 Robert David Henderson
Robert David Henderson Kasper Planeta Kepp
Kasper Planeta Kepp Andrew Eisen
Andrew Eisen- 1Department of Neurology, Royal Brisbane and Women's Hospital, Brisbane, QLD, Australia
- 2Department of Chemistry, Technical University of Denmark, Kongens Lyngby, Denmark
- 3Division of Neurology, Department of Medicine, Faculty of Medicine, University of British Columbia, Vancouver, BC, Canada
Amyotrophic lateral sclerosis and frontotemporal dementia (ALS/FTD) are neurodegenerations with evolutionary underpinnings, expansive clinical presentations, and multiple genetic risk factors involving a complex network of pathways. This perspective considers the complex cellular pathology of aging motoneuronal and frontal/prefrontal cortical networks in the context of evolutionary, clinical, and biochemical features of the disease. We emphasize the importance of evolution in the development of the higher cortical function, within the influence of increasing lifespan. Particularly, the role of aging on the metabolic competence of delicately optimized neurons, age-related increased proteostatic costs, and specific genetic risk factors that gradually reduce the energy available for neuronal function leading to neuronal failure and disease.
Introduction
Finely controlled fractionated muscle movement enables humans to perform complex activities that require precise voluntary execution of force and speed of movement (1). The anatomical basis for this is the corticomotoneuronal system, which in humans connects monosynaptically with all motor neuron pools except those innervating the ocular and sphincter muscles (2, 3).
Both advanced cognition and a versatile motor repertoire were critical to the success of human evolution, which involved a rapid expansion of cerebral network connectivity occurring within the constraints of a bony cranium (4). The relatively rapidly evolving brain incurred increased metabolic demands (5), and selection pressures relating to human migration within and out of Africa.
Since the mid-nineteenth century, recognition of devastating diseases, including amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), have emerged. These predominantly involve frontal and prefrontal neurons, but their pathology extends beyond these regions. Clinical, genetical, and biochemical features of these diseases converge on protein misfolding and metabolic dysfunction as common end points associated with impaired neuronal dysfunction (6).
This article briefly (1) outlines the evolution of the frontal and prefrontal cortex with respect to communication and language, and motor function specific to humans; (2) considers the roles played by evolutionary changes, cerebral metabolism, and senescence in an era of increasing lifespan; (3) raises the concept of possible neuron exhaustion, with proteomic cost minimization as a selective force challenged with increasing age; and (4) links this idea to ALS and FTD.
We hypothesize that these disorders and other neurodegenerations reflect in part a mismatch between evolved neocortical cellular and metabolic processes at a protein level, in the context of rapid and ever-increasing complexity of human interaction, and the relatively recent increased human lifespan. For conciseness, we do not attempt to detail genetic components of these diseases (7), nor do we consider their overlap with other neurodegenerative diseases (8).
A unified etiology of ALS/FTD is proposed that implicates evolutionary optimized neurons, metabolically challenged by RNA/protein turnover in certain risk phenotypes leading to neuronal exhaustion and disease. We suggest that the hypothesis can be expanded with more data and point toward metabolism and protein turnover as potentially key targets for efficient treatment paradigms.
Evolution of Cognitive and Motor Function
The evolution of cognitive function and brain development has resulted from the complex interplay of nature and nurture, where development seems to be driven by genes and shaped by the environment (9, 10). Modern humans have enhanced cognitive functioning, especially in the domains of cooperation, theory of mind, language, and culture (11), and are capable of processing vast information and solving abstract problems (12–14). Higher order cognitive skills of humans evolved through the separation of humans from earlier hominid lineages (15). Whether this was through adaptations of existing systems or the creation of new ones is undetermined (12, 16).
Language probably evolved out of gesture as a protolanguage (17–20). Gesture is universal to the animal kingdom. Some gestures are individual, but many are common to a specific language and others are common to all humans. Newborns and infants largely communicate with gestures accompanied by non-verbal vocalization, and children learn language through social interaction and gain practice using sentence constructions that have been created by linguistic communities over time.
Complex forms of communication, especially human language, defines one of the most difficult problems for evolutionary biology (21–23). Language is a particularly remarkable outcome of the evolution of cognitive complexity and requires perceiving the external world in terms of objects and actions and naming them using a set of signals. Even though human communication (gestures and language) is far more structured and complex than seen in other animals, there are no specific physiological, neurological, or genetic traits that explain the human communication, executive functioning, and abstract thinking skills that have evolved during the latest 100,000 years (12). But it has been suggested that speech could be more cost-effective compared to gesture and developed progressively as group size increased (24).
The basic layout of the larynx and vocal tract is highly conserved and virtually homologous in both form and function among all terrestrial mammals, including humans (25). Indeed, the macaque vocal apparatus is “speech ready,” capable of producing an adequate range of speech sounds to support spoken language (26). However, only humans have developed voluntary control of the larynx (27, 28). This required the unique expansion of fast-conducting monosynaptic corticomotoneuronal connections which in humans occurs for all motor neuron pools, except those of the external ocular muscles and bladder wall (29).
The greatly expanded corticomotoneuronal system with associated neo-cortical networks underlies finely tuned motor control of hand function (and thus use tool, play musical instruments, and paint), the ability to traverse uneven terrain, and for example, to skate, ski, and play professional football, and employ diversified vocalization enabling variable pitch, tone, velocity of speech and loudness, in a complex association with respiratory function (29, 30).
Vocal cues are a rich source of information about a speaker's emotional state. The term “prosody” refers to the changes in pitch, loudness, rhythm, and voice quality corresponding to a person's emotional state (31–33). The relationship between vocal complexity and brain architecture across non-human primates also has relevance to the evolution of human speech. A positive correlation has been recently observed between vocal repertoire scope and the relative size of cortical association areas which governs voluntary control (28).
The motor and premotor areas of the human and non-human primate cortex are engaged not only in preparation and execution of voluntary movement but also perform fundamental computations associated with executive function and other cognitive aspects of behavior (34). Furthermore, the incremental diversification of motor areas in humans is accompanied by the emergence of new cognitive abilities. In particular, primate motor regions not only control the low-level aspects of planning and control of movements but also participate in the perceptual and motor aspects of sophisticated cognitive functions such as decision-making, action understanding/imitation, and language (10, 35–37).
Humans have evolved a finely tuned pincer grip, utilizing the thumb and index finger. Impairment of this can be an early, unique feature of ALS, referred to as the split hand (29). Similarly, early loss of foot dorsiflexion and elbow flexion, referred to as split foot and elbow, may also be early impairments in ALS (38). The motor units subserving these movements have the strongest corticomotoneuronal drive, and it has been proposed that in ALS, there is loss of muscle synergies subserved by motor units with the strongest corticomotoneuronal drive (3).
It has been hypothesized that the evolution of cognition increased the returns from cooperating to the point where the benefits to self were sufficient for cooperation to remain stable as the group size increased and the relatedness decreased (39), with the higher cognitive needs of expanded, non-kin cooperation developing slowly. This change to co-operative behavior could be accomplished with more versatile communication (40).
Through evolution, hominin brain sizes smaller than homo sapiens remained stable at 400–500 cc until about 2 million years ago (41). The human brain size reaches its adult dimensions by 3–4 years, similar to chimpanzees yet human frontal lobe development continues until at least the early 20s (42–45). At a macro level, as human species developed improved frontal lobe function, including theory of mind, any change in cranial size was comparatively minor (46) and roughly scaled to the body size of primates (47). Within this fixed cranial capacity, cortical size could be increased by folding (48) while also allowing shorter axonal distances for the rapid connectivity needed in the frontal lobes.
At a micro level, the functional capacity of a neuronal structure is inherently limited by its neural architecture and signal processing time (48). An important component of the frontal lobe development was the discrete modifications in local circuitry and interconnectivity of selected parts of the brain which became highly organized in humans (44). The scaling of the number and distribution of neurons is an important component through evolution (41), with a greater scaling of the number of cortical motor neurons through primate evolution (49).
Environmental Factors at a Macro Level—Evolution, Aging, and Energy Metabolism of Motor Neurons
As modern humans migrated out of the African sub-continent into the colder habitats, there was pressure to modify cerebral energy metabolism in a brain that was progressively increasing its metabolic demands in comparison to other body organs (50). Within the constraints on cranium size, most changes in cognitive function were probably associated with altered neuronal networks (5). However, neurons with many synaptic connections and high-synaptic activity are very energy-demanding, and thus, vulnerable to energy-deficiency induced by genetic and environmental risk factors (51, 52).
Complex variations in the dietary intake associated with the habitat's wildlife and foraging options and diverse cultural and technological impacts (e.g., cooking) contribute to this evolutionary history (5, 53, 54). Also, metabolic adaptations in response to human migration to colder environments may have occurred more recently. For example, in the Scandinavia human settlement occurred perhaps 5,000–10,000 years ago as the polar ice cap receded (55).
In contrast to human evolution occurring over tens of thousands of years, the recent reduced mortality and associated increased longevity has been rapid, experienced predominantly by the last four generations of humans that have ever lived (56, 57). Progress in lowering human mortality is on par with or exceeds that made in the laboratory via various selection and dietary restriction experiments and endocrine pathway mutations (56). The change has been largely achieved by removing environmental challenges, making injuries, and illnesses less fatal, by improving nutrition and reducing disease at younger ages and also enhancing health in the elderly (58). As a result, there has been a considerable increase in humans reaching senescent ages with vulnerability to neurodegenerative diseases (57).
Intrinsic to aging is a slowing of cerebral metabolism (8, 59–62). Recent findings suggest that disruption of neuronal homeostasis, mainly due to deficient energy metabolism, underlies neurodegeneration (63–65). Although senescent neurons may remain metabolically active and continue to function within the neuronal network, their reduced metabolic efficiency will plausibly impact overall network integrity and ultimately cognitive performance. In addition, the senescent neurons excrete a plethora of molecules that affect the function of nearby cells and provoke local inflammation potentiating the destruction of the human brain networks (65).
The Molecular Level—Proteostasis of Neuronal Networks
Following DNA transcription, RNA molecules within a cell are bound by distinct sets of RNA-binding proteins that have the task of regulating the correct processing, transport, stability, and function/translation of proteins up to its final degradation. Proteins reach a native state but can change their folded structure if the environment changes (protein misfolding) leading to aggregates (66, 67). Mutations can also induce conformational changes and aggregation (68).
These cellular processes that maintain normal neuronal physiology throughout life are diverse, and exponentially fail with increasing age (69). Increasing age is associated with accumulation of protein aggregates characteristic of neurodegenerations but with different protein signaling pathways affected, depending upon the unfolded protein response (69, 70). Therefore, it is not surprising that the recent increase in human lifespan has been associated with increasingly prevalent cerebral protein aggregation (71).
It has been widely assumed that protein misfolding is pathogenic via some specific molecular toxicity as larger aggregates (i.e., protein deposits) or as more recently accepted, in the intracellular oligomeric state, with suggestions including interaction with other proteins and cell membranes (68). However, so far, the direct pathogenic species and mode of action remains obscure and therapeutic approaches that have been developed toward targeting the misfolded proteins directly, have so far met with the little clinical success (72).
Indeed, it has been proposed that the protein deposits may in some cases be beneficial as they contribute to reducing the pool of intracellular pathogenic oligomers, regardless of the mechanism of their pathogenicity (molecular toxicity or turnover costs), and outside the cells, these deposits will be less likely to interfere with cellular functions, and also less costly as they would be less targeted (and less accessible) to the intracellular proteases (73). In studies of TAR DNA binding protein 43 (TDP-43), beneficial effects of protein aggregation has also been observed (66, 68), suggesting that the toxicity occurs via a non-aggregated state, which we propose below is a state that is more easily subject to costly turnover.
Another recently suggested mechanism of protein-misfolding pathology employs the general metabolic cost (in ATP) of misfolded protein turnover within the cells (51). This effectively reduces energy available for basic housekeeping and cell-signaling purposes. Since the human body uses ~20% of its total energy budget on protein turnover (74), the energy costs of handling the misfolded proteins, rather than the protein's molecular toxicity per se—could be pathogenic. This is particularly relevant in the context of the most energy-demanding cells such as neurons, where the ATP costs of proteostasis would be first felt due to the large energy demands for inter-neuronal signaling via ion pumps (perhaps 50% of energy budget) (75).
The energy costs of maintaining the proteome (translation, transcription, and RNA and protein turnover) defines 20–70% of the cellular energy budget of various organisms and thus has probably been under heavy selection pressure for minimization (76). First in simple organisms to maximize energy available for cell maintenance and reproduction, and later to maximize survival of higher organisms, e.g., via reduced foraging needs and cognitive capacity.
Whereas, energy costs are not normally problematic, neuronal networks harbor some of the most energy-requiring cells in the human body (51, 77). It is plausible that selectively vulnerable networks of motor, or other, neurons, evolutionarily optimized for delicate metabolic competency, become challenged by lifestyle or genetic risk factors. They then may be subject to additional exhaustion caused by elevated RNA or protein turnover resulting in neuronal necrosis and network malfunction. The high-proteome turnover required to keep homeostasis in the presence of a highly abundant, repeatedly synthesized molecule via a repeat expansion or an unstable protein product, could be envisioned to contribute ATP costs to neurons operating near maximal capacity, and possibly accelerating aging-induced deterioration of involved networks (51).
A lack of energy could conceivably cause both depolarization of neurons, promoting excitotoxicity, a recognized event in ALS (77), but also contribute to longer-term oxidative stress and chemical imbalances that could further aggravate disease. Subsequent neuronal rewiring, which itself costs ATP as it is tightly coupled to aerobic glycolysis in energy cost-benefit tradeoffs (78), would accelerate disease progression given the metabolic deficiency cascade. Furthermore, the buildup of aggregated RNA molecules or protein copies could reflect the lack of sufficient energy available to maintain proteostasis as other energy costs increase, but also reflect the possible direct contribution of these turnover costs to the disease state. The recent studies linking proteopathy to elevated aerobic glycolysis in the human brain supports such a proteopathy-energy–cost relationship (79).
Relevance to ALS and FTD Pathology
ALS/FTD is recognized as complex polygenic disorders (80), the genetic component perhaps contributing ~50−60% of the risk (81). The known susceptibility genes appear to have different frequencies according to race (82). The most common ALS/FTD gene, is the c9orf72 mutation which links sporadic and genetic forms of ALS and FTD (83, 84). It has a higher prevalence in the far northern European population; an evolutionary recent migration. It has been proposed that the c9orf72 expansion occurred only once in the past, with estimates varying from 1,500 to 6,000 years ago (85–87). In the USA, the median age of onset of ALS/FTD patients with the C9orf72 expansion is 58 years, an age rarely reached until after 1900 (85).
The common risk genes identified in frontal lobe pathophysiology function in molecular pathways related to RNA-metabolism and proteostasis including autophagy/proteasome, vesicle trafficking and RNA-metabolism/homeostasis (88–90). In particular, the cellular accumulation of the DNA/RNA binding protein TDP-43 found in 98% of ALS cases highlights the importance of DNA/RNA-homeostasis in the neurons (91–93).
Aging is a major risk factor for ALS/FTD and other neurodegenerations (69, 94), and there is a substantial overlap of the genetic changes in the frontal lobe diseases and the genes regulating different pathways relevant in aging. These include autophagy, inflammation, nuclear-cytoplasmic transport, and RNA processing (69, 95, 96). The recent work has identified changes in the cerebral metabolism intrinsic to both ALS and FTD (8, 59–62) with an apparent selective vulnerability of the motor neurons to energetic stress (90, 97, 98).
The central role of proteopathy and RNA homeostasis in ALS/FTD is well-indicated by the genetic risk factors (6). SOD1 mutations are a risk factor for fALS that have been shown to induce loss of protein stability and protein misfolding consistent with increased costs of managing this protein which is one of the most highly expressed in the human body with important functions in anti-oxidant stress (51). Overexpression of wild-type SOD1 contributes to the mitochondrial dysfunction and motor defects in mice due to fALS SOD1 mutations, providing support for protein abundance/turnover rather than a specific molecular toxicity of mutants being pathogenic, and bridging phenotypes of sporadic (patients harboring wild-type proteins) and fALS (patients harboring inherited additionally severe mutations).
The GGGGCC hexanucleotide repeat expansion on chromosome 9, C9orf72, is the most common genetic risk factor for fALS and is associated with abnormal protein/RNA processing (81). Since these RNA management costs would consume energy, it is plausible that they could contribute to exhaustion of the motor neurons if the hexanucleotide expansion is continuously produced and degraded (99). TDP-43, another risk factor for ALS is an important protein in transcription control known to aggregate in ALS, and thus, consistent with such a mechanism. Other genetic risk factors such as FUS/TLS and ubiquilin-2 have also been associated with proteostasis (6).
In support of the protein/RNA turnover contributing to the disease state, proteasome inhibition has been found to prevent the pathogenicity of a fALS-causing SOD1 variant, whereas removal of the inhibitor (which would reinstate protein turnover) was associated with aggravation of disease (100).
At the clinical level, ALS has been associated with a hypermetabolic presentation that could suggest elevated metabolic costs during pathogenesis (62, 94, 101). It is also notable that low body mass index has been consistently identified as a risk factor for ALS, and high BMI has been associated with lower risk of ALS (102). Although it is not clear if this association is causative, these clinical features of ALS would be consistent with a hypothesis that evolutionary-optimized metabolic demands being exhausted by age-induced proteostasis costs in energy-demanding motor neurons. In our opinion, this etiology, which we have summarized in Figure 1, integrates and rationalizes both the evolutionary history of human cognition and aging, the senescence-induced proteostatic and metabolic challenges associated with this evolutionary process, and its relationship with the clinical state of ALS/FTD.
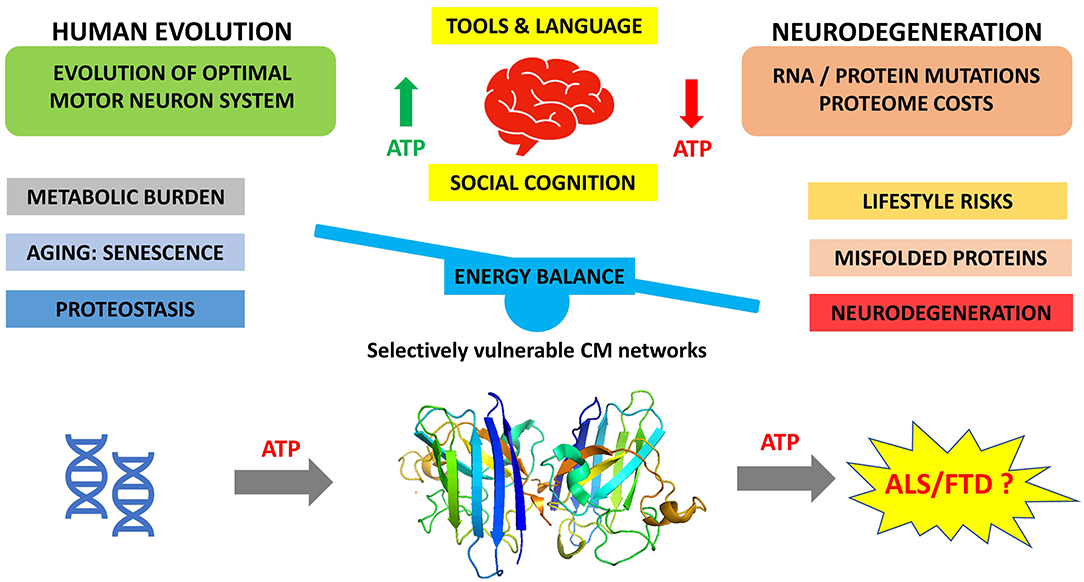
Figure 1. Cost-saving balance of human neural circuits resulting from evolution of human cognition and challenged by increasing age of modern humans.
Conclusion
Frontal and prefrontal lobe diseases are predominantly disorders of the aging nervous system. With the recent increase in longevity, largely determined by adequate shelter, good nutrition, medical advances, and reduced mortality in early life, the incidence of these neurodegenerations has increased. Extended longevity in the recent generations is unlikely to simply reflect the Darwinian natural selection, nor the Hamiltonian inclusive fitness (103). As humans age, neocortical neurons are particularly vulnerable to the effects of senescence, which include impaired energy metabolism homeostasis. This results in functional cellular failure and ultimately clinical disease. The cascade of events that determine cellular senescence are poorly defined, but among other factors, a genetic predisposition is likely relevant.
We propose that protein aggregation, the hallmark of neurodegenerations such as ALS and FTD, occurs because of the increasing metabolic burden accompanying neuronal proteostasis. This in turn is a consequence of the intersection of the evolving human brain in response to evolutionary and environmental pressures, and increasing age, which over time leads to metabolic exhaustion of energy-demanding neocortical neurons (Figure 1).
A possible mechanism for the protein aggregation lies in the energy costs of misfolded protein turnover, but other possibilities exist. While general proteasome inhibition is not a valid therapeutic strategy for these diseases, it does suggest that the burden of RNA/protein turnover could be a contributing factor in the etiology, consistent with the perspective provided earlier.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
RH conceived the concept. KK created the Figure. All authors contributed to writing and reviewing the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Maier MA, Bennett KM, Hepp-Reymond MC, Lemon RN. Contribution of the monkey corticomotoneuronal system to the control of force in precision grip. J Neurophysiol. (1993) 69:772–85. doi: 10.1152/jn.1993.69.3.772
2. Porter R, Lemon R. Corticospinal Function and Voluntary Movement. Oxford: Oxford University Press (1993).
3. Eisen A, Lemon R. The motor deficit of ALS reflects failure to generate muscle synergies for complex motor tasks, not just muscle strength. Neurosci Lett. (2021) 762:136171. doi: 10.1016/j.neulet.2021.136171
4. Neubauer S, Hublin JJ, Gunz P. The evolution of modern human brain shape. Sci Adv. (2018) 4:eaao5961. doi: 10.1126/sciadv.aao5961
5. Herculano-Houzel S. Scaling of brain metabolism with a fixed energy budget per neuron: implications for neuronal activity, plasticity and evolution. PLoS ONE. (2011) 6:e17514. doi: 10.1371/journal.pone.0017514
6. Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. (2013) 79:416–38. doi: 10.1016/j.neuron.2013.07.033
7. Henderson RD, Garton FC, Kiernan MC, Turner MR, Eisen A. Human cerebral evolution and the clinical syndrome of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. (2019) 90:570–75. doi: 10.1136/jnnp-2017-317245
8. Ahmed RM, Devenney EM, Irish M, Ittner A, Naismith S, Ittner LM, et al. Neuronal network disintegration: common pathways linking neurodegenerative diseases. J Neurol Neurosurg Psychiatry. (2016) 87:1234–41. doi: 10.1136/jnnp-2014-308350
10. Butcher LM, Plomin R. The nature of nurture: a genomewide association scan for family chaos. Behav Genet. (2008) 38:361–71. doi: 10.1007/s10519-008-9198-z
11. Lee YS, Silva AJ. The molecular and cellular biology of enhanced cognition. Nat Rev Neurosci. (2009) 10:126–40. doi: 10.1038/nrn2572
12. Heyes C. New thinking: the evolution of human cognition. Philos Trans R Soc Lond B Biol Sci. (2012) 367:2091–6. doi: 10.1098/rstb.2012.0111
13. Heyes C. Enquire within: cultural evolution and cognitive science. Philos Trans R Soc Lond B Biol Sci. (2018) 373:20170051. doi: 10.1098/rstb.2017.0051
14. Fernald RD. Cognitive skills and the evolution of social systems. J Exp Biol. (2017) 220(Pt 1):103–13. doi: 10.1242/jeb.142430
15. Srinivasan S, Bettella F, Frei O, Hill WD, Wang Y, Witoelar A, et al. Enrichment of genetic markers of recent human evolution in educational and cognitive traits. Sci Rep. (2018) 8:12585. doi: 10.1038/s41598-018-30387-9
16. Stout D. Stone toolmaking and the evolution of human culture and cognition. Philos Trans R Soc Lond B Biol Sci. (2011) 366:1050–9. doi: 10.1098/rstb.2010.0369
17. Corballis MC. From mouth to hand: gesture, speech, and the evolution of right-handedness. Behav Brain Sci. (2003) 26:199–208. Discussion 08–60. doi: 10.1017/S0140525X03000062
18. Pollick AS, de Waal FB. Ape gestures and language evolution. Proc Natl Acad Sci USA. (2007) 104:8184–9. doi: 10.1073/pnas.0702624104
19. Willems RM, Hagoort P. Neural evidence for the interplay between language, gesture, and action: a review. Brain Lang. (2007) 101:278–89. doi: 10.1016/j.bandl.2007.03.004
20. Willems RM, Ozyurek A, Hagoort P. When language meets action: the neural integration of gesture and speech. Cereb Cortex. (2007) 17:2322–33. doi: 10.1093/cercor/bhl141
21. Bednarik RG. An aetiology of hominin behaviour. Homo. (2012) 63:319–35. doi: 10.1016/j.jchb.2012.07.004
22. Pinker S. Colloquium paper: the cognitive niche: coevolution of intelligence, sociality, and language. Proc Natl Acad Sci USA. (2010) 107(Suppl. 2):8993–9. doi: 10.1073/pnas.0914630107
23. Donald M. Key cognitive preconditions for the evolution of language. Psychon Bull Rev. (2017) 24:204–08. doi: 10.3758/s13423-016-1102-x
24. Corballis MC. How language evolved from manual gestures. Gesture. (2012) 12:200–26. doi: 10.1075/gest.12.2.04cor
25. Fitch WT. The evolution of speech: a comparative review. Trends Cogn Sci. (2000) 4:258–67. doi: 10.1016/S1364-6613(00)01494-7
26. Fitch WT, de Boer B, Mathur N, Ghazanfar AA. Monkey vocal tracts are speech-ready. Sci Adv. (2016) 2:e1600723. doi: 10.1126/sciadv.1600723
27. Zhang Z. Mechanics of human voice production and control. J Acoust Soc Am. (2016) 140:2614. doi: 10.1121/1.4964509
28. Dunn JC, Smaers JB. Neural correlates of vocal repertoire in primates. Front Neurosci. (2018) 12:534. doi: 10.3389/fnins.2018.00534
29. Eisen A, Turner MR, Lemon R. Tools and talk: an evolutionary perspective on the functional deficits associated with amyotrophic lateral sclerosis. Muscle Nerve. (2014) 49:469–77. doi: 10.1002/mus.24132
30. Eisen A, Braak H, Del Tredici K, Lemon R, Ludolph AC, Kiernan MC. Cortical influences drive amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. (2017) 88:917–24. doi: 10.1136/jnnp-2017-315573
31. Liebenthal E, Silbersweig DA, Stern E. The language, tone and prosody of emotions: neural substrates and dynamics of spoken-word emotion perception. Front Neurosci. (2016) 10:506. doi: 10.3389/fnins.2016.00506
32. Filippi P. Emotional and interactional prosody across animal communication systems: a comparative approach to the emergence of language. Front Psychol. (2016) 7:1393. doi: 10.3389/fpsyg.2016.01393
33. Mol C, Chen A, Kager RWJ, ter Haar SM. Prosody in birdsong: a review and perspective. Neurosci Biobehav Rev. (2017) 81(Pt B):167–80. doi: 10.1016/j.neubiorev.2017.02.016
34. Mendoza G, Merchant H. Motor system evolution and the emergence of high cognitive functions. Prog Neurobiol. (2014) 122:73–93. doi: 10.1016/j.pneurobio.2014.09.001
35. Romo R. Conversion of sensory signals into perceptions, memories and decisions. Prog Neurobiol. (2013) 103:1–2. doi: 10.1016/j.pneurobio.2013.03.003
36. Romo R, de Lafuente V. Conversion of sensory signals into perceptual decisions. Prog Neurobiol. (2013) 103:41–75. doi: 10.1016/j.pneurobio.2012.03.007
37. Bak TH, Chandran S. What wires together dies together: verbs, actions and neurodegeneration in motor neuron disease. Cortex. (2012) 48:936–44. doi: 10.1016/j.cortex.2011.07.008
38. Henderson RD, Eisen A. ALS split phenotypes - to what extent do they exist? Clin Neurophysiol. (2020) 131:847–49. doi: 10.1016/j.clinph.2019.12.417
39. Dos Santos M, West SA. The coevolution of cooperation and cognition in humans. Proc Biol Sci. (2018) 285:20180723. doi: 10.1098/rspb.2018.0723
40. Derungs C, Kohl M, Weibel R, Bickel B. Environmental factors drive language density more in food-producing than in hunter-gatherer populations. Proc Biol Sci. (2018) 285:20172851. doi: 10.1098/rspb.2017.2851
41. Kaas JH. The evolution of mammalian brains from early mammals to present-day primates. In: Watanabe S. HM, Shimizu T, editors. Evolution of the Brain, Cognition, and Emotion in Vertebrates. Tokyo: Springer (2017). doi: 10.1007/978-4-431-56559-8_3
42. Kretschmann HJ, Schleicher A, Wingert F, Zilles K, Löblich HJ. Human brain growth in the 19th and 20th century. J Neurol Sci. (1979) 40:169–88. doi: 10.1016/0022-510X(79)90202-8
43. Irurtzun A. The “Globularization Hypothesis” of the language-ready brain as a developmental frame for prosodic bootstrapping theories of language acquisition. Front Psychol. (2015) 6:1817. doi: 10.3389/fpsyg.2015.01817
44. Teffer K, Buxhoeveden DP, Stimpson CD, Fobbs AJ, Schapiro SJ, Baze WB, et al. Developmental changes in the spatial organization of neurons in the neocortex of humans and common chimpanzees. J Comp Neurol. (2013) 521:4249–59. doi: 10.1002/cne.23412
45. Silbereis JC, Pochareddy S, Zhu Y, Li M, Sestan N. The cellular and molecular landscapes of the developing human central nervous system. Neuron. (2016) 89:248–68. doi: 10.1016/j.neuron.2015.12.008
46. Robson SL, Wood B. Hominin life history: reconstruction and evolution. J Anat. (2008) 212:394–425. doi: 10.1111/j.1469-7580.2008.00867.x
47. Sousa AMM, Meyer KA, Santpere G, Gulden FO, Sestan N. Evolution of the human nervous system function, structure, and development. Cell. (2017) 170:226–47. doi: 10.1016/j.cell.2017.06.036
48. Hofman MA. Evolution of the human brain: when bigger is better. Front Neuroanat. (2014) 8:15. doi: 10.3389/fnana.2014.00015
49. Herculano-Houzel S, Kaas JH, de Oliveira-Souza R. Corticalization of motor control in humans is a consequence of brain scaling in primate evolution. J Comp Neurol. (2016) 524:448–55. doi: 10.1002/cne.23792
50. Pontzer H, Brown MH, Raichlen DA, Dunsworth H, Hare B, Walker K, e al. Metabolic acceleration and the evolution of human brain size and life history. Nature. (2016) 533:390–2. doi: 10.1038/nature17654
51. Kepp KP. A quantitative model of human neurodegenerative diseases involving protein aggregation. Neurobiol Aging. (2019) 80:46–55. doi: 10.1016/j.neurobiolaging.2019.04.001
52. Kepp KP. Genotype-property patient-phenotype relations suggest that proteome exhaustion can cause amyotrophic lateral sclerosis. PLoS ONE. (2015) 10:e0118649. doi: 10.1371/journal.pone.0118649
53. Anton SC, Potts R, Aiello LC. Human evolution. Evolution of early Homo: an integrated biological perspective. Science. (2014) 345:1236828. doi: 10.1126/science.1236828
54. Cornelio AM, de Bittencourt-Navarrete RE, de Bittencourt Brum R, Queiroz CM, Coasta MR. Human brain expansion during evolution is independent of fire control and cooking. Front Neurosci. (2016) 10:167. doi: 10.3389/fnins.2016.00167
55. Bergman I, Olofsson A, Hörnberg G, Zackrisson O, Hellberg E. Deglaciation and colonization: pioneer settlements in northern fennoscandia J World Prehistory. (2004) 18:155–77. doi: 10.1007/s10963-004-2880-z
56. Burger O, Baudisch A, Vaupel JW. Human mortality improvement in evolutionary context. Proc Natl Acad Sci USA. (2012) 109:18210–4. doi: 10.1073/pnas.1215627109
57. Finch CE. Evolution in health and medicine Sackler colloquium: evolution of the human lifespan and diseases of aging: roles of infection, inflammation, and nutrition. Proc Natl Acad Sci USA. (2010) 107(Suppl. 1):1718–24. doi: 10.1073/pnas.0909606106
58. Gutwinski S, Loscher A, Mahler L, Kalbitzer J, Heinz A, Bermpohl F. Understanding left-handedness. Dtsch Arztebl Int. (2011) 108:849–53. doi: 10.3238/arztebl.2011.0849
59. Ahmed RM, Ke YD, Vucic S, Ittner LM, Seeley W, Hodges JR, et al. Physiological changes in neurodegeneration - mechanistic insights and clinical utility. Nat Rev Neurol. (2018) 14:259–71. doi: 10.1038/nrneurol.2018.23
60. Ahmed RM, Highton-Williamson E, Caga J, Thornton N, Ramsey E, Zoing M, et al. Lipid metabolism and survival across the frontotemporal dementia-amyotrophic lateral sclerosis spectrum: relationships to eating behavior and cognition. J Alzheimers Dis. (2018) 61:773–83. doi: 10.3233/JAD-170660
61. Tracey TJ, Steyn FJ, Wolvetang EJ, Ngo ST. Neuronal lipid metabolism: multiple pathways driving functional outcomes in health and disease. Front Mol Neurosci. (2018) 11:10. doi: 10.3389/fnmol.2018.00010
62. Steyn FJ, Ioannides ZA, van Eijk RPA, Heggie S, Thorpe KA, Ceslis A, et al. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J Neurol Neurosurg Psychiatry. (2018) 89:1016–23. doi: 10.1136/jnnp-2017-317887
63. Athauda D, Foltynie T. The ongoing pursuit of neuroprotective therapies in Parkinson disease. Nat Rev Neurol. (2015) 11:25–40. doi: 10.1038/nrneurol.2014.226
64. Johnson S, Imai SI. NAD (+) biosynthesis, aging, and disease. F1000Res. (2018) 7:132. doi: 10.12688/f1000research.12120.1
65. Quansah E, Peelaerts W, Langston JW, Simon DK, Colca J, Brundin P. Targeting energy metabolism via the mitochondrial pyruvate carrier as a novel approach to attenuate neurodegeneration. Mol Neurodegener. (2018) 13:28. doi: 10.1186/s13024-018-0260-x
66. Aprile FA, Temussi PA, Pastore A. Man does not live by intrinsically unstructured proteins alone: the role of structured regions in aggregation. Bioessays. (2021) 43:e2100178. doi: 10.1002/bies.202100178
68. Monti M, Armaos A, Fantini M, Pastore A, Tartaglia GG. Aggregation is a context-dependent constraint on protein evolution. Front Mol Biosci. (2021) 8:678115. doi: 10.3389/fmolb.2021.678115
69. Niccoli T, Partridge L, Isaacs AM. Ageing as a risk factor for ALS/FTD. Hum Mol Genet. (2017) 26:R105–13. doi: 10.1093/hmg/ddx247
70. Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. (2006) 313:1604–10. doi: 10.1126/science.1124646
71. Villar-Pique A VS. Villar-Pique A, Ventura S. Marseilles, France: Springer (2011). p. 103–23. doi: 10.1007/978-3-642-30425-5_6
72. Chiti F, Dobson CM. Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu Rev Biochem. (2017) 86:27–68. doi: 10.1146/annurev-biochem-061516-045115
73. Treusch S, Cyr DM, Lindquist S. Amyloid deposits: protection against toxic protein species? Cell Cycle. (2009) 8:1668–74. doi: 10.4161/cc.8.11.8503
74. Waterlow JC. Whole-body protein turnover in humans–past, present, and future. Annu Rev Nutr. (1995) 15:57–92. doi: 10.1146/annurev.nu.15.070195.000421
75. Belanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. (2011) 14:724–38. doi: 10.1016/j.cmet.2011.08.016
76. Kepp KP. Survival of the cheapest: how proteome cost minimization drives evolution. Q Rev Biophys. (2020) 53:e7. doi: 10.1017/S0033583520000037
77. Blizzard CA, Southam KA, Dawkins E, Lewis KE, King AE, Clark JA, et al. Identifying the primary site of pathogenesis in amyotrophic lateral sclerosis - vulnerability of lower motor neurons to proximal excitotoxicity. Dis Model Mech. (2015) 8:215–24. doi: 10.1242/dmm.018606
78. Chen Y, Lin Q, Liao X, He Y. Association of aerobic glycolysis with the structural connectome reveals a benefit-risk balancing mechanism in the human brain. Proc Natl Acad Sci USA. (2021) 118:e2013232118. doi: 10.1073/pnas.2013232118
79. Goyal MS, Gordon BA, Couture LE, Flores S, Xiong C, Morris JC, et al. Spatiotemporal relationship between subthreshold amyloid accumulation and aerobic glycolysis in the human brain. Neurobiol Aging. (2020) 96:165–75. doi: 10.1016/j.neurobiolaging.2020.08.019
80. Al-Chalabi A, Hardiman O, Kiernan MC, Chio A, Rix-Brooks B, van den Berg LH. Amyotrophic lateral sclerosis: moving towards a new classification system. Lancet Neurol. (2016) 15:1182–94. doi: 10.1016/S1474-4422(16)30199-5
81. Al-Chalabi A, Calvo A, Chio A, Colville S, Ellis CM, Hardiman O, et al. Analysis of amyotrophic lateral sclerosis as a multistep process: a population-based modelling study. Lancet Neurol. (2014) 13:1108–13. doi: 10.1016/S1474-4422(14)70219-4
82. Veldink JH. ALS genetic epidemiology 'How simplex is the genetic epidemiology of ALS?'. J Neurol Neurosurg Psychiatry. (2017) 88:537. doi: 10.1136/jnnp-2016-315469
83. Renton AE, Majounie E, Waite A, Simon-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. (2011) 72:257–68. doi: 10.1016/j.neuron.2011.09.010
84. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. (2011) 72:245–56. doi: 10.1016/j.neuron.2011.09.011
85. Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. (2012) 11:323–30. doi: 10.1016/S1474-4422(12)70043-1
86. Smith BN, Newhouse S, Shatunov A, Vance C, Topp S, Johnson L, et al. The C9ORF72 expansion mutation is a common cause of ALS+/-FTD in Europe and has a single founder. Eur J Hum Genet. (2013) 21:102–8. doi: 10.1038/ejhg.2012.98
87. Pliner HA, Mann DM, Traynor BJ. Searching for Grendel: origin and global spread of the C9ORF72 repeat expansion. Acta Neuropathol. (2014) 127:391–6. doi: 10.1007/s00401-014-1250-x
88. Weishaupt JH, Hyman T, Dikic I. Common molecular pathways in amyotrophic lateral sclerosis and frontotemporal dementia. Trends Mol Med. (2016) 22:769–83. doi: 10.1016/j.molmed.2016.07.005
89. Zhao M, Kim JR, Bruggen RV, Park J. RNA-binding proteins in amyotrophic lateral sclerosis. Mol Cells. (2018) 41:818–29. doi: 10.14348/molcells.2018.0243
90. Tank EM, Figueroa-Romero C, Hinder LM, Bedi K, Archbold HC, Li X, et al. Abnormal RNA stability in amyotrophic lateral sclerosis. Nat Commun. (2018) 9:2845. doi: 10.1038/s41467-018-05049-z
91. Zhang YJ, Gendron TF, Grima JC, Sasaguri H, Jansen-West K, Xu Y-F, et al. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat Neurosci. (2016) 19:668–77. doi: 10.1038/nn.4272
92. Gendron TF, Bieniek KF, Zhang YJ, Jansen-West K, Ash PEA, Caulfield T, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. (2013) 126:829–44. doi: 10.1007/s00401-013-1192-8
93. Taylor JP, Brown RH Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. (2016) 539:197–206. doi: 10.1038/nature20413
94. Ingre C, Roos PM, Piehl F, Kamel F, Fang F. Risk factors for amyotrophic lateral sclerosis. Clin Epidemiol. (2015) 7:181–93. doi: 10.2147/CLEP.S37505
95. Tsuiji H, Inoue I, Takeuchi M, Furuya A, Yamakage Y, Watanabe S, et al. TDP-43 accelerates age-dependent degeneration of interneurons. Sci Rep. (2017) 7:14972. doi: 10.1038/s41598-017-14966-w
96. Xu D, Jin T, Zhu H, Chen H, Ofengeim D, Zou C, et al. TBK1 Suppresses RIPK1-driven apoptosis and inflammation during development and in aging. Cell. (2018) 174:1477–91.e19. doi: 10.1016/j.cell.2018.07.041
97. Vandoorne T, De Bock K, Van Den Bosch L. Energy metabolism in ALS: an underappreciated opportunity? Acta Neuropathol. (2018) 135:489–509. doi: 10.1007/s00401-018-1835-x
98. Ahmed RM, Irish M, Piguet O, Halliday GM, Ittner LM, Farooqi S, et al. Amyotrophic lateral sclerosis and frontotemporal dementia: distinct and overlapping changes in eating behaviour and metabolism. Lancet Neurol. (2016) 15:332–42. doi: 10.1016/S1474-4422(15)00380-4
99. Jaarsma D, Haasdijk ED, Grashorn JA, Hawkins R, van Duijn W, Verspaget HW, et al. Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiol Dis. (2000) 7(6 Pt B):623–43. doi: 10.1006/nbdi.2000.0299
100. Kitamura A, Inada N, Kubota H, Matsumoto G, Kinjo M, Morimoto RI et al. Dysregulation of the proteasome increases the toxicity of ALS-linked mutant SOD1. Genes Cells. (2014) 19:209–24. doi: 10.1111/gtc.12125
101. O'Reilly EJ, Wang H, Weisskopf MG, Fitzgerald KC, Falcone G, McCullough ML, et al. Premorbid body mass index and risk of amyotrophic lateral sclerosis. Amyotrophic Lateral Scler Frontotemp Degen. (2013) 14:205–11. doi: 10.3109/21678421.2012.735240
102. Nakken O, Meyer HE, Stigum H, Holmøy T. High BMI is associated with low ALS risk: a population-based study. Neurology. (2019) 93:e424–32. doi: 10.1212/WNL.0000000000007861
Keywords: motor neuron disease, amyotrophic lateral sclerosis, evolution, protein, metabolism, aging
Citation: Henderson RD, Kepp KP and Eisen A (2022) ALS/FTD: Evolution, Aging, and Cellular Metabolic Exhaustion. Front. Neurol. 13:890203. doi: 10.3389/fneur.2022.890203
Received: 05 March 2022; Accepted: 19 April 2022;
Published: 27 May 2022.
Edited by:
Vincenzo La Bella, University of Palermo, ItalyReviewed by:
Mario Sabatelli, Catholic University of the Sacred Heart, ItalyNicola Ticozzi, University of Milan, Italy
Copyright © 2022 Henderson, Kepp and Eisen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert David Henderson, cm9iZXJ0LmhlbmRlcnNvbkBoZWFsdGgucWxkLmdvdi5hdQ==