 Peter Sparber
Peter Sparber Svetlana Mikhaylova2†
Svetlana Mikhaylova2† Mikhail Skoblov
Mikhail Skoblov- 1Laboratory of Functional Genomics, Research Centre for Medical Genetics, Moscow, Russia
- 2Medical Genetics Department, Russian Children's Clinical Hospital, Moscow, Russia
- 3Clinical Department, Research Centre for Medical Genetics, Moscow, Russia
- 4Laboratory of Inherited Metabolic Disorders, Research Centre for Medical Genetics, Moscow, Russia
Pathogenic variants in the SCN1A gene are associated with a spectrum of epileptic disorders ranging in severity from familial febrile seizures to Dravet syndrome. Large proportions of reported pathogenic variants in SCN1A are annotated as missense variants and are often classified as variants of uncertain significance when no functional data are available. Although loss-of-function variants are associated with a more severe phenotype in SCN1A, the molecular mechanism of single nucleotide variants is often not clear, and genotype-phenotype correlations in SCN1A-related epilepsy remain uncertain. Coding variants can affect splicing by creating novel cryptic splicing sites in exons or by disrupting exonic cis-regulation elements crucial for proper pre-mRNA splicing. Here, we report a novel case of Dravet syndrome caused by an undescribed missense variant, c.4852G>A (p.(Gly1618Ser)). By midigene splicing assay, we demonstrated that the identified variant is in fact splice-affecting. To our knowledge, this is the first report on the functional investigation of a missense variant affecting splicing in Dravet syndrome.
Introduction
Dravet syndrome (DS), also known as severe myoclonic epilepsy (SMEI) in infancy, is a developmental and epileptic encephalopathy (DEE) characterized by intractable childhood-onset seizures, neurodevelopmental delay, and other neurological impairments (1). With overall incidence ranging from 1:15,700 in the United States and 1:40,000 according to the Orphanet database (2), DS is considered to be one of the most common causes of genetic epilepsy in infants (3). The main cause of DS is heterozygous loss-of-function (LoF) pathogenic variants in the SCN1A gene that encodes the alpha subunit of type I voltage-gated sodium channel (Nav1.1) (4). In a small fraction of patients with DS phenotype, pathogenic variants in other genes, including PCDH19, GABRA1, and STXBP1, were shown to be causative (5). SCN1A expression is restricted to GABA-ergic inhibitory interneurons that control neuronal excitability, and loss of Nav1.1 is assumed to lead to uncontrolled firing and epileptic activity (6–8).
Given the fact that SCN1A haploinsufficiency is the main molecular mechanism leading to DS, protein-truncation variants (nonsense, frameshift, and splice-site variants) are, in general, associated with a more severe epileptic phenotype (9). However, there are hundreds of missense variants that are associated with DS in the literature lacking any functional data (10). The absence of functional data for the majority of missense variants makes it challenging for performing valid genotype-phenotype studies in SCN1A-related epileptic disorders.
Here, we report a novel case of a 2-year-old patient who was clinically diagnosed with DS. Genetic analysis revealed an undescribed missense variant in the SCN1A gene, c.4852G>A (p.(Gly1618Ser)). We showed by midigene splicing assay that the variant is in fact splice-affecting, leading to two independent splicing alterations and LoF effects, thus confirming the diagnosis.
Case Description
The proband, a 2-year-old Russian girl at the time of the last clinical evaluation, was admittedto the neurological department with repeated, prolonged myoclonic, and generalized seizures responsive only to intravenous injection of diazepam. She was the fourth child of healthy parents from a non-consanguineous marriage. The proband has three healthy siblings (Figure 1A). During the pregnancy, a risk of miscarriage was observed at 12 weeks of gestation. Delivery and neonatal period were unremarkable. At the age of 4 months, during obstructive bronchitis, she developed an absence seizure with apnea 3–5 s long that repeated daily afterward. At the age of 5 months, during hot water bathing, the proband had a prolonged generalized myoclonic seizure for 40 min that was responsive only to diazepam injection. Similar episodes repeated every 7–10 days without any provoking factors.
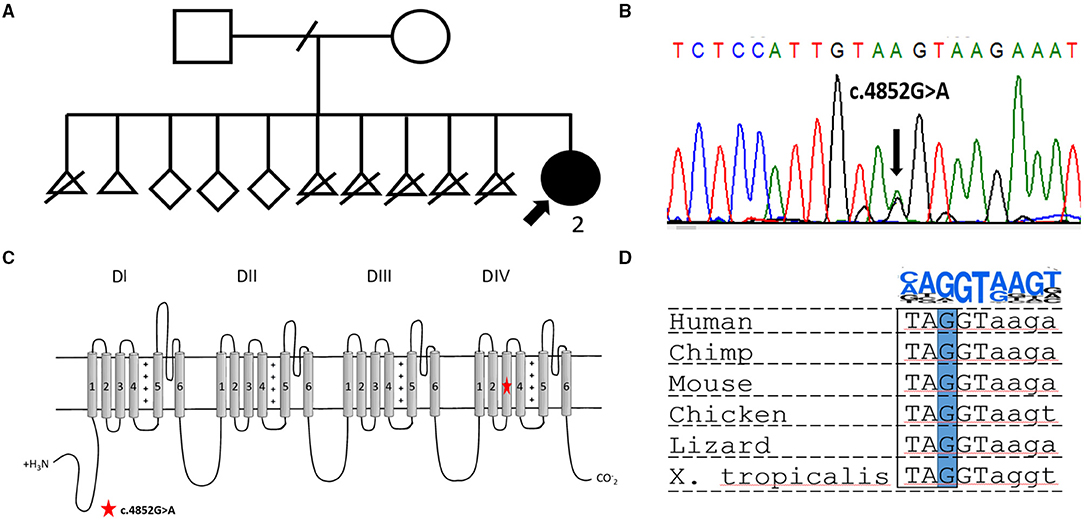
Figure 1. (A) Pedigree of the reported family. The proband is indicated by a bold arrow. (B) Electropherogram showing the heterozygous variant NM_001165963.2:c.4852G>A [p.(Gly1618Ser)] confirmed in the proband by Sanger sequencing. (C) Scheme of Nav1.1 channel structure. The location of the novel missense variant is indicated by a red star. (D) Multiply alignment showing the conservation of the affected nucleotide in different vertebrates. On the top is a position weight matrix of the site of the splicing donor.
On brain magnetic resonance imaging (MRI), periventricular leukomalacia was noted and was considered as a result of ischemic brain injury with no relevance to the epileptic phenotype of the patient. Routine electroencephalography (EEG) did not show any epileptiform activity in the interictal period. Antiepileptic therapy included carbamazepine (300 mg/day), topiramate (87.5 mg/day), and clonazepam (0.75 mg/day), which had no effect on seizure frequency and duration. In the evaluation, the proband had prolonged myoclonic, tonic-clonic, and atonic seizures once a week. Early motor milestones were normal, but language development was delayed. Neurological examination at the age of 2 revealed moderate hypotonia with brisk tendon reflexes and mild gait ataxia. Based on the clinical picture, the proband was diagnosed with DS.
Genetic and Bioinformatic Analysis
Targeted sequencing of 587 genes associated with neurodevelopmental disorders, such as the SCN1A gene, along with other DS associated genes, PCDH19 and STXBP1, was performed. DNA analysis revealed a previously undescribed missense variant in the SCN1A gene, NM_001165963.2:c.4852G>A (p.(Gly1618Ser)). Sanger sequencing confirmed the variant in the proband (Figure 1B), but segregation analysis was not performed because of the fact that the father was unavailable for biological sample collection and the mother declined genetic testing for herself and other healthy siblings. The variant is absent in the gnomAD database and predicted to be deleterious by several bioinformatic predictors, such as PolyPhen2, SIFT, and BadMut (11–13). According to the ACMG/AMP guidelines (14), it was classified as a variant of uncertain significance (VUS).
The undescribed missense variant is predicted to change a conservative amino acid residue located in the S3 transmembrane segment in domain IV of Nav1.1 (Figure 1B). Previous studies have shown that missense substitutions located in the pore-forming part are frequently associated with complete LoF and, therefore, lead to a severe epileptic phenotype (15). In other cases, for missense variants, it is nearly impossible to predict the phenotypic outcome based only on genetic data. At the DNA level, the c.4852G>A variant changes the last nucleotide in exon 28 of the SCN1A gene (Figures 1C,D). Therefore, we assumed that the variant could affect splicing. Bioinformatic analysis using SpliceAI (16) predicted donor loss (Δ score.53) and activation of a cryptic exonic donor site at position −4 (Δ score.83).
Functional Analysis
In order to investigate the potential impact of the c.4852G>A variant on splicing, we performed a midigene splicing assay. The genomic region containing exon 28 with 146 nucleotides of intron 27, intron 28, and exon 29 of the SCN1A gene was amplified from the genomic DNA of the proband and cloned into a pSpl3-Flu splicing vector (Figure 2A) (17). Wild-type (WT) and mutant (MUT) plasmids were transfected separately to HEK293T, and 48 h post transfection RT-PCR was performed. The WT plasmids demonstrated a normal splicing pattern with the inclusion of both exons 28 and 29 (Figure 2B).
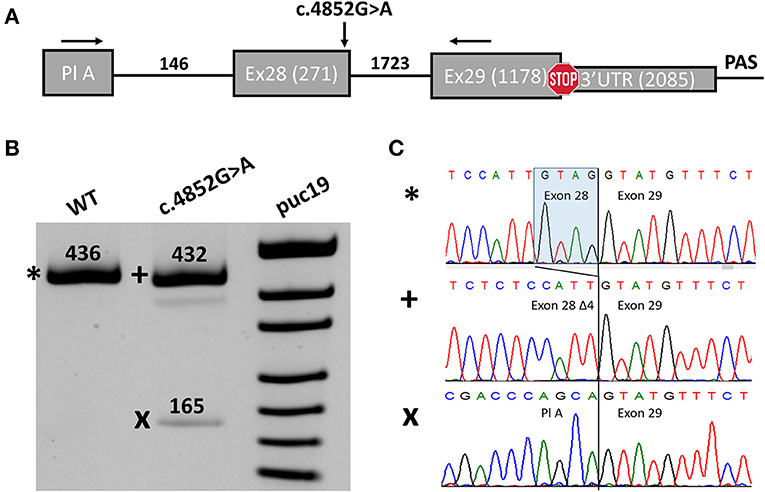
Figure 2. Midigene splicing assay results. (A) Scheme of the midigene plasmid used for splicing analysis with numbers indicating the length of exons and introns. The location of primers used for RT-PCR and the investigated variant are shown with black arrows. Pl A, plasmid exon A. (B) PAGE urea electrophoresis of RT-PCR products. *, Wild type isoform; +, isoform with four-nucleotide deletions of exon 28; x, isoform with exon 28 skipping. WT, wild type. (C) Sanger sequencing of the corresponding isoforms.
We detected two abnormal isoforms when analyzing the splicing pattern of the MUT plasmid (Figure 2B). In the major isoform, we observed a four-nucleotide deletion due to the activation of a cryptic donor splicing site (Figure 2C). The deletion caused frameshift and formation of a premature termination codon (PTC) (p.(Gly1618Cysfs*6)). Sanger sequencing of the second isoform revealed skipping of exon 28 (Figure 2C), which also leads to the frameshift in CDS (p.(Asn1528Valfs*7)). The splicing alteration affected the penultimate exon of the SCN1A gene. In both cases, the PTC is formed in the last exon and, therefore, is not predicted to induce non-sense-mRNA decay (NMD) (18, 19). Nevertheless, the predicted truncated protein, even if translated, could not function properly as it lacks the majority of the IV domain of Nav1.1 including pore-forming residues. Therefore, we conclude that the c.4852G>A variant leads to complete LoF and could be reclassified as likely pathogenic (PM2, PS3).
Discussion
Pathogenic variants in the SCN1A gene are associated with a spectrum of epilepsy phenotypes ranging in severity from familial febrile seizures (FSs) on the mild end to DS on the other. According to the SCN1A mutation database, almost 50% of reported pathogenic variants in this gene are missense substitutions (10). LoF variants (non-sense, frameshift, and splice-site variants) are frequently found in patients with more severe phenotypes and are evenly distributed in different exons, as they are predicted to activate NMD and, therefore, lead to loss of expression from one allele. On the other hand, missense variants are more often found in patients with milder phenotypes, such as mild generalized epilepsy (GE) and/or FS. In contrast, with LoF variants, the frequency of missense variants decreases with an increase in severity. Analysis of the distribution of missense variants across different Nav1.1 domains shows that variants linked to DS are enriched in the pore-forming region (S5–S6) (10). Such substitutions completely disturb the kinetics of the Nav1.1 channel, as they prevent sodium current from entering the cell (20).
The introduction of next-generation sequencing (NGS) to clinical practice has revolutionized the field of genetic-based epilepsy and has improved the diagnostic yield dramatically (21, 22). However, with more genetic tests performed, the interpretation has become more and more challenging for newly described variants. With more than 2,000 pathogenic variants in SCN1A reported in the professional version of the HGMD database, for the majority of missense variants, there is no functional characterization. The gold standard for functional tests in channelopathies is the patch clamp technique where ionic currents are measured in the context of the variant of interest (20, 23). Although very informative for the investigation of channel function, patch clamp remains low-scaled, time-consuming, and is not routinely performed.
Several studies have demonstrated that a large fraction of all disease-causing variants may influence normal pre-mRNA splicing (24, 25). A recent high-throughput study revealed that ~10% out of 5,000 pathogenic missense variants reported in the literature leads to exon skipping (26). Taking into account that only exon skipping was investigated and that other splicing alterations, such as cryptic splice-site activation and intron retention, could not be assessed, the true rate of missense variants affecting splicing could even be higher, which means that many missense variants reported in the literature could, in fact, affect splicing and are misannotated.
Ribonucleic acid (RNA) analysis using patient-derived samples is the most informative approach for the investigation of splicing alterations (27). Unfortunately, in many cases, when analyzing variants in a gene with a tissue-specific expression, such as SCN1A, the collection of a relevant biological sample is impossible. In such cases, the minigene splicing assay could be performed as an alternative. Minigene splicing assay is a well-established method that could be routinely used and has demonstrated high concordance with RT-PCR results (28). Functional analysis is of great importance given the fact that several phenotypes are linked to the SCN1A gene with different molecular mechanisms of channel dysfunction (29, 30). Moreover, in the absence of segregation data, functional tests are often the only option for the reclassification of an undescribed variant. Proper understanding of the molecular defect is crucial not only for proper genetic counseling but is also necessary for developing tailored therapeutic approaches in genetic epilepsy.
In this report, the diagnosis of DS was made based solely on the severe epileptic phenotype with poor response to antiepileptic medications. When performing a genetic test, a novel missense variant in SCN1A was observed. However, the predicted amino acid substitution in the lack of segregation data was not sufficient enough to explain or to confirm the diagnosis of the patient. Bioinformatic analysis predicted a possible effect on splicing that was shown by using midigene splicing assay. The reported patient fully fits the diagnostic criteria of DS, and the epileptic phenotype is similar to that of patients with other reported LoF variants (2). Thereby, we exclude the inconsistency between the genotype and phenotype data and help the clinician in performing straightforward genetic counseling and proper risk prognosis in the family.
Overall, we report a newly described missense variant in a patient with a clinical diagnosis of DS. Midigene splicing assay confirmed that the c.4852G>A variant affects splicing and leads to complete LoF of Nav1.1. This study expands the mutational spectrum of SCN1A related DS and delineated the patient phenotype on the molecular level. This report highlights the importance of functional validation in clinical practice and its role in robust genotype-phenotype correlations. To our knowledge, this is the first report on a splice-affecting missense variant in DS.
Materials and Methods
Subjects
The proband underwent a detailed clinical examination at the Russian Children's Clinical Hospital in Moscow. Genetic analysis was performed at the Research Center for Medical Genetics, Russia.
All research participants gave informed consent (or responsible consent form for infant proband) for the clinical examination and publication of their anonymized data. The study was performed in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of the Research Center for Medical Genetics., Russia.
Genetic Analysis
DNA was extracted from peripheral blood mononuclear cells using the standard phenol-chloroform method. Genetic analysis was performed at the Research Center for Medical Genetics by the next-generation sequencing of a gene panel containing 587 genes. The sequencing library was prepared with The Ion AmpliSeq™ Library Kit 2.0 and sequenced on an Ion Torrent S5 system with a minimum coverage of 140X. Reads were aligned to the human reference genome (Hg19) with BWA and filtered based on frequency and annotation. The candidate variant in the SCN1A gene identified in the proband was confirmed by Sanger sequencing. For the SCN1A gene, RefSeq accession numbers NG_011906.1 and NM_001165963.4 were used.
Midigene Splicing Assay
The last two exons of the SCN1A (28 and 29) gene along with intron 28 and the adjust 146 nucleotides of intron 27 were amplified from genomic DNA using a high-fidelity DNA polymerase. The PCR product was cloned to a pSpl3-Flu splicing vector as previously described (31). Three polyadenylation signals were introduced after the end of exon 29 for transcription termination. Due to the large insertion size (5,257 bp), we named the resultant vector a “midigene” as in Sangermano et al. (32). HEK293T cells were plated in a 24-well plate and transfected with the SCN1A midigene at ~80% cell confluency with 0.75 μg per well by using the calcium phosphate method (33). Forty-eight hours post transfection, the cells were harvested, and total RNA was extracted using the TRIzol-based method and reverse transcribed. RT-PCR using primers aligned to the plasmid exon and exon 29 of the SCN1A gene was performed, and PCR products were visualized by denaturing urea PAGE with subsequent Sanger sequencing.
Data Availability Statement
The datasets presented in this article are not readily available due to ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Review Board of the Research Center for Medical Genetics. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
PS: conceptualization, methodology, design, writing—original draft preparation. SM, VG, and YI: methodology, data curation. MS: conceptualization, methodology, supervision, writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was carried out within the state assignment of the Ministry of Science and Higher Education of the Russian Federation for RCMG. The reported study was funded by RFBR. Project number: 20-315-90042.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the reported family for participation in this study.
References
1. Anwar A, Saleem S, Patel UK, Arumaithurai K, Malik P. Dravet syndrome: an overview. Cureus. (2019) 11:e5006. doi: 10.7759/cureus.5006
2. Wu YW, Sullivan J, Mcdaniel SS, Meisler MH, Walsh EM, Li SX, et al. Incidence of dravet syndrome in a US population. Pediatrics. (2015) 136:e1310–5. doi: 10.1542/peds.2015-1807
3. Lindy AS, Stosser MB, Butler E, Downtain-Pickersgill C, Shanmugham A, Retterer K, et al. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia. (2018) 59:1062–71. doi: 10.1111/epi.14074
4. Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. (2001) 68:1327–32. doi: 10.1086/320609
5. Carvill GL, Weckhuysen S, Mcmahon JM, Hartmann C, Moller RS, Hjalgrim H, et al. GABRA1 and STXBP1: novel genetic causes of dravet syndrome. Neurology. (2014) 82:1245–53. doi: 10.1212/WNL.0000000000000291
6. Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, et al. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. (2007) 27:5903–14. doi: 10.1523/JNEUROSCI.5270-06.2007
7. Bender AC, Morse RP, Scott RC, Holmes GL, Lenck-Santini PP. SCN1A mutations in Dravet syndrome: impact of interneuron dysfunction on neural networks and cognitive outcome. Epilepsy Behav. (2012) 23:177–86. doi: 10.1016/j.yebeh.2011.11.022
8. Chopra R, Isom LL. Untangling the dravet syndrome seizure network: the changing face of a rare genetic epilepsy. Epilepsy Curr. (2014) 14:86–9. doi: 10.5698/1535-7597-14.2.86
9. Zuberi SM, Brunklaus A, Birch R, Reavey E, Duncan J, Forbes GH. Genotype-phenotype associations in SCN1A-related epilepsies. Neurology. (2011) 76:594–600. doi: 10.1212/WNL.0b013e31820c309b
10. Meng H, Xu HQ, Yu L, Lin GW, He N, Su T, et al. The SCN1A mutation database: updating information and analysis of the relationships among genotype, functional alteration, and phenotype. Hum Mutat. (2015) 36:573–80. doi: 10.1002/humu.22782
11. Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. (2012) 40:W452–7. doi: 10.1093/nar/gks539
12. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. (2013) Chanpter 7:Unit 20. doi: 10.1002/0471142905.hg0720s76
13. Korvigo I, Afanasyev A, Romashchenko N, Skoblov M. Generalising better: Applying deep learning to integrate deleteriousness prediction scores for whole-exome SNV studies. PLoS ONE. (2018) 13:e0192829. doi: 10.1371/journal.pone.0192829
14. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
15. Wallace R. A plethora of SCN1A mutations: what can they tell us? Epilepsy Curr. (2005) 5:17–20. doi: 10.1111/j.1535-7597.2005.05105.x
16. Jaganathan K, Kyriazopoulou Panagiotopoulou S, Mcrae JF, Darbandi SF, Knowles D, Li YI, et al. Predicting splicing from primary sequence with deep learning. Cell. (2019) 176:535–48.e24. doi: 10.1016/j.cell.2018.12.015
17. Sparber P, Marakhonov A, Filatova A, Sharkova I, Skoblov M. Novel case of neurodegeneration with brain iron accumulation 4 (NBIA4) caused by a pathogenic variant affecting splicing. Neurogenetics. (2018) 19:257–60. doi: 10.1007/s10048-018-0558-4
18. Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci. (1998) 23:198–9. doi: 10.1016/S0968-0004(98)01208-0
19. Lykke-Andersen S, Jensen TH. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol. (2015) 16:665–77. doi: 10.1038/nrm4063
20. Kluckova D, Kolnikova M, Lacinova L, Jurkovicova-Tarabova B, Foltan T, Demko V, et al. A study among the genotype, functional alternations, and phenotype of 9 SCN1A mutations in epilepsy patients. Sci Rep. (2020) 10:10288. doi: 10.1038/s41598-020-67215-y
21. Jang SS, Kim SY, Kim H, Hwang H, Chae JH, Kim KJ, et al. Diagnostic yield of epilepsy panel testing in patients with seizure onset within the first year of life. Front Neurol. (2019) 10:988. doi: 10.3389/fneur.2019.00988
22. Sanchez Fernandez I, Loddenkemper T, Gainza-Lein M, Sheidley BR, Poduri A. Diagnostic yield of genetic tests in epilepsy: a meta-analysis and cost-effectiveness study. Neurology. (2019) 92:e418–28. doi: 10.1212/WNL.0000000000006850
23. Kornreich BG. The patch clamp technique: principles and technical considerations. J Vet Cardiol. (2007) 9:25–37. doi: 10.1016/j.jvc.2007.02.001
24. Lopez-Bigas N, Audit B, Ouzounis C, Parra G, Guigo R. Are splicing mutations the most frequent cause of hereditary disease? FEBS Lett. (2005) 579:1900–3. doi: 10.1016/j.febslet.2005.02.047
25. Soukarieh O, Gaildrat P, Hamieh M, Drouet A, Baert-Desurmont S, Frebourg T, et al. Exonic splicing mutations are more prevalent than currently estimated and can be predicted by using in silico tools. PLoS Genet. (2016) 12:e1005756. doi: 10.1371/journal.pgen.1005756
26. Soemedi R, Cygan KJ, Rhine CL, Wang J, Bulacan C, Yang J, et al. Pathogenic variants that alter protein code often disrupt splicing. Nat Genet. (2017) 49:848–55. doi: 10.1038/ng.3837
27. Wai HA, Lord J, Lyon M, Gunning A, Kelly H, Cibin P, et al. Blood RNA analysis can increase clinical diagnostic rate and resolve variants of uncertain significance. Genet Med. (2020) 22:1005–14. doi: 10.1038/s41436-020-0766-9
28. Van Der Klift HM, Jansen AM, Van Der Steenstraten N, Bik EC, Tops CM, Devilee P, et al. Splicing analysis for exonic and intronic mismatch repair gene variants associated with lynch syndrome confirms high concordance between minigene assays and patient RNA analyses. Mol Genet Genomic Med. (2015) 3:327–45. doi: 10.1002/mgg3.145
29. Escayg A, Goldin AL. Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia. (2010) 51:1650–8. doi: 10.1111/j.1528-1167.2010.02640.x
30. Dhifallah S, Lancaster E, Merrill S, Leroudier N, Mantegazza M, Cestele S. Gain of Function for the SCN1A/hNav1.1-L1670W mutation responsible for familial hemiplegic migraine. Front Mol Neurosci. (2018) 11:232. doi: 10.3389/fnmol.2018.00232
31. Filatova AY, Vasilyeva TA, Marakhonov AV, Voskresenskaya AA, Zinchenko RA, Skoblov MY. Functional reassessment of PAX6 single nucleotide variants by in vitro splicing assay. Eur J Hum Genet. (2019) 27:488–93. doi: 10.1038/s41431-018-0288-y
32. Sangermano R, Khan M, Cornelis SS, Richelle V, Albert S, Garanto A, et al. ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in stargardt disease. Genome Res. (2018) 28:100–10. doi: 10.1101/gr.226621.117
Keywords: epilepsy, Dravet syndrome, medical genetics, functional analysis, splicing, molecular pathogenesis
Citation: Sparber P, Mikhaylova S, Galkina V, Itkis Y and Skoblov M (2021) Case Report: Functional Investigation of an Undescribed Missense Variant Affecting Splicing in a Patient With Dravet Syndrome. Front. Neurol. 12:761892. doi: 10.3389/fneur.2021.761892
Received: 20 August 2021; Accepted: 02 November 2021;
Published: 06 December 2021.
Edited by:
Matthew James Farrer, University of Florida, United StatesReviewed by:
Meng-Han Tsai, Kaohsiung Chang Gung Memorial Hospital, TaiwanLi Cao, Shanghai Jiao Tong University, China
Copyright © 2021 Sparber, Mikhaylova, Galkina, Itkis and Skoblov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Sparber, cHNwYXJiZXI5M0BnbWFpbC5jb20=
†ORCID: Svetlana Mikhaylova orcid.org/0000-0002-2115-985X