 Eveljn Scarian1,2†
Eveljn Scarian1,2† Giuseppe Fiamingo1†
Giuseppe Fiamingo1† Luca Diamanti3
Luca Diamanti3 Ilaria Palmieri4,5
Ilaria Palmieri4,5 Stella Gagliardi6*
Stella Gagliardi6* Orietta Pansarasa2
Orietta Pansarasa2- 1Department of Brain and Behavioral Sciences, University of Pavia, Pavia, Italy
- 2Cellular Models and Neuroepigenetics Unit, IRCCS Mondino Foundation, Pavia, Italy
- 3Neuroncology Unit, IRCCS Mondino Foundation, Pavia, Italy
- 4Department of Molecular Medicine, University of Pavia, Pavia, Italy
- 5Neurogenetics Research Center, IRCCS Mondino Foundation, Pavia, Italy
- 6Molecular Biology and Transcriptomics Unit, IRCCS Mondino Foundation, Pavia, Italy
Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD) are two neurological diseases which, respectively, and primarily affect motor neurons and frontotemporal lobes. Although they can lead to different signs and symptoms, it is now evident that these two pathologies form a continuum and that hallmarks of both diseases can be present within the same person in the so-called ALS-FTD spectrum. Many studies have focused on the genetic overlap of these pathologies and it is now clear that different genes, such as C9orf72, TARDBP, SQSTM1, FUS, and p97/VCP can be mutated in both the diseases. VCP was one of the first genes associated with both FTD and ALS representing an early example of gene overlapping. VCP belongs to the type II AAA (ATPases Associated with diverse cellular activities) family and is involved in ubiquitinated proteins degradation, autophagy, lysosomal clearance and mitochondrial quality control. Since its numerous roles, mutations in this gene lead to different pathological features, first and foremost TDP-43 mislocalization. This review aims to outline recent findings on VCP roles and on how its mutations are linked to the neuropathology of ALS and FTD.
Introduction
Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD) are two neurodegenerative diseases which affect motor neurons and frontotemporal lobes, respectively. Traditionally ALS was considered as limited to the motor neuron system; in the last decades a wide spectrum of possible cognitive and behavioral deficits, similar to the ones mostly seen in the behavioral variant of FTD (bvFTD), have been highlighted. Further evidence of the overlap between the syndromes has become evident through genetic, pathological, radiological and neuropsychological studies (1). It is now evident that the two diseases represent the opposite poles of a phenotypic continuum. The mixed phenotypes, constituted by a variable burden of motor and extra-motor deficits, are probably as frequent as the pure forms. The possible phenotypes related to ALS and FTD are still expanding due to the association of rarer genes already known to be causative of other systemic diseases (2, 3). One such example is Valosin Containing Protein (VCP), on which this review will be focused.
Clinical Aspects of ALS and FTD
ALS is a rare neurodegenerative disease primarily affecting the corticospinal tract and leading to progressive muscular paresis. Its peak age of onset is between 50 and 70 years and it is characterized by the simultaneous presence of symptoms and signs of both upper (UMN) and lower motor neuron (LMN). Common histopathological features are transactivation response DNA-binding protein 43 kDA (TDP-43)-positive cytoplasmic inclusions and neuronal degeneration with variable distribution and spreading (4). The diagnosis, according to El Escorial Criteria [first established in 1994 (5) and later revised (6, 7)] is clinical and supported by needle electromyography (EMG). As regards treatment, a few pharmacological options are available with only scant efficacy in slowing down the neurodegenerative process (8). An upcoming promising therapeutic approach is targeted-gene silencing through antisense oligonucleotide drugs intrathecally delivered (9).
FTD is the third leading cause of dementia and the second most common early-onset dementia after Alzheimer's Disease (AD). FTD encompasses different clinical and neuropathological subtypes according to consensus criteria (10–12). The most frequent FTD syndrome is bvFTD (nearly 70%), characterized by personality changes, dishinibition, apathy, hyperorality and executive dysfunction. Less represented language predominant variants are Semantic Dementia (SD) and Progressive Non-Fluent Agrammatic variant (PNFA) (13, 14). A third primary progressive aphasia (PPA) syndrome, the logopenic variant (lvPPA), is mainly attributed to AD pathology and the beta-amyloidogenic pathway (15). In bvFTD, the neurodegenerative process is predominantly focused on the prefrontal cortex, while temporal lobes are involved later on. On the contrary, PPA neurodegeneration seems to start primarily in the temporal lobes, with asymmetrical pattern (16, 17). Nearly all cases of frontotemporal lobar degeneration (FTLD) are driven by microtubule-associated protein tau (MAPT), TDP-43 or the fused-in-sarcoma (FUS) protein accumulation (18). FTD lacks disease-modifying therapies; behavioral symptoms can benefit from antidepressant and antipsychotic agents (19).
Phenotypic Variability of ALS and ALS-FTD Overlap
The clinicopathological variability of ALS is remarkable. The classical depiction of UMN and LMN concomitant involvement (Charcot's disease) is seldom present at the onset of disease; this motor variability reflects the site of onset of the neurodegenerative process, its rate of progression and subsequent spreading to different neuroanatomic areas. This heterogeneity actually represents a continuum that makes patients classification troublesome. However, some phenotypes are widely accepted because of reproducible clear-cut features (20). The predominance of LMN system is called progressive muscular atrophy (PMA) (21), while the predominance of UMN system configures primary lateral sclerosis (PLS) (22). According to the predominant muscle territory involved or the site of onset, ALS can manifest a bulbar or a spinal onset. A respiratory phenotype, with diaphragmatic failure since the onset of the disease, is rarely seen (23). ALS phenotypic differentiation is more easily accounted at disease onset. Later on, motor neuron degeneration spreads in adjacent segments or along the corticospinal tract (24). Population-based studies have highlighted different epidemiological distributions in the above-mentioned phenotypes, according to sex, age, prognostic value, and cognitive profile (20, 25).
Concerning neuropsychological (NPS) impairment, first reports of ALS patients with frontal lobe deficits or dementia date back to the nineties (26, 27). Already then, the cognitive and behavioral profile in ALS population was described as similar to the ones typical for bvFTD. Further evidence of ALS-FTD overlap came from the observation of motor deficits and/or signs in FTD patient (28) and the identification of common TDP-43 neuropathologic inclusions in both syndromes (29). The discovery of a shared FTD and ALS causative gene mutation, C9orf72 hexanucleotide GGGGCC (G4C2) repeat expansion, definitely confirmed previous hypotheses of a common pathogenic pathway (30). Overall, ALS is most typically associated with bvFTD, and the ALS-FTD overlap syndrome shows the strongest association with TDP-associated FTLD (FTLD-TDP) (18).
The ever-growing evidence of an ALS-FTD disease-spectrum led to operational consensus criteria aimed at classifying ALS according to motor features as well as extra-motor NPS features (31, 32). Through standardized NPS assessment, different shades of cognitive and behavioral impairment are categorized as follow: ALS-behavioral impairment (ALSbi); ALS-cognitive impairment (ALSci), with typical either executive and/or language dysfunction; ALS-cognitive and behavioral impairment (ALScbi), when meeting both criteria for ALSci and ALSbi (33); ALS-FTD, when ALS patients also show the presence of bvFTD or PPA, according to the respective diagnostic criteria (10, 12). Generally speaking, cognitive and/or behavioral impairment occurs in up to 50% of ALS patients, while nearly 15% have a full-blown bvFTD (34–36). Taking into account dementia-first patients, almost 15% of bvFTD patients develop ALS during the course of the disease, while signs of motor neuron impairment are observed in about 40% of cases (37, 38).
Both cognitive and behavioral impairment, with special consideration to apathy, negatively correlate with survival (39). A recent study shows that ALS-FTD patients' survival is significantly shorter than pure bvFTD patients'; in addition, among ALS-FTD patients, motor-onset patients' survival is much shorter than cognitive-onset ones, meaning that disease progression is accelerated when motor deficit come first (40). To better elucidate the reciprocal interplay between ALS and FTD, genetics has progressively shed some light.
Genetics of ALS, FTD, ALS + FTD
From the discovery of the first missense mutations in superoxide dismutase 1 (SOD1) in 1993 (41) to our date, over 50 genetic mutations have been linked to ALS (3), thanks to genome-wide association studies and “next-generation” sequencing techniques (42). Yet, these advancements have allowed to better explain only a minor fraction of cases so far. About 10% of ALS patients are considered familiar (fALS), as they have at least one other affected family member. However, one-third of fALS cases remain genetically unexplained (43). Considering the remaining 90–95% sporadic ALS (sALS), just about 10% carry a mutated causative gene, accordingly to current knowledge. This missing heritability in ALS may be due to technical issues as well as the inherent complexity of the disease (3).
Four genes, SOD1, C9orf72, FUS, and TARDBP, account for the majority of both fALS and sALS cases (44) (Table 1).
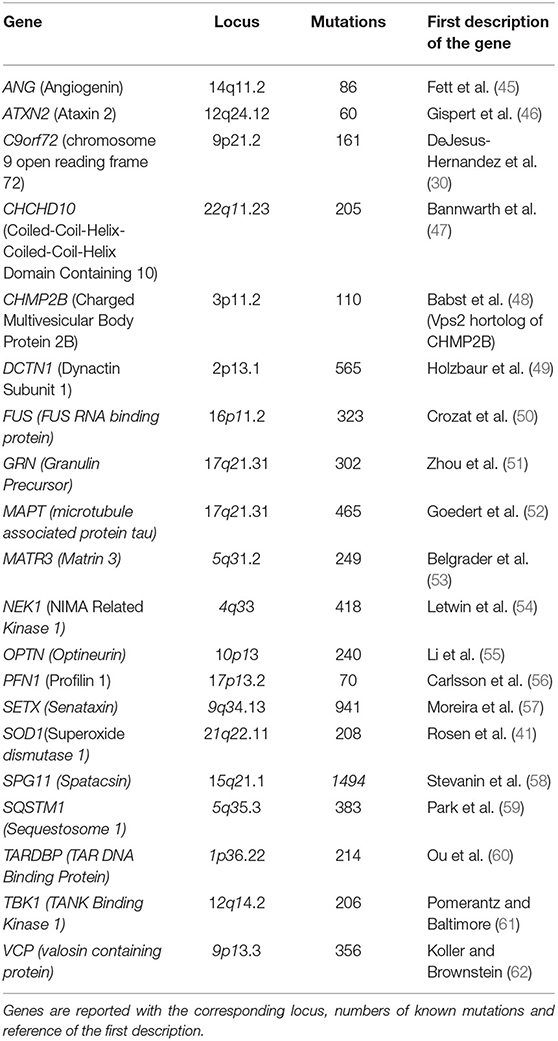
Table 1. List of genes involved in ALS and FTD pathologies.
SOD1 encodes a superoxide dismutase enzyme which resides in the cytosol and the intermembrane space of mitochondria. This enzyme catalyzes the production of oxygen and hydrogen peroxide from the superoxide species produced during cellular respiration, providing an antioxidant mechanism (63). SOD1 mutations are considered responsible for 15–30% of fALS and fewer than 2% of sALS cases (64). To date, 217 disease-associated variations in SOD1 have been identified,1 they are usually missense and lead to a decrease in enzyme activity and related oxidative stress and mitochondrial dysfunction (65). The pathology of SOD1 ALS seems distinct from that of all other types of ALS, in that it lacks the TDP-43 and/or FUS pathology hallmarks (66).
The first genetic link between ALS and FTD was the discovery of mutations in TARDBP, which encodes TDP-43 (67). TDP-43 is a DNA/RNA binding protein which shuttles back and forth between the nucleus and cytoplasm (68) and regulates gene expression and RNA processing (69).
To date, 69 variants in TARDBP have been linked to ALS,2 and they can variably lead to the loss or the overexpression of TDP-43, both causing disease. Indeed, TDP-43 homeostasis is critical for cell survival and it is tightly regulated: when excessive, it can form inclusion bodies in the cytoplasm; when depleted, it may result in mRNA metabolism dysregulation (70). Furtherly, a prion-like spreading mechanism has been shown for TDP-43 mutated products (71). Despite a central role in ALS pathogenesis, the frequency of the gene mutation remains much lower than the occurrence of TDP-43 neuropathologic neuronal inclusions (about 4% of fALS) (3).
Shortly after TARDBP, missense mutations of FUS were identified (72). FUS protein shares functional homology with TDP-43, playing a role in RNA metabolism and nucleocytoplasmatic transport (73). FUS is also involved in DNA repair (74) and paraspeckles formation against stressful noxa (75). One hundred twenty-six autosomal dominant FUS mutations are known to cause FUS cytoplasmatic aggregation,3 which occurs almost exclusively in association with FUS pathogenic variants (76, 77), while TDP-43 aggregation is seldom present in FUS-ALS patients.
As already mentioned, the major contribution in ALS-FTD genetic linkage has been the discovery of the hexanucleotide repeat expansion (GGGGCC) in the non-coding region of the C9orf72 gene (30). C9orf72 mutation is the most common inherited cause of fALS (approximately 34%) worldwide and in the European population (64). The exact C9orf72 function is still poorly understood, as is its broad phenotypic expression. Endosomal trafficking, autophagy (78) and immune dysregulation (79) are proposed functions. C9orf72 expansion, seems to confer a loss of function (80). Toxic gain of function is also theorized, since the expanded transcript forms stable secondary structures unable to effectively interact with other proteins, leading ultimately to impair RNA processing (81).
Next-generation sequencing has allowed the identification of numerous rarer genetic variants in many other genes related to ALS, whose function has been linked to RNA processing, protein homeostasis, cytoskeletal dynamic, mitochondrial function, or still unknown (Table 1). Although rare, their discovery is contributing to better understanding ALS pathogenic mechanisms (3, 82).
As regards FTD genetics, familiar hereditability and mutation rates are high in comparison to ALS, with great variability across the clinical phenotypes. Almost 50% of bvFTD patients have a strong family history; conversely, PPA occurs in a familiar fashion only in 12% of people (83). C9orf72, as for ALS, is the commonest cause of genetic FTD worldwide (about 25%), followed by GRN (10–20%) and MAPT (10–20%) (2). These three genes cover the vast majority of familiar FTD cases and are inherited in an autosomal dominant fashion (Table 1).
MAPT is the first gene associated with FTD, whose main function is microtubule stabilization. Mutations in protein tau result in hyperphosphorylated tau deposits, which are toxic for cell homeostasis without amyloid pathology, as seen in AD. To date, over 50 pathogenic mutations are known (84). MAPT accounts for 10–20% of familiar FTD and 0–3% of sporadic FTD (85).
GRN is a glycoprotein mainly expressed in myeloid cells and a subset of neurons, especially cerebral cortical neurons, motor neurons, Purkinye and hippocampal cells (86). It plays an important role in inflammation modulation, tissue repair and neuronal survival (87). GRN mutations are heterozygous and result in 75% loss of GRN (88). They are responsible for 5–20% of familial and 1–5% of sporadic FTD cases. GRN-related FTD is characterized by TDP43 proteinopathy, although it is not clear how GRN mutation impairs TARDBP43 metabolism.
Multiple other FTD-linked genes are known, although cumulatively accounting for <5% of cases (2). Among such genes, VCP, CHMP2B, TARDBP, FUS, SQSTM1, ANG, CHCHD10, TBK1, OPTN, NEK1 have also been described in ALS patients (2), providing further evidence of a genetic overlap in ALS-FTD clinical continuum.
Genotype-Phenotype Correlations
SOD1
Over 185 SOD1 disease-associated variations have been identified. Some of them have been linked to faster disease progression (A4V, H43R, L84V, G85R N86S, and G93A), longer life expectancy (G93C, D90A, or H46R) (89) and specific genotype-phenotype correlations. A4V variant determines a relentless fast limb-onset disease with an average of 1 year survival (90). D90 variant acts differently in relation to is mutational state: if homozygous, the disease is generally slow, with survival up to 14 years, and ascending from inferior to superior limbs (91); if heterozygous, the disease is more severe, with bulbar or upper limb onset and faster progression (92). Overall, SOD1 typically confers more LMN than UMN involvement, and cognitive impairment is not generally reported in SOD1 disease.
TARDBP
TARDBP patients have an early onset with upper limb predominance and longer disease-duration compared to sALS, non-mutated fALS and SOD1+ ALS (93). Mutational state can strikingly affect survival, from 27 months with G298S to over than 100 in M337 carriers (93). TARDBP is seldom associated with cognitive impairment or full-blown FTD (94). When cognition is affected, language deficits and temporal atrophic changes on imaging are frequently seen (95).
FUS
FUS variants are associated with early onset and juvenile ALS (96), and FUS genotype correlates with faster disease progression in comparison with SOD1+ and TARDBP+ patients (97). Dormann et al. (98) demonstrated that point mutations (R521G, R522G, R524S, P525L) in the FUS C-terminal domain impair nuclear import and enhance FUS cytosolic accumulation at a varying degree according to the mutation. This finding is in accordance with the evidence of shorter survival for fALS patients with P525L and R522G mutations, which determine the highest degree of FUS cytosolic inclusions (72, 99). As for TARDBP, FUS does not express prominent FTD overlap (94); remarkably, FTD patients with FUS pathology never carry FUS or other known mutations, lack motor or language involvement and manifest a predominant obsessive-repetitive behavioral impairment (100).
C9orf72
The whole phenotypic ALS-FTD spectrum has been linked to C9orf72 expansions. ALS motor phenotypes can be all expressed in C9orf72 carriers, with a slight higher incidence of bulbar onset compared to other fALS (101). Among FTD variants, bvFTD with executive dysfunction is the most frequent, though language-predominant phenotypes meeting SD and PNFA are also possible. Median survival in C9orf72-carriers is shorter than non-carriers (102). In a French study the disease duration of C9orf72-related ALS was significantly shorter than in patients with mutations in SOD1, TARDBP or other familial ALS cases, while disease onset was significantly later in C9orf72-related ALS compared to SOD1 and FUS-ALS (101).
C9orf72 expansion carriers may have an atypical neuropsychiatric presentation with associated hallucinations or delusions and a greater risk of psychiatric disorders (103).
Finally, patients co-expressing ATXN2 intermediate repeats are more likely to have a pure form of ALS. The striking variability in the phenotype associated with the C9orf72 expansion suggests that multiple modifiers may exist, either genetic and/or environmental. The other two main FTD-associated genes, GRN and MAPT, are never associated with motor features suggesting an ALS-FTD overlap (104).
The fourth most frequent FTD-associated gene, TBK1, has been linked to prominent PPA features as well as isolated ALS and ALS-FTD (105).
Phenotypic Pleiotropy: The Case of VCP
Other genetic diseases, affecting organs and systems seemingly unrelated to the nervous system, have been linked to ALS-FTD continuum (3). This is the case of genes of minor frequency and involvement such as OPTN, SQSTM1, and VCP, which show remarkable phenotypic pleiotropy.
Mutations in VCP were already known to underlie an unusual clinical syndrome characterized by inclusion body myopathy, Paget's disease of the bone and FTD (IBMPFD), or multisystem proteinopathy (106). In IBMPFD, a myopathic pattern with progressive weakness and atrophy of proximal skeletal muscles is the most common feature (90%). Histologically, rimmed vacuoles and TDP-43 positive inclusion are seen in the involved muscles (107).
VCP-associated Paget's disease typically involves the spine, pelvis, scapulae and skull, leading to their structural disruption by bone remodeling blockage. VCP-related FTD is characterized by TDP-43 inclusions (108). The relative low frequency of FTD (nearly 30%) in IBMPFD is thought to be a consequence of the early age at death in most patients. When present, FTD reduces life span to an average of 6 years (109).
Mutations in VCP have also been associated with pure ALS (110), hereditary spastic paraplegia (111), Charcot-Marie-Tooth type 2 disease (112), and multiple dystrophic syndromes (113). Hence, VCP mutations result in pathology on both sides of the neuromuscular junction and show that extra-neurologic tissues, such as muscle and bone, may be affected. Most cases of IBMPFD and less typical VCP-related diseases are caused by heterozygous missense mutations (106).
Mehta et al. (114) studied genotypic-phenotypic relations in 27 families (190 subjects, 45 carriers and 145 symptomatic) by grouping them according to VCP variants: 91% of patients had myopathy, 51.7% showed Paget's disease and 30.3% had FTD. R155C mutation was associated with shorter survival, while intergroup analysis was limited by the small sample size of each group. ALS was reported in 13 subjects (8.9%) from 6 different families, 10 of them carrying R155H mutation; furthermore, 24% of the symptomatic subjects had neurogenic changes on the EMG (114). The coexistence of myopathic and neurogenic findings has been reported by other authors, as well (115). Similar results for phenotypic distribution were later confirmed by the same group of authors (116), who also reported autopsy data on one individual with VCP-related ALS showing typical Bunina bodies and TDP-43 immunostaining within nervous tissue. In their cohort, the classic IBMPFD triad of symptoms was manifest in only 10% of cases (116). A very small percentage of cases was diagnosed as Parkinson's disease or AD. VCP-related ALS and FTD are phenotypically indistinguishable from sporadic forms. The only remarkable feature of VCP-attributed FTD is a younger onset (114). Although rare, both VCP-related FTD and ALS without ALS-FTD overlap or features of the IBMPFD spectrum have been described (110, 117). No specific genetic mutational signatures have been highlighted and the molecular mechanisms which distinguish one syndrome to the other are not clear (114).
Due to its highly variable phenotype, VCP disease is underdiagnosed and, whether present, genotype-phenotype correlations are not seen because of small sample size even in large cohorts. As awareness increases the scientific community is realizing that VCP disease is not as rare as previously considered. Hence, the experts suggest VCP molecular testing when two or more features are present for a prompt diagnosis, the adoption of surveillance protocols and hopefully the administration of a therapeutic option in the near future (114). As VCP-related phenotype has been expanding, IBMPFD term is currently misused in favor of the most inclusive and recently coined multisystem proteinopathy (MSP) (118). MSP is not restricted to VCP mutations, but other genes with similar functions have been shown to cause this severe disease (119).
For these reasons, it is even more crucial to understand the importance of VCP gene and its roles in the maintenance of homeostasis.
The VCP Gene and Its Roles
VCP, also called p97, is a well-conserved protein among all eukaryotes (120, 121). It is encoded by the VCP gene, located on chromosome 9p13.3 in humans. VCP belongs to the type II AAA (ATPases Associated with diverse cellular activities) family and is composed of a N-terminal domain and a C-terminal domain, which interact with substrates and cofactors, and by two AAA ATPase domains, D1 and D2, forming a hexameric double-ring structures. D1 plays a role in hexamerization and regulates the heat-induced ATPase activity, whereas D2 acts better at physiological temperatures. Different studies showed that the N-terminal domain can assume two positions: above the plane of the ring (up position) (Figure 1A) or in the plane formed by the D1 ring (down position) (Figure 1B), with ATP or ADP in D1, respectively (124).
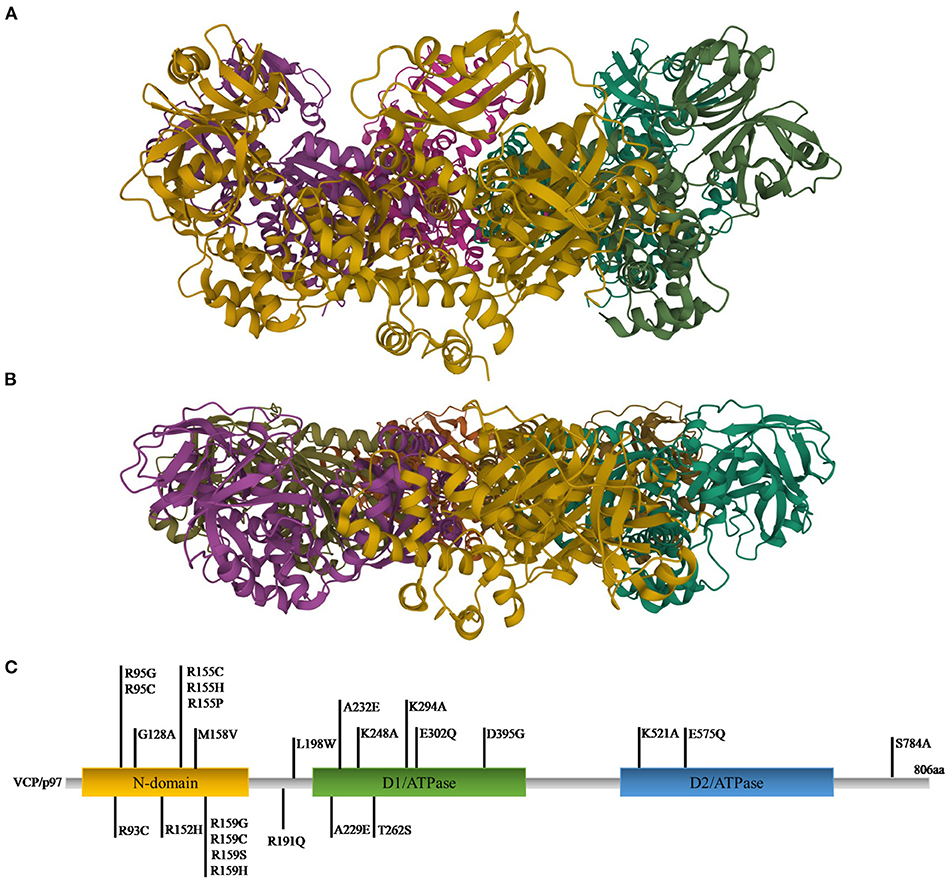
Figure 1. Ribbon diagram representations of X-ray structures of VCP in the up (A) and down (B) states [PDB ID code 4KO8 (122) for the up state and 1E32 (123) for the down state]. (C) Gene representation with the indication of the three main domains and the relative mutations (C-terminal domain was omitted).
VCP has multiple localizations: it is expressed in brain, skeletal muscles, ovary, testis, kidney, liver, heart, lung, lymph nodes, and whole blood, both in nucleus and cytoplasm (120, 121, 125). VCP uses the energy provided by the hydrolysis of ATP to change the conformations of target proteins, but it is also involved in different cellular processes, such as the reassembly of Golgi, endoplasmic reticulum (ER) and nuclear membranes, the ubiquitin-proteasome system, the regulation of cell cycle, the autophagosome maturation and the mitophagic process (121, 125) (Figure 2).
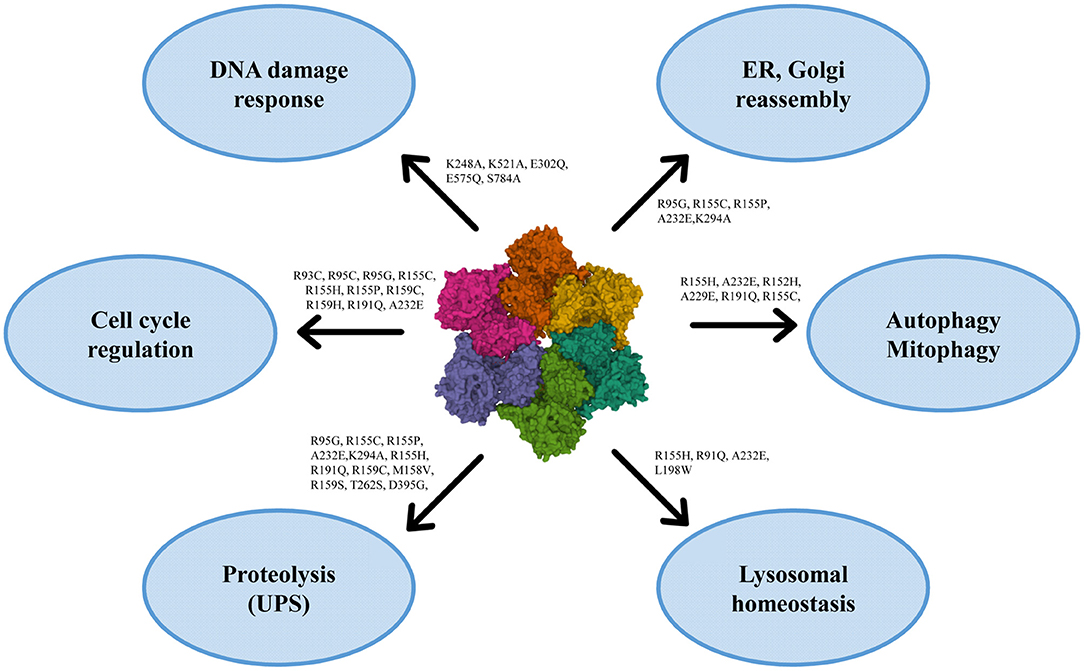
Figure 2. Roles of VCP in the cell with the principal mutations. VCP is involved in ER and Golgi reassembly, autophagy, mitophagy, lysosomal homeostasis, proteolysis, cell cycle regulation and DNA damage response [PDB ID code 3CF3 (126)].
To exert its roles, VCP interacts with many cofactors, most of which bind the N-terminal domain, whereas a small number interacts with the C-terminal domain. Among the cofactors the most important are Npl4 (nuclear protein localization homolog 4), Ufd1 (ubiquitin fusion degradation 1) heterodimers and an arrangement of 13 UBX (ubiquitin regulatory X) domain cofactors. Mutations in VCP are causes of many pathologies (127) (Figure 1C). The first six mutations (R95G, R155H, R155P, R155C, R191Q, and A232E) were found in 2004 to be causative of the IBMPFD. This disease causes muscle weakness, cardiac problems, bone deformity but also amnesic and other cognitive deficits associated with TDP-43 inclusions (106, 128). Many studies revealed that the mutations are located in those domains important for cofactor binding and for VCP conformational changes (125).
In addition, mutations in VCP were found in other neurodegenerative diseases, such as Huntington Disease, ALS and FTD (125). In 2010 Johnson et al. identified the R191Q mutation in an Italian family affected by familial ALS. Moreover, further investigation in a wide range of ALS patients discovered four additional mutations, including the R155H one (110). More recently, a study on a cohort of 231 individuals with 15 different VCP mutations demonstrated that FTD was present in 30% of the patients, whereas 9% had an ALS phenotype, 4% had PD and 2% AD (116). More than 30 VCP mutations are till now reported in ALS and FTD and it is now accepted that in FTD they cause the unique subtype FTLD-TDP (type D), characterized by numerous neuronal intranuclear inclusions and dystrophic neurites in the neocortex (129, 130).
Mutations lead inevitably to an improper regulation of the VCP gene in exert its roles, among us in protein clearance, autophagy, lysosomal homeostasis maintenance and mitochondrial quality control.
VCP and the Protein Clearance
One of the most important roles of VCP is its involvement in the clearance of proteins, especially in the ubiquitin-mediated protein degradation (125). In this process different cofactors are involved including the VCP–Ufd1–Npl4 complex which regulates different degradation processes, among which ERAD (endoplasmic reticulum-associated degradation), involved in the degradation of ER proteins and the maintaining of ER integrity. The VCP–Ufd1–Npl4 complex is also involved in the translocation of ubiquitinated ER proteins in the cytosol, where they are degraded by the proteasome, by the retro-traslocation (131, 132). With the same mechanism, the VCP–Vms1–Npl4 complex extracts the ubiquitinated substrates from mitochondria and transports them to the proteasome to be degraded (133, 134). Finally, VCP–p47 and VCP–UBXD1 complexes are involved in ubiquitinated protein degradation proteasome-independent; the two cofactors allow the transport of proteins to endosomes and the lysosomal degradation (125, 135). Moreover, it was demonstrated that Npl4 is required for normal microtubule and neuromuscular junction organization (136).
Mutations in VCP lead to a dysregulation of protein homeostasis with protein aggregation and accumulation, especially of TDP-43 and tau protein (134). Many studies demonstrated that down-regulation of VCP or mutations in this gene cause cytosolic TDP-43 aggregations followed by ER stress and cell death (137, 138). In 2010, Ritson et al. demonstrated that mutation in VCP causes TDP-43 redistribution in cytoplasm, both in vitro and in Drosophila melanogaster, gaining a cytotoxic function (139). Moreover, the knockdown of the cofactor Npl4 in vivo causes locomotor dysfunctions, reduced lifespan and inclusions of TDP-43 (136).
FTDL with ubiquitin-positive inclusions (FTDL-U) includes both sporadic and familial forms characterized by mutations in various genes, among which VCP. These inclusions are characterized by hyperphosphorylation, ubiquitination and fragmentation of the C-terminal (109, 140). Involvement of cortical and occipital lobes is more extensive in FTDL-U patients associated with VCP, and they were classified in the FTLD-TDP type 4, later renamed as type D (141). In 2018, a study on a cohort of FTD patients identified two different mutations, both characterized by the presence of diffuse TDP-43 immunoreactive neuronal intranuclear inclusions and dystrophic neurites and rare VCP inclusions (142), in contrast to a previous case report in which it was demonstrated the presence of VCP- and ubiquitin-positive cytoplasmic and nuclear aggregates in an Italian patients carrying the R159C mutation, and manifesting both body myopathy and FTD (143).
Inclusions of TDP-43 and its mislocalization in the cytoplasm was also observed in VCP-mutated iPSCs-derived MNs (144, 145) and in neuropathological examination of ALS patients (146, 147). TDP-43 was found accumulated also in cytoplasm of circulating lymphomonocytes as well in lymphocytes and monocytes separately evaluated in ALS patients (148).
A recent case study reported that the p.Asp395Gly mutation of VCP is associated in the occipital neocortex with aggregates immunoreactive with antibodies specific to phosphorylated tau and ubiquitin, characteristics similar to those of neurofibrillary tangles of AD (149). Moreover, the TDP-43 inclusions were often associated with FUS and splicing factor proline and glutamine rich (SFPQ) mislocalization, providing further evidence that VCP mutations cause an impairment of protein homeostasis (150). Often, the inclusions are associated with reactive microglia and astrocytosis and with cytokine imbalance (144, 149).
VCP and Autophagy
Directly linked with the clearance of proteins is the role of VCP in autophagy. Autophagy is an essential process for all the eukaryotes cells. This process has two different purposes, the supply of amino acids for the cells in poor environmental conditions, the so called “adaptive autophagy”, and the degradation of proteins and damaged organelles, the “basal or constitutive autophagy” (151, 152). Many studies focused on the role of VCP in both the autophagic pathways, finding an involvement in both the types of process, cooperating with many different cofactors (127, 138, 153–156). First evidence highlighted the role of VCP exclusively in the maturation of phagosomes. In 2009 Ju et al. demonstrated that loss of the activity of VCP leads to autophagy impairment, causing the impossibility for vacuoles to mature in autolysosomes and the accumulation of autophagosomes. (138). These data were confirmed in 2010 by a study that found that knockdown of VCP or the overexpression of dominant-negative VCP (VCPR155H and VCPA232E mutations, involved in ALS/IBMPFD pathogenesis) lead to an accumulation of immature autophagic vesicles, often containing ubiquitin-positive contents and with acidified autophagosomes, defects found also when disease-associated VCP mutations were expressed (154). VCP regulates autophagosomes maturation in many different ways, such as interacting with clathrin and regulating caveolin trafficking or governing the size and the trafficking of endosomes regulating the assembly of EEA1 oligomers (135, 157, 158). VCP has been associated with the removal of damaged lysosomes and of stress granules (156, 159, 160). However, a recent study, using specific VCP inhibitors, discovered the role of VCP also in the first step of autophagy pathway, namely autophagy initiation. VCP can stabilize Beclin-1 acting on ataxin-3, which is responsible for Beclin-1 deubiquitination, and can also regulate the activity of the Beclin-1-containing phosphatidylinositol-3-kinase (PI3K) complex I which is responsible for the recruitment of downstream autophagy factors (161). Finally, a study using a VCPR155H murine model, found a reduction in the mTOR targets EIF4EBP1 and RPS6KB1/p70S6 and as result an increasing autophagic activation and autophagosomes biogenesis (162).
Further studies focused on the VCP mutations typical of ALS and FTDL pathologies. In 2009 Ju et al. analyzed IBMPFD muscles and found degenerating fibers, rimmed vacuoles, which co-localized with autophagosomes-associated proteins such as Map1-LC3 (LC3) and p62/sequestosome, ubiquitin and TDP-43 inclusions. Moreover, they used U20S cells to silence VCP or to express an inactive VCP, confirming that it causes autophagosomes accumulation, which are unable to mature in autolysosomes, and to the accumulation of vacuoles and TDP-43 (138). Furthermore, a VCPR155H/R155H homozygote mouse model showed growth retardation, weakness and pathological abnormalities of muscle fibers and brain. A histopathological analysis revealed TDP-43 positive sarcoplasmic inclusions, ubiquitin aggregates and a higher expression of the autophagosome marker LC3 when compared to wild type mice. In addition, p62 was detected in the cytoplasm and nuclear areas. These data were also confirmed in the brains of mutant mice; a histological analysis revealed an increase of LC3-I/II immunoreactivity and increased levels of TDP-43, ubiquitin aggregates and LC3-I/II proteins (163). More recently, Kustermann et al. demonstrated that loss of VCP in vivo, as can happen in ALS/FTD mutations, compromised protein degradation via autophagic pathway (164).
VCP and Lysosomal Homeostasis
VCP has an important role in maintaining lysosomal homoeostasis. Lysosome dysfunction has been linked to many neurodegenerative diseases (165). Accumulation of lysosomes has been observed in muscle and myoblasts in IBMPFD/ALS patients carrying VCP mutations (138, 154). The endolysosomal pathway can be affected by membrane rupture and by lysosomal membrane permeabilization. Moreover, reactive oxygen species or protein aggregates can induce lysosomal damage (166). Damaged lysosomes are removed through the autophagic pathway in the so-called lysophagy process. The autophagosomal membrane associates the damaged organelle thanks to the ubiquitination of organelles components, which allows the interaction of the LC3-adapter proteins, such as p62/SQSTM1, and of the autophagy machinery (156).
In 2017 Papadopoulos and colleagues demonstrated the involvement of VCP in the endolysosomal damage response in Hela cells. They found that in cells depleted of VCP by siRNA, the clearance of damaged lysosomes was dramatically inhibited with an accumulation of the organelles. Moreover, VCP is essential for restoration after lysosomal damage, having regard to the fact that its silencing leads to cell death. The authors confirmed the data also in mice carrying the mutations typical of ALS and FTD, such as R155H and A232E. After the treatment to induce lysosomal damage, mouse embryonic fibroblasts showed an increase of LC3-II and an accumulation of lysosomes, suggesting an impairment of the lysophagic process. In the process UBXD1, PLAA, and YOD1 cofactors are importantly involved; they target specifically lysin 48-linked ubiquitin conjugates on lysin 63-decorated damaged lysosomes (156). The inactivation of VCP in skeletal muscle of adult mice leads to a necrotic myopathy, preceded by upregulation of LGALS3/Galectin-3, a typical marker of damaged lysosomes, and to the accumulation of autophagic proteins and of damaged lysosomes (167).
In 2019 Koerver et al. found that the ubiquitin-conjugating enzyme UBE2QL1 is essential for the coordinating of lysophagy process, associating with lysophagy effectors. They demonstrated that its knockdown abrogates the recruitment of VCP and consequently lysosomes clearance (168).
Moreover, a recent study defined the role of a specific VCP cofactor, the Small VCP-Interacting Protein (SVIP) in an animal model. It recruits VCP to lysosomes and its loss causes the disruption of the lysosomal network with an impairment of the autophagosome-lysosome fusion (169).
VCP and Mitochondrial Quality Control
Finally, VCP is important in maintaining mitochondrial function and consequently calcium homeostasis. As previously explained, VCP is involved in the extraction of misfolded proteins from both ER and mitochondria and in their degradation. Moreover, it participates in the regulation of calcium homeostasis through mitochondria-associated ER membranes (MAMs) and in regulating mitochondrial calcium intake acting on the mitochondrial calcium uptake (MICU) proteins (134). Furthermore, experiments on Drosophila larvae revealed that VCP regulates the axonal transport of mitochondria directly interacting with Dynein (170); downregulation of VCP or its mutation decreased mitochondrial density in axons and increased their time of retrograde transport (170).
Numerous studies revealed signs of mitochondrial dysfunctions and aberrations in neurodegenerative diseases (134, 171). Damaged mitochondria have a depolarization of the inner membrane leading to their autophagic degradation called mitophagy. Mitophagy is mediated by the E3 ubiquitin ligase Parkin in a VCP-mediated manner. The inhibition of VCP prevents mitochondrial fusion and consequently mitochondrial elimination (155). These data were further confirmed in a study which demonstrated in vivo that VCP is required for the degradation of mitofusins and for the subsequent clearance of the damaged mitochondria. Moreover, the expression of A232E VCP mutation leads to a failure in mitochondria clearance and to a mitochondrial aggregation (172). Furthermore, in 2017 Zhang and colleagues demonstrated that the inhibition of VCP mutants in vivo suppresses mitochondrial defects, cell death and muscle damage (173). In this function VCP is assisted by different cofactors: UBXD1 translocates to mitochondria and promotes the VCP recruitment (174), whereas the cofactor UBXN1/SAKS1 facilitates Mfn2 degradation from mitochondria (175).
It was demonstrated that fibroblast, cortical neurons and motor neurons carrying the VCP mutations R191Q and R155C exhibited mitochondrial dysfunction and oxidative stress. Mitochondria had a decrease in membrane potential consistent with uncoupling of oxidative phosphorylation, as already found in fibroblasts from IBMPFD patients (176), and a higher rate of ROS production (144). The uncoupling results in a reduction in ATP production and consequently in a more vulnerable state of the cells (176, 177).
Finally, it was also demonstrated that VCPR155H/R155H mice manifest striking mitochondrial abnormalities. Electron microscopy revealed abnormal mitochondrial structures with megaconia and disrupted cristae. Moreover, an increase in oxidative fibers and higher density of mitochondria were found out in muscle tissues (163).
Therapeutic Approaches
Despite many studies focusing on neurodegenerative pathogenesis and the advances in the characterization of the involved pathways, there are no available therapies that are able to revert or halt the pathological process. However, some evidence highlighted possible therapeutic approaches for VCP mutation diseases, such as ALS and FTD.
In recent years, many different VCP inhibitors have been described (178). In 2017 Zhang et al. tested the inhibitors NMS-873 and ML240, drugs developed for the treatment of cancer, on an IBMPFD Drosophila model. They discovered that the administration of these drugs in wild type animals results in mitochondrial elongation, similar to what happens with VCP RNAi, and in a reversion of mitochondrial defects in wild type and mutant animals. These molecules prevent muscle cell death and restore the muscle integrity, the mitochondrial size, the structure of cristae and the myofibril organization in VCP mutants. Moreover, the treatment increases the levels of mitofusins and of the oxygen consumption rate of patient's fibroblasts (173). Later on, it was demonstrated that the treatment with ML240 can reverse the mislocalization of TDP-43, FUS and SFPQ in mutant motor neurons (131).
Another well-studied drug used for the treatment of VCP-related diseases is rapamycin. In cells with accumulation of TDP-43, the administration of 0.5 g/ml rapamycin reduces TDP-43 mislocalization acting on mTOR, a negative regulator of autophagy (179). Moreover, the LC3-II/LC3-I ratio was increased, revealing an increase in autophagosomes formation (179). Similar results were obtained in 2015 by treating VCPR155H/+ mice with 3 mg/kg body weight rapamycin, three times a week for 8 weeks. Treated mice showed improvement in muscle performance and autophagy markers, reduced levels of apoptosis and rescue of ubiquitin and TDP-43 pathology (180). However, further studies in VCP-RH mutant mice revealed that a chronic treatment with rapamycin can worsen the degenerative phenotype by decreasing muscle strength, and increasing vacuolated and atrophic fibers (162), as what happens after treatment with chloroquine (180).
Finally, in 2015 Nalbandian et al. excised the R155H mutation in a mouse model and developed Cre-ERTM-VCPR155H/+ tamoxifen-inducible mice. These recombinant animals improved muscle strength, autophagy pathway and decreased apoptosis (181).
Conclusive Remarks
ALS and FTD have been considered for many years as two distinct pathologies with different pathological mechanisms. However, in recent years many studies have revealed that these diseases belong to a continuum, the so-called ALS-FTD spectrum. Several causative genes are shared in both diseases; one intriguing gene is ATPase VCP, already known for its relation to other muscle and neuropathic disorders, whose recent characterization and function determination has been shedding light into the dysfunctional molecular pathways in both ALS and FTD patients. In physiological conditions, VCP is involved in many different biological functions, namely protein degradation, autophagy, lysosomal and mitochondrial homeostasis, whose dysfunction hampers cell survival. Hence, its mutation can lead to severe or lethal consequences for the affected people. This review summarized the latest findings about the roles of VCP and about ALS-FTD mutations in this gene. Finally, we reported some evidence of possible therapeutic approaches targeting the VCP pathway.
Author Contributions
ES and GF wrote the manuscript. IP generated figures. LD, OP, and SG helped write and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by the Italian Ministry of Health (Ricerca Corrente 2020-2021 and Ricerca Finalizzata Giovani Ricercatori 2016) and by Agenzia di Ricerca per la Sclerosi Laterale Amiotrofica (AriSLA) Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
1. Couratier P, Corcia P, Lautrette G, Nicol M, Marin B. ALS and frontotemporal dementia belong to a common disease spectrum. Rev Neurol. (2017) 173:273–9. doi: 10.1016/j.neurol.2017.04.001
2. Greaves CV, Rohrer JD. An update on genetic frontotemporal dementia. J Neurol. (2019) 266:2075–86. doi: 10.1007/s00415-019-09363-4
3. Mejzini R, Flynn LL, Pitout IL, Fletcher S, Wilton SD, Akkari PA. ALS genetics, mechanisms, and therapeutics: where are we now? Front Neurosci. (2019) 13:1310. doi: 10.3389/fnins.2019.01310
4. Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M, et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol. (2013) 74:20–38. doi: 10.1002/ana.23937
5. Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. J Neurol Sci. (1994) 124:96–107. doi: 10.1016/0022-510X(94)90191-0
6. Brooks BR, Miller RG, Swash M, Munsat TL, Diseases WFoNRGoMN. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. (2000) 1:293–9. doi: 10.1080/146608200300079536
7. Ludolph A, Drory V, Hardiman O, Nakano I, Ravits J, Robberecht W, et al. A revision of the El Escorial criteria - 2015. Amyotroph Lateral Scler Frontotemporal Degener. (2015) 16:291–2. doi: 10.3109/21678421.2015.1049183
8. Jaiswal MK. Riluzole and edaravone: a tale of two amyotrophic lateral sclerosis drugs. Med Res Rev. (2019) 39:733–48. doi: 10.1002/med.21528
9. Miller T, Cudkowicz M, Shaw PJ, Andersen PM, Atassi N, Bucelli RC, et al. Phase 1-2 trial of antisense oligonucleotide tofersen for. N Engl J Med. (2020) 383:109–19. doi: 10.1056/NEJMoa2003715
10. Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, et al. Classification of primary progressive aphasia and its variants. Neurology. (2011) 76:1006–14. doi: 10.1212/WNL.0b013e31821103e6
11. Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. (1998) 51:1546–54. doi: 10.1212/WNL.51.6.1546
12. Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. (2011) 134:2456–77. doi: 10.1093/brain/awr179
13. Chare L, Hodges JR, Leyton CE, McGinley C, Tan RH, Kril JJ, et al. New criteria for frontotemporal dementia syndromes: clinical and pathological diagnostic implications. J Neurol Neurosurg Psychiatry. (2014) 85:865–70. doi: 10.1136/jnnp-2013-306948
14. Finger EC. Frontotemporal dementias. Continuum. (2016) 22:464–89. doi: 10.1212/CON.0000000000000300
15. Mesulam M, Wicklund A, Johnson N, Rogalski E, Léger GC, Rademaker A, et al. Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Ann Neurol. (2008) 63:709–19. doi: 10.1002/ana.21388
16. Rohrer JD, Warren JD, Modat M, Ridgway GR, Douiri A, Rossor MN, et al. Patterns of cortical thinning in the language variants of frontotemporal lobar degeneration. Neurology. (2009) 72:1562–9. doi: 10.1212/WNL.0b013e3181a4124e
17. Zhang Y, Tartaglia MC, Schuff N, Chiang GC, Ching C, Rosen HJ, et al. MRI signatures of brain macrostructural atrophy and microstructural degradation in frontotemporal lobar degeneration subtypes. J Alzheimers Dis. (2013) 33:431–44. doi: 10.3233/JAD-2012-121156
18. Josephs KA, Hodges JR, Snowden JS, Mackenzie IR, Neumann M, Mann DM, et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol. (2011) 122:137–53. doi: 10.1007/s00401-011-0839-6
19. Tsai RM, Boxer AL. Treatment of frontotemporal dementia. Curr Treat Options Neurol. (2014) 16:319. doi: 10.1007/s11940-014-0319-0
20. Chiò A, Calvo A, Moglia C, Mazzini L, Mora G, Group Ps. Phenotypic heterogeneity of amyotrophic lateral sclerosis: a population based study. J Neurol Neurosurg Psychiatry. (2011) 82:740–6. doi: 10.1136/jnnp.2010.235952
21. Visser J, van den Berg-Vos RM, Franssen H, van den Berg LH, Wokke JH, de Jong JM, et al. Disease course and prognostic factors of progressive muscular atrophy. Arch Neurol. (2007) 64:522–8. doi: 10.1001/archneur.64.4.522
22. Finegan E, Chipika RH, Shing SLH, Hardiman O, Bede P. Primary lateral sclerosis: a distinct entity or part of the ALS spectrum? Amyotroph Lateral Scler Frontotemporal Degener. (2019) 20:133–45. doi: 10.1080/21678421.2018.1550518
23. Shoesmith CL, Findlater K, Rowe A, Strong MJ. Prognosis of amyotrophic lateral sclerosis with respiratory onset. J Neurol Neurosurg Psychiatry. (2007) 78:629–31. doi: 10.1136/jnnp.2006.103564
24. Ravits JM, La Spada AR. ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology. (2009) 73:805–11. doi: 10.1212/WNL.0b013e3181b6bbbd
25. Chiò A, Moglia C, Canosa A, Manera U, D'Ovidio F, Vasta R, et al. ALS phenotype is influenced by age, sex, and genetics: a population-based study. Neurology. (2020) 94:e802–10. doi: 10.1212/WNL.0000000000008869
26. Mitsuyama Y. Presenile dementia with motor neuron disease. Dementia. (1993) 4:137–42. doi: 10.1159/000107312
27. Neary D, Snowden JS, Mann DM, Northen B, Goulding PJ, Macdermott N. Frontal lobe dementia and motor neuron disease. J Neurol Neurosurg Psychiatry. (1990) 53:23–32. doi: 10.1136/jnnp.53.1.23
28. Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. (2002) 59:1077–9. doi: 10.1212/WNL.59.7.1077
29. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. (2006) 314:130–3. doi: 10.1126/science.1134108
30. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. (2011) 72:245–56. doi: 10.1016/j.neuron.2011.09.011
31. Strong MJ, Grace GM, Freedman M, Lomen-Hoerth C, Woolley S, Goldstein LH, et al. Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. (2009) 10:131–46. doi: 10.1080/17482960802654364
32. Strong MJ, Abrahams S, Goldstein LH, Woolley S, Mclaughlin P, Snowden J, et al. Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener. (2017) 18:153–74. doi: 10.1080/21678421.2016.1267768
33. Witgert M, Salamone AR, Strutt AM, Jawaid A, Massman PJ, Bradshaw M, et al. Frontal-lobe mediated behavioral dysfunction in amyotrophic lateral sclerosis. Eur J Neurol. (2010) 17:103–10. doi: 10.1111/j.1468-1331.2009.02801.x
34. Consonni M, Iannaccone S, Cerami C, Frasson P, Lacerenza M, Lunetta C, et al. The cognitive and behavioural profile of amyotrophic lateral sclerosis: application of the consensus criteria. Behav Neurol. (2013) 27:143–53. doi: 10.1155/2013/126010
35. Phukan J, Elamin M, Bede P, Jordan N, Gallagher L, Byrne S, et al. The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatry. (2012) 83:102–8. doi: 10.1136/jnnp-2011-300188
36. Benbrika S, Desgranges B, Eustache F, Viader F. Cognitive, emotional and psychological manifestations in amyotrophic lateral sclerosis at baseline and overtime: a review. Front Neurosci. (2019) 13:951. doi: 10.3389/fnins.2019.00951
37. Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet. (2015) 386:1672–82. doi: 10.1016/S0140-6736(15)00461-4
38. Burrell JR, Kiernan MC, Vucic S, Hodges JR. Motor neuron dysfunction in frontotemporal dementia. Brain. (2011) 134:2582–94. doi: 10.1093/brain/awr195
39. Unglik J, Bungener C, Delgadillo D, Salachas F, Pradat PF, Bruneteau G, et al. Emotional feeling in patients suffering from amyotrophic lateral sclerosis. Geriatr Psychol Neuropsychiatr Vieil. (2018) 16:414–22. doi: 10.1684/pnv.2018.0762
40. Ahmed RM, Devenney EM, Strikwerda-Brown C, Hodges JR, Piguet O, Kiernan MC. Phenotypic variability in ALS-FTD and effect on survival. Neurology. (2020) 94:e2005–13. doi: 10.1212/WNL.0000000000009398
41. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. (1993) 362:59–62. doi: 10.1038/364362c0
42. Boylan K. Familial amyotrophic lateral sclerosis. Neurol Clin. (2015) 33:807–30. doi: 10.1016/j.ncl.2015.07.001
43. Talbott EO, Malek AM, Lacomis D. The epidemiology of amyotrophic lateral sclerosis. Handb Clin Neurol. (2016) 138:225–38. doi: 10.1016/B978-0-12-802973-2.00013-6
44. Kim G, Gautier O, Tassoni-Tsuchida E, Ma XR, Gitler AD. ALS genetics: gains, losses, and implications for future therapies. Neuron. (2020) 108:822–42. doi: 10.1016/j.neuron.2020.08.022
45. Fett JW, Strydom DJ, Lobb RR, Alderman EM, Bethune JL, Riordan JF, et al. Isolation and characterization of angiogenin, an angiogenic protein from human carcinoma cells. Biochemistry. (1985) 24:5480–6. doi: 10.1021/bi00341a030
46. Gispert S, Twells R, Orozco G, Brice A, Weber J, Heredero L, et al. Chromosomal assignment of the second locus for autosomal dominant cerebellar ataxia (SCA2) to chromosome 12q23-24.1. Nat Genet. (1993) 4:295–9. doi: 10.1038/ng0793-295
47. Bannwarth S, Ait-El-Mkadem S, Chaussenot A, Genin EC, Lacas-Gervais S, Fragaki K, et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain. (2014) 137:2329–45. doi: 10.1093/brain/awu138
48. Babst M, Katzmann DJ, Estepa-Sabal EJ, Meerloo T, Emr SD. Escrt-III: an endosome-associated heterooligomeric protein complex required for mvb sorting. Dev Cell. (2002) 3:271–82. doi: 10.1016/S1534-5807(02)00220-4
49. Holzbaur EL, Hammarback JA, Paschal BM, Kravit NG, Pfister KK, Vallee RB. Homology of a 150K cytoplasmic dynein-associated polypeptide with the Drosophila gene Glued. Nature. (1991) 351:579–83. doi: 10.1038/351579a0
50. Crozat A, Aman P, Mandahl N, Ron D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature. (1993) 363:640–4. doi: 10.1038/363640a0
51. Zhou J, Gao G, Crabb JW, Serrero G. Purification of an autocrine growth factor homologous with mouse epithelin precursor from a highly tumorigenic cell line. J Biol Chem. (1993) 268:10863–9. doi: 10.1016/S0021-9258(18)82064-6
52. Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci USA. (1988) 85:4051–5. doi: 10.1073/pnas.85.11.4051
53. Belgrader P, Dey R, Berezney R. Molecular cloning of matrin 3. A 125-kilodalton protein of the nuclear matrix contains an extensive acidic domain. J Biol Chem. (1991) 266:9893–9. doi: 10.1016/S0021-9258(18)92902-9
54. Letwin K, Mizzen L, Motro B, Ben-David Y, Bernstein A, Pawson T. A mammalian dual specificity protein kinase, Nek1, is related to the NIMA cell cycle regulator and highly expressed in meiotic germ cells. EMBO J. (1992) 11:3521–31. doi: 10.1002/j.1460-2075.1992.tb05435.x
55. Li Y, Kang J, Horwitz MS. Interaction of an adenovirus E3 14.7-kilodalton protein with a novel tumor necrosis factor alpha-inducible cellular protein containing leucine zipper domains. Mol Cell Biol. (1998) 18:1601–10. doi: 10.1128/MCB.18.3.1601
56. Carlsson L, Nyström LE, Sundkvist I, Markey F, Lindberg U. Actin polymerizability is influenced by profilin, a low molecular weight protein in non-muscle cells. J Mol Biol. (1977) 115:465–83. doi: 10.1016/0022-2836(77)90166-8
57. Moreira MC, Klur S, Watanabe M, Németh AH, Le Ber I, Moniz JC, et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet. (2004) 36:225–7. doi: 10.1038/ng1303
58. Stevanin G, Santorelli FM, Azzedine H, Coutinho P, Chomilier J, Denora PS, et al. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet. (2007) 39:366–72. doi: 10.1038/ng1980
59. Park I, Chung J, Walsh CT, Yun Y, Strominger JL, Shin J. Phosphotyrosine-independent binding of a 62-kDa protein to the src homology 2 (SH2) domain of p56lck and its regulation by phosphorylation of Ser-59 in the lck unique N-terminal region. Proc Natl Acad Sci USA. (1995) 92:12338–42. doi: 10.1073/pnas.92.26.12338
60. Ou SH, Wu F, Harrich D, García-Martínez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. (1995) 69:3584–96. doi: 10.1128/jvi.69.6.3584-3596.1995
61. Pomerantz JL, Baltimore D. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. (1999) 18:6694–704. doi: 10.1093/emboj/18.23.6694
62. Koller KJ, Brownstein MJ. Use of a cDNA clone to identify a supposed precursor protein containing valosin. Nature. (1987) 325:542–5. doi: 10.1038/325542a0
63. Fridovich I. Biological effects of the superoxide radical. Arch Biochem Biophys. (1986) 247:1–11. doi: 10.1016/0003-9861(86)90526-6
64. Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. (2017) 88:540–9. doi: 10.1136/jnnp-2016-315018
65. Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, et al. Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science. (1993) 261:1047–51. doi: 10.1126/science.8351519
66. Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. (2007) 61:427–34. doi: 10.1002/ana.21147
67. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. (2008) 319:1668–72. doi: 10.1126/science.1154584
68. Ayala YM, Zago P, D'Ambrogio A, Xu YF, Petrucelli L, Buratti E, et al. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci. (2008) 121:3778–85. doi: 10.1242/jcs.038950
69. Neelagandan N, Gonnella G, Dang S, Janiesch PC, Miller KK, Küchler K, et al. TDP-43 enhances translation of specific mRNAs linked to neurodegenerative disease. Nucleic Acids Res. (2019) 47:341–61. doi: 10.1093/nar/gky972
70. Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci. (2019) 22:167–79. doi: 10.1038/s41593-018-0300-4
71. Mompeán M, Baralle M, Buratti E, Laurents DV. An amyloid-like pathological conformation of TDP-43 is stabilized by hypercooperative hydrogen bonds. Front Mol Neurosci. (2016) 9:125. doi: 10.3389/fnmol.2016.00125
72. Kwiatkowski TJ, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. (2009) 323:1205–8. doi: 10.1126/science.1166066
73. Ratti A, Buratti E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J Neurochem. (2016) 138 (Suppl. 1):95–111. doi: 10.1111/jnc.13625
74. Mastrocola AS, Kim SH, Trinh AT, Rodenkirch LA, Tibbetts RS. The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J Biol Chem. (2013) 288:24731–41. doi: 10.1074/jbc.M113.497974
75. Hennig S, Kong G, Mannen T, Sadowska A, Kobelke S, Blythe A, et al. Prion-like domains in RNA binding proteins are essential for building subnuclear paraspeckles. J Cell Biol. (2015) 210:529–39. doi: 10.1083/jcb.201504117
76. Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. (2009) 323:1208–11. doi: 10.1126/science.1165942
77. Deng H, Gao K, Jankovic J. The role of FUS gene variants in neurodegenerative diseases. Nat Rev Neurol. (2014) 10:337–48. doi: 10.1038/nrneurol.2014.78
78. Nassif M, Woehlbier U, Manque PA. The enigmatic role of C9ORF72 in autophagy. Front Neurosci. (2017) 11:442. doi: 10.3389/fnins.2017.00442
79. Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee HC, et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep. (2016) 6:23204. doi: 10.1038/srep23204
80. Xiao S, MacNair L, McGoldrick P, McKeever PM, McLean JR, Zhang M, et al. Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann Neurol. (2015) 78:568–83. doi: 10.1002/ana.24469
81. Fratta P, Mizielinska S, Nicoll AJ, Zloh M, Fisher EM, Parkinson G, et al. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci Rep. (2012) 2:1016. doi: 10.1038/srep01016
82. Chia R, Chiò A, Traynor BJ. Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. (2018) 17:94–102. doi: 10.1016/S1474-4422(17)30401-5
83. Wood EM, Falcone D, Suh E, Irwin DJ, Chen-Plotkin AS, Lee EB, et al. Development and validation of pedigree classification criteria for frontotemporal lobar degeneration. JAMA Neurol. (2013) 70:1411–7. doi: 10.1001/jamaneurol.2013.3956
84. Olszewska DA, Lonergan R, Fallon EM, Lynch T. Genetics of frontotemporal dementia. Curr Neurol Neurosci Rep. (2016) 16:107. doi: 10.1007/s11910-016-0707-9
85. Benussi A, Padovani A, Borroni B. Phenotypic heterogeneity of monogenic frontotemporal dementia. Front Aging Neurosci. (2015) 7:171. doi: 10.3389/fnagi.2015.00171
86. Daniel R, He Z, Carmichael KP, Halper J, Bateman A. Cellular localization of gene expression for progranulin. J Histochem Cytochem. (2000) 48:999–1009. doi: 10.1177/002215540004800713
87. Karamysheva ZN, Tikhonova EB, Karamyshev AL. Granulin in frontotemporal lobar degeneration: molecular mechanisms of the disease. Front Neurosci. (2019) 13:395. doi: 10.3389/fnins.2019.00395
88. Nicholson AM, Gass J, Petrucelli L, Rademakers R. Progranulin axis and recent developments in frontotemporal lobar degeneration. Alzheimers Res Ther. (2012) 4:4. doi: 10.1186/alzrt102
89. Yamashita S, Ando Y. Genotype-phenotype relationship in hereditary amyotrophic lateral sclerosis. Transl Neurodegener. (2015) 4:13. doi: 10.1186/s40035-015-0036-y
90. Juneja T, Pericak-Vance MA, Laing NG, Dave S, Siddique T. Prognosis in familial amyotrophic lateral sclerosis: progression and survival in patients with glu100gly and ala4val mutations in Cu, Zn superoxide dismutase. Neurology. (1997) 48:55–7. doi: 10.1212/WNL.48.1.55
91. Andersen PM, Forsgren L, Binzer M, Nilsson P, Ala-Hurula V, Keränen ML, et al. Autosomal recessive adult-onset amyotrophic lateral sclerosis associated with homozygosity for Asp90Ala CuZn-superoxide dismutase mutation. A clinical and genealogical study of 36 patients. Brain. (1996) 119:1153–72. doi: 10.1093/brain/119.4.1153
92. Li HF, Wu ZY. Genotype-phenotype correlations of amyotrophic lateral sclerosis. Transl Neurodegener. (2016) 5:3. doi: 10.1186/s40035-016-0050-8
93. Corcia P, Valdmanis P, Millecamps S, Lionnet C, Blasco H, Mouzat K, et al. Phenotype and genotype analysis in amyotrophic lateral sclerosis with TARDBP gene mutations. Neurology. (2012) 78:1519–26. doi: 10.1212/WNL.0b013e3182553c88
94. Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat. (2013) 34:812–26. doi: 10.1002/humu.22319
95. Floris G, Borghero G, Cannas A, Di Stefano F, Murru MR, Corongiu D, et al. Clinical phenotypes and radiological findings in frontotemporal dementia related to TARDBP mutations. J Neurol. (2015) 262:375–84. doi: 10.1007/s00415-014-7575-5
96. Gromicho M, Oliveira Santos M, Pinto A, Pronto-Laborinho A, De Carvalho M. Young-onset rapidly progressive ALS associated with heterozygous FUS mutation. Amyotroph Lateral Scler Frontotemporal Degener. (2017) 18:451–3. doi: 10.1080/21678421.2017.1299762
97. Millecamps S, Salachas F, Cazeneuve C, Gordon P, Bricka B, Camuzat A, et al. SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: genotype-phenotype correlations. J Med Genet. (2010) 47:554–60. doi: 10.1136/jmg.2010.077180
98. Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. (2010) 29:2841–57. doi: 10.1038/emboj.2010.143
99. Chiò A, Restagno G, Brunetti M, Ossola I, Calvo A, Mora G, et al. Two Italian kindreds with familial amyotrophic lateral sclerosis due to FUS mutation. Neurobiol Aging. (2009) 30:1272–5. doi: 10.1016/j.neurobiolaging.2009.05.001
100. Snowden JS, Hu Q, Rollinson S, Halliwell N, Robinson A, Davidson YS, et al. The most common type of FTLD-FUS (aFTLD-U) is associated with a distinct clinical form of frontotemporal dementia but is not related to mutations in the FUS gene. Acta Neuropathol. (2011) 122:99–110. doi: 10.1007/s00401-011-0816-0
101. Millecamps S, Boillée S, Le Ber I, Seilhean D, Teyssou E, Giraudeau M, et al. Phenotype difference between ALS patients with expanded repeats in C9ORF72 and patients with mutations in other ALS-related genes. J Med Genet. (2012) 49:258–63. doi: 10.1136/jmedgenet-2011-100699
102. Cooper-Knock J, Hewitt C, Highley JR, Brockington A, Milano A, Man S, et al. Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain. (2012) 135:751–64. doi: 10.1093/brain/awr365
103. Devenney EM, Landin-Romero R, Irish M, Hornberger M, Mioshi E, Halliday GM, et al. The neural correlates and clinical characteristics of psychosis in the frontotemporal dementia continuum and the. Neuroimage Clin. (2017) 13:439–45. doi: 10.1016/j.nicl.2016.11.028
104. Lattante S, Millecamps S, Stevanin G, Rivaud-Péchoux S, Moigneu C, Camuzat A, et al. Contribution of ATXN2 intermediary polyQ expansions in a spectrum of neurodegenerative disorders. Neurology. (2014) 83:990–5. doi: 10.1212/WNL.0000000000000778
105. Caroppo P, Camuzat A, De Septenville A, Couratier P, Lacomblez L, Auriacombe S, et al. Semantic and nonfluent aphasic variants, secondarily associated with amyotrophic lateral sclerosis, are predominant frontotemporal lobar degeneration phenotypes in TBK1 carriers. Alzheimers Dement. (2015) 1:481–6. doi: 10.1016/j.dadm.2015.10.002
106. Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. (2004) 36:377–81. doi: 10.1038/ng1332
107. Kimonis VE, Kovach MJ, Waggoner B, Leal S, Salam A, Rimer L, et al. Clinical and molecular studies in a unique family with autosomal dominant limb-girdle muscular dystrophy and Paget disease of bone. Genet Med. (2000) 2:232–41. doi: 10.1097/00125817-200007000-00006
108. van der Zee J, Pirici D, Van Langenhove T, Engelborghs S, Vandenberghe R, Hoffmann M, et al. Clinical heterogeneity in 3 unrelated families linked to VCP p.Arg159His. Neurology. (2009) 73:626–32. doi: 10.1212/WNL.0b013e3181b389d9
109. Forman MS, Mackenzie IR, Cairns NJ, Swanson E, Boyer PJ, Drachman DA, et al. Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J Neuropathol Exp Neurol. (2006) 65:571–81. doi: 10.1097/00005072-200606000-00005
110. Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. (2010) 68:857–64. doi: 10.1016/j.neuron.2010.11.036
111. van de Warrenburg BP, Schouten MI, de Bot ST, Vermeer S, Meijer R, Pennings M, et al. Clinical exome sequencing for cerebellar ataxia and spastic paraplegia uncovers novel gene-disease associations and unanticipated rare disorders. Eur J Hum Genet. (2016) 24:1460–6. doi: 10.1038/ejhg.2016.42
112. Gonzalez MA, Feely SM, Speziani F, Strickland AV, Danzi M, Bacon C, et al. A novel mutation in VCP causes Charcot-Marie-tooth type 2 disease. Brain. (2014) 137:2897–902. doi: 10.1093/brain/awu224
113. Kimonis VE, Mehta SG, Fulchiero EC, Thomasova D, Pasquali M, Boycott K, et al. Clinical studies in familial VCP myopathy associated with Paget disease of bone and frontotemporal dementia. Am J Med Genet A. (2008) 146A:745–57. doi: 10.1002/ajmg.a.31862
114. Mehta SG, Khare M, Ramani R, Watts GD, Simon M, Osann KE, et al. Genotype-phenotype studies of VCP-associated inclusion body myopathy with Paget disease of bone and/or frontotemporal dementia. Clin Genet. (2013) 83:422–31. doi: 10.1111/cge.12000
115. Kazamel M, Sorenson EJ, McEvoy KM, Jones LK, Leep-Hunderfund AN, Mauermann ML, et al. Clinical spectrum of valosin containing protein (VCP)-opathy. Muscle Nerve. (2016) 54:94–9. doi: 10.1002/mus.24980
116. Al-Obeidi E, Al-Tahan S, Surampalli A, Goyal N, Wang AK, Hermann A, et al. Genotype-phenotype study in patients with valosin-containing protein mutations associated with multisystem proteinopathy. Clin Genet. (2018) 93:119–25. doi: 10.1111/cge.13095
117. Saracino D, Clot F, Camuzat A, Anquetil V, Hannequin D, Guyant-Maréchal L, et al. Novel VCP mutations expand the mutational spectrum of frontotemporal dementia. Neurobiol Aging. (2018) 72:187.e11–4. doi: 10.1016/j.neurobiolaging.2018.06.037
118. Evangelista T, Weihl CC, Kimonis V, Lochmüller H, Consortium Vrd. 215th ENMC international workshop VCP-related multi-system proteinopathy (IBMPFD) 13-15 November 2015, Heemskerk, The Netherlands. Neuromuscul Disord. (2016) 26:535–47. doi: 10.1016/j.nmd.2016.05.017
119. Korb M, Kimonis V, Mozaffar T. Multisystem proteinopathy: where myopathy and motorneuron disease converge. Muscle Nerve. (2020) 63:442–54. doi: 10.1002/mus.27097
120. Yamanaka K, Sasagawa Y, Ogura T. Recent advances in p97/VCP/Cdc48 cellular functions. Biochim Biophys Acta. (2012) 1823:130–7. doi: 10.1016/j.bbamcr.2011.07.001
121. Meyer H, Weihl CC. The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J Cell Sci. (2014) 127:3877–83. doi: 10.1242/jcs.093831
122. Tang WK, Xia D. Altered intersubunit communication is the molecular basis for functional defects of pathogenic p97 mutants. J Biol Chem. (2013) 288:36624–35. doi: 10.1074/jbc.M113.488924
123. Zhang X, Shaw A, Bates PA, Newman RH, Gowen B, Orlova E, et al. Structure of the AAA ATPase p97. Mol Cell. (2000) 6:1473–84. doi: 10.1016/S1097-2765(00)00143-X
124. Schütz AK, Rennella E, Kay LE. Exploiting conformational plasticity in the AAA+ protein VCP/p97 to modify function. Proc Natl Acad Sci USA. (2017) 114:E6822–E9. doi: 10.1073/pnas.1707974114
125. Yeo BK, Yu S-W. Valosin-containing protein (VCP): structure, functions, and implications in neurodegenerative diseases. Anim Cells Syst. (2016) 20:303–9. doi: 10.1080/19768354.2016.1259181
126. Davies JM, Brunger AT, Weis WI. Improved structures of full-length p97, an AAA ATPase: implications for mechanisms of nucleotide-dependent conformational change. Structure. (2008) 16:715–26. doi: 10.1016/j.str.2008.02.010
127. Zhang X, Gui L, Bulfer SL, Sanghez V, Wong DE, Lee Y, et al. Altered cofactor regulation with disease-associated p97/VCP mutations. Proc Natl Acad Sci USA. (2015) 112:E1705–14. doi: 10.1073/pnas.1418820112
128. Weihl CC, Temiz P, Miller SE, Watts G, Smith C, Forman M, et al. TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J Neurol Neurosurg Psychiatry. (2008) 79:1186–9. doi: 10.1136/jnnp.2007.131334
129. Abramzon YA, Fratta P, Traynor BJ, Chia R. The overlapping genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Front Neurosci. (2020) 14:42. doi: 10.3389/fnins.2020.00042
130. Mackenzie IR, Neumann M. Molecular neuropathology of frontotemporal dementia: insights into disease mechanisms from postmortem studies. J Neurochem. (2016) 138 (Suppl. 1):54–70. doi: 10.1111/jnc.13588
131. Bodnar N, Rapoport T. Toward an understanding of the Cdc48/p97 ATPase. F1000Res. (2017) 6:1318. doi: 10.12688/f1000research.11683.1
132. Lan B, Chai S, Wang P, Wang K. VCP/p97/Cdc48, a linking of protein homeostasis and cancer therapy. Curr Mol Med. (2017) 17:608–18. doi: 10.2174/1566524018666180308111238
133. Guo X, Qi X. VCP cooperates with UBXD1 to degrade mitochondrial outer membrane protein MCL1 in model of Huntington's disease. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:552–9. doi: 10.1016/j.bbadis.2016.11.026
134. Sun X, Qiu H. Valosin-containing protein, a calcium-associated ATPase protein, in endoplasmic reticulum and mitochondrial function and its implications for diseases. Int J Mol Sci. (2020) 21:3842. doi: 10.3390/ijms21113842
135. Ritz D, Vuk M, Kirchner P, Bug M, Schütz S, Hayer A, et al. Endolysosomal sorting of ubiquitylated caveolin-1 is regulated by VCP and UBXD1 and impaired by VCP disease mutations. Nat Cell Biol. (2011) 13:1116–23. doi: 10.1038/ncb2301
136. Byrne DJ, Harmon MJ, Simpson JC, Blackstone C, O'Sullivan NC. Roles for the VCP co-factors Npl4 and Ufd1 in neuronal function in Drosophila melanogaster. J Genet Genomics. (2017) 44:493–501. doi: 10.1016/j.jgg.2017.06.003
137. Gitcho MA, Strider J, Carter D, Taylor-Reinwald L, Forman MS, Goate AM, et al. VCP mutations causing frontotemporal lobar degeneration disrupt localization of TDP-43 and induce cell death. J Biol Chem. (2009) 284:12384–98. doi: 10.1074/jbc.M900992200
138. Ju JS, Fuentealba RA, Miller SE, Jackson E, Piwnica-Worms D, Baloh RH, et al. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol. (2009) 187:875–88. doi: 10.1083/jcb.200908115
139. Ritson GP, Custer SK, Freibaum BD, Guinto JB, Geffel D, Moore J, et al. TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J Neurosci. (2010) 30:7729–39. doi: 10.1523/JNEUROSCI.5894-09.2010
140. Neumann M, Mackenzie IR, Cairns NJ, Boyer PJ, Markesbery WR, Smith CD, et al. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol. (2007) 66:152–7. doi: 10.1097/nen.0b013e31803020b9
141. Van Mossevelde S, Engelborghs S, van der Zee J, Van Broeckhoven C. Genotype-phenotype links in frontotemporal lobar degeneration. Nat Rev Neurol. (2018) 14:363–78. doi: 10.1038/s41582-018-0009-8
142. Wong TH, Pottier C, Hondius DC, Meeter LHH, van Rooij JGJ, Melhem S, et al. Three VCP mutations in patients with frontotemporal dementia. J Alzheimers Dis. (2018) 65:1139–46. doi: 10.3233/JAD-180301
143. Bersano A, Del Bo R, Lamperti C, Ghezzi S, Fagiolari G, Fortunato F, et al. Inclusion body myopathy and frontotemporal dementia caused by a novel VCP mutation. Neurobiol Aging. (2009) 30:752–8. doi: 10.1016/j.neurobiolaging.2007.08.009
144. Hall CE, Yao Z, Choi M, Tyzack GE, Serio A, Luisier R, et al. Progressive motor neuron pathology and the role of astrocytes in a human stem cell model of VCP-related ALS. Cell Rep. (2017) 19:1739–49. doi: 10.1016/j.celrep.2017.05.024
145. Pirie E, Oh CK, Zhang X, Han X, Cieplak P, Scott HR, et al. S-nitrosylated TDP-43 triggers aggregation, cell-to-cell spread, and neurotoxicity in hiPSCs and in vivo models of ALS/FTD. PNAS. (2021) 118:e2021368118. doi: 10.1073/pnas.2021368118
146. Ayaki T, Ito H, Fukushima H, Inoue T, Kondo T, Ikemoto A, et al. Immunoreactivity of valosin-containing protein in sporadic amyotrophic lateral sclerosis and in a case of its novel mutant. Acta Neuropathol Commun. (2014) 2:172. doi: 10.1186/s40478-014-0172-0
147. Surampalli A, Nalbandian A, Donkervoort S, Khare M, Wang A. A clinicopathologic case report of a female with valosin-containing protein (VCP) gene mutation related disease. Int J Neurodegener Dis. (2018) 1:006. doi: 10.23937/ijnd-2017/1710006
148. De Marco G, Lomartire A, Calvo A, Risso A, De Luca E, Mostert M, et al. Monocytes of patients with amyotrophic lateral sclerosis linked to gene mutations display altered TDP-43 subcellular distribution. Neuropathol Appl Neurobiol. (2017) 43:133–53. doi: 10.1111/nan.12328
149. Darwich NF, Phan JM, Kim B, Suh E, Papatriantafyllou JD, Changolkar L, et al. Autosomal dominant VCP hypomorph mutation impairs disaggregation of PHF-tau. Science. (2020) 370: eaay8826. doi: 10.1126/science.aay8826
150. Harley J, Hagemann C, Serio A, Patani R. TDP-43 and FUS mislocalization in VCP mutant motor neurons is reversed by pharmacological inhibition of the VCP D2 ATPase domain. Brain Commun. (2021) 3:fcab166. doi: 10.1093/braincomms/fcab166
151. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. (2011) 147:728–41. doi: 10.1016/j.cell.2011.10.026
152. Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, et al. Autophagy in malignant transformation and cancer progression. EMBO J. (2015) 34:856–80. doi: 10.15252/embj.201490784
153. Krick R, Bremer S, Welter E, Schlotterhose P, Muehe Y, Eskelinen EL, et al. Cdc48/p97 and Shp1/p47 regulate autophagosome biogenesis in concert with ubiquitin-like Atg8. J Cell Biol. (2010) 190:965–73. doi: 10.1083/jcb.201002075
154. Tresse E, Salomons FA, Vesa J, Bott LC, Kimonis V, Yao TP, et al. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy. (2010) 6:217–27. doi: 10.4161/auto.6.2.11014
155. Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. (2010) 191:1367–80. doi: 10.1083/jcb.201007013
156. Papadopoulos C, Kirchner P, Bug M, Grum D, Koerver L, Schulze N, et al. VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J. (2017) 36:135–50. doi: 10.15252/embj.201695148
157. Pleasure IT, Black MM, Keen JH. Valosin-containing protein, VCP, is a ubiquitous clathrin-binding protein. Nature. (1993) 365:459–62. doi: 10.1038/365459a0
158. Ramanathan HN, Ye Y. The p97 ATPase associates with EEA1 to regulate the size of early endosomes. Cell Res. (2012) 22:346–59. doi: 10.1038/cr.2011.80
159. Custer SK, Neumann M, Lu H, Wright AC, Taylor JP. Transgenic mice expressing mutant forms VCP/p97 recapitulate the full spectrum of IBMPFD including degeneration in muscle, brain and bone. Hum Mol Genet. (2010) 19:1741–55. doi: 10.1093/hmg/ddq050
160. Buchan JR, Kolaitis RM, Taylor JP, Parker R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell. (2013) 153:1461–74. doi: 10.1016/j.cell.2013.05.037
161. Hill SM, Wrobel L, Ashkenazi A, Fernandez-Estevez M, Tan K, Bürli RW, et al. VCP/p97 regulates Beclin-1-dependent autophagy initiation. Nat Chem Biol. (2021) 17:448–55. doi: 10.1038/s41589-020-00726-x
162. Ching JK, Weihl CC. Rapamycin-induced autophagy aggravates pathology and weakness in a mouse model of VCP-associated myopathy. Autophagy. (2013) 9:799–800. doi: 10.4161/auto.23958
163. Nalbandian A, Llewellyn KJ, Kitazawa M, Yin HZ, Badadani M, Khanlou N, et al. The homozygote VCP(R55H/R55H) mouse model exhibits accelerated human VCP-associated disease pathology. PLoS One. (2012) 7:e46308. doi: 10.1371/journal.pone.0046308
164. Kustermann M, Manta L, Paone C, Kustermann J, Lausser L, Wiesner C, et al. Loss of the novel Vcp (valosin containing protein) interactor Washc4 interferes with autophagy-mediated proteostasis in striated muscle and leads to myopathy in vivo. Autophagy. (2018) 14:1911–27. doi: 10.1080/15548627.2018.1491491
165. Lie PPY, Nixon RA. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol Dis. (2019) 122:94–105. doi: 10.1016/j.nbd.2018.05.015
166. Papadopoulos C, Kravic B, Meyer H. Repair or lysophagy: dealing with damaged lysosomes. J Mol Biol. (2020) 432:231–9. doi: 10.1016/j.jmb.2019.08.010
167. Arhzaouy K, Papadopoulos C, Schulze N, Pittman SK, Meyer H, Weihl CC. VCP maintains lysosomal homeostasis and TFEB activity in differentiated skeletal muscle. Autophagy. (2019) 15:1082–99. doi: 10.1080/15548627.2019.1569933
168. Koerver L, Papadopoulos C, Liu B, Kravic B, Rota G, Brecht L, et al. The ubiquitin-conjugating enzyme UBE2QL1 coordinates lysophagy in response to endolysosomal damage. EMBO Rep. (2019) 20:e48014. doi: 10.15252/embr.201948014
169. Johnson AE, Orr BO, Fetter RD, Moughamian AJ, Primeaux LA, Geier EG, et al. SVIP is a molecular determinant of lysosomal dynamic stability, neurodegeneration and lifespan. Nat Commun. (2021) 12:513. doi: 10.1038/s41467-020-20796-8
170. Gonzalez AE, Wang X. VCP/p97 mediates dynein-dependent retrograde mitochondrial motility in axons. Front Cell Dev Biol. (2020) 8:256. doi: 10.3389/fcell.2020.00256
171. Jankovic M, Novakovic I, Gamil Anwar Dawod P, Gamil Anwar Dawod A, Drinic A, Abdel Motaleb FI, et al. Current concepts on genetic aspects of mitochondrial dysfunction in amyotrophic lateral sclerosis. Int J Mol Sci. (2021) 22:9832. doi: 10.3390/ijms22189832
172. Kim NC, Tresse E, Kolaitis RM, Molliex A, Thomas RE, Alami NH, et al. VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron. (2013) 78:65–80. doi: 10.1016/j.neuron.2013.04.020
173. Zhang T, Mishra P, Hay BA, Chan D, Guo M. Valosin-containing protein (VCP/p97) inhibitors relieve Mitofusin-dependent mitochondrial defects due to VCP disease mutants. Elife. (2017) 6:e17834. doi: 10.7554/eLife.17834
174. Bento AC, Bippes CC, Kohler C, Hemion C, Frank S, Neutzner A. UBXD1 is a mitochondrial recruitment factor for p97/VCP and promotes mitophagy. Sci Rep. (2018) 8:12415. doi: 10.1038/s41598-018-30963-z
175. Mengus C, Neutzner M, Bento ACPF, Bippes CC, Kohler C, Decembrini S, et al. VCP/p97 cofactor UBXN1/SAKS1 regulates mitophagy by modulating MFN2 removal from mitochondria. Autophagy. (2021). doi: 10.1080/15548627.2021.1922982. [Epub ahead of print].
176. Bartolome F, Wu HC, Burchell VS, Preza E, Wray S, Mahoney CJ, et al. Pathogenic VCP mutations induce mitochondrial uncoupling and reduced ATP levels. Neuron. (2013) 78:57–64. doi: 10.1016/j.neuron.2013.02.028
177. Ludtmann MHR, Arber C, Bartolome F, de Vicente M, Preza E, Carro E, et al. Mutations in valosin-containing protein (VCP) decrease ADP/ATP translocation across the mitochondrial membrane and impair energy metabolism in human neurons. J Biol Chem. (2017) 292:8907–17. doi: 10.1074/jbc.M116.762898
178. Chapman E, Maksim N, de la Cruz F, La Clair JJ. Inhibitors of the AAA+ chaperone p97. Molecules. (2015) 20:3027–49. doi: 10.3390/molecules20023027
179. Caccamo A, Majumder S, Deng JJ, Bai Y, Thornton FB, Oddo S. Rapamycin rescues TDP-43 mislocalization and the associated low molecular mass neurofilament instability. J Biol Chem. (2009) 284:27416–24. doi: 10.1074/jbc.M109.031278
180. Nalbandian A, Llewellyn KJ, Nguyen C, Yazdi PG, Kimonis VE. Rapamycin and chloroquine: the in vitro and in vivo effects of autophagy-modifying drugs show promising results in valosin containing protein multisystem proteinopathy. PLoS ONE. (2015) 10:e0122888. doi: 10.1371/journal.pone.0122888
Keywords: ALS, FTD, VCP, protein clearance, autophagy, lysophagy, mitophagy
Citation: Scarian E, Fiamingo G, Diamanti L, Palmieri I, Gagliardi S and Pansarasa O (2022) The Role of VCP Mutations in the Spectrum of Amyotrophic Lateral Sclerosis—Frontotemporal Dementia. Front. Neurol. 13:841394. doi: 10.3389/fneur.2022.841394
Received: 22 December 2021; Accepted: 28 January 2022;
Published: 22 February 2022.
Edited by:
Francesca Luisa Conforti, University of Calabria, ItalyReviewed by:
Caroline Vance, King's College London, United KingdomJoy Mitra, Houston Methodist Research Institute, United States
Copyright © 2022 Scarian, Fiamingo, Diamanti, Palmieri, Gagliardi and Pansarasa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stella Gagliardi, c3RlbGxhLmdhZ2xpYXJkaUBtb25kaW5vLml0
†These authors have contributed equally to this work and share first authorship