 Sam Amin1*
Sam Amin1* Marie Monaghan1
Marie Monaghan1 Angel Aledo-Serrano2
Angel Aledo-Serrano2 Nadia Bahi-Buisson3
Nadia Bahi-Buisson3 Richard F. Chin4
Richard F. Chin4 Angus J. Clarke5
Angus J. Clarke5 J. Helen Cross6Scott Demarest7
J. Helen Cross6Scott Demarest7 Orrin Devinsky8Jenny Downs9,10
Orrin Devinsky8Jenny Downs9,10 Elia M. Pestana Knight11Heather Olson12
Elia M. Pestana Knight11Heather Olson12 Carol-Anne Partridge13Graham Stuart14
Carol-Anne Partridge13Graham Stuart14 Marina Trivisano15Sameer Zuberi16,17
Marina Trivisano15Sameer Zuberi16,17 Tim A. Benke18
Tim A. Benke18- 1Department of Paediatric Neurology, Bristol Royal Hospital for Children, Bristol, United Kingdom
- 2Epilepsy Program, Department of Neurology, Ruber Internacional Hospital, Madrid, Spain
- 3Pediatric Neurology, Necker Enfants Malades, Université de Paris, Paris, France
- 4Royal Hospital for Sick Children, University of Edinburgh, Edinburgh, United Kingdom
- 5University Hospital of Wales, Cardiff University, Cardiff, United Kingdom
- 6Developmental Neurosciences, UCL NIHR BRC Great Ormond Street Institute of Child Health, London, United Kingdom
- 7Departments of Pediatrics and Neurology, University of Colorado School of Medicine, Children's Hospital Colorado, Aurora, CO, United States
- 8Department of Neurology, New York University, New York, NY, United States
- 9Telethon Kids Institute, The University of Western Australia, Perth, WA, Australia
- 10School of Physiotherapy and Exercise Science, Curtin University, Perth, WA, Australia
- 11Cleveland Clinic Epilepsy Center, Cleveland Clinic Learner College of Medicine, Cleveland, OH, United States
- 12Division of Epilepsy and Clinical Neurophysiology, Department of Neurology, Boston Children's Hospital, Boston, MA, United States
- 13CDKL5 UK, Somerset, United Kingdom
- 14Bristol Heart Institute, Bristol Royal Hospital for Children, University of Bristol, Bristol, United Kingdom
- 15Rare and Complex Epilepsy Unit, Department of Neuroscience, Bambino Gesù Children's Hospital, IRCCS, Rome, Italy
- 16Paediatric Neurosciences Research Group, Royal Hospital for Children, Glasgow, United Kingdom
- 17College of Medical, Veterinary and Life Sciences, University of Glasgow, United Kingdom
- 18Department of Pediatrics, Pharmacology, Neurology, and Otolaryngology, University of Colorado School of Medicine, Children's Hospital Colorado, Aurora, CO, United States
CDKL5 Deficiency Disorder (CDD) is a rare, X-linked dominant condition that causes a developmental and epileptic encephalopathy (DEE). The incidence is between ~ 1:40,000 and 1:60,000 live births. Pathogenic variants in CDKL5 lead to seizures from infancy and severe neurodevelopmental delay. During infancy and childhood, individuals with CDD suffer impairments affecting cognitive, motor, visual, sleep, gastrointestinal and other functions. Here we present the recommendations of international healthcare professionals, experienced in CDD management, to address the multisystem and holistic needs of these individuals. Using a Delphi method, an anonymous survey was administered electronically to an international and multidisciplinary panel of expert clinicians and researchers. To provide summary recommendations, consensus was set, a priori, as >70% agreement for responses. In the absence of large, population-based studies to provide definitive evidence for treatment, we propose recommendations for clinical management, influenced by this proposed threshold for consensus. We believe these recommendations will help standardize, guide and improve the medical care received by individuals with CDD.
Introduction
CDKL5 deficiency disorder (CDD) is a rare and X-linked dominant condition (1, 2), with many aliases, including Developmental Epileptic Encephalopathy 2 (DEE2) (3, 4). It is caused by loss-of-function variants in the CDKL5 gene (5) which maps to Xp22.13, a gene with 20 coding exons (6, 7). The gene codes for Cyclin-Dependent Kinase-like 5 (CDKL5) protein, previously known as Serine-Threonine Kinase 9 (STK9) (8). CDKL5 was first mapped by Montini et al. (9) before subsequently seeing an update to its described genomic structure in 2003 (6) by Kalscheuer and colleagues. It was at this time that CDKL5 was reported as the second cause of X-linked infantile spasms (ISSX), for the first time highlighting genetic heterogeneity in this clinical syndrome. Further genetic reports followed, describing CDKL5 variants as disease causing while also being genetically and clinically distinct from Rett syndrome (10–12).
As an X-linked dominant condition, CDD is more frequently found in females, with a varying report of female-to-male ratio of between 4:1 (2) up to 12:1 (13). Males are described as displaying a more severe phenotype. The incidence is estimated at between ~ 1:40,000 and 1:60,000 live births, approximating to one-third of the frequency of Dravet syndrome (1:20,000–1:50,000) (14, 15) or one-quarter of the frequency of Rett syndrome (1:10,000) (16). It is detected in 10–20% of females with early-onset DEEs presenting within the first 6 months of life and should be considered as part of a differential diagnosis for children, females and males, presenting with severe, early-onset epilepsy (17).
CDD presents with a broad phenotype that includes intellectual disability, and impairments in speech, gross and fine motor abilities (18), sleep, gastrointestinal function (19) and vision. Approximately 75% of individuals have cortical visual impairment (20, 21). Seizures typically present in early infancy, with a wide spectrum of semiologies, and are often refractory to treatment (22, 23). Criteria for recognition and diagnosis have been proposed to guide clinicians (2). Evidence is emerging of genotype phenotype correlations for CDD gene variants (24). Evidence-based guidelines have recently been suggested for another DEE, Rett syndrome (25) but there is currently a paucity of evidence and no published consensus to guide clinical management in CDD. Given the broad phenotype, unique features and rarity of CDD, an initial document describing comprehensive care is needed to assist specialist and primary care practitioners caring for individuals diagnosed with CDD. Accordingly, we reviewed the literature and used consensus methods to establish recommendations for clinical management in CDD.
Methods
Study design: Delphi method.
Literature review and initial guideline development: We performed a literature search and considered mortality, morbidities, diagnosis, treatment, and surveillance of CDD. We used Medline/Pubmed and the Cochrane Library to perform the search.
The main search terms were: “CDKL5” and “Cyclin-dependent Kinase-Like 5.” Other associated search terms, as relevant to the topics of interest, were also searched for in combination with the main search terms.
Searching of the Cochrane Library (26) yielded no articles featuring either of the two main search terms. Searching Medline (Pubmed) (27) for the presence of either of the two main search terms, within title or abstract, yielded 452 articles. The search term, “CDD” was not used, to avoid unrelated terms or conditions with the shared abbreviation e.g., “cervical degenerative disease” (CDD). To inform the questions of the survey, the evidence on each topic was reviewed by filtering the main search term results (n = 452) to identify topics of interest as included in the title or abstract (Figure 1).
Figure 1. Identification of studies.
Based on these findings, we identified 84 questions for consideration in the Delphi process. The items queried all aspects of CDD including initial assessments, diagnosis, treatment options, follow-up, and surveillance.
The questions were formed by a core committee and reviewed by a subcommittee. Families and caregivers also contributed by review of the design. The Delphi results were analyzed by committees, which consisted of experts in different aspects of CDD from the US, Europe, and the UK. Patient advocacy groups were also part of this process.
Delphi consensus method: The Delphi process provides consensus guidance for the delivery of best clinical care. It is important that the participants are selected carefully. One potential pitfall in a consensus process is that when questions address issues without an evidence base, some respondents may provide answers despite a lack of specific knowledge. It is crucial, therefore, that the respondents are experts in the field. It is generally believed that 15–20 subjects could be sufficient to take part in a Delphi process but the higher the number of the subjects and homogeneity of response, the better the outcome. Some papers have cited that consensus thresholds can be accepted even low as 51%, “in keeping with most other Delphi studies” (28) with others recommending that consensus thresholds should be higher or require unanimous agreement, depending on the gravity of the decisions being made (29). Reviews of Delphi methodology describe the varying nature of the consensus thresholds but all note the importance of having a pre-defined threshold level for consensus, to avoid author bias upon review of responses (30). For this project, a priori consensus was defined as 70% agreement for all areas.
General pediatricians, pediatric neurologists, ophthalmologists, developmental pediatricians, geneticists, orthopedic surgeons, adult neurologists, rehabilitation clinicians, allied health professionals, gastroenterologists and nutritionists were invited to take part. All the people surveyed were based in the US, Europe, and the UK. Clinicians were identified through CDD clinics and Centers of Excellence, and researchers were identified through publications. The surveys were conducted over 6 months from August 2020 to January 2021. A weekly electronic reminder was sent to the responders. Forty-seven experts responded to the survey. The responders were pediatric neurologists (n = 30), epileptologists (n = 10), geneticists (n = 2) as well as a general pediatrician, a development/community pediatrician and an allied health professional. Two of the 47 respondents did not describe their specialty. The number of years of experience within their current specialty favored highly experienced professionals, with 58% (n= 27) having >15 years, followed by 34% (n = 16) with >5 years of experience. Professionals had a mixed range of experience in managing CDD; under half had managed <5 individuals (n = 22, 47.8%) followed by nearly a fifth that had managed 6–10 individuals (n = 9, 19.6%) with nearly a third (32%) having managed >10 individuals with CDD (n = 15). While CDD exposure had been mixed, most of the surveyed professionals (n = 46, 97.9%) had significant (>20 individuals) experience in managing DEEs. Many members of the core and subcommittee had a wealth of expertise in managing patients with CDD, leading their national centers of excellence in their practicing countries.
Results
The survey contained questions relating to current practice in CDD and was sent to the respondents. Answers where respondents did not feel they had relevant experience to be able to answer, indicated by selection of “I am not qualified to answer” or “I do not know,” were excluded, for the purposes of assessing the degree of consensus of opinion among experienced responders for each particular topic. Questions referring to “at baseline” were in reference to where the diagnosis of CDD had already been made, with the exception of genetic testing (CDD is considered a genetic diagnosis).
Genetic Screening and Counseling
ACMG (American College of Medical Genetics and Genomics) guidelines state that genetic counseling should be “offered at all stages of genetic testing”.
Survey
There was consensus in favor (45 responses, 97.8%) of offering a genetic test, before diagnosis was established, to all individuals with DEE. There was no consensus when asked when they would offer genetic counseling; with responses nearly equally divided into “Prior to genetic testing” (17 responses, 44.8%) and “After genetic testing” (21 responses, 55.2%). There was no agreement between the geneticists who responded to this question.
Neurological Assessment
Clinical Management
CDD is a disorder associated with DEE. Seizures often take the form of spasms, with or without hypsarrhythmia demonstrated on electroencephalogram (2, 11, 20, 31, 32), tonic seizures, and hypermotor (mixed) seizures (20). In addition, individuals may present with hypotonia (33). Male children with CDKL5 mutations are believed to be more severely affected and have a higher frequency of epileptic (infantile) spasms and brain atrophy (34).
Survey
Regarding formal assessments by a pediatric neurologist, 97.6% (40 responses) of respondents felt individuals should be seen at baseline, and thereafter regularly. Asked whether individuals should be seen by a pediatric epilepsy specialist at baseline and regularly, the response was the same with 95.2% (40 responses) in agreement.
While Sudden Unexpected Death in Epilepsy (SUDEP) is reported to occur in individuals with CDD, data from large cohort studies suggests the frequency of SUDEP within the CDD population is lower than for Dravet syndrome or SCN8A-DEE, given the frequencies of these disorders (35–37). The annual risk of SUDEP among individuals with CDD remains uncertain due to the limited data.
Survey
Respondents were asked whether families should be informed about SUDEP at baseline. The responses were mainly in favor (86.5%, 32 responses), meeting the threshold for consensus.
Survey
The respondents were asked which laboratory tests should be carried out at baseline. Mixed responses included: “blood count” (18 responses, 64.3%), “vitamin D” (18 responses, 64.3%) and “urea and creatinine” (16 responses, 57.1%). Twenty-five percent (7 responses) felt no blood tests should be routinely performed. Similarly, when asked which should be carried out annually, leading responses were “blood count,” “vitamin D” (each having 16 responses, 59.3% each) and “metabolic profile with urea and creatinine” (14 responses, 51.9%). The predominance of these basic profiles and a vitamin D level suggest that the purpose of such tests is not for diagnostic benefit but to reduce the risk from associated comorbidities e.g., from gastrointestinal dysfunction or reduced mobility with associated fracture risk, as in other DEEs. There were 7 respondents who believed no annual laboratory requests should be performed (25.9%).
Neuroimaging
In terms of neuroimaging, there are limited, non-quantitative reports on the findings associated with CDD. One study (38) reported “cortical atrophy” in 13 of 20 girls, associated with areas of increased T2 signal in the white matter, especially in the temporal lobes in some.
Survey
Respondents were asked whether all individuals should have a brain MRI scan at baseline for those who have not been investigated with an MRI previously. The responses did reach a consensus with 70.3% (25 responses) responding “Yes.” As a follow-on, those who had responded “Yes” were asked whether all individuals should have a DTI brain MRI scan at baseline. Currently DTI is an area of research interest with no reports published in relation to CDD. The majority did not feel this was required; “No” (76.7%, 23 responses).
Electroclinical Findings and Use of Electroencephalogram
Individuals with CDD typically present with epileptic spasms within the first 4 months of life and subsequently manifest epileptic encephalopathy (32, 38, 39). Electroclinical findings in the first year of life include a peculiar seizure pattern with “prolonged” generalized tonic-clonic events, lasting 2–4 min, consisting of a tonic-vibratory contraction, followed by a clonic phase with a series of spasms, gradually transitioning into repetitive distal myoclonic jerks (40). The EEG during these seizures shows a bilateral, synchronous initial flattening, followed by repetitive sharp waves and spikes. Atypical hypsarrhythmia is often seen in infancy, developing into multifocal abnormalities in older individuals (38). Typical EEG findings develop over time and are not manifest in young infants. This likely reflects limited functional cortical organization in young infants, necessary to propagate and sustain an electrical discharge, and limited interhemispheric transmission from commissural immaturity (41, 42). Early EEG findings can vary from normal background to moderate slowing, with superimposed focal or multifocal interictal discharges and rarely, a burst-suppression pattern (40). In a follow-up of children older than 3 years, about half experienced seizure remission while continuing anti-seizure drugs, with the other half continuing to have intractable spasms, often associated with multifocal and myoclonic seizures (38, 39).
Survey
Most (86.0%, 37) respondents supported an EEG at baseline, regardless of clinical seizures. Most (76.9%, 30) respondents favored EEG performed to capture epileptic spasms before treatment. For less typical seizure-like events, respondents were asked whether an EEG should be repeated to capture and classify spells of unclear clinical significance. Responses were in favor, with 97.6% recommending this (40 responses). There was no consensus when asked what duration of an EEG to request. The leading response was “Routine (under 2 h)” (18 responses, 51.4%). The variation of responses may reflect the availability of prolonged EEG.
Seizure Management–Use of Antiseizure Drugs and Ketogenic Diet
Seizures associated with CDD typically present in early infancy, with a wide spectrum of semiologies, and are often refractory to treatment (22, 23). The most common seizure types in CDD are epileptic spasms (often without hypsarrhythmia) and tonic seizures that may cluster (20). It is uncertain what proportion of epileptic spasms are attributable to CDD, however one study identified 3 patients with pathological variants in CDKL5 among 73 patients with epileptic spasms (43). Other seizure types have been described including atonic, atypical absence, focal with motor components, myoclonic, typical absence and tonic-clonic (44). To have pathological variants in CDKL5 without associated seizures is extremely rare but has been reported (22) although this is unlikely to affect CDD being considered a DEE.
The treatment of epileptic spasms encompasses aspects of seizure control, side-effects and longer-term neurodevelopmental outcomes. O'Callaghan et al. performed a multicentre, open-label randomized controlled trial to investigate the effect of treatment options, either oral prednisolone (10 mg four times a day) or intramuscular tetracosactide (0.5 mg (40 IU) on alternate days), with or without oral vigabatrin (100 mg/kg per day) (45). The primary outcomes at 18 months, independently assessed, were neurodevelopmental outcomes and the frequency of seizures. While this study was not focussed on the epileptic spasms associated with CDD, it identified that earlier seizure control was a predictor of better developmental and epilepsy outcomes at 18 months. While earlier seizure control was obtained in the combination therapy group, it was surprising that there was no statistically significant difference in developmental or epilepsy outcomes at 18 months between the two groups (combination therapy or hormonal therapies alone). The authors explained this incongruity with the suggestion that those who had not responded to hormonal therapy alone would have rapidly received additional vigabatrin and therefore received combination therapy. Furthermore, any improvement in development associated with earlier cessation of seizures with combination treatment, may be undermined by the potential negative side-effects of vigabatrin such as drowsiness and visual field defects, as listed among others in the British National Formulary for Children. Studies assessing neurodevelopmental and seizure outcomes would be welcome for individuals with epileptic spasms associated with CDD, in light of reports of worse seizure outcomes with hormonal therapy for individuals with CDD. One study (22) assessed seizure variables in relation to CDD genotype and found that with a median age of questionnaire completion at 5 years, those who had previously been treated with corticosteroids had more frequent seizures than those who had never been treated, irrespective of a history of epileptic spasms.
Studies looking at the efficacy of anti-seizure drugs in the treatment of CDD-related epilepsy have frequently shown only temporary and frequently paradoxical (exacerbation) responses to various anti-seizure drugs, despite the use of medications with different mechanisms of action (23). In one study looking at the effect of anti-seizure drugs in 39 individuals with CDD (23), the highest, but still very low, responder rate after 12 months was reported with sodium valproate (9%, 3 individuals) whereas there was a very low number of individuals that responded to phenytoin, felbamate, carbamazepine and clonazepam. Drug response was defined as a more than 50% reduction in the preceding 4 weeks, compared to 4 weeks in the baseline period before starting the new anti-seizure drug. In this study, steroids/ACTH had a 19% (5) responder rate at 3 months but 0% response rate at 12 months. Similarly, vigabatrin had a 32% (8) responder rate at 3 months but just a 4% (1) responder rate at 12 months (23). For patients with earlier onset epilepsy with focal epileptiform activity, there is evidence supporting the use of sodium channel blockers, such as oxcarbazepine, carbamazepine and lacosamide (46).
Initial apparent benefit with subsequent loss of anti-seizure drug efficacy over time in the management of epilepsy associated with CDD has been described as the “honeymoon effect” (2, 22). This was first described following analysis of caregiver reports on the effects of anti-seizure medication on seizures from caregivers of 163 individuals with CDD with epilepsy registered in the CDKL5 Disorder Database (22). It was found that fewer than half (43%, 71/163) of caregivers reported ever having had more than 2 months of seizure freedom. Typically the honeymoon period had a median onset of 2 years (for 74%, 52/70) and a median duration of 6 months (for 84%, 59/70).
Survey
Respondents were asked to rank their first, second, third and fourth-line therapies for epileptic spasms associated with CDD. There was no consensus for any of the first, second, third or fourth line suggested therapies, although the standard treatments of vigabatrin, steroids and the combination of these featured most strongly. For first line therapy, 37.5% (15 responses) favored combination therapy (steroids and vigabatrin), 35% (14 responses) favored steroids alone and 27.5% (11 responses) favored vigabatrin alone. No responder suggested use of ketogenic diet as a first line therapeutic option. Similarly, there was no consensus among second line therapy options, however among a choice of steroids, vigabatrin, combination of these or the ketogenic diet, the ketogenic diet was selected by nearly a quarter (23.1%, 9 responses) as a second line therapeutic option. The ketogenic diet similarly made up an increasing preference (17 (54.8%) and 10 (41.7%) responses) for third and fourth line therapy preferences. The ketogenic diet was considered by respondents as early in the management of seizures as a second or third line therapy option, with few other epilepsies, e.g., SLC2A1 mutation (47), prompting such early consideration.
Lim et al. (48) studied the use of the ketogenic diet to manage refractory epilepsy associated with CDD. They found that of the approximately half of individuals with CDD who have tried the ketogenic diet, some 59% of individuals experienced improvement in seizure frequency, duration, or intensity. However, none of the individuals on the ketogenic diet became seizure-free. This lack of complete resolution of seizures, along with side-effects of the diet, led to poor long-term adherence (median duration 17 months). In a study on quality of life domains for individuals with CDD, 20% (5 of the 25 surveyed) were currently on a ketogenic diet (49).
Survey
The respondents were asked whether individuals should be treated with a ketogenic diet as soon as they fail their first line treatment for epileptic spasms. The responses were mixed with most in favor (23 responses, 53.5%). This response may be interpreted as encouragement for starting a ketogenic diet at the soonest moment that a first line therapy has proven inadequate for controlling epileptic spasms and that the diet may be in addition to a second line medication option (differentiating this nuance from the preceding survey responses).
While several studies looking at the use of CBD for the treatment of drug-resistant epilepsy have shown promising results, few have provided specific results for the performance of CBD in the CDD subpopulation (50). Devinsky et al. (51) undertook an open-label study exploring the use of CBD in individuals with severe, treatment-resistant, childhood-onset epilepsy including CDD, among other disorders. In individuals with CDD, the median monthly convulsive seizure frequency decreased from baseline (66.4 [n = 17], IQR: 25.9-212.0 to week 12 (35.8 [n = 11], IQR: 8.9-141.6) which was found to be statistically significant (p = 0.032). Further placebo-controlled randomized trials in a larger population sample are necessary to formally assess the safety and efficacy of cannabis-based products in CDD.
Survey
There was consensus on whether CBD (Epidiolex) should be offered for epilepsy in CDD. The responses provided strong support for this option with 92.6% (25 responses) in favor with 7.4% against (2 responses). This reflects an increasingly positive view of CBD for medicinal uses, including in the pursuit of reducing seizure burden among populations of children with mixed etiologies of drug-resistant epilepsy (52, 53).
Ganaxalone is a synthetic methyl derivative of allopregnanolone, a neurosteroid, which acts as a high-affinity allosteric modulator of GABAA receptors. Ganaxalone has been trialed for epilepsies including epileptic spasms, status epilepticus and protocadherin 19 related epilepsy (2). The Marigold Study (NCT03572933) is the first Phase 3, randomized, placebo-controlled trial that evaluated adjunctive ganaxolone in patients with refractory epilepsy associated with CDD. Patients on ganaxolone experienced a median of 30.7% reduction in major motor seizure frequency compared to a 6.9% reduction in the placebo group during the treatment period relative to baseline (p = 0.0036, Wilcoxon Rank-Sum Test). Ganaxolone demonstrated improving trends but did not achieve statistical significance in the key secondary endpoints. Adverse events occurred in 86% of ganaxolone patients and 88% of placebo patients. Ganaxolone was generally well-tolerated with a <5% discontinuation rate in the treatment arm, with somnolence being the most frequent adverse event (36% of patients) (54).
Survey
Respondents were asked whether Ganaxolone should be offered, if available (dependent on regulatory approval). The unanimous response was “Yes” (27 responses, 100%), meeting the threshold for consensus. The FDA has just approved ganaxolone (Ztalmy; Marinus Pharmaceuticals) for the treatment of seizures associated with CDD, in patients aged 2 years and older.
Epilepsy Surgery
The effects of vagus nerve stimulation (VNS) for the treatment of refractory epilepsy for CDD has been studied (55). Of 222 patients with CDKL5 variants where there was adequate information, 38, the equivalent of 1/6 or 17% had previous or current use of VNS. Improvement in seizure control was reported in 69% (25/36) and of them, this related to improvements in frequency in 68% (17/25), duration in 72% (18/25) or intensity in 60% (15/25). No patient with a VNS became seizure-free and termination of VNS occurred in 1 in 10 cases.
Survey
Respondents were asked whether individuals should be considered for VNS insertion if seizures are refractory to medications. The responses were mainly in favor (89.7%, 35 responses).
Patients with non-resectable, drug-resistant seizures with spread between hemispheres, i.e., generalization, may be considered for corpus callosotomy. In a meta-analysis of the effects of corpus callosotomy in epilepsy surgery, analyzing the impact of corpus callosotomy on 1,742 children and adults from 58 studies, it has been shown to be associated with drop attack freedom in 55.3% and complete seizure freedom in 18.8% (56). For those achieving complete seizure freedom, this favored patients whose etiology included infantile spasms (OR 3.86, 95%. CI 1.13-13.23), normal MRI (OR 4.63, 95%. CI 1.75-12.25), and a shorter epilepsy duration of <15 years (OR 2.57, 95%. CI 1.23-5.38). Interestingly, neither the presence of lateralising EEG abnormalities nor the selection of complete vs. partial corpus callosotomy made a significant impact on the outcome, unlike in the analysis of patients with drop attacks where these were associated with improved outcomes.
Survey
Respondents were asked whether individuals should be considered for corpus callosotomy if seizures were refractory to medications. The leading response was 71.0% in favor (22 responses), meeting the threshold for consensus.
Stereotypes and Movement Disorders
Hand stereotypies are reported in 80% of individuals and can negatively affect functional hand movements in 59% of females and 12.5% of males with CDD (1). Olson and colleagues (unpublished) describe self-stimulatory hand movement syndrome and repetitive leg crossing in CDD patients. Unquantified episodes of persistent, occasionally severe, choreoathetosis, akathisia, dystonia and parkinsonian features have been reported, potentially having been unmasked during temporary periods of improved seizure control or potentially secondary to polytherapy with antiseizure drugs (2).
Survey
Respondents were asked whether individuals should be screened for movement disorders at baseline. The responses were: “Yes” (39 responses, 100%), achieving consensus. The respondents were also asked whether individuals should be screened for movement disorders at regular clinical appointments, annually, with 100% in favor (38 responses). Respondents were 100% in favor with regard to movement disorders being treated if causing problems. Asked what would be the most suitable option, the leading responses were: “Gabapentin” (15 responses, 62.5%), “Clonidine” (13 responses, 54.2%) and “Benzodiazepines” (10 responses, 41.7%).
International Registry
With increased attention on therapies for CDD, prospective, randomized, and double-blind clinical trials are considered essential to establish statistical significance and thus will necessitate international collaboration (57).
Survey
When asked whether individuals should be offered to be enrolled in an international registry or other research studies, 100% were in favor (46 responses).
Neuropsychological Assessment
Survey
When asked whether individuals should have a neuropsychology assessment at baseline (where the diagnosis has already been made), there were mixed responses with 59.4% in favor (19 responses). Similarly, when asked whether individuals should have a neuropsychology assessment regularly, responses were: “Yes” (26 responses, 68.4%). This did not meet the threshold for a consensus of opinion.
Somnology
Sleep-related difficulties are reported in over 85% of individuals with CDD, sometimes dubbed “all night parties” with problematic night-waking reported in up to 58.5% (1, 2, 19) and males more severely affected (19). Sleep apnoeas have been documented in both individuals and mouse models of CDD (58, 59). The odds of reported sleep difficulties was higher in the 5–10 year age group than the under 5 year group (19).
Survey
Respondents in our survey were asked whether individuals should have their sleep assessed at baseline. The leading response met the threshold for consensus with 92.3% (36 responses) in favor. Similarly, there was consensus when respondents were asked whether individuals should have their sleep assessed annually with 85.7% (30 responses) in favor. When respondents were asked which drug or drugs could be used to help with sleep, the leading response, “Melatonin” (35 responses, 53.8%), did not meet the threshold for consensus of recommended first choice, however, was more popular than the second most selected answer, “Clonidine” (16 responses, 24.6).
Therapy Assessments and Interventions
Neuro-Rehabilitation Assessment
Neuro-rehabilitation services, sometimes referred to neuro-developmental or neuro-disability services, are part of the care of individuals with CDD. Assessing function and response to therapies is important in guiding and interpreting the findings of future research into therapies for CDD (60). A collaborative professional and caregiver-based standardized assessment method was designed using four cycles of a Delphi process, the CDD Clinical Severity Assessment (CCSA). This involved clinicians from the International Foundation for CDKL5 Research Centers of Excellence (COE) consortium and the National Institutes of Health' Rett Syndrome, MECP2 Duplication Disorder, and Rett- Related Disorders Natural History study consortium (U54 HD061222; ClinicalTrials.gov: NCT00299312/ NCT02738281). Initial consensus was provided by clinicians, researchers, industry, patient advisory groups and the parents of a child. The CCSA reviewed 53 items, 27 reported by parents and 26 reported by clinicians. It has recently been developed (61) and validated to enable its implementation for the assessment of outcome measures, as per FDA requirements (62, 63).
The final CCSA will be 50% clinician assessment of motor, cognition, behavior, vision, speech and autonomic function domains. The other 50% will be parent-led assessment, complimentary to the design and structure of the clinician assessment. The aims of the CCSA are to support design and interpretation of research, evidence-based management choices in CDD and identification of current patient needs. Specific items capture levels of functioning in the gross motor, hand function, communication and behavior domains.
Survey
We asked whether individuals should be offered a referral to a neuro-rehabilitation service at baseline, to assess equipment needs and diagnose or improve problems with mobility and hand function and to prevent contractures. There was strong support for this with 91.9% of respondents (34 responses) in favor. Similarly, when asked whether individuals should be offered a referral to a neurorehabilitation service annually for the same purpose, 92.1% (35 responses) were in favor.
Development Assessments
CDD is associated with global developmental delay including intellectual disability. Most individuals are severely impaired. In one study (18), data for 108 females and 16 males, registered with the International CDKL5 Disorder Database, were collected. Over half of females could sit on the floor and nearly a quarter could walk 10 steps. Most females and few males were able to pick up a large object. Those with a late truncating variant displayed better levels of ability than those with no functional protein. Subsequent research has expanded the correlations of the genotype-phenotype (20).
This work was also performed using an expanded cohort from the same International CDKL5 Disorder Database (24). The study looked at genotype-phenotype findings for 385 individuals with CDD. They then assessed genotype-phenotype relationships for 13 recurrent CDKL5 variants and compared these with previously analyzed historic variant groups. Developmental scores and severity assessments were performed using the CDKL5 Developmental Score (CDS) and an adapted CDKL5 Clinical Severity Assessment (CCSA). Individuals with the missense variant, p.Arg178Trp, had the highest mean adapted CCSA and lowest mean developmental scores. They also found that p.Arg559* and p.Arg178Gln produced severed phenotypes whereas p.Arg134*, pArg550* and p.Glu55Argfs*20 produced milder phenotypes. This study identified trends between variants and phenotypes and updated historic genotype-phenotype reports.
Regression, if encountered, is often related to worsening of seizure control and the presumed effect of epileptic encephalopathy (1, 18, 32, 33, 64). In girls, walking is attained by 22%, raking grasp by 49% by 5 years and pincer grasp by only 13% at any point (18, 65).
Survey
We asked whether individuals with CDD should have developmental assessments and 100% were in favor (44 responses), with 75% (24 responses) proposing, “Soon after diagnosis,” meeting the threshold for consensus. Nearly all (95.3%, 41 responses) of respondents felt developmental status assessment should be repeated. Nearly all (92.3%, 36 responses) felt the assessments ought to occur at key developmental points and periods of transition, proposed as during infancy (0–3 years), preschool age (3–6 years), pre-middle school age (6–9 years), adolescent age (12–16 years, early adulthood (18–25 years) and as needed thereafter.
Ophthalmology
CDD is associated with cortical visual impairment (CVI) with approximately 75% having cortical visual impairment (20).
Survey
The respondents were asked whether individuals should have a detailed vision assessment at baseline. The responses were: “Yes” (38 responses, 100%). Similarly, respondents felt individuals should have an annual vision assessment with all in favor (29 responses, 100%). When asked whether individuals with CDD should be referred to an ophthalmology specialist familiar with cortical visual impairment, for assessment, the responses were strongly (100%, 37 responses) in favor. For management by an ophthalmology specialist familiar with CVI, the responses were also 97.1% (34 responses) in favor.
Speech and Language Assessment and Communication Aids
As part of global developmental delay and associated cortical visual impairment, individuals with CDD experience difficulties with communication (18). In one study (65), it was found that under half of individuals could babble by the age of six (43/97, 44%) and under a quarter could say single words by the age of seven (17/105, 16%). Only 7.5% of females achieve speaking in full sentences (18) with males 80% less likely than females to be able to use advanced communication methods (OR 0.17, 95% CI 0.04–0.71). Upon assessment and categorization of highest communication ability, it was found that 26% were able to use spoken language, sign language and abstract symbols, followed by 39% who were able to use complex gestures, vocalizations and concrete symbols with 33% able to use only simple communication alone (such as body language, early sounds, facial expressions and simple gestures). While speech difficulties can present with other features suggestive of autism, this diagnosis is infrequently made while in the context of severe global developmental delay (2).
There have been few studies published reviewing the use of non-verbal communication aids for individuals with CDD. Unpublished data by Olson et al., reviewed the use of devices such as switches and eye gaze technology-based communication aids. They found that in those unaffected or mildly affected by cortical visual impairment, such devices provided assistance for some with CDD. A recent systematic review has investigated outcomes and uptake barriers for the pediatric population with complex disabilities using eye gaze assistive technology (66). This analysis reviewed the use of eye gaze technology on the World Health Organisation's International Classification of Functioning, Disability and Health Framework. There were 11 articles suitable for review, of which eight assessed communication and of which six reported enhanced communication outcomes. The review highlighted poor methodological quality and/or low level evidence, limiting the review's findings and reflecting a need for further published and high-quality evidence.
Survey
When asked whether individuals with CDD should be checked and assessed for augmentative and assistive communication aids such as switches, touch pads or eye gaze aids, respondents were unanimously in favor (41 responses, 100%).
Orthopedic, Physiotherapy and Occupational Therapy Assessments
Orthopedic concerns are a potential consequence of hypotonia and can lead to scoliosis, with 68.5% of individuals affected by 10 years (1, 19).
Survey
Asked whether individuals should have a hip and spine X-ray, most responses were: “If there is a clinical concern” (31 responses, 77.5%), reaching the threshold required for consensus. Respondents did not favor individuals with CDD having a routine orthopedic (specialist surgeon) review at baseline, with the leading response being not in favor (22 responses, 73.3%). Equally, when asked whether individuals should have a routine yearly orthopedic review, the responses leaned toward not being in favor (15 responses, 53.6%). Whether reflecting concerns (e.g., pertaining to reduced mobility or a ketogenic diet) when asked whether individuals with CDD should be offered a screening test for osteopenia (such as wrist X-ray or DEXA scan), the leading responses was: “If clinically indicated” (28 responses, 82.4%).
Consensus guidelines for the approach to screening and management of scoliosis and osteopenia are not available for CDD however a consensus of routine management for optimal bone health in Rett syndrome has been developed and is likely relevant to individuals with CDD until higher level evidence becomes available (25, 67–69).
Fu et al. provided observational data for 913 females with classic Rett Syndrome. They identified that severe scoliosis was found in 251 participants (27%), 113 of whom developed severe scoliosis during follow-up assessments with 168 (18%) having surgical correction. The study proposed the implementation of spinal bracing when spinal curvature reaches 25°, in the hope of retarding or minimizing further progression. Beyond 40°, the authors strongly promoted surgical intervention. Each study suggests annual evaluations for both of these issues along with guidelines for management and referrals.
Survey
There was consensus in favor when asked individuals with CDD should be offered Physical Therapy (PT) assessment at baseline (where diagnosis has already been made) with 97.8% of respondents in favor (44 responses). Equally 97.8% (44 responses) felt that individuals with CDD should have access to PT regularly for ongoing issues.
Survey
Asked whether individuals should be offered an occupational therapy (OT) assessment at baseline (where diagnosis has already been made), the responses strongly in favor (38 responses, 92.7%). Similarly, when asked whether individuals with CDD should have access to OT regularly for ongoing issues, the responses were strongly in favor (42 responses, 100%).
Educational
Individuals with CDD face difficulties such as communication difficulties and cortical visual impairment. Interventions, such as visual attention tracker, may assist in informing the wider team whether educational interventions are providing benefit (70).
Survey
Asked whether educational accommodations for visual impairment should be provided, 97.6% (41 responses) were in favor. More broadly, 92.1% (35 responses) were in favor when asked whether educational support provided in formal educational plans should be reviewed at baseline. Similarly, respondents felt a review of these should be performed annually, with 94.9% (37 responses) in favor.
Systemic
Auxology
Five individuals with CDD were reported to have normal head circumferences at birth and over the subsequent 2 years develop postnatal microcephaly (64). Similarly, deceleration of head growth has been described in 11 out of 20 (55%) individuals with CDD (33). Microcephaly has been associated with an increased degree of functional impairment (71).
Survey
When asked whether head circumference, weight, height should be each checked at baseline, respondents were in favor; 100% (46 responses), 97.8% (45 responses) and 97.6% (42 responses), respectively. Similarly, when asked whether height and weight should be checked annually, 100% (43 responses) were in favor.
Gastrointestinal Management Including Assessment and Management of Feeding
Patients with CDD may experience dysphagia and require gastrostomy (2). Evidence suggests that gastrostomy tube feeding for pediatric patients with neurological impairments may reduce the risk of death although associated with an increased the risk of severe pneumonia (72). Guidelines produced by the European Society for Pediatric Gastroenterology, Hepatology and Nutrition, for the evaluation and treatment of gastrointestinal and nutritional complications in children with neurological impairment, recommends the use of enteral tube feeding in cases of unsafe of inefficient oral feeding, preferably before the development of undernutrition, and that a gastrostomy is the preferred way to provide intragastric access for long-term tube feeding for this population. Aside from nutritional difficulties affecting growth, a gastrostomy tube may improve caregiver quality of life, assist in the administration of fluids and/or a ketogenic diet and, through compliance with medications and/or ketogenic diet, may reduce seizure burden (73, 74). A review of patients from the CDKL5 Disorder Database found that 20.7% of individuals were fed exclusively by gastrostomy or nasogastric tube (19) but this prevalence may be as high as 43% among individuals with CDD, following analysis of patients based in the United States of America (75) (154 individuals identified from data held by Centers of Excellence and 40 identified from the NIH's Natural History of Rett and Related Disorders database). In a smaller study on quality of life domains for those with CDD, as many as 56% (14/25 surveyed from the CDKL5 international registry) had a gastrostomy (49).
Survey
Respondents were asked whether gastrointestinal complications such as constipation, air swallowing and acid reflux should be assessed at each clinic visit annually. The responses were strongly in favor (43 responses, 97.7%). Asked whether individuals should be referred to a Gastrointestinal specialist, responses were in favor (92.0%, 23 responses). When asked whether individuals should be referred to a Nutrition specialist, responses were also in favor (30 responses, 96.8%). When asked when swallowing coordination should be formally assessed (i.e., by Speech and Language Specialists) most felt this should be, “Only if there are concerns” (25 responses, 61.0%). Respondents were more strongly in favor of individuals being offered an informal speech therapy assessment at baseline (where diagnosis has already been made) (38 responses, 92.7%). Similarly, a large majority felt that non-specialist feeding, and swallowing should be assessed at annual clinical reviews (36 responses, 90.0%). Respondents were asked when a gastrostomy should be considered, with responses meeting consensus in the selection of, Either (including, “When weight or BMI inappropriately plateaus or tails” or “When swallowing is considered unsafe”) (31 respondents, 72.1%). A third of respondents (14 responses, 32.6%) felt this should be limited to “When swallowing is considered unsafe”.
Respiratory Assessment
Breathing abnormalities with CDD have been reported and include hyperventilation in 13.6%, breath holding in 26.4% and aspiration in 22.6% (19). The respondents were asked whether a formal respiratory review should be offered routinely at baseline, including a sleep study, to all individuals. There was no consensus however the lead response was “Only if clinically indicated” (28 responses, 66.7%). Similarly, when asked whether individuals should be referred to a pulmonologist/respiratory clinician, 81.0% reported “Only if clinically indicated” (34 responses). However, when respondents were asked whether a non-specialist assessment for breathing disorders, including hyperventilation, breath-holding and other conditions should be offered at each clinic visit annually, the leading response met the threshold for consensus with 90.5% (38 responses) in favor.
Cardiovascular Assessment
Parents of children with CDD may have concerns about the risk of cardiac arrhythmias and, in one caregiver survey, arrhythmia was reported in 11 out of 29 individuals with CDD who had been investigated with electrocardiogram (ECG) (76). Despite parental reports of arrhythmias, there is a lack of data on the rates of arrhythmia among individuals with CDD [from published reviews based on a cohort of 93 individuals published from the International Foundation for CDKL5's Research Centers of Excellence (2)].
Survey
When asked whether individuals should be routinely screened for cardiac issues at baseline (where the diagnosis has already been made), the most common responses were: “Yes” (26 responses, 78.8%), meeting the threshold for consensus. Similarly, when asked whether individuals should have an ECG at baseline (where the diagnosis has already been made) the most cited response was “Yes” (31 responses, 86.1%) achieving consensus. However, there was a lack of consensus when respondents were asked whether individuals should have a routine annual ECG, the leading responses were “Yes” (19 responses, 63.3%). Equally, when respondents were asked whether the individuals should have an echocardiogram at baseline (where the diagnosis has already been made), the leading responses were: “No” (15 responses, 57.7%) with fewer in favor of this (11 responses, 42.3%). Furthermore, when respondents were asked whether individuals should have a routine annual echocardiogram, leading responses were: “No” (23 responses, 88.5%). Lastly, when asked whether individuals should have a routine annual cardiological review by a cardiology specialist, the leading response was “No” (17 responses, 73.9%).
Dermatology
Survey
The respondents were asked whether individuals should have a routine check for pressure ulcers and skin breakdown at baseline (where the diagnosis has already been made). The lead response was in favor (38 responses, 90.5%). Asked whether individuals should have a regular skin check at their annual clinic review, responses were similarly in favor (38 responses, 95%).
Urinary Tract Care
Survey
When respondents were asked whether bladder-related issues should be checked regularly (e.g., urinary retention and urinary tract infections), it was felt this was appropriate with 94.1% of respondents in favor (32 responses).
Audiological
Survey
All survey respondents were in favor of individuals with CDD having an audiological assessment in the form of Automated Auditory Brainstem Response (AABR) screening (100%, 36 responses).
Dental Care
Survey
All survey respondents were in favor that individuals should have baseline and regular dental checks upon diagnosis of CDD (100%, 40 responses).
Financial
Survey
Respondents were asked whether financial support options should be explored as a baseline assessment upon diagnosis of CDD and annually, during clinic reviews. The responses were 100% with 43 responses and 39 responses respectively, both in favor.
Summary of Areas Not Meeting Threshold for Consensus
While there was no consensus in the current study regarding the timing of genetic counseling, the ACMG has provided recommendations for genetic counseling prior to and following genetic testing (77).
Notably, for a condition predominantly regarded as an epileptic encephalopathy in the domain of epilepsy management, there was no consensus on the first, second or third line choices of anti-seizure drug. This may reflect varying clinician preferences or clinicians individually tailoring management to meet the specific needs and varying seizure types of their patients. Nevertheless, vigabatrin, steroids and the combination of these featured most strongly, favoring combination therapy as first line (37.5%, 15 responses) for the management of epileptic spasms.
Summary of Areas Meeting Threshold for Consensus
The following table (Table 1) outlines the responses in the survey which met the pre-defined 70% requirement for consensus status, and their recommended timepoints (“baseline,” “annually” or “if clinically indicated”).
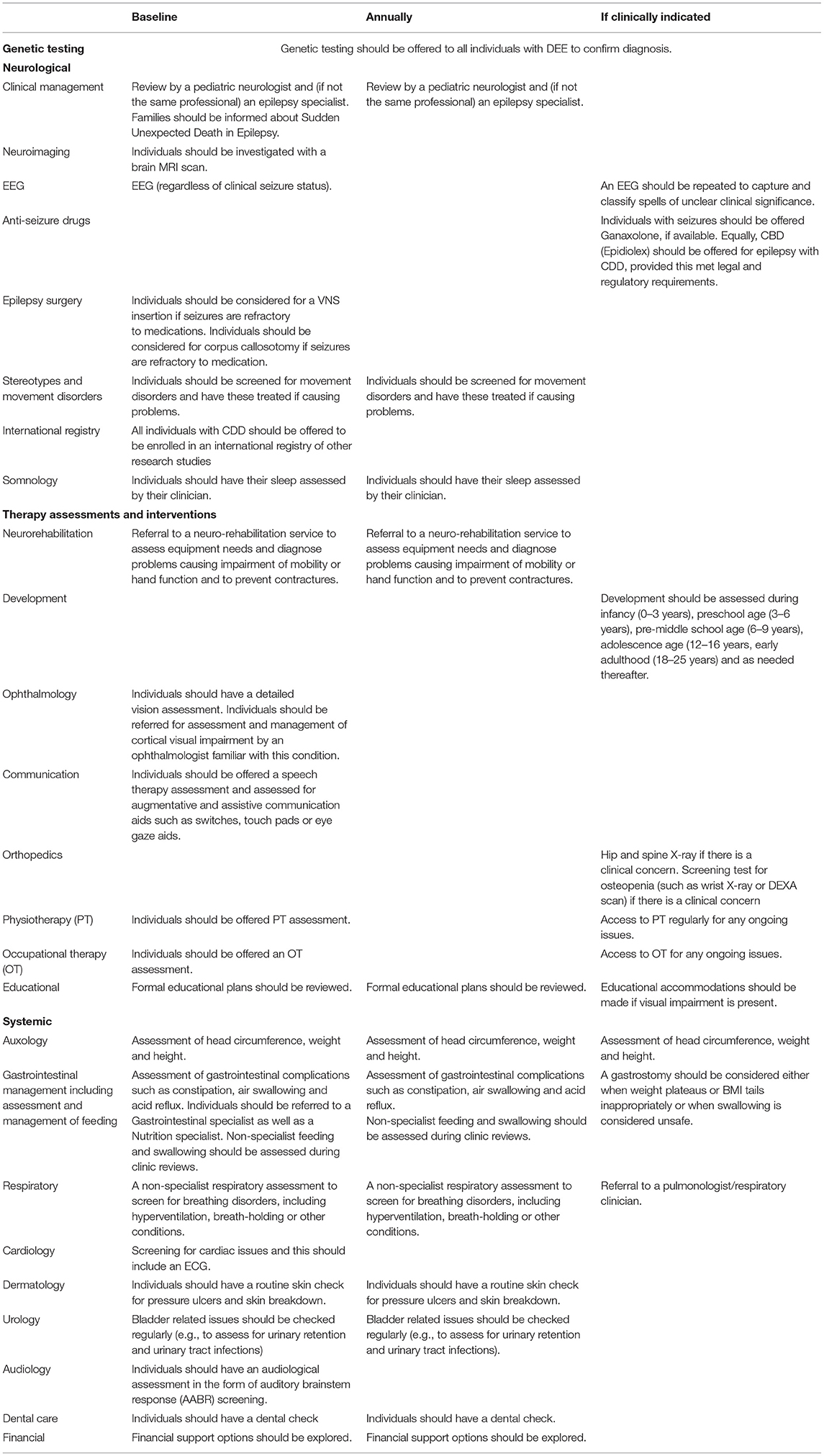
Table 1. Recommendations for the management of individuals with CDD with suggested timepoints for completion.
There were many areas of consensus recommendations identified. The majority of these are for completion at baseline. There is an emphasis upon holistic care, such as the monitoring of systemic functions and educational needs, with certain areas recommended to be reviewed, not only at baseline, but also annually and if clinically indicated. These included the monitoring of growth, the need for a regular review of feeding and swallowing, and non-specialist screening for respiratory difficulties.
A comprehensive neurological assessment is encouraged at baseline. The consensus recommendations are for the individual with CDD to be reviewed by a pediatric neurologist with experience in managing epilepsy, clinician discussion to inform families about the risk of SUDEP, completion of a baseline MRI and EEG, consideration for epilepsy surgery, screening for the presence of a movement disorder, registration with the CDKL5 international registry and a review of the individual's sleep. Despite limited published evidence on the use of novel antiseizure drugs for CDD in the literature, Ganaxolone and Epidiolex are encouraged to be offered for epilepsy associated with CDD, if clinically indicated, dependent on FDA and EMA approvals and legal and regulatory requirements, respectively.
Discussion
CDD is a debilitating condition where there is an urgent need for further development of management options. To achieve these necessary advances will require large scale and international, collaborative efforts to evaluate potentially effective interventions in sufficiently powered clinical trials. Progress will rely heavily on cooperation between international medical and scientific professionals, affected families, industry and funding organizations (57). The extensive experience of the author group includes those with direct experience in CDD management including authors of a clinically relevant CDD severity assessment tool (78). We hope that this survey adds to the current knowledge base concerning clinical aspects of care and provides a useful proposed standard of care elucidated by the agreed areas of consensus. These recommendations can support clinicians with less experience of CDD and act as a catalyst for further research that would aim to increase capacity for evidence-based management in CDD.
Limitations of Survey
In the survey there were occasions when incomplete responses were obtained, ie. fewer than 47 responses per question. This could represent difficulties in selecting the options available (for example, when no “other” option for selecting preferred first-, second- or third-line antiseizure drug preferences) or technical difficulties with the online survey.
For answers where respondents did not have experience in this area, answering “I am not qualified to answer” or “I do not know,” responses were excluded from analysis which led to a reduced number of responses included in the analysis. This was notable for certain technical questions, such as whether an MRI with DTI should be performed at baseline (8 respondents selected “Do not know/Do not feel strongly” and 6 selected “I am not qualified to answer”) and also for evolving areas of research interest, such as whether CBD (Epidiolex) should be offered for epilepsy in patients with CDD, where 6 respondents selected “Do not know/Do not feel strongly” and 7 selected “I am not qualified to answer.”
Certain answers provided professional discretion and may have been subject to personal interpretation, for example, in the use of screening tests for osteopenia, the leading response was ‘If clinically indicated' however the indications (e.g., poor mobility, fracture, poor height velocity, bony malformations) in this and other situations were not directly specified.
We invited respondents to provide additional feedback on areas of CDD management that were not covered in the survey. While the survey was designed and constructed with broad support at the outset, we acknowledge that some detail may have been overlooked and therefore we invited comments and suggestions for any missed areas at the end of the survey. There were few responses (4 out of 47) possibly suggesting the survey was felt to be sufficient by the majority. Of the responses, the feedback included a need to explore access to support groups and the contacting of other families. Another responder questioned whether mosaicism should be discussed within genetic counseling. This response may be in reference to reported findings of somatic mosaicism in patients with CDD (79, 80) or germline mosaicism with CDKL5 which was described in one family with two daughters with CDD found to have the same CDKL5 variant (c.283-3_290del) with parents that tested negative for CDKL5 variants in all tissues (81).
Further comments included reference to gynecological needs, not described in the survey. The responder queried whether clinicians should consider screening for precocious puberty or referring to gynecology, in the event of problems with menses. This suggestion addresses the unaddressed gynecological facet of CDD holistic care but may also be in reference to precocious puberty which has been described with CDD (82).
Reflective of increasing literature on CDD, one of the respondents suggested whether individuals should have an “anticipatory care plan” and whether this should be reviewed at least annually. This countered another piece of feedback: a concern that being too prescriptive with a potentially “exhaustive” list of management recommendations could heighten parental anxiety (if they feel they or those looking after their child are not fulfilling it). Clinicians managing CDD may need to decide whether to be “anticipatory” or, conversely, more “problem-driven” and which approach may be more appropriate for the individual and their family.
As with other work aiming to bring consensus to the understanding and management of CDD, our project lacks an objective “gold standard,” instead being designed with the topics and subtopic questions selected through limited published data, Delphi consensus and expert opinion. In the absence of a high level of evidence, Delphi consensus is considered the best available guidance. We recognize that despite our collective experiences, we are each limited by these experiences and the field still has much to learn regarding the breadth of patient experiences, potential treatments and outcomes. The concept of an “expert” is quite relative with regards to rare disorders such as CDD.
Given these shortcomings, additional discussion and study is needed regarding several issues. While our panel was equivocal, ACMG guidelines that genetic counseling should be provided at all phases of genetic testing (77) seems most prudent. Similarly, an approach toward scoliosis and osteopenia similar to that proposed for Rett Syndrome (25, 67–69) should be provided. All treatments carry risk of potentially significant morbidity and mortality that should be carefully reviewed with families so that informed treatment decisions should be made. Addressing a complete algorithm for use of anti-seizure medications, including variations with age and seizures types, was beyond the scope of our approach, but should be considered as a completely separate effort. A standard approach to epilepsy management in CDD including avoidance of polypharmacy should be considered, even though the literature indicates significant medical resistance (22).
Consistent with this, our survey indicates that medication and surgical options that may be offered to other individuals with medically resistant epilepsy, due to other causes, should also be offered to individuals with CDD. There has not been strong evidence until recently to support any specific treatment interventions in this population including steroids, surgery or any other specific anti-seizure medications. However, following the large international placebo controlled trial of ganaxolone, the FDA has just approved ganaxolone (Ztalmy; Marinus Pharmaceuticals) for the treatment of seizures associated with CDD, in patients aged 2 years and older.
Families should be part of the decision-making process and presented with both the clinician's experience and that of the broader community and literature. Our approach has been that management in rare diseases should be a “team sport.” This study was prompted by frequent emails to each other to discuss potential approaches to increase our collective pool of experience; the community is encouraged to join us.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
HO was supported by NINDS K23 NS107646-04, PI Olson. TB was supported by the Ponzio Family Chair in Neurology Research to the Children's Hospital Colorado Foundation.
Conflict of Interest
JD Consultancy for Marinus, Ultragenyx, Avexis, Anavex, and Newron; any remuneration went to Telethon Kids Institute. MM works as a pediatric researcher with investigator initiated studies funded through industry (PTC Therapeutics). EP is on the advisory board of Marinus Pharmaceuticals and has consulted for Biomarin Pharmaceuticals and Zogenix. JC has acted as an investigator for studies with GW Pharma, Zogenix, Vitaflo, Ovid, Marinius and Stoke Therapeutics. She has been a speaker and on advisory boards for GW Pharma, Biocodex, Zogenix, and Nutricia; all remuneration has been paid to her department. Her research is supported by the National Institute of Health Research (NIHR) Biomedical Research Centre at Great Ormond Street Hospital. She holds as endowed chair at UCL Great Ormond Street Institute of Child Health; she holds grants from NIHR, EPSRC, GOSH Charity, ERUK, the Waterloo Foundation and the Great Ormond Street Hospital Biomedical Research Centre. SA has received funding from GW Pharmaceuticals, Norvartis, PTC Therapeutics, Boston Scientific, Nutricia, UCB, BioMarin, LivaNova, Medtronic, Desitin, Ipsen, CDKL5 UK, TSA and the National Institute for Health Research. HO received consulting fees from Takeda Pharmaceuticals and Zogenix regarding clinical trial design, Ovid Therapeutics regarding clinical trial results, Marinus Pharmaceuticals regarding CDKL5 Deficiency Disorder, and has done consulting for the FOXG1 Research Foundation. TB performed consultancy for Ovid, GW Pharmaceuticals, International Rett Syndrome Foundation, Takeda, Neurogene, Ultragenyx, Zogenix, GrinTherapeutics, Alcyone, Acadia, Neuren and Marinus; Clinical Trials with Acadia, Ovid, GW Pharmaceuticals, Marinus and RSRT; all remuneration has been made to his department.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
CBD, Cannabidiol; CDD, CDKL5 Deficiency Disorder; CDKL5, Cyclin Dependent Kinase-like 5; DEE, Developmental Epileptic Encephalopathy; DEXA, Dual-Energy X-ray Absorptiometry; DTI, Diffusion Tension Imaging; ECG, Electrocardiogram; EEG, Electroencephalogram; MRI, Magnetic Resonance Imaging; SUDEP, Sudden Unexpected Death in Epilepsy.
References
1. Fehr S, Wilson M, Downs J, Williams S, Murgia A, Sartori S, et al. The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur J Hum Genet. (2013) 21:266–73. doi: 10.1038/ejhg.2012.156
2. Olson HE, Demarest ST, Pestana-Knight EM, Swanson LC, Iqbal S, Lal D, et al. Cyclin-dependent kinase-like 5 deficiency disorder: clinical review. Pediatr Neurol. (2019) 97:18–25. doi: 10.1016/j.pediatrneurol.2019.02.015
3. Paciorkowski AR, Seltzer LE, Neul JL. Developmental encephalopathies (2017). doi: 10.1016/B978-0-323-37101-8.00032-1
4. Rosas-Vargas H, Bahi-Buisson N, Philippe C, Nectoux J, Girard B, N'Guyen Morel M. A, Gitiaux C, et al. Impairment of CDKL5 nuclear localisation as a cause for severe infantile encephalopathy. J. Med. Genet. (2008) 45:172–78. doi: 10.1136/jmg.2007.053504
5. Hector RD, Kalscheuer VM, Hennig F, Leonard H, Downs J, Clarke A, et al. Variants: improving our understanding of a rare neurologic disorder. Neurol Genet. (2017) 3:e200. doi: 10.1212/NXG.0000000000000200
6. Kalscheuer VM, Tao J, Donnelly A, Hollway G, Schwinger E, Kübart S, et al. Disruption of the serine/threonine kinase 9 gene causes severe X-linked infantile spasms and mental retardation. Am J Hum Genet. (2003) 72:1401–11. doi: 10.1086/375538
7. Fichou Y, Nectoux J, Bahi-Buisson N, Chelly J, Bienvenu T. An isoform of the severe encephalopathy-related CDKL5 gene, including a novel exon with extremely high sequence conservation, is specifically expressed in brain. J. Hum. Genet. (2011) 56:52–57. doi: 10.1038/jhg.2010.143
8. Scala E, Ariani F, Mari F, Caselli R, Pescucci C, Longo I, et al. CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. J Med Genet. (2005) 42:103–7. doi: 10.1136/jmg.2004.026237
9. Montini E, Andolfi G, Caruso A, Buchner G, Walpole SM, Mariani M, et al. Identification and characterization of a novel serine-threonine kinase gene from the Xp22 region. Genomics. (1998) 51:427–33. doi: 10.1006/geno.1998.5391
10. Tao J, Van Esch H, Hagedorn-Greiwe M, Hoffmann K, Moser B, Raynaud M, et al. Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am J Hum Genet. (2004) 75:1149–54. doi: 10.1086/426460
11. Weaving LS, Christodoulou J, Williamson SL, Friend KL, McKenzie OLD, Archer H, et al. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet. (2004) 75:1079–93. doi: 10.1086/426462
12. White R, Ho G, Schmidt S, Scheffer IE, Fischer A, Yendle SC, et al. Cyclin-dependent kinase-like 5 (CDKL5) mutation screening in rett syndrome and related disorders. Twin Res Hum Genet. (2010) 13:168–78. doi: 10.1375/twin.13.2.168
13. Gürsoy S, Erçal D. Diagnostic Approach to genetic causes of early-onset epileptic encephalopathy. J Child Neurol. (2016) 31:523–32. doi: 10.1177/0883073815599262
14. Rosander C, Hallböök T. Dravet syndrome in Sweden: a population-based study. Dev Med Child Neurol. (2015) 57:628–33. doi: 10.1111/dmcn.12709
15. Wu YW, Sullivan J, McDaniel SS, Meisler MH, Walsh EM Li SX, et al. Incidence of dravet syndrome in a US population. Pediatrics. (2015) 136:e1310–5. doi: 10.1542/peds.2015-1807
16. Fehr S, Bebbington A, Nassar N, Downs J, Ronen GM, DE Klerk N, et al. Trends in the diagnosis of Rett syndrome in Australia. Pediatr Res. (2011) 70:313–9. doi: 10.1203/PDR.0b013e3182242461
17. Symonds JD, Zuberi SM, Stewart K, McLellan A, O‘Regan M, MacLeod S, et al. Incidence and phenotypes of childhood-onset genetic epilepsies: a prospective population-based national cohort. Brain. (2019) 142:2303–18. doi: 10.1093/brain/awz195
18. Fehr S, Downs J, Ho G, de Klerk N, Forbes D, Christodoulou J, et al. Functional abilities in children and adults with the CDKL5 disorder. Am J Med Genet A. (2016) 170:2860–9. doi: 10.1002/ajmg.a.37851
19. Mangatt M, Wong K, Anderson B, Epstein A, Hodgetts S, Leonard H, et al. Prevalence and onset of comorbidities in the CDKL5 disorder differ from Rett syndrome. Orphanet J Rare Dis. (2016) 11:39. doi: 10.1186/s13023-016-0418-y
20. Demarest ST, Olson HE, Moss A, Pestana-Knight E, Zhang X, Parikh S, et al. CDKL5 deficiency disorder: relationship between genotype, epilepsy, cortical visual impairment, and development. Epilepsia. (2019) 60:1733–42. doi: 10.1111/epi.16285
21. Brock D, Fidell A, Thomas J, Juarez-Colunga E, Benke TA, Demarest S. Cerebral visual impairment in CDKL5 deficiency disorder correlates with developmental achievement. J Child Neurol. (2021) 36:974–80. doi: 10.1177/08830738211019284
22. Fehr S, Wong K, Chin R, Williams S, de Klerk N, Forbes D, et al. Seizure variables and their relationship to genotype and functional abilities in the CDKL5 disorder. Neurology. (2016) 87:2206–13. doi: 10.1212/WNL.0000000000003352
23. Müller A, Helbig I, Jansen C, Bast T, Guerrini R, Jähn J, et al. Retrospective evaluation of low long-term efficacy of antiepileptic drugs and ketogenic diet in 39 patients with CDKL5-related epilepsy. Eur J Paediatr Neurol. (2016) 20:147–51. doi: 10.1016/j.ejpn.2015.09.001
24. MacKay CI, Wong K, Demarest ST, Benke TA, Downs J, Leonard H. Exploring genotype-phenotype relationships in the CDKL5 deficiency disorder using an international dataset. Clin Genet. (2021) 99:157–65. doi: 10.1111/cge.13862
25. Fu C, Armstrong D, Marsh E, Lieberman D, Motil K, Witt R, et al. Consensus guidelines on managing Rett syndrome across the lifespan. BMJ Paediatr Open. (2020) 4:e000717. doi: 10.1136/bmjpo-2020-000717
26. Cochrane Reviews. Available online at: http://www.cochranelibrary.com (accessed March 17, 2022).
27. “PubMed,” PubMed. Available online at: http://www.pubmed.ncbi.nlm.nih.gov.
28. Loughlin KG, Moore LF. Using Delphi to achieve congruent objectives and activities in a pediatrics department. J Med Educ. (1979) 54:101–6. doi: 10.1097/00001888-197902000-00006
29. Keeney S, Hasson F, McKenna H. Consulting the oracle: ten lessons from using the Delphi technique in nursing research. J Adv Nurs. (2006) 53:205–12. doi: 10.1111/j.1365-2648.2006.03716.x
30. Barrett D, Heale R. What are Delphi studies? Evid Based Nurs. (2020) 23:68–9. doi: 10.1136/ebnurs-2020-103303
31. Artuso R, Mencarelli MA, Polli R, Sartori S, Ariani F, Pollazzon M, et al. Early-onset seizure variant of Rett syndrome: definition of the clinical diagnostic criteria. Brain Dev. (2010) 32:17–24. doi: 10.1016/j.braindev.2009.02.004
32. Archer HL, Evans J, Edwards S, Colley J, Newbury-Ecob R, O'Callaghan F, et al. CDKL5 mutations cause infantile spasms, early onset seizures, and severe mental retardation in female patients. J Med Genet. (2006) 43:729–34. doi: 10.1136/jmg.2006.041467
33. Bahi-Buisson N, Nectoux J, Rosas-Vargas H, Milh M, Boddaert N, Girard B, et al. Key clinical features to identify girls with CDKL5 mutations. Brain. (2008) 131:2647–61. doi: 10.1093/brain/awn197
34. Liang JS, Huang H, Wang JS, Lu JF. Phenotypic manifestations between male and female children with CDKL5 mutations. Brain Dev. (2019) 41:783–9. doi: 10.1016/j.braindev.2019.05.003
35. Cooper MS, Mcintosh A, Crompton DE, McMahon JM, Schneider A, Farrell K, et al. Mortality in Dravet syndrome. Epilepsy Res. (2016) 128:43–7. doi: 10.1016/j.eplepsyres.2016.10.006
36. Johannesen KM, Gardella E, Scheffer I, Howell K, Smith DM, Helbig I, et al. Early mortality in SCN8A -related epilepsies. Epilepsy Res. (2018) 143:7981. doi: 10.1016/j.eplepsyres.2018.04.008
37. Verducci C, Hussain F, Donner E, Moseley BD, Buchhalter J, Hesdorffer D, et al. SUDEP in the North American SUDEP Registry: the full spectrum of epilepsies. Neurology. (2019) 93:e227–36. doi: 10.1212/WNL.0000000000007778
38. Bahi-Buisson N, Kaminska A, Boddaert N, Rio M, Afenjar A, Gérard M, et al. The three stages of epilepsy in patients with CDKL5 mutations. Epilepsia. (2008) 49:1027–37. doi: 10.1111/j.1528-1167.2007.01520.x
39. Mei D, Marini C, Novara F, Bernardina BD, Granata T, Fontana E, et al. Xp223 genomic deletions involving the CDKL5 gene in girls with early onset epileptic encephalopathy. Epilepsia. (2010) 51:647–54. doi: 10.1111/j.1528-1167.2009.02308.x
40. Melani F, Mei D, Pisano T, Savasta S, Franzoni E, Ferrari AR, et al. CDKL5 gene-related epileptic encephalopathy: electroclinical findings in the first year of life. Dev Med Child Neurol. (2011) 53:354–60. doi: 10.1111/j.1469-8749.2010.03889.x
41. Hirsch E, Velez A, Sellal F, Maton B, Grinspan A, Malafosse A, et al. Electroclinical signs of benign neonatal familial convulsions. Ann Neurol. (1993) 34:835–41. doi: 10.1002/ana.410340613
42. Guerrini R, Parrini E. Epilepsy in Rett syndrome, and CDKL5- and FOXG1-gene-related encephalopathies. Epilepsia. (2012) 53:2067–78. doi: 10.1111/j.1528-1167.2012.03656.x
43. Boutry-Kryza N, Labalme A, Ville D, de Bellescize J, Touraine R, Prieur F, et al. Molecular characterization of a cohort of 73 patients with infantile spasms syndrome. Eur J Med Genet. (2015) 58:51–8. doi: 10.1016/j.ejmg.2014.11.007
44. Devinsky O, King L, Schwartz D, Conway E, Price D. Effect of fenfluramine on convulsive seizures in CDKL5 deficiency disorder. Epilepsia. (2021) 62:e98–e102. doi: 10.1111/epi.16923
45. O'Callaghan FJK, Edwards SW, Alber FD, Cortina Borja M, Hancock E, Johnson AL, et al. Vigabatrin with hormonal treatment versus hormonal treatment alone (ICISS) for infantile spasms: 18-month outcomes of an open-label, randomised controlled trial. Lancet Child Adolesc Health. (2018) 2:715–25. doi: 10.1016/S2352-4642(18)30244-X
46. Aledo-Serrano Á, Gómez-Iglesias P, Toledano R, Garcia-Peñas JJ, Garcia-Morales I, Anciones C, et al. Sodium channel blockers for the treatment of epilepsy in CDKL5 deficiency disorder: findings from a multicenter cohort. Epilepsy Behav. (2021) 118:107946. doi: 10.1016/j.yebeh.2021.107946
47. Leary LD, Wang D, Nordli DR Jr, Engelstad K, De Vivo DC. Seizure characterization and electroencephalographic features in Glut-1 deficiency syndrome. Epilepsia. (2003) 44:701–7. doi: 10.1046/j.1528-1157.2003.05302.x
48. Lim Z, Wong K, Olson HE, Bergin AM, Downs J, Leonard H. Use of the ketogenic diet to manage refractory epilepsy in CDKL5 disorder: experience of >100 patients. Epilepsia. (2017) 58:1415–22. doi: 10.1111/epi.13813
49. Tangarorang J, Leonard H, Epstein A, Downs J. A framework for understanding quality of life domains in individuals with the CDKL5 deficiency disorder. Am J Med Genet A. (2019) 179:249–56. doi: 10.1002/ajmg.a.61012
50. Dale T, Downs J, Olson H, Bergin AM, Smith S, Leonard H. Cannabis for refractory epilepsy in children: a review focusing on CDKL5 deficiency disorder. Epilepsy Res. (2019) 151:31–9. doi: 10.1016/j.eplepsyres.2019.02.001
51. Devinsky O, Verducci C, Thiele EA, Laux LC, Patel AD, Filloux F, et al. Open-label use of highly purified CBD (Epidiolex®;) in patients with CDKL5 deficiency disorder and Aicardi, Dup15q, and Doose syndromes. Epilepsy Behav. (2018) 86:131–7. doi: 10.1016/j.yebeh.2018.05.013
52. Elliott J, DeJean D, Clifford T, Coyle D, Potter BK, Skidmore B, et al. Cannabis-based products for pediatric epilepsy: an updated systematic review. Seizure. (2020) 75:18–22. doi: 10.1016/j.seizure.2019.12.006
53. Perucca E. Cannabinoids in the treatment of epilepsy: hard evidence at last? J Epilepsy Res. (2017) 7:61–76. doi: 10.14581/jer.17012
54. Knight E, Amin S, Bahi-Buisson N, Benke TA, Cross JH, Demarest ST, et al. Safety and efficacy of ganaxolone in patients with CDKL5 deficiency disorder: results from the double-blind phase of a randomised, placebo-controlled, phase 3 trial. Lancet Neurol (2022) 21:417–27. doi: 10.1016/S1474-4422(22)00077-1
55. Lim Z, Wong K, Downs J, Bebbington K, Demarest S, Leonard H. Vagus nerve stimulation for the treatment of refractory epilepsy in the CDKL5 deficiency disorder. Epilepsy Res. (2018) 146:36–40. doi: 10.1016/j.eplepsyres.2018.07.013
56. Chan AY, Rolston JD, Lee B, Vadera S, Englot DJ. Rates and predictors of seizure outcome after corpus callosotomy for drug-resistant epilepsy: a meta-analysis. J Neurosurg. (2018). doi: 10.3171/2017.12.JNS172331
57. Kadam SD, Sullivan BJ, Goyal A, Blue ME, Smith-Hicks C. Rett syndrome and CDKL5 deficiency disorder: from bench to clinic. Int J Mol Sci. (2019) 20:5098. doi: 10.3390/ijms20205098
58. Hagebeuk EEO, Duran M, Abeling NGGM, Vyth A. Poll-The BTS-adenosylmethionine and S-adenosylhomocysteine in plasma and cerebrospinal fluid in Rett syndrome and the effect of folinic acid supplementation. J Inherit Metab Dis. (2013) 36:967–72. doi: 10.1007/s10545-013-9590-6
59. Martire V, Alvente S, Bastianini S, Berteotti C, Silvani A, et al. CDKL5 deficiency entails sleep apneas in mice. J Sleep Res. (2017) 26:495–97. doi: 10.1111/jsr.12512
60. Brod M, Tesler LE, Christensen TL. Qualitative research and content validity: developing best practices based on science and experience. Qual Life Res. (2009) 18:1263–78. doi: 10.1007/s11136-009-9540-9
61. Saldaris J, Weisenberg J, Pestana-Knight E, Marsh ED, Suter B, Rajaraman R, et al. Content validation of clinician-reported items for a severity measure for CDKL5 deficiency disorder. J Child Neurol. (2021) 36:998–1006. doi: 10.1177/08830738211019576
62. Bjorner JB, Gandek B, Cole J, Kosinski M. Response to the FDA Draft Guidance for Industry document: Patient-Reported Outcome Measures: Use in Medical Product Development to Support Labeling Claims (Docket 2006D-0044). Population and Quantitative Health Sciences Publications. Retrieved from https://escholarship.umassmed.edu/qhs_pp/611
63. Leidy NK, Vernon M. Perspectives on patient-reported outcomes. Pharmacoeconomics. (2008) 26:363–70. doi: 10.2165/00019053-200826050-00002
64. Russo S, Marchi M, Cogliati F, Bonati MT, Pintaudi M, Veneselli E, et al. Novel mutations in the CDKL5 gene, predicted effects and associated phenotypes. Neurogenetics. (2009) 10:241–50. doi: 10.1007/s10048-009-0177-1
65. Fehr S, Leonard H, Ho G, Williams S, de Klerk N, Forbes D, et al. There is variability in the attainment of developmental milestones in the CDKL5 disorder. J Neurodev Disord. (2015) 7:2. doi: 10.1186/1866-1955-7-2
66. Perfect E, Hoskin E, Noyek S, Davies TC. A systematic review investigating outcome measures and uptake barriers when children and youth with complex disabilities use eye gaze assistive technology. Dev Neurorehabil. (2020) 23:145–59. doi: 10.1080/17518423.2019.1600066
67. Killian JT, Lane JB, Lee HS, Skinner SA, Kaufmann WE, Glaze DG, et al. Scoliosis in Rett syndrome: progression, comorbidities, and predictors. Pediatr Neurol. (2017) 70:20–5. doi: 10.1016/j.pediatrneurol.2017.01.032
68. Downs J, Bergman A, Carter P, Anderson A, Palmer GM, Roye D, et al. Guidelines for management of scoliosis in Rett syndrome patients based on expert consensus and clinical evidence. Spine. (2009) 34:E607–17. doi: 10.1097/BRS.0b013e3181a95ca4
69. Jefferson A, Leonard H, Siafarikas A, Woodhead H, Fyfe S, Ward LM, et al. Clinical guidelines for management of bone health in rett syndrome based on expert consensus and available evidence. PLoS ONE. (2016) 11:e0146824. doi: 10.1371/journal.pone.0146824
70. Benson-Goldberg S, Erickson K. Eye-trackers, digital-libraries, and print-referencing: a single case study in CDKL5. Res Dev Disabil. (2021) 112:103913. doi: 10.1016/j.ridd.2021.103913
71. Cutri-French C, Armstrong D, Saby J, Gorman C, Lane J, Fu C, et al. Comparison of core features in four developmental encephalopathies in the rett natural history study. Ann Neurol. (2020) 88:396–406. doi: 10.1002/ana.25797
72. Lin JL, Rigdon J, Van Haren K, Buu M, Saynina O, Bhattacharya J, et al. Gastrostomy tubes placed in children with neurologic impairment: associated morbidity and mortality. J Child Neurol. (2021) 36:727–34. doi: 10.1177/08830738211000179
73. Howard C, Macken WL, Connolly A, Keegan M, Coghlan D, Webb DW. Percutaneous endoscopic gastrostomy for refractory epilepsy and medication refusal. Arch Dis Child. (2019) 104:690–2. doi: 10.1136/archdischild-2018-315629
74. Hosain SA, La Vega-Talbott M, Solomon GE. Ketogenic diet in pediatric epilepsy patients with gastrostomy feeding. Pediatr Neurol. (2005) 32:81–3. doi: 10.1016/j.pediatrneurol.2004.09.006
75. Olson HE, Daniels CI, Haviland I, Swanson LC, Greene CA, Denny AMM, et al. Current neurologic treatment and emerging therapies in CDKL5 deficiency disorder. J Neurodev Disord. (2021) 13:40. doi: 10.1186/s11689-021-09384-z
76. Amin S, Majumdar A, Mallick AA, Patel J, Scatchard R, Partridge CA, et al. Caregiver's perception of epilepsy treatment, quality of life and comorbidities in an international cohort of CDKL5 patients. Hippokratia. (2017) 21:130–5. doi: 10.1016/j.ejpn.2017.04.1141
77. Gregg AR, Aarabi M, Klugman S, Leach NT, Bashford MT, Goldwaser T, et al. Correction to: Screening for autosomal recessive and X-linked conditions during pregnancy and preconception: a practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. (2021) 23:2015. doi: 10.1038/s41436-021-01300-z
78. Demarest S, Pestana-Knight EM, Olson HE, Downs J, Marsh ED, Kaufmann WE, et al. Severity assessment in CDKL5 deficiency disorder. Pediatr Neurol. (2019) 97:38–42. doi: 10.1016/j.pediatrneurol.2019.03.017
79. Masliah-Plachon J, Auvin S, Nectoux J, Fichou Y, Chelly J, Bienvenu T. Somatic mosaicism for a CDKL5 mutation as an epileptic encephalopathy in males. Am J Med Genet A. (2010) 152A:2110–1. doi: 10.1002/ajmg.a.33037
80. Kato T, Morisada N, Nagase H, Nishiyama M, Toyoshima D, Nakagawa T, et al. Somatic mosaicism of a CDKL5 mutation identified by next-generation sequencing. Brain Dev. (2015) 37:911–5. doi: 10.1016/j.braindev.2015.03.002
81. Hagebeuk EEO, Marcelis CL, Alders M, Kaspers A, de Weerd AW. Two siblings with a CDKL5 mutation: genotype and phenotype evaluation. J Child Neurol. (2015) 30:1515–9. doi: 10.1177/0883073815573317
Keywords: CDKL5 deficiency disorder, cyclin-dependent kinase-like 5, developmental and epileptic encephalopathy, care guideline, consensus methods, Delphi methods
Citation: Amin S, Monaghan M, Aledo-Serrano A, Bahi-Buisson N, Chin RF, Clarke AJ, Cross JH, Demarest S, Devinsky O, Downs J, Pestana Knight EM, Olson H, Partridge C-A, Stuart G, Trivisano M, Zuberi S and Benke TA (2022) International Consensus Recommendations for the Assessment and Management of Individuals With CDKL5 Deficiency Disorder. Front. Neurol. 13:874695. doi: 10.3389/fneur.2022.874695
Received: 12 February 2022; Accepted: 05 April 2022;
Published: 20 June 2022.
Edited by:
Wang-Tso Lee, National Taiwan University Hospital, TaiwanReviewed by:
Bruria Ben-Zeev, Sheba Medical Center, IsraelJuan Xiong, Central South University, China
Steffen Syrbe, Heidelberg University Hospital, Germany
Copyright © 2022 Amin, Monaghan, Aledo-Serrano, Bahi-Buisson, Chin, Clarke, Cross, Demarest, Devinsky, Downs, Pestana Knight, Olson, Partridge, Stuart, Trivisano, Zuberi and Benke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sam Amin, c2FtYW1pbkBuaHMubmV0