 Adina N. MacMahon Copas
Adina N. MacMahon Copas Sarah F. McComish
Sarah F. McComish Jean M. Fletcher
Jean M. Fletcher Maeve A. Caldwell
Maeve A. Caldwell- 1Department of Physiology, School of Medicine, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland
- 2Trinity College Institute of Neuroscience, Trinity College Dublin, Dublin, Ireland
- 3School of Biochemistry and Immunology, Trinity Biomedical Sciences Institute, Trinity College Dublin, Dublin, Ireland
Parkinson's disease (PD), the second most common neurodegenerative disease, is characterised by the motor symptoms of bradykinesia, rigidity and resting tremor and non-motor symptoms of sleep disturbances, constipation, and depression. Pathological hallmarks include neuroinflammation, degeneration of dopaminergic neurons in the substantia nigra pars compacta, and accumulation of misfolded α-synuclein proteins as intra-cytoplasmic Lewy bodies and neurites. Microglia and astrocytes are essential to maintaining homeostasis within the central nervous system (CNS), including providing protection through the process of gliosis. However, dysregulation of glial cells results in disruption of homeostasis leading to a chronic pro-inflammatory, deleterious environment, implicated in numerous CNS diseases. Recent evidence has demonstrated a role for peripheral immune cells, in particular T lymphocytes in the pathogenesis of PD. These cells infiltrate the CNS, and accumulate in the substantia nigra, where they secrete pro-inflammatory cytokines, stimulate surrounding immune cells, and induce dopaminergic neuronal cell death. Indeed, a greater understanding of the integrated network of communication that exists between glial cells and peripheral immune cells may increase our understanding of disease pathogenesis and hence provide novel therapeutic approaches.
Introduction
Parkinson's disease (PD) is the second most common neurodegenerative disease affecting 1–2% of the population over the age of 65 (1) and it is estimated that the number of cases will exceed 12 million individuals in 2040 (2). PD is characterised by the degeneration of dopamine neurons in the substantia nigra (SN) of the midbrain, with concomitant loss of their axons that project to the striatum along the nigrostriatal pathway. This results in loss of the neurotransmitter dopamine which leads to the primary motor symptoms of PD, which were first described by James Parkinson in 1817 as a heterogeneous manifestation (3). These include bradykinesia, ataxia, tremor, rigidity, and postural instability which present themselves clinically once the levels of striatal dopamine decrease by 70% (4). Another key pathological feature of PD is the presence of protein inclusions known as Lewy bodies (5). The protein α-synuclein (α-syn) is a major component of Lewy bodies (6) and its mutant forms can cause familial PD (7–10).
In the central nervous system (CNS), the continuous interactions of neurons, glia, and the microenvironment are key for the maintenance of neural homeostasis and failures in this homeostatic state leads to neurodegenerative conditions such as PD. In recent years the role of inflammatory processes in the death of dopamine neurons has come to the fore and is now considered vital to this process (11–13). Gliosis is a typical pathological feature of many neurodegenerative diseases and emerging evidence indicates that sustained activation of microglia and astrocytes is central to dopaminergic degeneration in PD (14). Indeed, activation of microglia in people with PD was described for the first time in 1988 (15), and this phenomenon has also been described in animal models (16, 17). Wilson et al. have recently described a PET ligand 11C-BU99088 which is expressed on reactive astroglia, and as such, was reflective of astroglial pathology in people with PD and demonstrated a role for astroglia in the initiation and progression of PD (18). Astrocyte reactivity was also detected in the SN of patients with this condition [for review see (19)] and post-mortem studies of nigral tissue homogenates revealed multiple alterations in biochemical parameters attributed to astrocyte dysfunction including a global reduction of glutathione levels, mitochondrial damage, and accumulation of extracellular toxins (20). In addition, clinical and basic research have revealed a role for both the innate and adaptive immune system in PD [for review see (21, 22)]. In fact, the available data suggests that in PD there is a response not only by glial cells but also by peripheral immune cells, suggesting that an interplay between these cell types contributes to pathophysiology. We will review the role of astrocytes and microglia in PD, taking into account the emerging role of peripheral immune cells and its implications for disease pathogenesis.
Astrocytes and Their Role in Parkinson's Disease
Astrocytes are the most abundant non-neuronal cell type in the vertebrate nervous system (23) with the critical task of maintaining structural support and homeostasis. Their roles include provision of metabolic support, encapsulation of neuronal synapses (24), promotion of synaptogenesis (25), and control of the permeability of the blood brain barrier (BBB) (26). Classically they are recognised for their ability to secrete a number of neurotrophic factors such as glial derived neurotrophic factor (GDNF) and mesencephalic astrocyte derived neurotrophic factor (MANF); both of which have been shown to offer a degree of neuroprotection to dopamine neurons both in vitro and in vivo (27–29). However, in addition to this there is a growing appreciation of the role astrocytes play in neuroinflammation (30) in many neurodegenerative conditions including PD. Recently it has been shown that astrocytes become reactive in response to activated microglial secreted signals such as IL-1α, TNF-α, and C1q and as such adopt a pro-inflammatory phenotype (31, 32). Indeed, this astrocyte phenotype has been shown to exist in the post-mortem brain tissue of people with PD (31). Furthermore, astrocytes can also adopt a pro-inflammatory phenotype by endocytosis of α-syn released by neurons; they secrete cytokines, IL-1α, IL-1β, and IL-6 which are correlated to α-syn load (33). Moreover, α-syn accumulation in human astrocytes in vitro resulted in severe cellular stress which included mitochondrial, lysosomal, and endoplasmic reticulum deficiencies (34, 35). Indeed, these astrocytes responded to this stress by sending out nanotubes, which behaved like tunnels, and enabled the transfer of intracellular α-syn inclusions to nearby cells, indicating that astrocytes are critically important in the pathogenesis of PD (35). Interestingly, Yun et al., demonstrated that NLY101, a GLP-1R agonist is neuroprotective in the pre-formed α-syn fibril model of PD. It acts to prevent astrocyte stimulation by activated microglia and in so doing protects dopamine neurons and prevents behavioural deficits (36). Taken together, these studies would suggest that astrocyte dysfunction is a very strong contributor to the pathogenesis of PD.
Impact of Parkinson's Disease Related Genetics on Astrocyte Function
There are monogenic mutations identified in 20 genes that have been implicated in the pathogenesis of PD (37). Interestingly, a study by Zhang et al. (38) compared the transcriptome of human astrocytes to neurons, and found upregulation of some of these monogenic mutations in astrocytes was to a similar level and sometimes higher than that of neurons [for review see (39)]. This would strongly support the potential contribution of astrocytes to the pathogenesis of these familial forms of PD. Altered levels of these genes lead to many changes in astrocyte function including impaired glutamate uptake, liposomal homeostasis, lysosomal, and mitochondrial dysfunction and inflammatory response (Table 1).
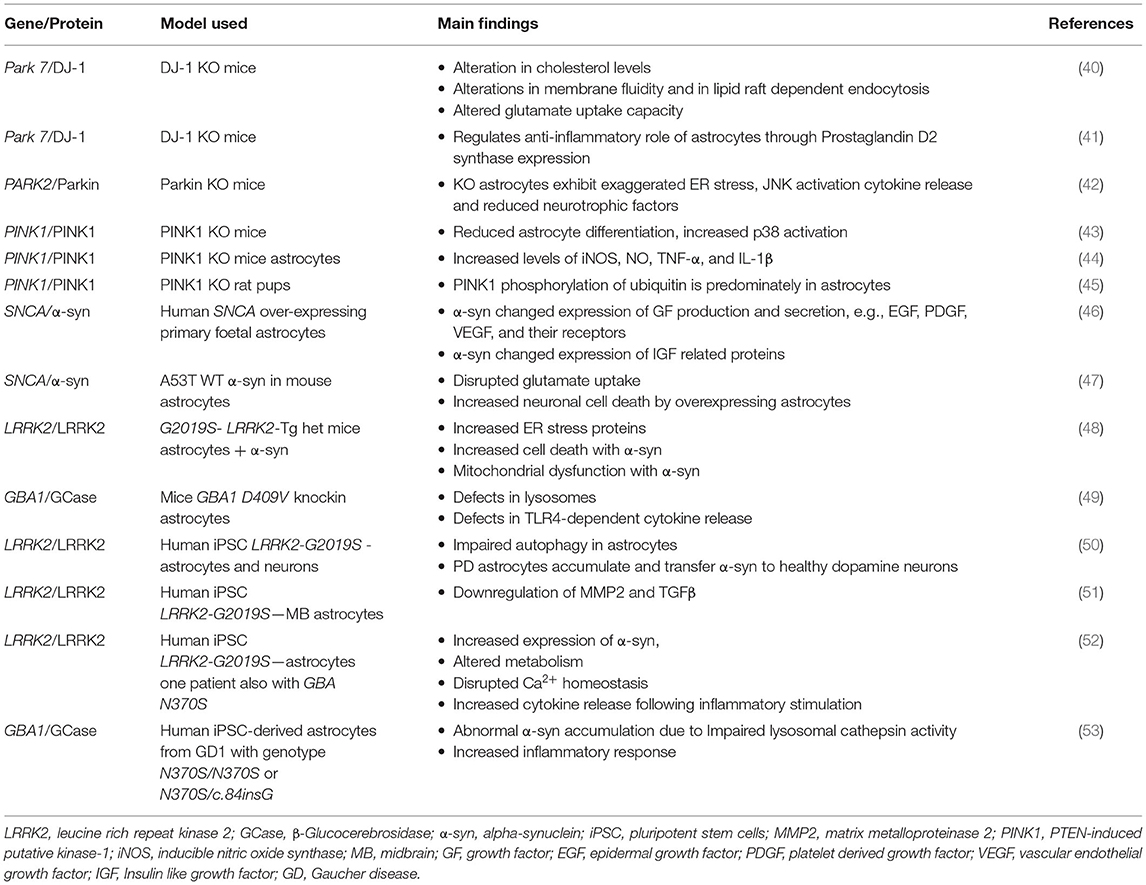
Table 1. The role of genes that are causative in Parkinson's disease pathogenesis and their implications in astrocytes.
DJ-1/PARK7
Some of these genes have been studied to a greater extent than others with respect to their roles in astrocyte biology. DJ-1, encoded by the PARK7 gene, causes early onset autosomal recessive PD (54) and is probably the most extensively studied. Mullett and Hinkle utilised neuron-astrocyte co-cultures to demonstrate that siRNA knockdown of DJ-1 in mouse astrocytes impairs their ability to protect against neurotoxins such as rotenone relative to wild type control astrocytes (55). In addition, studies using DJ-1 knockout mice astrocytes from postnatal day 1 cerebral cortices have also shown that loss of this gene can cause alterations in cholesterol levels and glutamate uptake via regulation of the expression of flotillin-1 and caveolin-1 (40). Furthermore, Choi et al. demonstrated using astrocytes from DJ-1 knockout mice that its deficiency in astrocytes reduced expression of prostaglandin D2 synthase and subsequent secretion of prostaglandin D2 demonstrating that DJ-1 is involved in the regulation of the anti-inflammatory role of astrocytes through prostaglandin D2 synthase expression (41). Moreover, a study from the Kahle laboratory found that DJ-1 knockout mice astrocytes produced 10 times more nitric oxide (NO) than littermate controls when treated with lipopolysaccharide (LPS), a TLR4 agonist, and interestingly lentiviral reintroduction of DJ-1 restored the response to LPS (56). Taken together, these studies demonstrate that DJ-1 is an important regulator of the pro-inflammatory response and that its knockout in astrocytes deregulates inflammatory associated damage.
PARK2 and PINK1
Both PARK2 and PINK1 are expressed at similar levels in both astrocytes and neurons (38, 39, 57). Interestingly, astrocytes deficient in Parkin, encoded by the PARK2 gene, demonstrated a stress induced increase in NOD2 expression; a receptor which integrates ER stress and inflammation and these astrocytes exhibited increased cytokine release and decreased secretion of neurotrophic factors (42). Parkin has also been shown to be involved in the response of astrocytes to an inflammatory signal; activation with TNF-α results in Parkin upregulation whereas activation with IL-1β results in Parkin downregulation (58). PINK1 expression, which encodes the protein PTEN-induced putative kinase 1 (PINK1), is a loss of function mutation that is associated with early onset PD (59). Its expression increases during embryonic development and it was also shown to affect the development of GFAP-positive astrocytes (43). Indeed when PINK1 is knocked out in astrocytes, this causes them to exhibit reduced differentiation (43), reduced neurotrophic factor release, endoplasmic reticulum (ER) stress, JNK activation (42), and generate elevated levels of iNOS, NO, TNF-α, and IL-1β via NF-κB signalling (44). Interestingly, the steady state levels of PINK1 protein are very low even in cells that express PINK1, because PINK1 is normally targeted for degradation after mitochondrial import by a process that is dependent on mitochondrial membrane potential (60). While the high penetrance of PINK1 mutations establish its critical function for maintaining neurons the activity of PINK1 in primary neurons has been difficult to detect. However, Barodia et al. determined the levels of PINK1 in neurons, astrocytes, microglia, and oligodendrocyte progenitor cells (OPCs) cultured from wild type and PINK1 knockout rat pups and showed that PINK1-dependent ubiquitin phosphorylation is predominately in astrocytes suggesting that the contribution of astrocyte dysfunction to PD pathogenesis warrants further investigation (45).
SNCA
SNCA encodes for the protein α-syn and missense mutations as well as duplication or triplication of this gene have been shown to lead to the development of rare hereditary forms of PD [for review see (61)]. Interestingly, one such mutation of the SNCA gene is also associated with an increase in α-syn aggregation in astrocytes; Gu et al. demonstrated that when A53T α-syn was selectively expressed in murine astrocytes it led to disrupted glutamate uptake and increased neuronal death (47). Indeed, SNCA is one of the PD associated genes that is expressed at higher levels in neurons than in astrocytes (38, 57). In 1997, Spillantini et al. reported that α-syn was the major component of Lewy bodies in the brains of people with idiopathic PD (6) and inclusions of α-syn have been found in astrocytes as well as neurons in post-mortem brains of people with PD (62, 63). Interestingly, it has been shown that neuronal α-syn can be directly transferred to astrocytes through sequential exocytosis and endocytosis (33, 64). In addition, overexpression of human α-syn in human primary foetal astrocytes resulted in significant changes in the profile of growth factor expression and release. The most remarkable changes were in epidermal growth factor (EGF), platelet derived growth factor (PDGF), vascular endothelial growth factor (VEGF), and their receptors as well as in insulin like growth factor (IGF) related proteins (46). Further analysis, using bioinformatics, revealed possible interactions between α-syn and EGFR and GDNF (46).
LRRK2 and GBA
LRRK2 encodes the protein leucine-rich repeat kinase 2 (LRRK2) and is causative for a dominantly inherited form of PD (65). Mutations in LRRK2 have been recognised as genetic risk factors for both familial and idiopathic forms of PD (66). When the pathogenic mutation in LRRK2, LRRK2-G2019S, was expressed in mice it resulted in increased ER stress and cell death in astrocytes that was exacerbated by the addition of α-syn (48). GBA encodes an enzyme important in glycolipid metabolism, beta-glucocerebrosidase (GCase). The effects of a mutation in GCase encoded by the GBA1 gene in astrocytes has been studied in mice with knockin of GBA D490V. This resulted in defects in lysosomes and in TLR4-dependent release of cytokines (49).
It is noteworthy that the vast majority of the studies carried out to date on the possible implications of the expression of PD related genes in astrocytes have been conducted in rodents. However, in the last 2 years a small number of studies have begun to emerge in the literature where human induced pluripotent stem cells (iPSC) carrying PD related mutations have been differentiated into astrocytes. It is not surprising that the majority of these are studying the LRRK2-G2019S mutation as this is the most common PD related mutation (67). A study by di Domenico et al. has shown that there is impaired autophagy in astrocytes with a LRRK2-G2019S mutation and that they can accumulate and transfer α-syn to healthy dopamine neurons (50). Another study patterned regional midbrain astrocytes from iPSC containing familial LRRK2-G2019S mutation and from healthy controls. RNAseq analysis revealed downregulation of genes such as TGFβ1 and MMP2 (51). TGFβ1 has previously been shown to inhibit the inflammatory microglial response in a rat model of PD (68), and MMP2 has been shown to be capable of degrading aggregates of α-syn (69). This suggests that these PD astrocytes have a reduced neuroprotective capacity and so may contribute to pathology (51). Further evidence for an increased expression of α-syn in astrocytes has been provided by Sonninen et al. using astrocytes differentiated from iPSC also with LRRK2-G2019S mutation. These astrocytes exhibited altered metabolism and increased cytokine release following inflammatory stimulation (52). iPSC-derived astrocytes with two different GBA1 mutations (N370S/N370S and N370S/c.84insG) have also demonstrated abnormal α-syn accumulation due to impaired lysosomal cathepsin activity and had an enhanced inflammatory response (53). Taken together these studies are consistent with the role for both LRRK2 and GBA1 mutations in accumulation of α-syn and an increased inflammatory response in astrocytes as contributors to PD pathology.
Other Parkinson's Disease Risk Genes
In addition to the six PD risk genes mentioned above there are a number of other risk genes in which there is very high confidence that they are actual PD genes; these are PLA2G6, ATP13A2, FBXO7, and VPS35 [for review see (37)]. Interestingly, the levels of PLA2G6, ATP13A2, and FBXO7 gene expression has been shown to be the same in neurons as in astrocytes but that of VPS35 is greater in astrocytes than in neurons (38, 39, 57). PLA2G6 encodes Ca2+-independent phospholipase A2 (iPLA2), an enzyme responsible for catalysing the release of fatty acids from phospholipids. A recent study by Strokin and Reiser in 2017 demonstrated that astrocytes from mice with a mutation in the Pla2g6 gene, that were treated with Ru360 (a blocker of mitochondrial Ca2+ uniporter), or with rotenone, had a reduced rate of glutamate-induced Ca2+ influx, which was ~2-fold lower than in wild type controls (70). The ATP13A2 gene encodes a transmembrane lysosomal P5-type ATPase and its missense or truncation mutation leads to lysosomal dysfunction (71). A study using primary astrocytes from the mouse midbrain with a deficiency in ATP13A2 demonstrated intense inflammation, which exacerbated dopamine neuron damage following MPP+ exposure (71). Furthermore, this same study showed that astrocytes lacking ATP13A2 had increased lysosomal membrane permeabilisation and cathepsin B release, which in turn led to the activation of the NLRP3 inflammasome. This led to increased production of IL-1β and suggested that there is a direct link between the astrocyte lysosome and neuroinflammation in PD (71). Lysosomal degradation was also shown using human iPSC, from healthy controls or from patients carrying a mutation in lysosomal ATP13A2, which were differentiated into midbrain dopamine neurons and astrocytes (72). This study showed that astrocytes rapidly internalised α-syn and when they were co-cultured with neurons, this led to a decreased accumulation of α-syn in neurons and as a consequence diminished interneuronal transfer of α-syn. Interestingly, loss of this protective function of astrocytes was seen with ATP13A2 deficiency, suggesting that this gene function in astrocytes, contributes partially to PD pathology (72).
Mutations in the FBXO7 (PARK15) gene have been implicated in a juvenile form of PD. When this gene was deleted in tyrosine hydroxylase neurons this resulted in motor deficits, similar to the phenotype of PARK15 patients (73). Vingill et al. also showed that the loss of FBXO7 affected the assembly of proteosomes leading to reduced proteosome activity. This is in keeping with dysfunction of the ubiquitin proteosome system being central to neurodegeneration (74, 75). VPS35 (PARK17) has recently come to the fore as a cause of late-onset familial PD (76). How this gene contributes to human PD is unclear however it has been suggested that VPS35 mutations lead to mitochondrial dysfunction (77), and impaired lysosomal and autophagy function [for review see (76)], all of which contribute to PD pathogenesis. To date the potential effects that mutations in either FBXO7 or VPS35 might have on astrocyte function remain unknown; further experiments would be required to elucidate this.
Microglia and Their Role in Parkinson's Disease
Microglia are the resident immune cells of the CNS. They originate in the yolk sac where they develop from early myeloid precursor cells. During embryonic development, primitive microglia migrate into the developing neural tube where they proliferate and populate the CNS (78). Due to the BBB, microglia lead a relatively sheltered existence compared to peripheral macrophages, although their functions remain the same. Their role is to continuously survey the microenvironment and respond to both physiological and pathological changes. In their capacity as the first line of defence in the CNS, they identify and remove unwanted material such as cellular debris.
The physiological roles of microglia include synaptic pruning and phagocytosis of apoptotic cells (79, 80). They may also aid synapse formation by releasing neurotrophic factors (81, 82). However, these cells do not escape the disruption that occurs during PD; they become activated and assume an inflammatory phenotype. Their expression of pattern-recognition receptors (PRR) allows them to respond to the presence of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) in the microenvironment thereby resulting in microglial activation. Sustained pro-inflammatory activation of microglia, i.e., microgliosis, has been implicated in the pathogenesis of many neurodegenerative disorders including PD. These activated microglia assume an amoeboid morphology, coupled with increased phagocytic capacity. They are also highly reactive and are associated with expression of inflammatory molecules, such as pro-inflammatory mediators and reactive species, in addition to receptors for antigen recognition such as TLR2 (2). Their physiological cellular processes are also thought to be disrupted as a result, further contributing to disease pathogenesis.
While the clinical characteristics of PD have been well-defined, the aetiology of idiopathic PD is still unknown and under investigation. Among the potential causes of PD which are being considered are inflammation and microglia, whose role in PD was first posited in 1988 (15). There is evidence of microgliosis in the SN of PD patients as a result of post-mortem studies where significant CD68 and Iba1 immunoreactivity was detected. Since Iba1 immunoreactivity was co-localised with TLR2 it is thought that this receptor may play a key role in microgliosis (83). This localised microgliosis is also present in the MPTP mouse model of PD (84), the A53T α-syn transgenic mouse model of PD following administration of LPS (85) and the L-dopa-induced dyskinesia rat model of PD (86). Blocking microglial activation with a combination of matrix metalloproteinase inhibitor 1-DNJ plus ibuprofen is protective against dopamine neuronal loss (87) suggesting that microglia are key contributors to PD pathology.
The Impact of Parkinson's Disease Genes on Microglial Function
PD-associated genes are expressed by microglia, not just neurons and astrocytes. The products of these gene mutations are thought to affect the functioning of these cells (14), and can exacerbate microgliosis (Table 2). The dominant PD-risk genes SNCA and LRRK2 promote neuroinflammation via activation of microglia and inflammatory signalling pathways such as NF-κB. Besides cell activation, PD-associated genes also disrupt other microglial processes including mitochondrial respiration and autophagy (99). Since autophagy is involved in regulating microglia inflammatory status (100, 101), its disruption has been reported to play a critical role in inflammation (102) and may also affect some of the key functions of microglia including phagocytosis (101). Furthermore, autophagy failure has been shown to promote intercellular propagation of α-syn (103) which in turn drives microglial activation and neuroinflammation. Mitochondrial dysfunction is another pathological feature in PD, as such the mitochondrial toxin MPTP, is commonly used to induce PD pathology in rodents in order to model disease pathology. Furthermore, pesticides paraquat and rotenone can also be used to induce parkinsonism via disruption of the respiratory chain in mitochondria (104). A number of genes associated with increased PD risk are linked to mitochondrial homeostasis. Among these are SNCA, PARK2, PINK1, PARK7, and LRRK2 (104) (Table 2).
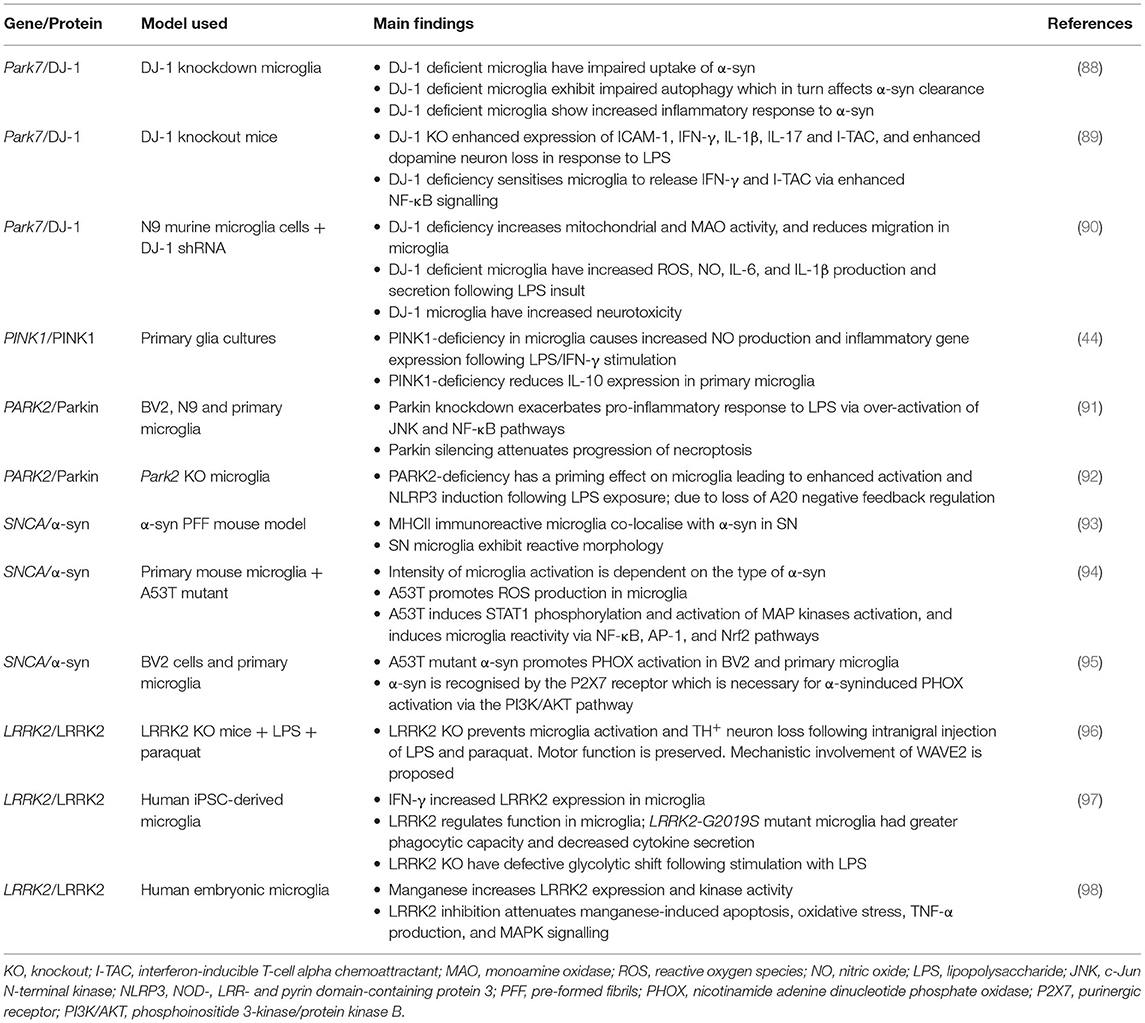
Table 2. The role of microglia and Parkinson's disease risk genes in Parkinson's disease pathology.
DJ-1/PARK7
DJ-1 is a sensor for oxidative stress which localises to the mitochondria when the cell is exposed to oxidative stress (90). DJ-1 mutations which lead to protein deficiency have been linked to PD, with 1% of hereditary cases of PD linked to this gene (88). DJ-1 deficient microglia demonstrate impaired uptake and degradation of α-syn, as well as impaired autophagy (88), and increased sensitivity to pro-inflammatory signals such as LPS (105, 106). This impairment of microglia function combined with increased sensitivity to inflammatory signals may leave PD patients with mutant DJ-1 prone to neuroinflammation. Monoamine oxidase is an enzyme which breaks down amine neurotransmitters such as dopamine. Monoamine oxidase inhibitors have been applied in combination therapy for PD and interestingly one of these, Rasagiline, has been shown to reduce the pro-inflammatory phenotype in microglia and subsequently reduce neurotoxicity in a DJ-1 knockout model (90). This suggests that Rasagiline may be particularly successful in the treatment of PD patients with PARK7 mutations.
SNCA
Encoded by the SNCA gene, α-syn is the main component of Lewy bodies in PD. Physiological roles of α-syn include synaptic vesicle trafficking and formation of the SNARE complex for exocytosis of synaptic vesicle contents into the synapse (14, 107). In PD, excess and/or mutant α-syn aggregates form fibrils. These neurotoxic fibrils act as an endogenous DAMP causing activation of microglia via TLR2 (2, 94) resulting in the activation of NF-κB and MAPK pathways and the production and release of pro-inflammatory mediators such as TNF-α, IL-6, CCL5, and IL-1β (2, 36). Interestingly, microglia activation in response to α-syn has been demonstrated to be mutant specific (94) (Table 2). While α-syn and Lewy bodies are characteristic of PD, it is possible that patients with SNCA mutations are vulnerable to enhanced levels of microgliosis and neuroinflammation. α-syn is now thought to aggregate in microglia as well as in neurons. α-syn binding to surface FcγRIIB receptors on microglia inhibits phagocytosis (108), furthermore α-syn accumulation within microglia can disrupt phagocytosis and lead to activation of microglia (109). TLR2 has been identified as a receptor for α-syn, capable of transporting secreted α-syn into the cytoplasm of microglia (110). Numerous studies have demonstrated a role for α-syn in the activation of microglia, via TLR2 (111–113) and TLR4 (114). Besides TLR2 and TLR4, uptake of α-syn by microglia is facilitated by FYN kinase and CD36 (115). Internalised α-syn can act as a priming signal for the NLRP3 inflammasome, and also disrupts mitochondria function leading to the production of ROS which are capable of activating the NLRP3 inflammasome (115).
LRRK2
LRRK2 encodes the leucine-rich repeat serine/threonine-protein kinase 2. LRRK2 is an incompletely penetrant gene associated with increased PD risk (116, 117). The LRRK2-G2019S mutation has a variable penetrance and is associated with both sporadic and familial PD. There is evidence that LRRK2 is capable of regulating macrophage and microglial motility and phagocytosis (118), mediated by RAB10 (119, 120). WAVE2, a novel interacting partner with LRRK2, regulates branched actin and the Rac1 effector molecule thereby promoting actin polymerisation and cytoskeletal reorganisation. This rearrangement of the cytoskeleton is crucial for the phagocytotic functions of myeloid cells (121). WAVE2 is proposed to be regulated by LRRK2, therefore LRRK2 deficiency can compromise this protein and subsequently reduce the phagocytic capacity of microglia (96, 121) (Table 2). Mutated LRRK2 is observed to alter mitochondrial morphology and increase mitochondrial fission in microglia. This was demonstrated via increased levels of Drp1, a mitochondrial fission marker, CD68, a microglia activation marker and TNF-α in LRRK2 mutant mice (122). Furthermore, inhibition of LRRK2 has been shown to attenuate microglial inflammatory response to TLR4 stimulation (123), and activation of microglia by extracellular α-syn (124, 125).
Other Parkinson's Disease Risk Genes
Besides the main culprits, there are many other genes associated with PD which are proposed to affect microglia function. GBA encodes the lysosomal hydrolase GCase; an incompletely penetrant gene associated with increased PD risk. There is limited literature pertaining to GBA microglia mutants in the PD context, the majority of papers discuss Gaucher disease. For example, in zebrafish, GBA knockout leads to early microglial activation, reduced motor activity, loss of dopaminergic neurons, and ubiquitin inclusions (126). Furthermore, GBA mutations can result in accumulation of glucosylceramide and complement activation which in turn drive inflammation (127).
Recent studies have revealed that autophagy dysfunction can be closely linked with PD pathogenesis. ATG5 (autophagy-related protein 5) is an essential component of the autophagosome, and deficiency results in impaired autophagy. A number of ATG5 variants have been found in the PD patient population (99, 128).
PINK1 and PARK2 have been linked to autosomal recessive forms of PD (116). PINK1 deficiency in microglia has been linked to increased production and secretion of pro-inflammatory mediators, coupled with a reduction in the production and secretion of anti-inflammatory factors thereby favouring a reactive phenotype in microglia (44). The same phenotype is also associated with PARK2 (Table 2). Deficiency in the associated protein Parkin, achieved via knockdown in microglial cells, has a priming effect and results in an enhanced response to the pro-inflammatory stimulus LPS (91, 92). PD patients with these genes may have greater levels of microgliosis as a result.
There is a clear role of microglia in the pathogenesis of PD and the major PD risk genes are a driving force behind this. However, it is becoming clear that microglia are not the only immune cells involved in PD. It is very well-established that the BBB is compromised in PD therefore allowing immune cells from the periphery to infiltrate the CNS.
An Emerging Role for the Peripheral Immune System in Parkinson's Disease
Evidence of an important role for inflammation in the pathogenesis of PD is emerging and the data suggests that peripheral immune cells may contribute to this inflammation. In this section of the review we cover the latest studies demonstrating a role for the peripheral immune system and in particular T cells in PD. By way of introduction however a brief explanation of innate and adaptive immunity is outlined below.
The immune system is divided into the innate and adaptive arms which cooperate to defend against infection, however when dysregulated, immune responses can be important contributors to diseases. The innate immune system represents the body's first line of defence against invading pathogens, it is comprised of numerous cell types which carry out functions such as phagocytosis and antigen presentation. Innate immune cells include dendritic cells, macrophages and also microglia, which can be activated via PRR recognising not only PAMPS but also endogenous DAMPS including α-syn (129). On the other hand, adaptive immunity consisting of B and T lymphocytes provides a highly specific, targeted response capable of dealing with a variety of different intracellular or extracellular infections. T cells, which are either CD4+ or CD8+ are initially naïve until their T cell receptor (TCR) recognises its specific antigen presented by antigen presenting cells via MHC molecules (130). In general, endogenous antigens such as those from viruses are presented via MHCI to CD8+ T cells whereas exogenous antigens are presented via MHCII to CD4+ T cells (130). All cells in the body express MHCI and can activate CD8+ T cells whereas only professional antigen presenting cells including dendritic cells, macrophages, B cells and microglial cells express MHCII and have the capacity to activate CD4 T cells. Once initially activated in the secondary lymphoid organs, naïve T cells differentiate into effector cells with specific functionalities tailored to the infection. CD4+ T cells differentiate into one of the T-helper subtypes; Th1, Th2, Th17 cells which produce various cytokines and provide help to B cells (130). CD8+ T cells differentiate into cytotoxic T lymphocytes which induce apoptosis of infected cells without affecting adjacent healthy cells. Naïve B cells, once activated by their specific antigen and with help from T helper cells, differentiate into plasma cells, producing antibodies which specifically target the antigen and promote clearance via phagocytosis (130). Once activated and differentiated in the lymphoid organs T and B cells traffic to the tissues where they become reactivated upon encounter of an antigen and carry out their effector functions (130). Thus, the immune system has evolved to deal effectively with infection, however various genetic and other factors can conspire to result in inappropriate or chronic inflammation in response to altered proteins or self-antigens that manifests in autoimmune and inflammatory disease (131). Neuroinflammation associated with disorders such as Alzheimer's disease and PD attracts peripheral immune cells to the CNS, and the disruption of the BBB which is a pathological feature of these diseases means that there is a pathway for these cells to enter the brain parenchyma (132–134).
A Potential Role for T Cells in Parkinson's Disease
In the last decade there has been mounting evidence of a role for T cells in the pathogenesis of PD (Table 3). Despite their important role as part of the adaptive immune system there is little evidence of the involvement of B cells in PD. Brochard et al., identified both CD8+ and CD4+ T cells but not B cells or natural killer cells in the post-mortem brain tissue of PD patients (138). The presence of both CD8+ and CD4+ T cells was also evident in the MPTP and α-syn overexpressing mouse models of PD (138, 147). However, Theodore et al., observed both infiltrating B and T cells following injection with α-syn overexpressing adeno-associated viral vector (AAV) into the SN of mice (151). The differences with respect to the possible role of B cells in these studies may be attributed to the study model and human PD tissue vs. animal model, however more research is required before a definitive conclusion can be drawn.
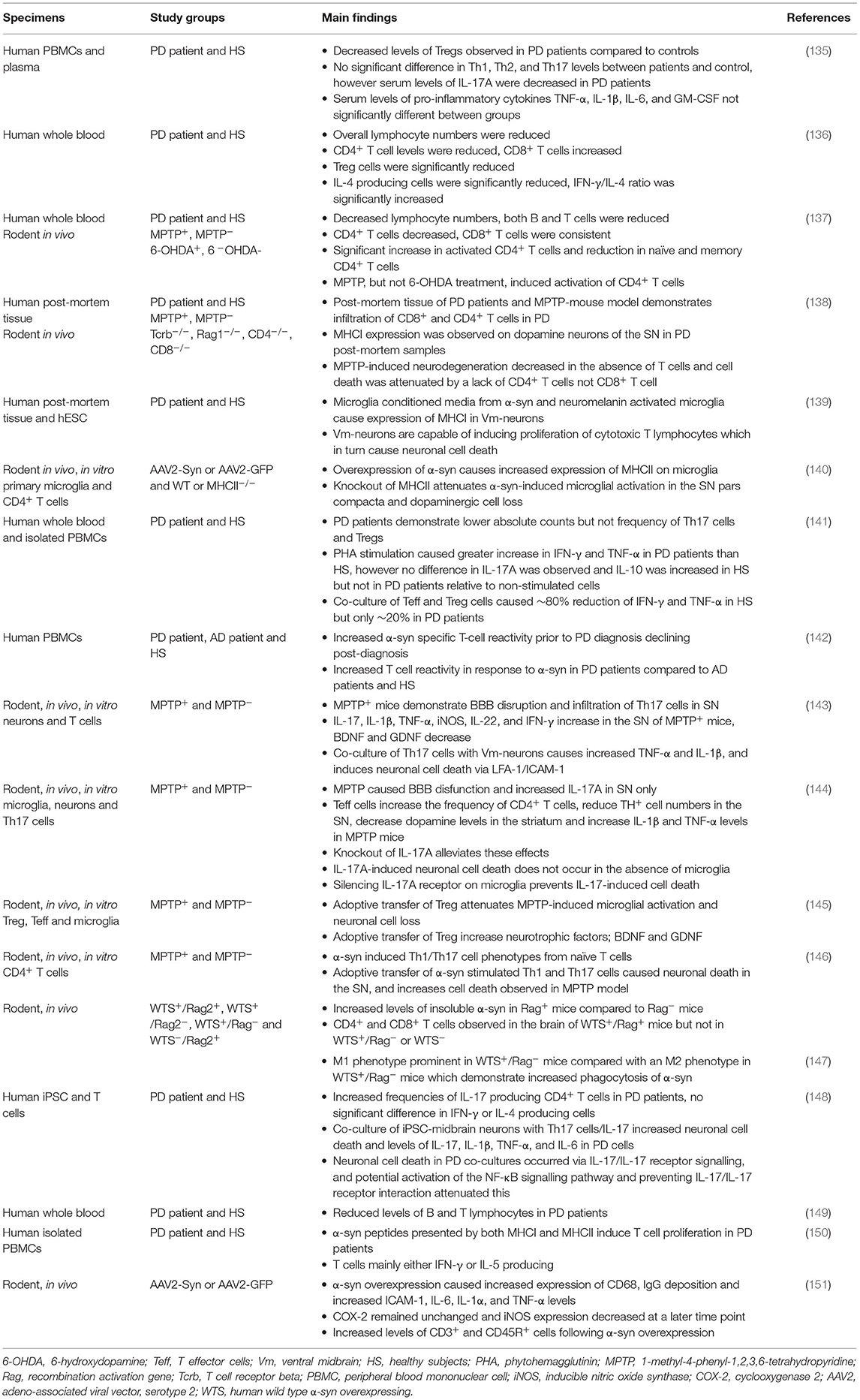
Table 3. Evidence for the role of T lymphocytes in Parkinson's disease.
Disruption of the BBB is commonly observed in PD pathogenesis, presenting an opportunity for peripheral immune cells to infiltrate into the brain. Infiltration of T cells from the periphery into the CNS has been observed in multiple PD studies (132, 144). Liu et al., demonstrated using MPTP treatment that BBB disruption increases the frequency of CD4+ T cells in the SN of mice (144). The infiltration of CD4+ T cells reduced the number of TH+ neurons, decreased dopamine levels and increased the production of IL-1β and TNF-α. Importantly, Brochard et al., observing both CD8+ and CD4+ T cell infiltrates, determined using CD4−/− and CD8−/− mice that CD4+ T cells, rather than CD8+ T cells, were responsible for the neurodegeneration associated with their infiltration (138). As discussed above, the accumulation of misfolded α-syn protein contributes to dysregulation and inflammation in PD (152). Studies have demonstrated that T cell infiltration is associated with α-syn overexpression (147, 151), additionally the knockout of lymphocytes reduced α-syn protein aggregates in this PD model (147). These studies indicate a role for T cells in PD, however given that T cells are activated in an antigen-specific manner via MHC molecules, it is essential to understand the antigen specificity of T cells involved in PD and the possible contribution of different MHC haplotypes.
The Role of MHC and Antigen Presentation in Parkinson's Disease
In addition to the evidence above showing the presence of T cells in the SN of PD patients and mouse models, a role for T cells is also implied by the identification of specific MHC haplotypes and non-coding SNPs in MHC genes as risk factors for PD. Such MHC associations are a common feature of autoimmune diseases driven by auto-reactive T or B cells and are thought to result from preferential presentation of dominant self-epitopes by particular MHC molecules to auto-reactive T cells that have escaped thymic selection. Interestingly, Sulzer et al., demonstrated significantly enhanced T cell responses to immunodominant α-syn peptides in PD patients, relative to healthy controls (150). Furthermore, an immunodominant peptide was shown to bind with high affinity to the DRB1*15:01 and DRB5*01:01 alleles which were part of the MHC haplotype previously associated with PD (150). In a subsequent study, Lindestam Arlehamn et al., determined in a single longitudinal case study that this α-syn-specific T cell response was elevated prior to diagnosis and decreased thereafter (142). In the studies above, not all PD patients exhibited α-syn-specific T cell responses and responses were also observed in some healthy controls, indicating that in both PD and healthy controls these auto-reactive T cells escaped deletion during thymic selection. However, such auto-reactive T cells only become pathogenic if peripheral tolerance mechanisms are overcome and they become activated in the secondary lymphoid organs upon recognition of α-syn peptides presented via MHC molecules on activated dendritic cells (Figure 1). Tolerance can be overcome when self-antigens become post-translationally modified and then appear as neoantigens to which the immune system is not yet tolerised. Indeed, T cell responses to both native and post-translationally modified α-syn were observed in PD patients (150). The α-syn that activates naïve T cells in PD could either be peripherally derived or have drained from the CNS as suggested by the proposed body-first vs. brain-first subtypes, respectively (153). In addition, activation of the antigen presenting cell via PRR is required in order to induce the necessary co-stimulation signals required for naïve T cell activation and the source of this signal is unclear but could possibly be provided via DAMPs including α-syn.
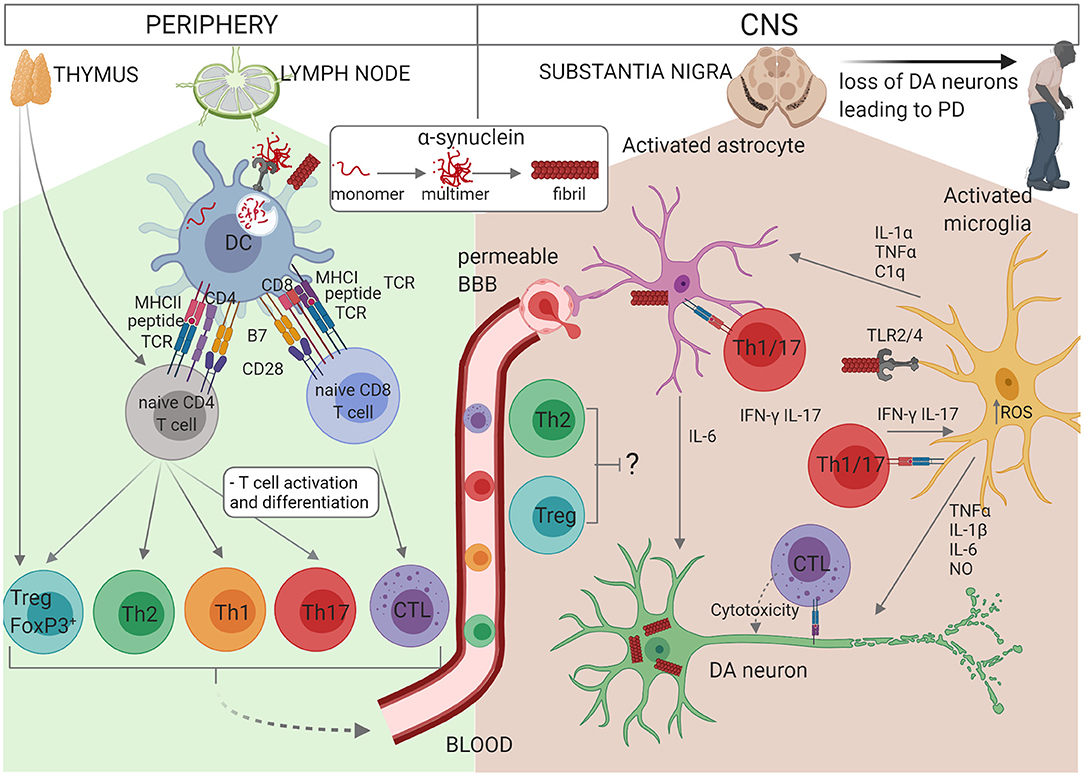
Figure 1. Schematic representation of the interactions between glial cells and immune cells in PD. Self-reactive α-syn-specific naïve T cells may have escaped thymic selection, or alternatively post-translationally modified α-syn may be recognised by naïve T cells as a neoantigen. In individuals with PINK1/PARK2 mutations, mitochondrial antigen presentation may also result in T cell activation. Autoreactive CD4+ or CD8+ T cells circulate through the lymph nodes where they may become activated by a dendritic cell (DC) presenting α-syn antigen via MHCII or MHCI, respectively. α-syn or other DAMPs activate the DC via PRR to express co-stimulatory molecules (B7) and cytokines which drive the proliferation and differentiation of effector T cell subsets. CD4+ T cells differentiate into Th1, Th2, Th17, or Treg cells depending on the cytokine milieu and CD8+ T cell differentiate into cytotoxic lymphocytes (CTL). Effector T cells traffic via the blood and extravasate through a permeable BBB into the CNS, where they re-encounter their α-syn antigens presented via MHCI on neurons or MHCII on astrocytes or microglia. Th1 and Th17 cells produce pro-inflammatory cytokines IFN-γ and IL-17 which contribute to the activation of astrocytes and microglia in synergy with other cytokines such as TNF-α, and CTL induce apoptosis in DA neurons. On the other hand, Treg and Th2 cells may protect against neuroinflammation. The accumulation of modified or aggregated α-syn is thought to be a key initiator of PD. α-syn accumulates within DA neurons and can also be secreted where it activates astrocytes and microglia. Activation of TLR2 or TLR4 by α-syn together with increased intracellular ROS activates the inflammasome. Activated microglia secrete TNF-α, IL-1β, IL-6, and NO which promote DA neuron degeneration. IL-1α, TNF-α, and C1q from activated microglia also activate astrocytes which in turn secrete IL-6, IL-1α, IL-1β, and NO. In addition, activated astrocytes exhibit decreased release of protective neurotrophic factors and impaired glutamate uptake. Thus, T cells, astrocytes and microglia cooperate to perpetuate neuroinflammation and loss of DA neurons in PD.
Mitochondrial Antigen Presentation in Parkinson's Disease
Recent studies in a mouse model have identified an intriguing link between the PD-associated genes PINK1 and PARK2 and antigen presentation. PARK2 and PINK1, which cause autosomal recessive forms of PD (116), maintain mitochondrial homeostasis through mitophagy (110). Loss of function mutations in the PINK1 gene (a mitochondrial kinase) lead to disturbances in mitophagy, and mitochondrial fusion and fission (154). PINK1 deficiency is associated with increased ROS production, oxidative stress, abnormal mitochondrial function, and altered morphology (44). Mitochondrial dysfunction can result in the release of numerous pro-inflammatory factors such as mitochondrial DNA, RNA, ATP, and cytochrome c (110). The presence of ROS and DNA from damaged mitochondria can act as danger signals for activation of the NLRP3 inflammasome which is involved in neuroinflammation in PD (2, 155, 156). Mitochondrial dysfunction has been shown to over-activate the NLRP3 inflammasome in microglia resulting in dopaminergic neuron cell death (157). In its capacity as an activator of ubiquitin PINK1 cooperates with Parkin a ubiquitin ligase (158, 159). In addition to these established roles in maintaining mitochondrial homeostasis, PINK1 and Parkin have more recently been shown to play a role in suppressing immune responses by actively inhibiting the presentation of self-mitochondrial antigens via MHCI (160). Mitochondrial derived vesicles can arise by budding from the mitochondria and then fuse with lysosomes, processes that are dependent on the proteins SXN9 and RAB7 GTPase. Mitochondrial antigens are then processed and presented via the MHCI pathway to activate autoreactive CD8+ T cells. PINK1 and Parkin however inhibited the activity of SXN9/RAB7 and thereby mitochondrial antigen presentation (160). These data suggest that familial PD associated with loss of function of PINK1/Parkin could involve an autoimmune component, whereby the presentation of self-mitochondrial antigens is enhanced in the absence of PINK1 or Parkin resulting in the activation of autoreactive CD8+ T cells. These mechanisms were demonstrated in dendritic cells, macrophages, and fibroblasts; however, a key outstanding question is whether lack of PINK1/Parkin exerts similar effects on mitochondrial antigen presentation in astrocytes, microglia and neurons. If so, then naïve CD8+ T cells could be initially activated in the periphery by dendritic cells, traffic to the CNS and cross the BBB where they would kill dopaminergic neurons upon recognition of their cognate mitochondrial antigen presented via MHCI. Furthermore, it is also unknown whether the absence of PINK1/Parkin could result in mitochondrial antigen presentation via MHCII to activate CD4 T cells. This is an important question given that neurodegeneration was shown to be dependent on CD4 T cells in a mouse model of PD (138). Interestingly mitochondrial antigen presentation was shown to be enhanced by an inflammatory insult and indeed in a follow up study, Matheoud et al. demonstrated that intestinal infection in PINK1 deficient mice promoted mitochondrial antigen presentation, the activation of CD8+ T cells specific for mitochondrial antigens and motor impairment that was reversed by L-DOPA (161). Given that loss of function of PINK1 or Parkin results in symptoms of PD in humans but not mice (162), this suggests that it is a combination of the genetic mutation together with an inflammatory insult that leads to PD in individuals with mutations in PINK1/PARK2. Importantly, a role for the gut-brain axis in PD is implicated by these findings and supported by a study that showed that the gut microbiome was required to induce disease in an α-syn mouse model of PD. Furthermore, transfer of microbiota from PD patients into α-syn overexpressing mice resulted in exacerbation of disease symptoms when compared with transfer of microbiota from healthy controls (163). These studies suggest that dysbiosis could be responsible for triggering or exacerbating disease at least in some familial cases of PD and raise the interesting possibility of preventative or therapeutic targeting of the microbiome.
In summary, the evidence from post-mortem and MHC association studies in PD patients together with that from murine studies, suggests involvement of T cells in the pathogenesis of PD. However, there are still many unanswered questions and more extensive research is required. For example, to translate the autoimmune hypothesis of PD described above it will be important to identify mitochondrial antigen-specific T cells in PD patients with PINK1/PARK2 mutations. The possible role of different T cell subtypes in PD is discussed below.
Th1 Cells in Parkinson's Disease
Th1 cells differentiate under the influence of cytokines IFN-γ and IL-12 released by antigen presenting cells, in combination with activation of the T-bet transcription factor. The main cytokines released by Th1 cells are IFN-γ and TNF-α, these cells are important in activation of B cells and increasing phagocytosis of microbes (130). PD studies have observed both increased frequencies of IFN-γ-producing Th1 cells in circulating blood from PD patients (136, 141), and no significant difference in Th1 levels (135). Kustrimovic et al., discovered decreased levels of the T-bet encoding gene, TBX21 (141, 164) in PD patients compared with controls. However, naive CD4+ T cells treated with α-syn polarised to a Th1 or Th17 phenotype causing cell death of TH+ neurons in the SN and exacerbated MPTP-induced cell death (146). The evidence above suggests pathogenic potential for Th1 cells in PD, however the evidence is also sparse and conflicting, which clearly highlights the need for further research in this area.
Th17 Cells in Parkinson's Disease
A large amount of research exists into the role of Th17 cells in PD. TGF-β, IL-6, IL-1, and IL-23 along with the RORγt transcription factor are important in inducing differentiation of Th17 cells (130, 165). The signature cytokine produced by Th17 cells is IL-17. Upon binding to its receptor, which is widely expressed on epithelial and other cell types, it induces the release of chemo-attractants such as CXCL1 to recruit additional immune cells, particularly neutrophils. IL-17 also induces the secretion of anti-microbial peptides such as S100A8/A9 (166). Sommer et al., observed increased frequencies of IL-17 producing CD4+ T cells in the peripheral blood of PD patients compared with control subjects (148) and infiltration of Th17 cells into the SN has been demonstrated in an MPTP mouse model of PD (143). In contrast however, a study using peripheral blood mononuclear cells (PBMCs) isolated from PD patients and healthy subjects discovered lower absolute counts but not frequency of Th17 cells in PD (141). A separate study observed no difference in the levels of Th17 cells between PD patients and controls, with reduced IL-17A levels in PD patients (135, 141).
Although the conflicting studies above indicate that further work is required to determine whether enrichment of peripheral Th17 cells is a consistent finding in PD, there is experimental evidence to suggest a pathogenic role for Th17 cells in the context of PD. Stimulation of Th17 cells with α-syn was found to cause neuronal cell death in the SN in an MPTP mouse model of PD (146). Furthermore, co-culture of MPTP-treated neurons with Th17 cells further exacerbated neuronal cell death and increased IL-1α and TNF-α levels observed with MPTP-treatment alone (143, 146). Liu et al., determined that these effects were mediated via lymphocyte function-associated antigen 1 (LFA-1) and intracellular adhesion molecule-1 (ICAM-1) interactions and ablation of either ICAM-1 or LFA-1 attenuated the dopaminergic cell death observed in this model (143).
Importantly, Sommer et al., demonstrated that the co-culture of autologous iPSC-midbrain neurons and Th17 cells led to increased neuronal cell death in cells derived from PD patients compared with controls and this effect was not observed with non-autologous cell cultures (148). This suggested that antigens were presented in an MHC-restricted manner by PD neurons to Th17 cells which then induced neuronal death. It was determined that either IL-17 or the IL-17 receptor signalling and a potential activation of NF-κB signalling was responsible for the neuronal cell death observed in the co-cultures. Furthermore, attenuation of neuronal cell death in the PD co-cultures was achieved by blocking the IL-17 or the IL-17 receptor. Taken together the above studies indicate a role for Th17 cells in the pathogenesis of PD. Although much of the research to date has been undertaken in animal models, future studies like that of Sommer et al., could utilise human-based models of PD, and patient-derived models to determine whether these interactions translate to the human disease phenotype.
Th2 Cells in Parkinson's Disease
Th2 cells differentiate from naïve T cells under the influence of IL-4, and the activation of the GATA3 transcription factor. These cells produce IL-4, IL-5, and IL-13 (130). IL-4 activates IgE production causing mast cell degranulation, releasing histamines, and other pro-inflammatory cytokines. Additionally, IL-4 as well as IL-13 increase mucous production and IL-5 activates eosinophils, releasing various pro-inflammatory and cytotoxic proteins (167). In the context of neuroinflammation unlike inflammatory Th1 and Th17 cells, Th2 cells are usually considered to be anti-inflammatory and protective. Alvarez-Luquin et al., demonstrated no significant difference in Th2 cell counts in PD patients compared with controls, however there was a significant increase in IL-13 observed (135). In contrast, studies have observed lower absolute numbers and frequency of Th2 cells compared with the healthy subjects (136, 141), however, increased GATA3 levels were identified (141). Sulzer et al., observed that α-syn peptides activated mainly IFN-γ-producing Th1 and IL-5-producing Th2 cells. As anti-inflammatory, protective immune cells Th2 cells have the potential to alter the progression of PD pathology. However, as observed with Th1 and Th17 cells the existing evidence is often contradictory, and therefore inconclusive, indicating the necessity for further research into this area before any concrete conclusions can be drawn.
T Cell Regulation in Parkinson's Disease
It is well-established that an overactive, aberrant immune response is rooted in the pathogenesis of numerous inflammatory diseases. Therefore, proper functioning of the regulatory mechanisms is key to preventing pathology. CTLA-4 is a crucial regulator of T cell activation, upon activation T cells upregulate the expression of CTLA-4, its role is to preferentially bind the co-stimulatory B7 in place of CD28, having an inhibitory, rather than stimulatory effect (130, 168, 169). Importantly, Cook et al., discovered a reduction in CTLA-4 expression on the surface of T cells from PD patients following stimulation (169), indicating the possibility of a potentially unregulated T cell response occurring in PD.
Regulatory (Treg) cells play a key role in constraining effector T cells and prevent excessive inflammation and autoimmunity. Treg cells are either directly generated early in life in the thymus during T cell development or differentiate in the periphery from naïve CD4+ T cells under the influence of TGF-β (130, 165) and they express the transcription factor FOXP3 which is necessary for their function. These cells produce IL-10 and TGF-β, which are important anti-inflammatory cytokines crucial to the regulation of the immune response and can also suppress effector T cells via various other mechanisms including via CTLA-4 (130). Reduced absolute numbers but not frequency of Treg cells have been observed in PD patients (135, 136, 141). Kustrimovic et al., also observed increased mRNA levels of FOXP3. Interestingly, T effector cells co-cultured with Treg cells only reduced IFN-γ and TNF-α by ~20% in PD patients, compared to an ~80% reduction observed in healthy controls, suggesting that Treg suppression in PD may be impaired (141). Reynolds et al., demonstrated a neuroprotective role for Treg cells in the MPTP mouse model of PD. The Treg cells were adoptively transferred into MPTP treated mice, a reduction in neuronal cell death, microglial activation, and increased production of both BDNF and GDNF were observed (145). Thus, Treg cells are likely to be protective in the context of PD and although there are some discrepancies, these data suggest that Treg cells may be numerically and or functionally impaired in PD and this may contribute to neuroinflammation.
The literature supporting a role for T cells in PD is growing, although there still remains evidence which contradicts this, as numerous studies have observed reduced levels of circulating T cells in the blood of PD patients compared with healthy subjects (136, 137, 141, 149). Similarly, most studies on T cell subsets presented contrasting findings. The discrepancies between studies may be explained in part by different methodologies; some studies have identified Th subsets via their production of signature cytokines whereas others used a combination of chemokine receptors. Similarly, the markers used to identify Treg cells were not always comparable between studies and identification of Treg cells is notoriously difficult as some of the markers used can also be induced on non-Treg cells during inflammation. For this reason, a full panel of markers including CD4, CD25, CD127, and FOXP3 is required to most reliably identify Treg cells. Finally, it is worth noting that alterations in the frequency of peripheral T cell subsets may not reflect changes occurring in the inflamed tissues and a reduction observed in the periphery may be due to trafficking to the tissue. Thus, in summary, this area requires a greater amount of research before the precise role of these cells can be elucidated.
This review has outlined evidence demonstrating the role of microglia, astrocytes and even peripheral immune cells in PD pathogenesis. Logically, this leads to the question of whether there is potential interplay between these infiltrating peripheral immune cells and the resident microglia and astrocytes of the CNS in PD.
The Interplay Between Glia and Peripheral Immune Cells in Parkinson's Disease
Glial cells are crucial to the maintenance of homeostasis within the CNS and the disruption of this is linked to numerous CNS diseases. There appears to be little evidence available outlining potential interactions between peripheral immune cells and glia in PD, however the research into this area is increasing and is summarised in Figure 1. A recent study demonstrated antigen presenting capabilities in astrocytes. MHCII expressing astrocytes were identified in close proximity to CD4+ T cells in the post-mortem brain tissue of PD patients and cultured human astrocytes exposed to pre-formed fibrils of α-syn expressed the T cell co-stimulatory structures, CD80, CD86, and CD40 (170), suggesting the capacity to activate CD4+ T cells. Although Rostami et al., determined that cultured human microglia demonstrated poor antigen presenting capabilities, Harms et al., determined in mice that microglia exposed to α-syn increased their expression of MHCII, became activated and induced proliferation of CD4+ T cells. Furthermore, the knockout of MHCII prevented microglial activation and dopaminergic cell loss in these mice (140).
ICAM-1 is present on immune cells and its binding to LFA-1 is key to the migration, extravasation and even activation of immune cells (171). Prajeeth et al., discovered that it is possible for Th1 and Th17 cells to induce reactive, pro-inflammatory astrocytes which microglia migrate toward and increase their phagocytic ability. In addition to this, the T cell activated astrocytes enhanced the infiltration of Th17 cells (172). In other neurodegenerative diseases, such as Alzheimer's disease, astrocytes expressing ICAM-1 have been observed in close proximity to LFA-1 positive microglia (173). Miklossey et al., discovered the occurrence of these key immune interactions in post-mortem brain tissue from PD patients. ICAM-1 expression by astrocytes in the SN of PD patients was observed, and co-localised to these areas were LFA-1 positive microglia and LFA-1 positive leukocytes (174), highlighting the presence of interactions between glia and immune cells in PD.
The ability of these immune cells to infiltrate into the brain in PD is demonstrated in numerous studies. BBB disruption in PD has been observed in both post-mortem studies and animal models of PD (132, 144) and is linked to increased infiltration of peripheral immune cells (144). Once the immune cells enter the brain parenchyma their functions include the release of various cytokines, which are capable of activating glial cells and causing neuronal cell death (175). Liu et al., observed increased levels of IL-17A following BBB disruption in the MPTP mouse model of PD. Addition of IL-17A to co-cultures of microglia and neurons resulted in microglial activation, TH+ neuronal cell death and a decrease in dopamine levels and the inhibition of the IL-17A receptor on microglia was sufficient to attenuate these effects (144). The synergistic and additive effect of IL-17 with factors commonly secreted by astrocytes and microglia, such as IL-6, IL-1β, and TNF-α has been an area of great interest and numerous studies have demonstrated this phenomenon (176–180). The studies discussed in this review support the idea that inflammation is a key factor in the pathogenesis of PD and may be driven or compounded by T cells. However, there are important outstanding questions regarding the specificity of T cells involved in PD and whether auto-reactive T cells are involved only in certain subsets of PD patients such as those with genetic mutation, or whether they are more widely involved in sporadic PD. The role of the adaptive immune system in PD is a novel area of research and as such requires deeper investigation before a complete picture can be drawn of the role of these cells in PD pathogenesis and their therapeutic potential can be fully realised.
To aid in this, novel human-based models such as patient-derived iPSC and 3D culture systems should be utilised to determine if these findings from animal models translate to the human disease. As well as this, more patient studies will be required to determine what is occurring physiologically within PD patients and if this corresponds to what is observed within animal and in vitro human models.
Implications for Therapeutic Targeting
Our increased understanding of the critical role the immune system plays in PD has stimulated research into the viable therapeutic targets it presents; so as to allow determination of their potential as disease modifying therapies. As mentioned earlier, Yun et al., demonstrated that targeting the GLP-1 receptor with NLY01 prevented conversion of astrocytes to a pro-inflammatory phenotype by activated microglia and inevitably reduced neuronal cell death (36). As a long-acting GLP-1 receptor agonist, with the ability to cross the BBB, NLY01 is currently undergoing phase 2 clinical trials to investigate its efficacy in early PD (NCT04232969). Although results have yet to be published for NLY01, another GLP-1 receptor agonist previously FDA approved for treatment of type 2 diabetes mellitus, Exenatide, is currently in phase 3 clinical trials (NCT04154072) having successfully completed early phase 1 and 2 trials (181).
A second newly emerging PD therapeutic target is that of monoclonal antibodies, specifically those targeting α-syn. PRX002/RG7935, an IgG 1 monoclonal antibody targeting aggregated α-syn, has demonstrated safety and tolerability in an initial phase 1 clinical trial and in a further phase 1b trial of multiple ascending-doses with a reduction in free serum α-syn observed (182). Phase 2 clinical trials are currently ongoing (NCT6868). Furthermore, BIIB054 another IgG 1 monoclonal antibody targeting α-syn has concluded phase 1 clinical trials (183) and is currently undergoing phase 2 (NCT03318523). However, these therapies involve regular injection with monoclonal antibodies to maintain an immune response, otherwise known as passive immunisation, another therapeutic avenue is the use of vaccines, or active immunisation in the treatment of PD. PD01A is an anti-α-syn vaccine, which maintains a higher affinity for aggregated rather than monomeric forms of α-syn. The results for the phase 1 clinical trial of this vaccine in PD patients demonstrated that it is safe and well-tolerated among treatment groups (184). Although these two strategies involve the use of antibodies to target the specified antigen, vaccination brings with it the added benefit of long-term immunisation however, passive immunisation enables treatment to cease if adverse side effects occur.
A phase 2 clinical trial is currently ongoing which investigates the use of the immunosuppressant drug, Azathioprine for treatment of PD. Azathioprine is an FDA approved, purine analogue and is used to treat numerous diseases, including multiple sclerosis. It acts by reducing B and T cell proliferation via nucleic acid synthesis inhibition (185). However, an important consideration for this therapy is the fact that it is an immunosuppressant, which exposes the patient to increased risk of infection and further illness. Another immunomodulatory therapy, Sargramostim, is an FDA approved GM-CSF that stimulates myeloid cell production and induces Treg cells. The phase 1 clinical trial demonstrated increased numbers and functionality of Treg cells while maintaining levels of T effector cells (186). Potentially enabling increased regulation of the immune response in PD.
As mentioned above, the gut-brain axis is being increasingly researched and studies have demonstrated a potential role for this pathway in PD. Importantly, gut microbiome alterations have been observed in PD patients (187) and faecal microbiome transplantation has been investigated as a therapeutic intervention for PD. A case report (188) and a preliminary study (187) have demonstrated potential benefits for this as a therapeutic strategy in the treatment of PD. Additionally, a clinical trial (NCT03808389) is ongoing in hopes that restoration of microbiome homeostasis will improve symptoms in PD patients.
These trials represent many varied and promising approaches targeting the immune system as a means of PD therapy which together may lead to future disease-modifying therapeutic strategies for the treatment of PD.
Future Directions to Increase Our Understanding of Neuro-Immune Cross Talk
The crosstalk between the brain and the immune system in PD is clearly complex with many questions remaining to be answered. Some of these many interesting questions include the following: (1) Is the increase or reduction of various T cell subsets in the blood of PD patients reflective of the situation in the CNS, or could for example a reduction in the blood vs. healthy controls merely indicate reciprocal trafficking to the CNS? (2) What role might the different PD associated genetic mutations have on crosstalk between glial and immune cells and how that crosstalk affects dopamine neuronal survival? (3) Is the peripheral activation of α-syn reactive T cells a primary event, or is it secondary to the aggregation/alteration/mutation of α-syn or other PD associated proteins in the CNS which are then released into the periphery to activate antigen specific naïve T cells? (4) Does T cell involvement only occur in a subset of PD patients who are genetically predisposed to T cell involvement as a result of their MHC haplotype, and are certain mutated peptides preferentially presented by particular MHC haplotypes? (5) What role might PD associated genetic mutations have on mitochondrial antigen presentation and could carriers of PINK1 or PARK2 mutations present with an autoimmune type of PD? (6) How might all of the queries above be influenced by PD medications?
The development of new research tools in recent years, including animal and various stem cell models, has already allowed a better understanding of astrocyte and microglial function and as these methods continue to improve, we can probe more deeply into how these cells communicate with one another and also begin to answer the questions posed above. Such tools include rapid and more cost-effective sequencing at single cells resolution at various stages of the progression of pathology (189). Others that have also become available are iPSC-derived neurons, astrocytes, and microglia (190), cerebral organoids (191), cerebral organoids containing microglia (192), CRISPR screening, and high content imaging (193). The major advantage offered by iPSC is that they allow the study of the known PD associated genetic mutations where genes are expressed at native levels without any forced genetic manipulation. To gain a better understanding of the mechanisms involved in glial and immune cell crosstalk will require a deeper knowledge of the proteome of T cell subsets, astrocytes and microglia and integration with multi-omic datasets. Research will also need to focus on functional studies to gain a better understanding of dysfunctional pathways (189) that may allow development of novel therapeutic targets and the continuing development of animal models will be critical to development of our increased understanding in this area. Many exciting discoveries have been made in this field in recent years; more are sure to follow.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the Health Research Board, Ireland, grant number 16287.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Tanner CM, Goldman SM. Epidemiology of Parkinson's disease. Neurol Clin. (1996) 14:317–35. doi: 10.1016/S0733-8619(05)70259-0
2. de Araujo FM, Cuenca-Bermejo L, Fernandez-Villalba E, Costa SL, Silva VDA, Herrero MT. Role of microgliosis and NLRP3 inflammasome in Parkinson's disease pathogenesis and therapy. Cell Mol Neurobiol. (2021) doi: 10.1007/s10571-020-01027-6
3. Parkinson J. An essay on the shaking palsy 1817. J Neuropsychiatry Clin Neurosci. (2002) 14:223–36; discussion 222. doi: 10.1176/jnp.14.2.223
4. Fearnley JM, Lees AJ. Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain. (1991) 114:2283–301. doi: 10.1093/brain/114.5.2283
5. Holdorff B. Friedrich heinrich lewy (1885-1950) and his work. J Hist Neurosci. (2002) 11:19–28. doi: 10.1076/jhin.11.1.19.9106
6. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. (1997) 388:839–40. doi: 10.1038/42166
7. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. (1997) 276:2045–7. doi: 10.1126/science.276.5321.2045
8. Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. (1998) 18:106–8. doi: 10.1038/ng0298-106
9. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. (2003) 302:841. doi: 10.1126/science.1090278
10. Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. (2004) 55:164–73. doi: 10.1002/ana.10795
11. Williams-Gray CH, Wijeyekoon R, Yarnall AJ, Lawson RA, Breen DP, Evans JR, et al. Serum immune markers and disease progression in an incident Parkinson's disease cohort (ICICLE-PD). Mov Disord. (2016) 31:995–1003. doi: 10.1002/mds.26563
12. Gelders G, Baekelandt V, Van der Perren A. Linking neuroinflammation and neurodegeneration in Parkinson's disease. J Immunol Res. (2018) 2018:4784268. doi: 10.1155/2018/4784268
13. Hall S, Janelidze S, Surova Y, Widner H, Zetterberg H, Hansson O. Cerebrospinal fluid concentrations of inflammatory markers in Parkinson's disease and atypical parkinsonian disorders. Sci Rep. (2018) 8:13276. doi: 10.1038/s41598-018-31517-z
14. Kam TI, Hinkle JT, Dawson TM, Dawson VL. Microglia and astrocyte dysfunction in parkinson's disease. Neurobiol Dis. (2020) 144:105028. doi: 10.1016/j.nbd.2020.105028
15. McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. (1988) 38:1285–91. doi: 10.1212/WNL.38.8.1285
16. Gao HM, Jiang J, Wilson B, Zhang W, Hong JS, Liu B. Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson's disease. J Neurochem. (2002) 81:1285–97. doi: 10.1046/j.1471-4159.2002.00928.x
17. Sanchez-Guajardo V, Annibali A, Jensen PH, Romero-Ramos M. alpha-Synuclein vaccination prevents the accumulation of parkinson disease-like pathologic inclusions in striatum in association with regulatory T cell recruitment in a rat model. J Neuropathol Exp Neurol. (2013) 72:624–45. doi: 10.1097/NEN.0b013e31829768d2
18. Wilson H, Dervenoulas G, Pagano G, Tyacke RJ, Polychronis S, Myers J, et al. Imidazoline 2 binding sites reflecting astroglia pathology in Parkinson's disease: an in vivo11C-BU99008 PET study. Brain. (2019) 142:3116–28. doi: 10.1093/brain/awz260
19. Forno LS, DeLanney LE, Irwin I, Di Monte D, Langston JW. Astrocytes and Parkinson's disease. Prog Brain Res. (1992) 94:429–36. doi: 10.1016/S0079-6123(08)61770-7
20. Jenner P, Olanow CW. Oxidative stress and the pathogenesis of Parkinson's disease. Neurology. (1996) 47:S161–70. doi: 10.1212/WNL.47.6_Suppl_3.161S
21. Martin HL, Santoro M, Mustafa S, Riedel G, Forrester JV, Teismann P. Evidence for a role of adaptive immune response in the disease pathogenesis of the MPTP mouse model of Parkinson's disease. Glia. (2016) 64:386–95. doi: 10.1002/glia.22935
22. Tansey MG, Romero-Ramos M. Immune system responses in Parkinson's disease: early and dynamic. Eur J Neurosci. (2019) 49:364–83. doi: 10.1111/ejn.14290
23. Azevedo FA, Carvalho LR, Grinberg LT, Farfel JM, Ferretti RE, Leite RE, et al. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol. (2009) 513:532–41. doi: 10.1002/cne.21974
24. Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. (1999) 22:208–15. doi: 10.1016/S0166-2236(98)01349-6
25. Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Control of synapse number by glia. Science. (2001) 291:657–61. doi: 10.1126/science.291.5504.657
26. Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. (2006) 7:41–53. doi: 10.1038/nrn1824
27. Clarkson ED, Zawada WM, Freed CR. GDNF reduces apoptosis in dopaminergic neurons in vitro. Neuroreport. (1995) 7:145–9. doi: 10.1097/00001756-199512000-00035
28. Tomac A, Lindqvist E, Lin LF, Ogren SO, Young D, Hoffer BJ, et al. Protection and repair of the nigrostriatal dopaminergic system by GDNF in vivo. Nature. (1995) 373:335–9. doi: 10.1038/373335a0
29. Voutilainen MH, Back S, Porsti E, Toppinen L, Lindgren L, Lindholm P, et al. Mesencephalic astrocyte-derived neurotrophic factor is neurorestorative in rat model of Parkinson's disease. J Neurosci. (2009) 29:9651–9. doi: 10.1523/JNEUROSCI.0833-09.2009
30. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. (2010) 119:7–35. doi: 10.1007/s00401-009-0619-8
31. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. (2017) 541:481–7. doi: 10.1038/nature21029
32. Clarke LE, Liddelow SA, Chakraborty C, Munch AE, Heiman M, Barres BA. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci USA. (2018) 115:E1896–905. doi: 10.1073/pnas.1800165115
33. Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, et al. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem. (2010) 285:9262–72. doi: 10.1074/jbc.M109.081125
34. Loria F, Vargas JY, Bousset L, Syan S, Salles A, Melki R, et al. alpha-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol. (2017) 134:789–808. doi: 10.1007/s00401-017-1746-2
35. Rostami J, Holmqvist S, Lindstrom V, Sigvardson J, Westermark GT, Ingelsson M, et al. Human astrocytes transfer aggregated alpha-synuclein via tunneling nanotubes. J Neurosci. (2017) 37:11835–53. doi: 10.1523/JNEUROSCI.0983-17.2017
36. Yun SP, Kam TI, Panicker N, Kim S, Oh Y, Park JS, et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson's disease. Nat Med. (2018) 24:931–8. doi: 10.1038/s41591-018-0051-5
37. Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson's disease. Lancet Neurol. (2020) 19:170–8. doi: 10.1016/S1474-4422(19)30287-X
38. Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. (2016) 89:37–53. doi: 10.1016/j.neuron.2015.11.013
39. Booth HDE, Hirst WD, Wade-Martins R. The role of astrocyte dysfunction in Parkinson's disease pathogenesis. Trends Neurosci. (2017) 40:358–70. doi: 10.1016/j.tins.2017.04.001
40. Kim JM, Cha SH, Choi YR, Jou I, Joe EH, Park SM. DJ-1 deficiency impairs glutamate uptake into astrocytes via the regulation of flotillin-1 and caveolin-1 expression. Sci Rep. (2016) 6:28823. doi: 10.1038/srep28823
41. Choi DJ, An J, Jou I, Park SM, Joe EH. A Parkinson's disease gene, DJ-1, regulates anti-inflammatory roles of astrocytes through prostaglandin D2 synthase expression. Neurobiol Dis. (2019) 127:482–91. doi: 10.1016/j.nbd.2019.04.003
42. Singh K, Han K, Tilve S, Wu K, Geller HM, Sack MN. Parkin targets NOD2 to regulate astrocyte endoplasmic reticulum stress and inflammation. Glia. (2018) 66:2427–37. doi: 10.1002/glia.23482
43. Choi I, Choi DJ, Yang H, Woo JH, Chang MY, Kim JY, et al. PINK1 expression increases during brain development and stem cell differentiation, and affects the development of GFAP-positive astrocytes. Mol Brain. (2016) 9:5. doi: 10.1186/s13041-016-0186-6
44. Sun L, Shen R, Agnihotri SK, Chen Y, Huang Z, Bueler H. Lack of PINK1 alters glia innate immune responses and enhances inflammation-induced, nitric oxide-mediated neuron death. Sci Rep. (2018) 8:383. doi: 10.1038/s41598-017-18786-w
45. Barodia SK, McMeekin LJ, Creed RB, Quinones EK, Cowell RM, Goldberg MS. PINK1 phosphorylates ubiquitin predominantly in astrocytes. NPJ Parkinsons Dis. (2019) 5:29. doi: 10.1038/s41531-019-0101-9
46. Sengul B, Dursun E, Verkhratsky A, Gezen-Ak D. Overexpression of alpha-synuclein reorganises growth factor profile of human astrocytes. Mol Neurobiol. (2021) 58:184–203. doi: 10.1007/s12035-020-02114-x
47. Gu XL, Long CX, Sun L, Xie C, Lin X, Cai H. Astrocytic expression of Parkinson's disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol Brain. (2010) 3:12. doi: 10.1186/1756-6606-3-12
48. Lee JH, Han J-H, Kim H, Park SM, Joe E-H, Jou I. Parkinson's disease-associated LRRK2-G2019S mutant acts through regulation of SERCA activity to control ER stress in astrocytes. Acta Neuropathol Commun. (2019) 7:68. doi: 10.1186/s40478-019-0716-4
49. Sanyal A, DeAndrade MP, Novis HS, Lin S, Chang J, Lengacher N, et al. Lysosome and inflammatory defects in GBA1-mutant astrocytes are normalized by LRRK2 inhibition. Mov Disord. (2020) 35:760–73. doi: 10.1002/mds.27994
50. di Domenico A, Carola G, Calatayud C, Pons-Espinal M, Munoz JP, Richaud-Patin Y, et al. Patient-specific iPSC-derived astrocytes contribute to non-cell-autonomous neurodegeneration in Parkinson's disease. Stem Cell Rep. (2019) 12:213–29. doi: 10.1016/j.stemcr.2018.12.011
51. Booth HDE, Wessely F, Connor-Robson N, Rinaldi F, Vowles J, Browne C, et al. RNA sequencing reveals MMP2 and TGFB1 downregulation in LRRK2 G2019S Parkinson's iPSC-derived astrocytes. Neurobiol Dis. (2019) 129:56–66. doi: 10.1016/j.nbd.2019.05.006
52. Sonninen TM, Hamalainen RH, Koskuvi M, Oksanen M, Shakirzyanova A, Wojciechowski S, et al. Metabolic alterations in Parkinson's disease astrocytes. Sci Rep. (2020) 10:14474. doi: 10.1038/s41598-020-71329-8
53. Aflaki E, Stubblefield BK, McGlinchey RP, McMahon B, Ory DS, Sidransky E. A characterization of Gaucher iPS-derived astrocytes: potential implications for Parkinson's disease. Neurobiol Dis. (2020) 134:104647. doi: 10.1016/j.nbd.2019.104647
54. Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. (2003) 299:256–9. doi: 10.1126/science.1077209
55. Mullett SJ, Hinkle DA. DJ-1 deficiency in astrocytes selectively enhances mitochondrial Complex I inhibitor-induced neurotoxicity. J Neurochem. (2011) 117:375–87. doi: 10.1111/j.1471-4159.2011.07175.x
56. Waak J, Weber SS, Waldenmaier A, Gorner K, Alunni-Fabbroni M, Schell H, et al. Regulation of astrocyte inflammatory responses by the Parkinson's disease-associated gene DJ-1. FASEB J. (2009) 23:2478–89. doi: 10.1096/fj.08-125153
57. Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. (2014) 34:11929–47. doi: 10.1523/JNEUROSCI.1860-14.2014
58. Khasnavis S, Pahan K. Cinnamon treatment upregulates neuroprotective proteins Parkin and DJ-1 and protects dopaminergic neurons in a mouse model of Parkinson's disease. J Neuroimmune Pharmacol. (2014) 9:569–81. doi: 10.1007/s11481-014-9552-2
59. Kawajiri S, Saiki S, Sato S, Hattori N. Genetic mutations and functions of PINK1. Trends Pharmacol Sci. (2011) 32:573–80. doi: 10.1016/j.tips.2011.06.001
60. Koyano F, Okatsu K, Ishigaki S, Fujioka Y, Kimura M, Sobue G, et al. The principal PINK1 and Parkin cellular events triggered in response to dissipation of mitochondrial membrane potential occur in primary neurons. Genes Cells. (2013) 18:672–81. doi: 10.1111/gtc.12066
61. Riederer P, Berg D, Casadei N, Cheng F, Classen J, Dresel C, et al. alpha-Synuclein in Parkinson's disease: causal or bystander? J Neural Transm. (2019) 126:815–40. doi: 10.1007/s00702-019-02025-9
62. Wakabayashi K, Hayashi S, Yoshimoto M, Kudo H, Takahashi H. NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson's disease brains. Acta Neuropathol. (2000) 99:14–20. doi: 10.1007/PL00007400
63. Braak H, Sastre M, Del Tredici K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson's disease. Acta Neuropathol. (2007) 114:231–41. doi: 10.1007/s00401-007-0244-3
64. Lee HJ, Kim C, Lee SJ. Alpha-synuclein stimulation of astrocytes: potential role for neuroinflammation and neuroprotection. Oxid Med Cell Longev. (2010) 3:283–7. doi: 10.4161/oxim.3.4.12809
65. Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. (2004) 44:601–7. doi: 10.1016/j.neuron.2004.11.005
66. Mata IF, Wedemeyer WJ, Farrer MJ, Taylor JP, Gallo KA. LRRK2 in Parkinson's disease: protein domains and functional insights. Trends Neurosci. (2006) 29:286–93. doi: 10.1016/j.tins.2006.03.006
67. Tolosa E, Vila M, Klein C, Rascol O. LRRK2 in Parkinson disease: challenges of clinical trials. Nat Rev Neurol. (2020) 16:97–107. doi: 10.1038/s41582-019-0301-2
68. Chen X, Liu Z, Cao BB, Qiu YH, Peng YP. TGF-beta1 neuroprotection via inhibition of microglial activation in a rat model of Parkinson's disease. J Neuroimmune Pharmacol. (2017) 12:433–46. doi: 10.1007/s11481-017-9732-y
69. Oh SH, Kim HN, Park HJ, Shin JY, Kim DY, Lee PH. The cleavage effect of mesenchymal stem cell and its derived matrix Metalloproteinase-2 on extracellular alpha-synuclein aggregates in parkinsonian models. Stem Cells Transl Med. (2017) 6:949–61. doi: 10.5966/sctm.2016-0111
70. Strokin M, Reiser G. Neurons and astrocytes in an infantile neuroaxonal dystrophy (INAD) mouse model show characteristic alterations in glutamate-induced Ca(2+) signaling. Neurochem Int. (2017) 108:121–32. doi: 10.1016/j.neuint.2017.03.004
71. Qiao C, Yin N, Gu HY, Zhu JL, Ding JH, Lu M, et al. Atp13a2 deficiency aggravates astrocyte-mediated neuroinflammation via NLRP3 inflammasome activation. CNS Neurosci Ther. (2016) 22:451–60. doi: 10.1111/cns.12514
72. Tsunemi T, Ishiguro Y, Yoroisaka A, Valdez C, Miyamoto K, Ishikawa K, et al. Astrocytes protect human dopaminergic neurons from alpha-synuclein accumulation and propagation. J Neurosci. (2020) 40:8618–28. doi: 10.1523/JNEUROSCI.0954-20.2020
73. Vingill S, Brockelt D, Lancelin C, Tatenhorst L, Dontcheva G, Preisinger C, et al. Loss of FBXO7 (PARK15) results in reduced proteasome activity and models a parkinsonism-like phenotype in mice. EMBO J. (2016) 35:2008–25. doi: 10.15252/embj.201593585
74. Bedford L, Hay D, Devoy A, Paine S, Powe DG, Seth R, et al. Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J Neurosci. (2008) 28:8189–98. doi: 10.1523/JNEUROSCI.2218-08.2008
75. Walden H, Muqit MM. Ubiquitin and Parkinson's disease through the looking glass of genetics. Biochem J. (2017) 474:1439–51. doi: 10.1042/BCJ20160498
76. Sassone J, Reale C, Dati G, Regoni M, Pellecchia MT, Garavaglia B. The role of VPS35 in the pathobiology of Parkinson's disease. Cell Mol Neurobiol. (2020) 41:199–227. doi: 10.1007/s10571-020-00849-8
77. Chiu CC, Weng YH, Huang YZ, Chen RS, Liu YC, Yeh TH, et al. (D620N) VPS35 causes the impairment of Wnt/beta-catenin signaling cascade and mitochondrial dysfunction in a PARK17 knockin mouse model. Cell Death Dis. (2020) 11:1018. doi: 10.1038/s41419-020-03228-9
78. Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. (2018) 18:225–42. doi: 10.1038/nri.2017.125
79. Cunningham CL, Martínez-Cerdeño V, Noctor SC. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci. (2013) 33:4216–33. doi: 10.1523/JNEUROSCI.3441-12.2013
80. Arandjelovic S, Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol. (2015) 16:907–17. doi: 10.1038/ni.3253
81. Parkhurst CN, Yang G, Ninan I, Savas JN, Yates J. R. III., et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. (2013) 155:1596–609. doi: 10.1016/j.cell.2013.11.030
82. Miyamoto A, Wake H, Ishikawa AW, Eto K, Shibata K, Murakoshi H, et al. Microglia contact induces synapse formation in developing somatosensory cortex. Nat Commun. (2016) 7:12540. doi: 10.1038/ncomms12540
83. Doorn KJ, Moors T, Drukarch B, van de Berg W, Lucassen PJ, van Dam AM. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson's disease patients. Acta Neuropathol Commun. (2014) 2:90. doi: 10.1186/PREACCEPT-2025829283135633
84. Lu J, Dou F, Yu Z. The potassium channel KCa3.1 represents a valid pharmacological target for microgliosis-induced neuronal impairment in a mouse model of Parkinson's disease. J Neuroinflammation. (2019) 16:273. doi: 10.1186/s12974-019-1682-2
85. La Vitola P, Balducci C, Baroni M, Artioli L, Santamaria G, Castiglioni M, et al. Peripheral inflammation exacerbates alpha-synuclein toxicity and neuropathology in Parkinson's models. Neuropathol Appl Neurobiol. (2021) 47:43–60. doi: 10.1111/nan.12644
86. Boi L, Pisanu A, Greig NH, Scerba MT, Tweedie D, Mulas G, et al. Immunomodulatory drugs alleviate l-dopa-induced dyskinesia in a rat model of Parkinson's disease. Mov Disord. (2019) 34:1818–30. doi: 10.1002/mds.27799
87. Costa T, Fernandez-Villalba E, Izura V, Lucas-Ochoa AM, Menezes-Filho NJ, Santana RC, et al. Combined 1-Deoxynojirimycin and Ibuprofen treatment decreases microglial activation, phagocytosis and dopaminergic degeneration in MPTP-treated mice. J Neuroimmune Pharmacol. (2020) 16:390–402. doi: 10.1007/s11481-020-09925-8
88. Nash Y, Schmukler E, Trudler D, Pinkas-Kramarski R, Frenkel D. DJ-1 deficiency impairs autophagy and reduces alpha-synuclein phagocytosis by microglia. J Neurochem. (2017) 143:584–94. doi: 10.1111/jnc.14222
89. Chien CH, Lee MJ, Liou HC, Liou HH, Fu WM. Microglia-derived cytokines/chemokines are involved in the enhancement of LPS-induced loss of nigrostriatal dopaminergic neurons in DJ-1 Knockout mice. PLoS ONE. (2016) 11:e0151569. doi: 10.1371/journal.pone.0151569
90. Trudler D, Weinreb O, Mandel SA, Youdim MB, Frenkel D. DJ-1 deficiency triggers microglia sensitivity to dopamine toward a pro-inflammatory phenotype that is attenuated by rasagiline. J Neurochem. (2014) 129:434–47. doi: 10.1111/jnc.12633
91. Dionísio PEA, Oliveira SR, Amaral J, Rodrigues CMP. Loss of microglial parkin inhibits necroptosis and contributes to neuroinflammation. Mol Neurobiol. (2019) 56:2990–3004. doi: 10.1007/s12035-018-1264-9
92. Mouton-Liger F, Rosazza T, Sepulveda-Diaz J, Ieang A, Hassoun SM, Claire E, et al. Parkin deficiency modulates NLRP3 inflammasome activation by attenuating an A20-dependent negative feedback loop. Glia. (2018) 66:1736–51. doi: 10.1002/glia.23337
93. Duffy MF, Collier TJ, Patterson JR, Kemp CJ, Luk KC, Tansey MG, et al. Lewy body-like alpha-synuclein inclusions trigger reactive microgliosis prior to nigral degeneration. J Neuroinflammation. (2018) 15:129. doi: 10.1186/s12974-018-1171-z
94. Hoenen C, Gustin A, Birck C, Kirchmeyer M, Beaume N, Felten P, et al. Alpha-Synuclein proteins promote pro-inflammatory cascades in microglia: stronger effects of the A53T mutant. PLoS ONE. (2016) 11:e0162717. doi: 10.1371/journal.pone.0162717
95. Jiang T, Hoekstra J, Heng X, Kang W, Ding J, Liu J, et al. P2X7 receptor is critical in α-synuclein–mediated microglial NADPH oxidase activation. Neurobiol Aging. (2015) 36:2304–18. doi: 10.1016/j.neurobiolaging.2015.03.015
96. Dwyer Z, Rudyk C, Thompson A, Farmer K, Fenner B, Fortin T, et al. Leucine-rich repeat kinase-2 (LRRK2) modulates microglial phenotype and dopaminergic neurodegeneration. Neurobiol Aging. (2020) 91:45–55. doi: 10.1016/j.neurobiolaging.2020.02.017
97. Panagiotakopoulou V, Ivanyuk D, De Cicco S, Haq W, Arsić A, Yu C, et al. Interferon-γ signaling synergizes with LRRK2 in neurons and microglia derived from human induced pluripotent stem cells. Nat Commun. (2020) 11:5163. doi: 10.1038/s41467-020-18755-4
98. Kim J, Pajarillo E, Rizor A, Son DS, Lee J, Aschner M, et al. LRRK2 kinase plays a critical role in manganese-induced inflammation and apoptosis in microglia. PLoS ONE. (2019) 14:e0210248. doi: 10.1371/journal.pone.0210248
99. Miki Y, Shimoyama S, Kon T, Ueno T, Hayakari R, Tanji K, et al. Alteration of autophagy-related proteins in peripheral blood mononuclear cells of patients with Parkinson's disease. Neurobiol Aging. (2018) 63:33–43. doi: 10.1016/j.neurobiolaging.2017.11.006
100. Hou X, Watzlawik JO, Fiesel FC, Springer W. Autophagy in Parkinson's disease. J Mol Biol. (2020) 432:2651–72. doi: 10.1016/j.jmb.2020.01.037
101. Qin Y, Qiu J, Wang P, Liu J, Zhao Y, Jiang F, et al. Impaired autophagy in microglia aggravates dopaminergic neurodegeneration by regulating NLRP3 inflammasome activation in experimental models of Parkinson's disease. Brain Behav Immun. (2021) 91:324–38. doi: 10.1016/j.bbi.2020.10.010
102. Cheng J, Liao Y, Dong Y, Hu H, Yang N, Kong X, et al. Microglial autophagy defect causes parkinson disease-like symptoms by accelerating inflammasome activation in mice. Autophagy. (2020) 16:2193–205. doi: 10.1080/15548627.2020.1719723
103. Lee HJ, Cho ED, Lee KW, Kim JH, Cho SG, Lee SJ. Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp Mol Med. (2013) 45:e22. doi: 10.1038/emm.2013.45
104. Grünewald A, Kumar KR, Sue CM. New insights into the complex role of mitochondria in Parkinson's disease. Prog Neurobiol. (2019) 177:73–93. doi: 10.1016/j.pneurobio.2018.09.003
105. Ji YJ, Wang HL, Yin BL, Ren XY. Down-regulation of DJ-1 augments neuroinflammation via Nrf2/Trx1/NLRP3 axis in MPTP-induced Parkinson's disease mouse model. Neuroscience. (2020) 442:253–63. doi: 10.1016/j.neuroscience.2020.06.001
106. Wang T, Zhao N, Peng L, Li Y, Huang X, Zhu J, et al. DJ-1 regulates microglial polarization through P62-Mediated TRAF6/IRF5 signaling in cerebral ischemia-reperfusion. Front Cell Dev Biol. (2020) 8:593890. doi: 10.3389/fcell.2020.593890
107. Burré J, Sharma M, Südhof TC. Cell biology and pathophysiology of α-Synuclein. Cold Spring Harb Perspect Med. (2018) 8:a024091. doi: 10.1101/cshperspect.a024091
108. Choi YR, Kang SJ, Kim JM, Lee SJ, Jou I, Joe EH, et al. FcγRIIB mediates the inhibitory effect of aggregated α-synuclein on microglial phagocytosis. Neurobiol Dis. (2015) 83:90–9. doi: 10.1016/j.nbd.2015.08.025
109. Lim S, Kim HJ, Kim DK, Lee SJ. Non-cell-autonomous actions of α-synuclein: implications in glial synucleinopathies. Prog Neurobiol. (2018) 169:158–71. doi: 10.1016/j.pneurobio.2018.06.010
110. Yang L, Mao K, Yu H, Chen J. Neuroinflammatory responses and Parkinson' disease: pathogenic mechanisms and therapeutic targets. J Neuroimmune Pharmacol. (2020) 15:830–7. doi: 10.1007/s11481-020-09926-7
111. Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. (2013) 4:1562. doi: 10.1038/ncomms2534
112. Qiao H, Zhang Q, Yuan H, Li Y, Wang D, Wang R, et al. Elevated neuronal alpha-synuclein promotes microglia activation after spinal cord ischemic/reperfused injury. Neuroreport. (2015) 26:656–61. doi: 10.1097/WNR.0000000000000406
113. Qiao H, He X, Zhang Q, Zhang N, Li L, Hui Y, et al. Alpha-synuclein induces microglial cell migration through stimulating HIF-1alpha accumulation. J Neurosci Res. (2017) 95:1809–17. doi: 10.1002/jnr.24012
114. Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, et al. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia. (2013) 61:349–60. doi: 10.1002/glia.22437
115. Panicker N, Sarkar S, Harischandra DS, Neal M, Kam TI, Jin H, et al. Fyn kinase regulates misfolded α-synuclein uptake and NLRP3 inflammasome activation in microglia. J Exp Med. (2019) 216:1411–30. doi: 10.1084/jem.20182191
116. Deng H, Wang P, Jankovic J. The genetics of Parkinson disease. Ageing Res Rev. (2018) 42:72–85. doi: 10.1016/j.arr.2017.12.007
117. Galet B, Cheval H, Ravassard P. Patient-derived midbrain organoids to explore the molecular basis of Parkinson's disease. Front Neurol. (2020) 11:1005. doi: 10.3389/fneur.2020.01005
118. Marker DF, Puccini JM, Mockus TE, Barbieri J, Lu SM, Gelbard HA. LRRK2 kinase inhibition prevents pathological microglial phagocytosis in response to HIV-1 Tat protein. J Neuroinflammation. (2012) 9:261. doi: 10.1186/1742-2094-9-261
119. Lee H, Flynn R, Sharma I, Haberman E, Carling PJ, Nicholls FJ, et al. LRRK2 is recruited to phagosomes and co-recruits RAB8 and RAB10 in human pluripotent stem cell-derived macrophages. Stem Cell Rep. (2020) 14:940–55. doi: 10.1016/j.stemcr.2020.04.001
120. Liu Z, Xu E, Zhao HT, Cole T, West AB. LRRK2 and Rab10 coordinate macropinocytosis to mediate immunological responses in phagocytes. EMBO J. (2020) 39:e104862. doi: 10.15252/embj.2020104862
121. Kim KS, Marcogliese PC, Yang J, Callaghan SM, Resende V, Abdel-Messih E, et al. Regulation of myeloid cell phagocytosis by LRRK2 via WAVE2 complex stabilization is altered in Parkinson's disease. Proc Natl Acad Sci USA. (2018) 115:E5164–73. doi: 10.1073/pnas.1718946115
122. Ho DH, Je AR, Lee H, Son I, Kweon HS, Kim HG, et al. LRRK2 kinase activity induces mitochondrial fission in microglia via Drp1 and modulates neuroinflammation. Exp Neurobiol. (2018) 27:171–80. doi: 10.5607/en.2018.27.3.171
123. Moehle MS, Webber PJ, Tse T, Sukar N, Standaert DG, DeSilva TM, et al. LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci. (2012) 32:1602–11. doi: 10.1523/JNEUROSCI.5601-11.2012
124. Russo I, Kaganovich A, Ding J, Landeck N, Mamais A, Varanita T, et al. Transcriptome analysis of LRRK2 knock-out microglia cells reveals alterations of inflammatory- and oxidative stress-related pathways upon treatment with α-synuclein fibrils. Neurobiol Dis. (2019) 129:67–78. doi: 10.1016/j.nbd.2019.05.012
125. Kim C, Beilina A, Smith N, Li Y, Kim M, Kumaran R, et al. LRRK2 mediates microglial neurotoxicity via NFATc2 in rodent models of synucleinopathies. Sci Transl Med. (2020) 12:eaay0399. doi: 10.1126/scitranslmed.aay0399
126. Keatinge M, Bui H, Menke A, Chen YC, Sokol AM, Bai Q, et al. Glucocerebrosidase 1 deficient Danio rerio mirror key pathological aspects of human Gaucher disease and provide evidence of early microglial activation preceding alpha-synuclein-independent neuronal cell death. Hum Mol Genet. (2015) 24:6640–52. doi: 10.1093/hmg/ddv369
127. Pandey MK, Burrow TA, Rani R, Martin LJ, Witte D, Setchell KD, et al. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature. (2017) 543:108–12. doi: 10.1038/nature21368
128. Chen D, Zhu C, Wang X, Feng X, Pang S, Huang W, et al. A novel and functional variant within the ATG5 gene promoter in sporadic Parkinson's disease. Neurosci Lett. (2013) 538:49–53. doi: 10.1016/j.neulet.2013.01.044
129. Beutler B. Innate immunity: an overview. Mol Immunol. (2004) 40:845–59. doi: 10.1016/j.molimm.2003.10.005
130. Pennock ND, White JT, Cross EW, Cheney EE, Tamburini BA, Kedl RM. T cell responses: naive to memory and everything in between. Adv Physiol Educ. (2013) 37:273–83. doi: 10.1152/advan.00066.2013
131. Kuchroo VK, Ohashi PS, Sartor RB, Vinuesa CG. Dysregulation of immune homeostasis in autoimmune diseases. Nat Med. (2012) 18:42–7. doi: 10.1038/nm.2621
132. Gray MT, Woulfe JM. Striatal blood-brain barrier permeability in Parkinson's disease. J Cereb Blood Flow Metab. (2015) 35:747–50. doi: 10.1038/jcbfm.2015.32
133. Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. (2018) 14:133–50. doi: 10.1038/nrneurol.2017.188
134. Al-Bachari S, Naish JH, Parker GJM, Emsley HCA, Parkes LM. Blood-brain barrier leakage is increased in Parkinson's disease. Front Physiol. (2020) 11:593026. doi: 10.3389/fphys.2020.593026
135. Alvarez-Luquin DD, Arce-Sillas A, Leyva-Hernandez J, Sevilla-Reyes E, Boll MC, Montes-Moratilla E, et al. Regulatory impairment in untreated Parkinson's disease is not restricted to Tregs: other regulatory populations are also involved. J Neuroinflammation. (2019) 16:212. doi: 10.1186/s12974-019-1606-1
136. Baba Y, Kuroiwa A, Uitti RJ, Wszolek ZK, Yamada T. Alterations of T-lymphocyte populations in Parkinson disease. Parkinsonism Relat Disord. (2005) 11:493–8. doi: 10.1016/j.parkreldis.2005.07.005
137. Bas J, Calopa M, Mestre M, Mollevi DG, Cutillas B, Ambrosio S, et al. Lymphocyte populations in Parkinson's disease and in rat models of parkinsonism. J Neuroimmunol. (2001) 113:146–52. doi: 10.1016/S0165-5728(00)00422-7
138. Brochard V, Combadiere B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. (2009) 119:182–92. doi: 10.1172/JCI36470
139. Cebrian C, Zucca FA, Mauri P, Steinbeck JA, Studer L, Scherzer CR, et al. MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. Nat Commun. (2014) 5:3633. doi: 10.1038/ncomms4633
140. Harms AS, Cao S, Rowse AL, Thome AD, Li X, Mangieri LR, et al. MHCII is required for alpha-synuclein-induced activation of microglia, CD4 T cell proliferation, dopaminergic neurodegeneration. J Neurosci. (2013) 33:9592–600. doi: 10.1523/JNEUROSCI.5610-12.2013
141. Kustrimovic N, Comi C, Magistrelli L, Rasini E, Legnaro M, Bombelli R, et al. Parkinson's disease patients have a complex phenotypic and functional Th1 bias: cross-sectional studies of CD4+ Th1/Th2/T17 and Treg in drug-naive and drug-treated patients. J Neuroinflammation. (2018) 15:205. doi: 10.1186/s12974-018-1248-8
142. Lindestam Arlehamn CS, Dhanwani R, Pham J, Kuan R, Frazier A, Rezende Dutra J, et al. alpha-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson's disease. Nat Commun. (2020) 11:1875. doi: 10.1038/s41467-020-15626-w
143. Liu Z, Huang Y, Cao BB, Qiu YH, Peng YP. Th17 cells induce dopaminergic neuronal death via LFA-1/ICAM-1 interaction in a mouse model of Parkinson's disease. Mol Neurobiol. (2017) 54:7762–76. doi: 10.1007/s12035-016-0249-9
144. Liu Z, Qiu AW, Huang Y, Yang Y, Chen JN, Gu TT, et al. IL-17A exacerbates neuroinflammation and neurodegeneration by activating microglia in rodent models of Parkinson's disease. Brain Behav Immun. (2019) 81:630–45. doi: 10.1016/j.bbi.2019.07.026
145. Reynolds AD, Banerjee R, Liu J, Gendelman HE, Mosley RL. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson's disease. J Leukoc Biol. (2007) 82:1083–094. doi: 10.1189/jlb.0507296
146. Reynolds AD, Stone DK, Hutter JA, Benner EJ, Mosley RL, Gendelman HE. Regulatory T cells attenuate Th17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson's disease. J Immunol. (2010) 184:2261–71. doi: 10.4049/jimmunol.0901852
147. Sommer A, Fadler T, Dorfmeister E, Hoffmann AC, Xiang W, Winner B, et al. Infiltrating T lymphocytes reduce myeloid phagocytosis activity in synucleinopathy model. J Neuroinflammation. (2016) 13:174. doi: 10.1186/s12974-016-0632-5
148. Sommer A, Marxreiter F, Krach F, Fadler T, Grosch J, Maroni M, et al. Th17 lymphocytes induce neuronal cell death in a human iPSC-based model of Parkinson's disease. Cell Stem Cell. (2018) 23:123–31 e126. doi: 10.1016/j.stem.2018.06.015
149. Stevens CH, Rowe D, Morel-Kopp M-C, Orr C, Russell T, et al. Reduced T helper and B lymphocytes in Parkinson's disease. J Neuroimmunol. (2012) 252:95–9. doi: 10.1016/j.jneuroim.2012.07.015
150. Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin-Liebes J, et al. T cells from patients with Parkinson's disease recognize alpha-synuclein peptides. Nature. (2017) 546:656–61. doi: 10.1038/nature22815
151. Theodore S, Cao S, McLean PJ, Standaert DG. Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J Neuropathol Exp Neurol. (2008) 67:1149–58. doi: 10.1097/NEN.0b013e31818e5e99
152. Balestrino R, Schapira AHV. Parkinson disease. Eur J Neurol. (2020) 27:27–42. doi: 10.1111/ene.14108
153. Horsager J, Andersen KB, Knudsen K, Skjaerbaek C, Fedorova TD, Okkels N, et al. Brain-first versus body-first Parkinson's disease: a multimodal imaging case-control study. Brain. (2020) 143:3077–88. doi: 10.1093/brain/awaa238
154. Morais VA, Haddad D, Craessaerts K, De Bock PJ, Swerts J, Vilain S, et al. PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science. (2014) 344:203–7. doi: 10.1126/science.1249161
155. Zhang P, Shao XY, Qi GJ, Chen Q, Bu LL, Chen LJ, et al. Cdk5-dependent activation of neuronal inflammasomes in Parkinson's disease. Mov Disord. (2016) 31:366–76. doi: 10.1002/mds.26488
156. Chatterjee K, Roy A, Banerjee R, Choudhury S, Mondal B, Halder S, et al. Inflammasome and α-synuclein in Parkinson's disease: a cross-sectional study. J Neuroimmunol. (2020) 338:577089. doi: 10.1016/j.jneuroim.2019.577089
157. Sarkar S, Malovic E, Harishchandra DS, Ghaisas S, Panicker N, Charli A, et al. Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson's disease. NPJ Parkinsons Dis. (2017) 3:30. doi: 10.1038/s41531-017-0032-2
158. Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and tbk1 activation to promote mitophagy. Mol Cell. (2015) 60:7–20. doi: 10.1016/j.molcel.2015.08.016
159. Torres-Odio S, Key J, Hoepken HH, Canet-Pons J, Valek L, Roller B, et al. Progression of pathology in PINK1-deficient mouse brain from splicing via ubiquitination, ER stress, and mitophagy changes to neuroinflammation. J Neuroinflammation. (2017) 14:154. doi: 10.1186/s12974-017-0928-0
160. Matheoud D, Sugiura A, Bellemare-Pelletier A, Laplante A, Rondeau C, Chemali M, et al. Parkinson's disease-related proteins PINK1 and parkin repress mitochondrial antigen presentation. Cell. (2016) 166:314–27. doi: 10.1016/j.cell.2016.05.039
161. Matheoud D, Cannon T, Voisin A, Penttinen AM, Ramet L, Fahmy AM, et al. Intestinal infection triggers Parkinson's disease-like symptoms in Pink1(-/-) mice. Nature. (2019) 571:565–9. doi: 10.1038/s41586-019-1405-y
162. Oliveras-Salva M, Van Rompuy AS, Heeman B, Van den Haute C, Baekelandt V. Loss-of-function rodent models for parkin and PINK1. J Parkinsons Dis. (2011) 1:229–51. doi: 10.3233/JPD-2011-11041
163. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson's disease. Cell. (2016) 167:1469–80 e1412. doi: 10.1016/j.cell.2016.11.018
164. Kanhere A, Hertweck A, Bhatia U, Gokmen MR, Perucha E, Jackson I, et al. T-bet and GATA3 orchestrate Th1 and Th2 differentiation through lineage-specific targeting of distal regulatory elements. Nat Commun. (2012) 3:1268. doi: 10.1038/ncomms2260
165. Storelli E, Cassina N, Rasini E, Marino F, Cosentino M. Do Th17 Lymphocytes and IL-17 contribute to Parkinson's disease? A systematic review of available evidence. Front Neurol. (2019) 10:13. doi: 10.3389/fneur.2019.00013
166. Kolls JK, McCray PB Jr, Chan YR. Cytokine-mediated regulation of antimicrobial proteins. Nat Rev Immunol. (2008) 8:829–35. doi: 10.1038/nri2433
167. Walker JA, McKenzie ANJ. TH2 cell development and function. Nat Rev Immunol. (2018) 18:121–33. doi: 10.1038/nri.2017.118
168. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. (2016) 39:98–106. doi: 10.1097/COC.0000000000000239
169. Cook DA, Kannarkat GT, Cintron AF, Butkovich LM, Fraser KB, Chang J, et al. LRRK2 levels in immune cells are increased in Parkinson's disease. NPJ Parkinsons Dis. (2017) 3:11. doi: 10.1038/s41531-017-0010-8
170. Rostami J, Fotaki G, Sirois J, Mzezewa R, Bergstrom J, Essand M, et al. Astrocytes have the capacity to act as antigen-presenting cells in the Parkinson's disease brain. J Neuroinflammation. (2020) 17:119. doi: 10.1186/s12974-020-01776-7
171. Walling BL, Kim M. LFA-1 in T cell migration and differentiation. Front Immunol. (2018) 9:952. doi: 10.3389/fimmu.2018.00952
172. Prajeeth CK, Kronisch J, Khorooshi R, Knier B, Toft-Hansen H, Gudi V, et al. Effectors of Th1 and Th17 cells act on astrocytes and augment their neuroinflammatory properties. J Neuroinflammation. (2017) 14:204. doi: 10.1186/s12974-017-0978-3
173. Akiyama H, Kawamata T, Yamada T, Tooyama I, Ishii T, McGeer PL. Expression of intercellular adhesion molecule (ICAM)-1 by a subset of astrocytes in Alzheimer disease and some other degenerative neurological disorders. Acta Neuropathol. (1993) 85:628–34. doi: 10.1007/BF00334673
174. Miklossy J, Doudet DD, Schwab C, Yu S, McGeer EG, McGeer PL. Role of ICAM-1 in persisting inflammation in Parkinson disease and MPTP monkeys. Exp Neurol. (2006) 197:275–83. doi: 10.1016/j.expneurol.2005.10.034
175. You T, Bi Y, Li J, Zhang M, Chen X, Zhang K, et al. IL-17 induces reactive astrocytes and up-regulation of vascular endothelial growth factor (VEGF) through JAK/STAT signaling. Sci Rep. (2017) 7:41779. doi: 10.1038/srep41779
176. Chabaud M, Fossiez F, Taupin JL, Miossec P. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J Immunol. (1998) 161:409–4.
177. Katz Y, Nadiv O, Beer Y. Interleukin-17 enhances tumor necrosis factor alpha-induced synthesis of interleukins 1,6, and 8 in skin and synovial fibroblasts: a possible role as a “fine-tuning cytokine” in inflammation processes. Arthritis Rheum. (2001) 44:2176–84. doi: 10.1002/1529-0131(200109)44:9<2176::AID-ART371>3.0.CO;2-4
178. Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. (2009) 9:556–67. doi: 10.1038/nri2586
179. Ma X, Reynolds SL, Baker BJ, Li X, Benveniste EN, Qin H. IL-17 enhancement of the IL-6 signaling cascade in astrocytes. J Immunol. (2010) 184:4898–906. doi: 10.4049/jimmunol.1000142
180. Noack M, Beringer A, Miossec P. Additive or synergistic interactions between IL-17A or IL-17F and TNF or IL-1beta depend on the cell type. Front Immunol. (2019) 10:1726. doi: 10.3389/fimmu.2019.01726
181. Athauda D, Maclagan K, Skene SS, Bajwa-Joseph M, Letchford D, Chowdhury K, et al. Exenatide once weekly versus placebo in Parkinson's disease: a randomised, double-blind, placebo-controlled trial. Lancet. (2017) 390:1664–75. doi: 10.1016/S0140-6736(17)31585-4
182. Jankovic J, Goodman I, Safirstein B, Marmon TK, Schenk DB, Koller M, et al. Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti-alpha-synuclein monoclonal antibody, in patients with parkinson disease: a randomized clinical trial. JAMA Neurol. (2018) 75:1206–14. doi: 10.1001/jamaneurol.2018.1487
183. Brys M, Fanning L, Hung S, Ellenbogen A, Penner N, Yang M, et al. Randomized phase I clinical trial of anti-alpha-synuclein antibody BIIB054. Mov Disord. (2019) 34:1154–63. doi: 10.1002/mds.27738
184. Volc D, Poewe W, Kutzelnigg A, Luhrs P, Thun-Hohenstein C, Schneeberger A, et al. Safety and immunogenicity of the alpha-synuclein active immunotherapeutic PD01A in patients with Parkinson's disease: a randomised, single-blinded, phase 1 trial. Lancet Neurol. (2020) 19:591–600. doi: 10.1016/S1474-4422(20)30136-8
185. Greenland JC, Cutting E, Kadyan S, Bond S, Chhabra A, Williams-Gray CH. Azathioprine immunosuppression and disease modification in Parkinson's disease (AZA-PD): a randomised double-blind placebo-controlled phase II trial protocol. BMJ Open. (2020) 10:e040527. doi: 10.1136/bmjopen-2020-040527
186. Gendelman HE, Zhang Y, Santamaria P, Olson KE, Schutt CR, Bhatti D, et al. Evaluation of the safety and immunomodulatory effects of sargramostim in a randomized, double-blind phase 1 clinical Parkinson's disease trial. NPJ Parkinsons Dis. (2017) 3:10. doi: 10.1038/s41531-017-0013-5
187. Xue LJ, Yang XZ, Tong Q, Shen P, Ma SJ, Wu SN, et al. Fecal microbiota transplantation therapy for Parkinson's disease: a preliminary study. Medicine. (2020) 99:e22035. doi: 10.1097/MD.0000000000022035
188. Huang H, Xu H, Luo Q, He J, Li M, Chen H, et al. Fecal microbiota transplantation to treat Parkinson's disease with constipation: a case report. Medicine. (2019) 98:e16163. doi: 10.1097/MD.0000000000016163
189. Liddelow SA, Marsh SE, Stevens B. Microglia and astrocytes in disease: dynamic duo or partners in crime? Trends Immunol. (2020) 41:820–35. doi: 10.1016/j.it.2020.07.006
190. McComish SF, Caldwell MA. Generation of defined neural populations from pluripotent stem cells. Philos Trans R Soc Lond B Biol Sci. (2018) 373:20170214. doi: 10.1098/rstb.2017.0214
191. Lancaster MA, Knoblich JA. Organogenesis in a dish: modeling development and disease using organoid technologies. Science. (2014) 345:1247125. doi: 10.1126/science.1247125
192. Ormel PR, Vieira de Sa R, van Bodegraven EJ, Karst H, Harschnitz O, Sneeboer MAM, et al. Microglia innately develop within cerebral organoids. Nat Commun. (2018) 9:4167. doi: 10.1038/s41467-018-06684-2
Keywords: Parkinson's disease, astrocyte, microglia, T lymphocytes, Th17 cell, neuroinflammation
Citation: MacMahon Copas AN, McComish SF, Fletcher JM and Caldwell MA (2021) The Pathogenesis of Parkinson's Disease: A Complex Interplay Between Astrocytes, Microglia, and T Lymphocytes? Front. Neurol. 12:666737. doi: 10.3389/fneur.2021.666737
Received: 10 February 2021; Accepted: 20 April 2021;
Published: 26 May 2021.
Edited by:
Andras Lakatos, University of Cambridge, United KingdomReviewed by:
Ayse Ulusoy, Helmholtz Association of German Research Centers (HZ), GermanyMichela Deleidi, Helmholtz Association of German Research Centers (HZ), Germany
Copyright © 2021 MacMahon Copas, McComish, Fletcher and Caldwell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maeve A. Caldwell, bWFldmUuY2FsZHdlbGxAdGNkLmll