 Shuang Li
Shuang Li Guangjian Li
Guangjian Li Xia Luo
Xia Luo Yan Huang
Yan Huang Lan Wen
Lan Wen Jinglun Li
Jinglun Li- 1Department of Neurology, The Affiliated Hospital of Southwest Medical University, Luzhou, China
- 2Laboratory of Neurological Diseases and Brain Function, The Affiliated Hospital of Southwest Medical University, Luzhou, China
- 3Department of Neurology, Southwest Hospital, Third Military Medical University (Army Medical University), Chongqing, China
- 4Department of Neurosurgery, West China Hospital, Sichuan University, Chengdu, China
Cerebral small vessel disease (cSVD)—a common cause of stroke and vascular dementia—is a group of clinical syndromes that affects the brain's small vessels, including arterioles, capillaries, and venules. Its pathogenesis is not fully understood, and effective treatments are limited. Increasing evidence indicates that an elevated total serum homocysteine level is directly and indirectly associated with cSVD, and endothelial dysfunction plays an active role in this association. Hyperhomocysteinemia affects endothelial function through oxidative stress, inflammatory pathways, and epigenetic alterations at an early stage, even before the onset of small vessel injuries and the disease. Therefore, hyperhomocysteinemia is potentially an important therapeutic target for cSVD. However, decreasing the homocysteine level is not sufficiently effective, possibly due to delayed treatment, which underlying reason remains unclear. In this review, we examined endothelial dysfunction to understand the close relationship between hyperhomocysteinemia and cSVD and identify the optimal timing for the therapy.
Introduction
Cerebral small vessel disease (cSVD) refers to a series of clinical, imaging, and pathological syndromes caused by various etiological factors affecting small arteries, capillaries, and venules in the brain. As an age-dependent disease, the cSVD incidence is predicted to increase with the aging population (1). Several factors can contribute to the occurrence of cSVD, such as age, hypertension, diabetes, hyperlipidemia, small artery sclerosis, cerebrovascular amyloid, gene mutations, immune-mediated vasculitis, and certain infections (2); arteriolar sclerosis is the most common factor in older individuals (3, 4).
Globally, cSVD causes up to 45% of dementia and accounts for approximately 20% of all strokes. Twenty-five percent of these stroke cases are of the ischemic (or lacunar) type, of which in about 20% patients left disabled (3), putting an enormous burden on the society and families of these patients. Evidence regarding the efficacy of treatment with antiplatelets, anticoagulants, lipid-regulating drugs, and other methods in cSVD is lacking (5–7). Therefore, early detection and intervention of the risk factors is currently a beneficial strategy (8). The characteristic magnetic resonance imaging (MRI) markers are white matter hyperintensities (WMH), lacunes of presumed vascular origin, cerebral microbleeds (CMBs), enlarged perivascular spaces (EPVS), and cerebral atrophy (9, 10). Apart from imaging markers, sensitive, specific, and simple serum markers, such as homocysteine (Hcy), intercellular adhesion molecule-1 (ICAM-1), C-reactive protein (CRP), and interleukin-6 (IL-6) are also valuable in diagnosing cSVD (11).
Homocysteine (Hcy) is also an age-dependent indicator, and an elevated serum total Hcy (tHcy) level is associated with cardiovascular diseases, atherosclerosis, cerebrovascular diseases, and neurodegenerative disorders (12). Therefore, Hcy-lowering treatment is gradually becoming a therapeutic target for these diseases. However, contradictory results have been reported in cSVD. Several trials reported that primary prevention with Hcy-lowering therapy could reduce the incidence of cerebrovascular diseases (13–15). Unfortunately, a few trials using vitamin B supplementation to reduce Hcy levels failed to demonstrate the benefit of reducing stroke and cognitive impairment events (16–18). Understanding the mechanism behind Hcy-lowering therapy could help elucidate these findings. Endothelial dysfunction (ED) is a core mechanism of hyperhomocysteinemia (HHcy) affecting cSVD. Reducing Hcy levels alleviates ED, and early intervention of ED could reverse cSVD progression. Therefore, it is critical to identify the correct timing of lowering serum Hcy levels for treating ED and improving cSVD patient's prognosis.
Hyperhomocysteinemia
Homocysteine Metabolism and HHcy
Hcy is an intermediate metabolite in the methionine cycle, and the only Hcy sources are methionine-containing foods, such as eggs, sesame seeds, nuts, and meat. Plasma Hcy concentrations increase with age and are higher in men than in women (12, 19, 20). Hcy is primarily involved in methyl transfer (or one-carbon metabolism) and can undergo three metabolic pathways (21–23) (Figure 1): (a) Remethylation: Hcy is remethylated with the participation of 5-methyltetrahydrofolate and vitamin B12 (cobalamin) and catalyzed by methionine synthase (MS) to form methionine. Alternatively, betaine provides methyl to produce methionine catalyzed by betaine-homocysteine methyltransferase (BHMT); (b) Transsulfuration Pathway: Hcy is formed as cysteine is catalyzed by cystathionine beta-synthase (CBS) and its coenzyme, vitamin B6; (c) a small amount of Hcy is released directly into the extracellular fluid to perform physiological functions.
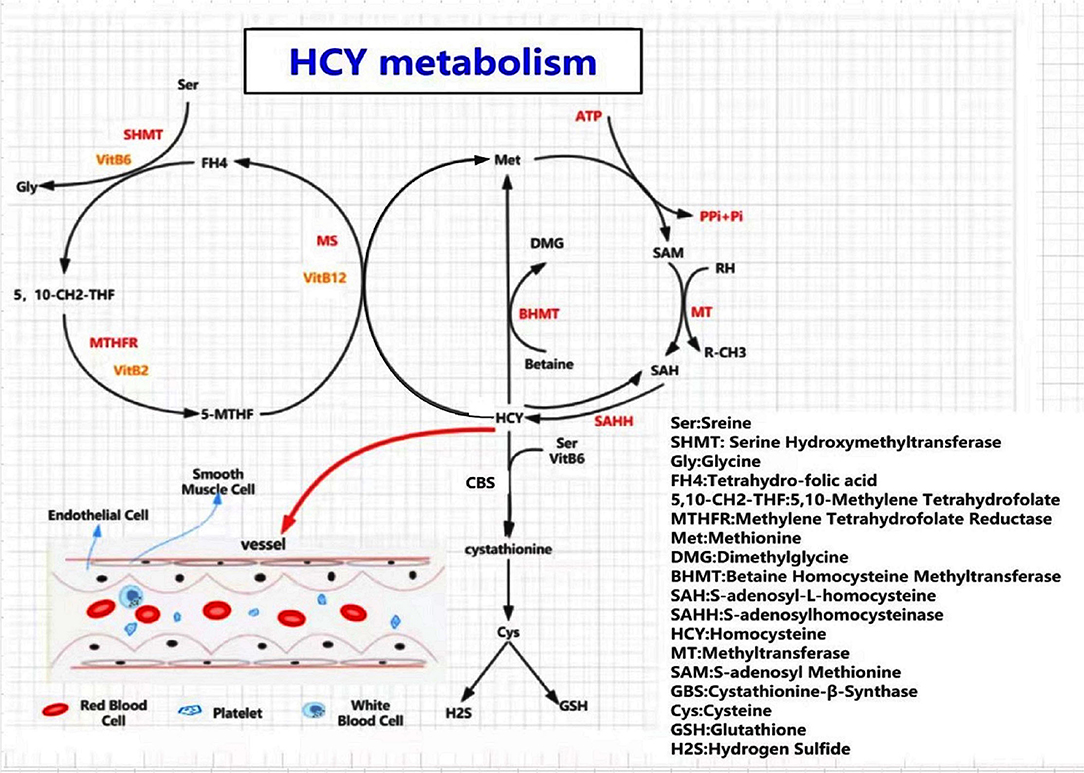
Figure 1. A diagram of homocysteine (Hcy) and methionine metabolism. Three pathways involved in Hcy meatabolism, including remethylation pathway, transsulfuration pathway, and a small amount of Hcy released directly into the extracellular fluid. When a deficiency of MTHFR, CBS, MS, or B-vitamins occurs, an excess of homocysteine enters the blood, causing HHcy, resulting in damage to the corresponding target organs. SAM is the only donor of methyl in all of the metabolic pathways. HHcy leads to hypomethylation of biomolecules, such as proteins, lipids, and nucleic acids, which contributes to metabolic abnormalities and promotes disease occurrence.
Vitamins B2, B6, B9 (folic acid), and B12 are indispensable coenzymes in the metabolism of the amino acid Hcy, and their deficiency could disturb the aforementioned pathways, leading to Hcy accumulation and elevated levels of Hcy in the plasma that triggers HHcy (24).
HHcy is defined by the plasma Hcy level of more than 15 μmol/L, and can be categorized into mild (Hcy = 15–30 μmol/L), moderate (Hcy = 30–100 μmol/L), and severe (Hcy >100 μmol/L) (24). HHcy can be classified as primary or secondary. Primary HHcy is mainly caused by mutations in the MTHFR (C677T or A1298C) and MTRR (A2756G) genes (25, 26). A higher MTHFR mutation rate is observed in the Chinese population than the others (27). The frequency of C677T mutation varies between 23.2 and 45.2% in the Chinese population (25), contributing to higher plasma-Hcy levels in Chinese than in Americans and Koreans (28–30). Secondary HHcy is mainly caused by impaired Hcy metabolism due to the deficiency of vitamin B2, B6, B9 (folic acid), or B12, renal failure, hypothyroidism, tumors, certain drugs, among several other causes (31). Dietary overdose may also lead to HHcy.
HHcy and Disease Condition
As a characteristic feature of aging, HHcy is causally linked to the development of age-associated disorders, such as ischemic heart diseases, atherosclerosis, cerebrovascular diseases, neurodegeneration, glomerular filtration rate decline, bone fractures, and venous thrombosis (12, 32). In particular, the focus has been on cardiocerebrovascular events. Several studies have shown that interventions to reduce tHcy, such as B-vitamin supplementation alone or in combination, had no effect on myocardial infarction and death from any cause or adverse event; however, statistical differences were observed in the stroke group (33, 34). These results suggested that the nervous system may be more sensitive to the toxic effects of HHcy than the cardiovascular system. Similarly, a study on the secondary prevention of stroke found that the use of B-vitamin supplements to lower Hcy significantly reduced stroke events and recurrence (13–15). Regardless of a history of acute ischemic stroke, patients with higher plasma-Hcy concentrations are more likely to develop cognitive impairments and depression than patients with normal levels (35, 36). Consequently, HHcy is a significant risk factor for nervous system disorders, especially cerebrovascular diseases.
HHcy Damages Brain Small Vessel Through Affecting Endothelial Function
There is an abundance of evidence that HHcy can damage brain small vessel, which increases the incidence of cSVD. The mechanisms include inflammation, atherosclerotic plaque formation, endothelial dysfunction, smooth muscle cell proliferation and oxidative stress response (37, 38). Among these pathological injury mechanisms, endothelial dysfunction plays a major role and determines the occurrence, development, and prognosis of cSVD (39). Hcy accumulation can interfere with the normal functioning of endothelial cells (ECs) through many pathological pathways (40–50). These include (a) oxidative stress resulting from the uncoupling of nitric oxide (NO) synthase, upregulation of the oxidation system, and decline of the antioxidant system; (b) competitive inhibition of NO synthase due to accumulation of asymmetric dimethylarginine (AMDA), resulting in decreased production of NO; (c) reduced hydrogen sulfide production, leading to abnormal vascular relaxation; (d) endoplasmic reticulum stress and unfolded protein response with eventual EC apoptosis; (e) chronic inflammation and prothrombotic conditions; and (f) epigenetic alterations, including hypomethylation and protein N-homocysteinylation. These mechanisms result in endothelial dysfunction and apoptosis, broken intercellular tight connections, and chronic hypoperfusion, which could be conducive to the formation and progression of cSVD.
Endothelial Dysfunction and cSVD
ED' Relevance in cSVD
Pathological changes in the small vessels vary during different periods of the cSVD process, including ED, blood-brain-barrier (BBB) injury, inflammatory response, smooth muscle proliferation, and small vessel sclerosis (3). Although different pathological changes play different roles in cSVD, they overlap and affect each other. ED triggers the first step of small blood vessel damage and plays a crucial role during the entire disease development process (39, 51). The significance of ED in cSVD has been well-established by population studies. A case-control study showed a worsening cerebral vasoreactivity in symptomatic lacunar stroke patients compared to that in matched controls, while cerebrovascular reactivity was used as a marker of endothelial function, indicating that ED severity in the cerebral artery may be a predisposing factor of symptomatic lacunar stroke in cSVD patients (51). Abnormal endothelial secretion of soluble ICAM-1, promotes the adhesion of leucocytes, which causes small vessels to occlude, and leads to incidence of silent brain infarctions and periventricular white matter lesions, implying ED in relation to progression of ischemic cerebral small vessel disease in type 2 diabetes (52). Markers of endothelial activation and damage, such as ICAM-1, thrombomodulin (TM), tissue factor (TF), and tissue factor pathway inhibitor (TFPI), were associated with the number of lacunes and severity of leukoaraiosis in the cSVD group in a prospective series of patients suffering from a lacunar strokes (53).
How ED Affects cSVD
ECs in the brain maintain BBB's integrity and regulate transmembrane transport functions. In addition, ECs are functionally active in controlling normal physiological processes, such as blood clotting, matching blood flow to neural activity, inflammatory reaction, and angiogenesis (54). When ischemic attacks, immune inflammation, or oxidative stress damage the endothelium, various abnormally released molecules (e.g., vasomotor factors, adhesion molecules, inflammatory cytokines, factors involved in the coagulation process, and enzymes) from ECs lead to impaired endothelium-dependent vasodilation, BBB destruction, and pre-thrombotic state, and exacerbates cSVD progression (11).
ED is the first step in cSVD pathogenesis. It is associated with leukoaraiosis, lacuna infarctions, and microbleeds through multiple underlying mechanisms (Figure 2). Firstly, the imbalance of vasomotor factors leads to pathological vasoconstriction and blood flow reduction, eventually leading to ischemia in the corresponding regions. Endothelially-derived NO, a key factor of vasodilation, is produced by L-arginine in a reaction catalyzed by endothelial-type NO synthase (eNOS), released into smooth muscle cells, wherein it activates guanylate cyclase to relax the vessel mediated by the cGMP pathway (55). In eNOS-deficient ED mice, NO reduction leads to pathological vasoconstriction and cerebral hypoperfusion, which exhibits similar MRI findings to those of cSVD, such as microhemorrhage, microinfarction, and white matter changes (56). In a cSVD rat model, ED, subsequent NO reduction, and BBB injury were associated with WMH burden (57). In addition, dysfunctional ECs can increase the secretion of endothelin-1, a vasoconstrictor, resulting in the pathological process of vasoconstriction (39).
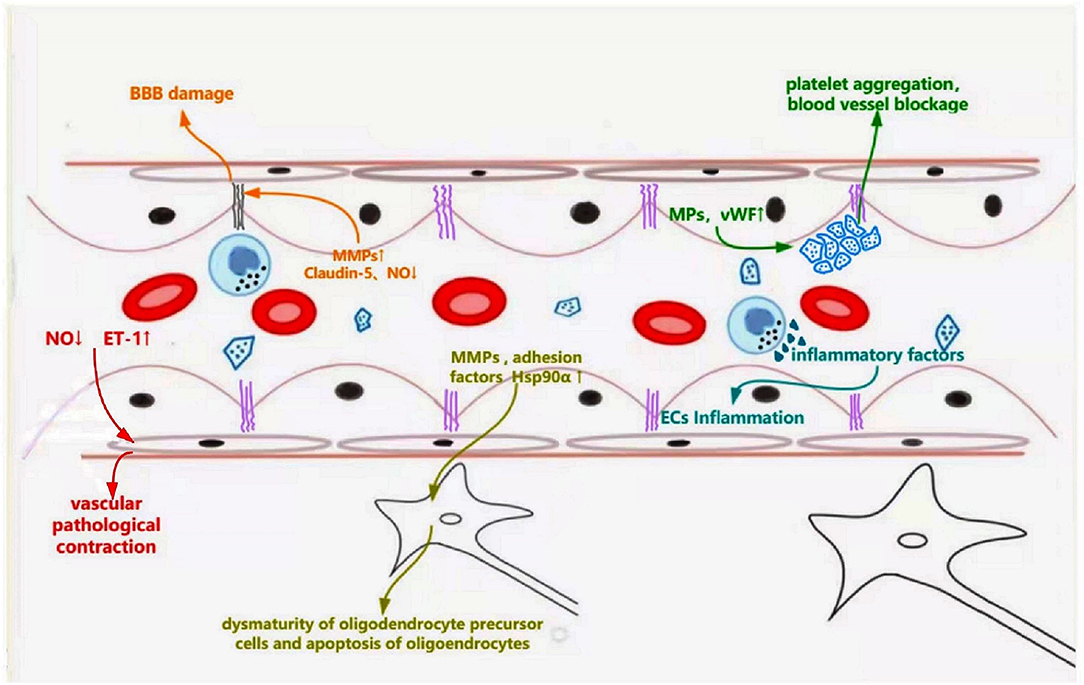
Figure 2. Schematic representation of dysfunctional ECs linking to specific features of cerebral small vessel disease. Dysfunctional ECs induce abnormal secretion of NO and endothelin-1, which leads pathological vascular contraction. Increased secretion of MMPs and decreased production of claudin-5 destroy the tight connections, leading to blood-brain barrier leakage. Abnormally released MMPs, adhesion factors (ICAM-1, VCAM-1), and Hsp90α result in dysmaturity of oligodendrocyte precursor cells and apoptosis of oligodendrocytes, which affects the myelination. Endothelial microparticles and vWF are released from the damaged ECs, causing platelet aggregation and inflammatory response.
Secondly, ECs and their tight junction (TJ) protein complexes are required to form a complete BBB. TJ protein complexes between ECs maintain BBB integrity and control the passage of cells and molecules. Damaged ECs and broken tight junctions can decrease BBB's integrity and increase its penetrability, leading to inflammation and edema of the surrounding tissue; corresponding signal changes can be observed on an MRI (58). The destruction of TJs caused by ECs dysfunction mainly results from the abnormal secretion of NO, matrix metalloproteinases (MMPs), and claudin proteins, inhibiting the modification of TJ proteins via nitrosylation or nitrosation, promoting TJ loss and accelerating BBB leakage (59). MMP-2 and MMP-9 also show increased expression, leading to decreased TJ proteins and transmembrane protein, claudin-5, which increases BBB's permeability (60). In addition, ED reduces the release of claudin-5, which adversely affects the tight connections between ECs and causes BBB injuries, which can impair brain function (61).
Thirdly, the survival and maturation of oligodendrocytes could be affected by ED and eventually interfere with white matter myelination. In an ATP11B-knockout rat model of cSVD, dysfunctional vascular endothelium secretes MMP chaperone protein, heat shock protein 90α (HSP90α), which leads to the maturation of oligodendrocyte precursor cells, contributing to white matter changes (57). Excessive MMPs, especially MMP-3 and MMP-9, secreted from dysfunctional ECs into the brain tissue cause and accelerate myelin breakdown (62). Moreover, dysfunctional ECs secrete increased CRP, IL-6, ICAM1, and e-selectin, leading to oligodendrocyte damage, and even apoptosis (4).
Finally, ED can participate in cSVD pathogenesis in multiple ways. Elevated plasma adhesion factors (ICAM-1 and vascular cell adhesion molecule-1 [VCAM-1]) and endothelial microparticles result from ED activating circulating inflammatory cells and causing vascular inflammation (63–65). Increased release of von Willebrand factor (vWF) caused by ED, which acts as a bridge to connect the process of platelets and inflammation, could lead to small vessel occlusion and further stimulate inflammation (66). These changes affect the structure and function of small vessels, contributing to the occurrence of cSVD and participating in its progression.
ED and Disease Reversal in the Early cSVD Stage
ED could be reversed through therapeutic interventions targeting the risk factors and inhibiting endothelial oxidative stress and inflammation. Previously, relieving eNOS uncoupling has been shown to reverse ED (54). This theory is also suitable for cSVD. Drug treatment to stabilize ED reverses the endothelial and oligodendroglial pathologies. Improved endothelial function can reverse ED itself, WMH, and oligodendroglial changes in the early disease state (57). Evidence suggests that in the early stage of cSVD, punctate lesions and a small number of white matter lesions caused by impaired interstitial fluid circulation may be quiescent and reversible. In contrast, fused and massive white matter lesions caused by demyelination and chronic hypoperfusion at later stages generally appear progressive and irreversible (67, 68). Thus, reversal of ED is a crucial therapeutic mechanism of cSVD, and the time window is notable and worth considering.
HHcy and cSVD
Several population-based experiments confirmed that elevated baseline Hcy levels are positively associated with cSVD incidence and the subsequent severity and disease progression, independent of vascular risk factors and severity of the atherosclerotic disease. The relevance of the presence and number of lacunar infarcts, WMH volume, and progression to high baseline Hcy levels (32, 69–71) provides a more intuitive view of the correlation between HHcy and cSVD. In patients with dementia caused by small vessel lesions, higher tHcy levels were used to predict WMH progression, and the level was associated with deep CMBs and lacunes (32). In cSVD-related stroke, serum Hcy levels were significantly higher in patients with large WMH volumes than those with small WMH volumes (69, 70). The association between Hcy and WMH burden suggested that the degree of ED may be greater in patients with increased WMH volume and may, in part, explain that ED acts as a bridge between HHcy and cSVD (72). Similar results were obtained in recent observational studies, where WMH volume and EPVS number were positively correlated with plasma Hcy concentrations. EPVS may be specifically associated with BBB leakage led by injured ECs and is considered as an important marker of BBB disruption (68, 73, 74).
ED in Hcy-Linked cSVD
As mentioned earlier, ED is a major mechanism of HHcy-linked cSVD, which mainly manifests as NO reduction, TJ breakage, BBB leakage, platelet aggregation, and pre-thrombotic state (Figure 3). The decrease in NO production and bioavailability predominates in Hcy-induced endothelial injury models, which exhibits vasoconstriction, vascular smooth muscle proliferation, and small arteriosclerosis; inadequate perfusion eventually develops cerebral white matter damage, acute small infarctions, and hemorrhages (44, 45). In addition, HHcy damages ECs and induces abnormal secretion of MMPs and transmembrane proteins, disrupting endothelial junctions, leading to BBB leakage, providing an important pathological basis for WMH, EPVS, lacunes, and CMB (75, 76). Furthermore, HHcy can also induce a pre-thrombotic state by modifying fibrinogen or enhancing platelet activation and coagulation and weakening fibrinolysis, which is prone to thrombosis and leads to ischemic stroke (42, 77). Naturally, Hcy-lowering therapy acts by ameliorating ED. AVE3085, a specific eNOS-targeting enhancer, prevents Hcy-induced ED by increasing NO production and reversing eNOS reduction in human ECs. The protective effects of AVE3085 translate into improved endothelium-dependent relaxation in the aorta of spontaneously hypertensive rats and significantly improved endothelium-dependent vasorelaxation in the Hcy-exposed human internal mammary artery (54). However, these studies indicate that ameliorating ED could reverse disease progression, which seems to be working just at early stage.
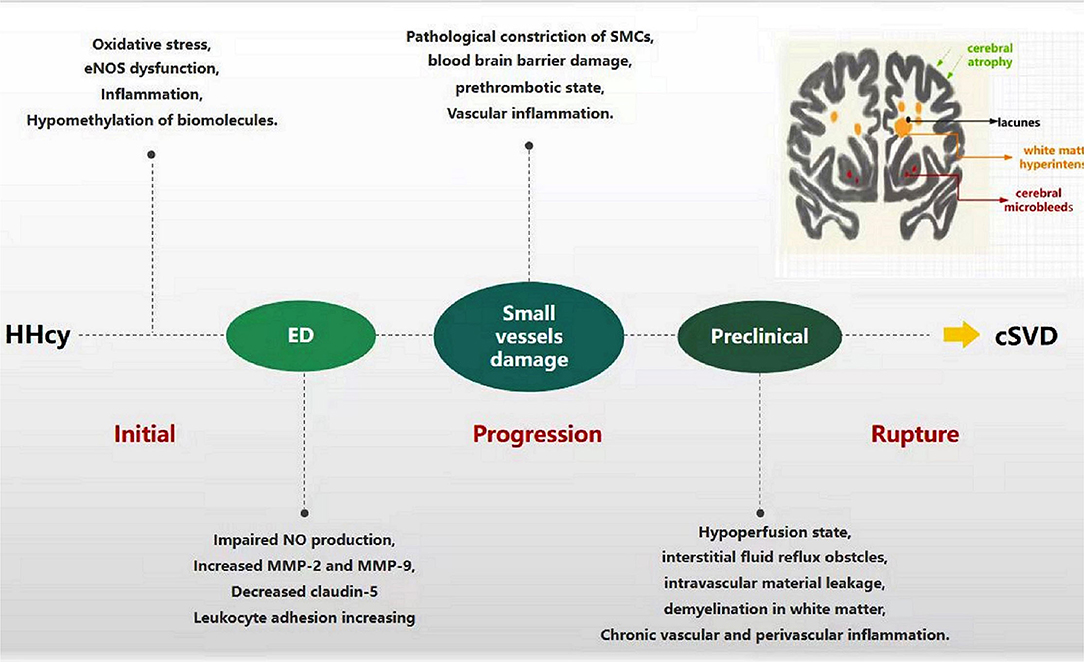
Figure 3. Progression of HHcy-induced cSVD through ED. In the beginning, HHcy induces endothelial cell injury through oxidative stress, eNOS dysfunction, inflammation, and hypomethylation of biomolecules. Dysfunctional endothelia release the molecules abnormally, such as impaired NO production, increased MMPs and adhesion factors, decreased claudin-5. The process facilitates the damage of small vessels, including pathological vasoconstriction, SMCs injury, blood-brain barrier damage, pre-thrombotic state and vascular inflammation. Along with these pathological changes progression, hypoperfusion, intravascular material leakage, demyelination, chronic vascular and perivascular inflammation appear successively, which eventually develop into cSVD.
Hcy-Lowering Therapy in cSVD
In HHcy patients, several trials, reviews, and meta-analyses evaluating primary prevention with treatment, have shown that Hcy-lowering therapy with B-vitamin supplementation reduces the incidence of cerebrovascular diseases (13–15), in contrast to studies that have shown no such association (16–18). Thus, the main question might be at what stage of the disease can cSVD lesions be reversed or stopped from progressing using proper Hcy-lowering treatment. Some studies did not observe positive results due to malabsorption and other individual factors. Patients with chronic kidney disease and those on antiplatelet therapy had reduced effects of B-vitamin supplementation on Hcy-associated cerebrovascular events (78). Older patients with gastrointestinal diseases failed to reach therapeutic concentrations of B-vitamins due to malabsorption and Hcy-lowering therapy may not be effective for hereditary cSVD (79). Moreover, delayed treatment may be a significant cause of a poor response to treatment. Nam et al. showed that if an ischemic stroke occurs in cSVD patients, disease progression might not be reversed by treatments meant to lower the tHcy levels, which suggest that patients with early or preclinical cSVD are more likely to benefit from Hcy-lowering therapy, and the curative effect was closely related to the timing of the treatment (68). In the stroke prevention trial, patients with low baseline Hcy levels had better outcomes than those with high levels (80). It indicates that Hcy-lowering treatment is similar to statin therapy for lowering lipid levels, and the underlying disease mechanisms are independent of the original baseline Hcy level. Considering that Hcy levels are associated with disease severity, it is also possible that patients with lower levels are more likely to be at an early stage of the disease and are more likely to benefit from treatment.
Discussion
Both cSVD and HHcy are age-dependent diseases, and as a risk factor of cSVD, HHcy contributes to its onset and progression. Nonetheless, the use of Hcy-lowering therapy is controversial. Therefore, it is unclear whether Hcy can be considered as an early indicator of cSVD and whether reducing Hcy levels is necessary for the treatment. Considering the prevalence of HHcy in older individuals and their vulnerability to cSVD, monitoring of tHcy levels is recommended. In consideration of the limited treatment time window, active measures should be taken sooner rather than later. ECs were injured when first exposed to elevated tHcy conditions; once Hcy is elevated, damage to the endothelium begins, which precedes cerebral small vessel injury years before the onset of cSVD. As mentioned above, ameliorating ED could reverse the progression of the disease in its early stage, which is the primary purpose of Hcy-lowering therapy. Since effective therapy relies on early intervention, immediate steps should be taken upon detection of HHcy. Currently, most pathological states are described as early or later stages; therefore, the critical point of the irreversible stage remains unclear. Further studies are needed to evaluate the relationship between different manifestations of endothelial injury and determine the timeline of brain damage in patients with cSVD.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and got involved in the process of preparation, correction, and modification of the manuscript. All of them approved it for publication.
Funding
LW was a post-doctoral fellow from the West China Hospital of Sichuan University and was supported by Postdoctoral Research Found of China (2021M692285).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Editage (www.editage.com) for English language editing.
References
1. De Silva TM, Faraci FM. Contributions of aging to cerebral small vessel disease. Annu Rev Physiol. (2020) 82:275–95. doi: 10.1146/annurev-physiol-021119-034338
2. Cannistraro RJ, Badi M, Eidelman BH, Dickson DW, Middlebrooks EH, Meschia JF. CNS small vessel disease: a clinical review. Neurology. (2019) 92:1146–56. doi: 10.1212/WNL.0000000000007654
3. Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. (2010) 9:689–701. doi: 10.1016/S1474-4422(10)70104-6
4. Mustapha M, Nassir C, Aminuddin N, Safri AA, Ghazali MM. Cerebral small vessel disease (CSVD) - lessons from the animal models. Front Physiol. (2019) 10:1317. doi: 10.3389/fphys.2019.01317
5. Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, et al. Safety and efficacy of recombinant human α-galactosidase a replacement therapy in Fabry's disease. N Engl J Med. (2001) 345:9–16. doi: 10.1056/NEJM200107053450102
6. Du H, Wilson D, Ambler G, Banerjee G, Shakeshaft C, Cohen H, et al. Small vessel disease and ischemic stroke risk during anticoagulation for atrial fibrillation after cerebral ischemia. Stroke. (2021) 52:91–9. doi: 10.1161/STROKEAHA.120.029474
7. Amarenco P, Benavente O, Goldstein LB, Callahan A 3rd, Sillesen H, Hennerici MG, et al. Results of the stroke prevention by aggressive reduction in cholesterol levels (SPARCL) trial by stroke subtypes. Stroke. (2009) 40:1405–9. doi: 10.1161/STROKEAHA.108.534107
8. Hilal S, Mok V, Youn YC, Wong A, Ikram MK, Chen CL. Prevalence, risk factors and consequences of cerebral small vessel diseases: data from three Asian countries. J Neurol Neurosurg Psychiatry. (2017) 88:669–74. doi: 10.1136/jnnp-2016-315324
9. Cuadrado-Godia E, Dwivedi P, Sharma S, Ois Santiago A, Roquer Gonzalez J, Balcells M, et al. Cerebral small vessel disease: a review focusing on pathophysiology, biomarkers, and machine learning strategies. J Stroke. (2018) 20:302–20. doi: 10.5853/jos.2017.02922
10. Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. (2013) 12:822–38. doi: 10.1016/S1474-4422(13)70124-8
11. Poggesi A, Pasi M, Pescini F, Pantoni L, Inzitari D. Circulating biologic markers of endothelial dysfunction in cerebral small vessel disease: a review. J Cereb Blood Flow Metab. (2016) 36:72–94. doi: 10.1038/jcbfm.2015.116
12. Ostrakhovitch EA, Tabibzadeh S. Homocysteine and age-associated disorders. Ageing Res Rev. (2019) 49:144–64. doi: 10.1016/j.arr.2018.10.010
13. Ji Y, Tan S, Xu Y, Chandra A, Shi C, Song B, et al. Vitamin B supplementation, homocysteine levels, and the risk of cerebrovascular disease: a meta-analysis. Neurology. (2013) 81:1298–307. doi: 10.1212/WNL.0b013e3182a823cc
14. Huo Y, Li J, Qin X, Huang Y, Wang X, Gottesman RF, et al. Efficacy of folic acid therapy in primary prevention of stroke among adults with hypertension in China: the CSPPT randomized clinical trial. JAMA. (2015) 313:1325–35. doi: 10.1001/jama.2015.2274
15. Park JH, Saposnik G, Ovbiagele B, Markovic D, Towfighi A. Effect of B-vitamins on stroke risk among individuals with vascular disease who are not on antiplatelets: a meta-analysis. Int J Stroke. (2016) 11:206–11. doi: 10.1177/1747493015616512
16. Lee M, Hong KS, Chang SC, Saver JL. Efficacy of homocysteine-lowering therapy with folic acid in stroke prevention: a meta-analysis. Stroke. (2010) 41:1205–12. doi: 10.1161/STROKEAHA.109.573410
17. Hsu CY, Chiu SW, Hong KS, Saver JL, Wu YL, Lee JD, et al. Folic acid in stroke prevention in countries without mandatory folic acid food fortification: a meta-analysis of randomized controlled trials. J Stroke. (2018) 20:99–109. doi: 10.5853/jos.2017.01522
18. McMahon JA, Green TJ, Skeaff CM, Knight RG, Mann JI, Williams SM. A controlled trial of homocysteine lowering and cognitive performance. N Engl J Med. (2006) 354:2764–72. doi: 10.1056/NEJMoa054025
19. Dankner R, Chetrit A, Lubin F, Sela BA. Life-style habits and homocysteine levels in an elderly population. Aging Clin Exp Res. (2004) 16:437–42. doi: 10.1007/BF03327398
20. Feng C, Bai X, Xu Y, Hua T, Huang J, Liu XY. Hyperhomocysteinemia associates with small vessel disease more closely than large vessel disease. Int J Med Sci. (2013) 10:408–12. doi: 10.7150/ijms.5272
21. Stipanuk MH, Ueki I. Dealing with methionine/homocysteine sulfur: cysteine metabolism to taurine and inorganic sulfur. J Inherit Metab Dis. (2011) 34:17–32. doi: 10.1007/s10545-009-9006-9
22. Castro R, Rivera I, Blom HJ, Jakobs C, Tavares de Almeida I. Homocysteine metabolism, hyperhomocysteinaemia and vascular disease: an overview. J Inherit Metab Dis. (2006) 29:3–20. doi: 10.1007/s10545-006-0106-5
23. Tchantchou F. Homocysteine metabolism and various consequences of folate deficiency. J Alzheimers Dis. (2006) 9:421–7. doi: 10.3233/JAD-2006-9408
24. Huang YC, Chang SJ, Chiu YT, Chang HH, Cheng CH. The status of plasma homocysteine and related B-vitamins in healthy young vegetarians and nonvegetarians. Eur J Nutr. (2003) 42:84–90. doi: 10.1007/s00394-003-0387-5
25. Yang B, Liu Y, Li Y, Fan S, Zhi X, Lu X, et al. Geographical distribution of MTHFR C677T, A1298C and MTRR A66G gene polymorphisms in China: findings from 15357 adults of han nationality. PLoS ONE. (2013) 8:e57917. doi: 10.1371/journal.pone.0057917
26. Cao Y, Su N, Zhang D, Zhou L, Yao M, Zhang S, et al. Correlation between total homocysteine and cerebral small vessel disease: a mendelian randomization study. Eur J Neurol. (2021) 28:1931–8. doi: 10.1111/ene.14708
27. Pepe G, Camacho Vanegas O, Giusti B, Brunelli T, Marcucci R, Attanasio M, et al. Heterogeneity in world distribution of the thermolabile C677T mutation in 5,10-methylenetetrahydrofolate reductase. Am J Hum Genet. (1998) 63:917–20. doi: 10.1086/302015
28. Xu R, Huang F, Wang Y, Liu Q, Lv Y, Zhang Q. Gender- and age-related differences in homocysteine concentration: a cross-sectional study of the general population of China. Sci Rep. (2020) 10:17401. doi: 10.1038/s41598-020-74596-7
29. Lim HS, Heo YR. Plasma total homocysteine, folate, and vitamin B12 status in Korean adults. J Nutr Science Vitaminol. (2002) 48:290–7. doi: 10.3177/jnsv.48.290
30. Jacques PF, Rosenberg IH, Rogers G, Selhub J, Bowman BA, Gunter EW, et al. Serum total homocysteine concentrations in adolescent and adult Americans: results from the third national health and nutrition examination survey. Am J Clin Nutr. (1999) 69:482–9. doi: 10.1093/ajcn/69.3.482
31. Selhub J. Homocysteine metabolism. Annu Rev Nutr. (1999) 19:217–46. doi: 10.1146/annurev.nutr.19.1.217
32. Miwa K, Tanaka M, Okazaki S, Yagita Y, Sakaguchi M, Mochizuki H, et al. Increased total homocysteine levels predict the risk of incident dementia independent of cerebral small-vessel diseases and vascular risk factors. J Alzheimers Dis. (2016) 49:503–13. doi: 10.3233/JAD-150458
33. Spence JD. Homocysteine-lowering therapy: a role in stroke prevention? Lancet Neurol. (2007) 6:830–8. doi: 10.1016/S1474-4422(07)70219-3
34. Marti-Carvajal AJ, Sola I, Lathyris D, Dayer M. Homocysteine-lowering interventions for preventing cardiovascular events. Cochrane Database Syst Rev. (2017) 8:CD006612. doi: 10.1002/14651858.CD006612.pub5
35. Refsum H, Nurk E, Smith AD, Ueland PM, Gjesdal CG, Bjelland I, et al. The hordaland homocysteine study: a community-based study of homocysteine, its determinants, and associations with disease. J Nutr. (2006) 136:1731S40. doi: 10.1093/jn/136.6.1731S
36. Kwon HM, Lee YS, Bae HJ, Kang DW. Homocysteine as a predictor of early neurological deterioration in acute ischemic stroke. Stroke. (2014) 45:871–3. doi: 10.1161/STROKEAHA.113.004099
37. Balint B, Jepchumba VK, Gueant JL, Gueant-Rodriguez RM. Mechanisms of homocysteine-induced damage to the endothelial, medial and adventitial layers of the arterial wall. Biochimie. (2020) 173:100–6. doi: 10.1016/j.biochi.2020.02.012
38. Hainsworth AH, Oommen AT, Bridges LR. Endothelial cells and human cerebral small vessel disease. Brain Pathol. (2015) 25:44–50. doi: 10.1111/bpa.12224
39. Quick S, Moss J, Rajani RM, Williams A. A vessel for change: endothelial dysfunction in cerebral small vessel disease. Trends Neurosci. (2021) 44:289–305. doi: 10.1016/j.tins.2020.11.003
40. Lai WK, Kan MY. Homocysteine-induced endothelial dysfunction. Ann Nutr Metab. (2015) 67:1–12. doi: 10.1159/000437098
41. Esse R, Barroso M, Tavares de Almeida I, Castro R. The contribution of homocysteine metabolism disruption to endothelial dysfunction: state-of-the-art. Int J Mol Sci. (2019) 20:867. doi: 10.3390/ijms20040867
42. Dayal S, Wilson KM, Leo L, Arning E, Bottiglieri T, Lentz SR. Enhanced susceptibility to arterial thrombosis in a murine model of hyperhomocysteinemia. Blood. (2006) 108:2237–43. doi: 10.1182/blood-2006-02-005991
43. Di Minno MN, Tremoli E, Coppola A, Lupoli R, Di Minno G. Homocysteine and arterial thrombosis: challenge and opportunity. Thromb Haemost. (2010) 103:942–61. doi: 10.1160/TH09-06-0393
44. Kaur K, Singh N, Dhawan RK. Exploring the role of dimethylarginine dimethylaminohydrolase-mediated reduction in tissue asymmetrical dimethylarginine levels in cardio-protective mechanism of ischaemic postconditioning in rats. Iran J Basic Med Sci. (2019) 22:1415–23. doi: 10.22038/IJBMS.2019.14067
45. Stanger O, Weger M. Interactions of homocysteine, nitric oxide, folate and radicals in the progressively damaged endothelium. Clin Chem Lab Med. (2003) 41:1444–54. doi: 10.1515/CCLM.2003.222
46. Yang Q, He GW. Imbalance of homocysteine and h2s: significance, mechanisms, and therapeutic promise in vascular injury. Oxid Med Cell Longev. (2019) 2019:7629673. doi: 10.1155/2019/7629673
47. Wang R, Szabo C, Ichinose F, Ahmed A, Whiteman M, Papapetropoulos A. The role of H2S bioavailability in endothelial dysfunction. Trends Pharmacol Sci. (2015) 36:568–78. doi: 10.1016/j.tips.2015.05.007
48. Chan SH, Hung CH, Shih JY, Chu PM, Cheng YH, Lin HC, et al. Exercise intervention attenuates hyperhomocysteinemia-induced aortic endothelial oxidative injury by regulating SIRT1 through mitigating NADPH oxidase/LOX-1 signaling. Redox Biol. (2018) 14:116–25. doi: 10.1016/j.redox.2017.08.016
49. Xiao Y, Xia J, Cheng J, Huang H, Zhou Y, Yang X, et al. Inhibition of S-adenosylhomocysteine hydrolase induces endothelial dysfunction via epigenetic regulation of p66shc-mediated oxidative stress pathway. Circulation. (2019) 139:2260–77. doi: 10.1161/CIRCULATIONAHA.118.036336
50. Jamaluddin MD, Chen I, Yang F, Jiang X, Jan M, Liu X, et al. Homocysteine inhibits endothelial cell growth via DNA hypomethylation of the cyclin A gene. Blood. (2007) 110:3648–55. doi: 10.1182/blood-2007-06-096701
51. Deplanque D, Lavallee PC, Labreuche J, Gongora-Rivera F, Jaramillo A, Brenner D, et al. Cerebral and extracerebral vasoreactivity in symptomatic lacunar stroke patients: a case-control study. Int J Stroke. (2013) 8:413–21. doi: 10.1111/j.1747-4949.2011.00755.x
52. Umemura T, Kawamura T, Umegaki H, Mashita S, Kanai A, Sakakibara T, et al. Endothelial and inflammatory markers in relation to progression of ischaemic cerebral small-vessel disease and cognitive impairment: a 6-year longitudinal study in patients with type 2 diabetes mellitus. J Neurol Neurosurg Psychiatry. (2011) 82:1186–94. doi: 10.1136/jnnp.2010.217380
53. Hassan A, Hunt BJ, O'Sullivan M, Parmar K, Bamford JM, Briley D, et al. Markers of endothelial dysfunction in lacunar infarction and ischaemic leukoaraiosis. Brain. (2003) 126:424–32. doi: 10.1093/brain/awg040
54. Xu S, Ilyas I, Little PJ, Li H, Kamato D, Zheng X, et al. Endothelial dysfunction in atherosclerotic cardiovascular diseases and beyond: from mechanism to pharmacotherapies. Pharmacol Rev. (2021) 73:924–67. doi: 10.1124/pharmrev.120.000096
55. Vanhoutte PM, Shimokawa H, Feletou M, Tang EH. Endothelial dysfunction and vascular disease - a 30th anniversary update. Acta Physiol. (2017) 219:22–96. doi: 10.1111/apha.12646
56. Liao FF, Lin G, Chen X, Chen L, Zheng W, Raghow R, et al. Endothelial nitric oxide synthase-deficient mice: a model of spontaneous cerebral small vessel disease. Am J Pathol. (2021) 191:1932–45. doi: 10.1016/j.ajpath.2021.02.022
57. Rajani RM, Quick S, Ruigrok SR, Graham D, Harris SE, Verhaaren BF, et al. Reversal of endothelial dysfunction reduces white matter vulnerability in cerebral small vessel disease in rats. Sci Transl Med. (2018) 10:eaam9507. doi: 10.1126/scitranslmed.aam9507
58. Zhang CE, Wong SM, van de Haar HJ, Staals J, Jansen JF, Jeukens CR, et al. Blood–brain barrier leakage is more widespread in patients with cerebral small vessel disease. Neurology. (2017) 88:426–32. doi: 10.1212/WNL.0000000000003556
59. Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb Perspect Biol. (2015) 7:a020412. doi: 10.1101/cshperspect.a020412
60. Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. (2007) 27:697–709. doi: 10.1038/sj.jcbfm.9600375
61. Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, et al. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. (2003) 161:653–60. doi: 10.1083/jcb.200302070
62. Jalal FY, Yang Y, Thompson J, Lopez AC, Rosenberg GA. Myelin loss associated with neuroinflammation in hypertensive rats. Stroke. (2012) 43:1115–22. doi: 10.1161/STROKEAHA.111.643080
63. Huang Y, Zhang W, Lin L, Feng J, Chen F, Wei W, et al. Is endothelial dysfunction of cerebral small vessel responsible for white matter lesions after chronic cerebral hypoperfusion in rats? J Neurol Sci. (2010) 299:72–80. doi: 10.1016/j.jns.2010.08.035
64. Porro C, Trotta T, Panaro MA. Microvesicles in the brain: biomarker, messenger or mediator? J Neuroimmunol. (2015) 288:70–8. doi: 10.1016/j.jneuroim.2015.09.006
65. Vion AC, Ramkhelawon B, Loyer X, Chironi G, Devue C, Loirand G, et al. Shear stress regulates endothelial microparticle release. Circ Res. (2013) 112:1323–33. doi: 10.1161/CIRCRESAHA.112.300818
66. Denorme F, Vanhoorelbeke K, De Meyer SF. von Willebrand factor and platelet glycoprotein ib: a thromboinflammatory axis in stroke. Front Immunol. (2019) 10:2884. doi: 10.3389/fimmu.2019.02884
67. Shi Y, Wardlaw JM. Update on cerebral small vessel disease: a dynamic whole-brain disease. Stroke Vasc Neurol. (2016) 1:83–92. doi: 10.1136/svn-2016-000035
68. Nam KW, Kwon HM, Jeong HY, Park JH, Kwon H, Jeong SM. Serum homocysteine level is related to cerebral small vessel disease in a healthy population. Neurology. (2019) 92:e317–25. doi: 10.1212/WNL.0000000000006816
69. Wong A, Mok V, Fan YH, Lam WW, Liang KS, Wong KS. Hyperhomocysteinemia is associated with volumetric white matter change in patients with small vessel disease. J Neurol. (2006) 253:441–7. doi: 10.1007/s00415-005-0022-x
70. Kloppenborg RP, Geerlings MI, Visseren FL, Mali WP, Vermeulen M, van der Graaf Y, et al. Homocysteine and progression of generalized small-vessel disease: the SMART-MR study. Neurology. (2014) 82:777–83. doi: 10.1212/WNL.0000000000000168
71. Naka H, Nomura E, Takahashi T, Wakabayashi S, Kajikawa H, Kohriyama T, et al. Plasma total homocysteine levels are associated with advanced leukoaraiosis but not with asymptomatic microbleeds on T2*-weighted MRI in patients with stroke. Eur J Neurol. (2006) 13:261–5. doi: 10.1111/j.1468-1331.2006.01205.x
72. Cloonan L, Fitzpatrick KM, Kanakis AS, Furie KL, Rosand J, Rost NS. Metabolic determinants of white matter hyperintensity burden in patients with ischemic stroke. Atherosclerosis. (2015) 240:149–53. doi: 10.1016/j.atherosclerosis.2015.02.052
73. Wardlaw JM, Doubal F, Armitage P, Chappell F, Carpenter T, Munoz Maniega S, et al. Lacunar stroke is associated with diffuse blood-brain barrier dysfunction. Ann Neurol. (2009) 65:194–202. doi: 10.1002/ana.21549
74. Wardlaw JM. Blood-brain barrier and cerebral small vessel disease. J Neurol Sci. (2010) 299:66–71. doi: 10.1016/j.jns.2010.08.042
75. Beard RS Jr., Reynolds JJ, Bearden SE. Hyperhomocysteinemia increases permeability of the blood-brain barrier by NMDA receptor-dependent regulation of adherens and tight junctions. Blood. (2011) 118:2007–14. doi: 10.1182/blood-2011-02-338269
76. Muradashvili N, Tyagi R, Metreveli N, Tyagi SC, Lominadze D. Ablation of MMP9 gene ameliorates paracellular permeability and fibrinogen-amyloid beta complex formation during hyperhomocysteinemia. J Cereb Blood Flow Metab. (2014) 34:1472–82. doi: 10.1038/jcbfm.2014.102
77. Wardlaw JM, Smith C, Dichgans M. Small vessel disease: mechanisms and clinical implications. Lancet Neurol. (2019) 18:684–96. doi: 10.1016/S1474-4422(19)30079-1
78. Vermeer SE, Van Dijk EJ, Koudstaal PJ, Oudkerk M, Hofman A, Clarke R, et al. Homocysteine, silent brain infarcts, and white matter lesions: the rotterdam scan study. Ann Neurol. (2002) 51:285–9. doi: 10.1002/ana.10111
79. Rutten-Jacobs LC, Traylor M, Adib-Samii P, Thijs V, Sudlow C, Rothwell PM, et al. Association of MTHFR C677T genotype with ischemic stroke is confined to cerebral small vessel disease subtype. Stroke. (2016) 47:646–51. doi: 10.1161/STROKEAHA.115.011545
Keywords: cerebral small vessel disease, homocysteine, hyperhomocysteinemia, endothelial dysfunction, homocysteine-lowering therapy
Citation: Li S, Li G, Luo X, Huang Y, Wen L and Li J (2021) Endothelial Dysfunction and Hyperhomocysteinemia-Linked Cerebral Small Vessel Disease: Underlying Mechanisms and Treatment Timing. Front. Neurol. 12:736309. doi: 10.3389/fneur.2021.736309
Received: 09 July 2021; Accepted: 01 November 2021;
Published: 24 November 2021.
Edited by:
Tao Liu, Hainan General Hospital, ChinaReviewed by:
Ivan V. Brak, State Scientific Research Institute of Physiology and Basic Medicine, RussiaYuzhen Xu, Tongji University, China
Copyright © 2021 Li, Li, Luo, Huang, Wen and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lan Wen, bF93ZW4xMjI1QDE2My5jb20=; Jinglun Li, bGpsMDMxNjExQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship