 Kimberly Goodspeed1
Kimberly Goodspeed1 Rachel M. Bailey1,2
Rachel M. Bailey1,2 Suyash Prasad3
Suyash Prasad3 Chanchal Sadhu3
Chanchal Sadhu3 Jessica A. Cardenas3
Jessica A. Cardenas3 Mary Holmay3
Mary Holmay3 Deborah A. Bilder4
Deborah A. Bilder4 Berge A. Minassian1*
Berge A. Minassian1*- 1Division of Child Neurology, Department of Pediatrics, University of Texas Southwestern, Dallas, TX, United States
- 2Center for Alzheimer's and Neurodegenerative Diseases, University of Texas Southwestern, Dallas, TX, United States
- 3Department of Research and Development, Taysha Gene Therapies, Dallas, TX, United States
- 4Division of Child and Adolescent Psychiatry, Department of Psychiatry, Huntsman Mental Health Institute, University of Utah, Salt Lake City, UT, United States
Genetic epilepsies are a spectrum of disorders characterized by spontaneous and recurrent seizures that can arise from an array of inherited or de novo genetic variants and disrupt normal brain development or neuronal connectivity and function. Genetically determined epilepsies, many of which are due to monogenic pathogenic variants, can result in early mortality and may present in isolation or be accompanied by neurodevelopmental disability. Despite the availability of more than 20 antiseizure medications, many patients with epilepsy fail to achieve seizure control with current therapies. Patients with refractory epilepsy—particularly of childhood onset—experience increased risk for severe disability and premature death. Further, available medications inadequately address the comorbid developmental disability. The advent of next-generation gene sequencing has uncovered genetic etiologies and revolutionized diagnostic practices for many epilepsies. Advances in the field of gene therapy also present the opportunity to address the underlying mechanism of monogenic epilepsies, many of which have only recently been described due to advances in precision medicine and biology. To bring precision medicine and genetic therapies closer to clinical applications, experimental animal models are needed that replicate human disease and reflect the complexities of these disorders. Additionally, identifying and characterizing clinical phenotypes, natural disease course, and meaningful outcome measures from epileptic and neurodevelopmental perspectives are necessary to evaluate therapies in clinical studies. Here, we discuss the range of genetically determined epilepsies, the existing challenges to effective clinical management, and the potential role gene therapy may play in transforming treatment options available for these conditions.
Introduction
While 20–30% of epilepsies are acquired nongenetically, 70–80% are due to 1 or more genetic factors (1). Developmental and epileptic encephalopathies (DEE) are rare disorders characterized by early-onset, refractory seizures that occur in the context of developmental regression or plateauing. DEE are severe and difficult to treat and may result from a single gene mutation that causes gain-of-function (2) or loss-of-function epilepsy (3, 4). Monogenic epilepsies may be autosomal recessive (e.g., EPM2A/B or SLC13A5), autosomal dominant (e.g., CHRNA4), autosomal haploinsufficiency (e.g., SLC6A1), or X-linked (e.g., ARHGEF9) (5–10). Further, different pathogenic variants in the same gene may result in different epilepsy phenotypes, as seen in the KCNQ2 gene, where the R213W variant causes benign familial neonatal seizures, and the R213Q variant causes neonatal epileptic encephalopathy with severe pharmacoresistant seizures (11).
Precision medicine describes a rational treatment strategy that is highly specific and aims to address the underlying cause of disease (12). One avenue of precision medicine involves the selection of a therapy that is directed toward modulating or bypassing the dysfunction caused by the underlying genetic defect (12). In the era of gene therapy, avenues that may be applied to epilepsy syndromes include treatments that aim to restore cellular function such as gene replacement therapy (GRT) for disorders due to loss-of-function pathogenic variants (13, 14); genetic substrate reduction therapy (gSRT) [reviewed in Coutinho et al. (15)] to reduce the overproduction of substrates; or transcriptional enhancement, designed to upregulate endogenous expression of a given gene via the introduction of regulatory elements (16, 17). Monogenic epilepsies are of particular interest for precision medicine, as simplified GRT, gSRT, and transcriptional enhancement therapies are promising in ameliorating disease. Here, we will focus specifically on Lafora disease, SLC13A5 deficiency disorder (SDD), and SLC6A1-related disorder (SRD).
Clinical Care
Current treatment approaches focus on treating the epilepsy syndrome via antiseizure medications, diet, and/or neurostimulation, rather than the underlying genetic basis of disease (9). Combinations of antiseizure medications may be necessary to achieve adequate seizure control. Further, patients may become refractory to antiseizure medications over time (18) and for some patients, specific antiseizure medications are contraindicated, as they may exacerbate neurodevelopmental disability associated with their specific epilepsy syndrome (19). Ketogenic (high fat/low carbohydrate) diets and vagus nerve stimulation approaches also have been attempted in patients with inadequate seizure control, however, with limited success (20–25). Notably, there are no currently approved treatments that address the underlying cause of disease for genetic epilepsies, presenting an urgent need for the community and an opportunity for novel approaches such as GRT and gSRT.
Historical Context
Advances in Genetic Diagnosis
Prior to modern genetic approaches, epilepsies were examined for their genetic basis in families using gene mapping and applied linkage analysis. The first discoveries in the 1990s identified ion channels and led to the “channelopathy” hypothesis that suggested that ion channel defects were a common underlying cause of epilepsy (1). Additionally, it is now recognized that other single-gene pathogenic variants contribute to seizure disorders.
Starting in the late 2000s, next-generation sequencing has increasingly led to discovery of pathogenic variants in specific genes and microdeletions resulting in epilepsies (26). Commercially available epilepsy panels are available to test for many genetic epilepsies.
Still, many genetic epilepsies and their natural histories are not well understood. The prognosis for genetic epilepsies is often not promising, and there is a need for innovative solutions to improve patient outcomes. In addition to the development of novel pharmaceuticals, genetic epilepsies may be approached via gene therapy.
Advances in Gene Therapy
The first successful human trial of gene therapy occurred in 1990 (27). The field has rapidly expanded in the twenty-first century. One approach is GRT, which utilizes a vector such as adeno-associated virus (AAV) serotype 9 (AAV9), to deliver a functional copy of a gene to correct loss-of-function pathogenic variants, including recessive disorders (e.g., SLC13A5) and haploinsufficiencies (e.g., SLC6A1) (7, 9, 13). One example is the recent FDA approval of a gene therapy product to treat spinal muscular atrophy—a rare disease that causes infant mortality—which was the first gene therapy approval for children >2 years of age (28). There are also AAV9-based gene therapies in neurodevelopmental disorders in clinical trials (NCT02362438) following promising preclinical results (13).
The gSRT approach may utilize an AAV vector to deliver small interfering RNA that will reduce the overproduction of substrates (15). For example, the GYS1 gene may be knocked down to prevent the overproduction of the substrate glycogen, which accumulates to cause Lafora disease (16, 29). Transcriptional enhancement approaches may be effective in haploinsufficiencies such as Dravet syndrome, where 1 allele of the SCN1A gene possesses loss-of-function pathogenic variants, and the other normal endogenous allele can be modified to increase its expression levels (17). These approaches have the potential to address the underlying cause of disease in inherited epilepsies that are the result of loss-of-function pathogenic variants and provide significant seizure relief to patients.
AAV vectors have been extensively studied for treatment of central nervous system (CNS) diseases (30). AAV9, specifically, is a vector with great potential for treating neurological disorders, as it crosses the blood-brain barrier and targets CNS neurons (31). While other viral vectors transduce neurons, AAV9 is the most studied AAV vector for CNS disorders, and there is more clinical evidence of safety, efficacy, and stability of gene transfer to the CNS with this serotype than with other vectors (32).
Further, to aid in the development of next-generation gene therapy technologies for diagnosis of genetic epilepsies, a better understanding of natural history of disease will be required and is addressed in the next section. These studies inform clinical development and help identify outcome measures for clinical investigation.
Clinical Trial Readiness
Studies into the natural history of disease are essential to understanding how diseases progress and to inform drug development so that researchers and clinicians can have strong metrics available to evaluate how best to demonstrate efficacy and, ultimately, improve patients' quality of life. Regulatory agencies are increasingly acknowledging the importance of natural history data in the context of rare disease and gene therapy drug development, having released draft guidance on the topics in recent years (33). Natural history studies, although informative, may not accurately represent disease populations due to factors such as study design, variability of supportive care practices, changes in medical care or terminology over time, selection bias, etc. In monogenic epilepsies, due to the relatively recent identification of genetic causes, may particularly be lacking in a detailed and longitudinal understanding of the disease course. Animal models, therefore, also have an important role to play in understanding disease progression. Animal models currently exist for some, but not all, of the recessive and haploinsufficient epilepsies (see Table 1 for examples of available models) but may not fully replicate the clinical phenotype, which represents a challenge to characterizing the outcomes of potentially disease-modifying investigational drugs. While electroencephalography findings in animal models are comparable to those in humans, neurologic and motor deficits do not always correspond well with the human disease. GRT and gSRT approaches utilizing AAV vector technology may address diseases resulting from pathogenic variants in single genes (13, 15). In particular, AAV9 has shown promise for treating neurological disorders as it crosses into the brain and infects neurons (31). In the following sections, this review will highlight 3 monogenic inherited diseases, areas of active research by our groups: Lafora disease, SDD, and SRD, as well as their clinical picture, mouse models, and approaches to gene therapy for each condition.
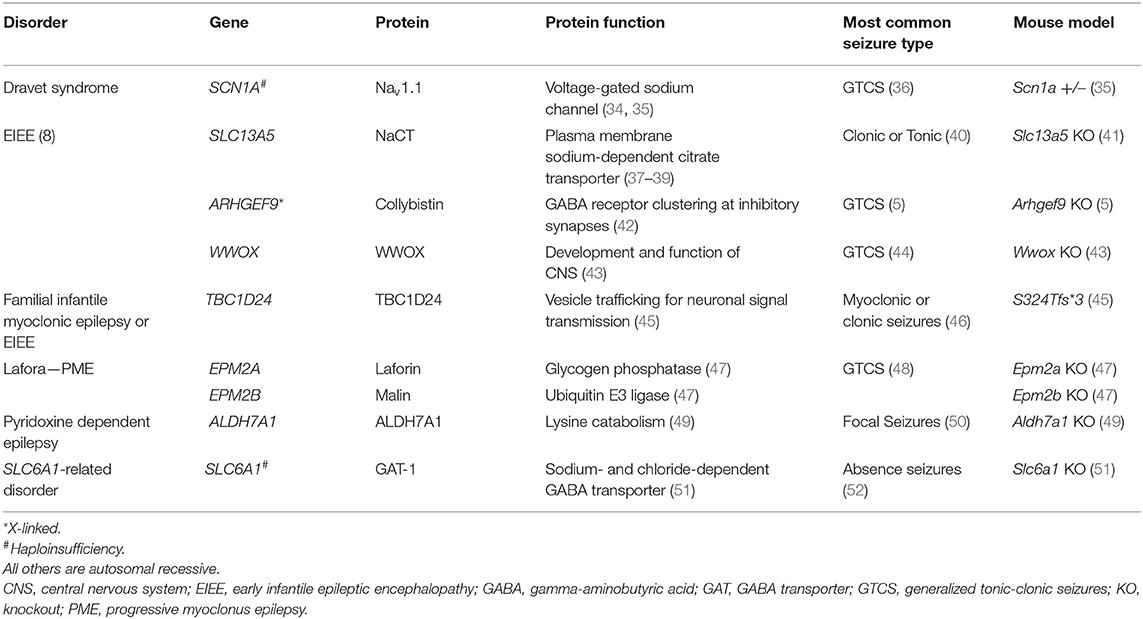
Table 1. Potential monogenic epilepsy candidates for gene therapy.
Lafora Disease
Lafora disease is a severe, fatal, autosomal recessive progressive myoclonus epilepsy (PME) that results from accumulation of Lafora bodies, abnormal glycogen aggregates (6). Two genes are now known to be involved in Lafora disease: EPM2A and EPM2B (48, 53–56). Loss-of-function pathogenic variants in EPM2A or EPM2B lead to an accumulation of Lafora bodies (an abnormal form of glycogen that cannot be metabolized) and subsequent Lafora disease (47).
Presentation and Progression
The mean age of Lafora disease onset is 13.4 years (57). Patients with classical Lafora disease develop normally until adolescence, when they present with action and stimulus-sensitive myoclonus, in addition to tonic-clonic and absence seizures (48). At presentation, it is challenging to distinguish Lafora disease from idiopathic generalized epilepsies (48). Thus genetic testing is critical, as it reveals pathogenic variants in the EPM2A and EPM2B genes (58).
Patients most often receive antiseizure medications, namely valproic acid, which is typically effective at suppressing seizure activity, however the treatment is palliative (59). Lafora patients quickly develop symptoms of dementia and intractable seizures (57). Patients tend to lose autonomy by 6 years after disease onset and die from status epilepticus, aspiration pneumonitis, or other complications of neurodegenerative disease within 10 years of disease onset (57).
To date, only one large-scale natural history study for Lafora disease exists, suggesting more studies are needed to describe the heterogenous disease and inform clinical investigation more fully (57).
Gene Therapy Development
It has been shown that Epm2a knockout (KO) and Epm2b KO mouse models replicate essential features of Lafora disease, such as neuronal degeneration and accumulation of Lafora bodies in muscle, liver, and brain (47, 60, 61). Recently, a proof-of-concept paper demonstrated that a viral vector carrying clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 with a guide RNA could be used to target and cut the Gys1 gene responsible for producing brain glycogen that leads to Lafora bodies and Lafora disease. In this study, neonatal Epm2a KO and Epm2b KO mice were injected intracerebroventricularly with an AAV9 vector targeting Gys1 that led to an editing rate of 17% of Gys1 alleles. The effect of this editing was a 50% reduction in GYS1 protein, decreased glycogen accumulation, and decreased neuroinflammatory markers (47). This approach addresses the underlying cause of disease using a gene editing strategy, but alternative approaches such as a simpler gene delivery system without CRISPR/Cas9 may have a better safety profile and greater clinical potential.
SLC13A5 Deficiency Disorder
Pathogenic variants in the gene SLC13A5 impair the sodium/citrate cotransporter, NaCT, with subsequent elevation in plasma and CSF citrate levels (62). These variants result in an autosomal recessive epileptic encephalopathy known as SLC13A5 deficiency disorder as SDD. SLC13A5 pathogenic variants were first identified in 2014 when whole-exome sequencing was performed in 3 individuals with similar clinical presentation of epileptic encephalopathy from 2 families (7). Whole-exome sequencing is one approach now used to detect SDD (63). Additionally, SLC13A5 is included in some commercially available epilepsy panels.
Presentation and Progression
Beginning within the first week of life most patients present with seizures and later often have status epilepticus (7, 64). However, there is phenotypic variability, and some patients have onset of seizures later in infancy. Patients with SDD may progress to lifelong drug-resistant epilepsy, with most seizures being convulsive (65). Seizure severity may decrease with age and some patients may even reach seizure freedom (40, 65). Broad-spectrum antiseizure medications often reduce seizure frequency, but targeted treatments are lacking and further innovation is needed.
Affected individuals show global developmental delay with intellectual disability and poor speech and communication (23). Patients often develop significant motor impairments and deficits in cognitive and expressive language (65). Patients typically have persistent neurological symptoms including ataxia, abnormal muscle tone, and abnormal involuntary movements (65). Additionally, patients with SDD may later develop dental enamel hypoplasia (65). It is possible for patients to live well into adulthood (65).
To date, there have been no published natural history studies for SDD. However, one natural history study of SDD is underway (NCT04681781), suggesting more studies may be needed to describe the disease state and inform clinical investigation more fully.
Gene Therapy Development
An Slc13a5 KO model has been utilized to investigate SLC13A5 disease pathology. It has demonstrated myoclonic and nonconvulsive focal seizures as seen in patients, but with no obvious behavior or pathological abnormalities (66). Recently, a self-complementary AAV9 vector carrying a SLC13A5 gene was developed (37). Preliminary data showed that delivery of this gene therapy to cerebrospinal fluid in young adult Slc13a5 KO mice resulted in rescue of epileptic activity. Additionally, treated KO mice had lower plasma citrate levels compared with KO mice that did not receive GRT (37). This approach addresses the underlying cause of the disease, and the clinical potential is under investigation.
SLC6A1-Related Disorder
SLC6A1 pathogenic variants were first identified in 2015 when 2 truncations and 4 missense pathogenic variants were found in patients with epileptic encephalopathies with myoclonic-atonic seizures (67). SLC6A1 is included in some commercially available epilepsy panels. SLC6A1 pathogenic variants cause a haploinsufficiency of sodium- and chloride-dependent gamma-aminobutyric acid transporter type-1 (GAT-1), resulting in SRD (65).
Presentation and Progression
The mean age of seizure onset is ~2.5 years of age in patients with SRD (9). Sixty percent of patients had developmental delay before seizure onset (9). The most prevalent epilepsy syndromes associated with SRD are myoclonic-atonic seizures (24%), genetic generalized epilepsy (23%), and non-acquired focal epilepsy (10%) (9). Further, it was found that absence seizures were the most common type of seizures in SRD (9). Common clinical features are epilepsy, developmental delay or cognitive impairment, and autistic traits (9). In addition patients may develop hypotonia, language disorder, and sleep issues.
Most patients require a care team consisting of neurologists, developmental pediatricians, genetic counselors, and speech and occupational therapists (9). Due to limited clinical data for SRD, treatment is determined based on the presenting clinical epilepsy syndrome and typically includes broad-spectrum antiseizure medications (9).
To date, there have been few natural history studies for SRD (52, 67, 68), indicating more studies are needed to describe the disease state and inform clinical investigation more fully.
Gene Therapy Development
Slc6a1 KO mice have been used to model seizure activity (51). These mice partially recapitulate human SRD as they have tremors, abnormal gait, reduced strength, absence seizures, anxious behavior, and cognitive impairment (9). SLC6A1 is a potential candidate for gene therapy because it results from pathogenic variants that cause haploinsufficiency, thereby allowing for gene replacement or transcriptional enhancement strategies to potentially alleviate the burden of disease. However, no gene therapy studies have been published on SRD.
Opportunity for Gene Therapy in Monogenic Epilepsies
GRT for CNS disorders has led to promising preliminary safety and efficacy data in clinical trials (31). gSRT has shown promising results preclinically, but additional work is needed in the clinic (16). Single-injection approaches of viral vectors may lead to a safe and effective strategy in the clinic (31). Importantly, these strategies address the underlying cause of disease and have the potential to stabilize the progression of the disease. However, there is still a need for preclinical proof-of-concept research for gene therapy applications for monogenic epilepsies in animal models. Important endpoints to track patient progress and measure success for gene therapy for genetic epilepsies are survival, seizure susceptibility, the number of recurrent seizures, biomarkers such as citrate levels in SDD, and adverse events (37). The development and application of appropriate outcome measures is vital to lead to the next generation of medicines for persons with monogenic epilepsies.
In contrast to targeting the gene underlying the monogenetic epilepsy, an alternative approach may be used, such as gene therapy delivering an AAV vector for an engineered voltage-gated potassium channel to drive down neuronal excitability and thereby reduce seizure (69). Another approach is to virally overexpress neuropeptide Y, which has been shown to suppress seizures in animal models (70). These approaches are not precision medicine addressing the underlying cause of disease, and their clinical applicability must be tested.
Remaining Challenges in The Clinical Development Path Forward For Gene Therapies
Seizure
By addressing the underlying cause of disease, gene therapy has the potential to impact disease course more than treating seizures alone. Seizure reduction will remain an important clinical goal for patients with epilepsy, yet clinicians rely upon patient and caregiver reports of seizure activity, which are known to have limited reliability (71). Furthermore, nocturnal seizure frequency is inherently difficult to capture through self- or parent-reporting. Reporting and monitoring of seizure activity is therefore often inadequate. Seizures themselves may not be the best target for genetic epilepsies, as they can vary in frequency and severity depending in part on the patient subpopulation. In some genetic epilepsies such as SDD, there may be a reduction in seizures, but continued morbidity due to developmental disabilities, including impairments in motor and cognitive abilities (65). Cognitive dysfunction may result from the underlying disease process itself, which gene therapies may address (72).
Developmental Concerns
In monogenic epilepsies, patients with DEE may miss or have delayed developmental milestones (7) that can negatively impact quality of life and capacity for achieving independent living. These motor and cognitive delays may affect functioning (7) and merit a means of systematic measurement and ongoing monitoring to inform the evaluation of treatment response. Early initiation of gene therapy for genetic epilepsies may mitigate or prevent the development of motor and cognitive manifestations of the diseases. For example, there is a growing body of evidence that patients with a degenerative motor neuron disease, spinal muscular atrophy, treated pre-symptomatically with GRT achieve improved motor outcomes compared to patients treated later in the disease course presumably by preventing or slowing neuronal loss (73).
Motor dysfunction such as hypotonia, stereotypies, and ataxia impair mobility and purposeful use of movement (7, 9). Motor impairment and global developmental delay may be apparent in infancy, such as in EIEE, or may manifest with severe, progressive deterioration following normal development, as experienced by children with Lafora disease (62, 74). It is therefore important to expand our understanding of the spectrum of motor impairments affecting patients with monogenic epilepsy and establish endpoints related to motor ability. Such endpoints would indicate clinical meaningful changes and be applicable across multiple monogenic epilepsy syndromes with early childhood onset.
Cognitive dysfunction, which can result from both recurrent seizure activity and the underlying disease process itself (72), has substantial impact on patient quality of life. It requires that clinicians consider metrics for improving not only seizure frequency and severity but also cognitive function. To this end, more research is needed to understand progressive cognitive decline in epilepsy, especially as the disease course in some genetic epilepsies shows a reduction in seizures, but a continued progression of cognitive decline.
Autism spectrum disorder may also accompany intellectual disability in patients with genetic epilepsies such as Dravet syndrome, and has a substantial impact on a patient's potential to achieve independence (75). There is a need for clearer neurodevelopmental/neurophysiological endpoints to track a patient's developmental abilities both accurately and efficiently over time. It will be important to identify endpoints that can characterize developmental trajectories associated with specific conditions. Such endpoints could subsequently provide an early indication of treatment response when patients' trajectories shift following intervention.
Discussion
Advances in genetic technologies have identified a growing number of monogenic genetic epilepsies potentially amenable to gene therapies. The state of AAV-based gene therapy has advanced a great deal with extensive study of AAV9 in preclinical models and in the clinic. Loss-of-function pathogenic variants may be highly amenable to gene therapy, namely by GRT and gSRT, which address the underlying cause of disease without the need for gene editing. However, there is still need for translational research to advance new therapeutics to the clinic. Understanding of disease progression through natural history studies may be an important precursor to interventional studies as meaningful clinical endpoints are highly dependent upon the severity and rapidity of clinical decline. Preclinical animal models may also be important to inform optimal timing of dosing relative to disease progression, as rapidly lethal diseases like Lafora disease may have a narrow therapeutic window. While SDD and SRD have different underlying pathology and less severe epilepsy outcomes than Lafora disease, early intervention may be critical in intervention strategies to improve cognitive, behavioral, and functional measures and the chance for good quality of life and greater independence from caregivers. Study design, clinical endpoints, dose selection, inclusion/exclusion criteria, and safety all need to be carefully considered in order to best serve patients.
Author Contributions
DB, SP, MH, and BM supported conceptualization of the paper, and reviewed and revised the manuscript. CS, JC, RB, and KG reviewed and revised the manuscript. All authors approved the final draft for submission.
Funding
This work was funded by the National Institutes of Health under award P01NS097197 and Taysha Gene Therapies. BM holds the University of Texas Southwestern Jimmy Elizabeth Westcott Chair in Pediatric Neurology and is Chief Medical Advisor at Taysha Gene Therapies. Medical writing and editorial support were provided by Kelly A. Hamilton, PhD, of AlphaScientia, LLC, and funded by Taysha Gene Therapies.
Conflict of Interest
DB is a consultant for Encoded Therapeutics, BioMarin Pharmaceuticals, and Synlogic Therapeutics. KG has provided consultation to Jaguar Gene Therapies. RB is an inventor on patents that have been licensed to various biopharmaceutical companies and for which she may receive payments. The authors declare that this study received funding from Taysha Gene Therapies. The funder had the following involvement in the study: MH, SP, CS, and JC are employees of Taysha Gene Therapies; KG and BM receive salary and research support from Taysha Gene Therapies; RB has sponsored research agreements with Taysha Gene Therapies; and DB is a member of the scientific advisory board for Taysha Gene Therapies. Each author was involved in the review, revision, and approval of the manuscript. UT Southwestern holds equity in Taysha Gene Therapies, which is a licensee of UTSW technology.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Myers CT, Mefford HC. Advancing epilepsy genetics in the genomic era. Genome Med. (2015) 7:91. doi: 10.1186/s13073-015-0214-7
2. Li M, Jancovski N, Jafar-Nejad P, Burbano LE, Rollo B, Richards K, et al. Antisense oligonucleotide therapy reduces seizures and extends life span in an SCN2A gain-of-function epilepsy model. J Clin Invest. (2021) 131:e152079. doi: 10.1172/JCI152079
3. Wagnon JL, Mencacci NE, Barker BS, Wengert ER, Bhatia KP, Balint B, et al. Partial loss-of-function of sodium channel SCN8A in familial isolated myoclonus. Hum Mutat. (2018) 39:965–9. doi: 10.1002/humu.23547
4. Hebbar M, Mefford HC. Recent advances in epilepsy genomics and genetic testing. F1000Res. (2020) 9:F1000. doi: 10.12688/f1000research.21366.1
5. Scala M, Zonneveld-Huijssoon E, Brienza M, Mecarelli O, van der Hout AH, Zambrelli E, et al. De novo ARHGEF9 missense variants associated with neurodevelopmental disorder in females: expanding the genotypic and phenotypic spectrum of ARHGEF9 disease in females. Neurogenetics. (2021) 22:87–94. doi: 10.1007/s10048-020-00622-5
6. Brewer MK, Putaux J-L, Rondon A, Uittenbogaard A, Sullivan MA, Gentry MS. Polyglucosan body structure in Lafora disease. Carbohyd Polym. (2020) 240:116260. doi: 10.1016/j.carbpol.2020.116260
7. Thevenon J, Milh M, Feillet F, St-Onge J, Duffourd Y, Jugé C, et al. Mutations in SLC13A5 cause autosomal-recessive epileptic encephalopathy with seizure onset in the first days of life. Am J Hum Genet. (2014) 95:113–20. doi: 10.1016/j.ajhg.2014.06.006
8. Mhanni AA, Hartley JN, Sanger WG, Chudley AE, Spriggs EL. Variable expressivity of a novel mutation in the SCN1A gene leading to an autosomal dominant seizure disorder. Seizure. (2011) 20:711–2. doi: 10.1016/j.seizure.2011.06.014
9. Goodspeed K, Pérez-Palma E, Iqbal S, Cooper D, Scimemi A, Johannesen KM, et al. Current knowledge of SLC6A1-related neurodevelopmental disorders. Brain Commun. (2020) 2:fcaa170. doi: 10.1093/braincomms/fcaa170
10. Chen Y, Wu L, Fang Y, He Z, Peng B, Shen Y, et al. A novel mutation of the nicotinic acetylcholine receptor gene CHRNA4 in sporadic nocturnal frontal lobe epilepsy. Epilepsy Res. (2009) 83:152–6. doi: 10.1016/j.eplepsyres.2008.10.009
11. Miceli F, Soldovieri MV, Ambrosino P, Barrese V, Migliore M, Cilio MR, et al. Genotype–phenotype correlations in neonatal epilepsies caused by mutations in the voltage sensor of Kv7.2 potassium channel subunits. Proc Natl Acad Sci USA. (2013) 110:4386–91. doi: 10.1073/pnas.1216867110
12. Striano P, Minassian BA. From genetic testing to precision medicine in epilepsy. Neurotherapeutics. (2020) 17:609–15. doi: 10.1007/s13311-020-00835-4
13. Bailey RM, Armao D, Nagabhushan Kalburgi S, Gray SJ. Development of intrathecal AAV9 gene therapy for giant axonal neuropathy. Mol Ther Methods Clin Dev. (2018) 9:160–71. doi: 10.1016/j.omtm.2018.02.005
14. Woodley E, Osmon KJL, Thompson P, Richmond C, Chen Z, Gray SJ, et al. Efficacy of a bicistronic vector for correction of Sandhoff disease in a mouse model. Mol Ther Methods Clin Dev. (2019) 12:47–57. doi: 10.1016/j.omtm.2018.10.011
15. Coutinho MF, Santos JI, Matos L, Alves S. Genetic substrate reduction therapy: a promising approach for lysosomal storage disorders. Diseases. (2016) 4:33. doi: 10.3390/diseases4040033
16. Dziedzic D, Wegrzyn G, Jakóbkiewicz-Banecka J. Impairment of glycosaminoglycan synthesis in mucopolysaccharidosis type IIIA cells by using siRNA: a potential therapeutic approach for Sanfilippo disease. Eur J Hum Genet. (2010) 18:200–5. doi: 10.1038/ejhg.2009.144
17. Belle A. ETX101, a GABAergic interneuron selective AAV-mediated gene therapy for the treatment of SCN1A+ dravet syndrome: biodistribution and safety in non-human primates. Am Epilepsy Soc Abstr. (2020) 391.
18. Xue-Ping W, Hai-Jiao W, Li-Na Z, Xu D, Ling L. Risk factors for drug-resistant epilepsy: A systematic review and meta-analysis. Medicine (Baltimore). (2019) 98:e16402. doi: 10.1097/MD.0000000000016402
19. de Lange IM, Gunning B, Sonsma ACM, van Gemert L, van Kempen M, Verbeek NE, et al. Influence of contraindicated medication use on cognitive outcome in Dravet syndrome and age at first afebrile seizure as a clinical predictor in SCN1A-related seizure phenotypes. Epilepsia. (2018) 59:1154–65. doi: 10.1111/epi.14191
21. González HFJ, Yengo-Kahn A, Englot DJ. Vagus nerve stimulation for the treatment of epilepsy. Neurosurg Clin N Am. (2019) 30:219–30. doi: 10.1016/j.nec.2018.12.005
22. D'Andrea Meira I, Romão TT, Pires do Prado HJ, Krüger LT, Pires MEP, da Conceição PO. Ketogenic diet and epilepsy: What we know so far. Front Neurosci. (2019) 13:5. doi: 10.3389/fnins.2019.00005
23. Klotz J, Porter BE, Colas C, Schlessinger A, Pajor AM. Mutations in the Na(+)/citrate cotransporter NaCT (SLC13A5) in pediatric patients with epilepsy and developmental delay. Mol Med. (2016) 22:310–21. doi: 10.2119/molmed.2016.00077
24. Boluk C, Ozkara C, Isler C, Uzan M. Vagus nerve stimulation in intractable epilepsy. Turk Neurosurg. (2022) 32:97–102. doi: 10.5137/1019-5149.JTN.33775-21.2
25. Lambrechts DA, de Kinderen RJ, Vles JS, de Louw AJ, Aldenkamp AP, Majoie HJ, et al. randomized controlled trial of the ketogenic diet in refractory childhood epilepsy. Acta Neurol Scand. (2017) 135:231–9. doi: 10.1111/ane.12737
26. Helbig I, Heinzen EL, Mefford HC. ILAE Genetics Commission. Primer Part 1-The building blocks of epilepsy genetics. Epilepsia. (2016) 57:861–8. doi: 10.1111/epi.13381
27. Onodera M, Ariga T, Kawamura N, Kobayashi I, Ohtsu M, Yamada M, et al. Successful peripheral T-lymphocyte-directed gene transfer for a patient with severe combined immune deficiency caused by adenosine deaminase deficiency. Blood. (1998) 91:30–6. doi: 10.1182/blood.V91.1.30
28. FDA Approves Innovative Gene Therapy to Treat Pediatric Patients with Spinal Muscular Atrophy, A Rare Disease and Leading Genetic Cause of Infant Mortality. U.S. Food & Drug Administration (2019). Available online at: https://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease
29. Duran J, Gruart A, García-Rocha M, Delgado-García JM, Guinovart JJ. Glycogen accumulation underlies neurodegeneration and autophagy impairment in Lafora disease. Hum Mol Genet. (2014) 23:3147–56. doi: 10.1093/hmg/ddu024
30. Mendell JR, Al-Zaidy SA, Rodino-Klapac LR, Goodspeed K, Gray SJ, Kay CN, et al. Current clinical applications of in vivo gene therapy with AAVs. Mol Ther. (2021) 29:464–88. doi: 10.1016/j.ymthe.2020.12.007
31. Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. (2017) 377:1713–22. doi: 10.1056/NEJMoa1706198
32. Lykken EA, Shyng C, Edwards RJ, Rozenberg A, Gray SJ. Recent progress and considerations for AAV gene therapies targeting the central nervous system. J Neurodev Disord. (2018) 10:16. doi: 10.1186/s11689-018-9234-0
33. Human Gene Therapy for Neurodegenerative Diseases. U.S. Food & Drug Administration (2021). Available online at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/human-gene-therapy-neurodegenerative-diseases
34. Gataullina S, Dulac O. From genotype to phenotype in Dravet disease. Seizure. (2017) 44:58–64. doi: 10.1016/j.seizure.2016.10.014
35. Hawkins NA, Calhoun JD, Huffman AM, Kearney JA. Gene expression profiling in a mouse model of Dravet syndrome. Exp Neurol. (2019) 311:247–56. doi: 10.1016/j.expneurol.2018.10.010
36. Genton P, Velizarova R, Dravet C. Dravet syndrome: the long-term outcome. Epilepsia. (2011) 52:44–9. doi: 10.1111/j.1528-1167.2011.03001.x
37. Bailey R, Bailey L, Schackmuth M, Garza I. scAAV9 gene replacement therapy for epileptic SLC13A5 deficiency. Mol Ther. (2021) 29:1–427.
38. Inoue K, Zhuang L, Maddox DM, Smith SB, Ganapathy V. Structure, function, and expression pattern of a novel sodium-coupled citrate transporter (NaCT) cloned from mammalian brain. J Biol Chem. (2002) 277:39469–76. doi: 10.1074/jbc.M207072200
39. Sauer DB, Song J, Wang B, Hilton JK, Karpowich NK, Mindell JA, et al. Structure and inhibition mechanism of the human citrate transporter NaCT. Nature. (2021) 591:157–61. doi: 10.1038/s41586-021-03230-x
40. Matricardi S, De Liso P, Freri E, Costa P, Castellotti B, Magri S, et al. Neonatal developmental and epileptic encephalopathy due to autosomal recessive variants in SLC13A5 gene. Epilepsia. (2020) 61:2474–85. doi: 10.1111/epi.16699
41. Birkenfeld AL, Lee HY, Guebre-Egziabher F, Alves TC, Jurczak MJ, Jornayvaz FR, et al. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. (2011) 14:184–95. doi: 10.1016/j.cmet.2011.06.009
42. Papadopoulos T, Soykan T. The role of collybistin in gephyrin clustering at inhibitory synapses: Facts and open questions. Front Cell Neurosci. (2011) 5:11. doi: 10.3389/fncel.2011.00011
43. Hussain T, Kil H, Hattiangady B, Lee J, Kodali M, Shuai B, et al. Wwox deletion leads to reduced GABA-ergic inhibitory interneuron numbers and activation of microglia and astrocytes in mouse hippocampus. Neurobiol Dis. (2019) 121:163–76. doi: 10.1016/j.nbd.2018.09.026
44. Aldaz CM, Hussain T. WWOX loss of function in neurodevelopmental and neurodegenerative disorders. Int J Mol Sci. (2020) 21:8922. doi: 10.3390/ijms21238922
45. Tona R, Chen W, Nakano Y, Reyes LD, Petralia RS, Wang Y-X, et al. The phenotypic landscape of a Tbc1d24 mutant mouse includes convulsive seizures resembling human early infantile epileptic encephalopathy. Hum Mol Genet. (2019) 28:1530–47. doi: 10.1093/hmg/ddy445
46. Balestrini S, Milh M, Castiglioni C, Luthy K, Finelli MJ, Verstreken P, et al. TBC1D24 genotype-phenotype correlation: epilepsies and other neurologic features. Neurology. (2016) 87:77–85. doi: 10.1212/WNL.0000000000002807
47. Gumusgoz E, Guisso DR, Kasiri S, Wu J, Dear M, Verhalen B, et al. Targeting gys1 with AAV-SaCas9 decreases pathogenic polyglucosan bodies and neuroinflammation in adult polyglucosan body and Lafora disease mouse models. Neurotherapeutics. (2021) 18:1414–25. doi: 10.1007/s13311-021-01040-7
48. Gentry MS, Guinovart JJ, Minassian BA, Roach PJ, Serratosa JM. Lafora disease offers a unique window into neuronal glycogen metabolism. J Biol Chem. (2018) 293:7117–25. doi: 10.1074/jbc.R117.803064
49. Al-Shekaili HH, Petkau TL, Pena I, Lengyell TC, Verhoeven-Duif NM, Ciapaite J, et al. A novel mouse model for pyridoxine-dependent epilepsy due to antiquitin deficiency. Hum Mol Genet. (2020) 29:3266–84. doi: 10.1093/hmg/ddaa202
50. Jiao X, Xue J, Gong P, Wu Y, Zhang Y, Jiang Y, et al. Clinical and genetic features in pyridoxine-dependent epilepsy: a Chinese cohort study. Dev Med Child Neurol. (2020) 62:315–21. doi: 10.1111/dmcn.14385
51. Cope DW, Di Giovanni G, Fyson SJ, Orbán G, Errington AC, Lorincz ML, et al. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med. (2009) 15:1392–8. doi: 10.1038/nm.2058
52. Kahen A, Kavus H, Geltzeiler A, Kentros C, Taylor C, Brooks E, et al. Neurodevelopmental phenotypes associated with pathogenic variants in SLC6A1. J Med Genet. (2021) jmedgenet-2021-107694. doi: 10.1136/jmedgenet-2021-107694
53. Minassian BA, Lee JR, Herbrick JA, Huizenga J, Soder S, Mungall AJ, et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet. (1998) 20:171–4. doi: 10.1038/2470
54. Chan EM, Young EJ, Ianzano L, Munteanu I, Zhao X, Christopoulos CC, et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet. (2003) 35:125–7. doi: 10.1038/ng1238
55. Lafora GR, Glueck B. Beitrag zur Histopathologie der myoklonischen Epilepsie: Bearbeitung des klinischen Teiles. Zeitschrift für die gesamte Neurologie und Psychiatrie. (1911) 6:1–14. doi: 10.1007/BF02863929
56. Nitschke F, Ahonen SJ, Nitschke S, Mitra S, Minassian BA. Lafora disease - from pathogenesis to treatment strategies. Nat Rev Neurol. (2018) 14:606–17. doi: 10.1038/s41582-018-0057-0
57. Pondrelli F, Muccioli L, Licchetta L, Mostacci B, Zenesini C, Tinuper P, et al. Natural history of Lafora disease: a prognostic systematic review and individual participant data meta-analysis. Orphanet J Rare Dis. (2021) 16:362. doi: 10.1186/s13023-021-01989-w
58. Brewer MK, Machio-Castello M, Viana R, Wayne JL, Kuchtova A, Simmons ZR, et al. An empirical pipeline for personalized diagnosis of Lafora disease mutations. iScience. (2021) 24:103276. doi: 10.1016/j.isci.2021.103276
59. Orsini A, Valetto A, Bertini V, Esposito M, Carli N, Minassian BA, et al. The best evidence for progressive myoclonic epilepsy: a pathway to precision therapy. Seizure. (2019) 71:247–57. doi: 10.1016/j.seizure.2019.08.012
60. Ganesh S, Delgado-Escueta AV, Sakamoto T, Avila MR, Machado-Salas J, Hoshii Y, et al. Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum Mol Genet. (2002) 11:1251–62. doi: 10.1093/hmg/11.11.1251
61. Turnbull J, Wang P, Girard JM, Ruggieri A, Wang TJ, Draginov AG, et al. Glycogen hyperphosphorylation underlies lafora body formation. Ann Neurol. (2010) 68:925–33. doi: 10.1002/ana.22156
62. Bainbridge MN, Cooney E, Miller M, Kennedy AD, Wulff JE, Donti T, et al. Analyses of SLC13A5 -epilepsy patients reveal perturbations of TCA cycle. Mol Genet Metab. (2017) 121:314–9. doi: 10.1016/j.ymgme.2017.06.009
63. Epi25 Collaborative. Ultra-rare genetic variation in the epilepsies: a whole-exome sequencing study of 17,606 individuals. Am J Hum Genet. (2019) 105:267–82. doi: 10.1016/j.ajhg.2019.05.020
64. Yang Q-Z, Spelbrink EM, Nye KL, Hsu ER, Porter BE. Epilepsy and EEG phenotype of SLC13A5 citrate transporter disorder. Child Neurol Open. (2020) 7:2329048X20931361. doi: 10.1177/2329048X20931361
65. Ozlu C, Bailey RM, Sinnett S, Goodspeed KD. Gene transfer therapy for neurodevelopmental disorders. Dev Neurosci. (2021) 43:230–40. doi: 10.1159/000515434
66. Henke C, Töllner K, van Dijk RM, Miljanovic N, Cordes T, Twele F, et al. Disruption of the sodium-dependent citrate transporter SLC13A5 in mice causes alterations in brain citrate levels and neuronal network excitability in the hippocampus. Neurobiol Dis. (2020) 143:105018. doi: 10.1016/j.nbd.2020.105018
67. Carvill GL, McMahon JM, Schneider A, Zemel M, Myers CT, Saykally J, et al. Mutations in the GABA transporter SLC6A1 cause epilepsy with myoclonic-atonic seizures. Am J Hum Genet. (2015) 96:808–15. doi: 10.1016/j.ajhg.2015.02.016
68. Johannesen KM, Gardella E, Linnankivi T, Courage C, de Saint Martin A, Lehesjoki AE, et al. Defining the phenotypic spectrum of SLC6A1 mutations. Epilepsia. (2018) 59:389–402. doi: 10.1111/epi.13986
69. Snowball A, Chabrol E, Wykes RC, Shekh-Ahmad T, Cornford JH, Lieb A, et al. Epilepsy gene therapy using an engineered potassium channel. J Neurosci. (2019) 39:3159–69. doi: 10.1523/JNEUROSCI.1143-18.2019
70. Richichi C, Lin E-JD, Stefanin D, Colella D, Ravizza T, Grignaschi G, et al. Anticonvulsant and antiepileptogenic effects mediated by adeno-associated virus vector neuropeptide Y expression in the rat hippocampus. J Neurosci. (2004) 24:3051–9. doi: 10.1523/JNEUROSCI.4056-03.2004
71. Brinkmann BH, Karoly PJ, Nurse ES, Dumanis SB, Nasseri M, Viana PF, et al. Seizure diaries and forecasting with wearables: epilepsy monitoring outside the clinic. Front Neurol. (2021) 12:690404. doi: 10.3389/fneur.2021.690404
72. Holmes GL. Cognitive impairment in epilepsy: the role of network abnormalities. Epileptic Disord. (2015) 17:101–16. doi: 10.1684/epd.2015.0739
73. Dangouloff T, Servais L. Clinical evidence supporting early treatment of patients with spinal muscular atrophy: current perspectives. Ther Clin Risk Manag. (2019) 15:1153–61. doi: 10.2147/TCRM.S172291
74. Dirani M, Nasreddine W, Abdulla F, Beydoun A. Seizure control and improvement of neurological dysfunction in Lafora disease with perampanel. Epilepsy Behav Case Rep. (2014) 2:164–6. doi: 10.1016/j.ebcr.2014.09.003
Keywords: genetic epilepsy, AAV9, Lafora, SLC13A5, SLC6A1, gene therapy (GT)
Citation: Goodspeed K, Bailey RM, Prasad S, Sadhu C, Cardenas JA, Holmay M, Bilder DA and Minassian BA (2022) Gene Therapy: Novel Approaches to Targeting Monogenic Epilepsies. Front. Neurol. 13:805007. doi: 10.3389/fneur.2022.805007
Received: 29 October 2021; Accepted: 20 April 2022;
Published: 21 June 2022.
Edited by:
Mario Mastrangelo, Umberto 1 Polyclinic, ItalyReviewed by:
Marina Trivisano, Bambino Gesù Children's Hospital (IRCCS), ItalyNicola Specchio, Bambino Gesù Children's Hospital (IRCCS), Italy
Copyright © 2022 Goodspeed, Bailey, Prasad, Sadhu, Cardenas, Holmay, Bilder and Minassian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Berge A. Minassian, YmVyZ2UubWluYXNzaWFuQHV0c291dGh3ZXN0ZXJuLmVkdQ==