 Robert Duba-Kiss
Robert Duba-Kiss Yosuke Niibori
Yosuke Niibori David R. Hampson
David R. Hampson- 1Department of Pharmacology and Toxicology, Faculty of Medicine, University of Toronto, Toronto, ON, Canada
- 2Department of Pharmaceutical Sciences, Leslie Dan Faculty of Pharmacy, University of Toronto, Toronto, ON, Canada
Several neurological and psychiatric disorders have been associated with impairments in GABAergic inhibitory neurons in the brain. Thus, in the current era of accelerated development of molecular medicine and biologically-based drugs, there is a need to identify gene regulatory sequences that can be utilized for selectively manipulating the expression of nucleic acids and proteins in GABAergic neurons. This is particularly important for the use of viral vectors in gene therapy. In this Mini Review, we discuss the use of various gene regulatory elements for targeting GABAergic neurons, with an emphasis on adeno-associated viral vectors, the most widely used class of viral vectors for treating brain diseases.
Adeno-Associated Viruses Used in Gene Therapy
Gene therapy based on the delivery of therapeutic transgenes to affected tissues by recombinant adeno-associated viruses (rAAVs) has been intensively studied in preclinical and clinical research. AAVs are small viruses (25 nm particle diameter) that exhibit several properties that make them attractive for therapeutic applications. They are non-pathogenic in humans and other mammals and elicit mild to moderate immune responses. rAAV genomes are maintained in an extrachromosomal state (1) and generally do not recombine into the host genome, minimizing the risk of insertion-based genotoxicity. Despite existing outside of the host genome, rAAV DNA persists in non-dividing host cells, such as most neurons, and mediates transgene expression over a protracted time frame – studies have demonstrated expression 8 and 15 years after treatment in the brains of non-human primates (2, 3).
Several natural AAV serotypes exist, each of which display different levels of tropism for different cells and tissues. Of these AAV serotypes 2, 5, 8, and 9 have shown the highest tropism in the central nervous system (CNS), and thus have been most broadly applied for CNS delivery (4). In addition, artificial serotypes have been created that convey more favorable properties, such as rAAV-PHP.B, which was designed to efficiently cross the blood-brain barrier after intravenous injection (5). However, the ability of various rAAV serotypes to selectively target individual cell types is currently still limited. Instead, cell-type specific promoter or enhancer gene regulatory sequences can be utilized to direct transgene expression to selected cell types.
The wild type AAV genome is single stranded in structure, consists of two genes encoding the capsid and reproduction proteins, and is flanked by inverted terminal repeats (6). rAAVs are constructed by removal of the native viral genome and insertion of the recombinant gene of interest and associated gene regulatory elements, along with two essential flanking inverted terminal repeats (7). A drawback of rAAVs is that the maximum size of the DNA they can carry is limited to the size of the native genome: ~4.8 kilobases (kb) for single stranded rAAVs, and ~2.4 kb for double stranded self-complementary rAAVs. This limited space must include the two inverted terminal repeat sequences, a promoter and/or other regulatory elements, and the polyadenylation region, and therefore restricts the size of the transgene open reading frame that can be packaged within an AAV vector. For example, rAAVs that utilize regulatory elements that are 2,000 bases in length would limit the inserted coding sequence for the transgene, polyadenylation sequence, and inverted terminal repeats to about 2,800 bases in single stranded rAAV vectors and only about 400 bases in self-complimentary rAAV vectors.
In some cases, the limited DNA packaging capacity of these vectors presents a challenge for the creation of therapeutic rAAVs. Certain disorders involve mutations in genes larger than the rAAV packaging limit, rendering a direct gene replacement strategy infeasible. Depending on the mechanism underlying the disorder, therapeutic strategies using smaller, alternate genes may be an option. Of course, the size of a gene that can be packaged in an rAAV can be increased by minimizing the size of promoter and other regulatory elements used to drive transgene expression. Thus, the creation of miniaturized promoter and enhancer elements that maintain cellular specificity and the ability to drive robust expression is an active area of research. A list of studies using GABA gene-based regulatory elements is shown in Table 1.
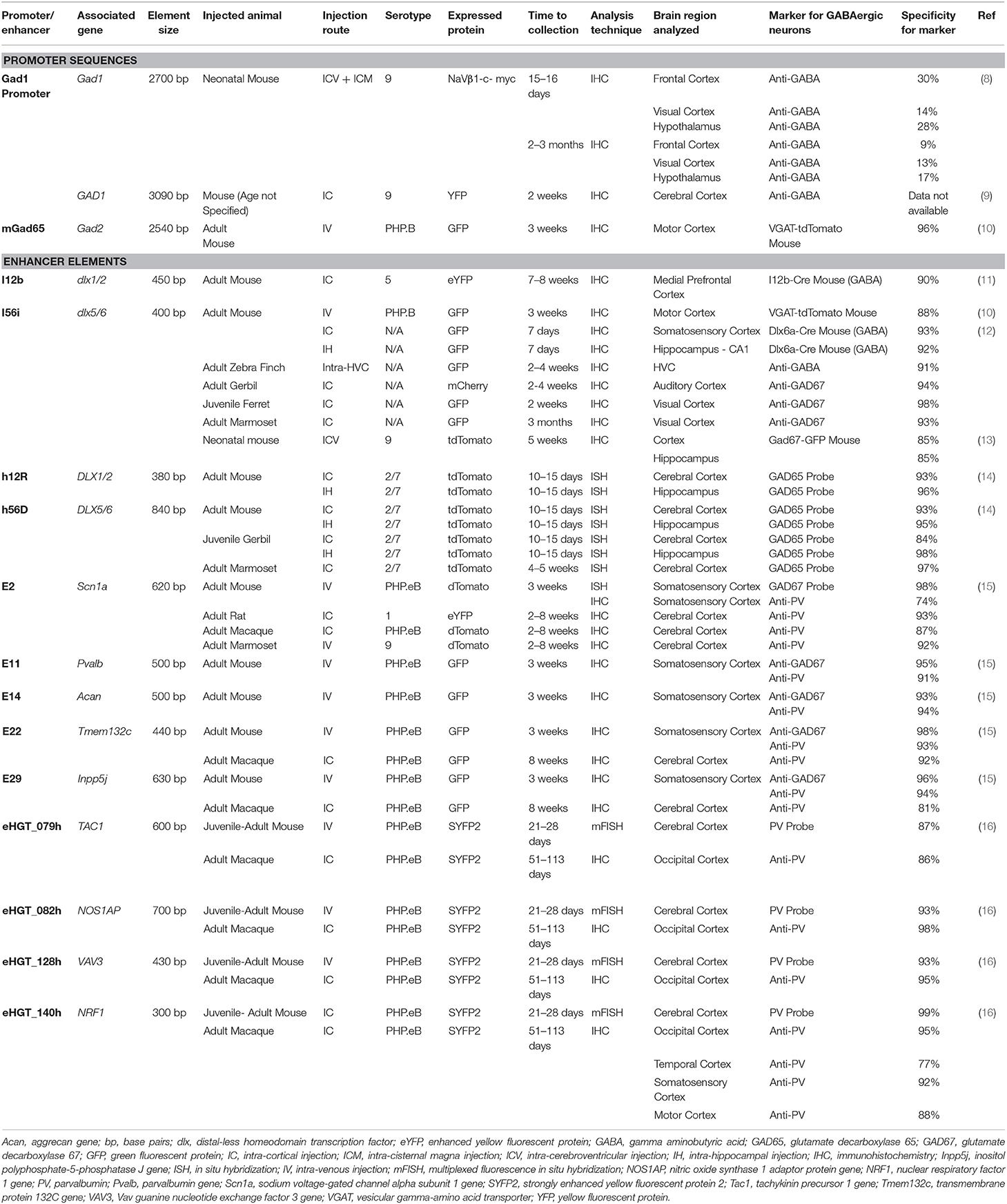
Table 1. GABAergic neuron-selective regulatory elements tested with rAAVs.
GABAergic Genes and GABA-Related Genetic Disorders
Brain cells can be broadly categorized into excitatory and inhibitory neurons, and glial cells such as astrocytes, oligodendrocytes, and microglia. Inhibitory neurons, also called GABAergic neurons that produce the inhibitory neurotransmitter gamma-aminobutyric acid (GABA), account for about 10–20 % of the total neuronal population in the mouse cerebral cortex (17). GABAergic neurons are required to maintain the balance between excitation and inhibition and the control of physiological activity in the brain (18).
Examples of genes that regulate GABAergic neuronal activity and are expressed specifically in GABAergic neurons include the GAD1, GAD2, and SLC32A1 genes. GAD1 and GAD2 encode the decarboxylase enzymes, GAD67 and GAD65, respectively, which catalyze the synthesis of GABA from L-glutamate. SLC32A1 encodes a vesicular GABA transporter (VGAT) that concentrates GABA into the synaptic vesicles. GABAergic neurons can also be divided into distinct subclasses such as those expressing the genes encoding the Ca2+-binding protein parvalbumin (PVALB), the neuropeptide somatostatin (SST), vasoactive intestinal peptide (VIP), and the ionotropic 5-hyroxytryptamine 3a serotonin receptor (HTR3A).
Impairment of GABA synthesis or GABA neuron activity is thought to lead to neurodevelopmental disorders, epilepsy, and psychiatric disorders. For instance, GABA synthesis failure by bi-allelic loss-of-function mutations in the GAD1 gene causes early infantile epilepsy, severe cleft palate, and neonatal death (19, 20). In addition, reduced GABAergic neuronal activity, abnormal brain activity, or GABA markers in postmortem studies have been observed in epilepsy (21), autism (22, 23), schizophrenia (24), Alzheimer's disease (25), Down's Syndrome (26), and bipolar disorder (27). Results from postmortem brain analyses of schizophrenia subjects have demonstrated decreased expression of GAD67 in the prefrontal cortex (28–30). Dravet syndrome, a genetic disorder which causes childhood epilepsy, occurs from mutations in the SCN1A gene encoding the Nav1.1 voltage-gated sodium channel. Experiments in mice have demonstrated that Nav1.1 is highly expressed in GABAergic neurons, and mice with mutations in Scn1a have an impaired ability to maintain the normal high frequency firing rate in inhibitory neurons (31, 32). Thus, overall, there is a need to develop an expanded toolbox of GABA-related gene regulatory elements for use in viral vectors. It should be noted that most of the constructs described in this review have only been analyzed using a single study protocol, and that their efficacies may vary under different experimental conditions.
The DLX Gene Enhancer Elements
The distal-less homeodomain transcription factor (DLX) family is a gene family highly conserved across vertebrates. DLX 1, 2, 5, and 6 are expressed in the CNS during brain development and promote the expansion and differentiation of GABAergic neurons. DLX1 and DLX2 are important in the migration of nascent cortical GABAergic neurons and in the embryonic expression of GABAergic neuron-specific genes such as GAD1, GAD2, and SLC32A1 (33, 34). The precise roles of DLX5 and DLX6 are less clear although they are known to be important for the differentiation and maturation of cortical parvalbumin (PV) GABAergic neurons (35). Similarly, DLX1 is important for the development of cortical SST, calretinin, and neuropeptide Y GABAergic neurons (36). Expression of the DLX proteins declines after the neonatal period, although they are still present at lower levels in the CNS during adulthood (37, 38).
The DLX genes are grouped into bicistronic gene clusters and are regulated by intergenic enhancer sequences with high degrees of homology between mice and humans. Dlx1/2 and Dlx5/6 each form such bicistronic clusters, and in mice are regulated by the enhancer domains I12b (39) and I56i (12, 40), respectively. Analogous enhancer sequences are found in human DLX genes: h12R (found in DLX1/2) and h56D (found in DLX5/6) (14). Each of these enhancers, when packaged with a minimal promoter onto rAAV vectors, effectively direct transgene expression to GABAergic neurons. Direct injection of rAAVs carrying I56i, h12R, or h56D into the murine cerebral cortex and hippocampus resulted in the transduction of 80–90% of cortical and hippocampal GABAergic neurons after each injection, respectively (12, 14). Intravenous injection of rAAV-I56i labeled 57% of murine cortical GABAergic neurons (10). All of the aforementioned DLX enhancers (I12b, I56i, h12R, and h56D) directed transgene expression to murine cortical and hippocampal GABAergic neurons with approximately 90% or greater specificity [(10–12, 14); see Table 1]. Similar levels of GABAergic neuronal specificity and coverage were observed in gerbils and in non-human primates (12, 14). Moreover, application of an rAAV-I56i, as well as a rAAV with a triplicate repeat of an I56i core sequence (rAAV-DLX2.0), to ex vivo human cortical tissue demonstrated that these vectors effectively transduced human GABAergic neurons (16). A rAAV using a mouse dlx5/6 enhancer was also used in Scn1a mutant mice, a mouse model of Dravet Syndrome. Neonatal injections into the ventricles resulted in transduction of about 20% of cortical GABAergic interneurons near the injection site, and caused normalization of GABA neuron firing and a reduction in febrile seizures (13).
Despite the excellent ability of these enhancers to target GABAergic neurons in anterior brain regions, neither intravenous injection of rAAV-I56i (10, 16), nor intra-inferior collicular injection of rAAV-h65D (14) induced transgene expression in posterior brain regions (such as the midbrain, cerebellum, and brainstem). The non-functionality of DLX enhancers in the posterior brain poses a limitation for studies of candidate therapies requiring posterior brain or CNS-wide GABAergic neuronal targeting; other regulatory elements will likely be necessary in these cases.
The GAD1 Promoter
The GAD1 and GAD2 genes are evolutionarily conserved across eukaryotes. In the human cerebral cortex, expression of both GAD1 and GAD2 rapidly increases after birth; GAD1 continues this upward trajectory for another 2–3 years after birth, while GAD2 reaches a plateau around 1 year after birth (41). In mice, deletion of Gad1 caused dramatically reduced GABA levels in the CNS, severe cleft palate, respiratory failure, and death shortly after birth (42). The expression of GAD1 is regulated by homeobox domain transcription factors DLX1 and DLX2. Multiple putative binding sites for DLX proteins have been identified in the 5' intergenic region of the Gad1 gene (34). In addition, the expression levels of GAD67 were decreased in Dlx1/2 knockout mice, suggesting that Dlx1/2 regulates Gad1 transcription (34, 43).
The 5' intergenic region between the GAD1 gene and the 5' upstream gene ERICH2, spans 23 kb. DNA sequences from a segment of the 5' intergenic region and exon 1 containing the transcription start site of GAD1 have been utilized in rAAVs (8, 9). Niibori et al. reported that an rAAV containing a 2.7 kb section of the mouse Gad1 promoter showed 80% overall neuronal specificity and 12–30% GABAergic neuronal selectivity, depending on the brain region examined; AAV-mediated transgene expression was found in the forebrain and midbrain, but not in the cerebellum or brainstem (8). By comparison, Liu et al. (9) reported that a 3.1 kb section of the human GAD1 promoter showed complete GABAergic neuronal specificity; however, no data were presented to support this statement. The difference in GABA specificity reported by Niibori et al. (8) and Liu et al. (9) could be due to the absence of a GABA specificity element in the shorter 2.7 kb construct vs. the longer 3.1 kb construct. However, it is also possible that differences in the experimental conditions of the two studies (i.e., age, route of injection, and dose of rAAV), rather than differences in DNA sequences, contributed to the difference in GABAergic neuronal specificity. For example, lowering the dose of an rAAV encoding a neuron-specific (Synapsin) promoter markedly increased its specificity for GABAergic neurons (44). Additionally, the GABA selectivity of rAAVs injected during the early postnatal period was reported to be lower than that of rAAVs injected into adult mice (15).
The GAD2 Promoter
Like GAD1, GAD2 expression begins during embryonic development and is regulated by the transcription factors DLX1 and DLX2. Unlike GAD1, the null mutation of GAD2 is not fatal, although Gad2−/− mice show increased fearful and anxious behavior, as well as elevated susceptibility to seizures (45, 46).
An rAAV carrying a 3 kb region of the murine Gad2 promoter (3,000 base pairs upstream from the start codon) was used to examine enhanced green fluorescent protein (eGFP) transgene expression in mouse brain; this rAAV displayed 20% specificity to excitatory neurons in the murine cerebral cortex (10). However, another construct carrying a deletion of Gad2 exon 1 abolished excitatory neuron expression and induced labeling of 60% of cortical GABAergic neurons with 96% GABA neuron specificity after intravenous injection of AAV-PHP.B (10). rAAVs carrying this truncated Gad2 promoter (denoted as rAAV-mGad65 by the authors of the study) showed broad expression in the mouse CNS, including in anterior areas such as the cerebral cortex, hippocampus, and striatum, and in posterior areas including the midbrain, cerebellum, and brainstem. In the cerebellum, this construct induced high transduction of eGFP in GABAergic neurons in the molecular layer (coverage = 80%), but low transduction of GABAergic Golgi cells in the granule layer (coverage = 8%) and of GABAergic Purkinje cells (coverage = 1%). This rAAV construct is the first to use sequences from the Gad2 promoter to direct rAAV-mediated transgene expression to GABAergic neurons. Despite limited research with Gad2 sequences, the broad CNS distribution of expression and high degree of specificity to GABAergic neurons demonstrates the potential utility in gene therapy. Additional testing in non-human primates would be useful to evaluate whether such Gad2 promoter constructs might be useful for clinical applications.
The VGAT Promoter
The vesicular gamma-amino acid transporter VGAT, previously known as the vesicular inhibitory amino acid transporter, is coded for by the SLC32A1 gene. Analysis of the mouse Slc32a1 promoter indicated that a minimal promoter region containing a CG rich sequence is sufficient for efficient expression in neural stem and precursor cells (47, 48). Deletion of this CG rich region greatly reduced its activity in neural precursor cells. It was suggested that the CG rich region may be acting as a core promoter element, mediating the activity of SP1 or other SP family transcription factors (47, 48).
To date, no studies have reported the use of the Slc32a1 promoter region in rAAV gene therapy experiments. However, a cell type-specific promoter-driven fluorescent reporter construct was developed that utilized the human vesicular GABA transporter SLC32A1 promoter to drive the expression of mCherry specifically in Vgat-expressing neurons. A 1,865 bp region of the SLC32A1 gene, including 262 bp downstream of the transcription start site and 1,603 bp upstream of the transcription start site, was PCR amplified from genomic DNA. This region overlaps with peaks for several markers of promoter activation, histone H3K4 monomethylation (H3K4me1), histone H3K4 trimethylation H3K4me3, and RNA polymerase II (Pol2) binding (49). The transduction of iPSC derived forebrain neuronal cultures with a hVGAT promoter-mCherry lentiviral reporter construct specifically labeled GABAergic neurons. Immunocytochemical analysis of hVGAT-mCherry expression showed prominant co-labeling with GABAergic neuronal markers including VGAT, GABA, and GAD67 (49).
The Somatostatin Promoter
SST is a peptide hormone that regulates the endocrine system, functions as a neuropeptide, and affects cellular proliferation. In the brain, SST is selectively expressed in a subset of GABAergic interneurons. Impaired function of this class of interneurons has been suggested to be involved in the pathophysiology of depression, bipolar disorder, and schizophrenia (50). Promoter sequences derived from putative orthologous fugu (Pufferfish) Sst have been generated and tested, as were composite Sst regulatory elements containing transcription factor binding sites (51). A 2,597 base pair fragment of the fugu Sst promoter was packaged into a recombinant rAAV2/1 (AAV2 backbone packaged with AAV1 capsid) in front of an eGFP reporter, and injections were made into the somatosensory cortex of adult mice. In the somatosensory cortex this rAAV displayed 55–88% coverage across subclasses of inhibitory neurons including SST, PV, neuropeptide Y, and VIP-positive neurons. In contrast, an attempt to use a segment containing the mouse SST promoter in a lentivirus construct was not successful (51).
An intersection strategy was used in rAAV vector design by Mehta et al. whereby rAAV Sst-Cre recombinase and a construct with DLX enhancers and eGFP with a double-floxed inverted open reading frame, were co-injected into the hippocampus of adult mice; eGFP was expressed only when both promoters were active in the same cell (14). The selected DNA sequence included a segment that was conserved between the mouse and human genes, extended 2,000 bases upstream of the mouse Sst start codon, and encompassed three conserved domains. This arrangement resulted in high GABA interneuron specificity and coverage in the hippocampus (about 90% for both).
Regulatory Elements in the Vasointestinal Peptide and HTR3A Genes
The VIP gene encoding the neurotransmitter vasointestinal peptide (VIP) is expressed in GABAergic neurons, especially in cell populations distinct from PV and SST expressing neurons (52). VIP is a neurotransmitter expressed in forebrain regions such as the amygdala, and in the midbrain including the suprachiasmatic nucleus (52, 53). The VIP pathway is associated with regulation of appetite and circadian rhythm (54, 55). The HTR3A gene encodes the 5-hydroxytryptamine 3A serotonin receptor (5HT3AR). Abnormalities in the 5HT3AR pathway have been associated with psychiatric disorders such as bipolar disorder, schizophrenia (56), and autism (57). In mice, Htr3a is also expressed primarily in GABAergic neurons distinct from Pvalb and Sst expressing neurons, and is not expressed in excitatory neurons or glial cells (52, 58). Vip is expressed in 40% of Htr3a expressing GABAergic neurons (58, 59). To date there have been no reports of development of rAAV or other viral vectors using the Htr3a or Vip promoters. However, identified functional promoters/enhancers in Vip-expressing neurons include Dlx enhancers from human and mouse (12, 14), mouse Scn1a enhancers (15), and Pvalb, Sst, and, Npy promoters from pufferfish (51).
The Parvalbumin Promoter and Expression in Parvalbumin Neurons
PV is a small, Ca2+-binding protein that is prominently expressed in a subset of GABAergic neurons throughout the CNS. Examples include cortical basket and chandelier cells (60), some hippocampal interneurons (61), and interneurons in the reticular thalamic nucleus (62). Dysfunction of PV-expressing GABAergic neurons have been implicated in several neurological and psychiatric disorders such as epilepsy (63, 64), autism (65–67), and schizophrenia (30, 68, 69). Thus, successful targeting of PV GABAergic neurons with rAAVs could have implications for gene therapy treatments of these diseases.
A region of the PVALB promoter with high homology between humans and mice was shown to be unable to restrict rAAV-mediated expression to PV GABAergic neurons (although the exact sequence and position of the sequence used was not reported in the study) (14). However, another study using a lentiviral vector and segments of the Pvalb promoter (−4 to −1,880 and −4 to −780) conserved between the mouse and the macaque, reported high specificity to PV GABAergic neurons after injection into the thalamic reticular nucleus of adult mice (70). However, these Pvalb promoter regions have not been tested in rAAVs.
Another method to restrict rAAV transgene expression to PV GABAergic neurons in mice is the intersectional strategy by using two rAAV constructs, one carrying the Cre-recombinase gene, and the other carrying a Cre-dependent reporter transgene (see the somatostain promoter section above). The Cre-recombinase-carrying rAAV used a promoter from the PaqR4 gene, which is active in PV GABAergic neurons, but also in non-GABAergic cells, and the reporter-carrying rAAV contained the h56D enhancer which expresses in GABAergic neurons. Reporter gene expression was therefore only induced in PV GABAergic neurons where both rAAVs were present (80% and 69% GABA specificity in mouse hippocampus and cortex, respectively) (14).
Numerous short, conserved enhancer elements have been identified by chromatin accessibility profiling that effectively restrict rAAV-mediated expression to PV GABAergic neurons (15, 16). These enhancers directed rAAV-mediated expression to cortical PV GABAergic neurons with 85–90% specificity after intravenous injection into mice, but varied with respect to the degree of distribution of transgene expression they induce in the murine CNS. Notably, an enhancer from the Inpp5j gene drives particularly robust and widespread CNS expression (transducing the cerebral cortex, hippocampus, striatum and cerebellum) (15). Another enhancer from the NOS1AP gene is particularly effective at transducing posterior regions such as the midbrain, brainstem and cerebellar nuclei, but shows comparatively limited expression in the forebrain (16). These PV GABAergic neuron-selective enhancers are also operative in non-human primates; direct intra-cortical injection of macaques with enhancer-carrying rAAVs directed reporter expression to cortical PV GABAergic neurons with 80% or greater specificity (15, 16). The PV GABAergic neuron selective enhancers that showed the highest specificity are listed in Table 1. The excellent PV GABAergic neuron targeting ability of these enhancer elements, as well as their small size and utility in non-human primates, make them attractive candidates for potential rAAV therapies.
Summary
Perturbations in GABAergic neurons cause an imbalance between excitatory and inhibitory brain activity and can lead to CNS disease. GABAergic neuron-specific rAAVs are expected to drive therapeutic protein expression in GABA neurons and improve symptoms by normalization of GABAergic neuronal function. The possibility exists that certain disorders may alter the transcriptional activities of GABAergic neurons and render promoter/enhancer elements that function in healthy neurons ineffective. Thus, when developing a candidate therapy, it will be important to evaluate the GABAergic neuronal targeting ability of the rAAV construct in the pertinent disease model to ensure its functionality is not altered. Although the discovery of GABA-based regulatory elements for use in rAAVs is in an early stage of development, the various GABA promoters and enhancers studied so far varied in length from about 300 bases to 2.5 kb and displayed a range of GABAergic neuronal specificities (about 30–90%). Among them, a fragment of the mouse Gad2 promoter appeared to operate in multiple GABA neuron subtypes and across many regions throughout the CNS (10). However, the majority of studies reported to date were based on the analysis of fluorescent reporter proteins and did not assess therapeutic efficacy in disease models. We expect this state-of-the-art to improve rapidly whereby GABA neuron-directed viral vectors will be tested in additional animal models of neurological and psychiatric disorders, with the expectation that some of these candidate gene regulatory elements in combination with therapeutic transgenes will advance to clinical testing.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that this study received funding from Regenxbio Inc. The funder had the following involvement in the study: study design, decision to publish, and preparation of the manuscript.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to Dravet Canada, The Rare Diseases Models and Mechanisms Network, and Regenxbio Inc. for financially supporting this work.
References
1. Schnepp BC, Jensen RL, Chen C-L, Johnson PR, Clark KR. Characterization of adeno-associated virus genomes isolated from human tissues. J Virol. (2005) 79:14793–803. doi: 10.1128/JVI.79.23.14793-14803.2005
2. Hadaczek P, Eberling JL, Pivirotto P, Bringas J, Forsayeth J, Bankiewicz KS. Eight years of clinical improvement in MPTP-lesioned primates after gene therapy with AAV2-hAADC. Mol Ther. (2010) 18:1458–61. doi: 10.1038/mt.2010.106
3. Sehara Y, Fujimoto K-I, Ikeguchi K, Katakai Y, Ono F, Takino N, et al. Persistent expression of dopamine-synthesizing enzymes 15 years after gene transfer in a primate model of Parkinson's disease. Hum Gene Ther Clin Dev. (2017) 28:74–9. doi: 10.1089/humc.2017.010
4. Lykken EA, Shyng C, Edwards RJ, Rozenberg A, Gray SJ. Recent progress and considerations for AAV gene therapies targeting the central nervous system. J Neurodev Disord. (2018) 10:16. doi: 10.1186/s11689-018-9234-0
5. Deverman BE, Pravdo PL, Simpson BP, Kumar SR, Chan KY, Banerjee A, et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol. (2016) 34:204–9. doi: 10.1038/nbt.3440
6. Naso MF, Tomkowicz B, Perry WL, Strohl WR. Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs. (2017) 31:317–34. doi: 10.1007/s40259-017-0234-5
7. Li C, Samulski RJ. Engineering adeno-associated virus vectors for gene therapy. Nat Rev Genet. (2020) 21:255–72. doi: 10.1038/s41576-019-0205-4
8. Niibori Y, Lee SJ, Minassian BA, Hampson DR. Sexually divergent mortality and partial phenotypic rescue after gene therapy in a mouse model of dravet syndrome. Hum Gene Ther. (2020) 31:339–51. doi: 10.1089/hum.2019.225
9. Liu Y-J, Ehrengruber MU, Negwer M, Shao H-J, Cetin AH, Lyon DC. Tracing inputs to inhibitory or excitatory neurons of mouse and cat visual cortex with a targeted rabies virus. Curr Biol. (2013) 23:1746–55. doi: 10.1016/j.cub.2013.07.033
10. Hoshino C, Konno A, Hosoi N, Kaneko R, Mukai R, Nakai J, et al. GABAergic neuron-specific whole-brain transduction by AAV-PHP.B incorporated with a new GAD65 promoter. Mol Brain. (2021) 14:33. doi: 10.1186/s13041-021-00746-1
11. Rubin AN, Malik R, Cho KKA, Lim KJ, Lindtner S, Robinson Schwartz SE, et al. Regulatory elements inserted into AAVs confer preferential activity in cortical interneurons. eNeuro. (2020) 7:ENEURO.0211-20.2020. doi: 10.1523/ENEURO.0211-20.2020
12. Dimidschstein J, Chen Q, Tremblay R, Rogers S, Saldi G, Guo L, et al. A viral strategy for targeting and manipulating interneurons across vertebrate species. Nat Neurosci. (2016) 19:1743–9. doi: 10.1038/nn.4430
13. Colasante G, Lignani G, Brusco S, Di Berardino C, Carpenter J, Giannelli S, et al. dCas9-based scn1a gene activation restores inhibitory interneuron excitability and attenuates seizures in dravet syndrome mice. Mol Ther. (2020) 28:235–53. doi: 10.1016/j.ymthe.2019.08.018
14. Mehta P, Kreeger L, Wylie DC, Pattadkal JJ, Lusignan T, Davis MJ, et al. Functional access to neuron subclasses in rodent and primate forebrain. Cell Rep. (2019) 26:2818–32.e8. doi: 10.1016/j.celrep.2019.02.011
15. Vormstein-Schneider D, Lin JD, Pelkey KA, Chittajallu R, Guo B, Arias-Garcia MA, et al. Viral manipulation of functionally distinct interneurons in mice, non-human primates and humans. Nat Neurosci. (2020) 23:1629–36. doi: 10.1038/s41593-020-0692-9
16. Mich JK, Graybuck LT, Hess EE, Mahoney JT, Kojima Y, Ding Y, et al. Functional enhancer elements drive subclass-selective expression from mouse to primate neocortex. Cell Rep. (2021) 34:108754. doi: 10.1016/j.celrep.2021.108754
17. Meyer HS, Schwarz D, Wimmer VC, Schmitt AC, Kerr JND, Sakmann B, et al. Inhibitory interneurons in a cortical column form hot zones of inhibition in layers 2 and 5A. Proc Natl Acad Sci U S A. (2011) 108:16807–12. doi: 10.1073/pnas.1113648108
18. Sohal VS, Rubenstein JLR. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol Psychiatry. (2019) 24:1248–57. doi: 10.1038/s41380-019-0426-0
19. Chatron N, Becker F, Morsy H, Schmidts M, Hardies K, Tuysuz B, et al. Bi-allelic GAD1 variants cause a neonatal onset syndromic developmental and epileptic encephalopathy. Brain. (2020) 143:1447–61. doi: 10.1093/brain/awaa085
20. Neuray C, Maroofian R, Scala M, Sultan T, Pai GS, Mojarrad M, et al. Early-infantile onset epilepsy and developmental delay caused by bi-allelic GAD1 variants. Brain. (2020) 143:2388–97. doi: 10.1093/brain/awaa178
21. Dravet C. The core dravet syndrome phenotype: core dravet syndrome. Epilepsia. (2011) 52:3–9. doi: 10.1111/j.1528-1167.2011.02994.x
22. Paluszkiewicz SM, Martin BS, Huntsman MM. Fragile X syndrome: the GABAergic system and circuit dysfunction. Dev Neurosci. (2011) 33:349–64. doi: 10.1159/000329420
23. Soghomonian J-J, Zhang K, Reprakash S, Blatt GJ. Decreased parvalbumin mRNA levels in cerebellar Purkinje cells in autism. Autism Res. (2017) 10:1787–96. doi: 10.1002/aur.1835
24. Lang UE, Puls I, Müller DJ, Strutz-Seebohm N, Gallinat J. Molecular mechanisms of schizophrenia. Cell Physiol Biochem. (2007) 20:687–702. doi: 10.1159/000110430
25. Xu Y, Zhao M, Han Y, Zhang H. GABAergic Inhibitory interneuron deficits in alzheimer's disease: implications for treatment. Front Neurosci. (2020) 14:660. doi: 10.3389/fnins.2020.00660
26. Contestabile A, Magara S, Cancedda L. The GABAergic hypothesis for cognitive disabilities in down syndrome. Front Cell Neurosci. (2017) 11:54. doi: 10.3389/fncel.2017.00054
27. Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacol. (2001) 25:1–27. doi: 10.1016/S0893-133X(01)00225-1
28. Akbarian S. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry. (1995) 52:258. doi: 10.1001/archpsyc.1995.03950160008002
29. Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA. Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical 3-aminobutyric acid neurons in subjects with schizophrenia. Arch Gen Psychiatry. (2000) 57:9. doi: 10.1001/archpsyc.57.3.237
30. Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, et al. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci. (2003) 23:6315–26. doi: 10.1523/JNEUROSCI.23-15-06315.2003
31. Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, et al. Nav11 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an scn1a gene mutation. J Neurosci. (2007) 27:5903–14. doi: 10.1523/JNEUROSCI.5270-06.2007
32. Dutton SB, Makinson CD, Papale LA, Shankar A, Balakrishnan B, Nakazawa K, et al. Preferential inactivation of scn1a in parvalbumin interneurons increases seizure susceptibility. Neurobiol Dis. (2013) 49:211–20. doi: 10.1016/j.nbd.2012.08.012
33. Anderson SA, Eisenstat DD, Shi L, Rubenstein JL. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. (1997) 278:474–6. doi: 10.1126/science.278.5337.474
34. Le TN, Zhou Q-P, Cobos I, Zhang S, Zagozewski J, Japoni S, et al. GABAergic interneuron differentiation in the basal forebrain is mediated through direct regulation of glutamic acid decarboxylase isoforms by dlx homeobox transcription factors. J Neurosci. (2017) 37:8816–29. doi: 10.1523/JNEUROSCI.2125-16.2017
35. Wang Y, Dye CA, Sohal V, Long JE, Estrada RC, Roztocil T, et al. Dlx5 and Dlx6 regulate the development of parvalbumin-expressing cortical interneurons. J Neurosci. (2010) 30:5334–45. doi: 10.1523/JNEUROSCI.5963-09.2010
36. Cobos I, Calcagnotto ME, Vilaythong AJ, Thwin MT, Noebels JL, Baraban SC, et al. Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat Neurosci. (2005) 8:1059–68. doi: 10.1038/nn1499
37. Saino-Saito S, Berlin R, Baker H. Dlx-1 and Dlx-2 expression in the adult mouse brain: relationship to dopaminergic phenotypic regulation. J Comp Neurol. (2003) 461:18–30. doi: 10.1002/cne.10611
38. de Lombares C, Heude E, Alfama G, Fontaine A, Hassouna R, Vernochet C, et al. Dlx5 and Dlx6 expression in GABAergic neurons controls behavior, metabolism, healthy aging and lifespan. Aging. (2019) 11:6638–56. doi: 10.18632/aging.102141
39. Potter GB, Petryniak MA, Shevchenko E, McKinsey GL, Ekker M, Rubenstein JLR. Generation of cre-transgenic mice using Dlx1/Dlx2 enhancers and their characterization in GABAergic interneurons. Mol Cell Neurosci. (2009) 40:167–86. doi: 10.1016/j.mcn.2008.10.003
40. Ghanem N, Jarinova O, Amores A, Long Q, Hatch G, Park BK, et al. Regulatory roles of conserved intergenic domains in vertebrate dlx bigene clusters. Genome Res. (2003) 13:533–43. doi: 10.1101/gr.716103
41. Xu G, Broadbelt KG, Haynes RL, Folkerth RD, Borenstein NS, Belliveau RA, et al. Late development of the GABAergic system in the human cerebral cortex and white matter. J Neuropathol Exp Neurol. (2011) 70:841–58. doi: 10.1097/NEN.0b013e31822f471c
42. Asada H, Kawamura Y, Maruyama K, Kume H, Ding R-G, Kanbara N, et al. Cleft palate and decreased brain - aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc Nat Acad Sci. (1997) 94:6496–9. doi: 10.1073/pnas.94.12.6496
43. Pla R, Stanco A, Howard MA, Rubin AN, Vogt D, Mortimer N, et al. Dlx1 and Dlx2 promote interneuron gaba synthesis, synaptogenesis, and dendritogenesis. Cerebral Cortex. (2018) 28:3797–815. doi: 10.1093/cercor/bhx241
44. Nathanson JL, Yanagawa Y, Obata K, Callaway EM. Preferential labeling of inhibitory and excitatory cortical neurons by endogenous tropism of adeno-associated virus and lentivirus vectors. Neuroscience. (2009) 161:441–50. doi: 10.1016/j.neuroscience.2009.03.032
45. Kash SF, Johnson RS, Tecott LH, Noebels JL, Mayfield RD, Hanahan D, et al. Epilepsy in mice deficient in the 65-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci U S A. (1997) 94:14060–5. doi: 10.1073/pnas.94.25.14060
46. Müller I, Çalişkan G, Stork O. The GAD65 knock out mouse – a model for GABAergic processes in fear- and stress-induced psychopathology. Genes Brain Behav. (2015) 14:37–45. doi: 10.1111/gbb.12188
47. Smale ST, Kadonaga JT. The RNA polymerase II core promoter. Annu Rev Biochem. (2003) 72:449–79. doi: 10.1146/annurev.biochem.72.121801.161520
48. Oh W-J, Noggle SA, Maddox DM, Condie BG. The mouse vesicular inhibitory amino acid transporter gene: expression during embryogenesis, analysis of its core promoter in neural stem cells and a reconsideration of its alternate splicing. Gene. (2005) 351:39–49. doi: 10.1016/j.gene.2005.01.009
49. DeRosa BA, Belle KC, Thomas BJ, Cukier HN, Pericak-Vance MA, Vance JM, et al. hVGAT-mCherry: a novel molecular tool for analysis of GABAergic neurons derived from human pluripotent stem cells. Mol Cell Neurosci. (2015) 68:244–57. doi: 10.1016/j.mcn.2015.08.007
50. Fee C, Banasr M, Sibille E. Somatostatin-positive GABA interneuron deficits in depression: cortical microcircuit and therapeutic perspectives. Biol Psychiatry. (2017) 82:549–59. doi: 10.1016/j.biopsych.2017.05.024
51. Nathanson JL, Jappelli R, Scheeff ED, Manning G, Obata K, Brenner S, et al. Short promoters in viral vectors drive selective expression in mammalian inhibitory neurons, but do not restrict activity to specific inhibitory cell-types. Front Neural Circuits. (2009) 3:19. doi: 10.3389/neuro.04.019.2009
52. Paul A, Crow M, Raudales R, He M, Gillis J, Huang ZJ. Transcriptional architecture of synaptic communication delineates gabaergic neuron identity. Cell. (2017) 171:522–39.e20. doi: 10.1016/j.cell.2017.08.032
53. Sims KB, Hoffman DL, Said SI, Zimmerman EA. Vasoactive intestinal polypeptide (VIP) in mouse and rat brain: an immunocytochemical study. Brain Res. (1980) 186:165–83. doi: 10.1016/0006-8993(80)90263-2
54. Bechtold DA, Brown TM, Luckman SM, Piggins HD. Metabolic rhythm abnormalities in mice lacking VIP-VPAC2 signaling. Am J Physiol Regul Integr Comp Physiol. (2008) 294:R344–51. doi: 10.1152/ajpregu.00667.2007
55. Vu JP, Larauche M, Flores M, Luong L, Norris J, Oh S, et al. Regulation of appetite, body composition, and metabolic hormones by vasoactive intestinal polypeptide (VIP). J Mol Neurosci. (2015) 56:377–87. doi: 10.1007/s12031-015-0556-z
56. Niesler B, Weiss B, Fischer C, Nöthen MM, Propping P, Bondy B, et al. Serotonin receptor gene HTR3A variants in schizophrenic and bipolar affective patients. Pharmacogenet Genomics. (2001) 11:21–7. doi: 10.1097/00008571-200102000-00003
57. Anderson BM, Schnetz-Boutaud NC, Bartlett J, Wotawa AM, Wright HH, Abramson RK, et al. Examination of association of genes in the serotonin system to autism. Neurogenetics. (2009) 10:209–16. doi: 10.1007/s10048-009-0171-7
58. Lee S, Hjerling-Leffler J, Zagha E, Fishell G, Rudy B. The Largest group of superficial neocortical gabaergic interneurons expresses ionotropic serotonin receptors. J Neurosci. (2010) 30:16796–808. doi: 10.1523/JNEUROSCI.1869-10.2010
59. Rudy B, Fishell G, Lee S, Hjerling-Leffler J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Devel Neurobio. (2011) 71:45–61. doi: 10.1002/dneu.20853
60. Tremblay R, Lee S, Rudy B. GABAergic interneurons in the neocortex: from cellular properties to circuits. Neuron. (2016) 91:260–92. doi: 10.1016/j.neuron.2016.06.033
61. Pelkey KA, Chittajallu R, Craig MT, Tricoire L, Wester JC, McBain CJ. Hippocampal GABAergic inhibitory interneurons. Physiol Rev. (2017) 97:1619–747. doi: 10.1152/physrev.00007.2017
62. Albéri L, Lintas A, Kretz R, Schwaller B, Villa AEP. The calcium-binding protein parvalbumin modulates the firing 1 properties of the reticular thalamic nucleus bursting neurons. J Neurophysiol. (2013) 109:2827–41. doi: 10.1152/jn.00375.2012
63. Tai C, Abe Y, Westenbroek RE, Scheuer T, Catterall WA. Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of dravet syndrome. Proc Natl Acad Sci U S A. (2014) 111:E3139–48. doi: 10.1073/pnas.1411131111
64. Jiang X, Lachance M, Rossignol E. Involvement of cortical fast-spiking parvalbumin-positive basket cells in epilepsy. Prog Brain Res. (2016) 226:81–126. doi: 10.1016/bs.pbr.2016.04.012
65. Wöhr M, Orduz D, Gregory P, Moreno H, Khan U, Vörckel KJ, et al. Lack of parvalbumin in mice leads to behavioral deficits relevant to all human autism core symptoms and related neural morphofunctional abnormalities. Transl Psychiatry. (2015) 5:e525. doi: 10.1038/tp.2015.19
66. Schwede M, Nagpal S, Gandal MJ, Parikshak NN, Mirnics K, Geschwind DH, et al. Strong correlation of downregulated genes related to synaptic transmission and mitochondria in post-mortem autism cerebral cortex. J Neurodev Disord. (2018) 10:18. doi: 10.1186/s11689-018-9237-x
67. Ariza J, Rogers H, Hashemi E, Noctor SC, Martínez-Cerdeño V. The number of chandelier and basket cells are differentially decreased in prefrontal cortex in autism. Cereb Cortex. (2018) 28:411–20. doi: 10.1093/cercor/bhw349
68. Curley AA, Arion D, Volk DW, Asafu-Adjei JK, Sampson AR, Fish KN, et al. Cortical deficits of glutamic acid decarboxylase 67 expression in schizophrenia: clinical, protein, and cell type-specific features. Am J Psychiatry. (2011) 168:921–9. doi: 10.1176/appi.ajp.2011.11010052
69. Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. (2012) 35:57–67. doi: 10.1016/j.tins.2011.10.004
Keywords: enhancer, GABA, gene therapy, interneuron, promoter, viral vector
Citation: Duba-Kiss R, Niibori Y and Hampson DR (2021) GABAergic Gene Regulatory Elements Used in Adeno-Associated Viral Vectors. Front. Neurol. 12:745159. doi: 10.3389/fneur.2021.745159
Received: 21 July 2021; Accepted: 06 September 2021;
Published: 04 October 2021.
Edited by:
Laura Cancedda, Italian Institute of Technology (IIT), ItalyReviewed by:
Lorena Zentilin, International Centre for Genetic Engineering and Biotechnology, ItalyAyumu Konno, Gunma University, Japan
Copyright © 2021 Duba-Kiss, Niibori and Hampson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David R. Hampson, ZC5oYW1wc29uQHV0b3JvbnRvLmNh