 Burak Arslan
Burak Arslan Gökçe Ayhan Arslan3†
Gökçe Ayhan Arslan3† Aslı Tuncer
Aslı Tuncer Aylin Sepici Dinçel
Aylin Sepici Dinçel- 1Department of Medical Biochemistry, Gazi University Faculty of Medicine, Ankara, Turkey
- 2Department of Medical Biochemistry, Erciş State Hospital, Van, Turkey
- 3Department of Neurology, Erciş State Hospital, Van, Turkey
- 4Department of Neurology, Hacettepe University Faculty of Medicine, Ankara, Turkey
Objectives: The aim of this pilot study was to evaluate dynamic thiol-disulfide homeostasis as a novel oxidative stress parameter in multiple sclerosis (MS), neuromyelitis optica spectrum disorders (NMOSD), and myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) to better understand the role of thiol homeostasis in neuroimmunological diseases.
Methods: A total of 85 participants were included in this study, consisting of 18 healthy controls, 52 patients diagnosed with MS, seven with NMOSD, and eight with MOGAD. We measured total thiol (–SH+-S–S–) and native thiol (–SH) levels in the serum of all the participants, and in a subset of patients (n = 11), these parameters were investigated in paired cerebrospinal fluid (CSF) and serum samples. Dynamic disulfide concentrations were calculated separately. Finally, we determined if there was any relationship between clinical features and dynamic thiol homeostasis.
Results: There was a statistically significant difference between serum and CSF levels of biomarkers of thiol homeostasis. Serum total thiol (317.88 ± 66.04) and native thiol (211.61 ± 44.15) levels were significantly lower in relapsed patients compared to those in remission (368.84 ± 150.36 vs. 222.52 ± 70.59, respectively).
Conclusions: Oxidative stress plays a crucial role in the physiopathology of neuroimmunological diseases. Thiol homeostasis may be useful for monitoring disease activity.
Introduction
Multiple sclerosis (MS) is a chronic autoimmune demyelinating disease of the central nervous system (CNS) that causes neuronal damage which underlies many of the clinical features. It was first described by Jean-Martin Charcot in 1868 and is the most common cause of physical and cognitive disability in young adults (1). It has been estimated that ~2.3 million people worldwide suffer from MS (2). The disease is thought to occur in genetically susceptible individuals as a result of environmental factors and potential triggers such as Epstein Barr virus seropositivity, obesity, smoking, and Vitamin D deficiency. However, the exact cause of MS remains unknown (3–7). Myelin sheaths, oligodendrocytes, and inevitably, axons and neurons are damaged. MS is characterized by the presence of demyelinated areas called plaques in the brain and spinal cord. In early stages of the disease, the permeability of the blood–brain barrier increases due to the migration of peripheral immune cells to the central nervous system, and a significant inflammation of the parenchyma is observed (8). Importantly, both neuroinflammation and oxidative stress, which are closely interrelated, play a crucial role in the pathophysiology of MS (9–11). MS can be diagnosed based on various clinical presentations ranging from motor and autonomic dysfunction to psychobehavioral deficits (12–14). Although the presentation of the disease is generally relapsing–remitting (85%), ~15% of it is progressive from the onset. Secondary progression occurs in the later stages of the disease in most patients with relapsing–remitting MS (9).
Neuromyelitis optica spectrum disorder (NMOSD) is a type of astrocytopathy in which aquaporine-4 (AQP4)-IgG antibodies bind to AQP4 water channels on the end feet of astrocytes, leading to immune-mediated inflammation and secondary demyelination that can occur in varying degrees (15, 16). This disease is typically characterized by clinically moderate to severe attacks, and secondary progression is not usually seen. It is more common in middle-aged women, and complete recovery after attacks is usually rare (17, 18).
Myelin oligodendrocyte glycoprotein (MOG)-Ab-associated disease (MOGAD) has been proposed to be distinctly different from seronegative NMOSD. In this context, anti-MOG antibodies are developed against MOG protein expressed on myelin sheath and oligodendrocytes (19). Although MOGAD can occur at any age, younger adults are more susceptible. The course of the disease is characterized by monophasic or relapsing features. Currently, the relationship of MOGAD with MS and NMOSD is being discussed. Although this disease has clinical and radiological similarities with NMOSD and MS, the current trend is to classify MOGAD as a different entity from MS and NMOSD (20–23).
Multiple sclerosis treatment has emerged in the last 15 years. Today, there are different choices of disease-modifying treatments for different phases of the disease and medications with different immune actions. Glatiramer acetate and interferon beta 1a, teriflunomide, and dimethyl fumarate are the first-line drugs, whereas there are anti-CD20 treatment options as ocrelizumab and barrier blockers as natalizumab and fingolimod. For neuromyelitis optica, there are guidelines for using rituximab, azathioprine, mycophenolate mofetil, and eculizumab (15, 24).
Autoimmune mediated inflammatory tissue damage increases the production of reactive oxygen molecules that cause cell damage ultimately leading to cell death. In order to maintain the balance at the cellular level, the antioxidant system is activated in response to the production of reactive oxygen molecules (25). If the balance between these systems is not maintained, oxidative stress can emerge at the cellular level. There are many antioxidant defense mechanisms at the cellular level to eliminate oxidative stress, such as the glutathione system, catalase, thioredoxin-peroxiredoxin, alpha-ketoglutarate dehydrogenases, and endogen and exogen antioxidant molecules (26, 27). However, the measurement of only one of these does not give us adequate information about oxidative balance. Thiol-disulfide homeostasis is one of the most common parameters used to evaluate the redox balance in organisms. Thiols are organic complexes that contain a sulfur and a hydrogen group attached to a carbon. Reactive oxygen species (ROS) transfer their excess electrons to thiols, thus oxidizing them. As a result of this process, disulfide bonds are formed. These disulfide bonds are then broken down depending on the oxidant-antioxidant status of organism. Thiols are considered to be antioxidant molecules due to their reducing properties. This chain of events is termed dynamic thiol-disulfide homeostasis (28–31). Therefore, by evaluating this dynamic homeostasis, we can determine the oxidative status of an individual without having to measure oxidant and antioxidant molecules separately with a single run.
Neuroinflammation is one of the most common pathways seen in diseases associated with neurodegeneration (32). Importantly, certain diseases, particularly those characterized by neuroinflammation of the CNS, can lead to increased risk of oxidative stress and high oxygen consumption in the CNS. Thiol-disulfide homeostasis has been shown to play a role in the pathogenesis of many diseases, including cardiovascular diseases, rheumatoid arthritis, Parkinson, Alzheimer's, amyotrophic lateral sclerosis, and MS (33–38).
Previous studies have reported on the association between MS and oxidative stress (39, 40). Moreover, many studies using blood, cerebrospinal fluid, and post-mortem brain samples from patients with MS have shown impairments in reduction-oxidation (redox) homeostasis (26). However, no study to date has assessed MS, NMOSD, and MOGAD and their relationship with thiol-homeostasis. Although there have been hypotheses about the pathophysiology of the inflammatory CNS diseases that can mimic MS in the last decade, there are still many gaps that need to be filled. Although the relationship between inflammation and oxidative stress has been demonstrated in the pathophysiology of MS, this relationship is not yet clear in diseases such as NMOSD and MOGAD (15). In this study, we tried to fill these gaps in the disorders.
Distinguishing MS from CNS diseases that can mimic MS is important in terms of therapeutic and prognostic approach. In this context, thiol-disulfide homeostasis would not only provide information about the oxidative status but perhaps distinguish these diseases from each other.
In this study, we aimed to evaluate dynamic thiol-disulfide homeostasis in different subgroups of MS, NMOSD, and MOGAD patients along with their clinical features compared to healthy controls (HCs).
Methods
Subjects
In total, seven patients with AQP4-Abs + NMOSD (seven serum samples), eight patients with MOG-Abs+MOGAD (eight serum samples), and 52 patients with seronegative MS (52 serum, 11 CSF samples, and 11 paired samples) were enrolled in the study. The MS group consisted of three subgroups: (1) relapsing-remitting MS (RRMS), n = 31; (2) secondary progressive MS (SPMS), n = 15; and (3) primary progressive MS (PPMS), n = 6. All patients with CSF samples were newly diagnosed by neurologists (RRMS = 11 CSF samples).
Serum samples were also collected from 18 HCs (18 serum samples) who were sex- and age-matched to the patient cohort. Both AQP4-Abs and MOG-Abs were verified twice (>1:40 titer) by a commercially available fixed cell-based assay (Euroimmun, Lubeck, Germany). Serum and CSF samples were collected at the Department of Neurology, Hacettepe University (Ankara, Turkey) and then stored at −80°C in the Department of Medical Biochemistry, Gazi University (Ankara, Turkey). Additional CSF samples were gathered within patients undergoing relapse. All samples were collected between January 1, 2019, and January 1, 2020 with a maximum of 2 months between the symptom onset of relapse and sample collection. The treatment-naive group consisted of patients (n = 30) who had not received steroid treatment in the last month and who had not been treated with immunomodulatory therapy in the last 3 months before sample collection. Our cohort also included patients who received different types of immune modulator therapy (n = 37) in addition to the treatment-naive group (all of the treatment-naive patients were in MS subtypes).
Diagnosis, Clinical Evaluation, and Data Collection
AQP4-Abs-positive NMOSD and MOG-Abs-positive MOGAD patients were diagnosed according to previously determined consensus criteria (18). MS diagnosis was made according to the modified McDonald criteria (9). Medical records, laboratory data, and magnetic resonance imaging (MRI) findings of the patients were reviewed retrospectively. We carried out clinical evaluations using the neurological examination and Expanded Disability Status Scale (EDSS). Relapse was defined as a monophasic clinical episode with patient-reported symptoms and objective findings typical of MS, reflecting a focal or multifocal inflammatory demyelinating event in the CNS, developing acutely or subacutely, with a duration of at least 24 h, with or without recovery, and in the absence of a fever or infection (9).
Demographics data (sex, age, age at onset of the disease, and time of first complaints), clinical features (number of relapses, presenting symptoms, disease duration, disease subtype for MS, acute and maintenance treatments, and neurological findings at last follow-up), laboratory findings (serum and cerebrospinal fluid examinations, MOG-IgG titer, AQP4-IgG titer, and biomarker levels), and radiological findings were examined retrospectively.
Standard Protocol Approvals, Registrations, and Patient Consents
Our study was approved and reviewed by the ethics committee of the faculty of Medicine, at Gazi University (ethical approval number: 33-14.01.2019). All individuals provided written informed consent.
Biomarker Analysis
CSF and serum samples were aliquoted into Eppendorf tubes and stored prior to conducting the biomarker studies which were performed at different times. Centrifuge protocols were used for CSF and blood sample biobanking as previously described (41). Briefly, CSF samples were centrifuged at 400 × g for 10min and blood samples at 2,000 × g for 10min at room temperature.
Cell-Based Assays
The MOG-IgG test was performed using a Euroimmun kit that utilizes a cell-based assay (CBA) employing formalin-fixed HEK293 cells transfected with full-length human MOG (reactivity at a dilution of 1:10 is positive). A baseline AQP4-IgG test was performed by a fixed-cell CBA (Euroimmun, titer >1:10 is positive) in all patients.
Evaluation of Thiol-Disulfide Homeostasis
Serum thiol-disulfide homeostasis was determined using a novel spectrophotometric method as previously described (29). Briefly, dynamic and reducible disulfide bonds (–S–S) in the samples were reduced to free functional thiol groups (–SH) using sodium borohydride (NaBH4). In order to avoid the reduction of unused reduced sNaBH4 to dithionite-2 nitrobenzoic (DTNB), NaBH4 was removed with formaldehyde. Then, the native thiol (NTL) and total thiol (TTL) levels were determined after reacting with DTNB. Half of the difference of the result obtained by the subtraction of NTL amount from TTL content indicated the disulfide (DS) level. Finally, the NTL/TTL (–SH/–SH+-S–S), disulfide/NTL (–S–S/–SH), and disulfide/TTL (–S–S/–SH+-S–S) ratios were calculated.
Statistics
All data were analyzed using the SPSS package program (ver. 21.0; IBM Corp., Armonk, NY, USA) with a 95% confidence level. Categorical variables are represented as frequency (n) and percentage (%), while numerical variables are represented as mean, standard deviation (SD), and median (M). The normality of the distribution of numerical variables was examined using Shapiro–Wilk tests. Ordinal variables were described by median and interquartile ranges (IQRs), mean and standard deviations (SD) in Gaussian distributed data, and categorical variables by counts and percentages. Spearman correlation coefficients were calculated to determine the relationship between serum and CSF biomarker levels and numeric clinical variables across all the patients and within each disease. The Mann Whitney test was used to compare groups in terms of a quantitative variables. The Kruskal–Wallis test was used to compare independent k groups (k > 2) in terms of quantitative variables. Demographic features of the participants at baseline were compared using the Fisher exact test or the Wilcoxon test. The GraphPad Prism software was used for some graphical demonstrations. Variables with two-tailed p < 0.05 were considered significant.
Results
The demographic and clinical characteristics of the patients are summarized in Table 1.

Table 1. Demographic and clinical characteristics of the patients.
There were significantly more females among the subgroups of diseases compared to males (p < 0.05), except for the PPMS (male = 66.7%) and MOGAD (male = 62.5%) groups. The SPMS group (median: 15.0, IQR: 12.0–20.0) had the highest median level of disease duration (p < 0.05) and the highest number of GDT2 + lesions (median: 70, IQR: 25–100) (p<0.05). The PPMS group (median: 5.7, IQR: 4.1–6.6) had the highest median EDSS level (p < 0.05).
There was a statistically significant difference between serum and CSF levels of biomarkers (p < 0.05). That is, serum levels of biomarkers were significantly higher than CSF values (p < 0.001; Figure 1 and Table 2). No significant relationship was found between serum and CSF values of biomarkers (p > 0.05). There was also no statistically significant difference in terms of serum biomarkers between subgroups (p > 0.05; Figure 2). MOGAD serum TTL levels (410.7 ± 281.5) and PPMS serum disulfide levels (75.8 ± 74.9) were comparable between the different disease subgroups and healthy controls (p > 0.05; Table 2). Serum TTL (317.88 ± 66.04) and NTL (211.61 ± 44.15) levels were significantly lower in relapsed patients (368.84 ± 150.36) compared to those in remission (222.52 ± 70.59; p < 0.05).
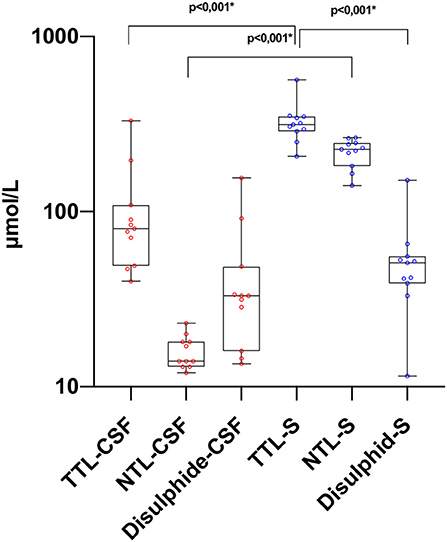
Figure 1. Thiol levels in paired CSF and serum samples. TTL, total thiol; NTL, native thiol; CSF, cerebrospinal fluid. *The statistical significance is marked with asterisks (Mann–Whitney U-test).
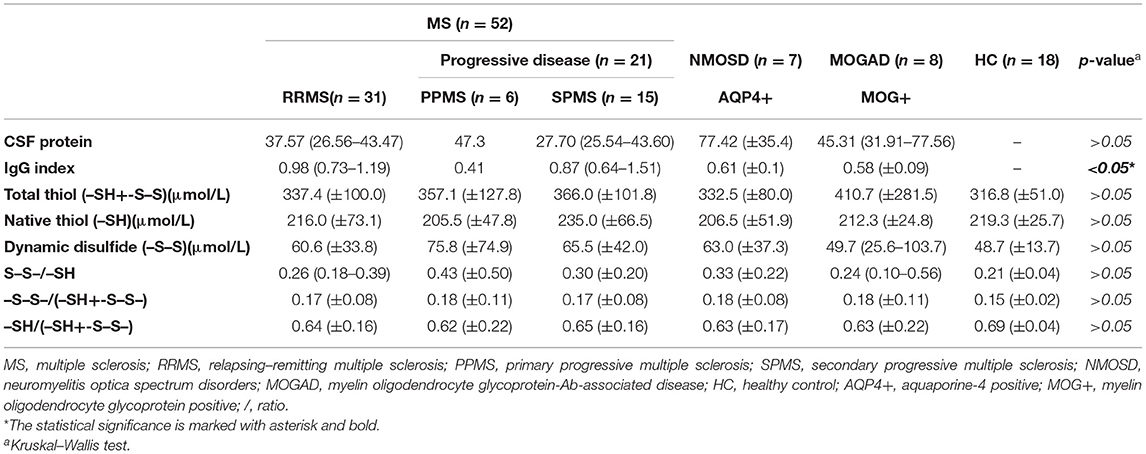
Table 2. Serum biomarker levels in different disease subgroups.
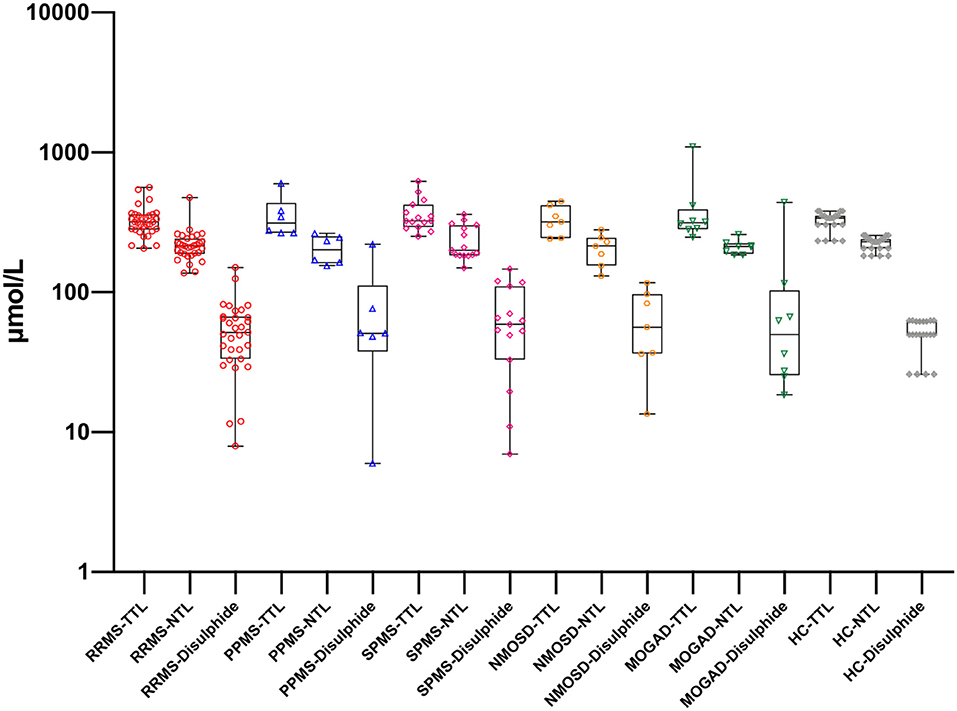
Figure 2. Thiol levels in serum samples with different disease subgroups and healthy controls. RRMS, relapsing-remitting multiple sclerosis; PPMS, primary progressive multiple sclerosis; SPMS, secondary progressive multiple sclerosis; NMOSD, neuromyelitis optica spectrum disorders; MOGAD, myelin oligodendrocyte glycoprotein-Ab-associated disease; HC, healthy control; TTL, total thiol; NTL, native thiol. There is no statistically significant difference for the same biomarker between groups.
Serum TTL was negatively correlated with the number of relapses in relapsed patients (p = 0.027, r = −0.521). Serum NTL levels were negatively correlated with age at onset and EDSS, respectively (p = 0.045, r = −0.477; p = 0.031, r = −0.508). The age at sampling and number of GDT2 + lesions were positively correlated (p = 0.018, r = 0.537). When all patient subgroups were evaluated, there was a negative correlation between serum NTL levels and age at sampling, age at onset (p = 0.024, r = −0.244; p = 0.031, r = −0,264; respectively). For the RRMS group, there was a negative correlation between serum TTL, NTL, and EDSS (p = 0.017, r = −0.439; p = 0.004, r = −0.522; respectively). For the PPMS group, there was a positive correlation between serum disulfide and disease duration (p = 0.024, r = 0.870).
Discussion
Previous studies have investigated the oxidative stress markers and antioxidant molecules that develop in MS patients (42–45). Furthermore, their relationships with disability and subtypes of disease have also been examined (46).
Although previous studies have assessed thiol homeostasis in MS (47, 48), to the best of our knowledge, this is the first study to evaluate it within MS subtypes (NMOSD and MOGAD) compared to HCs. Moreover, in these studies, very limited clinical information was used, and its relationship with clinical findings was largely unexplored. Interestingly, until now, no study has investigated dynamic thiol disulfide homeostasis in NMOSD and MOGAD.
We found that there was a higher ratio of women to men among the MS subgroups, except in the PPMS and NMOSD groups, which is consistent with the literature (11, 15). Furthermore, similar to previous studies, we found that the duration of the disease was longer in the progressive MS group. Likewise, the progressive group had a higher EDSS score than the RRMS group, even if there were naive patients in RRMS group.
Importantly, we are the first to evaluate CSF and serum thiol homeostasis together. The brain parenchyma produces a high rate of oxidative radicals due to its high oxygen consumption (49, 50). In our study, it was observed that TTL, NTL, and disulfide CSF levels were lower than serum values. Presumably, in this case, peripheral oxidative damage and dynamic thiol homeostasis that emerges in response also plays a role. These findings suggest that CSF TTL, NTL, and disulfide levels may be used to evaluate disease status in patients who undergo CSF sampling for diagnostic purposes.
When oxidative stress increases, thiol groups of proteins react with oxidants and turn into disulfide bonds. These formed bonds can then be reduced to thiol groups again, reestablishing thiol-disulfide homeostasis (51).
In our study, the SPMS group had the highest serum mean TTL levels among the MS group. The SPMS group also had the highest number of relapses and longest disease duration. This suggests that as new disulfide bonds are formed, more thiol groups can be reduced in this group. The MOGAD group had the highest TTL levels among the disease subtypes. This may actually be related to the pathophysiology of this disorder (15), as the main problem in this disease is the MOG-IgG Ab that develops against myelin sheath, which causes oxidative damage, thereby leading to an increase in TTL levels.
The PPMS group had the highest serum disulfide levels among the disease subtypes. This condition, which is indicative of oxidative damage, may also be associated with neuronal damage. In addition, the negative correlation between serum TTL, NTL, and EDSS in the RRMS group suggests that these values could be used in the assessment of disability.
Plasma membrane is like the primary sensor of the cells' extrinsic stressors. Excess amounts of ROS cause cell damage and death by disrupting plasma membrane functions. These mechanisms are believed to play a major role in the pathophysiology of many neurodegenerative diseases. However, low amounts of membrane-associated ROS may activate adaptive pathways against stress. There is increasing evidence for the hormetic role of membranes in states of low and transient oxidative stress. Under normal conditions, membrane lipid peroxidation is usually low. However, an increase in oxidative stressors in cells such as neurons with high energy demand and oxygen consumption may deteriorate the hormetic roles of the membrane (52–55). Perhaps, the low thiol levels in relapsed patients in our study can be explained by the deterioration in the hormetic role of the membrane. In addition, irrespective of the cause, the increase in cellular oxidative stress at subtoxic levels causes a neuroprotective effect known as “preconditioning.” Preconditioning signal provides cellular protection through hormesis, which is a dose-response phenomenon characterized by a high-dose inhibition and low-dose stimulation (56–59). Thanks to this chain of events, neuroprotection is also provided.
Protein thiols are important mediators of multiple metabolic, signaling, and transcriptional processes; they play a key role in the regulation of redox sensing and cellular redox status. “Vitagenes,” which play a key role in oxidative stress protection, are protective genes that control pro-survival pathways that must be activated in response to cellular stress. They encode cytoprotective heat shock proteins, thioredoxin, and sirtuins (60). Under normal physiological conditions, long-term health is maintained by protein homeostasis. When this homeostasis is disrupted, cellular stress is released and heat shock proteins, including chaperones, are produced and contribute to cell survival (61, 62). In this study, the SPMS group had the highest serum mean TTL levels among the MS groups. According to this result, we may assume that vitagen activity may have increased, and pro-survival mechanisms are activated for the continuity of protein homeostasis.
Our study had some limitations. Particularly, the number of patients in the NMOSD and MOGAD groups was lower than the other groups. While the numbers in this group reflected all of the patients in our tertiary center, all of them were being treated with immunomodulatory therapy. In addition, due to ethical considerations, we could not obtain CSF samples from HCs; thus, there were no CSF samples included in our HC group. Furthermore, the number of patients in our progressive MS group was not close to that of the RRMS group, either. In addition, our patient population did not include more rare MS subtypes, such as clinically isolated syndrome (CIS) and radiologically isolated syndrome (RIS). Although this study demonstrates the usefulness of assessing dynamic thiol homeostasis among neuroinflammatory diseases of the CNS, future studies incorporating larger patient cohorts are needed.
Conclusions
Oxidative stress plays a crucial role in the physiopathology of neuroimmunological diseases. This study is the first to assess thiol homeostasis among different subgroups of MS and NMOSD. Previously, thiol homeostasis has only been evaluated using serum. In future studies, CSF and serum values of thiols should be examined in detail using a larger number of paired serum and CSF samples. Perhaps in the near future, thiol status can be used by itself or in combination with other candidate biomarkers to monitor oxidative status and disease activity.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The study was approved and reviewed by the ethics committee of the faculty of Medicine, Gazi University. The number of the ethical approval is 33-14.01.2019. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all the patients, their families, and the researchers who participated in the studies.
References
1. Lehmann HC, Compston A, Hartung H-P. 150th anniversary of clinical description of multiple sclerosis: Leopold Ordenstein's legacy. Neurology. (2018) 90:1011–6. doi: 10.1212/WNL.0000000000005613
2. Vos T, Abajobir AA, Abate KH, Abbafati C, Abbas KM, Abd-Allah F, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study (2016). Lancet. (2017) 390:1211–59. doi: 10.1016/S0140-6736(17)32154-2
3. Münz C, Lünemann JD, Getts MT, Miller SD. Antiviral immune responses: triggers of or triggered by autoimmunity? Nat Rev Immunol. (2009) 9:246–58. doi: 10.1038/nri2527
4. Hedström AK, Åkerstedt T, Hillert J, Olsson T, Alfredsson L. Shift work at young age is associated with increased risk for multiple sclerosis. Ann Neurol. (2011) 70:733–41. doi: 10.1002/ana.22597
5. Nourbakhsh B, Mowry EM. Multiple sclerosis risk factors and pathogenesis. Continuum. (2019) 25:596–610. doi: 10.1212/CON.0000000000000725
6. Dobson R, Giovannoni G. Autoimmune disease in people with multiple sclerosis and their relatives: a systematic review and meta-analysis. J Neurol. (2013) 260:1272–85. doi: 10.1007/s00415-012-6790-1
7. Belbasis L, Bellou V, Evangelou E, Ioannidis JP, Tzoulaki I. Environmental risk factors and multiple sclerosis: an umbrella review of systematic reviews and meta-analyses. Lancet Neurol. (2015) 14:263–73. doi: 10.1016/S1474-4422(14)70267-4
8. Popescu BFG, Lucchinetti CF. Pathology of demyelinating diseases. Annu Rev Pathol. (2012) 7:185–217. doi: 10.1146/annurev-pathol-011811-132443
9. Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. (2018) 17:162–73. doi: 10.1016/S1474-4422(17)30470-2
10. Gilgun-Sherki Y, Melamed E, Offen D. The role of oxidative stress in the pathogenesis of multiple sclerosis: the need for effective antioxidant therapy. J Neurol. (2004) 251:261–8. doi: 10.1007/s00415-004-0348-9
11. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. (2015) 15:545–58. doi: 10.1038/nri3871
12. Boeschoten RE, Braamse AM, Beekman AT, Cuijpers P, van Oppen P, Dekker J, et al. Prevalence of depression and anxiety in multiple sclerosis: a systematic review and meta-analysis. J Neurol Sci. (2017) 372:331–41. doi: 10.1016/j.jns.2016.11.067
13. Tanaka M, Toldi J, Vécsei L. Exploring the etiological links behind neurodegenerative diseases: Inflammatory cytokines and bioactive kynurenines. Int J Mol Sci. (2020) 21:2431. doi: 10.3390/ijms21072431
14. Lehmann-Horn K, Kronsbein HC, Weber MS. Targeting B cells in the treatment of multiple sclerosis: recent advances and remaining challenges. Ther Adv Neurol Disord. (2013) 6:161–73. doi: 10.1177/1756285612474333
15. Flanagan EP. Neuromyelitis optica spectrum disorder and other non-multiple sclerosis central nervous system inflammatory diseases. Continuum. (2019) 25:815–44. doi: 10.1212/CON.0000000000000742
16. Lennon VA, Kryzer TJ, Pittock SJ, Verkman A, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. (2005) 202:473–7. doi: 10.1084/jem.20050304
17. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. (2004) 364:2106–12. doi: 10.1016/S0140-6736(04)17551-X
18. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. (2015) 85:177–89. doi: 10.1212/WNL.0000000000001729
19. Juryńczyk M, Jacob A, Fujihara K, Palace J. Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease: practical considerations. Pract Neurol. (2019) 19:187–95. doi: 10.1136/practneurol-2017-001787
20. Fujihara K, Cook LJ. Neuromyelitis optica spectrum disorders and myelin oligodendrocyte glycoprotein antibody-associated disease: current topics. Curr Opin Neurol. (2020) 33:300–8. doi: 10.1097/WCO.0000000000000828
21. Reindl M, Jarius S, Rostasy K, Berger T. Myelin oligodendrocyte glycoprotein antibodies: how clinically useful are they? Curr Opin Neurol. (2017) 30:295–301. doi: 10.1097/WCO.0000000000000446
22. Jurynczyk M, Messina S, Woodhall MR, Raza N, Everett R, Roca-Fernandez A, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. (2017) 140:3128–38. doi: 10.1093/brain/awx276
23. Hennes E-M, Baumann M, Schanda K, Anlar B, Bajer-Kornek B, Blaschek A, et al. Prognostic relevance of MOG antibodies in children with an acquired demyelinating syndrome. Neurology. (2017) 89:900–8. doi: 10.1212/WNL.0000000000004312
24. Freedman MS, Selchen D, Arnold DL, Prat A, Banwell B, Yeung M, et al. Treatment optimization in MS: Canadian MS Working Group updated recommendations. Can J Neurol Sci. (2013) 40:307–23. doi: 10.1017/S0317167100014244
25. Peng F, Yang Y, Liu J, Jiang Y, Zhu C, Deng X, et al. Low antioxidant status of serum uric acid, bilirubin and albumin in patients with neuromyelitis optica. Eur J Neurol. (2012) 19:277–83. doi: 10.1111/j.1468-1331.2011.03488.x
26. Tanaka M, Vécsei L. Monitoring the redox status in multiple sclerosis. Biomedicines. (2020) 8:406. doi: 10.3390/biomedicines8100406
27. Lalkovičová M, Danielisová V. Neuroprotection and antioxidants. Neural Regener Res. (2016) 11:865. doi: 10.4103/1673-5374.184447
28. Cremers CM, Jakob U. Oxidant sensing by reversible disulfide bond formation. J Biol Chem. (2013) 288:26489–96. doi: 10.1074/jbc.R113.462929
29. Erel O, Neselioglu S. A novel and automated assay for thiol/disulphide homeostasis. Clin Biochem. (2014) 47:326–32. doi: 10.1016/j.clinbiochem.2014.09.026
30. Turell L, Radi R, Alvarez B. The thiol pool in human plasma: the central contribution of albumin to redox processes. Free Radic Biol Med. (2013) 65:244–53. doi: 10.1016/j.freeradbiomed.2013.05.050
31. Jones DP. Redefining oxidative stress. Antioxid Redox Signal. (2006) 8:1865–79. doi: 10.1089/ars.2006.8.1865
32. Guzman-Martinez L, Maccioni RB, Andrade V, Navarrete LP, Pastor MG, Ramos-Escobar N. Neuroinflammation as a common feature of neurodegenerative disorders. Front Pharmacol. (2019) 10:1008. doi: 10.3389/fphar.2019.01008
33. Matteucci E, Giampietro O. Thiol signalling network with an eye to diabetes. Molecules. (2010) 15:8890–903. doi: 10.3390/molecules15128890
34. Go Y-M, Jones DP. Cysteine/cystine redox signaling in cardiovascular disease. Free Radic Biol Med. (2011) 50:495–509. doi: 10.1016/j.freeradbiomed.2010.11.029
35. Prabhu A, Sarcar B, Kahali S, Yuan Z, Johnson JJ, Adam K-P, et al. Cysteine catabolism: a novel metabolic pathway contributing to glioblastoma growth. Cancer Res. (2014) 74:787–96. doi: 10.1158/0008-5472.CAN-13-1423
36. Calabrese V, Lodi R, Tonon C, D'Agata V, Sapienza M, Scapagnini G, et al. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich's ataxia. J Neurol Sci. (2005) 233:145–62. doi: 10.1016/j.jns.2005.03.012
37. Smeyne M, Smeyne RJ. Glutathione metabolism and Parkinson's disease. Free Radic Biol Med. (2013) 62:13–25. doi: 10.1016/j.freeradbiomed.2013.05.001
38. Steele ML, Fuller S, Maczurek AE, Kersaitis C, Ooi L, Münch G. Chronic inflammation alters production and release of glutathione and related thiols in human U373 astroglial cells. Cell Mol Neurobiol. (2013) 33:19–30. doi: 10.1007/s10571-012-9867-6
39. Tasset I, Agüera E, Sánchez-López F, Feijóo M, Giraldo AI, Cruz AH, et al. Peripheral oxidative stress in relapsing–remitting multiple sclerosis. Clin Biochem. (2012) 45:440–4. doi: 10.1016/j.clinbiochem.2012.01.023
40. Gonsette R. Neurodegeneration in multiple sclerosis: the role of oxidative stress and excitotoxicity. J Neurol Sci. (2008) 274:48–53. doi: 10.1016/j.jns.2008.06.029
41. Teunissen C, Petzold A, Bennett J, Berven F, Brundin L, Comabella M, et al. A consensus protocol for the standardization of cerebrospinal fluid collection and biobanking. Neurology. (2009) 73:1914–22. doi: 10.1212/WNL.0b013e3181c47cc2
42. Karlík M, Valkovič P, Hančinová V, KríŽová L, Tóthová L, Celec P. Markers of oxidative stress in plasma and saliva in patients with multiple sclerosis. Clin Biochem. (2015) 48:24–8. doi: 10.1016/j.clinbiochem.2014.09.023
43. Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. (2010) 48:749–62. doi: 10.1016/j.freeradbiomed.2009.12.022
44. Koch M, Mostert J, Arutjunyan A, Stepanov M, Teelken A, Heersema D, et al. Peripheral blood leukocyte NO production and oxidative stress in multiple sclerosis. Mult Scler J. (2008) 14:159–65. doi: 10.1177/1352458507082075
45. HadŽović-DŽuvo A, Lepara O, Valjevac A, Avdagić N, Hasić S, Kiseljaković E, et al. Serum total antioxidant capacity in patients with multiple sclerosis. Bosnian J Basic Med Sci. (2011) 11:33. doi: 10.17305/bjbms.2011.2620
46. Miller E, Walczak A, Saluk J, Ponczek MB, Majsterek I. Oxidative modification of patient's plasma proteins and its role in pathogenesis of multiple sclerosis. Clin Biochem. (2012) 45:26–30. doi: 10.1016/j.clinbiochem.2011.09.021
47. Vural G, Gumusyayla S, Bektas H, Deniz O, Ergin M, Erel O. Dynamic thiol-disulphide homeostasis in patients with multiple sclerosis. World J Neurosci. (2016) 6:214–9. doi: 10.4236/wjns.2016.63026
48. Vural G, Gümüşyayla S, Deniz O, Neşelioglu S, Erel Ö. Relationship between thiol-disulphide homeostasis and visual evoked potentials in patients with multiple sclerosis. Neurol Sci. (2019) 40:385–91. doi: 10.1007/s10072-018-3660-3
49. Friedman J. Why is the nervous system vulnerable to oxidative stress? In: Gadoth N, Göbel H, editors. Oxidative Stress and Free Radical Damage in Neurology. Oxidative Stress in Applied Basic Research and Clinical Practice. Humana Press; Springer Nature Switzerland AG (2011). p. 19–27. doi: 10.1007/978-1-60327-514-9_2
50. Radak Z, Zhao Z, Koltai E, Ohno H, Atalay M. Oxygen consumption and usage during physical exercise: the balance between oxidative stress and ROS-dependent adaptive signaling. Antioxid Redox Signal. (2013) 18:1208–46. doi: 10.1089/ars.2011.4498
51. Jones DP, Liang Y. Measuring the poise of thiol/disulfide couples in vivo. Free Radic Biol Med. (2009) 47:1329–38. doi: 10.1016/j.freeradbiomed.2009.08.021
52. Calabrese V, Cornelius C, Dinkova-Kostova AT, Calabrese EJ, Mattson MP. Cellular stress responses, the hormesis paradigm, and vitagenes: novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid Redox Signal. (2010) 13:1763–811. doi: 10.1089/ars.2009.3074
53. Ragusa N, Sfogliano L, Calabrese V, Rizza V. Effects of multivitamin treatment on the activity of rat liver tryptophan pyrrolase during ethanol administration. Acta Vitaminol Enzymol. (1981) 3:199–204.
54. Siracusa R, Scuto M, Fusco R, Trovato A, Ontario ML, Crea R, et al. Anti-inflammatory and anti-oxidant activity of Hidrox® in rotenone-induced Parkinson's disease in mice. Antioxidants. (2020) 9:824. doi: 10.3390/antiox9090824
55. Calabrese V, Renis M, Calderone A, Russo A, Barcellona M, Rizza V. Stress proteins and SH-groups in oxidant-induced cell damage after acute ethanol administration in rat. Free Radic Biol Med. (1996) 20:391–7. doi: 10.1016/0891-5849(95)02095-0
56. Calabrese EJ, Bachmann KA, Bailer AJ, Bolger PM, Borak J, Cai L, et al. Biological stress response terminology: integrating the concepts of adaptive response and preconditioning stress within a hormetic dose–response framework. Toxicol Appl Pharmacol. (2007) 222:122–8. doi: 10.1016/j.taap.2007.02.015
57. Calabrese EJ, Baldwin LA. Hormesis: the dose-response revolution. Annu Rev Pharmacol Toxicol. (2003) 43:175–97. doi: 10.1146/annurev.pharmtox.43.100901.140223
58. Calabrese EJ, Blain R. The occurrence of hormetic dose responses in the toxicological literature, the hormesis database: an overview. Toxicol Appl Pharmacol. (2005) 202:289–301. doi: 10.1016/j.taap.2004.06.023
59. Calabrese EJ, Blain RB. The hormesis database: the occurrence of hormetic dose responses in the toxicological literature. Regul Toxicol Pharmacol. (2011) 61:73–81. doi: 10.1016/j.yrtph.2011.06.003
60. Calabrese V, Guagliano E, Sapienza M, Panebianco M, Calafato S, Puleo E, et al. Redox regulation of cellular stress response in aging and neurodegenerative disorders: role of vitagenes. Neurochem Res. (2007) 32:757–73. doi: 10.1007/s11064-006-9203-y
61. Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. (2008) 319:916–9. doi: 10.1126/science.1141448
Keywords: multiple sclerosis, neuromyelitis optica spectrum disease, myelin oligodendrocyte glycoprotein, thiol, oxidative stress
Citation: Arslan B, Arslan GA, Tuncer A, Karabudak R and Dinçel AS (2021) Evaluation of Thiol Homeostasis in Multiple Sclerosis and Neuromyelitis Optica Spectrum Disorders. Front. Neurol. 12:716195. doi: 10.3389/fneur.2021.716195
Received: 28 May 2021; Accepted: 12 July 2021;
Published: 30 August 2021.
Edited by:
Sally Ann Frautschy, University of California, Los Angeles, United StatesReviewed by:
Masaru Tanaka, University of Szeged, HungaryVittorio Calabrese, University of Catania, Italy
Copyright © 2021 Arslan, Arslan, Tuncer, Karabudak and Dinçel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Burak Arslan, YnVyYWthcnNsYW5tZEBnbWFpbC5jb20=
†These authors have contributed equally to this work and share first authorship