 Gergo Kovacs
Gergo Kovacs Lasse Reimer
Lasse Reimer Poul Henning Jensen
Poul Henning Jensen- 1Danish Research Institute of Translational Neuroscience – DANDRITE, Aarhus University, Aarhus, Denmark
- 2Department of Biomedicine, Aarhus University, Aarhus, Denmark
Neuronal calcium dyshomeostasis has been associated to Parkinson's disease (PD) development based on epidemiological studies on users of calcium channel antagonists and clinical trials are currently conducted exploring the hypothesis of increased calcium influx into neuronal cytosol as basic premise. We reported in 2018 an opposite hypothesis based on the demonstration that α-synuclein aggregates stimulate the endoplasmic reticulum (ER) calcium pump SERCA and demonstrated in cell models the existence of an α-synuclein-aggregate dependent neuronal state wherein cytosolic calcium is decreased due to an increased pumping of calcium into the ER. Inhibiting the SERCA pump protected both neurons and an α-synuclein transgenic C. elegans model. This models two cellular states that could contribute to development of PD. First the prolonged state with reduced cytosolic calcium that could deregulate multiple signaling pathways. Second the disease ER state with increased calcium concentration. We will discuss our hypothesis in the light of recent papers. First, a mechanistic study describing how variation in the Inositol-1,4,5-triphosphate (IP3) kinase B (ITPKB) may explain GWAS studies identifying the ITPKB gene as a protective factor toward PD. Here it was demonstrated that how increased ITPKB activity reduces influx of ER calcium to mitochondria via contact between IP3-receptors and the mitochondrial calcium uniporter complex in ER-mitochondria contact, known as mitochondria-associated membranes (MAMs). Secondly, it was demonstrated that astrocytes derived from PD patients contain α-synuclein accumulations. A recent study has demonstrated how human astrocytes derived from a few PD patients carrying the LRRK2-2019S mutation express more α-synuclein than control astrocytes, release more calcium from ER upon ryanodine receptor (RyR) stimulation, show changes in ER calcium channels and exhibit a decreased maximal and spare respiration indicating altered mitochondrial function in PD astrocytes. Here, we summarize the previous findings focusing the effect of α-synuclein to SERCA, RyR, IP3R, MCU subunits and other MAM-related channels. We also consider how the SOCE-related events could contribute to the development of PD.
Old and New Hypotheses on Ca2+ in Parkinson's Disease and Synucleinopathies
Parkinson's disease (PD) is the second most common neurodegenerative disorder with more than 6 million diagnosed cases, and more than 100 000 deaths/year (1, 2). It belongs to the group of neurodegenerative synucleinopathies, dominated by PD, dementia with Lewy bodies (DLB) and Multiple system atrophy (MSA). These diseases are hallmarked by the presence of inclusions in degenerating brain cells that contain aggregates of the presynaptic protein α-synuclein (α-syn). In PD and DLB inclusions are in neurons whereas they predominantly are located in oligodendrocytes in MSA.
Our insight into the pathophysiology of the synucleinopathies has evolved greatly since α-syn was identified as a presynaptic protein (3) and first associated to neurodegenerative diseases as a component of amyloid plaques in Alzheimer's disease (4). The breakthrough came in 1997 when a missense mutation in SNCA (the gene encoding the α-syn protein) was identified as causing autosomal dominant PD (5) and α-syn was identified as an unifying component of Lewy bodies in PD and DLB and in glial cytoplasmic inclusions in MSA (6, 7). Later, several missense α-syn mutations as well as multiplication of the SNCA gene has been demonstrated as causing autosomal dominant forms of PD and DLB (8). The progressive nature of the Lewy body pathology was described by Braak that proposed the so-called Braak hypothesis. It postulates pathology to spread from two places in the nervous system: neurons in the olfactory bulb and also neurons of the gut that via the Vagal nerve spread to the brain stem and further progress to the substantia nigra and neocortical areas (9, 10). Recent data suggest an opposite route of spreading also exists, so-called top down PD, from the central nervous system to the peripheral nervous system (11). These routes of spreading are hypothesized to be brought about by a prion-like spreading of α-syn aggregates from degenerating neurons to connected neurons where they seed the aggregation of native α-syn in the healthy recipient neurons.
The loss of the dopamine producing neurons of the substantia nigra pars compacta (SNpc) forms the basis for the motor symptoms that is diagnostic for PD. The demonstration of slow rhythmic calcium (Ca2+) oscillation in this population of pacemaking dopaminergic (DA) neurons driven by Ca2+ influx through CaV1 Ca2+ channels, along with their low Ca2+ buffering capacity and increased oxidative stress formed part of the basis for an almost 30-year-old calcium hypothesis (12–14). It gained strong support from epidemiological studies demonstrating a decreased risk for PD in users of L-type Ca2+ channel antagonists (15, 16), although the latter study rather interpreted their data as supportive for a symptomatic effect. Influenced by these findings, isradipine, a CaV1.3 channel antagonist with promising pre-clinical neuroprotective effects in some PD animal models (17–19), was administered to patients with early-stage PD for 36 months in a large clinical trial (20). Yet, despite encouraging pre-clinical studies, isradipine failed to slow the clinical progression of early-stage PD patients. This might be explained by insufficient dosing of the drug in the clinical study, but it could also reflect that this calcium hypothesis does not reflect the underlying disease mechanisms driving the development and progression of PD through the nervous system.
In 2018 we offered an alternative calcium hypothesis founded on progressive α-syn aggregate toxicity in neurons as a basis for the progression of PD (Figures 1A,B). It was based on our demonstration of a biphasic cytosolic Ca2+ response caused by α-syn aggregates that bind and stimulate the endoplasmic reticulum (ER) Ca2+ pump SERCA thereby enhancing its pumping of Ca2+ from the cytosol into the ER. This activation causes an initial prolonged phase characterized by a decrease in cytosolic Ca2+ along with an increased Ca2+ loading of the ER that is followed by a second degenerative phase with increased cytosolic Ca2+ prior to cell death (21). Both phases could be counteracted by treating the neurons and a transgenic C. elegans model with low doses of the SERCA inhibitor cyclopiazonic acid (CPA) (Figure 1C). The prodegenerative signaling pathways activated in the early and in the late phases are yet to be discovered. Given that PD is a slowly progressive disease hypothesized to be driven by a spreading of α-syn aggregates through the nervous system, we envision that at any given time there will be some neurons in the frontier of spreading that are affected by the first low Ca2+ phase and other areas affected for longer time by the late high Ca2+ phase. Treatment of the SERCA activation thus holds potential of reducing the dysfunction of the neurons, e.g., symptomatology, but also modification of the progression of the disease.
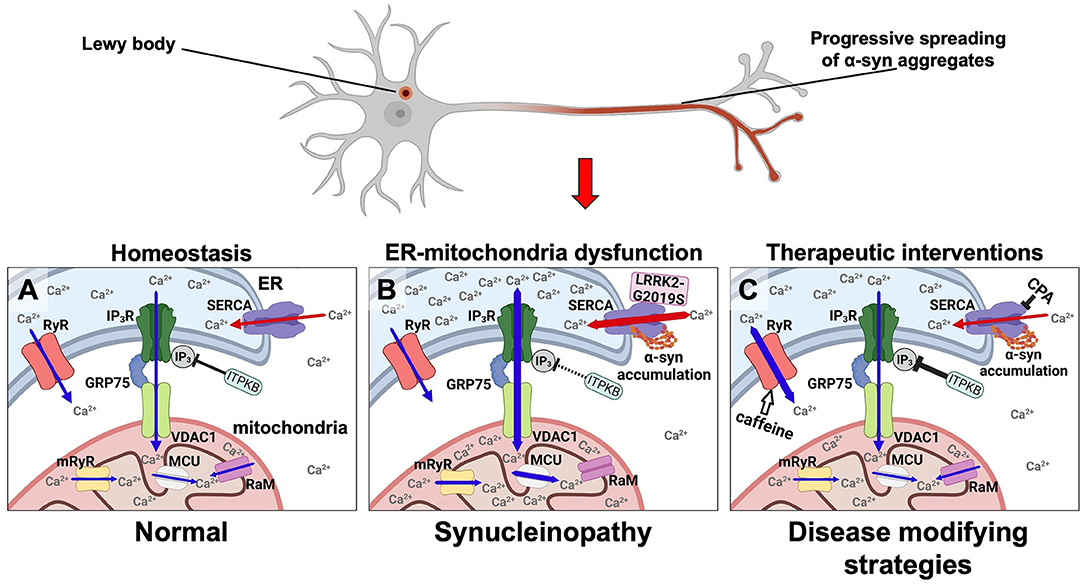
Figure 1. Disease modifying strategies for synucleinopathies by modulating aberrant Ca2+ fluxes in ER and mitochondria via targeting the SERCA pump, the RyR, and the ER-mitochondrial transport complex. α-syn is under normal conditions predominantly located in nerve terminals. In synucleinopathies, its initial aggregation is hypothesized to initiate at this site. From here, aggregates are transported retrograde through the axon to the cell body where Lewy bodies can form. (A) In healthy neurons the low resting Ca2+ level in the cytosol and the high Ca2+ levels in the ER and mitochondria are tuned by (1) the SERCA pump loading the ER with Ca2+ from the cytosol (red arrows), (2) the release of Ca2+ from ER to the cytosol by RyR and IP3R (blue arrows), (3) where the latter is part of the multi-molecular Ca2+ funnel between ER lumen and the mitochondrial matrix (IP3R, GRP75, VDAC1, mRyR, MCU, and RaM). (B) In synucleinopathies, the α-syn aggregates activate SERCA causing an increased Ca2+ uptake into the ER thereby deregulating ER functions and increasing the Ca2+ flux through the IP3R-GRP75-VDAC1 complex and MCU into mitochondria. These increased Ca2+ fluxes into ER and mitochondria along with a decreased cytosolic Ca2+ concentration result in a reduced functionality and viability of neurons in synucleinopathies. The same pathways can be activated by mutant LRRK2-G2019S that activates SERCA and by loss-of-function genetic risk variants in the ITPKB kinase that increases the IP3-dependent Ca2+ flux from ER to mitochondria. (C) These abnormal Ca2+ fluxes can be counteracted as a disease modifying strategy in synucleinopathies. The α-syn activated SERCA can be inhibited e.g., by CPA. The Ca2+ flux from ER to mitochondria can for example be attenuated by stimulating ITPKB or inhibiting the IP4 phosphatase. The Ca2+ flux from ER into the cytosol can be stimulated by caffeine (white arrow) or other RyR activators.
In the current review we will primarily focus on Ca2+ regulatory molecules in the ER in relation to synucleinopathy-related pathology and discuss how our SERCA activation calcium hypothesis align with recent publications implicating mitochondria in this pathway.
Organelles, Ca2+ Fluxes and their Interplay in PD
Neuronal Ca2+ influx is a vital component in the process of neurotransmission from the regulation of dendritic responses to neurotransmitter binding, propagation of action potential along axons and transmitter release from nerve terminals; and also serves as a second messenger in signaling pathways regulating multiple processes, including metabolism, protein phosphorylation, exocytosis, gene transcription and programmed cell death (22, 23). Each of these pathways can affect neuronal function and neurodegenerative processes, thus Ca2+ dyshomeostasis is proposed to play a vital part in complex brain processes like age-dependent cognitive decline and development of e.g., schizophrenia (24, 25). Maintenance of specific subcellular Ca2+ levels is therefore a prerequisite for a plethora of cellular processes and their integrated support of the mind. Neuronal Ca2+ signaling is highly advanced and mediated by various ion channels, exchangers, and pumps situated on the plasma membrane (PM) and membranes of different organelles such as ER, mitochondria, lysosomes, Golgi apparatus and others (23). In the following section we will briefly outline the role of the ER and mitochondria representing two central organelles in Ca2+ transport, buffering and whose proper function depend on their luminal Ca2+ levels (Figures 1A,B). This will include description of their interphases and the ER interphase with PM, and a discussion on how dysregulation of specific Ca2+ pumps and channels in these organelles might play a role in the etiology and pathology of PD and other synucleinopathies.
Endoplasmic Reticulum (ER)
ER forms an interconnected network of tubules throughout the neuron from dendrites to nerve terminals (26). It is in close contact with many other intracellular organelles and their communication has essential functions in maintaining Ca2+ homeostasis during physiology processes and disturbance herein holds potential for facilitating psychiatric symptoms and neurodegenerative processes (27). While the resting cytosolic Ca2+ concentration in neurons is in the nanomolar range (28), in the ER and in the extracellular compartments it is in the millimolar range (29, 30). To maintain such steep gradients, active transporters are necessary, and in the case of ER, the Sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) carries the load. Together with the PM Ca2+ ATPase (PMCA), SERCA, and Na+/Ca2+ exchangers (NCX) contribute to maintain a low resting cytosolic Ca2+ concentrations in neurons (31).
The Active Player for Ca2+ Uptake–The SERCA Pump
SERCA plays a crucial role in maintaining ~150 nM concentration of Ca2+ in the cytosol and do this by pumping the ions against a ~1 mM Ca2+ gradient into the ER at the expense of ATP hydrolysis. SERCA pumps has a quite complex biology (32): they are expressed ubiquitously from three genes, ATP2A1, ATP2A2, and ATP2A3; and taking alternative splicing into account, 13 different isoforms exist (33, 34). The molecular weight of SERCA is usually referred as 110 kDa however the different isoforms have different molecular weights between 95 and 115 kDa (35, 36). The different SERCA isoforms are expressed in a developmental and tissue specific manner with the SERCA2B and SERCA3 being dominant in the brain (37).
We have shown that α-syn aggregates bind to and stimulate SERCA resulting in an increased Ca2+-load in the ER-lumen and a reduced cytosolic Ca2+ level. Following a prolonged phase with reduced cytosolic Ca2+, yet unknown homeostatic mechanisms deteriorate resulting in increased cytosolic Ca2+ and cell death (21). These two cellular states may contribute to development and progression of PD where the first phase with reduced cytosolic Ca2+ could deregulate multiple signaling pathways affecting the circuitry functions of neurons and thus contribute to a range of neurological and psychiatric symptoms. The second phase with increased Ca2+ concentration will then initiate the irreversible phase with enhanced neuron loss. The latter phase may resemble aspects of the “old” calcium hypothesis. We demonstrated the specific SERCA inhibitor CPA was able to counteract both phases of Ca2+ deregulation caused by α-syn aggregate stress and as a result increased the viability of primary neuron models and protected against cell loss in a C. elegans model (21) (Figure 1C).
Astrocytes has been linked to α-syn pathology after Braak reported that astrocytes in PD patients of advanced stages were immunoreactive for α-syn (38) and such astrocytic α-syn causes proinflammatory stress from the astrocytes (39). Genetic and molecular evidence has recently linked the LRRK2-G2019S mutant, which is the most common dominant cause of familial PD (40, 41), to SERCA, ER and mitochondrial stress in astrocytes (42, 43).
Ca2+ Release Channels–IP3R and RyR
The release of Ca2+ ions from the ER in neurons is mediated through two distinct receptor channels, namely the Inositol-1,4,5-trisphosphate receptors (IP3Rs) and Ryanodine receptors (RyRs) (44, 45). Contrary to the RyRs, which only are expressed in excitable cells like neurons and muscle cells, the IP3Rs show ubiquitous expression patterns.
IP3R
The IP3R is a Ca2+ channel located in ER membrane that is activated by binding of Inositol-1,4,5-trisphosphate (IP3). The IP3 is generated by phospholipase C action on the membrane phospholipid Phosphatidylinositol 4,5-bisphosphate (PIP2) in a process often associated to G-protein coupled receptor activation. The IP3R is critically involved in the transport of Ca2+ from ER to mitochondria through its interaction with the major mitochondrial Ca2+ transport channel located in the outer mitochondrial membrane, called Voltage-dependent anion channel 1 (VDAC1) in a complex bridged by the chaperone GRP75 (75 kDa Glucose-regulated protein) (46, 47). This interaction occurs at specialized ER-mitochondrial interphases known as mitochondria-associated membranes (MAMs). The tethering complex of IP3R-GRP75-VDAC1 allows the efficient diffusion of Ca2+ from ER into mitochondria. Other chaperones, like calnexin and calreticulin can also interact with IP3R and SERCA2B to regulate mitochondrial Ca2+ homeostasis (48). There are 3 known isoforms of IP3R encoded by ITPR1, ITPR2, and ITPR3 genes, that are expressed as 312, 308, and 304 kDa protein products, respectively (49, 50). Although all three isoforms are expressed ubiquitously, there is also a heterogeneity in their expression pattern in different tissues and in their function (51).
The activation of IP3R is not just regulated by phospholipase C dependent release of IP3 but can also be modulated by inactivating IP3 via its phosphorylation to IP4 by IP3 kinases. The IP3 kinase B (ITPKB) is such an inactivating kinase (52). It is ubiquitously expressed and it is the most abundantly expressed IP3 kinase in the central nervous system across several brain regions related to PD (53). ITPKB is a risk gene for sporadic PD (54) and recently, Apicco et al. investigated how modulating ITPKB activity in neurons by genetic and pharmacological methods impacted the accumulation of pS129 labeled α-syn aggregates in human neuron and in vivo rodent models (55). They demonstrated that in neurons ITPKB activity negatively regulate the transfer of Ca2+ from ER stores to mitochondria. Here the increased Ca2+ is associated with functional changes in mitochondria, including increased respiration, ATP production, and the accumulation of reactive oxygen species (ROS). In summary, this molecular study indicated that the genetic PD risk contributed by the ITPKB locus is caused by its modulation of the Ca2+ flux from ER to mitochondria (55). We find it tempting to speculate that an increased Ca2+ load in ER caused by α-syn aggregates activation of SERCA likewise will favor a pathogenic Ca2+ flux from ER to mitochondria (Figure 1B).
RyR
The RyRs exists in three tissue-specific isoforms encoded by RyR1, RyR2, and RyR3 genes. While the 565 kDa RyR1 is mainly expressed in skeletal muscles and cerebellar Purkinje cells, the 565 kDa RyR2 is the predominant isoform in the brain and heart, and the 552 kDa RyR3 is brain-specific albeit with low expression levels (23). The RyR channels forms by homo-tetramerization, and thereby constitutes the largest known intracellular ion channel with a size that exceeds 2 MDa. Although the role of RyR in the brain is still incompletely understood, several lines of evidence point toward involvement in long term potentiation (LTP) and long term depression (LTD) (56, 57). A recent genome-wide association study (GWAS) suggests a role in PD, where a specific SNP (Single Nucleotide Polymorphism) variant of the RyR2 gene was associated with a lower cognitive score in PD patients (58).
Structurally, the transmembrane pore of the RyR, in which the Ca2+ ions are transported is formed by the C-terminal part of the receptor, while a large cytoplasmic region of the channel is the interaction site of most RyR modulators (23). One modulator of RyR is Ca2+ itself, which can act as an activating ligand. This also explains how an increase in cytosolic Ca2+ triggers further Ca2+ release in a mechanism known as Ca2+-induced Ca2+ release (CICR). On the other hand, a detrimental decrease in cytosolic Ca2+, e.g., due increased activation of SERCA by α-syn aggregates (21), could in theory be aggravated by a decrease in RyR activation due to the reduction of its Ca2+ ligand. Interestingly, an increase in cytosolic Ca2+ more easily activates the brain-predominant isoforms, RyR2 and RyR3, compared to RyR1 (59) suggesting a prominent role of CICR in brain. Besides Ca2+, another cellular activation pathway of RyR via cyclic adenosine diphosphate ribose (cADP-ribose) exists (60). Pharmacologically RyRs can be activated by nanomolar concentrations of the plant alkaloid ryanodine, whereas high concentrations are inhibitory (61). Caffeine is a well-known activator of RyR that sensitize RyR to its activation by cytosolic and luminal Ca2+ to facilitate spontaneous Ca2+ release (62–65). RyR was also identified in rat heart mitochondria (66), named as mitochondrial Ryanodine receptor (mRyR), where it is located in the inner mitochondrial membrane and partially is responsible for facilitating rapid mitochondrial Ca2+ uptake. mRyR has also been demonstrated in mitochondria of primary cultures of rat striatal neurons (67). Using subtype specific antibodies and RyR1 knockout mice tissue it was demonstrated that cardiac mRyR is the RyR1 isoform (68).
According to our calcium hypothesis, which postulates that a normalization of an otherwise decreased cytosolic Ca2+ level will alleviate PD-related pathophysiology, a caffeine-induced release of Ca2+ from the ER through RyR would be favorable. Interestingly, several epidemiological studies point toward a protective role of caffeine in relation to PD onset [for a review, see (69)]. A clinical study demonstrated improved objective motor measures in PD patients treated with caffeine (70), and another study found an association between coffee consumption and improved mood and cognition in PD patients (71). The short-term nature of the studies may have missed a positive disease modifying potential considering the slowly progressive nature of PD and lacking good biomarkers of disease progression. In line with this, plasma concentrations of caffeine and its metabolites, paraxanthine, theophylline and 1-methylxanthine, was significantly lower in both sporadic PD patients, and PD patients carrying an LRRK2 mutation, relative to unaffected controls (72). Chronic caffeine treatment attenuated α-syn inclusion formation and reverted autophagic defects caused by intrastriatal injection of preformed α-syn fibrils (PFFs) in an α-syn transgenic mouse model (73) (Figure 1C). Caffeine is a somewhat promiscuous drug with a complex pharmacological profile where antagonism of adenosine receptors is often considered the main pharmacological target, and the current leading hypothesis in the field is that caffeine exerts its protective effects through antagonizing adenosine A2A receptor (69). The adenosine A2A receptor antagonist istradefylline is approved for treatment of PD patients (74) but istradefylline did not, compared to caffeine, protect against the progressive neurodegeneration in our yet unpublished preclinical study using the A53T-α-syn transgenic M83 model (Betzer et al., unpublished). It is thus yet unclear how caffeine mechanistically decreased the progression of α-syn in preclinical models of synucleinopathies [(73) and Betzer et al., unpublished] but could in principle be via activation of RyR thus supporting our calcium hypothesis.
Another protein that may contribute to the interplay between molecules regulating Ca2+ in the ER is the Soluble resistance-related Ca2+-binding protein (SORCIN) that upon Ca2+ binding is able to modulate the function of SERCA and RyR (75).
Ca2+ Flux Across the Plasma Membrane Into the ER–Store Operated Ca2+ Entry (SOCE)
The ER can also form Ca2+ conducive membrane interphases with the PM (76) and this interaction is critical for the process of store operated Ca2+ entry (SOCE) into the ER and cytosol from the extracellular space with the tethering of the membranes mediated by the proteins ORAI and Stromal interaction molecules (STIMs) (76). SOCE plays an important role in the nerve terminals where α-syn is present in high concentrations (77). Upon Ca2+ depletion of ER, Ca2+-selective store-operated channels (SOCs) are assembled by interaction between ER Ca2+-sensing STIM1/2 proteins and plasma membrane-associated ORAI1/2/3 proteins, which together form the PM Ca2+ release-activated Ca2+ (CRAC) channels, thereby allowing restoration of ER Ca2+ stores by influx from the extracellular space (78). In addition to ORAI proteins, the transient receptor potential canonical channels (TRPC) have also been suggested to participate in CRAC channel formation (78, 79). SERCA participates in termination of SOCE by restoring ER its high Ca2+ levels (80).
By inducing Ca2+ influx into the cytoplasm and ER as a consequence of ER Ca2+ depletion, SOCE plays an important role in maintaining neuronal Ca2+ homeostasis, and SOCE has been linked to a multitude of biological processes, including transcription, exocytosis, and metabolism (78). The α-syn aggregate dependent activation of SERCA that increases ER Ca2+ levels holds potential for disturbing SOCE (21).
Interestingly, post-mortem SNpc samples from PD patients demonstrated a decreased presence of TRPC1 compared to non-PD patients, which could suggest a downregulation of SOCE (81). Human primary skin fibroblasts derived from patients with either familial PD, caused by mutations in PLA2G6 gene (PARK14), or idiopathic PD also show impaired SOCE relative to those of control patients (82). Moreover, Pla2g6 knockout mice displayed autophagic dysfunctions, loss of SNpc DA neurons and an age-dependent motor dysfunction (82).
ER Stress Responses
Apart from storing Ca2+ and securing a low cytosolic Ca2+ level, several functions of the ER is critically dependent on a regulated luminal Ca2+ level like folding of secretory proteins and membrane proteins for Golgi, lysosomes and the plasma membrane; and lipid and sterol biogenesis (83). Perturbation of these functions can cause the unfolded protein responses (UPR) (84). UPR can be induced by environmental or genetic insults that result in dysfunctional protein folding in the ER lumen (85, 86). Experimentally UPR has been induced by lowering the ER Ca2+ level by the SERCA inhibitor thapsigargin and the inhibitor of protein glycosylation tunicamycin (87, 88). The prototypic stress response aims at restoring the protein folding homeostasis in the ER and is dominated by three transmembrane ER proteins PERK (PKR-like ER kinase), IRE1α (Inositol-requiring transmembrane kinase/endoribonuclease 1α), and ATF6 (Activating transcription factor 6) that all become activated when the ER chaperones BiP and GRP78 (78 kDa Glucose-regulated protein) are released upon their interaction with misfolded proteins (89, 90). α-Syn aggregate stress has been linked to the UPR pathway (91, 92), and salubrinal, an eIF2α phosphatase inhibitor that attenuates ER stress delayed the onset of α-synucleinopathy in A53T transgenic mice (93). α-syn aggregates has been demonstrated to accumulate at ER in sick α-syn transgenic mice and in ER containing microsomes from PD brains (89, 93).
Missense mutations in the lysosomal enzyme glucocerebrosidase are responsible for Gaucher's disease that is a strong risk factor for PD. The pathophysiology for some mutations likely contains a component of ER stress as the RyR blocker Dantrolene was able to alleviate disease in a mouse model of Gaucher's disease (94).
GWAS studies have revealed that genetic risk factors for sporadic PD frequently encode for proteins involved in lysosomal function and autophagy, processes relying on ER function (54). This is for example demonstrated by the generation of the double membrane of autophagosomes that engulf cargo for degradation by macroautophagy. The double membrane is derived from the ER membrane or ER-mitochondrial contact sites (95). Attenuation of the α-syn aggregate activated SERCA pump in neurons decreased their cellular α-syn level suggesting a direct link between the ER dysfunction and α-syn regulating autophagy (21). Inhibition of ER in neurons by knockout of ATG5 surprisingly revealed that the reticular ER in axons is a primary target for neuronal autophagy and demonstrated the ER resident RyR Ca2+ channels as especially sensitive to autophagic dysfunction (96). The changes in ER by autophagy inhibition did change axonal and ER Ca2+ levels but did remarkably not elicit the conventional ER response driven by the PERK/IRE1a and ATF6 signaling axis (96) although it affected neuronal transmission as also observed for ER stress (97).
Ca2+ Fluxes From ER to Mitochondria
Although ER is considered the main Ca2+ store within cells, mitochondria also play a key function in buffering Ca2+ ions. Moreover, Ca2+ plays a pivotal role in mitochondria as oxidative phosphorylation requires mitochondrial Ca2+ and dysregulation hereof can generate oxidative stress associated with cell death in neurodegenerative diseases (98). Modest accumulation of Ca2+ in the mitochondria can lead to decreased respiration, while further increase in Ca2+-level results to the degradation of metabolic enzymes through the activation of mitochondrial Calpain 1, and also increases ROS generation by impairing of respiratory enzymes. Oxidative stress, Ca2+-overload in mitochondria and ROS-generation are amplifying each other's effect which leads to mitochondrial damage (99, 100). In neurons, increased cytosolic Ca2+ can inhibit both anterograde and retrograde transport of mitochondria to axons and dendrites, where mitochondria act as Ca2+ buffers to sustain synaptic activity (101).
The Ca2+ flow from ER to mitochondria relies on the proximity of their membranes in the MAMs structure (47). Here tethering of IP3R to VDAC1 is facilitated by the cytosolic GRP75 thereby creating a funnel facilitating the diffusion of the high concentration of Ca2+ in ER into the intermembranous mitochondrial space. The further diffusion into the mitochondrial matrix is facilitated via the mitochondrial Ca2+ uniporter (MCU), mRyR and the so-called rapid mode uptake channel (RaM) located in the inner mitochondrial membrane (67, 102, 103). RaM, which exact molecular identity is still unknown, facilitates a rapid mode of mitochondrial Ca2+ uptake with faster kinetics than MCU but, interestingly, high extra-mitochondrial Ca2+ level has inhibiting effect to RaM (102). The importance of this pathway is highlighted by the PD risk variants of the IP3 kinase ITPKB that increases the Ca2+ flux from ER to mitochondria through the IP3R and causes aggregation of α-syn in neurons (55), which is a hallmark of PD cytopathology.
Due to a low affinity of the MCU to Ca2+ ions, the local cytosolic Ca2+ concentration must reach ~5–10 μM in order for MCU to produce significant transport of Ca2+ into the mitochondria matrix (104). This may explain why up to 20% of the mitochondrial surface is situated in nanometer proximity to the ER (105–107).
An increased ER Ca2+ concentration, e.g., by an α-syn aggregate stimulated SERCA will favor the Ca2+ transport to mitochondria (Figure 1).
Conclusion
Recent evidence points to a critical role for dysregulation of Ca2+ fluxes into the ER from the cytosol and from ER into mitochondria in the development of PD and other synucleinopathies. The evidence is based on genetic, biochemical and molecular studies. Promising preclinical data demonstrate disease modifying potential of targeting the SERCA pump, Ca2+ channels RyR, IP3R and IP3 regulating enzymes. This holds promise for identification of a new lines of disease modifying drug targets that can be tested in the near future.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
The study was supported by Lundbeck Foundation grants R223-2015-4222 for PHJ, R248-2016-2518 for Danish Research Institute of Translational Neuroscience-DANDRITE, Nordic-EMBL Partnership for Molecular Medicine, Aarhus University, Denmark. LR has been supported by The Michael J Fox Foundation (grant ID 18283) and The EU Joint Programme-Neurodegenerative Disease Research (JPND) and The VELUX Foundations for the project OligoFIT. GK has been supported by the Novo Nordisk Foundation Distinguished Innovator 1-2020 award to Claus E. Olesen.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dorsey ER, Elbaz A, Nichols E, Abd-Allah F, Abdelalim A, Adsuar JC, et al. Global, regional, and national burden of Parkinson's disease, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. (2018) 17:939–53. doi: 10.1016/S1474-4422(18)30295-3
2. Bloem BR, Okun MS, Klein C. Parkinson's disease. Lancet. (2021) 397:2284–303. doi: 10.1016/S0140-6736(21)00218-X
3. Maroteaux L, Campanelli J, Scheller R. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. (1988) 8:2804–15. doi: 10.1523/JNEUROSCI.08-08-02804.1988
4. Uéda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, et al. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci USA. (1993) 90:11282–6. doi: 10.1073/pnas.90.23.11282
5. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science. (1997) 276:2045–7. doi: 10.1126/science.276.5321.2045
6. Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M. α-synuclein in lewy bodies. Nature. (1997) 388:839–40. doi: 10.1038/42166
7. Gai WP, Power JHT, Blumbergs PC, Blessing WW. Multiple-system atrophy: a new α-synuclein disease? Lancet. (1998) 352:547–8. doi: 10.1016/S0140-6736(05)79256-4
8. Stefanis L. α-synuclein in Parkinson's disease. Cold Spring Harb Perspect Med. (2012) 2:a009399. doi: 10.1101/cshperspect.a009399
9. Braak H, Rüb U, Gai WP, Del Tredici. K. Idiopathic Parkinson's disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm. (2003) 110:517–36. doi: 10.1007/s00702-002-0808-2
10. Braak H, Tredici KD, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. (2003) 24:197–211. doi: 10.1016/S0197-4580(02)00065-9
11. Horsager J, Andersen KB, Knudsen K, Skjærbæk C, Fedorova TD, Okkels N, et al. Brain-first versus body-first Parkinson's disease: a multimodal imaging case-control study. Brain. (2020) 143:3077–88. doi: 10.1093/brain/awaa238
12. Khachaturian ZS. Calcium, membranes, aging, Alzheimer's disease. Introduction and Overview. Ann N Y Acad Sci. (1989) 568:1–4. doi: 10.1111/j.1749-6632.1989.tb12485.x
13. Nedergaard S, Flatman JA, Engberg I. Nifedipine- and omega-conotoxin-sensitive Ca2+ conductances in guinea-pig substantia nigra pars compacta neurones. J Physiol. (1993) 466:727–47.
14. Surmeier DJ, Schumacker PT, Guzman JD, Ilijic E, Yang B, Zampese E. Calcium and Parkinson's disease. Biochem Biophys Res Commun. (2017) 483:1013–9. doi: 10.1016/j.bbrc.2016.08.168
15. Ritz B, Rhodes SL, Qian L, Schernhammer E, Olsen JH, Friis S. L-type calcium channel blockers and Parkinson disease in Denmark. Ann Neurol. (2010) 67:600–6. doi: 10.1002/ana.21937
16. Pasternak B, Svanström H, Nielsen NM, Fugger L, Melbye M, Hviid A. Use of calcium channel blockers and Parkinson's disease. Am J Epidemiol. (2012) 175:627–35. doi: 10.1093/aje/kwr362
17. Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, et al. ‘Rejuvenation' protects neurons in mouse models of Parkinson's disease. Nature. (2007) 447:1081–6. doi: 10.1038/nature05865
18. Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. (2010) 468:696–700. doi: 10.1038/nature09536
19. Ilijic E, Guzman JN, Surmeier DJ. The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson's disease. Neurobiol Dis. (2011) 43:364–71. doi: 10.1016/j.nbd.2011.04.007
20. ClinicalTrials_NCT02168842. Isradipine versus placebo in early Parkinson disease. Ann Intern Med. (2020) 172:591–8. doi: 10.7326/M19-2534
21. Betzer C, Lassen LB, Olsen A, Kofoed RH, Reimer L, Gregersen E, et al. Alpha-synuclein aggregates activate calcium pump SERCA leading to calcium dysregulation. EMBO Rep. (2018) 19:e44617. doi: 10.15252/embr.201744617
22. Gleichmann M, Mattson MP. Neuronal calcium homeostasis and dysregulation. Antioxidants Redox Signal. (2010) 14:1261–73. doi: 10.1089/ars.2010.3386
23. Brini M, Calì T, Ottolini D, Carafoli E. Neuronal calcium signaling: function and dysfunction. Cell Mol Life Sci. (2014) 71:2787–814. doi: 10.1007/s00018-013-1550-7
24. Yarlagadda A. Role of calcium regulation in pathophysiology model of schizophrenia and possible interventions. Med Hypotheses. (2002) 58:182–6. doi: 10.1054/mehy.2001.1511
25. Oliveira AM, Bading H. Calcium signaling in cognition and aging-dependent cognitive decline. BioFactors. (2011) 37:168–74. doi: 10.1002/biof.148
26. Wu Y, Whiteus C, Xu CS, Hayworth KJ, Weinberg RJ, Hess HF, et al. Contacts between the endoplasmic reticulum and other membranes in neurons. Proc Nat Acad Sci. (2017) 114:E4859–E67. doi: 10.1073/pnas.1701078114
27. Vallese F, Barazzuol L, Maso L, Brini M, Calì T. ER-Mitochondria calcium transfer, organelle contacts neurodegenerative diseases. Adv Exp Med Biol. (2020) 1131:719–46. doi: 10.1007/978-3-030-12457-1_29
28. Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. (2006) 86:369–408. doi: 10.1152/physrev.00004.2005
29. Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. (2003) 4:517–29. doi: 10.1038/nrm1155
30. Surmeier DJ, Schumacker PT. Calcium, bioenergetics, and neuronal vulnerability in Parkinson's disease. J Biol Chem. (2013) 288:10736–41. doi: 10.1074/jbc.R112.410530
31. Rcom-H'cheo-Gauthier A, Goodwin J, Pountney DL. Interactions between calcium and alpha-synuclein in neurodegeneration. Biomolecules. (2014) 4:795–811. doi: 10.3390/biom4030795
32. Primeau JO, Armanious GP, Fisher MLE, Young HS. The sarcoendoplasmic reticulum calcium ATPase. In: Harris JR, Boekema EJ, editors. Membrane Protein Complexes: Structure Function. Singapore: Springer Singapore (2018). p. 229–58. doi: 10.1007/978-981-10-7757-9_8
33. Wuytack F, Raeymaekers L, Missiaen L. Molecular physiology of the SERCA and SPCA pumps. Cell Calcium. (2002) 32:279–305. doi: 10.1016/S0143416002001847
34. Dally S, Corvazier E, Bredoux R, Bobe R, Enouf J. Multiple and diverse coexpression, location, and regulation of additional SERCA2 and SERCA3 isoforms in nonfailing and failing human heart. J Mol Cell Cardiol. (2010) 48:633–44. doi: 10.1016/j.yjmcc.2009.11.012
35. Lytton J, Westlin M, Burk SE, Shull GE, MacLennan DH. Functional comparisons between isoforms of the sarcoplasmic or endoplasmic reticulum family of calcium pumps. J Biol Chem. (1992) 267:14483–9. doi: 10.1016/S0021-9258(19)49738-X
36. Corvazier E, Bredoux R, Kovács T, Enouf J. Expression of sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) 3 proteins in two major conformational states in native human cell membranes. Biochim Biophys Acta. (2009) 1788:587–99. doi: 10.1016/j.bbamem.2008.12.004
37. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Science. (2015) 347:1260419. doi: 10.1126/science.1260419
38. Braak H, Sastre M, Del Tredici. K. Development of α-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson's disease. Acta Neuropathol. (2007) 114:231–41. doi: 10.1007/s00401-007-0244-3
39. Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, et al. Direct transfer of α-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem. (2010) 285:9262–72. doi: 10.1074/jbc.M109.081125
40. Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. (2004) 44:601–7. doi: 10.1016/j.neuron.2004.11.005
41. Martin I, Dawson VL, Dawson TM. Recent advances in the genetics of Parkinson's disease. Annu Rev Genomics Hum Genet. (2011) 12:301–25. doi: 10.1146/annurev-genom-082410-101440
42. Lee JH, Han JH, Kim H, Park SM, Joe EH, Jou I. Parkinson's disease-associated LRRK2-G2019S mutant acts through regulation of SERCA activity to control ER stress in astrocytes. Acta Neuropathol Commun. (2019) 7:68. doi: 10.1186/s40478-019-0716-4
43. Sonninen T-M, Hämäläinen RH, Koskuvi M, Oksanen M, Shakirzyanova A, et al. Metabolic alterations in Parkinson's disease astrocytes. Sci Rep. (2020) 10:14474. doi: 10.1038/s41598-020-71329-8
44. Go LO, Moschella MC, Watras J, Handa KK, Fyfe BS, Marks AR. Differential regulation of two types of intracellular calcium release channels during end-stage heart failure. J Clin Invest. (1995) 95:888–94. doi: 10.1172/JCI117739
45. Santulli G, Nakashima R, Yuan Q, Marks AR. Intracellular calcium release channels: an update. J Physiol. (2017) 595:3041–51. doi: 10.1113/JP272781
46. Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski MR, Cavagna D, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. (2006) 175:901–11. doi: 10.1083/jcb.200608073
47. Gómez-Suaga P, Bravo-San Pedro JM, González-Polo RA, Fuentes JM, Niso-Santano M. ER–mitochondria signaling in Parkinson's disease. Cell Death Dis. (2018) 9:337. doi: 10.1038/s41419-017-0079-3
48. Hayashi T, Rizzuto R, Hajnoczky G, Su T-P. MAM: more than just a housekeeper. Trends Cell Biol. (2009) 19:81–8. doi: 10.1016/j.tcb.2008.12.002
49. Bartok A, Weaver D, Golenár T, Nichtova Z, Katona M, Bánsághi S, et al. IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat Commun. (2019) 10:3726. doi: 10.1038/s41467-019-11646-3
50. Parys JB, Vervliet T. New insights in the IP3 receptor its regulation. Adv Exp Med Biol. (2020) 1131:243–70. doi: 10.1007/978-3-030-12457-1_10
51. Monkawa T, Miyawaki A, Sugiyama T, Yoneshima H, Yamamoto-Hino M, Furuichi T, et al. Heterotetrameric complex formation of inositol 1,4,5-trisphosphate receptor subunits. J Biol Chem. (1995) 270:14700–4. doi: 10.1074/jbc.270.24.14700
52. Thillaiappan NB, Chakraborty P, Hasan G, Taylor CW. IP3 receptors and Ca2+ entry. Biochim Biophys Acta. (2019) 1866:1092–100. doi: 10.1016/j.bbamcr.2018.11.007
53. Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. (2016) 89:37–53. doi: 10.1016/j.neuron.2015.11.013
54. Chang D, Nalls MA, Hallgrímsdóttir IB, Hunkapiller J, van der Brug M, Cai F, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson's disease risk loci. Nat Genet. (2017) 49:1511–6. doi: 10.1038/ng.3955
55. Apicco DJ, Shlevkov E, Nezich CL, Tran DT, Guilmette E, Nicholatos JW, et al. The Parkinson's disease-associated gene ITPKB protects against α-synuclein aggregation by regulating ER-to-mitochondria calcium release. Proc Nat Acad Sci USA. (2021) 118:e2006476118. doi: 10.1073/pnas.2006476118
56. Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol. (2010) 2:a003996. doi: 10.1101/cshperspect.a003996
57. Arias-Cavieres A, Barrientos GC, Sánchez G, Elgueta C, Muñoz P, Hidalgo C. Ryanodine receptor-mediated calcium release has a key role in hippocampal LTD induction. Front Cell Neurosci. (2018) 12:403. doi: 10.3389/fncel.2018.00403
58. Park KW, Jo S, Kim MS, Jeon SR, Ryu HS, Kim J, et al. Genomic association study for cognitive impairment in Parkinson's disease. Front Neurol. (2021) 11:1648. doi: 10.3389/fneur.2020.579268
59. Meissner G. The structural basis of ryanodine receptor ion channel function. J General Physiol. (2017) 149:1065–89. doi: 10.1085/jgp.201711878
60. Lee HC. Mechanisms of calcium signaling by cyclic ADP-ribose and NAADP. Physiol Rev. (1997) 77:1133–64. doi: 10.1152/physrev.1997.77.4.1133
61. Vervliet T. Ryanodine receptors in autophagy: implications for neurodegenerative diseases? Front Cell Neurosci. (2018) 12:89. doi: 10.3389/fncel.2018.00089
62. Rousseau E, Meissner G. Single cardiac sarcoplasmic reticulum Ca2+-release channel: activation by caffeine. Am J Physiol Heart Circul Physiol. (1989) 256:H328–33. doi: 10.1152/ajpheart.1989.256.2.H328
63. O'Neill SC, Eisner DA. A mechanism for the effects of caffeine on Ca2+ release during diastole and systole in isolated rat ventricular myocytes. J Physiol. (1990) 430:519–36. doi: 10.1113/jphysiol.1990.sp018305
64. Kong H, Jones PP, Koop A, Zhang L, Duff HJ, Chen SR, et al. Caffeine induces Ca2+ release by reducing the threshold for luminal Ca2+ activation of the ryanodine receptor. Biochem J. (2008) 414:441–52. doi: 10.1042/BJ20080489
65. Porta M, Zima AV, Nani A, Diaz-Sylvester PL, Copello JA, Ramos-Franco J, et al. Single ryanodine receptor channel basis of caffeine's action on Ca2+ sparks. Biophys J. (2011) 100:931–8. doi: 10.1016/j.bpj.2011.01.017
66. Beutner G, Sharma VK, Giovannucci DR, Yule DI, Sheu S-S. Identification of a ryanodine receptor in rat heart mitochondria. J Biol Chem. (2001) 276:21482–8. doi: 10.1074/jbc.M101486200
67. Jakob R, Beutner G, Sharma VK, Duan Y, Gross RA, Hurst S, et al. Molecular and functional identification of a mitochondrial ryanodine receptor in neurons. Neurosci Lett. (2014) 575:7–12. doi: 10.1016/j.neulet.2014.05.026
68. Beutner G, Sharma VK, Lin L, Ryu SY, Dirksen RT, Sheu SS. Type 1 ryanodine receptor in cardiac mitochondria: Transducer of excitation–metabolism coupling. Biochim Biophys Acta. (2005) 1717:1–0. doi: 10.1016/j.bbamem.2005.09.016
69. Ren X, Chen J-F. Caffeine and Parkinson's disease: multiple benefits and emerging mechanisms. Front Neurosci. (2020) 14:1334. doi: 10.3389/fnins.2020.602697
70. Postuma RB, Lang AE, Munhoz RP, Charland K, Pelletier A, Moscovich M, et al. Caffeine for treatment of Parkinson disease. A randomized controlled trial. Neurology. (2012) 79:651–8. doi: 10.1212/WNL.0b013e318263570d
71. Cho BH, Choi SM, Kim JT, Kim BC. Association of coffee consumption and non-motor symptoms in drug-naïve, early-stage Parkinson's disease. Parkinson Related Disord. (2018) 50:42–7. doi: 10.1016/j.parkreldis.2018.02.016
72. Crotty GF, Maciuca R, Macklin EA, Wang J, Montalban M, Davis SS, et al. Association of caffeine and related analytes with resistance to Parkinson disease among LRRK2 mutation carriers. A metabolomic study. Neurology. (2020) 95:e3428–37. doi: 10.1212/WNL.0000000000010863
73. Luan Y, Ren X, Zheng W, Zeng Z, Guo Y, Hou Z, et al. Chronic caffeine treatment protects against α-synucleinopathy by reestablishing autophagy activity in the mouse striatum. Front Neurosci. (2018) 12:301. doi: 10.3389/fnins.2018.00301
74. Chen J-F, Cunha RA. The belated US FDA approval of the adenosine A2A receptor antagonist istradefylline for treatment of Parkinson's disease. Purinergic Signal. (2020) 16:167–74. doi: 10.1007/s11302-020-09694-2
75. Genovese I, Giamogante F, Barazzuol L, Battista T, Fiorillo A, Vicario M, et al. Sorcin is an early marker of neurodegeneration, Ca2+ dysregulation and endoplasmic reticulum stress associated to neurodegenerative diseases. Cell Death Dis. (2020) 11:861. doi: 10.1038/s41419-020-03063-y
76. Li C, Qian T, He R, Wan C, Liu Y, Yu H. Endoplasmic reticulum–plasma membrane contact sites: regulators, mechanisms, physiological functions. Front Cell Dev Biol. (2021) 9:126. doi: 10.3389/fcell.2021.627700
77. de Juan-Sanz J, Holt GT, Schreiter ER, de Juan F, Kim DS, Ryan TA. Axonal endoplasmic reticulum Ca2+ content controls release probability in CNS nerve terminals. Neuron. (2017) 93:867–81. doi: 10.1016/j.neuron.2017.01.010
78. Zhang I, Hu H. Store-operated calcium channels in physiological and pathological states of the nervous system. Front Cell Neurosci. (2020) 14:400. doi: 10.3389/fncel.2020.600758
79. Bollimuntha S, Pani B, Singh BB. Neurological motor disorders: neuronal store-operated Ca2+ signaling: an overview its function. In: Groschner K, Graier WF, Romanin C, editors. Store-Operated Ca2? Entry (SOCE) Pathways: Emerging Signaling Concepts in Human (Patho)physiology. Cham: Springer International Publishing (2017). p. 535–56. doi: 10.1007/978-3-319-57732-6_27
80. Prakriya M, Lewis RS. Store-operated calcium channels. Physiol Rev. (2015) 95:1383–436. doi: 10.1152/physrev.00020.2014
81. Selvaraj S, Sun Y, Watt JA, Wang S, Lei S, Birnbaumer L, et al. Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J Clin Invest. (2012) 122:1354–67. doi: 10.1172/JCI61332
82. Zhou Q, Yen A, Rymarczyk G, Asai H, Trengrove C, Aziz N, et al. Impairment of PARK14-dependent Ca2+ signalling is a novel determinant of Parkinson's disease. Nat Commun. (2016) 7:10332. doi: 10.1038/ncomms10332
83. Wang WA, Liu WX, Durnaoglu S, Lee SK, Lian J, Lehner R, et al. Loss of calreticulin uncovers a critical role for calcium in regulating cellular lipid homeostasis. Sci Rep. (2017) 7:5941. doi: 10.1038/s41598-017-05734-x
84. Schröder M. Endoplasmic reticulum stress responses. Cell Mol Life Sci. (2008) 65:862–94. doi: 10.1007/s00018-007-7383-5
85. Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ. (2006) 13:385–92. doi: 10.1038/sj.cdd.4401778
86. Ghemrawi R, Khair M. Endoplasmic reticulum stress and unfolded protein response in neurodegenerative diseases. Int J Mol Sci. (2020) 21:6127. doi: 10.3390/ijms21176127
87. Sagara Y, Inesi G. Inhibition of the sarcoplasmic reticulum Ca2+ transport ATPase by thapsigargin at subnanomolar concentrations. J Biol Chem. (1991) 266:13503–6. doi: 10.1016/S0021-9258(18)92726-2
88. Helenius A. How N-linked oligosaccharides affect glycoprotein folding in the endoplasmic reticulum. Mol Biol Cell. (1994) 5:253–65. doi: 10.1091/mbc.5.3.253
89. Colla E, Jensen PH, Pletnikova O, Troncoso JC, Glabe C, Lee MK. Accumulation of toxic α-synuclein oligomer within endoplasmic reticulum occurs in α-synucleinopathy in vivo. J Neurosci. (2012) 32:3301–5. doi: 10.1523/JNEUROSCI.5368-11.2012
90. Carrara M, Prischi F, Nowak PR, Kopp MC, Ali MMU. Noncanonical binding of BiP ATPase domain to Ire1 and Perk is dissociated by unfolded protein CH1 to initiate ER stress signaling. Elife. (2015) 4:e03522. doi: 10.7554/eLife.03522
91. Bellucci A, Navarria L, Zaltieri M, Falarti E, Bodei S, Sigala S, et al. Induction of the unfolded protein response by α-synuclein in experimental models of Parkinson's disease. J Neurochem. (2011) 116:588–605. doi: 10.1111/j.1471-4159.2010.07143.x
92. Bellani S, Mescola A, Ronzitti G, Tsushima H, Tilve S, Canale C, et al. GRP78 clustering at the cell surface of neurons transduces the action of exogenous alpha-synuclein. Cell Death Differ. (2014) 21:1971–83. doi: 10.1038/cdd.2014.111
93. Colla E, Coune P, Liu Y, Pletnikova O, Troncoso JC, Iwatsubo T, et al. Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J Neurosci. (2012) 32:3306–20. doi: 10.1523/JNEUROSCI.5367-11.2012
94. Liou B, Peng Y, Li R, Inskeep V, Zhang W, Quinn B, et al. Modulating ryanodine receptors with dantrolene attenuates neuronopathic phenotype in Gaucher disease mice. Hum Mol Genet. (2016) 25:5126–41. doi: 10.1093/hmg/ddw322
95. Melia TJ, Lystad AH, Simonsen A. Autophagosome biogenesis: From membrane growth to closure. J Cell Biol. (2020) 219:2085. doi: 10.1083/jcb.202002085
96. Kuijpers M, Kochlamazashvili G, Stumpf A, Puchkov D, Swaminathan A, Lucht MT, et al. Neuronal autophagy regulates presynaptic neurotransmission by controlling the axonal endoplasmic reticulum. Neuron. (2021) 109:299–13. doi: 10.1016/j.neuron.2020.10.005
97. Bahar E, Kim H, Yoon H. ER stress-mediated signaling: action potential and Ca2+ as key players. Int J Mol Sci. (2016) 17:1558. doi: 10.3390/ijms17091558
98. Müller M, Ahumada-Castro U, Sanhueza M, Gonzalez-Billault C, Court FA, Cárdenas C. Mitochondria and calcium regulation as basis of neurodegeneration associated with aging. Front Neurosci. (2018) 12:470. doi: 10.3389/fnins.2018.00470
99. Llorente-Folch I, Rueda CB, Pardo B, Szabadkai G, Duchen MR, Satrustegui J. The regulation of neuronal mitochondrial metabolism by calcium. J Physiol. (2015) 593:3447–62. doi: 10.1113/JP270254
100. Mohsin AA, Thompson J, Hu Y, Hollander J, Lesnefsky EJ, Chen Q. Endoplasmic reticulum stress-induced complex I defect: Central role of calcium overload. Arch Biochem Biophys. (2020) 683:108299. doi: 10.1016/j.abb.2020.108299
101. Mandal A, Drerup CM. Axonal transport and mitochondrial function in neurons. Front Cell Neurosci. (2019) 13:373. doi: 10.3389/fncel.2019.00373
102. Ryu S.-Y., Beutner G, Dirksen RT, Kinnally KW, Sheu -S., et al. Mitochondrial ryanodine receptors and other mitochondrial Ca2+ permeable channels. FEBS Lett. (2010) 584:1948–55. doi: 10.1016/j.febslet.2010.01.032
103. Patron M, Raffaello A, Granatiero V, Tosatto A, Merli G, De Stefani D, et al. The Mitochondrial Calcium Uniporter (MCU): molecular identity and physiological roles. J Biol Chem. (2013) 288:10750–8. doi: 10.1074/jbc.R112.420752
104. Marchi S, Pinton P. The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. J Physiol. (2014) 592:829–39. doi: 10.1113/jphysiol.2013.268235
105. Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. (1998) 280:1763–6. doi: 10.1126/science.280.5370.1763
106. Csordás G, Renken C, Várnai P, Walter L, Weaver D, Buttle KF, et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol. (2006) 174:915–21. doi: 10.1083/jcb.200604016
Keywords: Parkinson's disease, α-synuclein, calcium, endoplasmic reticulum, SERCA, RyR, IP3R
Citation: Kovacs G, Reimer L and Jensen PH (2021) Endoplasmic Reticulum-Based Calcium Dysfunctions in Synucleinopathies. Front. Neurol. 12:742625. doi: 10.3389/fneur.2021.742625
Received: 16 July 2021; Accepted: 21 September 2021;
Published: 20 October 2021.
Edited by:
Johannes Haybaeck, Innsbruck Medical University, AustriaReviewed by:
Gennaro Pagano, King's College London, United KingdomFrancisco José Pan-Montojo, Ludwig Maximilian University of Munich, Germany
Copyright © 2021 Kovacs, Reimer and Jensen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Poul Henning Jensen, cGhqQGJpb21lZC5hdS5kaw==