 Elena Spronk1*
Elena Spronk1* Gina Sykes1
Gina Sykes1 Sarina Falcione1
Sarina Falcione1 Danielle Munsterman1Twinkle Joy1
Danielle Munsterman1Twinkle Joy1 Joseph Kamtchum-Tatuene2
Joseph Kamtchum-Tatuene2 Glen C. Jickling1
Glen C. Jickling1- 1Division of Neurology, Department of Medicine, Faculty of Medicine and Dentistry, University of Alberta, Edmonton, AB, Canada
- 2Neuroscience and Mental Health Institute, Faculty of Medicine and Dentistry, University of Alberta, Edmonton, AB, Canada
Hemorrhagic transformation (HT) is a common complication in patients with acute ischemic stroke. It occurs when peripheral blood extravasates across a disrupted blood brain barrier (BBB) into the brain following ischemic stroke. Preventing HT is important as it worsens stroke outcome and increases mortality. Factors associated with increased risk of HT include stroke severity, reperfusion therapy (thrombolysis and thrombectomy), hypertension, hyperglycemia, and age. Inflammation and the immune system are important contributors to BBB disruption and HT and are associated with many of the risk factors for HT. In this review, we present the relationship of inflammation and immune activation to HT in the context of reperfusion therapy, hypertension, hyperglycemia, and age. Differences in inflammatory pathways relating to HT are discussed. The role of inflammation to stratify the risk of HT and therapies targeting the immune system to reduce the risk of HT are presented.
Introduction
Hemorrhagic transformation (HT) is a common complication of ischemic stroke that is often exacerbated by reperfusion with alteplase (recombinant tissue plasminogen activator) or endovascular therapy (EVT) (1). It occurs when the blood-brain barrier (BBB) is sufficiently disrupted to permit extravasation of peripheral blood into the brain. When HT occurs, it increases stroke morbidity and mortality and thus is important to prevent (2).
HT can be identified in 3–40% of patients with ischemic stroke, depending on the definition used and the characteristics of the cohort studied. HT is classified using both clinical and radiological criteria as summarized in Table 1. Clinical classification distinguishes symptomatic intracranial hemorrhage (sICH) from asymptomatic intracranial hemorrhage (aICH). sICH is defined as a worsening of the National Institutes of Health Stroke Scale (NIHSS) by ≥4 points within the first 36 h of stroke onset that is attributable to HT. A limitation of the clinical classification is that HT may be missed if it does not result in a major worsening of the neurological status. This may be the case when reperfusion therapy improves the NIHSS and offsets potential worsening caused by HT. The radiological classification of HT arose from the European Cooperative Acute Stroke Study (ECASS) and distinguishes small petechial hemorrhagic infarction (HI1), confluent petechial hemorrhagic infarction (HI2), small parenchymal hemorrhage (PH1) (<30% of infarct, mild mass effect), and large parenchymal hemorrhage (PH2, >30% of infarct, marked mass effect) (3). The ECASS definition has fairly poor intra- and inter-rater agreement for all types/categories except large PH2 (4). A Heidelberg Bleeding Classification scale has been proposed to address some of the challenges and limitations of the ECASS classification.
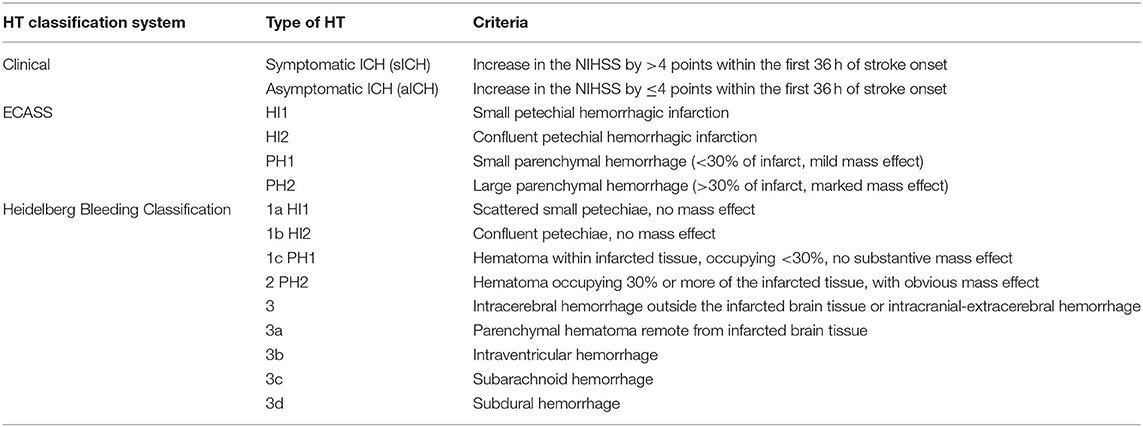
Table 1. Classification systems of HT in ischemic stroke.
A challenge with clinical and radiological classifications of HT is that they lack specificity to the underlying mechanism of BBB disruption and HT. Details of the mechanism of BBB disruption and the cause of HT might be included in future classification systems as our understanding of the processes involved in HT evolves. Figure 1 provides a schematic of the discussed immune pathways contributing to HT in ischemic stroke.
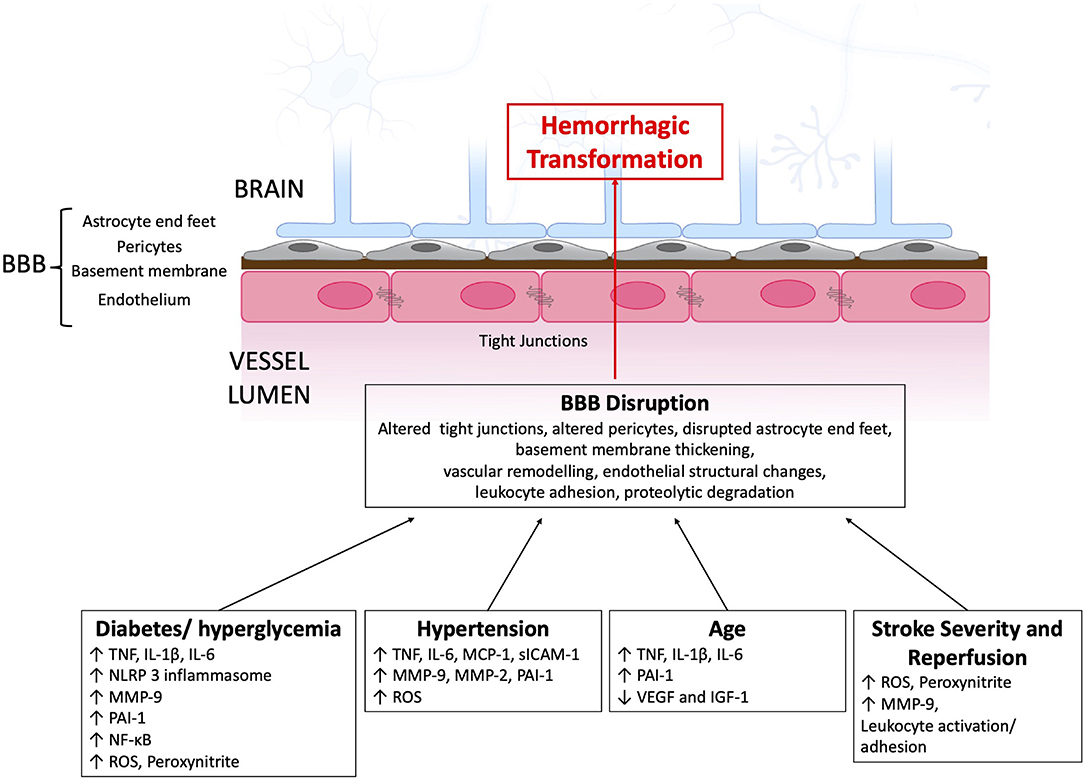
Figure 1. Immune pathways contributing to BBB disruption and subsequent HT after AIS. BBB, blood-brain barrier; IGF-1, insulin-like growth factor 1IL, interleukin; MCP-1, monocyte chemoattractant protein 1; MMP, matrix metalloproteinase; NF-κB, nuclear factor-κB; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; PAI-1, plasminogen activator inhibitor-1; ROS, reactive oxygen species; TLR, toll like receptor; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor.
Inflammation and HT
Inflammation and immune system activation are important contributors to BBB disruption and HT (5). In acute stroke activated neutrophils and monocytes produce reactive oxygen species (ROS) and matrix metalloproteinases (MMP-9, MMP-2) that contribute to BBB disruption and HT (1). Microglia, astrocytes, and endothelial cells also contribute to BBB disruption and HT through the production of MMPs, proteases, vascular remodeling, and neuroinflammation (Table 2). In the following sections, we discuss how the immune system contributes to HT specifically in the context of clinical factors related to HT, notably stroke severity, reperfusion, hypertension, diabetes, and age.
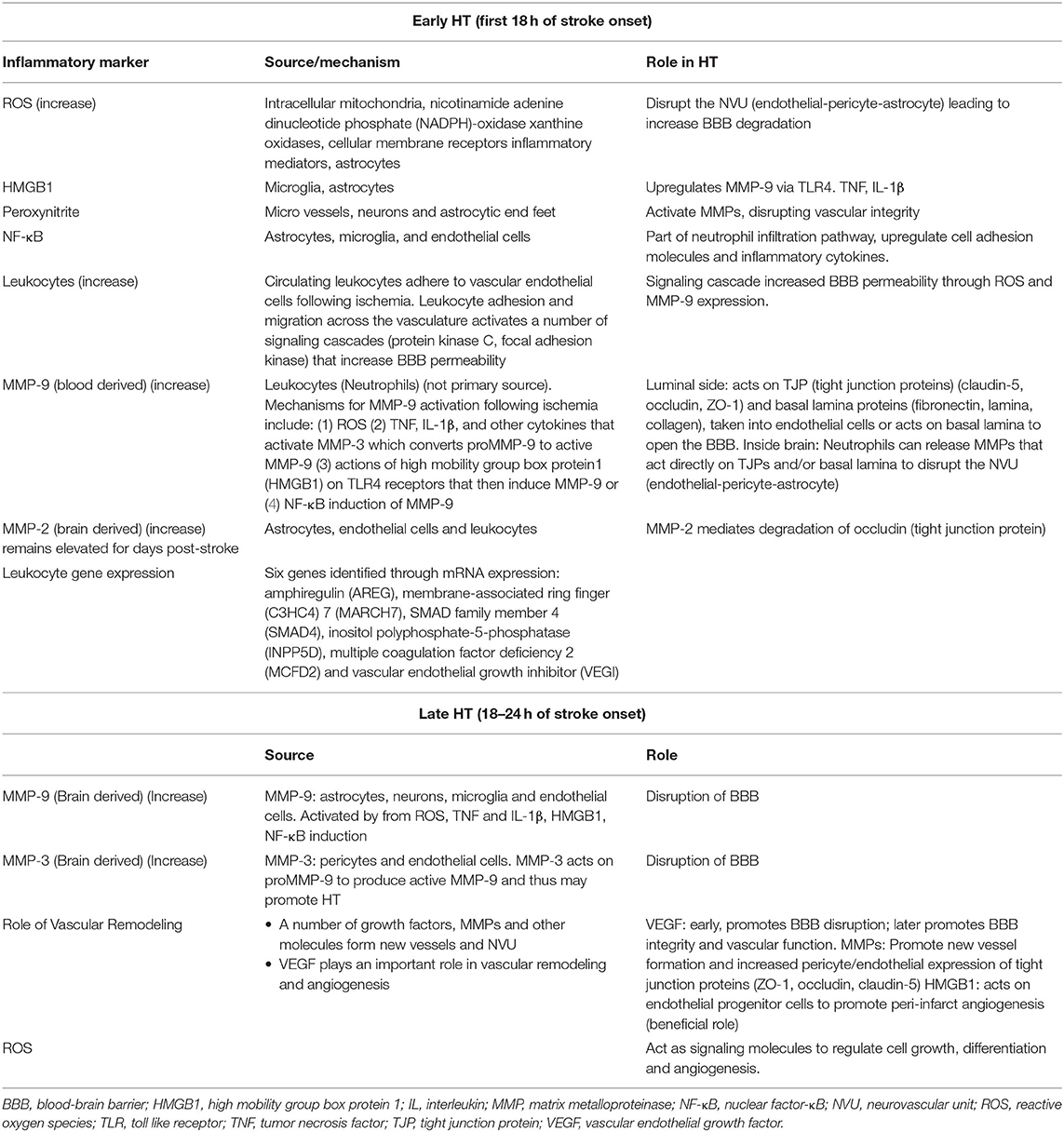
Table 2. Inflammatory markers associated with HT.
Several trials have focused on improving stroke outcomes through targeting immune factors. There has yet to be immunomodulator identified to reduce the risk of HT. A summary of discussed treatments and their molecular targets is provided in Table 3.

Table 3. Therapeutic targets in acute ischemic stroke focused on modulating HT.
Clinical Risk Factors for HT
Clinical features associated with an increased risk of HT in patients with ischemic stroke include advanced age, stroke severity (NIHSS), hypertension, hyperglycemia, poor collaterals, early infarction on brain imaging, low platelet count, use of antithrombotic drugs, and reperfusion therapy (15). Many of these factors are associated with inflammation and activation of the immune system, as discussed below. The clinical features have been summarized into scores to estimate the risk of HT and guide reperfusion therapy (Table 4). However, clinical characteristics and risk scores fail to identify all patients who experience HT, highlighting that a gap remains in our understanding of the pathophysiology of HT. Variables most commonly included in HT risk scores are stroke severity (admission NIHSS), hyperglycemia, hypertension, and age.
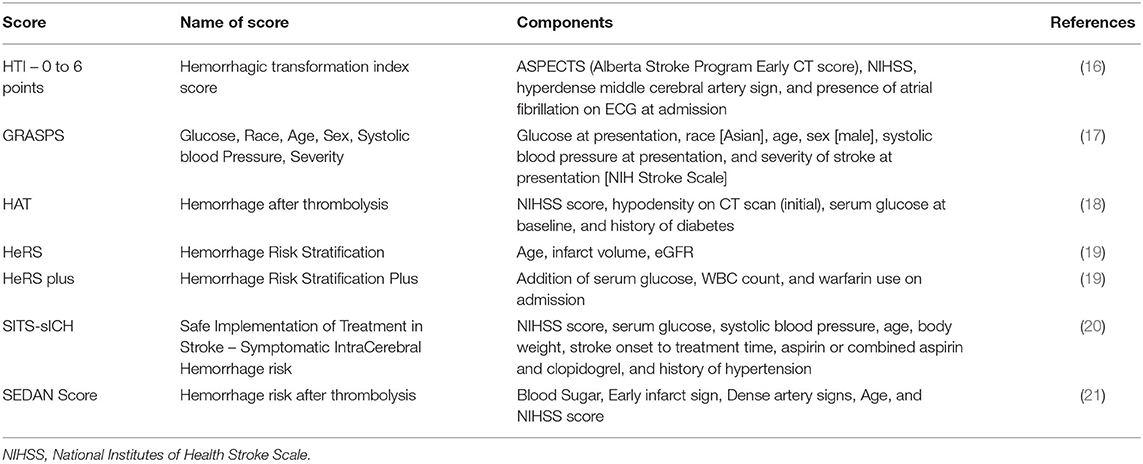
Table 4. Risk scores proposed for HT prediction with alteplase administration.
Hemorrhagic Transformation and Stroke Severity
There is a strong relationship between duration and severity of brain ischemia and the risk of HT in both patients with stroke and experimental stroke models. Increased time from stroke onset is associated with larger core volumes, a higher degree of vascular disruption, and therefore a higher risk of HT. Increased time from ischemia onset to reperfusion has been associated with an increased risk of HT whether they received thrombolytic therapy or not (1). Recanalization beyond 6 h of stroke onset is an independent predictor of HT in human stroke, while early reperfusion is associated with a reduced risk of HT. Increased NIHSS is also associated with risk of HT. Patients with an NIHSS < 10 had <13% rate of HT. In comparison, patients with NIHSS > 15 had >50% rate of HT (22). Activation of the immune system is related to severity of infarction. Whether the greater immune activation from a larger infarct increases risk of HT remains unclear and warrants further evaluation.
Thrombolytic Therapy and HT
Treatment with alteplase is associated with a 6–8% risk of sICH (23–27). This is mediated in part by the fact that thrombolytics breakdown blood clots and recanalize occluded cerebral vasculature. Recanalization of an occluded blood vessel can promote BBB disruption, contributing to reperfusion injury and increased risk of HT (1, 28). Activation of platelets, coagulation factors, and the innate and adaptive immune systems also contribute to injury following restoration of blood flow (29, 30).
Alteplase can also promote HT through non-fibrinolytic mechanisms, including activation of the immune system (31). Alteplase promotes neutrophil degranulation and release of MMP-9 (32). It acts on protease-activated receptor 1 (PAR-1) to increase MMP-9 expression and disrupt the BBB (33). Additionally, alteplase activates platelet-derived growth factor-CC (PDGF-CC), an agonist of platelet-derived growth factor receptor alpha (PDGFRa) on astrocyte end-feet (34). This can promote BBB disruption and HT, as PDGFRa activates MMPs (33). Alteplase also binds the lipoprotein receptor (LRP) on neurons and perivascular astrocytes to induce MMP-3 and MMP-6 expression (33). While inhibition of MMPs can reduce the risk of HT in animal stroke models, evidence in humans is lacking. Minocycline inhibits MMP-9 and has been evaluated in the MINOS trial (10). A phase 4 trial was later terminated due to lack of effect. MMP-9 inhibition, while potentially beneficial to reduce the risk of HT, may also impair post-stroke vascular remodeling and impair recovery. Indeed, inhibition of MMP-9 at 24–48 h after stroke worsens outcome in animal models (35). A phase 2 study, SAFE-TPA, evaluated different doses of Otaplimastat, a neuroprotectant inhibiting the MMP pathway (14). In animal models, Otaplimastat in combination with alteplase helped to reduce edema and HT. While Kim et al. (14) found the drug to be safe in patients, further investigation is required to determine effect in stroke.
Evidence is emerging regarding the use of Tenecteplase (TNK) in acute ischemic stroke (36, 37), a current treatment for acute myocardial infarction. TNK promotes the conversion of plasminogen to plasmin which degrades fibrin-based clots. It is a variant of recombinant tPA (tissue plasminogen activator), with higher resistance to plasminogen activator inhibitor-1 (PAI-1) and enhanced fibrin specificity (38). Studies of TNK in stroke are ongoing. In the EXTEND-IA TNK Trial, recanalization rates before thrombectomy were higher in the TNK group (22%, n = 101) compared with the alteplase group (10%, n = 101) with improved functional outcomes and similar risk of HT (1% sICH and 5–6% HT PH2) (37). As with alteplase, TNK may promote HT through several mechanisms that warrant further investigation.
Experimental evidence supports a role of reactive oxygen species (ROS) and reactive nitrogen species (RNS) in early HT. Superoxide and peroxynitrite can disrupt microvascular integrity and may contribute to HT (1). Cerbral ischemia and reperfusion produce reactive oxygen and nitrogen species including superoxide, nitric oxide (NO), and peroxynitrite (ONOO−) in microvessels, neurons and astrocyte end feet (39). Peroxynitrite can activate MMPs further disrupting vascular integrity. Hyperglycemia, as described in the section Hyperglycemia and HT can also enhance ROS and peroxynitrite.
High-mobility group box 1 (HMGB1) is released from injured cells of the central nervous system such as microglia and astrocytes (40). HMGB1 binds to its receptors (RAGE, toll-like receptor 2 (TLR2), and TLR4) and augments inflammation via the upregulation of tumor necrosis factor (TNF), interleukin 1β (IL-1β), and other cytokines (41). HMGB1 upregulates MMP-9 in neurons and astrocytes via TLR4. This could be a proposed target to help dampen the inflammatory response in cerebral ischemia and prevent HT as discussed in the section Hyperglycemia and HT.
Nuclear factor-κB (NF-κB) acts as a complex regulator of the innate immune system leading to activation of cellular adhesion molecules such as ICAM-1, VCAM-1 and E-selectin and inflammatory cytokines, mediating the neutrophil infiltration pathway (42, 43). A study targeting HIF-1, hypoxia-inducible factor 1, a transcription factor that works to maintain homeostasis under hypoxia, found that BBB integrity was protected through suppressing the HMGB1/TLR4/NF-κB pathway to reduce the risk of alteplase-induced HT (44).
In experimental studies, Enlimomab, an ICAM-1 antibody, reduced leukocyte adhesion and infarct size (7). In the Enlimomab Acute Stroke Trial, it was associated with poor outcomes and increased HT.
In the RHAPSODY phase 2 trial, 3K3A-APC (a modified APC), a recombinant variant of human activated protein C (part of the coagulation pathway), was evaluated as a neuroprotectant in acute ischemic stroke treated with alteplase and or mechanical thrombectomy (6). 3K4A-APC acts on the protease-activated receptor 1 (PAR1) to promote vascular integrity and reduce neurological injury. The study showed a trend toward reduced HT, but further confirmation is required. However, targeting the PAR1 receptor shows promise as a strategy to reduce HT in stroke patients treated with reperfusion therapy.
Endovascular Therapy and HT
Endovascular thrombectomy is of benefit in patients with acute large vessel occlusion and salvageable brain tissue (45). In a metanalysis of SWIFT PRIME, ESCAPE, EXTEND-IA, and REVASCAT the risk of sICH was 2.5% in EVT treated compared to 2.8% in non-EVT treated patients (p = 0.76), and the risk of PH was 8% in both EVT-treated patients and non-EVT patients (p = 0.96) (46). In the HERMES pooled analysis of MR CLEAN, ESCAPE, REVASCAT, SWIFT PRIME, and EXTEND IA stroke trials, the rate of sICH was 4.4% in the EVT group compared to 4.3% in the medical therapy group (45). Thus, the rate of HT is not significantly higher in the patients enrolled in EVT trials compared to controls receiving alteplase alone.
The DIRECT-MT trial compared EVT-only to EVT plus alteplase (47). The rates of HT were comparable between groups, with sICH occurring 4.3% in EVT group vs. 6.1% in EVT + alteplase (p = 0.30) and aICH occurring in 33.3% of the EVT group 36.2% in the EVT + alteplase group (p0.45). Similar rates of HT in EVT + alteplase vs. EVT alone have been reported by others (45, 48).
Thus, reperfusion with EVT may have lower rate of sICH than might be expected. EVT can provide a more rapid recanalization of large vessel occlusions which may reduce severity of ischemic brain injury, and thus reduce risk of HT. This is particularly true when EVT-treated patients are selected for small core and large penumbra by imaging. Core size does relate to risk of HT, with HT being higher in EVT-treated patients with larger core on baseline imaging (49). EVT may also produce differences in the type BBB disruption compared to alteplase. In an animal model, alteplase produced a more diffuse pattern of BBB disruption, whereas EVT had a more concentrated BBB disruption in central areas of ischemia (50). Additional evaluation is needed to understand HT in EVT-treated patients including differences in BBB disruption and potential benefits and risks of rt-PA (alteplase) use before EVT.
Hyperglycemia and HT
Hyperglycemia is present in approximately 28–40% of patients with acute ischemic stroke (51–53). It can relate to pre-existing diabetes mellitus (DM) or a stress response leading to a rise in cortisol, glucose and catecholamines. For both diabetic and non-diabetic patients, hyperglycemia increases the risk of HT and is associated with worse clinical outcomes (51). There are conflicting reports concerning infarct growth in a hyperglycemic setting relative to normal glycemic levels (54–56). For the most part, hyperglycemia is associated with more rapid infarct growth and worse outcomes. In a review of 426 patients, higher values of HbA1c were associated with a higher risk of HT in patients with fasting blood glucose > 7.8 mmol/L (27% higher risk) and in those with fasting blood glucose <7.8 mmol/L (36% higher risk) (57). Higher levels of HbA1c were also associated with a 48% higher risk of poor functional outcome. Treatment of hyperglycemia is recommended by stroke guidelines for the management of patients with acute ischemic stroke (58). Hyperglycemia is a risk factor for HT in both alteplase-treated patients and those not receiving thrombolysis. This association might be driven by hyperglycemia's effects on brain vasculature and the inflammatory response.
Hyperglycemia can potentiate inflammation resulting in increased BBB disruption and HT (59). Factors related to hyperglycemia that can increase the risk of HT are presented in Table 5. Diabetes is associated with an increase in plasma TNF, IL-1β, interleukin 6 (IL-6), interferon-g (IFNγ), and PAI-1, as well as alteration in immune cell response leading to chronic low-grade inflammation (60). An increase in PAI-1 may decrease the efficacy of alteplase and increase infarct size (67). This may result in increased risk of HT (68, 69). Hyperglycemia also activates nuclear factor-κB (NF-κB) in astrocytes, and enhances leukocyte adhesion through higher expression of cell adhesion molecules in cerebral blood vessels (ICAM1, intracellular adhesion molecule 1; VCAM-1, vascular cell adhesion molecule 1 and E-selectin) and the production of pro-inflammatory cytokines (TNF, IL-6, and IL-1β) (42, 70).
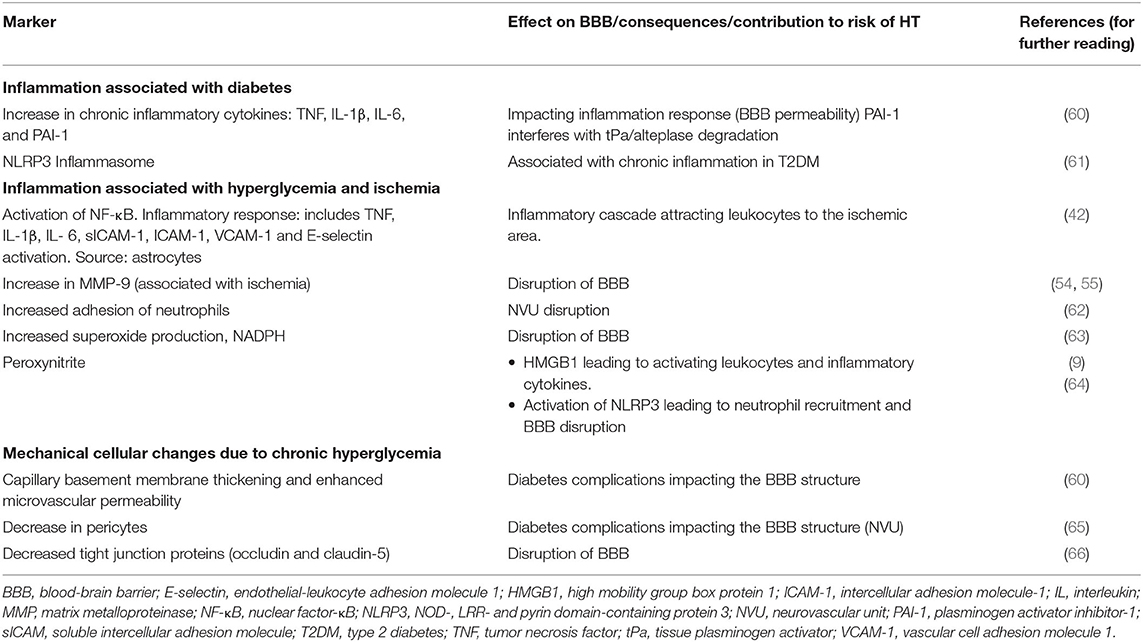
Table 5. Summary of factors in hyperglycemia that promote BBB disruption and risk of HT.
Neutrophils from patients with diabetes have enhanced capacity for endothelial adhesion (62). In stroke models using diabetic rats, hyperglycemia increases neutrophil adhesion and transmigration, and also increases expression of TNF, IL-1β, E-selectin, soluble intercellular adhesion molecules (sICAMs), endothelial NO synthase (eNOS) and inducible nitric oxide synthase (iNOS) (54, 71). In a rat model, hyperglycemia increases infarct volume, worsens outcome and increases risk of HT (55). Additionally, hyperglycemia increased the activity of MMP-9. Risk of HT is increased by glucose injection regardless of the time of ischemia (54, 55). Hyperglycemia can also increase superoxide production through NADPH oxidase, resulting in greater BBB disruption and HT. Apocynin, an NADPH oxidase inhibitor, prevents BBB disruption and HT in animal models of stroke (63).
In a rat model of stroke, hyperglycemia enhanced release of HMGB1 and the rate of alteplase related HT (9, 40, 56). Treatment with glycyrrhizin, a direct HMBG1 inhibitor, downregulated the expression of NADPH. This decreased superoxide and peroxynitrite(ONOO−), reduced TLR2, and MMP-9, and preservation of type IV collagen and claudin-5, thus attenuating HT and BBB damage. The role of an HMGB1 inhibitor in patients with stroke remains to be determined.
NLRP3 (NOD-like receptor protein 3) inflammasome can also promote BBB disruption and HT. In a MCAO rat model with acute hyperglycemia and increased expression of NADPH oxidase, iNOS induction lead to the production of superoxide, nitric oxide and peroxynitrite (56). It was suggested that peroxynitrite could activate NLRP3 inflammasome in hyperglycemia, a mediator in HT following reperfusion. Inhibiting the NLRP3 inflammasome minimized HT in vivo, and could potentially offer a treatment option.
NLRP3 inflammasome is also involved in chronic inflammation associated with type 2 diabetes (T2DM) (61). Primary activation of NLRP3 leads to transcription of pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18. Active IL-1β is then responsible for increased expression of adhesion molecules, leading to neutrophil recruitment and contributing to BBB disruption (64). Inhibiting NLRP3 may reduce the impact of stroke in patients with diabetes by regulating neutrophil recruitment. Hyperglycemia may upregulate TXNIP-NLRP3 inflammasome causing alteplase induced BBB disruption and HT (72). Thioredoxin interacting protein (TXNIP) mediates hyperglycemia-induced oxidative damage and inflammation in the brain, while reducing cerebral glucose uptake/utilization.
Other mechanisms by which hyperglycemia can promote BBB disruption and cause HT include a decrease in tight junction proteins occludin and claudin-5 (66), chronic oxidative stress and thickening of microvascular basement membrane, increase of microvascular permeability (60), and loss of pericytes (65).
Effect of Hyperglycemia on Treatment With Alteplase
Hyperglycemia impairs the fibrinolytic activity of alteplase, particularly in the acute phase of treatment, which may result in larger infarct size with more severe ischemia and thus, higher risk of HT (63, 73–75). Type 2 DM is associated with increased blood levels of PAI-1, an inhibitor of tPA (endogenous and exogenous) (76). Hyperglycemia also affects the coagulation system by increasing thrombin production and stimulating the intrinsic tissue factor pathway, thus affecting reperfusion and contributing to HT (77). In rats with Type 1 DM, treatment of acute stroke with alteplase increased MMP-9 activity, BBB breakdown, brain hemorrhage, and worsened functional outcomes (75).
Treatment of Hyperglycemia to Prevent HT
Several stroke trials have evaluated the benefit of treating hyperglycemia to reduce the risk of HT. Control of hyperglycemia through insulin infusion was tested in patients in the UK Glucose Insulin in Stroke Trial (GIST UK) (78) and the SHINE trial (51). In the SHINE trial, 80% of the patients had Type 2 DM. In the intensive treatment group, the average blood glucose was 6.6 mmol/L compared to 9.9 mmol/L in the standard treatment group. No net improvement in stroke outcome was identified, despite a significant reduction in blood glucose achieved with insulin infusion. Potential acute lowering of glucose does not fully mitigate downstream effects already initiated by hyperglycemia such as enhanced inflammation, endothelial cell dysfunction, neutrophil adhesion, MMP-9 activity, and ROS production. There is an ongoing secondary analysis of the effect of acute insulin therapy on the risk of HT, which may provide further insights into the link between hyperglycemia and HT from the SHINE trial.
A systematic review of 274 research articles aimed to identify the ideal target and strategy for blood glucose management (79). Glucose control was recommended to start within the first few hours of stroke onset. Tight glucose control was not recommended due to the risk of hypoglycemic events. Glycemic variability is to be avoided, as it is associated with adverse outcomes.
Hypertension and HT
An elevated blood pressure can increase the risk of HT through a variety of mechanisms, including direct mechanical effects on the cerebral/brain vasculature, exacerbated inflammation, and vascular remodeling affecting collateral circulation, endothelial function, and autoregulation (80). A summary of factors in hypertension that impacted HT through BBB modification is shown in Table 6.

Table 6. Summary of factors related to hypertension that promote BBB disruption and risk of HT.
Hypertension is common in patients with ischemic stroke and associated with increased risk of HT (20). Blood pressure is elevated in over 60% of patients with acute stroke (83). The elevated blood pressure may be caused by several components, including pre-existing or untreated hypertension, stress response (transient response), increased intracranial pressure, disrupted central autonomic regulation, activation of the neuroendocrine system, or inflammatory responses.
Impact of Hypertension on Inflammation and BBB Permeability
Hypertension is associated with disruption of the BBB which could increase risk of HT (84). In the spontaneous hypertensive rat, BBB disruption can be observed as early as 3 months of age (85). The renin-angiotensin system is involved in hypertension and contributes to BBB disruption. It activates MMPs, causing an increase in circulating and tissular MMP-2 and MMP-9 (81, 86). The increase of angiotensin II in hypertension induces perivascular macrophages to produce ROS and contribute to BBB disruption (82, 84). Hypertension is associated with an increase in blood levels of pro-inflammatory cytokines such as TNF, IL-6, monocyte chemoattractant protein-1(MCP-1), and sICAM-1 which can contribute to BBB disruption (80). Hypertension is associated with a decreased expression of cerebral endothelial tight junction proteins (claudin-3, claudin-5, and claudin-12), thus enhancing BBB permeability (84). Angiotensin II causes an increase in serum PAI-1 levels, thus reducing the effectiveness of alteplase (69).
Hypertension Related Vascular Remodeling
Chronic hypertension alters the cerebral vasculature, with microvascular rarefaction, arterial remodeling, and increased BBB permeability (80). Hypertension is associated with a decrease in lumen diameter, increase in vascular resistance, and impairment of endothelial function. These effects impair collateral circulation, reducing capacity to maintain adequate oxygenation when a cerebral artery occlusion occurs. Poor collateral circulation, which sustains the penumbra, results in faster expansion of infarct core, associated with risk of HT (87, 88).
Impact of Hypertension on HT Following Reperfusion Therapy
In a rabbit clot model, hypertension induced before and after alteplase administration increases the risk of HT (89). The size of HT also relates to the peak mean arterial blood pressure. Decreasing pre-stroke blood pressure in rats reduced the risk of HT (90). BBB changes may be mechanical or result from acute changes related to endothelial or inflammatory activation.
Maintaining the blood pressure below 185/110 mmHg is recommended to reduce the risk of HT in patients treated with alteplase (91). The risk of HT increases with each 10 mmHg rise in systolic blood pressure from 140 to 180 mmHg (92). Observational studies suggest lower blood pressure within the first 24 h is associated with more favorable outcomes and less frequent sICH (93). In addition, the risk of HT following alteplase is higher in patients with greater blood pressure variability, particularly in the first 6 h (94), and in patients with elevated systolic blood pressure (95). An elevated systolic blood pressure (SBP) is also associated with worse outcomes and higher risks of HT in patients treated with EVT (96, 97).
Hypertension Treatment to Prevent HT
The ENCHANTED trial compared the benefit of intensive blood pressure control (SBP target of 130–140 mmHg) to that of the standard of care (SBP <180 mmHg) (98). Intensive lowering of blood pressure was safe and decreased the incidence of HT from 18.7 to 14.8% (p = 0.0137). However, despite a reduction in HT, no net improvement in the 90-day Modified Rankin Scale (mRS) was observed. The best stroke outcomes are observed when BP is maintained between 140 and 180 mmHg (99). It is challenging to understand the ideal blood pressure for stroke treatment. In an acute setting, a rise in BP may be beneficial for preservation of brain perfusion in case of large vessel occlusion (opening of collateral vessels) while intensive BP lowering in the hyperacute phase (<48 h) may be detrimental in the context of impaired regulation of the cerebral circulation. The RIGHT-2 trial showed that functional outcomes were not improved with blood pressure reduction prior to stroke treatment (100). The Dutch ThRombolysis in Uncontrolled Hypertension (TRUTH) trial is an ongoing study evaluating whether actively lowering blood pressure below 185/110 mmHg affects stroke outcome in alteplase treated patients (101).
The BP-TARGET aimed to evaluate intense systolic blood pressure targets (100–129 mm Hg) and standard care systolic targets (130–185 mm Hg) for 24 h after reperfusion (102). Intensive blood pressure control was not superior to standard of care for the prevention of HT. A comment letter suggest that alternative variables should also be considered, such as collateral circulation, blood pressure variability at baseline and during monitoring (103).
Age and Risk of HT
Advancing age is associated with an increased risk of HT (20, 104, 105), as well as worse stroke outcomes (106, 107). Infarct size is not consistent between sources; however, most authors suggest an increase in infarct size. Despite an increased risk, patients of older age derive similar benefit from thrombolysis (108, 109). The rationale for age to increase the risk of HT is multifactorial, including an increase in systemic inflammation and BBB permeability. Additionally, elderly patients are also more likely to be on antithrombotic treatment, have a greater burden of cerebrovascular disease, and are more likely to have comorbid diseases such as hypertension and diabetes. These comorbidities contribute to inflammation, notably atherosclerosis, diabetes, hypertension, and hyperlipidemia. When stroke occurs, this age-associated inflammation may contribute to BBB disruption leading to an increased risk of HT. Table 7 summarizes the mechanisms through which age affects the risk of HT.
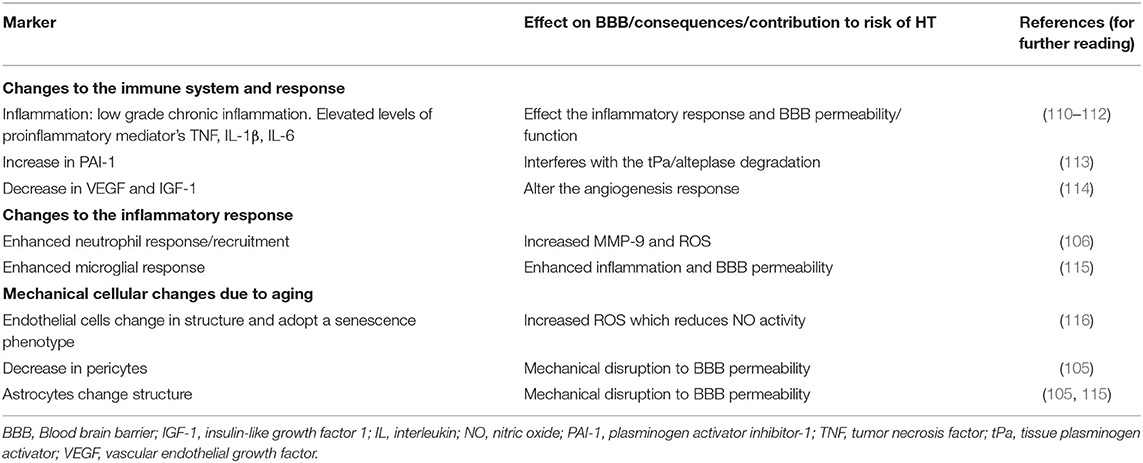
Table 7. Summary of factors related to age that impact HT and BBB.
Effect of Age on the Immune System
Advanced age is characterized by immunosenescence, gradual immune dysregulation and changes to cell function, leading to a reduced capacity to respond to antigen stimulation (110). Aging is also associated with inflammaging: chronic inflammation with elevated plasma concentrations of TNF, IL-1β, and IL-6 predictive of fragility, mortality, and functional disability (110–112). This chronic low-grade inflammation state alters the innate immunity of the brain and may contribute to the risk of HT.
Aging impacts both the innate and adaptive immune systems (110). Over time, the availability of hematopoietic stem cells declines, leading to decreased capacity to renew peripheral immune cells. Fewer naive B-lymphocytes are produced from the bone marrow, and T cell diversity is also reduced after age 70. Cells of the myeloid lineage are also affected by age, neutrophils have decreased phagocytic capacity and a reduced oxidative burst, leading to an increased inflammatory time period (110, 112). Moreover, there is reduced production of cytokines from macrophages which have decreased phagocytic capacity (106, 110). During stroke, young mice tend to recruit monocytes, whereas aged mice have greater recruitment of neutrophils. Additionally, higher levels of ROS and MMP-9 are associated with increased age. As discussed in the section Inflammatory Biomarkers and Prediction of HT, an increased neutrophil to leukocyte (NIL) ratio was independently associated with HT (117). Furthermore, with advancing age, microglia may become more prone to generate an inflammatory response when stroke occurs (115). With advancing age, there is a dysregulation in microglia pathways leading to a prolonged and amplified immune response, thus potentially increasing the risk of HT.
Age and the Neurovascular Unit
The neurovascular unit also undergoes age-related changes which may contribute to the risk of HT (104, 105). Aged cerebral endothelial cells have a smaller cytoplasm and dysfunctional mitochondria that adopt a senescent phenotype, promoting low-grade inflammation. Endothelial cells also produce more ROS with age, which reduces NO activity and limits the vasodilation capacity (116). Moreover, the number of pericytes, vital for BBB integrity, decreases with age, with a negative impact on the microcirculation, reducing the ability to maintain the BBB integrity after stroke (105). Astrocytes can become more inflammatory with age, which may also contribute to the risk of HT (115). Additionally, astrocytes undergo clasmatodendrosis with age and retract their end-feet, thus altering BBB integrity (105, 115). Finally, with aging, miRNA and RNA expression for genes associated with vascular tone, tight junction protein expression and cell adhesion are downregulated, impacting the BBB.
Age and Cerebral Vasculature
Angiogenesis is a necessary process to reduce infarct volume and support recovery after stroke. The most critical factors for supporting capillary angiogenesis include fibroblast growth factor (FGF), TNF, transforming growth factor-beta (TGF-β), and the angiopoietins (Ang) (118). With age, there is an attenuated pro-angiogenic response, decreased expression of vascular endothelial growth factor (VEGF) and insulin-like growth factor 1 (IGF-1) possibly contributing to the risk of HT (114). VEGF has an important role in vascular remodeling and angiogenesis and is associated with delayed HT and vascular remodeling (1). VEGF has a biphasic role in stroke. Early after stroke, VEGF promotes BBB disruption and HT but supportes vascular function and BBB integrity at a later stage. In rats, VEGF signal inhibition attenuates alteplase-related HT (119). Age is associated with a decrease in the number and diameter of native collaterals as well as impaired collateral remodeling in the brain (120). Collaterals influence the rate of HT after thrombolytic treatment (121). Thus, the increased risk of HT with age may also relate to an age-associated reduction in collaterals.
Inflammatory Biomarkers and Prediction of HT
Several protein and transcription biomarkers have been evaluated to predict the risk of HT. This includes plasma cellular-fibronectin (c-Fn), a major component of extracellular matrix involved in wound healing and cell adhesion (122); MMPs (122, 123); neutrophil and lymphocyte counts (117, 123); ferritin (123), vascular adhesion molecule−1 (VAP-1), and IL-10 that are associated with alteplase induced HT (124, 125). Additionally, inflammatory markers such as TNF, C-reactive protein and homocysteine are associated with the risk and severity of HT (126). Fibrinolysis inhibitors [PAI-1, lipoprotein(a), thrombin-activated fibrinolysis inhibitor (TAFI), and homocysteine] may help to predict sICH after thrombolysis (69, 122, 124).
Ischemic stroke elicits a strong local and peripheral inflammatory response. There is an increase in inflammatory cytokines (e.g., TNF, IL-1β, IL-6), cell adhesion molecules (e.g., ICAM-1, VCAM, P-selectin), damage-associated molecular patterns [DAMPs, e.g., high mobility group box protein 1 (HMGB1), peroxiredoxin, mitochondrial DNA], MMPs and ROS (127). Microglia, cerebral endothelial cells, and peripheral leukocytes are also activated during the immune response. This results in the recruitment, adhesion, and infiltration of the ischemic brain by peripheral leukocytes. Aspects of the inflammatory response alters the structure of the BBB, reorganizing tight junction proteins and the actin cytoskeleton, and contributing to HT (128).
A few trials have targeted ROS. NXY-059 targeting ROS was trialed to reduced alteplase-related intracranial hemorrhages (13). In animal studies, NXY-059 was associated with improved functional recovery and reduced infarction size. In the human trial, NXY-059 was found to be ineffective for preventing HT. Additionally, as part of a retrospective study in Japan, Edaravone administration with alteplase (rtPA) found that there may be a benefit in early stroke outcome (8). Edaravone suppresses ROS, inhibits vascular endothelial injuries, and protects the neurovascular unit. No benefit was found for HT occurrence.
Neutrophils are one of the first innate immune cells to reach the ischemic brain tissue (129). Neutrophils contribute to injury through inflammatory mediators and damage the BBB through the release of proteolytic enzymes, ROS, and interactions with the endothelium of cerebral blood vessels. In addition, neutrophils accumulate in higher numbers in regions of HT and correlate with disruption of the basal lamina. In experimental stroke studies, depletion of circulating neutrophils reduces thrombolysis-related hemorrhage (130). In 846 patients treated with thrombolysis, an increased neutrophil to leukocyte (NIL) ratio was independently associated with HT (117).
Neutrophils produce neutrophil extracellular traps (NETs), which can promote clot formation associated with resistance to thrombolysis (131). In cultured human neutrophils, incubation with alteplase induced neutrophil degranulation and an increase in MMP-9 activity in a dose-dependent manner (132). This may elevate the number of activated neutrophils, thus exacerbating the detrimental effects of neutrophils on the BBB to promote HT (129, 131). In mice with circulating leukocytes from MMP-9 null bone marrow, BBB disruption was decreased (133).
Leukocytes enter the parenchyma and adhere to the cells largely through adhesion molecules ICAM-1, P-selectin, E-selectin, and VCAM-1 (134). Cytokines released by cerebral ischemia such as TNF, IL-1, and IFN-γ up-regulate ICAM-1 on both cerebral endothelial cells and leukocytes. Leukocytes adhering the endothelium can block erythrocytes in the microvasculature, leading to further injury (135). Activated leukocytes can also lead to secondary injury by producing proteases, MMPs, and ROS that can disrupt blood vessels. Leukocyte derived MMP-9s are mediators of early HT (1). Leukocyte adhesion and migration across the vasculature activates a number of signaling cascades (protein kinase C, focal adhesion kinase) that increase the BBB permeability. Once in the CNS, activated leukocytes release inflammatory cytokines at the site of injury such as TNF, IL-1β, and IL-6 (136).
In both rat (11) and rabbit models (12), treated with alteplase plus N-tert-butyl-α-phenylnitrone (PBN) vs. alteplase alone, PBN was found to attenuate alteplase-induced HT. PBN is a spin trap agent, targeting ROS from leukocytes.
Inflammatory Genes and HT
In both patient and animal studies, the peripheral immune response in ischemic stroke can be characterized by RNA expression in circulating leukocytes (128). Using leukocyte RNA obtained before thrombolysis treatment in 44 patients with acute stroke (33 with HT), microarray gene expression studies have identified 6 genes that predict the occurrence of HT with 80% sensitivity and 70% specificity. The six-gene panel in Table 8 highlights differences in inflammation and coagulation processes. Factors involved in HT of human stroke include a shift in growth factor-beta signaling involving SMAD4, INPP5D, and IRAK3, a disruption of coagulation factors V and VIII, and amphiregulin, a growth factor-beta that regulates MMP-9. Additionally, increased expression of AKAP7 in peripheral lymphocytes is linked to post stroke complications and BBB permeability (137). Genes associated with increased HT and mortality risk include two single nucleotide polymorphisms rs669 in α-2-macroglobulin (A2M) and rs1801020 in coagulation Factor XII (138). Factors associated with the efficacy of alteplase include low fibrin levels and polymorphism of Factor XIII V/V genotype (137).
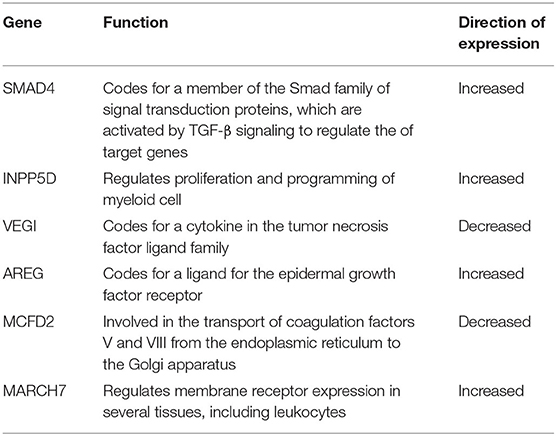
Table 8. Leukocyte genes associated with HT in patients with ischemic stroke.
The Genot-PA score, to predict HT in patients treated with alteplase, considered both genetics and clinical variables to establish patient risk (137). In the trial, 1,324 patients in a Spanish population, were risk-stratified and treated with either alteplase or a combination of alteplase and mechanical thrombectomy. The study was able to predict HT in patients treated with alteplase. External validation of the score is pending.
Conclusions
Inflammation contributes to BBB disruption and risk of HT, through an increase in ROS, MMPs and neutrophil activation. Clinical factors associated with inflammation and HT include hypertension, hyperglycemia, and age. Diabetes and hypertension lead to structural and functional changes of the neurovascular unit, alter collaterals, and elevate MMP-9, ROS, and proinflammatory cytokines (TNF, IL-1β, and IL-6). Advancing age is associated with a chronic elevation of inflammatory cytokine levels and changes in inflammatory response. These inflammatory markers provide insight to how clinical factors may increase the risk of HT. They may represent novel targets to reduce the risk of HT.
Further studies of the immune system and inflammation will be useful to better understand the risk of HT associated with reperfusion following thrombolysis and EVT and determine if modulation of the immune system could be useful to prevent HT. With increased use of reperfusion therapy and extension of eligibility criteria (e.g., inclusion of older patients, extended time windows), the importance of preventing HT will become more apparent. An improved safety profile with lower risk of HT could permit more widespread use of reperfusion therapy and greatly affect patients with stroke. Incorporating clinical features, genetics, biomarkers, and imaging to identify patients at risk for HT may help develop novel therapies to prevent HT in stroke.
Author Contributions
All authors meet the authorship criteria and contributed substantially to conception and design or acquisition of data (ES and GJ), or analysis and interpretation of data (all authors), drafted (ES) or revised (all authors) and gave final approval of the version to be published (all authors).
Funding
GJ received research grant support from CIHR, Heart and Stroke Foundation, University Hospital Foundation, Hypertension Canada, CFI, and NIH. JK-T was supported by a Faculty of Medicine and Dentistry Motyl Graduate Studentship in Cardiac Sciences and an Alberta Innovates Graduate Student Scholarship.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
A2M, a-2-macroglobulin; aICH, asymptomatic intracranial hemorrhage; AIS, acute ischemic stroke; Ang, angiopoietins; BBB, blood-brain barrier; c-Fn, cellular-fibronectin; DAMPs, damage associated molecular patterns; DM, diabetes mellitus; E-selectin, endothelial-leukocyte adhesion molecule 1; ECASS, European Cooperative Acute Stroke Study; eNOS, endothelial NO synthase; EVT, endovascular therapy; FGF, fibroblast growth factor; HgA1c, hemoglobin A1c; HI1, small petechial hemorrhagic infarction; HI2, confluent petechial hemorrhagic infarction; HIF-1, hypoxia-inducible factor 1; HMGB1, high mobility group box protein 1; HT, hemorrhagic transformation; ICAM-1, intercellular adhesion molecule-1; IFN, interferon; IGF-1, insulin-like growth factor 1; IL, interleukin; iNOS, inducible nitric oxide synthase; IV-tPA, intravenous tissue plasminogen activator; LPR, lipoprotein receptor; MCP-1, monocyte chemoattractant protein 1; MMP, matrix metalloproteinase; mRS, modified Rankin Scale; NETs, neutrophil extracellular traps; NFκB, nuclear factor-κB; NIHSS, National Institutes of Health Stroke Scale; NIL, neutrophil to leukocyte ratio; NLRP3, NOD-like receptor protein 3; NO, nitric oxide; NOX2, NADPH oxidase 2; NVU, neurovascular unit NXY-059, disodium 2, 4-sulphophenyl-N-tert-butylnitrone; ONOO−, peroxynitrite; PAI-1, plasminogen activator inhibitor-1; PAR-1, protease activated receptor 1; PDGF-CC, platelet derived growth factor-CC; PDGFRα, platelet derived growth factor receptor alpha; PH1, small parenchymal hemorrhage; PH2, large parenchymal hemorrhage; RAGE, receptor for advanced glycation end products; ROS, reactive oxygen species; RNS, reactive nitrogen species; SBP, systolic blood pressure; SHR, spontaneously hypertensive rat; sICAM, soluble intercellular adhesion molecule; sICH, symptomatic intracranial hemorrhage; TAFI, thrombin-activated fibrinolysis inhibitor; TGF-β, transforming growth factor-beta; TJP, tight junction proteins; TLR, toll like receptor; TNF, tumor necrosis factor; TNK, Tenecteplase; tPa, tissue plasminogen activator; TXNIP, thioredoxin interacting protein; VAP-1, vascular adhesion molecule-1; VCAM-1, vascular cell adhesion molecule 1; VEGF, vascular endothelial growth factor.
References
1. Jickling GC, Liu D, Stamova B, Ander BP, Zhan X, Lu A, et al. Hemorrhagic transformation after ischemic stroke in animals and humans. J Cereb Blood Flow Metab. (2014) 34:185–99. doi: 10.1038/jcbfm.2013.203
2. Kastrup A, Gröschel K, Ringer TM, Redecker C, Cordesmeyer R, Witte OW, et al. Early disruption of the bloodbrain barrier after thrombolytic therapy predicts hemorrhage in patients with acute stroke. Stroke. (2008) 39:2385–7. doi: 10.1161/STROKEAHA.107.505420
3. Fiorelli M, Bastianello S, von Kummer R, del Zoppo GJ, Larrue V, Lesaffre E, et al. Hemorrhagic transformation within 36 hours of a cerebral infarct: relationships with early clinical deterioration and 3-month outcome in the European Cooperative Acute Stroke Study I (ECASS I) cohort. Stroke. (1999) 30:2280–4. doi: 10.1161/01.STR.30.11.2280
4. Guenego A, Lecler A, Raymond J, Sabben C, Khoury N, Premat K, et al. Hemorrhagic transformation after stroke: inter- and intrarater agreement. Eur J Neurol. (2019) 26:476–82. doi: 10.1111/ene.13859
5. Kulik T, Kusano Y, Aronhime S, Sandler AL, Winn HR. Regulation of cerebral vasculature in normal and ischemic brain. Neuropharmacology. (2008) 55:281–8. doi: 10.1016/j.neuropharm.2008.04.017
6. Lyden, Pryor KE, Coffey CS, Cudkowicz M, Conwit R, Jadhav A, et al. Final results of the RHAPSODY trial: a multi-center, phase 2 trial using a continual reassessment method to determine the safety and tolerability of 3K3A-APC, a recombinant variant of human activated protein C, in combination with tissue plasminogen activator, mechanical thrombectomy or both in moderate to severe acute ischemic stroke. Ann Neurol. (2019) 85:125–36. doi: 10.1002/ana.25383
7. Sherman D, Bes A, J E. Use of anti-ICAM-1 therapy in ischemic stroke: results of the enlimomab acute stroke trial. Neurology. (2001) 57:1428–34. doi: 10.1212/WNL.57.8.1428
8. Wada T, Yasunaga H, Inokuchi R, Horiguchi H, Fushimi K, Matsubara T, et al. Effects of edaravone on early outcomes in acute ischemic stroke patients treated with recombinant tissue plasminogen activator. J Neurol Sci. (2014) 345:106–11. doi: 10.1016/j.jns.2014.07.018
9. Chen H, Guan B, Wang B, Pu H, Bai X, Chen X, et al. Glycyrrhizin prevents hemorrhagic transformation and improves neurological outcome in ischemic stroke with delayed thrombolysis through targeting peroxynitrite-mediated HMGB1 signaling. Transl Stroke Res. (2020) 11:967–82. doi: 10.1007/s12975-019-00772-1
10. Fagan SC, Waller JL, Nichols FT, Edwards DJ, Pettigrew LC, Clark WM, et al. Minocycline to improve neurologic outcome in stroke (MINOS): a dose-finding study. Stroke. (2010) 41:2283–7. doi: 10.1161/STROKEAHA.110.582601
11. Asahi M, Asahi K, Wang X, Lo EH. Reduction of tissue plasminogen activator-induced hemorrhage and brain injury by free radical spin trapping after embolic focal cerebral ischemia in rats. J Cereb Blood Flow Metab. (2000) 20:452–7. doi: 10.1097/00004647-200003000-00002
12. Lapchak PA, Chapman DF, Zivin JA. Pharmacological effects of the spin trap agents N-t-butyl-phenylnitrone (PBN) and 2,2,6, 6-tetramethylpiperidine-N-oxyl (TEMPO) in a rabbit thromboembolic stroke model: combination studies with the thrombolytic tissue plasminogen activator. Stroke. (2001) 32:147–53. doi: 10.1161/01.STR.32.1.147
13. Shuaib A, Lees KR, Lyden P, Grotta J, Davalos A, Davis SM, et al. NXY-059 for the treatment of acute ischemic stroke. N Engl J Med. (2007) 357:562–71. doi: 10.1056/NEJMoa070240
14. Kim, Lee KB, Park J-H, Sung SM, Oh K, Kim E-G, et al. Safety and efficacy of otaplimastat in patients with acute ischemic stroke requiring tPA (SAFE-TPA): a multicenter, randomized, double-blind, placebo-controlled phase 2 study. Ann Neurol. (2020) 87:233–45. doi: 10.1002/ana.25644
15. Whiteley WN, Slot KB, Fernandes P, Sandercock P, Wardlaw J. Risk factors for intracranial hemorrhage in acute ischemic stroke patients treated with recombinant tissue plasminogen activator: a systematic review and meta-analysis of 55 studies. Stroke. (2012) 43:2904–9. doi: 10.1161/STROKEAHA.112.665331
16. Kalinin MN, Khasanova DR, Ibatullin MM. The hemorrhagic transformation index score: a prediction tool in middle cerebral artery ischemic stroke. BMC Neurol. (2017) 17:177. doi: 10.1186/s12883-017-0958-3
17. Menon BK, Saver JL, Prabhakaran S, Reeves M, Liang L, Olson DM, et al. Risk score for intracranial hemorrhage in patients with acute ischemic stroke treated with intravenous tissue-type plasminogen activator. Stroke. (2012) 43:2293–9. doi: 10.1161/STROKEAHA.112.660415
18. Lou M, Safdar A, Mehdiratta M, Kumar S, Schlaug G, Caplan L, et al. The HAT score: a simple grading scale for predicting hemorrhage after thrombolysis. Neurology. (2008) 71:1417–23. doi: 10.1212/01.wnl.0000330297.58334.dd
19. Marsh EB, Llinas RH, Schneider ALC, Hillis AE, Lawrence E, Dziedzic P, et al. Predicting hemorrhagic transformation of acute ischemic stroke: prospective validation of the HeRS score. Medicine. (2016) 95:e2430. doi: 10.1097/MD.0000000000002430
20. Mazya M, Egido JA, Ford GA, Lees KR, Mikulik R, Toni D, et al. Predicting the risk of symptomatic intracerebral hemorrhage in ischemic stroke treated with intravenous alteplase: safe implementation of treatments in stroke (SITS) symptomatic intracerebral hemorrhage risk score. Stroke. (2012) 43:1524–31. doi: 10.1161/STROKEAHA.111.644815
21. Lyden PD. Stroke: Haemorrhage risk after thrombolysis–the SEDAN score. Nat Rev Neurol. (2012) 8:246–7. doi: 10.1038/nrneurol.2012.66
22. Kidwell CS, Saver JL, Carneado J, Sayre J, Starkman S, Duckwiler G, et al. Predictors of hemorrhagic transformation in patients receiving intra-arterial thrombolysis. Stroke. 33:717–24 (2002). doi: 10.1161/hs0302.104110
23. Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke t-PA Stroke Study Group. J Neurosurg Anesthesiol. (1996) 333:1581–7.
24. Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation. (2019) 139:e56–528.
25. Miller DJ, Simpson JR, Silver B, Silver B. Safety of thrombolysis in acute ischemic stroke: a review of complications, risk factors, and newer technologies. Neurohospitalist. (2011) 1:138–47. doi: 10.1177/1941875211408731
26. Sandercock P, Wardlaw JM, Lindley RI, Dennis M, Cohen G, Murray G, et al. The benefits and harms of intravenous thrombolysis with recombinant tissue plasminogen activator within 6 h of acute ischaemic stroke (the third international stroke trial [IST-3]): a randomised controlled trial. Lancet. (2012) 379:2352–63. doi: 10.1016/S0140-6736(12)60768-5
27. Whiteley WN, Emberson J, Lees KR, Blackwell L, Albers G, Bluhmki E, et al. Risk of intracerebral haemorrhage with alteplase after acute ischaemic stroke: a secondary analysis of an individual patient data meta-analysis. Lancet Neurol. (2016) 15:925–33. doi: 10.1016/S1474-4422(16)30076-X
28. Yaghi S, Willey JZ, Cucchiara B, Goldstein JN, Gonzales NR, Khatri P, et al. Treatment and outcome of hemorrhagic transformation after intravenous alteplase in acute ischemic stroke a scientific statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. (2017) 48:e343–61. doi: 10.1161/STR.0000000000000152
29. Nour M, Scalzo F, Liebeskind DS. Ischemia-reperfusion injury in stroke. Interv Neurol. (2013) 1:185–99. doi: 10.1159/000353125
30. Khatri R, McKinney AM, Swenson B, Janardhan V. Blood-brain barrier, reperfusion injury, and hemorrhagic transformation in acute ischemic stroke. Neurology. (2012) 79:S52–7. doi: 10.1212/WNL.0b013e3182697e70
31. Kaur J, Zhao Z, Klein GM, Lo EH, Buchan AM. The neurotoxicity of tissue plasminogen activator? J Cereb Blood Flow Metab. (2004) 24:945–63. doi: 10.1097/01.WCB.0000137868.50767.E8
32. Cuadrado E, Ortega L, Hernández-Guillamon M, Penalba A, Fernández-Cadenas I, Rosell A, et al. Tissue plasminogen activator (t-PA) promotes neutrophil degranulation and MMP-9 release. J Leukoc Biol. (2008) 84:207–14. doi: 10.1189/jlb.0907606
33. Lakhan SE, Kirchgessner A, Tepper D, Leonard A. Matrix metalloproteinases and blood-brain barrier disruption in acute ischemic stroke. Front Neurol. (2013) 4 APR:1–15. doi: 10.3389/fneur.2013.00032
34. Su EJ, Fredriksson L, Geyer M, Folestad E, Cale J, Andrae J, et al. Activation of PDGF-CC by tissue plasminogen activator impairs blood-brain barrier integrity during ischemic stroke. Nat Med. (2008) 14:731–7. doi: 10.1038/nm1787
35. Kyung-Pil P, Anna R, Christian F, Changhong X, Jean KW, Seoul L, et al. Plasma and brain matrix metalloproteinase-9 after acute focal cerebral ischemia in rats. Stroke. (2009) 40:2836–42. doi: 10.1161/STROKEAHA.109.554824
36. Huang X, MacIsaac R, Thompson JL, Levin B, Buchsbaum R, Haley ECJ, et al. Tenecteplase versus alteplase in stroke thrombolysis: an individual patient data meta-analysis of randomized controlled trials. Int J Stroke. (2016) 11:534–43. doi: 10.1177/1747493016641112
37. Campbell BCV, Mitchell PJ, Churilov L, Yassi N, Kleinig TJ, Dowling RJ, et al. Tenecteplase versus alteplase before thrombectomy for ischemic stroke. N Engl J Med. (2018) 378:1573–82. doi: 10.1056/NEJMoa1716405
38. Adivitiya KY. The evolution of recombinant thrombolytics: current status and future directions. Bioengineered. (2017) 8:331–58. doi: 10.1080/21655979.2016.1229718
39. Gürsoy-Özdemir Y, Can A, Dalkara T. Reperfusion-induced oxidative/nitrative injury to neurovascular unit after focal cerebral ischemia. Stroke. (2004) 35:1449–53. doi: 10.1161/01.STR.0000126044.83777.f4
40. Huang J, Liu B, Yang C, Chen H, Eunice D, Yuan Z. Acute hyperglycemia worsens ischemic stroke-induced brain damage via high mobility group box-1 in rats. Brain Res. (2013) 1535:148–155. doi: 10.1016/j.brainres.2013.08.057
41. Qiu J, Xu J, Zheng Y, Wei Y, Zhu X, Eng HL, et al. High-mobility group box 1 promotes metalloproteinase-9 upregulation through toll-like receptor 4 after cerebral ischemia. Stroke. (2010) 41:2077–82. doi: 10.1161/STROKEAHA.110.590463
42. Martini SR, Kent TA. Hyperglycemia in acute ischemic stroke: a vascular perspective. J Cereb Blood Flow Metab. (2006) 27:435–51. doi: 10.1038/sj.jcbfm.9600355
43. Harari OA, Liao JK. NF-κB and innate immunity in ischemic stroke. Ann N Y Acad Sci. (2010) 1207:32–40. doi: 10.1111/j.1749-6632.2010.05735.x
44. Kong L, Ma Y, Wang Z, Liu N, Ma G, Liu C, et al. Inhibition of hypoxia inducible factor 1 by YC-1 attenuates tissue plasminogen activator induced hemorrhagic transformation by suppressing HMGB1/TLR4/NF-κB mediated neutrophil infiltration in thromboembolic stroke rats. Int Immunopharmacol. (2021) 94:107507. doi: 10.1016/j.intimp.2021.107507
45. Goyal M, Menon BK, van Zwam WH, Dippel DWJ, Mitchell PJ, Demchuk AM, et al. Endovascular thrombectomy after large-vessel ischaemic stroke: a meta-analysis of individual patient data from five randomised trials. Lancet. (2016) 387:1723–31. doi: 10.1016/S0140-6736(16)00163-X
46. Campbell BC, Hill MD, Rubiera M, Menon BK, Demchuk A, Donnan GA, et al. Safety and efficacy of solitaire stent thrombectomy. Stroke. (2016) 47:798–806. doi: 10.1161/STROKEAHA.115.012360
47. Yang P, Zhang Y, Zhang L, Zhang Y, Treurniet KM, Chen W, et al. Endovascular thrombectomy with or without intravenous alteplase in acute stroke. N Engl J Med. (2020) 382:1981–93. doi: 10.1056/NEJMoa2001123
48. Chalos V, LeCouffe NE, Uyttenboogaart M, Lingsma HF, Mulder MJHL, Venema E, et al. Endovascular treatment with or without prior intravenous alteplase for acute ischemic stroke. J Am Heart Assoc. (2019) 8:e011592. doi: 10.1161/JAHA.118.011592
49. Langel C, Popovic KS. Infarct-core CT perfusion parameters in predicting post-thrombolysis hemorrhagic transformation of acute ischemic stroke. Radiol Oncol. (2018) 53:25–30. doi: 10.2478/raon-2018-0048
50. Aoki T, Sumii T, Mori T, Wang X, Lo EH. Blood-brain barrier disruption and matrix metalloproteinase-9 expression during reperfusion injury. Stroke. (2002) 33:2711–7. doi: 10.1161/01.STR.0000033932.34467.97
51. Johnston KC, Bruno A, Pauls Q, Hall CE, Barrett KM, Barsan W, et al. Intensive vs standard treatment of hyperglycemia and functional outcome in patients with acute ischemic stroke: the SHINE randomized clinical trial. J Am Med Assoc. (2019) 322:326–35. doi: 10.1001/jama.2019.9346
52. Garg R, Chaudhuri A, Munschauer F, Dandona P. Hyperglycemia, insulin, and acute ischemic stroke: a mechanistic justification for a trial of insulin infusion therapy. Stroke. (2006) 37:267–73. doi: 10.1161/01.STR.0000195175.29487.30
53. Williams LS, Rotich J, Qi R, Fineberg N, Espay A, Bruno A, et al. Effects of admission hyperglycemia on mortality and costs in acute ischemic stroke. Neurology. (2002) 59:67–71. doi: 10.1212/WNL.59.1.67
54. Ritter L, Davidson L, Henry M, Davis-Gorman G, Morrison H, Frye JB, et al. Exaggerated neutrophil-mediated reperfusion injury after ischemic stroke in a rodent model of type 2 diabetes. Microcirculation. (2011) 18:552–61. doi: 10.1111/j.1549-8719.2011.00115.x
55. Elgebaly MM, Ogbi S, Li W, Mezzetti EM, Prakash R, Johnson MH, et al. Neurovascular injury in acute hyperglycemia and diabetes: a comparative analysis in experimental stroke. Transl Stroke Res. (2011) 2:391–8. doi: 10.1007/s12975-011-0083-3
56. Chen H, Guan B, Chen S, Yang D, Shen J. Peroxynitrite activates NLRP3 inflammasome and contributes to hemorrhagic transformation and poor outcome in ischemic stroke with hyperglycemia. Free Radic Biol Med. (2021) 165:171–83. doi: 10.1016/j.freeradbiomed.2021.01.030
57. Zhang G, He M, Xu Y, Li X, Cai Z, Guo Z, et al. Hemoglobin A1c predicts hemorrhagic transformation and poor outcomes after acute anterior stroke. Eur J Neurol. (2018) 25:1432.e122. doi: 10.1111/ene.13747
58. Powers WJ, Alejandro R, Ackerson T, Adeoye OM, Bambakidis NC, Becker K, et al. 2018 guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. (2018) 49:e46–99. doi: 10.1161/STR.0000000000000172
59. Desilles JP, Meseguer E, Labreuche J, Lapergue B, Sirimarco G, Gonzalez-Valcarcel J, et al. Diabetes mellitus, admission glucose, and outcomes after stroke thrombolysis: a registry and systematic review. Stroke. (2013) 44:1915–23. doi: 10.1161/STROKEAHA.111.000813
60. Dokken BB. The pathophysiology of cardiovascular disease and diabetes: beyond blood pressure and lipids. Diabetes Spectr. (2008) 21:160–5. doi: 10.2337/diaspect.21.3.160
61. Hong P, Gu R-N, Li F-X, Xiong X-X, Liang W-B, You Z-J, et al. NLRP3 inflammasome as a potential treatment in ischemic stroke concomitant with diabetes. J Neuroinflam. (2019) 16:121. doi: 10.1186/s12974-019-1498-0
62. Desilles JP, Syvannarath V, Ollivier V, Journé C, Delbosc S, Ducroux C, et al. Exacerbation of thromboinflammation by hyperglycemia precipitates cerebral infarct growth and hemorrhagic transformation. Stroke. (2017) 48:1932–40. doi: 10.1161/STROKEAHA.117.017080
63. Won SJ, Tang XN, Suh SW, Yenari MA, Swanson RA. Hyperglycemia promotes tissue plasminogen activator-induced hemorrhage by increasing superoxide production. Ann Neurol. (2011) 70:583–90. doi: 10.1002/ana.22538
64. Guo Z, Yu S, Chen X, Zheng P, Hu T, Duan Z, et al. Suppression of NLRP3 attenuates hemorrhagic transformation after delayed rtPA treatment in thromboembolic stroke rats: involvement of neutrophil recruitment. Brain Res Bull. (2018) 137:229–40. doi: 10.1016/j.brainresbull.2017.12.009
65. Price TO, Eranki V, Banks WA, Ercal N, Shah GN. Topiramate treatment protects blood-brain barrier pericytes from hyperglycemia-induced oxidative damage in diabetic mice. Endocrinology. (2012) 153:362–72. doi: 10.1210/en.2011-1638
66. Yoo DY, Yim HS, Jung HY, Nam SM, Kim JW, Choi JH, et al. Chronic type 2 diabetes reduces the integrity of the blood-brain barrier by reducing tight junction proteins in the hippocampus. J Vet Med Sci. (2016) 78:957–62. doi: 10.1292/jvms.15-0589
67. Tjärnlund-Wolf A, Brogren H, Lo EH, Wang X. Plasminogen activator inhibitor-1 and thrombotic cerebrovascular diseases. Stroke. (2012) 43:2833–9. doi: 10.1161/STROKEAHA.111.622217
68. Dohgu S, Takata F, Matsumoto J, Oda M, Harada E, Watanabe T, et al. Autocrine and paracrine up-regulation of blood–brain barrier function by plasminogen activator inhibitor-1. Microvasc Res. (2011) 81:103–7. doi: 10.1016/j.mvr.2010.10.004
69. Ribo M, Montaner J, Molina CA, Arenillas JF, Santamarina E, Quintana M, et al. Admission fibrinolytic profile is associated with symptomatic hemorrhagic transformation in stroke patients treated with tissue plasminogen activator. Stroke. (2004) 35:2123–7. doi: 10.1161/01.STR.0000137608.73660.4c
70. Shukla V, Shakya AK, Perez-Pinzon MA, Dave KR. Cerebral ischemic damage in diabetes: an inflammatory perspective. J Neuroinflam. (2017) 14:21. doi: 10.1186/s12974-016-0774-5
71. Couret D, Bourane S, Catan A, Nativel B, Planesse C, Dorsemans AC, et al. A hemorrhagic transformation model of mechanical stroke therapy with acute hyperglycemia in mice. J Comp Neurol. (2018) 526:1006–16. doi: 10.1002/cne.24386
72. Ismael S, Nasoohi S, Yoo A, Ahmed HA, Ishrat T. Tissue plasminogen activator promotes TXNIP-NLRP3 inflammasome activation after hyperglycemic stroke in mice. Mol Neurobiol. (2020) 57:2495–508. doi: 10.1007/s12035-020-01893-7
73. Pandolfi A, Giaccari A, Cilli C, Alberta MM, Morviducci L, De Filippis EA, et al. Acute hyperglycemia and acute hyperinsulinemia decrease plasma fibrinolytic activity and increase plasminogen activator inhibitor type 1 in the rat. Acta Diabetol. (2001) 38:71–6. doi: 10.1007/s005920170016
74. Ribo M, Molina C, Montaner J, Rubiera M, Delgado-Mederos R, Arenillas JF, et al. Acute hyperglycemia state is associated with lower tPA-induced recanalization rates in stroke patients. Stroke. (2005) 36:1705–9. doi: 10.1161/01.STR.0000173161.05453.90.9f
75. Ning R, Chopp M, Yan T, Zacharek A, Zhang C, Roberts C, et al. Tissue plasminogen activator treatment of stroke in type-1 diabetes rats. Neuroscience. (2012) 222:326–32. doi: 10.1016/j.neuroscience.2012.07.018
76. Schneider DJ, Sobel BE. PAI-1 and diabetes: a journey from the bench to the bedside. Diabetes Care. (2012) 35:1961–7. doi: 10.2337/dc12-0638
77. Hafez S, Coucha M, Bruno A, Fagan SC, Ergul A. Hyperglycemia, acute ischemic stroke, and thrombolytic therapy. Transl Stroke Res. (2014) 5:442–53. doi: 10.1007/s12975-014-0336-z
78. Gray CS, Hildreth AJ, Sandercock PA, O'Connell JE, Johnston DE, Cartlidge NE, et al. Glucose-potassium-insulin infusions in the management of post-stroke hyperglycaemia: the UK glucose insulin in stroke trial (GIST-UK). Lancet Neurol. (2007) 6:397–406. doi: 10.1016/S1474-4422(07)70080-7
79. Palaiodimou L, Lioutas V-A, Lambadiari V, Paraskevas GP, Voumvourakis K, Tsivgoulis G. Glycemia management in acute ischemic stroke: current concepts and novel therapeutic targets. Postgrad Med. (2019) 131:423–37. doi: 10.1080/00325481.2019.1651206
80. Pires PW, Dams Ramos CM, Matin N, Dorrance AM. The effects of hypertension on the cerebral circulation. Am J Physiol Circ Physiol. (2013) 304:H1598–614. doi: 10.1152/ajpheart.00490.2012
81. Castro MM, Tanus-Santos JE, Gerlach RF. Matrix metalloproteinases: targets for doxycycline to prevent the vascular alterations of hypertension. Pharmacol Res. (2011) 64:567–72. doi: 10.1016/j.phrs.2011.04.002
82. Faraco G, Sugiyama Y, Lane D, Garcia-Bonilla L, Chang H, Santisteban MM, et al. Perivascular macrophages mediate the neurovascular and cognitive dysfunction associated with hypertension. J Clin Invest. (2016) 126:4674–89. doi: 10.1172/JCI86950
83. Qureshi AI. Acute hypertensive response in patients with stroke. Circulation. (2008) 118:176–87. doi: 10.1161/CIRCULATIONAHA.107.723874
84. Mohammadi MT, Dehghani GA. Acute hypertension induces brain injury and blood-brain barrier disruption through reduction of claudins mRNA expression in rat. Pathol Res Pract. (2014) 210:985–90. doi: 10.1016/j.prp.2014.05.007
85. Ueno M, Sakamoto H, Liao Y-J, Onodera M, Huang C-L, Miyanaka H, et al. Blood-brain barrier disruption in the hypothalamus of young adult spontaneously hypertensive rats. Histochem Cell Biol. (2004) 122:131–7. doi: 10.1007/s00418-004-0684-y
86. Flamant M, Placier S, Dubroca C, Esposito B, Lopes I, Chatziantoniou C, et al. Role of matrix metalloproteinases in early hypertensive vascular remodeling. Hypertens. (2007) 50:212–8. doi: 10.1161/HYPERTENSIONAHA.107.089631
87. Campbell, Christensen S, Tress BM, Churilov L, Desmond PM, Parsons MW, et al. Failure of collateral blood flow is associated with infarct growth in ischemic stroke. J Cereb Blood Flow Metab. (2013) 33:1168–72. doi: 10.1038/jcbfm.2013.77
88. Zhang, Yang Y, Sun H, Xing Y. Hemorrhagic transformation after cerebral infarction: current concepts and challenges. Ann Transl Med. (2014) 2:81. doi: 10.3978/j.issn.2305-5839.2014.08.08
89. Fagan SC, Bowes MP, Lyden PD, Zivin JA. Acute hypertension promotes hemorrhagic transformation in a rabbit embolic stroke model: effect of labetalol. Exp Neurol. (1998) 150:153–8. doi: 10.1006/exnr.1997.6756
90. Tejima E, Katayama Y, Suzuki Y, Kano T, Lo EH. Hemorrhagic transformation after fibrinolysis with tissue plasminogen activator. Stroke. (2001) 32:1336–40. doi: 10.1161/01.STR.32.6.1336
91. Powers WJ, Rabinstein AA, Ackerson T, Adeoye OM, Bambakidis NC, Becker K, et al. Guidelines for the early management of patients with acute ischemic stroke: 2019 update to the 2018 guidelines for the early management of acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke. Stroke. (2019) 50:e344–418. doi: 10.1161/STR.0000000000000211
92. Ahmed N, Wahlgren N, Brainin M, Castillo J, Ford GA, Kaste M, et al. Relationship of blood pressure, antihypertensive therapy, and outcome in ischemic stroke treated with intravenous thrombolysis: retrospective analysis from safe implementation of thrombolysis in stroke-international stroke thrombolysis register (SITS-IST). Stroke. (2009) 40:2442–9. doi: 10.1161/STROKEAHA.109.548602
93. Wu W, Huo X, Zhao X, Liao X, Wang C, Pan Y, et al. Relationship between blood pressure and outcomes in acute ischemic stroke patients administered lytic medication in the TIMS-China study. PLoS ONE. (2016) 11:e0144260. doi: 10.1371/journal.pone.0144260
94. Liu K, Yan S, Zhang S, Guo Y, Lou M. Systolic blood pressure variability is associated with severe hemorrhagic transformation in the early stage after thrombolysis. Transl Stroke Res. (2016) 7:186–91. doi: 10.1007/s12975-016-0458-6
95. Perini F, De Boni A, Marcon M, Bolgan I, Pellizzari M, Dionisio L. Systolic blood pressure contributes to intracerebral haemorrhage after thrombolysis for ischemic stroke. J Neurol Sci. (2010) 297:52–4. doi: 10.1016/j.jns.2010.06.025
96. Mulder MJHL, Ergezen S, Lingsma HF, Berkhemer OA, Fransen PSS, Beumer D, et al. Baseline blood pressure effect on the benefit and safety of intra-arterial treatment in MR CLEAN (multicenter randomized clinical trial of endovascular treatment of acute ischemic stroke in the Netherlands). Stroke. (2017) 48:1869–76. doi: 10.1161/STROKEAHA.116.016225
97. Matusevicius M, Cooray C, Bottai M, Mazya M, Tsivgoulis G, Nunes AP, et al. Blood pressure after endovascular thrombectomy: modeling for outcomes based on recanalization status. Stroke. (2020) 51:519–25. doi: 10.1161/STROKEAHA.119.026914
98. Anderson CS, Huang Y, Lindley RI, Chen X, Arima H, Chen G, et al. Intensive blood pressure reduction with intravenous thrombolysis therapy for acute ischaemic stroke (ENCHANTED): an international, randomised, open-label, blinded-endpoint, phase 3 trial. Lancet. (2019) 393:877–88. doi: 10.1016/S0140-6736(19)30038-8
99. Tikhonoff V, Zhang H, Richart T, Staessen JA. Blood pressure as a prognostic factor after acute stroke. Lancet Neurol. (2009) 8:938–48. doi: 10.1016/S1474-4422(09)70184-X
100. Bath PM, Scutt P, Anderson CS, Appleton JP, Berge E, Cala L, et al. Prehospital transdermal glyceryl trinitrate in patients with ultra-acute presumed stroke (RIGHT-2): an ambulance-based, randomised, sham-controlled, blinded, phase 3 trial. Lancet. (2019) 393:1009–20. doi: 10.1016/S0140-6736(19)30194-1
101. Zonneveld TP, Algra A, Dippel DWJ, Kappelle LJ, van Oostenbrugge RJ, Roos YBWEM, et al. The ThRombolysis in UnconTrolled Hypertension (TRUTH) protocol: an observational study on treatment strategy of elevated blood pressure in stroke patients eligible for IVT. BMC Neurol. (2015) 15:241. doi: 10.1186/s12883-015-0493-z
102. Mazighi M, Richard S, Lapergue B, Sibon I, Gory B, Berge J, et al. Safety and efficacy of intensive blood pressure lowering after successful endovascular therapy in acute ischaemic stroke (BP-TARGET): a multicentre, open-label, randomised controlled trial. Lancet Neurol (2021).
103. Ziai WC, Al-Kawaz M. Blood pressure management after endovascular therapy. Lancet Neurol. (2021) 20:248–49. doi: 10.1016/S1474-4422(21)00058-2
104. Sohrabji F, Bake S, Lewis DK. Age-related changes in brain support cells: implications for stroke severity. Neurochem Int. (2013) 63:291–301. doi: 10.1016/j.neuint.2013.06.013
105. Erickson MA, Banks WA. Age-associated changes in the immune system and blood−brain barrier functions. Int J Mol Sci. (2019) 20:1632. doi: 10.3390/ijms20071632
106. Ritzel RM, Lai Y-J, Crapser JD, Patel AR, Schrecengost A, Grenier JM, et al. Aging alters the immunological response to ischemic stroke. Acta Neuropathol. (2018) 136:89–110. doi: 10.1007/s00401-018-1859-2
107. Manwani B, Liu F, Scranton V, Hammond MD, Sansing LH, McCullough LD. Differential effects of aging and sex on stroke induced inflammation across the lifespan. Exp Neurol. (2013) 249:120–31. doi: 10.1016/j.expneurol.2013.08.011
108. Emberson J, Lees KR, Lyden P, Blackwell L, Albers G, Bluhmki E, et al. Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: a meta-analysis of individual patient data from randomised trials. Lancet. (2014) 384:1929–35. doi: 10.1016/S0140-6736(14)60584-5
109. Costello CA, Campbell BC V, Perez de la Ossa N, Zheng TH, Sherwin JC, Weir L, et al. Age over 80 years is not associated with increased hemorrhagic transformation after stroke thrombolysis. J Clin Neurosci. (2012) 19:360–3. doi: 10.1016/j.jocn.2011.08.014
110. Weiskopf D, Weinberger B, Grubeck-Loebenstein B. The aging of the immune system. Transpl Int. (2009) 22:1041–50. doi: 10.1111/j.1432-2277.2009.00927.x
111. Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. (2018) 15:505–22. doi: 10.1038/s41569-018-0064-2
112. Shaw AC, Joshi S, Greenwood H, Panda A, Lord JM. Aging of the innate immune system. Curr Opin Immunol. (2010) 22:507–13. doi: 10.1016/j.coi.2010.05.003
113. Chan SL, Bishop N, Li Z, Cipolla MJ. Inhibition of PAI (plasminogen activator inhibitor)-1 improves brain collateral perfusion and injury after acute ischemic stroke in aged hypertensive rats. Stroke. (2018) 49:1969–76. doi: 10.1161/STROKEAHA.118.022056
114. Petcu EB, Smith RA, Miroiu RI, Opris MM. Angiogenesis in old-aged subjects after ischemic stroke: a cautionary note for investigators. J Angiogenes Res. (2010) 2:26. doi: 10.1186/2040-2384-2-26
115. Norden DM, Godbout JP. Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol. (2013) 39:19–34. doi: 10.1111/j.1365-2990.2012.01306.x
116. Cui H, Kong Y, Zhang H. Oxidative stress, mitochondrial dysfunction, and aging. J Signal Transduct. (2012) 2012:646354. doi: 10.1155/2012/646354
117. Maestrini I, Strbian D, Gautier S, Haapaniemi E, Moulin S, Sairanen T, et al. Higher neutrophil counts before thrombolysis for cerebral ischemia predict worse outcomes. Neurology. (2015) 85:1408–16. doi: 10.1212/WNL.0000000000002029
118. Ucuzian AA, Gassman AA, East AT, Greisler HP. Molecular mediators of angiogenesis. J Burn Care Res. (2010) 31:158–75. doi: 10.1097/BCR.0b013e3181c7ed82
119. Kanazawa M, Igarashi H, Kawamura K, Takahashi T, Kakita A, Takahashi H, et al. Inhibition of VEGF signaling pathway attenuates hemorrhage after tPA treatment. J Cereb Blood Flow Metab. (2011) 31:1461–74. doi: 10.1038/jcbfm.2011.9
120. Faber JE, Zhang H, Lassance-Soares RM, Prabhakar P, Najafi AH, Burnett MS, et al. Aging causes collateral rarefaction and increased severity of ischemic injury in multiple tissues. Arterioscler Thromb Vasc Biol. (2011) 31:1748–56. doi: 10.1161/ATVBAHA.111.227314
121. Bang OY, Saver JL, Kim SJ, Kim G-M, Chung C-S, Ovbiagele B, et al. Collateral flow averts hemorrhagic transformation after endovascular therapy for acute ischemic stroke. Stroke. (2011) 42:2235–9. doi: 10.1161/STROKEAHA.110.604603
122. Ng GJL, Quek AML, Cheung C, Arumugam T V, Seet RCS. Stroke biomarkers in clinical practice: a critical appraisal. Neurochem Int. (2017) 107:11–22. doi: 10.1016/j.neuint.2017.01.005
123. Lu G, He Q, Shen Y, Cao F. Potential biomarkers for predicting hemorrhagic transformation of ischemic stroke. Int J Neurosci. (2018) 128:79–89. doi: 10.1080/00207454.2017.1349766
124. Piccardi B, Biagini S, Iovene V, Palumbo V. Blood biomarkers of parenchymal damage in ischemic stroke patients treated with revascularization therapies. Biomark Insights. (2019) 14:1177271919888225. doi: 10.1177/1177271919888225
125. Castellanos M, Leira R, Serena J, Blanco M, Pedraza S, Castillo J, et al. Plasma cellular-fibronectin concentration predicts hemorrhagic transformation after thrombolytic therapy in acute ischemic stroke. Stroke. (2004) 35:1671–6. doi: 10.1161/01.STR.0000131656.47979.39
126. Bernstein JE, Savla P, Dong F, Zampella B, Wiginton JG 4th, Miulli DE, et al. Inflammatory markers and severity of intracerebral hemorrhage. Cureus. (2018) 10:e3529. doi: 10.7759/cureus.3529
127. Shichita T, Ito M, Yoshimura A. Post-ischemic inflammation regulates neural damage and protection. Front Cell Neurosci. (2014) 8:1–8. doi: 10.3389/fncel.2014.00319
128. Jickling GC, Ander BP, Stamova B, Zhan X, Liu D, Rothstein L, et al. RNA in blood is altered prior to hemorrhagic transformation in ischemic stroke. Ann Neurol. (2013) 74:232–40. doi: 10.1002/ana.23883
129. Planas AM. Role of immune cells migrating to the ischemic brain. Stroke. (2018) 49:2261–7. doi: 10.1161/STROKEAHA.118.021474
130. Yenari MK MA. Inflammatory responses in brain ischemia. Curr Med Chem. (2015) 22:1258–77. doi: 10.2174/0929867322666150209154036
131. Ouk T, Potey C, Maestrini I, Petrault M, Mendyk A-M, Leys D, et al. Neutrophils in tPA-induced hemorrhagic transformations: main culprit, accomplice or innocent bystander? Pharmacol Ther. (2019) 194:73–83. doi: 10.1016/j.pharmthera.2018.09.005
132. Carbone F, Vuilleumier N, Bertolotto M, Burger F, Galan K, Roversi G, et al. Treatment with recombinant tissue plasminogen activator (r-TPA) induces neutrophil degranulation in vitro via defined pathways. Vascul Pharmacol. (2015) 64:16–27. doi: 10.1016/j.vph.2014.11.007
133. Wang G, Guo Q, Hossain M, Fazio V, Zeynalov E, Janigro D, et al. Bone marrow-derived cells are the major source of MMP-9 contributing to blood–brain barrier dysfunction and infarct formation after ischemic stroke in mice. Brain Res. (2009) 1294:183–92. doi: 10.1016/j.brainres.2009.07.070
134. Ramiro L, Simats A, García-Berrocoso T, Montaner J. Inflammatory molecules might become both biomarkers and therapeutic targets for stroke management. Ther Adv Neurol Disord. (2018) 11:1756286418789340. doi: 10.1177/1756286418789340
135. Jayaraj RL, Azimullah S, Beiram R, Jalal FY, Rosenberg GA. Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflam. (2019) 16:142. doi: 10.1186/s12974-019-1516-2
136. Kim JY, Park J, Chang JY, Kim S-H, Lee JE. Inflammation after ischemic stroke: the role of leukocytes and glial cells. Exp Neurobiol. (2016) 25:241–51. doi: 10.5607/en.2016.25.5.241
137. O'Connell GC, Treadway MB, Petrone AB, Tennant CS, Lucke-Wold N, Chantler PD, et al. Peripheral blood AKAP7 expression as an early marker for lymphocyte-mediated post-stroke blood brain barrier disruption. Sci Rep. (2017) 7:1172. doi: 10.1038/s41598-017-01178-5
Keywords: ischemic stroke, hemorrhagic transformation, inflammation, hypertension, diabetes, aging, reperfusion therapy
Citation: Spronk E, Sykes G, Falcione S, Munsterman D, Joy T, Kamtchum-Tatuene J and Jickling GC (2021) Hemorrhagic Transformation in Ischemic Stroke and the Role of Inflammation. Front. Neurol. 12:661955. doi: 10.3389/fneur.2021.661955
Received: 31 January 2021; Accepted: 29 March 2021;
Published: 14 May 2021.
Edited by:
Nishant K. Mishra, University of California, Los Angeles, United StatesReviewed by:
Georgios Tsivgoulis, National and Kapodistrian University of Athens, GreeceHansen Chen, Stanford University, United States
Copyright © 2021 Spronk, Sykes, Falcione, Munsterman, Joy, Kamtchum-Tatuene and Jickling. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena Spronk, ZXNwcm9ua0B1YWxiZXJ0YS5jYQ==