave sleep with a significantly longer slow-wave period (349). Norepinephrine induces astroglial process elongation and astroglial synaptic coverage during wakefulness (345, 350). Conversely, decreased levels of norepinephrine may be responsible for reduction of direct and indirect astroglial release of ATP/adenosine and D-serine, thus contributing to the overall decreased synaptic failure and increased release probability during sleep. Interestingly, norepinephrine level is particularly low during NREM sleep, due to suppression of noradrenergic neuronal firing (351).
Astroglial-dependent cerebrospinal fluid (CSF) flow is responsible for waste and interstitial fluid clearance during sleep (352) and inward flow of CSF through astroglial AQP4 occurs mainly during NREM sleep (343, 353). The CSF flow is under circadian control mediated by changes of AQP4 polarization (354). Recently, a haplotype of AQP4 carrying several single nucleotide polymorphisms (SNPs), among which some associated with reduced AQP4 expression, has been linked to altered slow-wave NREM sleep modulation (355). Moreover, astroglial gap junction coupling is likely to contribute to the regulation of the sleep/wake by means of modulation of both CSF flow and waste clearance. To date, most studies addressing GJ coupling in astrocytes are focused on the altered metabolite trafficking (namely glucose and lactate) resulting from GJ manipulation that impairs the fundamental role of astrocytes in synaptic energy support and brain energy metabolism (356). Astrocyte-specific conditional knock-out of Cx43 in mice resulted in enhanced sleepiness, fragmented wakefulness, and impaired neuromodulation of the sleep/wake cycle (357). Conversely, astroglial neurometabolic coupling impairment results from sleep deprivation that leads to astroglial upregulation of the transporters GLUT1, GLT1, the Na+/K+ pump as well as other components of the astrocyte-neuron lactate shuttle (358). Sleep deprivation could therefore possibly contribute to increased network activity also through enhanced lactate delivery to neurons as suggested by the fact that the anticonvulsant stiripentol is a lactate dehydrogenase inhibitor (359) in addition to being a positive allosteric modulator of GABAA receptors (360).
Impairment of astroglial exocytosis and gliotransmission using cell-specific expression of dnSNARE results in reduced tonic A1R adenosinergic signaling, altered sleep homeostasis and reduced slow-wave power, and reduced sleep pressure in mice (361), as confirmed by previous evidence suggesting the role of A1 receptors in augmented sleep pressure (362–364). The same genetic manipulation suppressed the LPS-induced increase in slow-wave power during NREM sleep, proving the astroglial contribution to inflammatory-derived increased sleep pressure (365). Although the role of A2 receptor (A2R) activation by astroglial adenosine is still controversial, A2Rs may play a role in sleep homeostasis through activation of A2AR-expressing neurons in the nucleus accumbens core involved in the induction of slow-wave sleep (366). In the cortex, altered gliotransmission resulted in reduced neuronal NMDA receptor activity and reduced slow oscillations (367), whereas astroglial specific activation induced neuronal transition to slow oscillations (368). In vivo characterization of Ca2+ signaling in both rat cortical astrocytes and neurons revealed that astroglial synchronized activity reliably precedes neuronal oscillations and that both astrocyte uncoupling and intracellular Ca2+ chelation reduced the fraction of astrocytes and neurons involved in the cortical slow waves. Remarkably, neurons closer to active astrocytes were more likely involved in the oscillations (369). Recently, simultaneously recording of BOLD fMRI and astroglial Ca2+ signaling in anesthetized rats revealed that a fraction of intrinsic cortical Ca2+ signals were associated with reduced EEG power and negative fMRI signal throughout the cortex (correlated with decreased neuronal activity) and that increased activity in the thalamus specifically preceded these signals (370). Conversely, reduction in the density of cortical astrocytes in the medial prefrontal cortex (and therefore putatively in their connectivity) has been linked to a decrease in δ (0.5–4 Hz) and α (8–12 Hz) spectrum power (371). Moreover, mice overexpressing an astrocyte-specific inositol triphosphate (IP3) phosphatase, and therefore displaying reduced IP3-dependent Ca2+ activity, spent more time in REM sleep and revealed more transitions to REM sleep from passive wakefulness (372). In line with that, mice lacking the inositol 1,4,5-triphosphate receptor type 2 exhibit reduced Ca2+ signaling, a more fragmented and shorter NREM sleep associated with decreased δ spectrum power and more frequent microarousals (373). Remarkably, astroglial intracellular Ca2+ increases precede the transition from NREM sleep to wakefulness but follow arousal from REM sleep (373), thus suggesting that astroglia could mediate norepinephrine-induced arousal from NREM sleep (374). Furthermore, it has been recently shown that in vitro application of an oscillatory electric field specifically in the slow-wave range (and not at higher frequencies) increased astroglial synaptic vesicle mobility (375), thus suggesting a positive feedback mechanism for slow-wave state perpetuation. Taken together, these data suggest that astroglial connectivity, astroglial Ca2+ waves and gliotransmitter release may initiate and/or support the initiation of cortical slow wave oscillations or favor this transition over others (e.g., passive wakefulness-REM).
Another piece of evidence supporting the contribution of astroglial synaptic plasticity in the modulation of sleep derives from studies focused on the extracellular matrix components in the synaptic cleft. In the mouse forebrain, sleep pressure after sleep deprivation decreased the expression of astroglial neuroligin-1 (376), a cell adhesion molecule binding to presynaptic neurexins (377, 378). Conversely, neuroligin-1 knock-out mice have increased slow-wave sleep and enhanced synchrony during sleep (379). Astrocytes contribute to glutamatergic synaptic plasticity of thalamo-cortical synapses through secretion of hevin, a synaptogenic protein inducing the interaction between non-canonical synaptic partners including neuroligin-1 (380). Knock-out mice lacking another member of the neuroligin family, neuroligin-2, develop SWDs and behavioral arrests, a phenotype blocked by ethosuximide and attenuated by expression of neuroligin-2 selectively in the thalamic neurons or optogenetic activation of GABAergic projections from the NRT (381). This could be due to an interaction with GABAA receptors as suggested by previous studies on sleep-deprived mice (382). Mice with a missense mutation in neuroligin-3 exhibit an altered EEG power spectrum (383). Moreover, variations in the copy number of the gene encoding the postsynaptic scaffolding protein Shank3, which interacts with both neuroligin and glutamate receptors thus regulating synaptic plasticity (384, 385), have been linked to epileptiform activity specifically arising during slow-wave sleep (386). Shank3 loss-of-function mutations have been associated with several different epileptic forms but most commonly with atypical absence seizures (387).
Finally, as mentioned already above, inflammation and sleep are tightly intertwined. TNFα and IL-1β increase during wakefulness and decrease during sleep both at mRNA and protein level (388–390) and their systemic injection selectively increase slow-wave sleep (389, 391). Mice lacking IL-1β and TNFα receptors are characterized by less slow-wave and REM sleep (392, 393). Notably, agonists of both P2X- and P2Y-type purinergic receptors are known to activate the astroglial release of TNFα and IL-1β (394) and pannexin-1 knock-out mice with impaired ATP release display altered slow-wave sleep (395). Interestingly, unilateral cortical TNFα injection induces state-specific EEG asymmetries during NREM sleep (396) and ipsilateral increase in the number of IL-1β positive cells (mainly astrocytes) in the cortex, the NRT and in the thalamic nuclei (397). Along with TNFα and IL-1β, reactive astroglia also produce nitric oxide (NO) via inducible NO synthetase (iNOS) (398, 399) and NO has been linked to the pathology of epilepsy (400, 401). NO affects both NREM and REM sleep (402, 403) but inhibiting iNOS in sleep-deprived mice specifically impairs NREM recovery, whereas inhibiting neuronal NOS (nNOS) impairs the recovery of REM sleep (404), thus suggesting a specific role of astrocyte in NREM physiology.
Astroglial homeostatic control of the extracellular space and synaptic transmission through K+ clearance mediates widespread neuronal hyperpolarization and decreased firing activity during sleep and impacts NREM sleep architecture. Astroglial regulation of the extracellular volume controls both waste and cerebrospinal fluid clearance essential for correct functioning of the brain and relies on AQP4-mediated water influx and GJ coupling. Adenosine released by astrocytes governs sleep homeostasis, sleep pressure and slow-wave power and astroglial gliotransmission contributes to neuronal slow oscillations in the cortex. Moreover, artificial lowering of astroglial Ca2+ oscillations and connectivity leads to reduced slow-wave power and relative shortening and fragmentation of NREM sleep. Finally, astroglial TNFα, IL-1β, and NO are likely to impact sleep architecture by selective increase of slow-wave sleep. These findings support the fact that astroglia play a fundamental role in the physiology of NREM sleep and therefore represent a promising target to study pathophysiological alterations inducing or sustaining the abnormal recruitment of the cortico-thalamo-cortical network during SWDs.
Conclusion
From a clinical, social and human point of view, epilepsy is probably one of the most heterogeneous neurological diseases. This variability partially relies on the fact that epilepsy may originate from a plethora of different conditions, among others traumatic injury, stroke, CNS infections or inflammation, brain tumor, genetic predisposition, and drug or alcohol abuse. In addition, in six out of 10 epilepsy cases the pathological origin is unknown. Yet, from a scientific point of view, epilepsy can ultimately be reduced to a local imbalance of excitation and inhibition and altered synchrony and functioning of neural networks in the brain. The heterogeneity of pathological outcomes associated with epilepsy arises from the variety and complexity of functions carried out by the human brain and the multiple layers of fine-tuning that each of them requires for reliable physiological functioning of the electrical activity in neural circuits. Given the existence of common molecular and cellular mechanisms at the basis of epilepsy and their nature as pathological transitions of altered physiological processes, both epilepsy research and clinical treatment benefit from the understanding of the inner functioning of neural networks.
Since spike and slow-wave discharges (SWDs) share some key anatomical and functional physiological brain oscillations naturally occurring during slow-wave sleep, absence seizure research could advance our understanding of both epilepsy and healthy brain mechanisms. In this review, we collected evidence supporting the functional and mechanistic relationship between slow-wave sleep and SWDs, thus providing insights into network alterations that contribute to the pathology of SWDs. Moreover, proving and characterizing the interdependency between epilepsy, sleep architecture and sleep/wake cycles possess an undeniable therapeutic value, since sleep is a pre-existing condition affecting any treatment outcome and efficacy.
We focused our attention on the role of astrocytes in the physiology of sleep and in their putative pathophysiological contribution to SWDs. Astroglial control on extracellular homeostasis in terms of ionic composition, volume regulation and transmitter clearance, astroglial connectivity, Ca2+ signaling and gliotransmission as well as cytokine release are hallmarks of astroglial function for physiological brain performance and were addressed in the context of SWDs and sleep research. Please note that it is insufficient and underestimating of the system complexity to label the astrocytic contribution to neural homeostasis as exclusively anti- or pro-epileptic. Many astroglial mechanisms may be beneficial or detrimental with respect to different forms of epilepsy, not to mention different network connectivities and states. Current research on astroglial contribution to epileptic brain functioning mostly relies on studies focused on convulsive seizures, possibly due to their lower remission rate and their clinical symptoms which appear more obvious and life threatening. Nevertheless, some clues suggest the mechanisms governing network excitability and synchrony may have a role in SWDs, too. This work was not intended to be and is far from being comprehensive neither of the role of astroglia in epilepsy nor of their contribution to sleep homeostasis and architecture but provides with significant associations in the tripartite synapse engaging astroglia, epilepsy and sleep in the context of the pathophysiology of cortico-thalamo-cortical oscillations. Understanding how astroglia contribute to the mechanisms underlying slow-wave sleep and how these are altered in pathology could possibly shine light on new therapeutical targets for a plethora of epileptic forms displaying SWDs, among which absence epilepsy, a condition that still affects 50 million people worldwide and is pharmacoresistant in almost one third of those.
Author Contributions
DG screened the literature, conceptualized the review focus, wrote the first draft, designed and realized the figures, and finalized the manuscript. AS and FK contributed to figure conceptualization, reviewed and finalized the manuscript. All authors approved on the final version of the manuscript.
Funding
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 722053 (EU-GliaPhD), ERANET Neuron (BrIE) and from the Deutsche Forschungsgemeinschaft DFG (SFB 894, SFB 1158, SPP 1757, and FOR 2289).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the members of the department for fruitful discussion and Jens Grosche (Effigos AG) for his contribution to Figures 2, 5.
References
1. Huff J, Murr N. Seizure. In: StatPearls. Treasure Island, FL: StatPearls Publishing (2021). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK430765/
2. Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. (2014) 55:475–82. doi: 10.1111/epi.12550
3. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. (2017) 58:512–21. doi: 10.1111/epi.13709
4. Panayiotopoulos CP. Typical absence seizures and related epileptic syndromes: assessment of current state and directions for future research. Epilepsia. (2008) 49:2131–9. doi: 10.1111/j.1528-1167.2008.01777.x
5. Guo JN, Kim R, Chen Y, Negishi M, Jhun S, Weiss S, et al. Impaired consciousness in patients with absence seizures investigated by functional MRI, EEG, and behavioural measures: a cross-sectional study. Lancet Neurol. (2016) 15:1336–45. doi: 10.1016/S1474-4422(16)30295-2
6. Panayiotopoulos CP, Koutroumanidis M, Giannakodimos S, Agathonikou A. Idiopathic generalised epilepsy in adults manifested by phantom absences, generalised tonic-clonic seizures, and frequent absence status. J Neurol Neurosurg Psychiatry. (1997) 63:622–7. doi: 10.1136/jnnp.63.5.622
7. Crunelli V, Leresche N. Childhood absence epilepsy: genes, channels, neurons and networks. Nat Rev Neurosci. (2002) 3:371–82. doi: 10.1038/nrn811
8. Blumenfeld H. Consciousness and epilepsy: why are patients with absence seizures absent? Prog Brain Res. (2005) 150:271–86. doi: 10.1016/S0079-6123(05)50020-7
9. Camfield C, Camfield P. Management guidelines for children with idiopathic generalized epilepsy. Epilepsia. (2005) 46(Suppl.9):112–6. doi: 10.1111/j.1528-1167.2005.00322.x
10. Gardiner M. Genetics of idiopathic generalized epilepsies. Epilepsia. (2005) 46(Suppl.9):15–20. doi: 10.1111/j.1528-1167.2005.00310.x
11. Matricardi S, Verrotti A, Chiarelli F, Cerminara C, Curatolo P. Current advances in childhood absence epilepsy. Pediatr Neurol. (2014) 50:205–12. doi: 10.1016/j.pediatrneurol.2013.10.009
12. Berg AT, Levy SR, Testa FM, Blumenfeld H. Long-term seizure remission in childhood absence epilepsy: might initial treatment matter? Epilepsia. (2014) 55:551–7. doi: 10.1111/epi.12551
13. Glauser TA, Cnaan A, Shinnar S, Hirtz DG, Dlugos D, Masur D, et al. Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy: initial monotherapy outcomes at 12 months. Epilepsia. (2013) 54:141–55. doi: 10.1111/epi.12028
14. Masur D, Shinnar S, Cnaan A, Shinnar RC, Clark P, Wang J, et al. Pretreatment cognitive deficits and treatment effects on attention in childhood absence epilepsy. Neurology. (2013) 81:1572–80. doi: 10.1212/WNL.0b013e3182a9f3ca
15. Holmes GL, Noebels JL. The epilepsy spectrum: targeting future research challenges. Cold Spring Harb Perspect Med. (2016) 6:a028043. doi: 10.1101/cshperspect.a028043
16. Cnaan A, Shinnar S, Arya R, Adamson PC, Clark PO, Dlugos D, et al. Second monotherapy in childhood absence epilepsy. Neurology. (2017) 88:182–90. doi: 10.1212/WNL.0000000000003480
17. Crunelli V, Lorincz ML, McCafferty C, Lambert RC, Leresche N, Di Giovanni G, et al. Clinical and experimental insight into pathophysiology, comorbidity and therapy of absence seizures. Brain. (2020) 143:2341–68. doi: 10.1093/brain/awaa072
18. Shi Q, Zhang T, Miao A, Sun J, Sun Y, Chen Q, et al. Differences between interictal and ictal generalized spike-wave discharges in childhood absence epilepsy: a MEG study. Front Neurol. (2019) 10:1359. doi: 10.3389/fneur.2019.01359
19. Cerminara C, Coniglio A, El-Malhany N, Casarelli L, Curatolo P. Two epileptic syndromes, one brain: childhood absence epilepsy and benign childhood epilepsy with centrotemporal spikes. Seizure. (2012) 21:70–4. doi: 10.1016/j.seizure.2011.09.005
20. Coenen AM, Van Luijtelaar EL. Genetic animal models for absence epilepsy: a review of the WAG/Rij strain of rats. Behav Genet. (2003) 33:635–55. doi: 10.1023/A:1026179013847
21. Panayiotopoulos CP. Treatment of typical absence seizures and related epileptic syndromes. Paediatr Drugs. (2001) 3:379–403. doi: 10.2165/00128072-200103050-00006
22. Bai X, Vestal M, Berman R, Negishi M, Spann M, Vega C, et al. Dynamic time course of typical childhood absence seizures: EEG, behavior, and functional magnetic resonance imaging. J Neurosci. (2010) 30:5884–93. doi: 10.1523/JNEUROSCI.5101-09.2010
23. Tangwiriyasakul C, Perani S, Centeno M, Yaakub SN, Abela E, Carmichael DW, et al. Dynamic brain network states in human generalized spike-wave discharges. Brain. (2018) 141:2981–94. doi: 10.1093/brain/awy223
24. von Krosigk M, Bal T, McCormick DA. Cellular mechanisms of a synchronized oscillation in the thalamus. Science. (1993) 261:361–4. doi: 10.1126/science.8392750
25. McCafferty C, David F, Venzi M, Lorincz ML, Delicata F, Atherton Z, et al. Cortical drive and thalamic feed-forward inhibition control thalamic output synchrony during absence seizures. Nat Neurosci. (2018) 21:744–56. doi: 10.1038/s41593-018-0130-4
26. Meyer J, Maheshwari A, Noebels J, Smirnakis S. Asynchronous suppression of visual cortex during absence seizures in stargazer mice. Nat Commun. (2018) 9:1938. doi: 10.1038/s41467-018-04349-8
27. Panayiotopoulos CP. Typical absence seizures and their treatment. Arch Dis Child. (1999) 81:351–5. doi: 10.1136/adc.81.4.351
28. Blumenfeld H. Cellular and network mechanisms of spike-wave seizures. Epilepsia. (2005) 46(Suppl.9):21–33. doi: 10.1111/j.1528-1167.2005.00311.x
29. Sitnikova E, van Luijtelaar G. Electroencephalographic characterization of spike-wave discharges in cortex and thalamus in WAG/Rij rats. Epilepsia. (2007) 48:2296–311. doi: 10.1111/j.1528-1167.2007.01250.x
30. Terlau J, Yang JW, Khastkhodaei Z, Seidenbecher T, Luhmann HJ, Pape HC, et al. Spike-wave discharges in absence epilepsy: segregation of electrographic components reveals distinct pathways of seizure activity. J Physiol. (2020) 598:2397–414. doi: 10.1113/JP279483
31. Aghakhani Y, Bagshaw AP, Bénar CG, Hawco C, Andermann F, Dubeau F, et al. fMRI activation during spike and wave discharges in idiopathic generalized epilepsy. Brain. (2004) 127:1127–44. doi: 10.1093/brain/awh136
32. Gotman J, Grova C, Bagshaw A, Kobayashi E, Aghakhani Y, Dubeau F. Generalized epileptic discharges show thalamocortical activation and suspension of the default state of the brain. Proc Natl Acad Sci USA. (2005) 102:15236–40. doi: 10.1073/pnas.0504935102
33. Hamandi K, Salek-Haddadi A, Laufs H, Liston A, Friston K, Fish DR, et al. EEG-fMRI of idiopathic and secondarily generalized epilepsies. Neuroimage. (2006) 31:1700–10. doi: 10.1016/j.neuroimage.2006.02.016
34. Moeller F, Siebner HR, Wolff S, Muhle H, Granert O, Jansen O, et al. Simultaneous EEG-fMRI in drug-naive children with newly diagnosed absence epilepsy. Epilepsia. (2008) 49:1510–9. doi: 10.1111/j.1528-1167.2008.01626.x
35. Moeller F, LeVan P, Muhle H, Stephani U, Dubeau F, Siniatchkin M, et al. Absence seizures: individual patterns revealed by EEG-fMRI. Epilepsia. (2010) 51:2000–10. doi: 10.1111/j.1528-1167.2010.02698.x
36. Bai X, Guo J, Killory B, Vestal M, Berman R, Negishi M, et al. Resting functional connectivity between the hemispheres in childhood absence epilepsy. Neurology. (2011) 76:1960–7. doi: 10.1212/WNL.0b013e31821e54de
37. Coenen AM, Drinkenburg WH, Inoue M, van Luijtelaar EL. Genetic models of absence epilepsy, with emphasis on the WAG/Rij strain of rats. Epilepsy Res. (1992) 12:75–86. doi: 10.1016/0920-1211(92)90029-S
38. Danober L, Deransart C, Depaulis A, Vergnes M, Marescaux C. Pathophysiological mechanisms of genetic absence epilepsy in the rat. Prog Neurobiol. (1998) 55:27–57. doi: 10.1016/S0301-0082(97)00091-9
39. Depaulis A, David O, Charpier S. The genetic absence epilepsy rat from Strasbourg as a model to decipher the neuronal and network mechanisms of generalized idiopathic epilepsies. J Neurosci Methods. (2016) 260:159–74. doi: 10.1016/j.jneumeth.2015.05.022
40. PitkäNen A, Buckmaster PS, Galanopoulou AS, Moshé SL. Models of Seizures and Epilepsy. Cambridge: Academic Press (2017).
41. Depaulis A, Charpier S. Pathophysiology of absence epilepsy: insights from genetic models. Neurosci Lett. (2018) 667:53–65. doi: 10.1016/j.neulet.2017.02.035
42. Depaulis A, Luijtelaar vG. Characteristics of genetic absence seizures in the rat. In: Pitkanen A, Schwartzkroin PA, Moshe S, editors, Models of Seizure and Epilepsy (London: International: Elsevier Academic Press). (2006). p. 233–48. doi: 10.1016/B978-012088554-1/50020-7
44. Frankel WN. Genetics of complex neurological disease: challenges and opportunities for modeling epilepsy in mice and rats. Trends Genet. (2009) 25:361–7. doi: 10.1016/j.tig.2009.07.001
45. Maheshwari A, Noebels JL. Monogenic models of absence epilepsy: windows into the complex balance between inhibition and excitation in thalamocortical microcircuits. Prog Brain Res. (2014) 213:223–52. doi: 10.1016/B978-0-444-63326-2.00012-0
46. Meeren HK, Pijn JP, Van Luijtelaar EL, Coenen AM, Lopes da Silva FH. Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. J Neurosci. (2002) 22:1480–95. doi: 10.1523/JNEUROSCI.22-04-01480.2002
47. Manning JP, Richards DA, Leresche N, Crunelli V, Bowery NG. Cortical-area specific block of genetically determined absence seizures by ethosuximide. Neuroscience. (2004) 123:5–9. doi: 10.1016/j.neuroscience.2003.09.026
48. Polack PO, Guillemain I, Hu E, Deransart C, Depaulis A, Charpier S. Deep layer somatosensory cortical neurons initiate spike-and-wave discharges in a genetic model of absence seizures. J Neurosci. (2007) 27:6590–9. doi: 10.1523/JNEUROSCI.0753-07.2007
49. Polack PO, Mahon S, Chavez M, Charpier S. Inactivation of the somatosensory cortex prevents paroxysmal oscillations in cortical and related thalamic neurons in a genetic model of absence epilepsy. Cereb Cortex. (2009) 19:2078–91. doi: 10.1093/cercor/bhn237
50. Studer F, Laghouati E, Jarre G, David O, Pouyatos B, Depaulis A. Sensory coding is impaired in rat absence epilepsy. J Physiol. (2019) 597:951–66. doi: 10.1113/JP277297
51. Snead OC. gamma-Hydroxybutyrate model of generalized absence seizures: further characterization and comparison with other absence models. Epilepsia. (1988) 29:361–8. doi: 10.1111/j.1528-1157.1988.tb03732.x
52. Snead OC. Pharmacological models of generalized absence seizures in rodents. J Neural Transm Suppl. (1992) 35:7–19. doi: 10.1007/978-3-7091-9206-1_2
53. Venzi M, Di Giovanni G, Crunelli V. A critical evaluation of the gamma-hydroxybutyrate (GHB) model of absence seizures. CNS Neurosci Ther. (2015) 21:123–40. doi: 10.1111/cns.12337
54. Cortez MA, Kostopoulos GK, Snead OC. Acute and chronic pharmacological models of generalized absence seizures. J Neurosci Methods. (2016) 260:175–84. doi: 10.1016/j.jneumeth.2015.08.034
55. Lee S, Hwang E, Lee M, Choi JH. Distinct topographical patterns of spike-wave discharge in transgenic and pharmacologically induced absence seizure models. Exp Neurobiol. (2019) 28:474–84. doi: 10.5607/en.2019.28.4.474
56. van Luijtelaar G, Sitnikova E. Global and focal aspects of absence epilepsy: the contribution of genetic models. Neurosci Biobehav Rev. (2006) 30:983–1003. doi: 10.1016/j.neubiorev.2006.03.002
57. Lüttjohann A, Zhang S, de Peijper R, van Luijtelaar G. Electrical stimulation of the epileptic focus in absence epileptic WAG/Rij rats: assessment of local and network excitability. Neuroscience. (2011) 188:125–34. doi: 10.1016/j.neuroscience,.2011.04.038
58. van Luijtelaar G, Behr C, Avoli M. Is there such a thing as “generalized” epilepsy? Adv Exp Med Biol. (2014) 813:81–91. doi: 10.1007/978-94-017-8914-1_7
59. Paz JT, Huguenard JR. Microcircuits and their interactions in epilepsy: is the focus out of focus? Nat Neurosci. (2015) 18:351–9. doi: 10.1038/nn.3950
60. Avoli M. A brief history on the oscillating roles of thalamus and cortex in absence seizures. Epilepsia. (2012) 53:779–89. doi: 10.1111/j.1528-1167.2012.03421.x
61. Lüttjohann A, van Luijtelaar G. Dynamics of networks during absence seizure's on- and offset in rodents and man. Front Physiol. (2015) 6:16. doi: 10.3389/fphys.2015.00016
62. Sorokin JM, Davidson TJ, Frechette E, Abramian AM, Deisseroth K, Huguenard JR, et al. Bidirectional control of generalized epilepsy networks via rapid real-time switching of firing mode. Neuron. (2017) 93:194–210. doi: 10.1016/j.neuron.2016.11.026
63. Lüttjohann A, Schoffelen JM, van Luijtelaar G. Peri-ictal network dynamics of spike-wave discharges: phase and spectral characteristics. Exp Neurol. (2013) 239:235–47. doi: 10.1016/j.expneurol.2012.10.021
64. Sysoeva MV, Lüttjohann A, van Luijtelaar G, Sysoev IV. Dynamics of directional coupling underlying spike-wave discharges. Neuroscience. (2016) 314:75–89. doi: 10.1016/j.neuroscience.2015.11.044
65. Lüttjohann A, Pape HC. Regional specificity of cortico-thalamic coupling strength and directionality during waxing and waning of spike and wave discharges. Sci Rep. (2019) 9:2100. doi: 10.1038/s41598-018-37985-7
66. Zhang W, Bruno RM. High-order thalamic inputs to primary somatosensory cortex are stronger and longer lasting than cortical inputs. Elife. (2019) 8:18. doi: 10.7554/eLife.44158.018
67. Bal T, von Krosigk M, McCormick DA. Role of the ferret perigeniculate nucleus in the generation of synchronized oscillations in vitro. J Physiol. (1995) 483:665–85. doi: 10.1113/jphysiol.1995.sp020613
68. Bal T, von Krosigk M, McCormick DA. Synaptic and membrane mechanisms underlying synchronized oscillations in the ferret lateral geniculate nucleus in vitro. J Physiol. (1995) 483:641–63. doi: 10.1113/jphysiol.1995.sp020612
69. McCormick DA, Contreras D. On the cellular and network bases of epileptic seizures. Annu Rev Physiol. (2001) 63:815–46. doi: 10.1146/annurev.physiol.63.1.815
70. Pinault D. Cellular interactions in the rat somatosensory thalamocortical system during normal and epileptic 5–9 Hz oscillations. J Physiol. (2003) 552:881–905. doi: 10.1113/jphysiol.2003.046573
71. Cain SM, Snutch TP. T-type calcium channels in burst-firing, network synchrony, and epilepsy. Biochim Biophys Acta. (2013) 1828:1572–8. doi: 10.1016/j.bbamem.2012.07.028
72. Richards DA, Lemos T, Whitton PS, Bowery NG. Extracellular GABA in the ventrolateral thalamus of rats exhibiting spontaneous absence epilepsy: a microdialysis study. J Neurochem. (1995) 65:1674–80. doi: 10.1046/j.1471-4159.1995.65041674.x
73. Cope DW, Hughes SW, Crunelli V. GABAA receptor-mediated tonic inhibition in thalamic neurons. J Neurosci. (2005) 25:11553–63. doi: 10.1523/JNEUROSCI.3362-05.2005
74. Cope DW, Di Giovanni G, Fyson SJ, Orbán G, Errington AC, Lorincz ML, et al. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med. (2009) 15:1392–8. doi: 10.1038/nm.2058
75. Errington AC, Cope DW, Crunelli V. Augmentation of tonic GABA(A) inhibition in absence epilepsy: therapeutic value of inverse agonists at extrasynaptic GABA(A) receptors. Adv Pharmacol Sci. (2011) 2011:790590. doi: 10.1155/2011/790590
76. Errington AC, Gibson KM, Crunelli V, Cope DW. Aberrant GABA(A) receptor-mediated inhibition in cortico-thalamic networks of succinic semialdehyde dehydrogenase deficient mice. PLoS ONE. (2011) 6:e19021. doi: 10.1371/journal.pone.0019021
77. Pirttimaki T, Parri HR, Crunelli V. Astrocytic GABA transporter GAT-1 dysfunction in experimental absence seizures. J Physiol. (2013) 591:823–33. doi: 10.1113/jphysiol.2012.242016
78. Huguenard JR. Perspective: is cortical hyperexcitability the only path to generalized absence epilepsy? Epilepsy Curr. (2020) 20:59S−61S. doi: 10.1177/1535759720959325
79. Bomben VC, Aiba I, Qian J, Mark MD, Herlitze S, Noebels JL. Isolated P/Q calcium channel deletion in layer VI. Corticothalamic neurons generates absence epilepsy. J Neurosci. (2016) 36:405–18. doi: 10.1523/JNEUROSCI.2555-15.2016
80. Halassa MM, Fellin T, Haydon PG. The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol Med. (2007) 13:54–63. doi: 10.1016/j.molmed.2006.12.005
81. von Bartheld CS, Bahney J, Herculano-Houzel S. The search for true numbers of neurons and glial cells in the human brain: a review of 150 years of cell counting. J Comp Neurol. (2016) 524:3865–95. doi: 10.1002/cne.24040
82. Allen NJ, Lyons DA. Glia as architects of central nervous system formation and function. Science. (2018) 362:181–5. doi: 10.1126/science.aat0473
83. Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. (1999) 22:208–15. doi: 10.1016/S0166-2236(98)01349-6
84. Bazargani N, Attwell D. Astrocyte calcium signaling: the third wave. Nat Neurosci. (2016) 19:182–9. doi: 10.1038/nn.4201
85. Poskanzer KE, Molofsky AV. Dynamism of an astrocyte in vivo: perspectives on identity and function. Annu Rev Physiol. (2018) 80:143–57. doi: 10.1146/annurev-physiol-021317-121125
86. Caudal LC, Gobbo D, Scheller A, Kirchhoff F. The paradox of astroglial Ca2+ signals at the interface of excitation and inhibition. Front Cell Neurosci. (2020) 14:609947. doi: 10.3389/fncel.2020.609947
87. Fellin T, Haydon PG. Do astrocytes contribute to excitation underlying seizures? Trends Mol Med. (2005) 11:530–3. doi: 10.1016/j.molmed.2005.10.007
88. Jabs R, Seifert G, Steinhäuser C. Astrocytic function and its alteration in the epileptic brain. Epilepsia. (2008) 49(Suppl.2):3–12. doi: 10.1111/j.1528-1167.2008.01488.x
89. Wetherington J, Serrano G, Dingledine R. Astrocytes in the epileptic brain. Neuron. (2008) 58:168–78. doi: 10.1016/j.neuron.2008.04.002
90. Coulter DA, Steinhäuser C. Role of astrocytes in epilepsy. Cold Spring Harb Perspect Med. (2015) 5:a022434. doi: 10.1101/cshperspect.a022434
91. Crunelli V, Carmignoto G, Steinhäuser C. Novel astrocyte targets: new avenues for the therapeutic treatment of epilepsy. Neuroscientist. (2015) 21:62–83. doi: 10.1177/1073858414523320
92. Robel S, Sontheimer H. Glia as drivers of abnormal neuronal activity. Nat Neurosci. (2016) 19:28–33. doi: 10.1038/nn.4184
93. Binder DK. Astrocytes: stars of the sacred disease. Epilepsy Curr. (2018) 18:172–9. doi: 10.5698/1535-7597.18.3.172
94. Boison D, Steinhäuser C. Epilepsy and astrocyte energy metabolism. Glia. (2018) 66:1235–43. doi: 10.1002/glia.23247
95. Patel DC, Tewari BP, Chaunsali L, Sontheimer H. Neuron-glia interactions in the pathophysiology of epilepsy. Nat Rev Neurosci. (2019) 20:282–97. doi: 10.1038/s41583-019-0126-4
96. Verhoog QP, Holtman L, Aronica E, van Vliet EA. Astrocytes as guardians of neuronal excitability: mechanisms underlying epileptogenesis. Front Neurol. (2020) 11:591690. doi: 10.3389/fneur.2020.591690
97. Buskila Y, Bellot-Saez A, Morley JW. Generating brain waves, the power of astrocytes. Front Neurosci. (2019) 13:1125. doi: 10.3389/fnins.2019.01125
98. Pacholko AG, Wotton CA, Bekar LK. Astrocytes-the ultimate effectors of long-range neuromodulatory networks? Front Cell Neurosci. (2020) 14:581075. doi: 10.3389/fncel.2020.581075
99. Wang F, Qi X, Zhang J, Huang JH. Astrocytic modulation of potassium under seizures. Neural Regen Res. (2020) 15:980–7. doi: 10.4103/1673-5374.270295
100. Ma B, Buckalew R, Du Y, Kiyoshi CM, Alford CC, Wang W, et al. Gap junction coupling confers isopotentiality on astrocyte syncytium. Glia. (2016) 64:214–26. doi: 10.1002/glia.22924
101. Steinhäuser C, Grunnet M, Carmignoto G. Crucial role of astrocytes in temporal lobe epilepsy. Neuroscience. (2016) 323:157–69. doi: 10.1016/j.neuroscience.2014.12.047
102. Bellot-Saez A, Kékesi O, Morley JW, Buskila Y. Astrocytic modulation of neuronal excitability through K. Neurosci Biobehav Rev. (2017) 77:87–97. doi: 10.1016/j.neubiorev.2017.03.002
103. Kiyoshi CM, Du Y, Zhong S, Wang W, Taylor AT, Xiong B, et al. Syncytial isopotentiality: a system-wide electrical feature of astrocytic networks in the brain. Glia. (2018) 66:2756–69. doi: 10.1002/glia.23525
104. Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Reichenbach A, et al. Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia. (2007) 55:274–81. doi: 10.1002/glia.20455
105. Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci. (2007) 27:11354–65. doi: 10.1523/JNEUROSCI.0723-07.2007
106. Haj-Yasein NN, Jensen V, Vindedal GF, Gundersen GA, Klungland A, Ottersen OP, et al. Evidence that compromised K+ spatial buffering contributes to the epileptogenic effect of mutations in the human Kir4.1 gene (KCNJ10). Glia. (2011) 59:1635–42. doi: 10.1002/glia.21205
107. Sicca F, Ambrosini E, Marchese M, Sforna L, Servettini I, Valvo G, et al. Gain-of-function defects of astrocytic Kir4.1 channels in children with autism spectrum disorders and epilepsy. Sci Rep. (2016) 6:34325. doi: 10.1038/srep34325
108. Scholl UI, Choi M, Liu T, Ramaekers VT, Häusler MG, Grimmer J, et al. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci USA. (2009) 106:5842–7. doi: 10.1073/pnas.0901749106
109. Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J. Med. (2009) 360:1960–70. doi: 10.1056/NEJMoa0810276
110. Papasavvas CA, Parrish RR, Trevelyan AJ. Propagating activity in neocortex, mediated by gap junctions and modulated by extracellular potassium. eNeuro. (2020) 7:2020. doi: 10.1523/ENEURO.0387-19.2020
111. Bazzigaluppi P, Weisspapir I, Stefanovic B, Leybaert L, Carlen PL. Astrocytic gap junction blockade markedly increases extracellular potassium without causing seizures in the mouse neocortex. Neurobiol Dis. (2017) 101:1–7. doi: 10.1016/j.nbd.2016.12.017
112. Mukai T, Kinboshi M, Nagao Y, Shimizu S, Ono A, Sakagami Y, et al. Antiepileptic drugs elevate astrocytic Kir4.1 expression in the rat limbic region. Front Pharmacol. (2018) 9:845. doi: 10.3389/fphar.2018.00845
113. Walch E, Murphy TR, Cuvelier N, Aldoghmi M, Morozova C, Donohue J, et al. Astrocyte-selective volume increase in elevated extracellular potassium conditions is mediated by the Na. ASN Neuro. (2020) 12:1759091420967152. doi: 10.1177/1759091420967152
114. Eid T, Lee TS, Thomas MJ, Amiry-Moghaddam M, Bjørnsen LP, Spencer DD, et al. Loss of perivascular aquaporin 4 may underlie deficient water and K+ homeostasis in the human epileptogenic hippocampus. Proc Natl Acad Sci USA. (2005) 102:1193–8. doi: 10.1073/pnas.0409308102
115. Binder DK, Steinhäuser C. Functional changes in astroglial cells in epilepsy. Glia. (2006) 54:358–68. doi: 10.1002/glia.20394
116. Binder DK, Yao X, Zador Z, Sick TJ, Verkman AS, Manley GT. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia. (2006) 53:631–6. doi: 10.1002/glia.20318
117. Strohschein S, Hüttmann K, Gabriel S, Binder DK, Heinemann U, Steinhäuser C. Impact of aquaporin-4 channels on K+ buffering and gap junction coupling in the hippocampus. Glia. (2011) 59:973–80. doi: 10.1002/glia.21169
118. Alvestad S, Hammer J, Hoddevik EH, Skare Ø, Sonnewald U, Amiry-Moghaddam M, et al. Mislocalization of AQP4 precedes chronic seizures in the kainate model of temporal lobe epilepsy. Epilepsy Res. (2013) 105:30–41. doi: 10.1016/j.eplepsyres.2013.01.006
119. Lauderdale K, Murphy T, Tung T, Davila D, Binder DK, Fiacco TA. Osmotic edema rapidly increases neuronal excitability through activation of NMDA receptor-dependent slow inward currents in juvenile and adult hippocampus. ASN Neuro. (2015) 7:1759091415605115. doi: 10.1177/1759091415605115
120. Szu JI, Patel DD, Chaturvedi S, Lovelace JW, Binder DK. Modulation of posttraumatic epileptogenesis in aquaporin-4 knockout mice. Epilepsia. (2020) 61:1503–14. doi: 10.1111/epi.16551
121. Murphy TR, Davila D, Cuvelier N, Young LR, Lauderdale K, Binder DK, et al. Hippocampal and cortical pyramidal neurons swell in parallel with astrocytes during acute hypoosmolar stress. Front Cell Neurosci. (2017) 11:275. doi: 10.3389/fncel.2017.00275
122. Fujiwara H, Tenney J, Kadis DS, Altaye M, Spencer C, Vannest J. Cortical and subcortical volume differences between Benign Epilepsy with Centrotemporal Spikes and Childhood Absence Epilepsy. Epilepsy Res. (2020) 166:106407. doi: 10.1016/j.eplepsyres.2020.106407
123. Kim EH, Shim WH, Lee JS, Yoon HM, Ko TS, Yum MS. Altered structural network in newly onset childhood absence epilepsy. J Clin Neurol. (2020) 16:573–80. doi: 10.3988/jcn.2020.16.4.573
124. Orkand RK. Glial-interstitial fluid exchange. Ann N Y Acad Sci. (1986) 481:269–72. doi: 10.1111/j.1749-6632.1986.tb27157.x
125. Jin MM, Chen Z. Role of gap junctions in epilepsy. Neurosci Bull. (2011) 27:389–406. doi: 10.1007/s12264-011-1944-1
126. Pannasch U, Vargová L, Reingruber J, Ezan P, Holcman D, Giaume C, et al. Astroglial networks scale synaptic activity and plasticity. Proc Natl Acad Sci USA. (2011) 108:8467–72. doi: 10.1073/pnas.1016650108
127. Pannasch U, Rouach N. Emerging role for astroglial networks in information processing: from synapse to behavior. Trends Neurosci. (2013) 36:405–17. doi: 10.1016/j.tins.2013.04.004
128. Mylvaganam S, Ramani M, Krawczyk M, Carlen PL. Roles of gap junctions, connexins, and pannexins in epilepsy. Front Physiol. (2014) 5:172. doi: 10.3389/fphys.2014.00172
129. Chever O, Dossi E, Pannasch U, Derangeon M, Rouach N. Astroglial networks promote neuronal coordination. Sci Signal. (2016) 9:ra6. doi: 10.1126/scisignal.aad3066
130. Lapato AS, Tiwari-Woodruff SK. Connexins and pannexins: at the junction of neuro-glial homeostasis and disease. J Neurosci Res. (2018) 96:31–44. doi: 10.1002/jnr.24088
131. Li Q, Li QQ, Jia JN, Liu ZQ, Zhou HH, Mao XY. Targeting gap junction in epilepsy: perspectives and challenges. Biomed Pharmacother. (2019) 109:57–65. doi: 10.1016/j.biopha.2018.10.068
132. Parpura V, Verkhratsky A. Homeostatic function of astrocytes: Ca(2+) and Na(+) signalling. Transl Neurosci. (2012) 3:334–44. doi: 10.2478/s13380-012-0040-y
133. Wallraff A, Köhling R, Heinemann U, Theis M, Willecke K, Steinhäuser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci. (2006) 26:5438–47. doi: 10.1523/JNEUROSCI.0037-06.2006
134. Bedner P, Dupper A, Hüttmann K, Müller J, Herde MK, Dublin P, et al. Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain. (2015) 138:1208–22. doi: 10.1093/brain/awv067
135. Manjarrez-Marmolejo J, Franco-Pérez J. Gap junction blockers: an overview of their effects on induced seizures in animal models. Curr Neuropharmacol. (2016) 14:759–71. doi: 10.2174/1570159X14666160603115942
136. Chang WP, Wu JJ, Shyu BC. Thalamic modulation of cingulate seizure activity via the regulation of gap junctions in mice thalamocingulate slice. PLoS ONE. (2013) 8:e62952. doi: 10.1371/journal.pone.0062952
137. Gigout S, Louvel J, Rinaldi D, Martin B, Pumain R. Thalamocortical relationships and network synchronization in a new genetic model “in mirror” for absence epilepsy. Brain Res. (2013) 1525:39–52. doi: 10.1016/j.brainres.2013.05.044
138. Gigout S, Louvel J, Pumain R. Effects in vitro and in vivo of a gap junction blocker on epileptiform activities in a genetic model of absence epilepsy. Epilepsy Res. (2006) 69:15–29. doi: 10.1016/j.eplepsyres.2005.12.002
139. Proulx E, Leshchenko Y, Kokarovtseva L, Khokhotva V, El-Beheiry M, Snead OC, et al. Functional contribution of specific brain areas to absence seizures: role of thalamic gap-junctional coupling. Eur J. Neurosci. (2006) 23:489–96. doi: 10.1111/j.1460-9568.2005.04558.x
140. Gareri P, Condorelli D, Belluardo N, Citraro R, Barresi V, Trovato-Salinaro A, et al. Antiabsence effects of carbenoxolone in two genetic animal models of absence epilepsy (WAG/Rij rats and lh/lh mice). Neuropharmacology. (2005) 49:551–63. doi: 10.1016/j.neuropharm.2005.04.012
141. Vincze R, Péter M, Szabó Z, Kardos J, Héja L, Kovács Z. Connexin 43 differentially regulates epileptiform activity in models of convulsive and non-convulsive epilepsies. Front Cell Neurosci. (2019) 13:173. doi: 10.3389/fncel.2019.00173
142. Venance L, Piomelli D, Glowinski J, Giaume C. Inhibition by anandamide of gap junctions and intercellular calcium signalling in striatal astrocytes. Nature. (1995) 376:590–4. doi: 10.1038/376590a0
143. Guan X, Cravatt BF, Ehring GR, Hall JE, Boger DL, Lerner RA, et al. The sleep-inducing lipid oleamide deconvolutes gap junction communication and calcium wave transmission in glial cells. J Cell Biol. (1997) 139:1785–92. doi: 10.1083/jcb.139.7.1785
144. Citraro R, Russo E, Scicchitano F, van Rijn CM, Cosco D, Avagliano C, et al. Antiepileptic action of N-palmitoylethanolamine through CB1 and PPAR-α receptor activation in a genetic model of absence epilepsy. Neuropharmacology. (2013) 69:115–26. doi: 10.1016/j.neuropharm.2012.11.017
145. Chemin J, Monteil A, Perez-Reyes E, Nargeot J, Lory P. Direct inhibition of T-type calcium channels by the endogenous cannabinoid anandamide. EMBO J.. (2001) 20:7033–40. doi: 10.1093/emboj/20.24.7033
146. Cravatt BF, Prospero-Garcia O, Siuzdak G, Gilula NB, Henriksen SJ, Boger DL, et al. Chemical characterization of a family of brain lipids that induce sleep. Science. (1995) 268:1506–9. doi: 10.1126/science.7770779
147. Lees G, Edwards MD, Hassoni AA, Ganellin CR, Galanakis D. Modulation of GABA(A) receptors and inhibitory synaptic currents by the endogenous CNS sleep regulator cis-9,10-octadecenoamide (cOA). Br J. Pharmacol. (1998) 124:873–82. doi: 10.1038/sj.bjp.0701918
148. Medina-Ceja L, Salazar-Sánchez JC, Ortega-Ibarra J, Morales-Villagrán A. Connexins-based hemichannels/channels and their relationship with inflammation, seizures and epilepsy. Int J. Mol Sci. (2019) 20:ijms20235976. doi: 10.3390/ijms20235976
149. Griemsmann S, Höft SP, Bedner P, Zhang J, von Staden E, Beinhauer A, et al. Characterization of panglial gap junction networks in the thalamus, neocortex, and hippocampus reveals a unique population of glial cells. Cereb Cortex. (2015) 25:3420–33. doi: 10.1093/cercor/bhu157
150. Aquilino MS, Whyte-Fagundes P, Zoidl G, Carlen PL. Pannexin-1 channels in epilepsy. Neurosci Lett. (2019) 695:71–5. doi: 10.1016/j.neulet.2017.09.004
151. Scemes E, Velíšek L, Velíšková J. Astrocyte and neuronal Pannexin1 contribute distinctly to seizures. ASN Neuro. (2019) 11:1759091419833502. doi: 10.1177/1759091419833502
152. Wang J, Ma M, Locovei S, Keane RW, Dahl G. Modulation of membrane channel currents by gap junction protein mimetic peptides: size matters. Am J Physiol Cell Physiol. (2007) 293:C1112–9. doi: 10.1152/ajpcell.00097.2007
153. Willebrords J, Maes M, Crespo Yanguas S, Vinken M. Inhibitors of connexin and pannexin channels as potential therapeutics. Pharmacol Ther. (2017) 180:144–60. doi: 10.1016/j.pharmthera.2017.07.001
154. Delvaeye T, Vandenabeele P, Bultynck G, Leybaert L, Krysko DV. Therapeutic targeting of connexin channels: new views and challenges. Trends Mol Med. (2018) 24:1036–53. doi: 10.1016/j.molmed.2018.10.005
155. Giaume C, Naus CC, Sáez JC, Leybaert L. Glial connexins and pannexins in the healthy and diseased brain. Physiol Rev. (2021) 101:93–145. doi: 10.1152/physrev.00043.2018
156. Dallérac G, Zapata J, Rouach N. Versatile control of synaptic circuits by astrocytes: where, when and how? Nat Rev Neurosci. (2018) 19:729–43. doi: 10.1038/s41583-018-0080-6
157. Savtchouk I, Volterra A. Gliotransmission: beyond black-and-white. J Neurosci. (2018) 38:14–25. doi: 10.1523/JNEUROSCI.0017-17.2017
158. Riquelme J, Wellmann M, Sotomayor-Zárate R, Bonansco C. Gliotransmission: a novel target for the development of antiseizure drugs. Neuroscientist. (2020) 26:293–309. doi: 10.1177/1073858420901474
159. Leal A, Vieira JP, Lopes R, Nunes RG, Gonçalves SI, Lopes da Silva F, et al. Dynamics of epileptic activity in a peculiar case of childhood absence epilepsy and correlation with thalamic levels of GABA. Epilepsy Behav Case Rep. (2016) 5:57–65. doi: 10.1016/j.ebcr.2016.03.004
160. Dikow N, Maas B, Karch S, Granzow M, Janssen JW, Jauch A, et al. 3p25.3 microdeletion of GABA transporters SLC6A1 and SLC6A11 results in intellectual disability, epilepsy and stereotypic behavior. Am J Med Genet A. (2014) 164A:3061–8. doi: 10.1002/ajmg.a.36761
161. Mattison KA, Butler KM, Inglis GAS, Dayan O, Boussidan H, Bhambhani V, et al. SLC6A1 variants identified in epilepsy patients reduce γ-aminobutyric acid transport. Epilepsia. (2018) 59:e135–41. doi: 10.1111/epi.14531
162. Galer PD, Ganesan S, Lewis-Smith D, McKeown SE, Pendziwiat M, Helbig KL, et al. Semantic similarity analysis reveals robust gene-disease relationships in developmental and epileptic encephalopathies. Am J Hum Genet. (2020) 107:683–97. doi: 10.1016/j.ajhg.2020.08.003
163. Goodspeed K, Pérez-Palma E, Iqbal S, Cooper D, Scimemi A, Johannesen KM, et al. Current knowledge of SLC6A1-related neurodevelopmental disorders. Brain Commun. (2020) 2:fcaa170. doi: 10.1093/braincomms/fcaa170
164. Johannesen KM, Gardella E, Linnankivi T, Courage C, de Saint Martin A, Lehesjoki A-E, et al. Defining the phenotypic spectrum of SLC6A1 mutations. Epilepsia. (2018) 59:389–402. doi: 10.1111/epi.13986
165. Jiménez-González C, Pirttimaki T, Cope DW, Parri HR. Non-neuronal, slow GABA signalling in the ventrobasal thalamus targets δ-subunit-containing GABA(A) receptors. Eur J. Neurosci. (2011) 33:1471–82. doi: 10.1111/j.1460-9568.2011.07645.x
166. Herd MB, Brown AR, Lambert JJ, Belelli D. Extrasynaptic GABA(A) receptors couple presynaptic activity to postsynaptic inhibition in the somatosensory thalamus. J Neurosci. (2013) 33:14850–68. doi: 10.1523/JNEUROSCI.1174-13.2013
167. Höft S, Griemsmann S, Seifert G, Steinhäuser C. Heterogeneity in expression of functional ionotropic glutamate and GABA receptors in astrocytes across brain regions: insights from the thalamus. Philos Trans R Soc Lond B Biol Sci. (2014) 369:20130602. doi: 10.1098/rstb.2013.0602
168. Merlo D, Mollinari C, Inaba Y, Cardinale A, Rinaldi AM, D'Antuono M, et al. Reduced GABAB receptor subunit expression and paired-pulse depression in a genetic model of absence seizures. Neurobiol Dis. (2007) 25:631–41. doi: 10.1016/j.nbd.2006.11.005
169. Inaba Y, D'Antuono M, Bertazzoni G, Biagini G, Avoli M. Diminished presynaptic GABA(B) receptor function in the neocortex of a genetic model of absence epilepsy. Neurosignals. (2009) 17:121–31. doi: 10.1159/000197864
170. Vergnes M, Marescaux C, Micheletti G, Depaulis A, Rumbach L, Warter JM. Enhancement of spike and wave discharges by GABAmimetic drugs in rats with spontaneous petit-mal-like epilepsy. Neurosci Lett. (1984) 44:91–4. doi: 10.1016/0304-3940(84)90226-X
171. Crunelli V, Leresche N. A role for GABAB receptors in excitation and inhibition of thalamocortical cells. Trends Neurosci. (1991) 14:16–21. doi: 10.1016/0166-2236(91)90178-W
172. Liu Z, Vergnes M, Depaulis A, Marescaux C. Involvement of intrathalamic GABAB neurotransmission in the control of absence seizures in the rat. Neuroscience. (1992) 48:87–93. doi: 10.1016/0306-4522(92)90340-8
173. Marescaux C, Vergnes M, Bernasconi R. GABAB receptor antagonists: potential new anti-absence drugs. J Neural Transm Suppl. (1992) 35:179–88. doi: 10.1007/978-3-7091-9206-1_12
174. Manning JP, Richards DA, Bowery NG. Pharmacology of absence epilepsy. Trends Pharmacol Sci. (2003) 24:542–9. doi: 10.1016/j.tips.2003.08.006
175. Beenhakker MP, Huguenard JR. Astrocytes as gatekeepers of GABAB receptor function. J Neurosci. (2010) 30:15262–76. doi: 10.1523/JNEUROSCI.3243-10.2010
176. Bortolato M, Frau R, Orrù M, Fà M, Dessì C, Puligheddu M, et al. GABAB receptor activation exacerbates spontaneous spike-and-wave discharges in DBA/2J mice. Seizure. (2010) 19:226–31. doi: 10.1016/j.seizure.2010.02.007
177. Gould T, Chen L, Emri Z, Pirttimaki T, Errington AC, Crunelli V, et al. GABA(B) receptor-mediated activation of astrocytes by gamma-hydroxybutyric acid. Philos Trans R Soc Lond B Biol Sci. (2014) 369:20130607. doi: 10.1098/rstb.2013.0607
178. D'Amore V, von Randow C, Nicoletti F, Ngomba RT, van Luijtelaar G. Anti-absence activity of mGlu1 and mGlu5 receptor enhancers and their interaction with a GABA reuptake inhibitor: effect of local infusions in the somatosensory cortex and thalamus. Epilepsia. (2015) 56:1141–51. doi: 10.1111/epi.13024
179. Ngomba RT, Biagioni F, Casciato S, Willems-van Bree E, Battaglia G, Bruno V, et al. The preferential mGlu2/3 receptor antagonist, LY341495, reduces the frequency of spike-wave discharges in the WAG/Rij rat model of absence epilepsy. Neuropharmacology. (2005) 49(Suppl.1):89–103. doi: 10.1016/j.neuropharm.2005.05.019
180. Ngomba RT, Ferraguti F, Badura A, Citraro R, Santolini I, Battaglia G, et al. Positive allosteric modulation of metabotropic glutamate 4 (mGlu4) receptors enhances spontaneous and evoked absence seizures. Neuropharmacology. (2008) 54:344–54. doi: 10.1016/j.neuropharm.2007.10.004
181. Celli R, Santolini I, Van Luijtelaar G, Ngomba RT, Bruno V, Nicoletti F. Targeting metabotropic glutamate receptors in the treatment of epilepsy: rationale and current status. Expert Opin Ther Targets. (2019) 23:341–51. doi: 10.1080/14728222.2019.1586885
182. Celli R, Wall MJ, Santolini I, Vergassola M, Di Menna L, Mascio G, et al. Pharmacological activation of mGlu5 receptors with the positive allosteric modulator VU0360172, modulates thalamic GABAergic transmission. Neuropharmacology. (2020) 178:108240. doi: 10.1016/j.neuropharm.2020.108240
183. Parri HR, Gould TM, Crunelli V. Sensory and cortical activation of distinct glial cell subtypes in the somatosensory thalamus of young rats. Eur J. Neurosci. (2010) 32:29–40. doi: 10.1111/j.1460-9568.2010.07281.x
184. Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. (1997) 276:1699–702. doi: 10.1126/science.276.5319.1699
185. Watanabe T, Morimoto K, Hirao T, Suwaki H, Watase K, Tanaka K. Amygdala-kindled and pentylenetetrazole-induced seizures in glutamate transporter GLAST-deficient mice. Brain Res. (1999) 845:92–6. doi: 10.1016/S0006-8993(99)01945-9
186. Coulter DA, Eid T. Astrocytic regulation of glutamate homeostasis in epilepsy. Glia. (2012) 60:1215–26. doi: 10.1002/glia.22341
187. Eid T, Lee TW, Patrylo P, Zaveri HP. Astrocytes and glutamine synthetase in epileptogenesis. J Neurosci Res. (2019) 97:1345–62. doi: 10.1002/jnr.24267
188. Dutuit M, Touret M, Szymocha R, Nehlig A, Belin MF, Didier-Bazès M. Decreased expression of glutamate transporters in genetic absence epilepsy rats before seizure occurrence. J Neurochem. (2002) 80:1029–38. doi: 10.1046/j.0022-3042.2002.00768.x
189. Ingram EM, Tessler S, Bowery NG, Emson PC. Glial glutamate transporter mRNAs in the genetically absence epilepsy rat from Strasbourg. Brain Res Mol Brain Res. (2000) 75:96–104. doi: 10.1016/S0169-328X(99)00301-0
190. Takano T, Kang J, Jaiswal JK, Simon SM, Lin JHC, Yu YF, et al. Receptor-mediated glutamate release from volume sensitive channels in astrocytes. Proc Natl Acad Sci USA. (2005) 102:16466–71. doi: 10.1073/pnas.0506382102
191. Héja L, Simon Á, Szabó Z., Kardos J. Feedback adaptation of synaptic excitability via Glu:Na. Neuropharmacology. (2019) 161:107629. doi: 10.1016/j.neuropharm.2019.05.006
192. Melø TM, Sonnewald U, Touret M, Nehlig A. Cortical glutamate metabolism is enhanced in a genetic model of absence epilepsy. J Cereb Blood Flow Metab. (2006) 26:1496–506. doi: 10.1038/sj.jcbfm.9600300
193. Dutuit M, Didier-Bazès M, Vergnes M, Mutin M, Conjard A, Akaoka H, et al. Specific alteration in the expression of glial fibrillary acidic protein, glutamate dehydrogenase, and glutamine synthetase in rats with genetic absence epilepsy. Glia. (2000) 32:15–24. doi: 10.1002/1098-1136(200010)32:1<15::AID-GLIA20>3.0.CO;2-#
194. Dufour F, Nalecz KA, Nalecz MJ, Nehlig A. Metabolic approach of absence seizures in a genetic model of absence epilepsy, the GAERS: study of the leucine-glutamate cycle. J Neurosci Res. (2001) 66:923–30. doi: 10.1002/jnr.10086
195. Bahi-Buisson N, El Sabbagh S, Soufflet C, Escande F, Boddaert N, Valayannopoulos V, et al. Myoclonic absence epilepsy with photosensitivity and a gain of function mutation in glutamate dehydrogenase. Seizure. (2008) 17:658–64. doi: 10.1016/j.seizure.2008.01.005
196. Bazzigaluppi P, Ebrahim Amini A, Weisspapir I, Stefanovic B, Carlen PL. Hungry neurons: metabolic insights on seizure dynamics. Int J Mol Sci. (2017) 18:112269. doi: 10.3390/ijms18112269
197. van Rijn CM, Gaetani S, Santolini I, Badura A, Gabova A, Fu J, et al. WAG/Rij rats show a reduced expression of CB1 receptors in thalamic nuclei and respond to the CB1 receptor agonist, R(+)WIN55,212-2, with a reduced incidence of spike-wave discharges. Epilepsia. (2010) 51:1511–21. doi: 10.1111/j.1528-1167.2009.02510.x
198. Citraro R, Russo E, Ngomba RT, Nicoletti F, Scicchitano F, Whalley BJ, et al. CB1 agonists, locally applied to the cortico-thalamic circuit of rats with genetic absence epilepsy, reduce epileptic manifestations. Epilepsy Res. (2013) 106:74–82. doi: 10.1016/j.eplepsyres.2013.06.004
199. Perescis MFJ, Flipsen NAR, van Luijtelaar G, van Rijn CM. Altered SWD stopping mechanism in WAG/Rij rats subchronically treated with the cannabinoid agonist R(+)WIN55,212-2. Epilepsy Behav. (2020) 102:106722. doi: 10.1016/j.yebeh.2019.106722
200. Nikolic L, Nobili P, Shen W, Audinat E. Role of astrocyte purinergic signaling in epilepsy. Glia. (2020) 68:1677–91. doi: 10.1002/glia.23747
201. Engel T, Alves M, Sheedy C, Henshall DC. ATPergic signalling during seizures and epilepsy. Neuropharmacology. (2016) 104:140–53. doi: 10.1016/j.neuropharm.2015.11.001
202. Rassendren F, Audinat E. Purinergic signaling in epilepsy. J Neurosci Res. (2016) 94:781–93. doi: 10.1002/jnr.23770
203. Weltha L, Reemmer J, Boison D. The role of adenosine in epilepsy. Brain Res Bull. (2019) 151:46–54. doi: 10.1016/j.brainresbull.2018.11.008
204. Ekonomou A, Angelatou F, Vergnes M, Kostopoulos G. Lower density of A1 adenosine receptors in nucleus reticularis thalami in rats with genetic absence epilepsy. Neuroreport. (1998) 9:2135–40. doi: 10.1097/00001756-199806220-00042
205. D'Alimonte I, D'Auro M, Citraro R, Biagioni F, Jiang S, Nargi E, et al. Altered distribution and function of A2A adenosine receptors in the brain of WAG/Rij rats with genetic absence epilepsy, before and after appearance of the disease. Eur J Neurosci. (2009) 30:1023–35. doi: 10.1111/j.1460-9568.2009.06897.x
206. Lakatos RK, Dobolyi Á, Todorov MI, Kékesi KA, Juhász G, Aleksza M, et al. Guanosine may increase absence epileptic activity by means of A2A adenosine receptors in Wistar Albino Glaxo Rijswijk rats. Brain Res Bull. (2016) 124:172–81. doi: 10.1016/j.brainresbull.2016.05.001
207. Ilbay G, Sahin D, Karson A, Ates N. Effects of adenosine administration on spike-wave discharge frequency in genetically epileptic rats. Clin Exp Pharmacol Physiol. (2001) 28:643–6. doi: 10.1046/j.1440-1681.2001.03499.x
208. Germé K, Faure JB, Koning E, Nehlig A. Effect of caffeine and adenosine receptor ligands on the expression of spike-and-wave discharges in Genetic Absence Epilepsy Rats from Strasbourg (GAERS). Epilepsy Res. (2015) 110:105–14. doi: 10.1016/j.eplepsyres.2014.11.022
209. Kovács Z, D'Agostino DP, Dobolyi A, Ari C. Adenosine A1 receptor antagonism abolished the anti-seizure effects of exogenous ketone supplementation in wistar albino glaxo rijswijk rats. Front Mol Neurosci. (2017) 10:235. doi: 10.3389/fnmol.2017.00235
210. Muhle H, Steinich I, von Spiczak S, Franke A, Weber Y, Lerche H, et al. A duplication in 1q21.3 in a family with early onset and childhood absence epilepsy. Epilepsia. (2010) 51:2453–6. doi: 10.1111/j.1528-1167.2010.02712.x
211. Chen CP, Lin SP, Chen M, Su YN, Chern SR, Wang TY, et al. Mosaic supernumerary r(1)(p13.2q23.3) in a 10-year-old girl with epilepsy facial asymmetry psychomotor retardation kyphoscoliosis dermatofibrosarcoma and multiple exostoses. Genet Couns. (2011) 22:273–80.
212. Sun QQ, Huguenard JR, Prince DA. Neuropeptide Y. receptors differentially modulate G-protein-activated inwardly rectifying K+ channels and high-voltage-activated Ca2+ channels in rat thalamic neurons. J Physiol. (2001) 531:67–79. doi: 10.1111/j.1469-7793.2001.0067j.x
213. Elms J, Powell KL, van Raay L, Dedeurwaerdere S, O'Brien TJ, Morris MJ. Long-term valproate treatment increases brain neuropeptide Y. expression and decreases seizure expression in a genetic rat model of absence epilepsy. PLoS ONE. (2013) 8:e73505. doi: 10.1371/journal.pone.0073505
214. Stroud LM, O'Brien TJ, Jupp B, Wallengren C, Morris MJ. Neuropeptide Y. suppresses absence seizures in a genetic rat model. Brain Res. (2005) 1033:151–6. doi: 10.1016/j.brainres.2004.11.022
215. Morris MJ, Gannan E, Stroud LM, Beck-Sickinger AG, O'Brien TJ. Neuropeptide Y. suppresses absence seizures in a genetic rat model primarily through effects on Y. receptors. Eur J. Neurosci. (2007) 25:1136–43. doi: 10.1111/j.1460-9568.2007.05348.x
216. van Raay L, Jovanovska V, Morris MJ, O'Brien TJ. Focal administration of neuropeptide Y. into the S2 somatosensory cortex maximally suppresses absence seizures in a genetic rat model. Epilepsia. (2012) 53:477–84. doi: 10.1111/j.1528-1167.2011.03370.x
217. Woldbye DP, Nanobashvili A, Sørensen AT, Husum H, Bolwig TG, Sørensen G, et al. Differential suppression of seizures via Y2 and Y5 neuropeptide Y. receptors. Neurobiol Dis. (2005) 20:760–72. doi: 10.1016/j.nbd.2005.05.010
218. Lin EJ, Young D, Baer K, Herzog H, During MJ. Differential actions of NPY on seizure modulation via Y1 and Y2 receptors: evidence from receptor knockout mice. Epilepsia. (2006) 47:773–80. doi: 10.1111/j.1528-1167.2006.00500.x
219. Powell KL, Fitzgerald X, Shallue C, Jovanovska V, Klugmann M, Von Jonquieres G, et al. Gene therapy mediated seizure suppression in Genetic Generalised Epilepsy: Neuropeptide Y. overexpression in a rat model. Neurobiol Dis. (2018) 113:23–32. doi: 10.1016/j.nbd.2018.01.016
220. Ramamoorthy P, Whim MD. Trafficking and fusion of neuropeptide Y-containing dense-core granules in astrocytes. J Neurosci. (2008) 28:13815–27. doi: 10.1523/JNEUROSCI.5361-07.2008
221. Schwarz Y, Zhao N, Kirchhoff F, Bruns D. Astrocytes control synaptic strength by two distinct v-SNARE-dependent release pathways. Nat Neurosci. (2017) 20:1529–39. doi: 10.1038/nn.4647
222. Gimpl G, Kirchhoff F, Lang RE, Kettenmann H. Identification of neuropeptide Y. receptors in cultured astrocytes from neonatal rat brain. J Neurosci Res. (1993) 34:198–205. doi: 10.1002/jnr.490340207
223. Barnea A, Aguila-Mansilla N, Bigio EH, Worby C, Roberts J. Evidence for regulated expression of neuropeptide Y. gene by rat and human cultured astrocytes. Regul Pept. (1998) 75–76:293–300. doi: 10.1016/S0167-0115(98)00081-0
224. Carmignoto G, Haydon PG. Astrocyte calcium signaling and epilepsy. Glia. (2012) 60:1227–33. doi: 10.1002/glia.22318
225. Shigetomi E, Saito K, Sano F, Koizumi S. Aberrant Calcium signals in reactive astrocytes: a key process in neurological disorders. Int J. Mol Sci. (2019) 20:40996. doi: 10.3390/ijms20040996
226. Parri HR, Gould TM, Crunelli V. Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nat Neurosci. (2001) 4:803–12. doi: 10.1038/90507
227. Angulo MC, Kozlov AS, Charpak S, Audinat E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci. (2004) 24:6920–7. doi: 10.1523/JNEUROSCI.0473-04.2004
228. Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron. (2004) 43:729–43. doi: 10.1016/j.neuron.2004.08.011
229. Tian GF, Azmi H, Takano T, Xu QW, Peng WG, Lin J, et al. An astrocytic basis of epilepsy. Nat Med. (2005) 11:973–81. doi: 10.1038/nm1277
230. Gómez-Gonzalo M, Losi G, Chiavegato A, Zonta M, Cammarota M, Brondi M, et al. An excitatory loop with astrocytes contributes to drive neurons to seizure threshold. PLoS Biol. (2010) 8:e1000352. doi: 10.1371/journal.pbio.1000352
231. Koizumi S. Synchronization of Ca2+ oscillations: involvement of ATP release in astrocytes. FEBS J. (2010) 277:286–92. doi: 10.1111/j.1742-4658.2009.07438.x
232. Sasaki T, Ishikawa T, Abe R, Nakayama R, Asada A, Matsuki N, et al. Astrocyte calcium signalling orchestrates neuronal synchronization in organotypic hippocampal slices. J Physiol. (2014) 592:2771–83. doi: 10.1113/jphysiol.2014.272864
233. Álvarez-Ferradas C, Morales JC, Wellmann M, Nualart F, Roncagliolo M, Fuenzalida M, et al. Enhanced astroglial Ca2+ signaling increases excitatory synaptic strength in the epileptic brain. Glia. (2015) 63:1507–21. doi: 10.1002/glia.22817
234. Wellmann M, Álvarez-Ferradas C, Maturana CJ, Sáez JC, Bonansco C. Astroglial Ca2+-dependent hyperexcitability requires P2Y1purinergic receptors and pannexin-1 channel activation in a chronic model of epilepsy. Front Cell Neurosci. (2018) 12:446. doi: 10.3389/fncel.2018.00446
235. Heuser K, Nome CG, Pettersen KH, Åbjørsbråten KS, Jensen V, Tang W, et al. Ca2+ Signals in astrocytes facilitate spread of epileptiform activity. Cereb Cortex. (2018) 28:4036–48. doi: 10.1093/cercor/bhy196
236. Seidel JL, Escartin C, Ayata C, Bonvento G, Shuttleworth CW. Multifaceted roles for astrocytes in spreading depolarization: a target for limiting spreading depolarization in acute brain injury? Glia. (2016) 64:5–20. doi: 10.1002/glia.22824
237. Parri HR, Crunelli V. Pacemaker calcium oscillations in thalamic astrocytes in situ. Neuroreport. (2001) 12:3897–900. doi: 10.1097/00001756-200112210-00008
238. Yu X, Taylor AM. W., Nagai J, Golshani P, Evans CJ, Coppola G, et al. Reducing astrocyte calcium signaling in vivo alters striatal microcircuits and causes repetitive behavior. Neuron. (2018) 99:1170–87.e1179. doi: 10.1016/j.neuron.2018.08.015
239. Pirttimaki TM, Hall SD, Parri HR. Sustained neuronal activity generated by glial plasticity. J Neurosci. (2011) 31:7637–47. doi: 10.1523/JNEUROSCI.5783-10.2011
240. Pirttimaki TM, Parri HR. Glutamatergic input-output properties of thalamic astrocytes. Neuroscience. (2012) 205:18–28. doi: 10.1016/j.neuroscience.2011.12.049
241. Copeland CS, Wall TM, Sims RE, Neale SA, Nisenbaum E, Parri HR, et al. Astrocytes modulate thalamic sensory processing via mGlu2 receptor activation. Neuropharmacology. (2017) 121:100–10. doi: 10.1016/j.neuropharm.2017.04.019
242. Christian CA, Huguenard JR. Astrocytes potentiate GABAergic transmission in the thalamic reticular nucleus via endozepine signaling. Proc Natl Acad Sci USA. (2013) 110:20278–83. doi: 10.1073/pnas.1318031110
243. Pirttimaki TM, Sims RE, Saunders G, Antonio SA, Codadu NK, Parri HR. Astrocyte-mediated neuronal synchronization properties revealed by false gliotransmitter release. J Neurosci. (2017) 37:9859–70. doi: 10.1523/JNEUROSCI.2761-16.2017
244. Kékesi O, Ioja E, Szabó Z, Kardos J, Héja L. Recurrent seizure-like events are associated with coupled astroglial synchronization. Front Cell Neurosci. (2015) 9:215. doi: 10.3389/fncel.2015.00215
245. Ujita S, Sasaki T, Asada A, Funayama K, Gao M, Mikoshiba K, et al. cAMP-dependent calcium oscillations of astrocytes: an implication for pathology. Cereb Cortex. (2017) 27:1602–14. doi: 10.1093/cercor/bhv310
246. Sofroniew MV. Astrogliosis. Cold Spring Harb Perspect Biol. (2014) 7:a020420. doi: 10.1101/cshperspect.a020420
247. Dossi E, Vasile F, Rouach N. Human astrocytes in the diseased brain. Brain Res Bull. (2018) 136:139–56. doi: 10.1016/j.brainresbull.2017.02.001
248. Escartin C, Guillemaud O, Carrillo-de Sauvage MA. Questions and (some) answers on reactive astrocytes. Glia. (2019) 67:2221–47. doi: 10.1002/glia.23687
249. Robel S, Buckingham SC, Boni JL, Campbell SL, Danbolt NC, Riedemann T, et al. Reactive astrogliosis causes the development of spontaneous seizures. J Neurosci. (2015) 35:3330–45. doi: 10.1523/JNEUROSCI.1574-14.2015
250. Robel S. Astroglial scarring and seizures: a cell biological perspective on epilepsy. Neuroscientist. (2017) 23:152–68. doi: 10.1177/1073858416645498
251. Noè F, Cattalini A, Vila Verde D, Alessi C, Colciaghi F, Figini M, et al. Epileptiform activity contralateral to unilateral hippocampal sclerosis does not cause the expression of brain damage markers. Epilepsia. (2019) 60:1184–99. doi: 10.1111/epi.15611
252. Çavdar S, Kuvvet Y, Sur-Erdem I, Özgür M, Onat F. Relationships between astrocytes and absence epilepsy in rat: an experimental study. Neurosci Lett. (2019) 712:134518. doi: 10.1016/j.neulet.2019.134518
253. Sitnikova E, Kulikova S, Birioukova L, Raevsky VV. Cellular neuropathology of absence epilepsy in the neocortex: a population of glial cells rather than neurons is impaired in genetic rat model. Acta Neurobiol Exp. (2011) 71:263–8.
254. Oberheim NA, Tian GF, Han X, Peng W, Takano T, Ransom B, et al. Loss of astrocytic domain organization in the epileptic brain. J Neurosci. (2008) 28:3264–76. doi: 10.1523/JNEUROSCI.4980-07.2008
255. van Vliet EA, Aronica E, Gorter JA. Blood-brain barrier dysfunction, seizures and epilepsy. Semin Cell Dev Biol. (2015) 38:26–34. doi: 10.1016/j.semcdb.2014.10.003
256. Rana A, Musto AE. The role of inflammation in the development of epilepsy. J Neuroinflammation. (2018) 15:144. doi: 10.1186/s12974-018-1192-7
257. Vezzani A, Balosso S, Ravizza T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat Rev Neurol. (2019) 15:459–72. doi: 10.1038/s41582-019-0217-x
258. Terrone G, Balosso S, Pauletti A, Ravizza T, Vezzani A. Inflammation and reactive oxygen species as disease modifiers in epilepsy. Neuropharmacology. (2020) 167:107742. doi: 10.1016/j.neuropharm.2019.107742
259. Akin D, Ravizza T, Maroso M, Carcak N, Eryigit T, Vanzulli I, et al. IL-1β is induced in reactive astrocytes in the somatosensory cortex of rats with genetic absence epilepsy at the onset of spike-and-wave discharges, and contributes to their occurrence. Neurobiol Dis. (2011) 44:259–69. doi: 10.1016/j.nbd.2011.05.015
260. van Luijtelaar G, Lyashenko S, Vastyanov R, Verbeek G, Oleinik A, van Rijn C, et al. Cytokines and Absence Seizures in a Genetic Rat Model. Neurophysiology. (2012) 43:478–86. doi: 10.1007/s11062-012-9252-6
261. Fine SM, Angel RA, Perry SW, Epstein LG, Rothstein JD, Dewhurst S, et al. Tumor necrosis factor alpha inhibits glutamate uptake by primary human astrocytes. Implications for pathogenesis of HIV-1 dementia. J Biol Chem. (1996) 271:15303–6. doi: 10.1074/jbc.271.26.15303
262. Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci. (2005) 25:3219–28. doi: 10.1523/JNEUROSCI.4486-04.2005
263. Kovács Z, Kékesi KA, Szilágyi N, Abrahám I, Székács D, Király N, et al. Facilitation of spike-wave discharge activity by lipopolysaccharides in Wistar Albino Glaxo/Rijswijk rats. Neuroscience. (2006) 140:731–42. doi: 10.1016/j.neuroscience.2006.02.023
264. Kovács Z, Czurkó A, Kékesi KA, Juhász G. Intracerebroventricularly administered lipopolysaccharide enhances spike-wave discharges in freely moving WAG/Rij rats. Brain Res Bull. (2011) 85:410–6. doi: 10.1016/j.brainresbull.2011.05.003
265. Russo E, Citraro R, Donato G, Camastra C, Iuliano R, Cuzzocrea S, et al. mTOR inhibition modulates epileptogenesis, seizures and depressive behavior in a genetic rat model of absence epilepsy. Neuropharmacology. (2013) 69:25–36. doi: 10.1016/j.neuropharm.2012.09.019
266. Russo E, Andreozzi F, Iuliano R, Dattilo V, Procopio T, Fiume G, et al. Early molecular and behavioral response to lipopolysaccharide in the WAG/Rij rat model of absence epilepsy and depressive-like behavior, involves interplay between AMPK, AKT/mTOR pathways and neuroinflammatory cytokine release. Brain Behav Immun. (2014) 42:157–68. doi: 10.1016/j.bbi.2014.06.016
267. Kovács Z, Dobolyi A, Juhász G, Kékesi KA. Lipopolysaccharide induced increase in seizure activity in two animal models of absence epilepsy WAG/Rij and GAERS rats and Long Evans rats. Brain Res Bull. (2014) 104:7–18. doi: 10.1016/j.brainresbull.2014.03.003
268. Leo A, Nesci V, Tallarico M, Amodio N, Gallo Cantafio EM, De Sarro G, et al. IL-6 receptor blockade by tocilizumab has anti-absence and anti-epileptogenic effects in the WAG/Rij rat model of absence epilepsy. Neurotherapeutics. (2020) 17:2004–14. doi: 10.1007/s13311-020-00893-8
269. Billiau AD, Witters P, Ceulemans B, Kasran A, Wouters C, Lagae L. Intravenous immunoglobulins in refractory childhood-onset epilepsy: effects on seizure frequency, EEG activity, and cerebrospinal fluid cytokine profile. Epilepsia. (2007) 48:1739–49. doi: 10.1111/j.1528-1167.2007.01134.x
270. Steinborn B, Zarowski M, Winczewska-Wiktor A, Wójcicka M, Młodzikowska-Albrecht J, Losy J. Concentration of Il-1β, Il-2, Il-6, TNFα in the blood serum in children with generalized epilepsy treated by valproate. Pharmacol Rep. (2014) 66:972–5. doi: 10.1016/j.pharep.2014.06.005
271. Nir Y, Massimini M, Boly M, Tononi G. Sleep and consciousness. In: Cavanna A, Nani A, Blumenfeld H, Laureys S, editors. Neuroimaging of Consciousness. Berlin; Heidelberg: Springer (2013). p. 133–82. doi: 10.1007/978-3-642-37580-4_9
272. Beenhakker MP, Huguenard JR. Neurons that fire together also conspire together: is normal sleep circuitry hijacked to generate epilepsy? Neuron. (2009) 62:612–32. doi: 10.1016/j.neuron.2009.05.015
273. Contreras D, Steriade M. Cellular basis of EEG slow rhythms: a study of dynamic corticothalamic relationships. J Neurosci. (1995) 15:604–22. doi: 10.1523/JNEUROSCI.15-01-00604.1995
274. Steriade M, Contreras D, Curró Dossi R, Nuñez A. The slow (<1 Hz) oscillation in reticular thalamic and thalamocortical neurons: scenario of sleep rhythm generation in interacting thalamic and neocortical networks. J Neurosci. (1993) 13:3284–99. doi: 10.1523/JNEUROSCI.13-08-03284.1993
275. Halász P, Terzano MG, Parrino L. Spike-wave discharge and the microstructure of sleep-wake continuum in idiopathic generalised epilepsy. Neurophysiol Clin. (2002) 32:38–53. doi: 10.1016/S0987-7053(01)00290-8
276. Steriade M, Contreras D. Relations between cortical and thalamic cellular events during transition from sleep patterns to paroxysmal activity. J Neurosci. (1995) 15:623–42. doi: 10.1523/JNEUROSCI.15-01-00623.1995
277. McCormick DA, Bal T. Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci. (1997) 20:185–215. doi: 10.1146/annurev.neuro.20.1.185
278. Steriade M. Neuronal Substrates of Sleep and Epilepsy. Cambridge: Cambridge University Press. (2003). p. 322–48.
279. Steriade M, Timofeev I. Neuronal plasticity in thalamocortical networks during sleep and waking oscillations. Neuron. (2003) 37:563–76. doi: 10.1016/S0896-6273(03)00065-5
280. Crunelli V, David F, Lorincz ML, Hughes SW. The thalamocortical network as a single slow wave-generating unit. Curr Opin Neurobiol. (2015) 31:72–80. doi: 10.1016/j.conb.2014.09.001
281. Krishnan GP, Chauvette S, Shamie I, Soltani S, Timofeev I, Cash SS, et al. Cellular and neurochemical basis of sleep stages in the thalamocortical network. Elife. (2016) 5:16. doi: 10.7554/eLife.18607.016
282. Halász P, Szucs A. Sleep and epilepsy link by plasticity. Front Neurol. (2020) 11:911. doi: 10.3389/fneur.2020.00911
283. Jin B, Aung T, Geng Y, Wang S. Epilepsy and its interaction with sleep and circadian rhythm. Front Neurol. (2020) 11:327. doi: 10.3389/fneur.2020.00327
284. Smyk MK, van Luijtelaar G. Circadian rhythms and epilepsy: a suitable case for absence epilepsy. Front Neurol. (2020) 11:245. doi: 10.3389/fneur.2020.00245
285. Xu C, Yu J, Ruan Y, Wang Y, Chen Z. Decoding circadian rhythm and epileptic activities: clues from animal studies. Front Neurol. (2020) 11:751. doi: 10.3389/fneur.2020.00751
286. Gloor P. Generalized cortico-reticular epilepsies. Some considerations on the pathophysiology of generalized bilaterally synchronous spike and wave discharge. Epilepsia. (1968) 9:249–63. doi: 10.1111/j.1528-1157.1968.tb04624.x
287. Kostopoulos GK. Spike-and-wave discharges of absence seizures as a transformation of sleep spindles: the continuing development of a hypothesis. Clin Neurophysiol. (2000) 111(Suppl.2):S27–38. doi: 10.1016/S1388-2457(00)00399-0
288. Kellaway P, Frost JD, Crawley JW. Time modulation of spike-and-wave activity in generalized epilepsy. Ann Neurol. (1980) 8:491–500. doi: 10.1002/ana.410080506
289. Nobili L, Baglietto MG, Beelke M, De Carli F, Veneselli E, Ferrillo F. Temporal relationship of generalized epileptiform discharges to spindle frequency activity in childhood absence epilepsy. Clin Neurophysiol. (2001) 112:1912–6. doi: 10.1016/S1388-2457(01)00624-1
290. Kostopoulos G, Gloor P, Pellegrini A, Gotman J. A study of the transition from spindles to spike and wave discharge in feline generalized penicillin epilepsy: microphysiological features. Exp Neurol. (1981) 73:55–77. doi: 10.1016/0014-4886(81)90045-5
291. Fan D, Liao F, Wang Q. The pacemaker role of thalamic reticular nucleus in controlling spike-wave discharges and spindles. Chaos. (2017) 27:073103. doi: 10.1063/1.4991869
292. Halász P, Bódizs R, Ujma PP, Fabó D, Szucs A. Strong relationship between NREM sleep, epilepsy and plastic functions - a conceptual review on the neurophysiology background. Epilepsy Res. (2019) 150:95–105. doi: 10.1016/j.eplepsyres.2018.11.008
293. Sitnikova E, Grubov V, Hramov AE. Slow-wave activity preceding the onset of 10–15-Hz sleep spindles and 5-9-Hz oscillations in electroencephalograms in rats with and without absence seizures. J Sleep Res. (2020) 29:e12927. doi: 10.1111/jsr.12927
294. Meeren HK, Veening JG, Möderscheim TA, Coenen AM, van Luijtelaar G. Thalamic lesions in a genetic rat model of absence epilepsy: dissociation between spike-wave discharges and sleep spindles. Exp Neurol. (2009) 217:25–37. doi: 10.1016/j.expneurol.2009.01.009
295. Leresche N, Lambert RC, Errington AC, Crunelli V. From sleep spindles of natural sleep to spike and wave discharges of typical absence seizures: is the hypothesis still valid? Pflugers Arch. (2012) 463:201–12. doi: 10.1007/s00424-011-1009-3
296. Kozák G, Földi T, Berényi A. Spike-and-wave discharges are not pathological sleep spindles, network-level aspects of age-dependent absence seizure development in rats. eNeuro. (2020) 7:201+9. doi: 10.1523/ENEURO.0253-19.2019
297. Steriade M, Nunez A, Amzica F. Intracellular analysis of relations between the slow (<1 Hz) neocortical oscillation and other sleep rhythms of the electroencephalogram. J Neurosci. (1993) 13:3266. doi: 10.1523/JNEUROSCI.13-08-03266.1993
298. Steriade M, Nuñez A, Amzica F. A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci. (1993) 13:3252–65. doi: 10.1523/JNEUROSCI.13-08-03252.1993
299. Crunelli V, Hughes SW. The slow (<1 Hz) rhythm of non-REM sleep: a dialogue between three cardinal oscillators. Nat Neurosci. (2010) 13:9–17. doi: 10.1038/nn.2445
300. Neske GT. The slow oscillation in cortical and thalamic networks: mechanisms and functions. Front Neural Circuits. (2015) 9:88. doi: 10.3389/fncir.2015.00088
301. Fiáth R, Kerekes BP, Wittner L, Tóth K, Beregszászi P, Horváth D, et al. Laminar analysis of the slow wave activity in the somatosensory cortex of anesthetized rats. Eur J. Neurosci. (2016) 44:1935–51. doi: 10.1111/ejn.13274
302. Lannes B, Micheletti G, Vergnes M, Marescaux C, Depaulis A, Warter JM. Relationship between spike-wave discharges and vigilance levels in rats with spontaneous petit mal-like epilepsy. Neurosci Lett. (1988) 94:187–91. doi: 10.1016/0304-3940(88)90293-5
303. Terzano MG, Parrino L, Anelli S, Halasz P. Modulation of generalized spike-and-wave discharges during sleep by cyclic alternating pattern. Epilepsia. (1989) 30:772–81. doi: 10.1111/j.1528-1157.1989.tb05337.x
304. Coenen AM, Drinkenburg WH, Peeters BW, Vossen JM, van Luijtelaar EL. Absence epilepsy and the level of vigilance in rats of the WAG/Rij strain. Neurosci Biobehav Rev. (1991) 15:259–63. doi: 10.1016/S0149-7634(05)80005-3
305. Drinkenburg WH, Coenen AM, Vossen JM, Van Luijtelaar EL. Spike-wave discharges and sleep-wake states in rats with absence epilepsy. Epilepsy Res. (1991) 9:218–24. doi: 10.1016/0920-1211(91)90055-K
306. Tucker DM, Waters AC, Holmes MD. Transition from cortical slow oscillations of sleep to spike-wave seizures. Clin Neurophysiol. (2009) 120:2055–62. doi: 10.1016/j.clinph.2009.07.047
307. Koutroumanidis M, Tsiptsios D, Kokkinos V, Kostopoulos GK. Focal and generalized EEG paroxysms in childhood absence epilepsy: topographic associations and distinctive behaviors during the first cycle of non-REM sleep. Epilepsia. (2012) 53:840–9. doi: 10.1111/j.1528-1167.2012.03424.x
308. Smyk MK, Sysoev IV, Sysoeva MV, van Luijtelaar G, Drinkenburg WH. Can absence seizures be predicted by vigilance states? advanced analysis of sleep-wake states and spike-wave discharges' occurrence in rats. Epilepsy Behav. (2019) 96:200–9. doi: 10.1016/j.yebeh.2019.04.012
309. Durazzo TS, Spencer SS, Duckrow RB, Novotny EJ, Spencer DD, Zaveri HP. Temporal distributions of seizure occurrence from various epileptogenic regions. Neurology. (2008) 70:1265–71. doi: 10.1212/01.wnl.0000308938.84918.3f
310. Gurkas E, Serdaroglu A, Hirfanoglu T, Kartal A, Yilmaz U, Bilir E. Sleep-wake distribution and circadian patterns of epileptic seizures in children. Eur J Paediatr Neurol. (2016) 20:549–54. doi: 10.1016/j.ejpn.2016.04.004
311. Loddenkemper T, Vendrame M, Zarowski M, Gregas M, Alexopoulos AV, Wyllie E, et al. Circadian patterns of pediatric seizures. Neurology. (2011) 76:145–53. doi: 10.1212/WNL.0b013e318206ca46
312. Zarowski M, Loddenkemper T, Vendrame M, Alexopoulos AV, Wyllie E, Kothare SV. Circadian distribution and sleep/wake patterns of generalized seizures in children. Epilepsia. (2011) 52:1076–83. doi: 10.1111/j.1528-1167.2011.03023.x
313. Halász P. Sleep, arousal and electroclinical manifestations of generalized epilepsy with spike wave pattern. Epilepsy Res Suppl. (1991) 2:43–8.
314. Minecan D, Natarajan A, Marzec M, Malow B. Relationship of epileptic seizures to sleep stage and sleep depth. Sleep. (2002) 25:899–904. doi: 10.1093/sleep/25.8.56
315. Seneviratne U, Lai A, Cook M, D'Souza W, Boston RC. “Sleep Surge”: the impact of sleep onset and offset on epileptiform discharges in idiopathic generalized epilepsies. Clin Neurophysiol. (2020) 131:1044–50. doi: 10.1016/j.clinph.2020.01.021
316. Van Luijtelaar EL, Coenen AM. Circadian rhythmicity in absence epilepsy in rats. Epilepsy Res. (1988) 2:331–6. doi: 10.1016/0920-1211(88)90042-3
317. Smyk MK, Coenen AM, Lewandowski MH, van Luijtelaar G. Endogenous rhythm of absence epilepsy: relationship with general motor activity and sleep-wake states. Epilepsy Res. (2011) 93:120–7. doi: 10.1016/j.eplepsyres.2010.11.003
318. Smyk MK, van Luijtelaar G, Huysmans H, Drinkenburg WH. Spike-wave discharges and sleep-wake states during circadian desynchronization: no effects of agomelatine upon re-entrainment. Neuroscience. (2019) 408:327–38. doi: 10.1016/j.neuroscience.2019.03.062
319. Smyk MK, Coenen A, Lewandowski MH, van Luijtelaar G. Internal desynchronization facilitates seizures. Epilepsia. (2012) 53:1511–8. doi: 10.1111/j.1528-1167.2012.03577.x
320. Kovács Z, Slézia A, Bali ZK, Kovács P, Dobolyi A, Szikra T, et al. Uridine modulates neuronal activity and inhibits spike-wave discharges of absence epileptic Long Evans and Wistar Albino Glaxo/Rijswijk rats. Brain Res Bull. (2013) 97:16–23. doi: 10.1016/j.brainresbull.2013.05.009
321. Honda K, Komoda Y, Nishida S, Nagasaki H, Higashi A, Uchizono K, et al. Uridine as an active component of sleep-promoting substance: its effects on nocturnal sleep in rats. Neurosci Res. (1984) 1:243–52. doi: 10.1016/S0168-0102(84)80003-6
322. Halász P, Filakovszky J, Vargha A, Bagdy G. Effect of sleep deprivation on spike-wave discharges in idiopathic generalised epilepsy: a 4 x 24 h continuous long term EEG monitoring study. Epilepsy Res. (2002) 51:123–32. doi: 10.1016/S0920-1211(02)00123-7
323. Giorgi FS, Perini D, Maestri M, Guida M, Pizzanelli C, Caserta A, et al. Usefulness of a simple sleep-deprived EEG protocol for epilepsy diagnosis in de novo subjects. Clin Neurophysiol. (2013) 124:2101–7. doi: 10.1016/j.clinph.2013.04.342
324. Rosenow F, Klein KM, Hamer HM. Non-invasive EEG evaluation in epilepsy diagnosis. Expert Rev Neurother. (2015) 15:425–44. doi: 10.1586/14737175.2015.1025382
325. Renzel R, Baumann CR, Poryazova R. EEG after sleep deprivation is a sensitive tool in the first diagnosis of idiopathic generalized but not focal epilepsy. Clin Neurophysiol. (2016) 127:209–13. doi: 10.1016/j.clinph.2015.06.012
326. Van Luijtelaar EL, Van der Werf SJ, Vossen JM, Coenen AM. Arousal, performance and absence seizures in rats. Electroencephalogr Clin Neurophysiol. (1991) 79:430–4. doi: 10.1016/0013-4694(91)90208-L
327. Osterhagen L, Breteler M, van Luijtelaar G. Does arousal interfere with operant conditioning of spike-wave discharges in genetic epileptic rats? Epilepsy Res. (2010) 90:75–82. doi: 10.1016/j.eplepsyres.2010.03.010
328. Sudbrack-Oliveira P, Lima Najar L, Foldvary-Schaefer N, da Mota Gomes M. Sleep architecture in adults with epilepsy: a systematic review. Sleep Med. (2019) 53:22–7. doi: 10.1016/j.sleep.2018.09.004
329. van Luijtelaar G, Bikbaev A. Midfrequency cortico-thalamic oscillations and the sleep cycle: genetic, time of day and age effects. Epilepsy Res. (2007) 73:259–65. doi: 10.1016/j.eplepsyres.2006.11.002
330. Yi PL, Chen YJ, Lin CT, Chang FC. Occurrence of epilepsy at different zeitgeber times alters sleep homeostasis differently in rats. Sleep. (2012) 35:1651–65. doi: 10.5665/sleep.2238
331. Krueger JM, Fang J, Taishi P, Chen Z, Kushikata T, Gardi J. Sleep. A physiologic role for IL-1 beta and TNF-alpha. Ann N Y Acad Sci. (1998) 856:148–59. doi: 10.1111/j.1749-6632.1998.tb08323.x
332. Györffy B, Kovács Z, Gulyássy P, Simor A, Völgyi K, Orbán G, et al. Brain protein expression changes in WAG/Rij rats, a genetic rat model of absence epilepsy after peripheral lipopolysaccharide treatment. Brain Behav Immun. (2014) 35:86–95. doi: 10.1016/j.bbi.2013.09.001
333. Lipton JO, Boyle LM, Yuan ED, Hochstrasser KJ, Chifamba FF, Nathan A, et al. Aberrant proteostasis of BMAL1 underlies circadian abnormalities in a paradigmatic mTOR-opathy. Cell Rep. (2017) 20:868–80. doi: 10.1016/j.celrep.2017.07.008
334. Cooper JM, Halter KA, Prosser RA. Circadian rhythm and sleep-wake systems share the dynamic extracellular synaptic milieu. Neurobiol Sleep Circadian Rhythms. (2018) 5:15–36. doi: 10.1016/j.nbscr.2018.04.001
335. Re CJ, Batterman AI, Gerstner JR, Buono RJ, Ferraro TN. The molecular genetic interaction between circadian rhythms and susceptibility to seizures and epilepsy. Front Neurol. (2020) 11:520. doi: 10.3389/fneur.2020.00520
336. Lananna BV, Nadarajah CJ, Izumo M, Cedeño MR, Xiong DD, Dimitry J, et al. Cell-autonomous regulation of astrocyte activation by the circadian clock protein BMAL1. Cell Rep. (2018) 25:1–9.e5. doi: 10.1016/j.celrep.2018.09.015
337. Brancaccio M, Edwards MD, Patton AP, Smyllie NJ, Chesham JE, Maywood ES, et al. Cell-autonomous clock of astrocytes drives circadian behavior in mammals. Science. (2019) 363:187–92. doi: 10.1126/science.aat4104
338. Brancaccio M, Patton AP, Chesham JE, Maywood ES, Hastings MH. Astrocytes control circadian timekeeping in the suprachiasmatic nucleus via glutamatergic signaling. Neuron. (2017) 93:1420–35.e1425. doi: 10.1016/j.neuron.2017.02.030
339. Hablitz LM, Gunesch AN, Cravetchi O, Moldavan M, Allen CN. Cannabinoid signaling recruits astrocytes to modulate presynaptic function in the suprachiasmatic nucleus. eNeuro. (2020) 7:2020. doi: 10.1523/ENEURO.0081-19.2020
340. Chrobok L, Palus K, Jeczmien-Lazur JS, Chrzanowska A, Kepczynski M, Lewandowski MH. Disinhibition of the intergeniculate leaflet network in the WAG/Rij rat model of absence epilepsy. Exp Neurol. (2017) 289:103–16. doi: 10.1016/j.expneurol.2016.12.014
341. Haydon PG. Astrocytes and the modulation of sleep. Curr Opin Neurobiol. (2017) 44:28–33. doi: 10.1016/j.conb.2017.02.008
342. Garofalo S, Picard K, Limatola C, Nadjar A, Pascual O, Tremblay M. Role of Glia in the regulation of sleep in health and disease. Compr Physiol. (2020) 10:687–712. doi: 10.1002/cphy.c190022
343. Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, et al. Sleep drives metabolite clearance from the adult brain. Science. (2013) 342:373–7. doi: 10.1126/science.1241224
344. Ding F, O'Donnell J, Xu Q, Kang N, Goldman N, Nedergaard M. Changes in the composition of brain interstitial ions control the sleep-wake cycle. Science. (2016) 352:550–5. doi: 10.1126/science.aad4821
345. Sherpa AD, Xiao F, Joseph N, Aoki C, Hrabetova S. Activation of β-adrenergic receptors in rat visual cortex expands astrocytic processes and reduces extracellular space volume. Synapse. (2016) 70:307–16. doi: 10.1002/syn.21908
346. Baskey G, Singh A, Sharma R, Mallick BN. REM sleep deprivation-induced noradrenaline stimulates neuronal and inhibits glial Na-K ATPase in rat brain: in vivo and in vitro studies. Neurochem Int. (2009) 54:65–71. doi: 10.1016/j.neuint.2008.10.006
347. Vyazovskiy VV, Olcese U, Lazimy YM, Faraguna U, Esser SK, Williams JC, et al. Cortical firing and sleep homeostasis. Neuron. (2009) 63:865–78. doi: 10.1016/j.neuron.2009.08.024
348. Watson BO, Levenstein D, Greene JP, Gelinas JN, Buzsáki G. Network homeostasis and state dynamics of neocortical sleep. Neuron. (2016) 90:839–52. doi: 10.1016/j.neuron.2016.03.036
349. Cucchiara F, Frumento P, Banfi T, Sesso G, Di Galante M, D'Ascanio P, et al. Electrophysiological features of sleep in children with Kir4.1 channel mutations and Autism-Epilepsy phenotype: a preliminary study. Sleep. (2020) 43:zsz255. doi: 10.1093/sleep/zsz255
350. Bellesi M, de Vivo L, Tononi G, Cirelli C. Effects of sleep and wake on astrocytes: clues from molecular and ultrastructural studies. BMC Biol. (2015) 13:66. doi: 10.1186/s12915-015-0176-7
351. DiNuzzo M, Nedergaard M. Brain energetics during the sleep-wake cycle. Curr Opin Neurobiol. (2017) 47:65–72. doi: 10.1016/j.conb.2017.09.010
352. Benveniste H, Lee H, Volkow ND. The glymphatic pathway: waste removal from the CNS via cerebrospinal fluid transport. Neuroscientist. (2017) 23:454–65. doi: 10.1177/1073858417691030
353. Hablitz LM, Vinitsky HS, Sun Q, Stæger FF, Sigurdsson B, Mortensen KN, et al. Increased glymphatic influx is correlated with high EEG delta power and low heart rate in mice under anesthesia. Sci Adv. (2019) 5:eaav5447. doi: 10.1126/sciadv.aav5447
354. Hablitz LM, Plá V, Giannetto M, Vinitsky HS, Stæger FF, Metcalfe T, et al. Circadian control of brain glymphatic and lymphatic fluid flow. Nat Commun. (2020) 11:4411. doi: 10.1038/s41467-020-18115-2
355. Ulv Larsen SM, Landolt HP, Berger W, Nedergaard M, Knudsen GM, Holst SC. Haplotype of the astrocytic water channel AQP4 is associated with slow wave energy regulation in human NREM sleep. PLoS Biol. (2020) 18:e3000623. doi: 10.1371/journal.pbio.3000623
356. Petit JM, Magistretti PJ. Regulation of neuron-astrocyte metabolic coupling across the sleep-wake cycle. Neuroscience. (2016) 323:135–56. doi: 10.1016/j.neuroscience.2015.12.007
357. Clasadonte J, Scemes E, Wang Z, Boison D, Haydon PG. Connexin 43-mediated astroglial metabolic networks contribute to the regulation of the sleep-wake cycle. Neuron. (2017) 95:1365–80.e1365. doi: 10.1016/j.neuron.2017.08.022
358. Petit JM, Gyger J, Burlet-Godinot S, Fiumelli H, Martin JL, Magistretti PJ. Genes involved in the astrocyte-neuron lactate shuttle (ANLS) are specifically regulated in cortical astrocytes following sleep deprivation in mice. Sleep. (2013) 36:1445–58. doi: 10.5665/sleep.3034
359. Sada N, Lee S, Katsu T, Otsuki T, Inoue T. Epilepsy treatment. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science. (2015) 347:1362–7. doi: 10.1126/science.aaa1299
360. Fisher JL. The anti-convulsant stiripentol acts directly on the GABA(A) receptor as a positive allosteric modulator. Neuropharmacology. (2009) 56:190–7. doi: 10.1016/j.neuropharm.2008.06.004
361. Halassa MM, Florian C, Fellin T, Munoz JR, Lee S.-Y, Abel T, et al. Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron. (2009) 61:213–9. doi: 10.1016/j.neuron.2008.11.024
362. Ursin R, Bjorvatn B. Sleep-wake and eeg effects following adenosine a1 agonism and antagonism: similarities and interactions with sleep-wake and eeg effects following a serotonin reuptake inhibitor in rats. Sleep Res Online. (1998) 1:119–27.
363. Thakkar MM, Winston S, McCarley RW. A1 receptor and adenosinergic homeostatic regulation of sleep-wakefulness: effects of antisense to the A1 receptor in the cholinergic basal forebrain. J Neurosci. (2003) 23:4278–87. doi: 10.1523/JNEUROSCI.23-10-04278.2003
364. Blutstein T, Haydon PG. The Importance of astrocyte-derived purines in the modulation of sleep. Glia. (2013) 61:129–39. doi: 10.1002/glia.22422
365. Nadjar A, Blutstein T, Aubert A, Laye S, Haydon PG. Astrocyte-derived adenosine modulates increased sleep pressure during inflammatory response. Glia. (2013) 61:724–31. doi: 10.1002/glia.22465
366. Zhou X, Oishi Y, Cherasse Y, Korkutata M, Fujii S, Lee CY, et al. Extracellular adenosine and slow-wave sleep are increased after ablation of nucleus accumbens core astrocytes and neurons in mice. Neurochem Int. (2019) 124:256–63. doi: 10.1016/j.neuint.2019.01.020
367. Fellin T, Halassa MM, Terunuma M, Succol F, Takano H, Frank M, et al. Endogenous non-neuronal modulators of synaptic transmission control cortical slow oscillations in vivo. Proc Natl Acad Sci USA. (2009) 106:15037–42. doi: 10.1073/pnas.0906419106
368. Poskanzer KE, Yuste R. Astrocytes regulate cortical state switching in vivo. Proc Natl Acad Sci USA. (2016) 113:E2675–84. doi: 10.1073/pnas.1520759113
369. Szabó Z, Héja L, Szalay G, Kékesi O, Füredi A, Szebényi K, et al. Extensive astrocyte synchronization advances neuronal coupling in slow wave activity in vivo. Sci Rep. (2017) 7:6018. doi: 10.1038/s41598-017-06073-7
370. Wang M, He Y, Sejnowski TJ, Yu X. Brain-state dependent astrocytic Ca2+ signals are coupled to both positive and negative BOLD-fMRI signals. Proc Natl Acad Sci USA. (2018) 115:E1647–56. doi: 10.1073/pnas.1711692115
371. Brockett AT, Kane GA, Monari PK, Briones BA, Vigneron PA, Barber GA, et al. Evidence supporting a role for astrocytes in the regulation of cognitive flexibility and neuronal oscillations through the Ca2+ binding protein S100β. PLoS ONE. (2018) 13:e0195726. doi: 10.1371/journal.pone.0195726
372. Foley J, Blutstein T, Lee S, Erneux C, Halassa MM, Haydon P. Astrocytic IP3/Ca2+ signaling modulates theta rhythm and REM sleep. Front Neural Circuits. (2017) 11:3. doi: 10.3389/fncir.2017.00003
373. Bojarskaite L, Bjørnstad DM, Pettersen KH, Cunen C, Hermansen GH, Åbjørsbråten KS, et al. Astrocytic Ca2+signaling is reduced during sleep and is involved in the regulation of slow wave sleep. Nat Commun. (2020) 11:3240. doi: 10.1038/s41467-020-17062-2
374. Takahashi K, Kayama Y, Lin JS, Sakai K. Locus coeruleus neuronal activity during the sleep-waking cycle in mice. Neuroscience. (2010) 169:1115–26. doi: 10.1016/j.neuroscience.2010.06.009
375. Wang Y, Burghardt TP, Worrell GA, Wang HL. The frequency-dependent effect of electrical fields on the mobility of intracellular vesicles in astrocytes. Biochem Biophys Res Commun. (2020) 22:111286. doi: 10.1101/2020.05.22.111286
376. El Helou J, Bélanger-Nelson E, Freyburger M, Dorsaz S, Curie T, La Spada F, et al. Neuroligin-1 links neuronal activity to sleep-wake regulation. Proc Natl Acad Sci USA. (2013) 110:9974–9. doi: 10.1073/pnas.1221381110
377. Reissner C, Runkel F, Missler M. Neurexins. Genome Biol. (2013) 14:213. doi: 10.1186/gb-2013-14-9-213
378. Rudenko G. Dynamic control of synaptic adhesion and organizing molecules in synaptic plasticity. Neural Plast. (2017) 2017:6526151. doi: 10.1155/2017/6526151
379. Massart R, Freyburger M, Suderman M, Paquet J, El Helou J, Belanger-Nelson E, et al. The genome-wide landscape of DNA methylation and hydroxymethylation in response to sleep deprivation impacts on synaptic plasticity genes. Transl Psychiatry. (2014) 4:e347. doi: 10.1038/tp.2013.120
380. Singh SK, Stogsdill JA, Pulimood NS, Dingsdale H, Kim YH, Pilaz LJ, et al. Astrocytes assemble thalamocortical synapses by bridging NRX1α and NL1 via Hevin. Cell. (2016) 164:183–96. doi: 10.1016/j.cell.2015.11.034
381. Cao F, Liu JJ, Zhou S, Cortez MA, Snead OC, Han J, et al. Neuroligin. 2 regulates absence seizures and behavioral arrests through GABAergic transmission within the thalamocortical circuitry. Nat Commun. (2020) 11:3744. doi: 10.1038/s41467-020-17560-3
382. Matsuki T, Takasu M, Hirose Y, Murakoshi N, Sinton CM, Motoike T, et al. GABAA receptor-mediated input change on orexin neurons following sleep deprivation in mice. Neuroscience. (2015) 284:217–24. doi: 10.1016/j.neuroscience.2014.09.063
383. Liu JJ, Grace KP, Horner RL, Cortez MA, Shao Y, Jia Z. Neuroligin. 3 R451C mutation alters electroencephalography spectral activity in an animal model of autism spectrum disorders. Mol Brain. (2017) 10:10. doi: 10.1186/s13041-017-0290-2
384. Iasevoli F, Tomasetti C, de Bartolomeis A. Scaffolding proteins of the post-synaptic density contribute to synaptic plasticity by regulating receptor localization and distribution: relevance for neuropsychiatric diseases. Neurochem Res. (2013) 38:1–22. doi: 10.1007/s11064-012-0886-y
385. O'Connor EC, Bariselli S, Bellone C. Synaptic basis of social dysfunction: a focus on postsynaptic proteins linking group-I mGluRs with AMPARs and NMDARs. Eur J. Neurosci. (2014) 39:1114–29. doi: 10.1111/ejn.12510
386. Lesca G, Rudolf G, Labalme A, Hirsch E, Arzimanoglou A, Genton P, et al. Epileptic encephalopathies of the Landau-Kleffner and continuous spike and waves during slow-wave sleep types: genomic dissection makes the link with autism. Epilepsia. (2012) 53:1526–38. doi: 10.1111/j.1528-1167.2012.03559.x
387. Holder JL, Quach MM. The spectrum of epilepsy and electroencephalographic abnormalities due to SHANK3 loss-of-function mutations. Epilepsia. (2016) 57:1651–9. doi: 10.1111/epi.13506
388. Imeri L, Opp MR. How (and why) the immune system makes us sleep. Nat Rev Neurosci. (2009) 10:199–210. doi: 10.1038/nrn2576
389. Krueger JM, Clinton JM, Winters BD, Zielinski MR, Taishi P, Jewett KA, et al. Involvement of cytokines in slow wave sleep. Prog Brain Res. (2011) 193:39–47. doi: 10.1016/B978-0-444-53839-0.00003-X
390. Irwin MR, Opp MR. Sleep health: reciprocal regulation of sleep and innate immunity. Neuropsychopharmacology. (2017) 42:129–55. doi: 10.1038/npp.2016.148
391. Del Gallo F, Opp MR, Imeri L. The reciprocal link between sleep and immune responses. Arch Ital Biol. (2014) 152:93–102. doi: 10.12871/000298292014234
392. Fang J, Wang Y, Krueger JM. Mice lacking the TNF55 kDa receptor fail to sleep more after TNFalpha treatment. J Neurosci. (1997) 17:5949–55. doi: 10.1523/JNEUROSCI.17-15-05949.1997
393. Fang J, Wang Y, Krueger JM. Effects of interleukin-1 beta on sleep are mediated by the type I. receptor. Am J. Physiol. (1998) 274:R655–60. doi: 10.1152/ajpregu.1998.274.3.R655
394. Krueger JM, Taishi P, De A, Davis CJ, Winters BD, Clinton J, et al. ATP and the purine type 2 X7 receptor affect sleep. J Appl Physiol. (2010) 109:1318–27. doi: 10.1152/japplphysiol.00586.2010
395. Kovalzon VM, Moiseenko LS, Ambaryan AV, Kurtenbach S, Shestopalov VI, Panchin YV. Sleep-wakefulness cycle and behavior in pannexin1 knockout mice. Behav Brain Res. (2017) 318:24–7. doi: 10.1016/j.bbr.2016.10.015
396. Yoshida H, Peterfi Z, García-García F, Kirkpatrick R, Yasuda T, Krueger JM. State-specific asymmetries in EEG slow wave activity induced by local application of TNFalpha. Brain Res. (2004) 1009:129–36. doi: 10.1016/j.brainres.2004.02.055
397. Churchill L, Yasuda K, Yasuda T, Blindheim KA, Falter M, Garcia-Garcia F, et al. Unilateral cortical application of tumor necrosis factor alpha induces asymmetry in Fos- and interleukin-1beta-immunoreactive cells within the corticothalamic projection. Brain Res. (2005) 1055:15–24. doi: 10.1016/j.brainres.2005.06.052
398. Murphy S, Simmons ML, Agullo L, Garcia A, Feinstein DL, Galea E, et al. Synthesis of nitric oxide in CNS glial cells. Trends Neurosci. (1993) 16:323–8. doi: 10.1016/0166-2236(93)90109-Y
399. Wong ML, Rettori V, al-Shekhlee A, Bongiorno PB, Canteros G, McCann SM, et al. Inducible nitric oxide synthase gene expression in the brain during systemic inflammation. Nat Med. (1996) 2:581–4. doi: 10.1038/nm0596-581
400. Banach M, Piskorska B, Czuczwar SJ, Borowicz KK. Nitric oxide, epileptic seizures, and action of antiepileptic drugs. CNS Neurol Disord Drug Targets. (2011) 10:808–19. doi: 10.2174/187152711798072347
401. Sharma S, Puttachary S, Thippeswamy T. Glial source of nitric oxide in epileptogenesis: a target for disease modification in epilepsy. J Neurosci Res. (2019) 97:1363–77. doi: 10.1002/jnr.24205
402. Brown RE, Basheer R, McKenna JT, Strecker RE, McCarley RW. Control of sleep and wakefulness. Physiol Rev. (2012) 92:1087–7. doi: 10.1152/physrev.00032.2011
403. Cespuglio R, Amrouni D, Meiller A, Buguet A, Gautier-Sauvigné S. Nitric oxide in the regulation of the sleep-wake states. Sleep Med Rev. (2012) 16:265–79. doi: 10.1016/j.smrv.2012.01.006
Keywords: astrocytes, sleep/wake cycle, NREM, network plasticity, cortico-thalamo-cortical oscillations, spike and wave discharges, sleep
Citation: Gobbo D, Scheller A and Kirchhoff F (2021) From Physiology to Pathology of Cortico-Thalamo-Cortical Oscillations: Astroglia as a Target for Further Research. Front. Neurol. 12:661408. doi: 10.3389/fneur.2021.661408
Received: 30 January 2021; Accepted: 11 May 2021;
Published: 09 June 2021.
Edited by:
Kjell Heuser, Oslo University Hospital, NorwayReviewed by:
Vincenzo Crunelli, Cardiff University, United KingdomHirokazu Oguni, TMG Asaka Medical Center, Japan
Copyright © 2021 Gobbo, Scheller and Kirchhoff. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frank Kirchhoff, ZnJhbmsua2lyY2hob2ZmQHVrcy5ldQ==
†ORCID: Davide Gobbo orcid.org/0000-0002-4076-2697
Anja Scheller orcid.org/0000-0001-8955-2634
Frank Kirchhoff orcid.org/0000-0002-2324-2761