Abstract
Traumatic peripheral nerve injury occurs frequently and is a major clinical and public health problem that can lead to functional impairment and permanent disability. Despite the availability of modern diagnostic procedures and advanced microsurgical techniques, active recovery after peripheral nerve repair is often unsatisfactory. Peripheral nerve regeneration involves several critical events, including the recreation of the microenvironment and remyelination. Results from previous studies suggest that the peripheral nervous system (PNS) has a greater capacity for repair than the central nervous system. Thus, it will be important to understand myelin and myelination specifically in the PNS. This review provides an update on myelin biology and myelination in the PNS and discusses the mechanisms that promote myelin clearance after injury. The roles of Schwann cells and macrophages are considered at length, together with the possibility of exogenous intervention.
Introduction
Injury to peripheral nerves leads to a series of molecular, cellular, and microstructural responses that promote their regeneration and functional recovery. Peripheral nerve regeneration may be hindered by slow organizational growth, degeneration of its distal segments, and impediments to its reinnervation. Regeneration is frequently insufficient and accompanied by negative clinical sequelae (1). While there has been substantial focus on clinical interventions designed to improve this process, the contributions associated with myelin clearance remain comparatively unexplored.
Lipid-rich myelin formed by Schwann cells (SCs) facilitates saltatory impulse transmission and provides trophic support to the peripheral nervous system (PNS) via radial sorting of its axons. SCs associate with and wrap around larger axons late in embryonic development to facilitate myelination that will be initiated postnatally. Regeneration of myelin is a critical aspect of any effective treatment for peripheral nerve injury (PNI). Remyelination is critical for effective repair after PNI and will be needed to provide new support for the PNS. Furthermore, myelin breakdown has been recognized as a key contributor to various peripheral nerve diseases.
Fragments generated by myelin breakdown obstruct axon regeneration. Myelin ovoids resulting from the destruction of the myelin sheath can be detected in SCs associated with degenerating axons (22). These ovoids contain numerous factors, including myelin-related glycoprotein (MAG) which can inhibit axon development (23, 24). Myelin debris at the lesion site results in local pressure and also impedes nerve repair (25). Jessen et al. reported that the PNS was able to undergo repair more effectively than the central nervous system (CNS) (26). This may be explained at least in part by its superior capacity for myelin removal (27). The results of several recent studies suggested that the removal of myelin debris is a prerequisite for effective nerve regeneration (28–30). Thus, an understanding of the mechanisms contributing to myelin removal from injured nerves will have broad implications for this process.
Earlier work has established that myelin breakdown occurs during the early stages immediately following nerve injury and is followed by the clearance of myelin debris. In this review, we will begin with a discussion of myelin formation. We will then discuss previous research and consider the results of recent studies that address the mechanisms involved in myelin removal. We will conclude with a discussion of the therapeutic implication of these findings.
The accumulation of myelin debris has a significant impact on nerve regeneration; this material is no longer useful and contains many inhibitory signals that prevent repair. Interestingly, and in contrast to the PNS, the CNS does not have satisfied mechanisms that promote effective myelin degradation (31). Myelin fragments can persist at the site of a CNS injury for months or even years where they serve to inhibit essential repair mechanisms (32). For example, Fujita et al. reported that MAG detected in CNS inhibits regeneration via its capacity to suppress tropomyosin receptor kinase (Trk) activity (33). In the nerve system, there is undoubtedly a balance between the clearance of myelin and detrimental consequences that impede regeneration, and this relationship seems to be better regulated in PNS.
In this review, we focus on new insights directed at the earliest responses to nerve injury (Figure 1); established mechanisms and conventional insights into this process have been reviewed elsewhere (37). The article highlights new findings that address various mechanisms that drive myelin removal and concentrates on new perspectives of peripheral nerve regeneration based on the construction of a suitable microenvironment. We also discuss the potential impact of myelin removal in other neurological diseases.
Figure 1
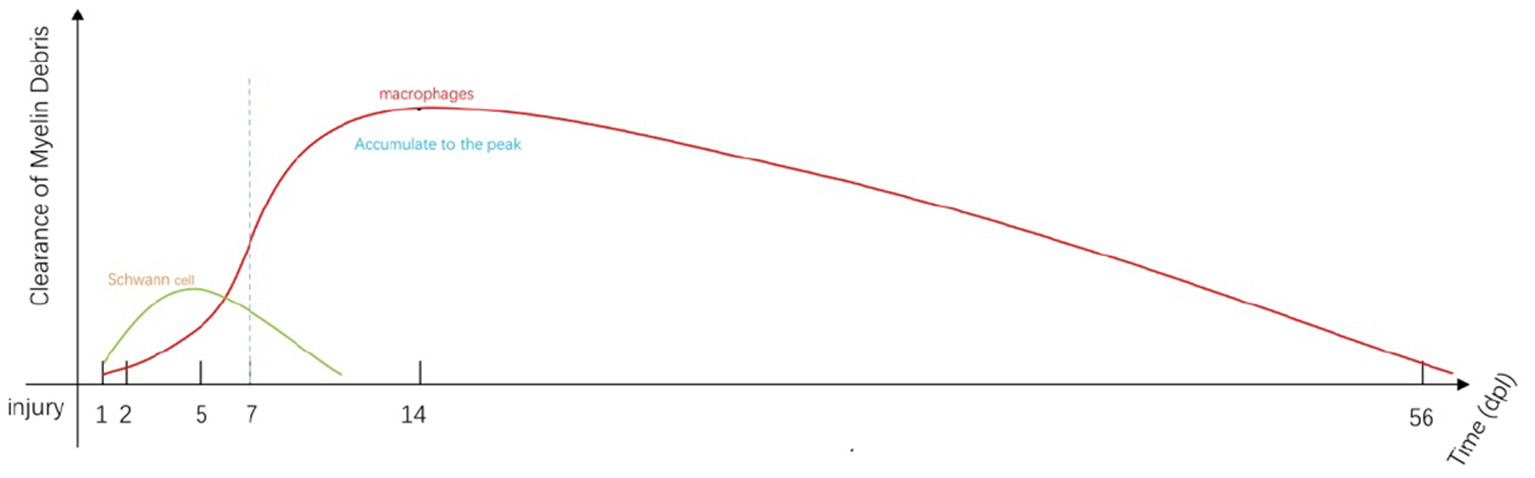
Immediately after injury, SCs and macrophages (including resident and early-stage infiltrating macrophages) begin to remove myelin debris (34). SC-mediated phagocytosis reaches a peak at approximately 5 days post-injury (dpi; green line). Most of the engulfment results from macrophage activity that increases from 5 to 7 dpi (red line). Macrophage-mediated phagocytosis reaches a peak at approximately 14 dpi and remains detectable at day 56 (35). Fibroblasts and neutrophils also contribute to myelin clearance at this stage (35, 36). The myelin fragments are ultimately cleared by localized apoptosis followed by transport by the lymphatics to the spleen.
The formation and function of myelin
Before discussing clearance, we need to have a full understanding of the mechanisms underlying myelin formation and function in PNS. Myelin is a lipid–protein complex derived from membranes of SCs (in the PNS) and oligodendrocytes (in the CNS) that forms a sheath that supports and protects individual axons (38), facilitates rapid transit of electrical signals, and provides critical nourishment. Myelin-providing (pro-myelin) SCs are generated from precursors that undergo three developmental shifts, including transitions from crest cells to SC precursors (SCPs), from SCPs to immature SCs, and from immature SCs to either pro-myelin SCs or non-myelinating (Remak) SCs. The pro-myelin SCs then undergo radial sorting; some of these cells go on to generate myelin sheaths (39, 40).
Myelin in the PNS is comprised of both lipids (70–80%) and proteins (20–30%) (41), the latter group including primarily myelin protein zero (MPZ), as well as myelin basic protein (MBP) and peripheral myelin proteins 2 and 22 (PMP2/PMP22). Those proteins undergo upregulation at the start of myelination and provide the myelin sheath with a unique membrane structure. The interactions between these proteins and the phospholipid bilayer result in compact cytoplasmic leaflets. Upregulated expression of these myelin genes is regulated by the POU-associated transcription factor, Oct6 (SCIP), Brn2, the high-mobility group protein Sox10, and the protein Krox20/Egr2 (42).
Both extrinsic and intracellular signals regulate the process of myelination. Among these signals, neuregulin (Nrg)1 regulates nearly all aspects and functions of SCs (40). Similarly, laminin and laminin receptors modulate myelination via their role in promoting essential autocrine signals (43). Likewise, Gpr126 provides critical signals that promote myelination via its role in regulating signaling by cyclic adenosine monophosphate (cAMP) (44, 45). Myelination also relies on several downstream intracellular signaling pathways involving PI3K, PLC-γ, focal adhesion kinase (FAK), mitogen-activated protein kinase (MAPK), and Wnt/β-Catenin (46–48). In addition, cAMP promotes and maintains myelination via both intracellular signaling as well as mechanisms that regulate its expression (49, 50).
Results from previous reports revealed that iron was required for myelin production and its maintenance in the CNS (51, 52). Results from more recent studies revealed that iron homeostasis is also required for myelin formation in the PNS; Santiago González et al. reported the divalent metal transporter 1 (DMT1) (53), heavy ferritin chain (Fth), and the transferrin receptor 1 (Tfr1), which are factors that regulate iron absorption and storage, provide crucial contributions to SC maturation and myelin formation. Iron outflow also has an impact on myelin formation. The ferroxidase enzyme, ceruloplasmin (Cp), protects the PNS and regulates iron efflux, thereby modulating the maturation of SCs and axon myelination (54). Disorders of iron homeostasis can induce oxidative stress and SC injury (53, 54).
A comprehensive understanding of the composition of myelin and the mechanisms involved in its formation will be essential for our assessment of myelin clearance that takes place in response to nervous system injuries. We note that the factors and signals described earlier also play critical roles in regulating myelin removal after injury. Furthermore, the connections linking myelin formation and removal have important implications with respect to the treatment of PNI.
Negative regulation of myelination
Demyelination is a characteristic response to a peripheral nerve injury. Pro-myelin SCs activate a cellular program that leads to the disintegration of the myelin sheath. Demyelination of injured peripheral nerves typically takes place via Wallerian degeneration (WD) that both negatively regulates myelination and leads to myelin degradation. Reduced expression of myelin-associated proteins may also contribute to rapid demyelination, as this response promotes SC proliferation, clearance of myelin debris, and nerve regeneration (9). We will thus review this process as the first step of the clearance program, as this approach can help us to understand the molecular mechanisms that promote the clearance of myelin sheath fragments (Figure 2).
Figure 2
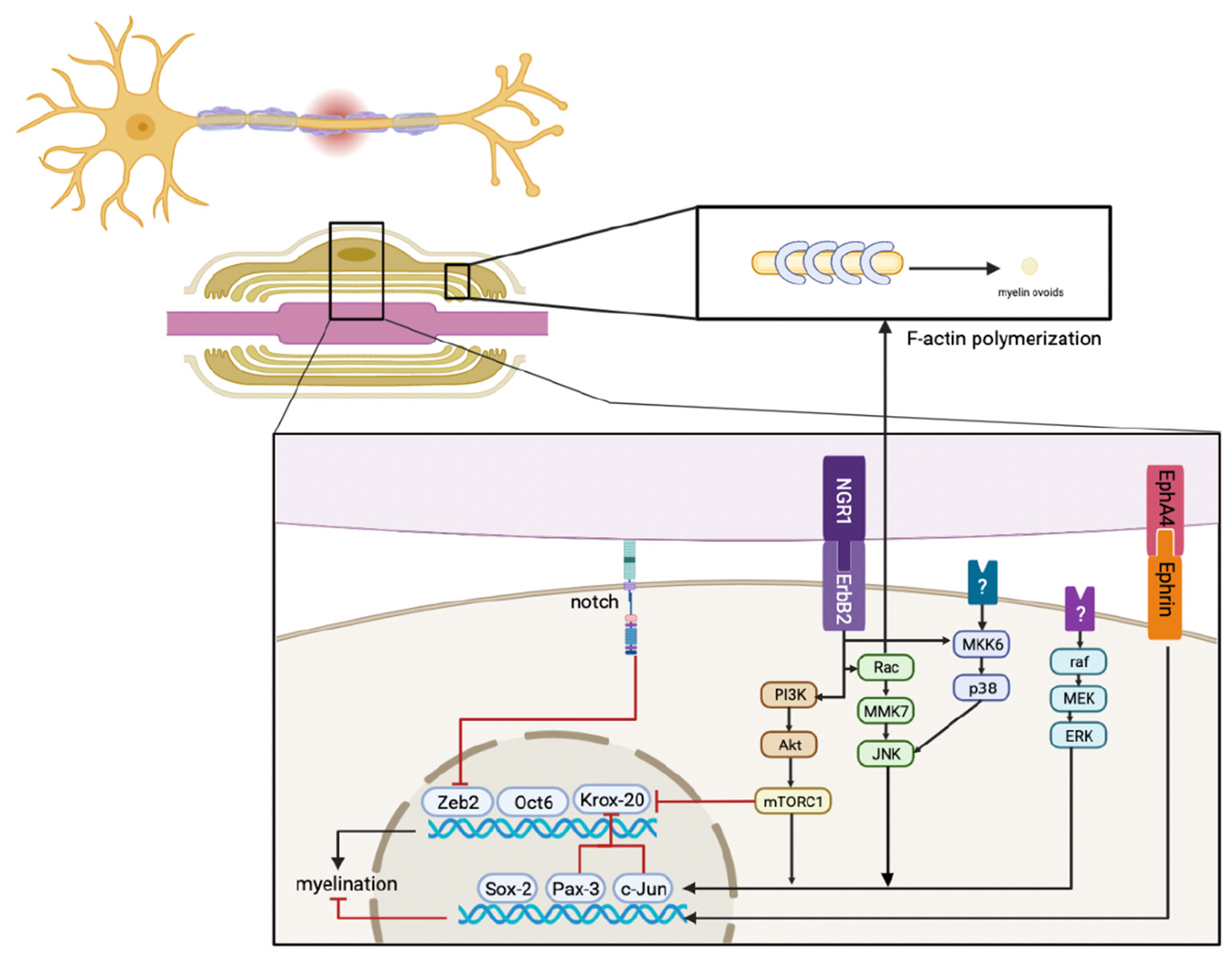
Immediately after injury, ErbB2, MAPK, PI3K, Notch, and EphA4 signals respond in SCs, their crosstalk, and the downstream signaling pathways that regulate factors that mediate myelination. This results in the downregulation of pro-myelinating genes, including Zeb2, Oct6, and Krox20, and the upregulation of negative regulatory factors, including Sox-2, Pax-3, and c-Jun. In addition, activated Rac promotes F-actin polymerization to generate myelin ovoids. Myelin is also cleared during this stage.
Factors that regulate myelination
c-Jun
The transcription factor, c-Jun, is found at the core of the AP-1 complex and is involved in the formation of the myelin sheath (55). More recent evidence has elucidated the role of c-Jun-mediated activities that regulate the responses of denervated SCs after injury, notably the formation of regeneration tracks, support of neuronal survival, promotion of axon regrowth, and myelin clearance (56). Furthermore, c-Jun deletion resulted in diminished clearance of myelin fragments and delayed regeneration (56). Additional evidence revealed that diminished quantities of O-GlcNAc transferase (OGT), which is an enzyme associated with post-translational modifications that suppress c-Jun, also impaired the process of remyelination (57); this result also suggested that activated c-Jun was an essential component of the regeneration process. Results from additional studies revealed that AMP-activated protein kinase (AMPK) attenuated myelin gene expression and thus reduced myelin formation, which is also a response to the activation of c-Jun signaling (58). SC dedifferentiation, myelin clearance, and regeneration were all impaired in the absence of c-Jun (56, 59). While overexpression of c-Jun can also harm the repair, process, and moderate activation is required (60).
Sox2
The Sox2 transcription factor regulates the activities of Schwann cells. Earlier in vitro studies revealed that Sox2 negatively regulated both crest and stem cells and thus regulated the formation of myelin (61). Recent findings have revealed that Sox2 may have a specific impact on myelin produced in the PNS. Interestingly, sustained expression of negative regulators, including Sox2 and Id2, protected mice modeling Charcot–Marie–Tooth (CMT) 1B disease from both dysmyelination and the deleterious impact of mutated myelin-related proteins, including MPZ (62). Activation of SC-autonomous and SC-nonautonomous protective functions may be a key mechanism underlying this result.
Pax-3
Pax-3 is a paired domain transcription factor expressed in the PNS and SCs that represses the induction of MBP by preventing the activation of the MBP promoter by cAMP (63, 64). Furthermore, Doddrell et al. (65) reported that Pax-3 strongly inhibited the cAMP-mediated induction of both Krox20 and Oct-6 in SCs as well as the expression of Sox-10. Interestingly, Pax-3 inhibited the expression of c-Jun and enhanced Notch-1 expression (65); collectively, these results suggested that Pax-3 inhibited Krox20 in a c-Jun-independent manner. However, while these factors are all involved in the repression of myelination, the precise mechanisms will need to be elucidated by further research.
Pathways regulating myelin gene expression after injury
MAPK and NRG1
ERK signaling is strongly and rapidly activated approximately 4 h after insult at both injured and distal sites (66, 67). Harrisingh et al. (67) described the role of the Raf-ERK pathway in promoting SC dedifferentiation. As a first step, Raf activation suppresses the expression of myelin-associated proteins induced by cAMP (67, 68). Moreover, results from another previous study revealed that Raf overexpression generated in response to tamoxifen strongly activates Raf-ERK signaling and downregulates myelin-related gene expression in an uninjured nerve (69). Sustained activation of ERK also resulted in morphological defects, restricted regeneration, and impaired functional recovery, although rapid demyelination was observed (70).
Crosstalk between NRG1 and MAPK signaling pathways promotes demyelination and is a crucial factor involved in the migration of crest cells and the generation of mature SCs, as well as maintaining appropriate myelin thickness (71–73). Although NRG1 promotes both proliferation and the formation of myelin (50), recent results suggest that NRG1 may also promote reductions in the concentration of myelin fragments (73). The role of Rac-MAPK in the processes underlying demyelination is somewhat better clarified and is regulated by Ngr1/ErbB2 signaling (74, 75). Its intracellular target, MAPKK7 (MKK7), is upstream of JNK and was reported to induce the expression of c-Jun as opposed to ERK (76). The net result was the downregulation of myelin gene expression at the distal site of an injured nerve.
Yang et al. reported that p38 MAPK was responsible for NRG1-mediated downregulation of myelin as well as the blockade of Krox20 expression. Results from this report also revealed that activated MKK6, which is a direct and specific upstream activator of p38 MAPK, also promoted demyelination (77).
Several additional factors related to this pathway were also found to delay myelination. Among these, bone morphogenetic protein (BMP)7 limited the formation of myelin sheath during development (78). Dummula et al. reported that the inhibition of BMP expression resulted in myelin preservation in the CNS; these events also had an immediate impact on the maturation of oligodendrocyte precursor cells and myelin formation (78). Recent evidence suggests that BMP family proteins exhibit a similar function in PNS. Results in this report revealed that BMP4 was upregulated beginning at 12 h after injury and reached peak expression at 24 h; expression then decreases continuously thereafter (79), paralleling demyelination. Further analysis revealed that BMP resulted in reductions in cAMP-induced expression of myelin genes, potentially due to its capacity to target the p38MAPK/c-Jun axis. Similarly, fibroblast growth factor 21 (FGF21) was originally identified as a factor that promoted remyelination in CNS (80). However, recent results suggest that FGF21 has completely different functions in PNS, specifically those resulting in the repression of myelin-related genes (81). The results from this report revealed that negative regulation secondary to FGF21 was mediated by the p38 mitogen-activated protein kinase (MAPK)/c-Jun axis. Zhang et al. reported that the expression of FGF21 in developing SCs was diminished in the presence of dibutyryl-cAMP, thus suggesting dynamic regulation of myelin production (81). SCs in the PNS can synthesize and secrete FGF21 which extends its impact beyond the paracrine observed in the liver. Therefore, FGF21 and BMP7 may be among the critical negative regulatory factors involved in myelination after nerve injury. Interestingly, Wang et al. reported the increased expression of genes associated with immature SCs and decreased expression of genes associated with mature SCs in FGF9-gene-deleted mice (82). Overall, these results suggest that FGF9 may also seem to be involved in the process of myelin degeneration.
The intracellular GTPase Rac regulates actin polymerization and is a component of an essential pathway involved in the fragmentation of myelin (74, 83). Myelin ovoids formed during the early stages of WD may be responsible for the SC-mediated removal of compact myelin sheath material (84). After an injury, WD begins with the stereotypic fragmentation of myelin sheath into myelin ovoids at the Schmidt–Lanterman cleft (SLC), which is an area that contains non-compact myelin sheath material. Then, the structure of the SC cytoskeleton changes considerably. Although the SLC features atypical adherens junctions (AJs) (48, 85), the molecular nature of SLC appears to parallel those, and polymerization of new actin filaments occurs after injury and promotes the degeneration of both AJs and E-cadherin, thereby inducing the myelin fragmentation (74). Activated Rac GTPase localizes to the SLC and regulates actin polymerization. By contrast, inhibition of actin polymerization also suppressed the formation of myelin ovoids and the degeneration of E-cadherin. Thus, we conclude that new F-actin that polymerizes after WD promotes the clearance of myelin sheath material (83).
Notch signaling
Notch is a transmembrane receptor protein that is cleaved to generate a Notch intracellular domain (NICD) after combining it with its ligand, Jagged-1. Notch signaling is a central pathway in developing invertebrates that are upregulated in response to Sox-2 and downregulated in response to Krox20 signaling. NICD preserves myelination and induces its expression (50).
Results from previous research revealed that activated Notch pathways regulated demyelination and proliferation of SCs (86); engineered overexpression of NCID results in rapid demyelination of injured nerves (87). Wang et al. reported that forced overexpression of NCID promotes peripheral nerve regeneration via pathways that promote accelerated demyelination (82). Moreover, Notch acts as the inhibitor of Zeb2 (88) and is thus involved in another essential pathway associated with myelin regeneration (89).
A recent study reported that post-translational sumoylation negatively regulates angiogenesis via its impact on Notch signaling (90). Accordingly, one might speculate that sumoylation might also have a negative impact on myelination via its capacity to target the Notch signaling pathway in SCs (91). Sumoylation may also inhibit Oct6 and Krox20 (11), although the impact of these modifications on myelination remains unexplored.
PI3K-Akt-mTORC1
mTORC1 is a central modulator of numerous anabolic reactions and promotes myelination in the PNS (60). Interestingly, mTORC1 undergoes transient activation after injury and suppresses myelination (92). mTORC1 is activated in SCs after injury via NRG1-PI3K-Akt signaling that suppresses myelination rather than promoting regeneration (53, 93, 94). Activated mTORC1 has no impact on macrophage recruitment (60). mTORC1 also promotes demyelination by inhibiting Krox20 and activating c-Jun (60, 93). However, mTORC1-mediated demyelination was not enhanced via the additional activation; this may be due to feedback inhibition on the PI3K-Akt pathway (94). Over-activation of mTORC1 also resulted in delayed regeneration; this dual function may be a new target for therapeutics designed to combat nervous system diseases.
Neuregulin1
Neuregulin1/ErbB2 signaling pathways are involved in crosstalk with MAPK and PI3K and regulate demyelination. NRG1 is crucial to the formation of glia and the migration of crest cells as well as the formation of mature SCs and mechanisms that control myelin thickness (71–73). The results of several previous studies revealed that NRG1/ErbB signaling could target the bZIP transcription factor Maf, thereby limiting the formation of myelin via its impact on cholesterol levels (57, 95). Therefore, NRG1 signaling is strongly connected to myelin clearance.
Recent reports have revealed that the expression of soluble NRG1 changes in response to peripheral nerve injury and ultimately downregulates the expression of myelin-related genes including Pmp22, Serinc5, Ndrg1, Fa2h, Mal, Rxrg, and Krox20, in response to injury and activation of c-Jun (96). NRG1 responses during the first stage of clearance regulated myelin-associated gene expression. By contrast, NRG1 promotes myelin regeneration during the following stages (and also during development); these results suggest that NRG1 also acts as a dynamic regulator that stabilizes these processes with respect to the pathophysiological responses of the PNS. The interactions between these mechanisms permit myelin clearance to proceed.
The NRG1 type III (NRG1 III) isoform positively regulates the formation of myelin and radial sorting together with input from laminin α2β1γ1 (Lm211) (97, 98). Recently, laminin α2β1γ1 was identified as playing a negative role in the regulation of myelin formation in SCs (99). Likewise, tumor necrosis factor (TNF)-α-converting enzyme (TACE; also known as ADAM17) was found to cleave NRG1 III within its epidermal growth factor domain which then negatively regulates the formation of the myelin sheath (100).
EphA4
Eph/Ephrin is an important neuronal mediator that is responsible for glial activation, axon guidance and regeneration, and synapse plasticity and formation (101), as well as the regulation of myelin formation (102). EphA4 regulates CNS myelination via the ephrin-A1-EphA4 pathway (103). More recent research suggests that Eph4 negatively regulates the formation of myelin in SCs (104). The results of this study revealed upregulation of EphA4 until 14 days after injury; expression of both MAG and MPZ decreased after peripheral nerve injury, consistent with a role for EphA4 in inhibiting myelin sheath formation. The observed suppression of remyelination may be the result of SC proliferation during the early phases after injury and suppression of the differentiation of SCs at a later stage (40, 104, 105). Although this mechanism inhibits regeneration for an extended period (until 28 days) after injury, its potential to accelerate myelin clearance must also be considered. Collectively, these results suggest that appropriate negative regulation of myelination is beneficial to the regenerative process. Although the mechanisms remain to be clarified, factors that regulate the expression of EphA4 may emerge as new targets for drugs designed to promote myelin removal.
Transient receptor potential vanilloid 4 channels
Transient receptor potential vanilloid 4 is a non-specific calcium-permeable cation channel that is expressed widely and activated throughout the nervous system. One recent study revealed that TRPV4 promoted regeneration via demyelination of static nerves 2 to 14 days after injury (106). Interestingly, Remak SCs express high levels of TRPV4. Notably, TRPV4 gene-deleted mice exhibit delayed demyelination, thereby repressing nerve regeneration and delaying functional recovery. Moreover, the expression of TRPV4 undergoes a significant increase and returns to normal levels at 21 days due to the rapid myelination that takes place during the stage to follow (106). TRPV4-mediated alterations in calcium flux may have an impact on this process (107, 108). Therefore, TRPV4 and/or related calcium channels may be new targets used to treat PNI.
Because demyelination and regeneration are temporally correlated to one another, demyelination may be one factor contributing to the mechanism underlying nerve regeneration. WD and inhibition of myelin regeneration at early stages both have a positive impact on myelin clearance. Furthermore, some signals are dual-functioning with respect to myelin generation; these signals might be explored as a means to promote more effective regeneration. However, the basis of these interactions and/or synergy needs to be investigated further to promote effective treatments of neurological diseases.
Autophagy and myelin clearance
Autophagy is an important pathway used to dispose of and ultimately recycle disordered proteins and discarded cellular structures and is critical for maintaining cell stability (109). At its most basic level, autophagy is necessary to preserve the integrity of cellular structures. The process involves five main steps, including (1) initiation and nucleation, (22) elongation of the membrane, (23) cargo sorting, (24) fusion of the mature autophagosome with lysosomes, and (25) nutrient recycling and renewal (110). Autophagy is one of the critical processes used by SCs to clear tissue debris that accumulates after PNI.
Autophagy in SCs: A crucial process
The results of a recent study by Jang et al. (111) revealed that the primary myelin ovoid contains a fluid-filled axoplasm with a compact myelin sheath and scanty abaxonal cytoplasm that form P (proximal)-fibers. The P-fibers were eventually overtaken by D(degeneration)-fibers, which have been described as hypertrophic SCs that contain several small myelin vesicles associated with the process of degeneration. Agents that inhibit the function of lysosomes delay the transition from P- to D-fibers. These findings revealed that autophagy is essential for the timely clearance of the myelin sheath after PNI; interestingly, this process cannot be completely accomplished by macrophages recruited from circulation. Thus, autophagic SCs are needed to promote the earliest stages of myelin clearance, myelin regeneration, and scar reduction (112).
Autophagy in SCs is activated during development and suppressed once the cells reach a mature stage (111, 113). Although autophagy will be suppressed at maturity, SCs still utilize this function to eliminate unnecessary cytoplasm, thereby maintaining homeostasis (Figure 3).
Figure 3
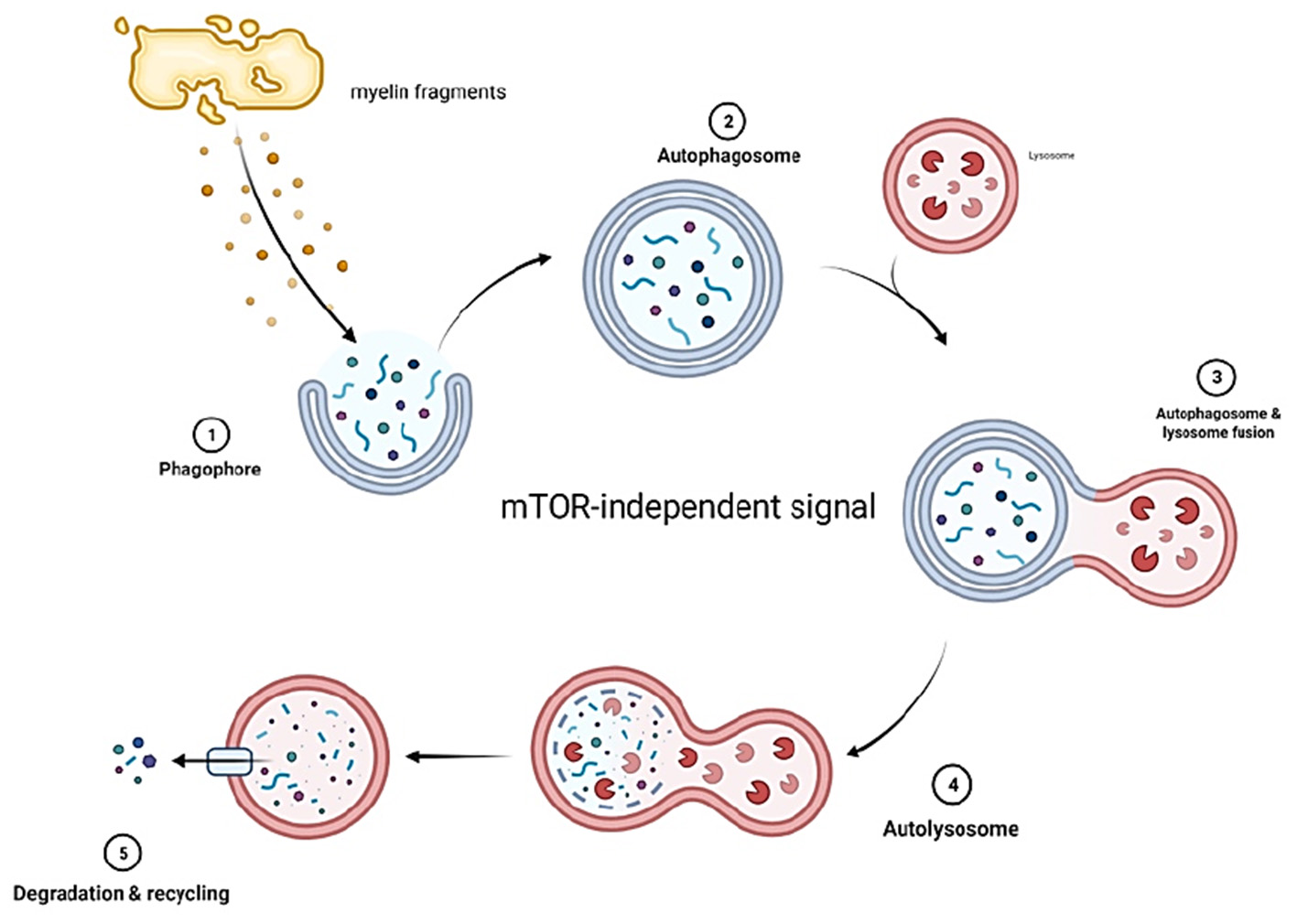
Myelin fragments at the site of injury form phagophores which develop into autophagosomes. The autophagosomes then fuse with lysosomes that contain enzymes capable of digesting myelin fragments. The decomposed fragments are then released into circulation. Autophagy in SCs is mTOR independent.
SCs will rapidly switch their transcriptional program to a repair state that promotes the early stages of myelin clearance in response to PNI. Activation of autophagy is a critical component of this transformation (114). Autophagic clearance mechanisms improve the microenvironment, provide basal energy for SC survival (104), and prevent the emergence and recurrence of neuropathic pain (115). Notably, lipids represent 70–80% of the composition of peripheral myelin (116) and lipid droplets have been detected as autophagic cargo (117). Therefore, autophagy is a crucial process involved in nerve repair; impaired autophagy results in reductions in both myelin degradation and remyelination (118).
Main regulators of autophagy and autophagic flux
Nerve growth factor
Li et al. reported that levels of autophagy-related (ATG)-7, ATG-5, Beclin-1, and LC3 proteins all increased in response to treatment with exogenous nerve growth factor (NGF) (119). In contrast to the traditional perspective, recent results suggest that the administration of NGF upregulates autophagic activity in SCs, most likely via the p75NTR/AMPK/mTOR pathway. In addition, NGF binds to p75NTR and activates small GTPases to increase the expression of c-Jun. Augmented expression of c-Jun might inhibit the myelination as described earlier. Therefore, the pathways linking NGF and autophagy specifically in SCs may represent novel therapeutic targets for drugs designed to promote nerve regeneration.
SIRT1/Hypoxia-inducible factor 1α
The NAD-dependent deacetylase, Sirt1, plays a critical role in regulating autophagy (120). Recent evidence suggests that overexpression of Sirt1 promotes regeneration and functional recovery via the activation of autophagy (121). Sirt1 promotes mTOR-independent autophagy via the activation of the downstream factor, Hif1α (121). Interestingly, these results suggest that Sirt1 might drive autophagy directly via its capacity to inhibit both mTOR and FOXO (122). Although the precise mechanism involved in this pathway requires further elucidation, it may also represent a target for one or more novel treatment strategies.
CXCL12
CXCL12 is one of the most widely studied chemokines and binds to its specific receptor, CXCR4/7. Administration of CXCL12 elicited positive therapeutic effects in various nervous system diseases (123–126). Of note, CXCL12 enhances autophagy after nerve injury and thus promotes nerve regeneration (126). Specifically, recombinant CXCL12 enhances autophagy in SCs in a time-dependent manner and inhibits the PI3K/AKT/mTOR pathway. Thus, the increased levels of autophagy induced by CXCL12 may be related to its capacity to inhibit mTOR (126).
Calcineurin
Calcineurin is a calcium-dependent serine/threonine protein phosphatase that is composed of calcineurin A (CnA) and calcineurin B (CnB). Cn was originally identified as a regulator of autophagy in lysosomal biogenesis (127). Recent findings reveal that Cn regulates autophagy via the activation of a calcium signaling pathway and transcriptional factor EB (TFEB) (127, 128). Additional evidence documented the role of exogenous trehalose in inducing autophagy in models of motor neuron degeneration (129); Reed et al. reported that CnB might play a critical role in this pathway. Furthermore, CnB gene-deleted mice exhibited a reduced autophagic influx, delayed myelination, and altered radial sorting (130). CnB ablation also impaired translocation to the nucleus, suggesting that Cn was capable of activating autophagy in SCs via the actions of TFEB.
Interestingly, Cn had no significant impact on the development of myelin (130); likewise, most of the other factors that regulate myelin clearance are also not closely associated with the process of myelin formation.
Vacuolar protein sorting family
PI3K vacuolar protein sorting 32 (Pik3c3) contributes to the synthesis of PI3P and is thus crucial for membrane transport and autophagy. The complex nature of PI3P regulation has been considered one of the mechanisms associated with CMT disease. In one recent study, Logan et al. found that mature autophagosomes could not be formed in Vps34-deficient SCs in the absence of PI3P, and thus the process of autophagy would be destroyed (131).
Wang et al. have recently suggested the existence of an ESCRT-Vps4-autophagy pathway that regulates the clearance of cellular debris (132). Vps4 is an ESCRT accessory protein that plays a vital role in regulating axonal autophagic flux. Cellular levels of Vps4 were found to influence the autophagic flux in experiments performed in Drosophila melanogaster. Subsequent studies performed in mammalian models revealed that Vsp4 provided support to the autophagic flux but was not critical for its induction (132). Vps4 was consumed rapidly after injury and may induce the accumulation of autophagosomes and thus impair cellular organization. Therefore, we speculate that Vps4 may have the capacity to promote nerve regeneration via the regulation of autophagy and thus the clearance of myelin debris.
FGF
The basic fibroblast growth factor (bFGF) is a major neurotrophic factor secreted by SCs as well as neuronal cell populations. High levels of bFGF expression can promote numerous functions associated with PNI, including neuroprotection, neurogenesis, and the generation of an appropriate microenvironment (133, 134). Notably, recent studies revealed that the upregulation of bFGF promotes myelin clearance in vitro via the maintenance of an effective autophagic flux at the early stages after PNI (135). Additional studies suggested that bFGF most likely targets the TFEB pathway (135).
In addition, recent evidence suggests that fibroblast growth factor 1 (FGF1) elicits its protective and regenerative effects on SCs due to its capacity to regulate autophagy via peroxiredoxin (PRDX1), possibly in association with the Wnt pathway (136).
Unlike the classical activation of autophagy, which relies on the inhibition of the mTOR pathway, autophagy in SCs is activated by an mTOR-independent mechanism that is regulated by the actions of ceramides on the JNK/c-Jun pathway (114, 118, 137). Activation of this pathway may be synergistic with the inhibitory effect of c-Jun on myelin-associated protein expression in response to acute injury. Interestingly, mTORC1 is necessary for myelin clearance during the post-injury period via its role in promoting the dedifferentiation of SCs and increasing the expression of c-Jun, thereby inhibiting the expression of myelin-related proteins (60). Collectively, these findings may explain how SC-mediated autophagy occurs via an mTOR-independent mechanism.
Other SC-associated mechanisms that promote myelin clearance
TAM receptor tyrosine kinases
Tyro3, AXL, and Mer are among the TAM receptors. Members of this family of receptor tyrosine kinases promote SC-mediated phagocytosis via mechanisms that are similar to those used by macrophages. Interestingly, mouse model strains with defective autophagy remain capable of debris clearance over an extended period. These observations imply the existence of other pathways that can compensate for one or more autophagic defects. Recent evidence from mouse models of PNI suggests that SCs may promote clearance via TAM receptor (Axl/Mertk)-mediated phagocytosis (138). Clearance of myelin debris was impaired in mice devoid of these receptors.
Transcription factor nuclear factor erythroid-2-related factor 2
Transcription factor Nrf2 is activated in response to oxidative stress that develops after PNI. Oxidative stress typically results from a crush injury or chronic neuronal constriction; reactive oxygen species (ROS) are also produced after a transection injury (139–141). Nrf2 is held in the cytoplasm by its Kelch-like ECH-associated protein 1 (Keap1) inhibitor domain. Gene expression is activated via its antioxidant response elements (AREs) (142). When compared to wild-type mice, Nrf2-deficient strains were present with more myelin residue, lower levels of macrophage infiltration, and weak recovery of the neuromuscular junction. Thus, there may be a critical role for a Keap1-Nrf2-ARE pathway (143), and Nrf2 may be a crucial factor involved in promoting the timely clearance of myelin fragments.
Macrophages
After PNI, SCs will call on local macrophages and recruit cells from the peripheral circulation to assist with cleaning the microenvironment. Both resident and infiltrating macrophages provide critical contributions to this process (144) (Figure 4).
Figure 4
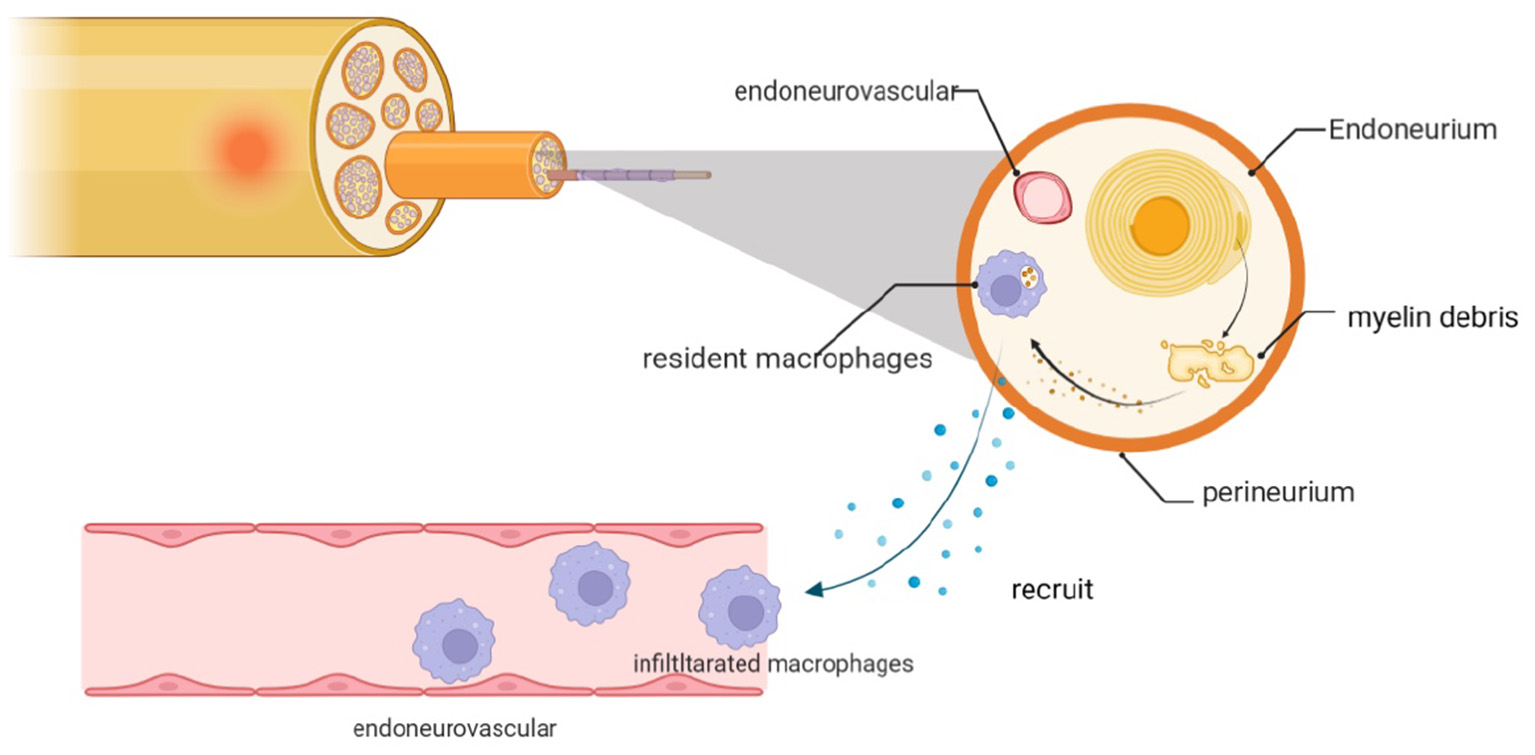
Myelin debris that accumulates after PNI is cleared by macrophages, including resident cells and those recruited locally from the neurovasculature. The infiltrating macrophages are recruited in response to chemokines and cytokines that are expressed by dedifferentiated SCs and resident macrophages.
Contribution of resident and infiltrating macrophages
Resident macrophages
Macrophages that are resident cells in peripheral nerves respond to injury and also play an active role in the early stages of peripheral nerve diseases (35, 145). Similar to SCs, resident macrophages are capable of phagocytosis, albeit to a more limited degree (23, 34). However, it is critical to recognize that all resident macrophages do not respond to all injuries. For example, a recent study by Ydens et al. revealed that primarily endoneurial macrophages respond to nerve crush injury (146). In addition to debris removal, resident macrophages secrete monocyte-attracting chemokines and can thus recruit additional macrophages and enhance debris clearance (36).
Infiltrating macrophages
More efficient macrophage recruitment and activation results in improved myelin clearance. Within the first 2 to 3 days after the disruption of the endoneurovascular, macrophages are recruited to and infiltrate the injury site. Macrophage accumulation reaches a peak at approximately 14 days post-injury (36). Circulating macrophages are attracted to the site of injury by inflammatory cytokines and chemokines, including TNF-α, interleukin (IL)-1α, IL-1β, chemokine C-C motif ligand 2 (CCL2), leukemia inhibitory factor (LIF), and pancreatitis-associated protein III (PAP-III) (36, 138, 147). Galectin-1 and galectin-3 are also critical factors involved in macrophage accumulation (147, 148); these mediators are expressed in SCs, nerves, and fibroblasts. Infiltrating macrophages also produce CCL2, TNFα, IL-1α, and IL-1β that collectively serve to augment recruitment and phagocytosis. Decomposition of the blood–nerve barrier (BNB) which isolates the nerve following the recruitment of macrophages is induced by Raf/ERK signaling in dedifferentiated SCs (69). A full discussion of several related mechanisms involved in the regulation of macrophage recruitment and activation is included in the sections to follow.
Macrophages associated mechanisms promoting myelin clearance
Complement system
The complement system plays a critical role in modulating the impact of macrophages responding to a PNI. After an injury, the complement system can be activated through either classical or alternative pathways (149, 150). Complement component 3 (C3) and its receptor, complement receptor 3 (CR3), are central mediators of myelin clearance. CR3 expressed on the macrophage surface binds to the degenerating myelin sheaths and initiates phagocytosis (151). Phagocytosis is delayed in cells that have been treated with soluble (s)CR1, which is an inhibitor of both the classical and alternative pathways of complement activation (150). Recruitment and activation of macrophages are also suppressed in C3- (152), C5-, and C6-deficit mice (150, 153); myelin seems to remain intact in the C6-deficient strain (150). Collectively, these results suggest that the complement system contributes directly to SC- and macrophage-mediated myelin clearance.
Calcium-binding proteins S100A8 and S100A9
Calcium-binding proteins S100A8 and S100A9 activate inflammation and may initiate an acute phase response and a chemotactic gradient that precedes the main inflammatory response (154). Both S100A8 and S100A9 are upregulated in response to injury, specifically in both proximal and distal segments in nerve transection models. As these proteins are both toll-like receptor (TLR)4 ligands (155), their expression results in the induction of additional pro-inflammatory genes in SCs, including partial chemokines, inflammatory cytokines, and matrix metalloproteinases. These factors result in the infiltration of circulating macrophages at the injured site (154).
Toll-like receptors
Toll-like receptors (TLRs) are strongly induced in response to acute injury and play essential roles in the PNS (29). TLRs are expressed on the surface of resident macrophages and can be activated by endogenous ligands that initiate an innate immune response. Mice devoid of TLR2, TLR4, or myeloid differentiation factor 88 (MyD88) exhibited diminished macrophage recruitment due to impaired expression of CCL2 and IL-1β compared with their respective wild-type strains (156). The activation and recruitment of macrophages are augmented in response to the administration of TLR agonists, although this mechanism is impaired in mice that are TLR or TLR-signaling deficient. For example, Vallières et al. (157) reported that systemic injections of the TLR4 agonist, lipopolysaccharide (LPS), increased both macrophage recruitment and phagocytosis. TLRs respond to both pathogen-associated molecular patterns (PAMPs) (158) and to nonpathogenic ligands, including necrotic cells, heat shock proteins (HSPs 60 and 70), and extracellular matrix (ECM) components (159) that typically accumulate and promote inflammation at the site of a nerve injury site (160). TLR3, TLR4, and TLR7 also contribute to the immune surveillance of the PNS (29). Overall, TLRs provide critical signals that permit the PNS to respond to injury by activating macrophages and initiating myelin clearance.
G protein-coupled receptor Gpr126/Adgrg6
Gpr126/Adgrg6 has been characterized as a critical mediator of nerve development (161) and a regulator of myelin sheath formation (162). However, recent evidence suggests that this receptor plays an essential role in the recruitment of macrophages and ultimately myelin regeneration. Mogha et al. reported that Gpr126 contributed to the regulation of chemokines and TNF-α; levels of downstream targets, including CCL2, CCL3, and CXCL10, are reduced in Gpr126 deficit mice (163). These results suggest that the relationship between Gpr126 and chemokines has a significant impact on the regulation of macrophage recruitment. Notably, c-Jun, a factor known to be involved in regeneration, is upregulated in Gpr126-deficient mice. Therefore, we speculate that efforts to regulate Gpr126 may lead to new strategies for the treatment of PNI (27, 164) and that the Gpr126/Adgrg6 receptor may be targeted to promote the recruitment of macrophages and clearance of myelin debris.
Serum amyloid A
Serum amyloid A is a major component of the acute phase response to injury and infection that induces the production of pro-inflammatory chemokines. While SAA is synthesized primarily in the liver, the generation of SAA at extrahepatic sites may serve to regulate local inflammatory responses (165). SAA can be synthesized in SCs in response to injury and can induce the production of chemokines CCL2 and CCL12. SAA also has functions that are similar to those of chemokines and provides crucial contributions to macrophage recruitment. Expression of SAA is suppressed in IL-6-deficit mice in association with reduced levels of CCL2; these results suggest that SAA may be a key modulator of IL-6-mediated induction of this chemokine. Results from recent reports reveal that SAA is upregulated in SCs 3 days after injury (166) and that this response is significantly diminished in IL-6 gene-deleted mice. Collectively, these results suggest that IL-6 plays an essential role in SAA expression induced in response to injury (166) and that the IL-6-SAA-CCL2 pathway may be a new target that can be exploited therapeutically to regulate macrophage recruitment.
Apolipoprotein D
Apolipoprotein D is a glia-derived apolipoprotein that is required for a timely and effective response to injury (167, 168). As reported in a previous study, the role played by ApoD in response to nerve injury is associated with the dedifferentiation SCs (169). Myelin clearance and regeneration are delayed in injured nerves from ApoD-deficient mice; these results suggest that ApoD may play a crucial role in promoting myelin clearance. The report further demonstrated that the number of macrophages at the injury site was related to levels of ApoD, which in turn had a substantial impact on myelin clearance (169). The rates of both macrophage recruitment and myelin clearance were reduced in ApoD gene-deleted mice. Expression of the effector molecule, galectin-3, was also influenced by ApoD (169). The results of this study suggested a negative feedback mechanism in which ApoD regulated the production of factors associated with TLR activation, including CCL2 and TNF-α, and thus controlled for the potential over-recruitment of circulating macrophages (169). ApoD also helps to promote efficient myelin degradation; while myelin-positive ApoD-deficient macrophages accumulate more rapidly, these cells accumulate larger myelin particles and require more time to degrade MBP and MAG (169). Overall, the main impact of ApoD on myelin clearance relates to its capacity to recruit and activate macrophages and to promote autophagy via Becn1 (168). Thus, ApoD is another potential target that might be developed for new treatment strategies.
Sox2
The transcription factor, Sox2, limits the myelination of SCs both in vitro and in vivo via its capacity to suppress Krox20 (170). Sox2 also promotes clearance of myelin debris via its capacity to promote macrophage recruitment of macrophages. Long-term expression of Sox2 results in a persistent inflammatory state. Interestingly, while overexpression of Sox2 promotes both proliferation and radial sorting of SCs, it also limits SC-mediated myelination and thus inhibits functional recovery in the PNS (171). While these findings may seem contradictory, they can be understood in light of the development of PNS. Thus, Sox2 may also be a critical therapeutic target via strategies that precisely regulate its expression based on the time elapsed after the acute injury.
Fibroblast growth factor 9
Fibroblast growth factor 9 contributes to the recruitment of M1 macrophages and macrophage recruitment is diminished in FGF9-deficient mice. Ablation of FGF9 also limited the accumulation of CD68-positive and CD86-positive macrophages (172). Consequently, we speculate that FGF9 is a critical factor involved in clearing myelin debris after a PNI.
Phospholipase A2 family
The phospholipase A2 enzyme family includes a calcium-dependent group IVA (GIVA cPLA2) and a calcium-independent group VIA (GVIA iPLA 2). PLA2s regulate phospholipid metabolism, membrane turnover, host defense, and signal transduction (61, 173); these activities have been characterized in the PNS (174). PLA2s play important roles in regulating the metabolic activities of infiltrated macrophages. SCs express both group IVA and group VIA PLAs approximately 6 h after injury. López-Vales et al. (175) reported diminished expression of IL-1β and CCL2 mRNA in nerves devoid of group IVA and group VIA PLA2s in association with a reduction in the number of macrophages. Therefore, this mechanism also needs to be considered as a way to regulate macrophages.
Adhesion molecules
Intercellular cell adhesion molecule-1 (ICAM-1) is a member of the immunoglobulin superfamily that is upregulated in SCs and venous endothelial cells in the nerve tissue that promotes macrophage recruitment (176). ICAM-1 is upregulated in the early stages immediately after injury. Ablation of ICAM-1 results in decreased recruitment of macrophages in response to a transection injury as well as the accumulation of more myelin debris (177). However, this result was not confirmed in experiments performed using a different strain of ICAM-1 gene-deleted mice (178). Further study will be needed to confirm or refute the original findings.
P-selectin is another cell adhesion molecule that is expressed both on blood vessel endothelial cells and activated platelets. The interaction between P-selectin and its ligand, P-selectin ligand (PSGL-1), may promote macrophage recruitment and sustain this cell population at a later stage after injury (179). The expression of TNF-α, IL-6, and other pro-inflammatory cytokines was significantly attenuated in injured nerves from P-selectin-deficient mice (179). These results imply that P-selectin may be an essential mediator of macrophage infiltration.
Microvascular endothelial cells
Results from several recent studies revealed that microvascular endothelial cells are also crucial for the recruitment of macrophages and engulfing myelin debris that accumulates in response to injury (35, 180).
Macrophages are the cells that are primarily responsible for clearing myelin debris that accumulates in response to acute PNI. The processes involved in macrophage recruitment and activation are complex and in need of further research. Among the points not considered in this review, additional consideration of macrophage classification and the unique roles of M1 and M2 macrophages might be warranted. However, the M1 and M2 macrophage phenotypes cannot be determined accurately in cells and tissues from experiments performed in vivo (146). Here, we focused our attention on the activation and recruitment of macrophages under conditions that have been closely associated with myelin clearance. Interestingly, although macrophages are typically the primary mode of myelin clearance, disruption of macrophage recruitment has no clear impact on this process. Results from several studies revealed that neutrophils can also clear myelin debris and can compensate for the absence of macrophages in these circumstances (35). Fibroblasts may also have the capacity to remove tissue debris along with SCs during the early stages of the injury (4, 181).
Exogeneous interventions
Numerous exogenous approaches, listed in Table 1, may be used to promote the clearance of myelin fragments. These approaches are based on the mechanisms associated with myelination (including negative regulation), autophagy, and the role of resident and infiltrating macrophages. These strategies will be discussed at length in the following article and may provide one or more new directions for research undertakings.
Table 1
| Name | Intervention ways | Mechanism | References |
|---|---|---|---|
| Nuclear factor κB (NF-κB) | Exogenous inhibition | Suppress myelination | (2, 3) |
| Ascorbic acid | Exogenous inhibition | Suppress myelination; Promote macrophage infiltration |
(4) |
| Wnt/β-catenin | Exogenous inhibition | Suppress myelination | (5, 6) |
| Oxysterols | Treat with LXRs | Suppress myelination | (7) |
| Low-density lipoprotein receptor-related protein 4(Lrp4) | Exogenous inhibition | Suppress myelination | (8, 9) |
| LKB1 Liver kinase B1(LKB1) | Exogenous inhibition | Suppress myelination | (10) |
| HDAC1/2 | Exogenous inhibition | Suppress myelination | (11) |
| SncRNA715 | Not mentioned in vivo | Suppress myelination | (12) |
| circRNA.2837 | Exogenous inhibition | Promote autophagy | (13) |
| Resveratrol (RSV) | Given exogenously | Promote autophagy | (14) |
| Rapamycin | Given exogenously | Promote autophagy | (15) |
| Epothilone B (EropB) | Given exogenously | Promote autophagy | (16) |
| Metformin | Given exogenously | Promote autophagy | (17) |
| β-Site amyloid precursor protein (APP) cleaving enzyme 1 (BACE1) | Exogenous inhibition | Promote the macrophages phagocytosis | (18, 19) |
| E6020 | Given exogenously | Promote the macrophages phagocytosis | (20) |
| Signal regulatory protein-α (SIRPα) | Exogenous inhibition | Promote the macrophages phagocytosis | (21) |
Exogeneous interventions.
Inhibition of NF-κB
Nuclear factor κB (NF-κB) is a master regulator of the inflammatory response and a mediator in many disease processes. While activation and translocation of NF-κB are essential to promote SC myelination and differentiation in vitro (182, 183), this factor is dispensable for myelination in vivo (3). Previous evidence suggested that NF-κB might be an essential mediator of PNI and that compact remyelination is delayed by short-term suppression of this transcription factor (2). Interestingly, NF-κB inhibition was found to promote regeneration after PNI (2). Therefore, factors that regulate NF-κB activation and translocation may be critical factors associated with the process of myelin removal.
Ascorbic acid
Ascorbic acid, also known as vitamin C, is an essential micronutrient. Recent reports suggest that AA was also important for repairing nerve injury, specifically, both morphological and functional recovery of injured peripheral nerves (184). Other researchers reported that the administration of AA resulted in enhanced levels of c-Jun level and diminished levels of MAG during the early stages of PNI; collectively, these responses all repress myelin formation (4). AA also plays a critical role in macrophage recruitment. The results revealed that AA could enhance macrophage migration and infiltration and thus provide support for a suitable microenvironment (4). Interestingly, AA was also capable of promoting myelination via the activation of DNA demethylation (185). Collectively, these results suggest that AA may be an effective means to promote repair throughout the entire period after nerve injury.
Inhibition of Wnt/β-catenin
The Wnt/β-catenin pathway regulates the expression of myelin-related genes (48). Wnt promotes the myelin gene expression by suppressing glycogen synthase kinase 3β (GSK3β); this will preserve β-catenin which then enters the nucleus. Once in the nucleus, β-catenin interacts with T-cell factor/lymphoid-enhancer factors (TCF/LEF or TCFs) that regulate the expression of myelin-related genes (79).
Notably, GSK3β is an essential factor in the pathway that regulates myelin-related gene expression. In addition, recent evidence suggests that early clearance of myelin debris is enhanced in response to lithium chloride (LiCl) which is a characterized inhibitor of the enzymatic activity of GSK3β (6). An evaluation of the subsequent phase revealed that myelin regeneration occurred in parallel with the findings described in the previous study (5). Thus, GSK3β may be another critical target for therapeutic strategies designed to promote the clearance of myelin debris and ultimately myelin regeneration. Accordingly, the GSK3β inhibitor LiCl may be applied in peripheral nerves as a bidirectional regulator of myelin synthesis. The roles of other GSK3β inhibitors remain to be explored.
Oxysterols
Oxysterol is known for its impact on cholesterol homeostasis and in neurodegenerative disorders including Alzheimer's disease (186) and multiple sclerosis (187, 188). Oxysterol produced in SCs of PNS suppresses the expression of myelin-related genes, Pmp22, and MPZ in experiments performed in vitro (5). Oxysterol treatment of cells expressing high levels of liver X receptors (LXRs) also resulted in significant suppression; these results suggested that these inhibitory effects may be mediated by LXR ligands. Additional evidence revealed that LXR-mediated suppression might be the result of inhibition of the classical Wnt pathway (7). LXRs are likely to be involved in many pathways that promote both physiological and pathological responses in the PNS (189). The aforementioned mechanism might be clarified with additional research.
Low-density lipoprotein receptor-related protein 4
Low-density lipoprotein receptor-related protein 4 is a critical protein that contributes directly to the Agrin-Lrp4-MuSK signaling pathway and is an essential regulator in nervous system development (8). Recent evidence revealed that Lrp4 expressed in SCs may be crucial for the regeneration of axons via an extrinsic mechanism (190). Moreover, Krox20 is downregulated in Lrp4 conditional gene-deleted mice together with low levels of MPZ (9). In this study, mutant mice exhibited superior repair properties and benefitted from the rapid clearance of myelin and proliferation of SCs that resulted from Krox20 downregulation after injury. Exploring a more defined mechanism of inhibition, Krox20 may support a new target for regenerating axons.
Inhibition of liver kinase B1
Liver kinase B1 is asymmetrically localized at the SC–axon interface; this specific localization is dependent on PKA-mediated phosphorylation at Ser-431. LKB1 may be the central regulator of cellular asymmetry in SCs which is a property required to initiate myelination. Accordingly, the expression of Krox20 and MPZ is suppressed in LKB1 gene-deleted mice in association with delayed myelination (10). The LKB1-mutant mice demonstrated hypomyelination and limb handicaps (10, 191). In addition, the LKB1 deficit limits the activation of the tricarboxylic acid cycle (TCA) and thus reduces the production of citrate. Citrate is a six-carbon precursor to many cellular lipids; thus, changes in its intracellular concentration will have an immediate impact on normal myelination (191, 192). Therefore, we hypothesize that in the early stages after injury, LKB1 localization might be modulated by targeting PKA-mediated phosphorylation of Ser431, thereby regulating the process of myelin removal.
Epigenetic modulators
Epigenetic modulators may have an impact on myelin-related gene expression and clearance. For example, the expression of histone deacetylase (HDAC)1/2 was strongly upregulated after PNI. The high expression levels of HDAC1/2 resulted in delayed demyelination and axon degeneration (11). Conversely, deletion of the genes encoding HDAC1/2 delayed the expression of Oct6 and enhanced c-Jun. These events helped to transform SCs into a pro-repair state and to reduce the expression of both Krox20 and MPZ. Moreover, recent results revealed that a brief period of HDAC1/2 inhibition promotes nerve regeneration together with the increased thickness of myelin. Thus, we conjecture that early inhibition of HDAC1/2 promotes regeneration by accelerating phenotype conversion and suppressing myelination during the early stages.
Recently, it has been found that histone deacetylase 3 (HDAC3, a histone-modifying enzyme) acts as an inhibitor of SC myelination (193). HDAC3 was proved with a high-level expression after injury in SCs to suppress the process of remyelination. This overexpression represses the activation of PI3K-AKT and ERK. HDAC3 cooperates with histone acetyltransferase (HAT) targeting TEAD4, described as a new inhibitor of SC myelin growth, which is shown by genomic occupancy analyses (193). Although partial inhibition of HDAC3 led to faster neurological and functional recovery, we currently do not know the contribution of HDAC3 to myelin clearance in the early phase after injury. Still, this possibility function cannot be denied. HDAC3 may play a bidirectional regulatory role in the process of neurological recovery, which needs to be demonstrated by further studies.
Noncoding RNA
Small non-coding RNA 715 (sncRNA 715) originates from ribosomal DNA (rDNA) and was identified as an early inhibitor of demyelination and MBP translation in the CNS (194). Recent results documented that sncRNA 715 was expressed in pro-myelin SCs in PNS where it regulates MBP translation via a mechanism similar to that described in the CNS (12). Although we are not aware of any alterations in sncRNA715 expression in response to injury, we hypothesize that myelin removal might be regulated by exogenous interventions that target sncRNA715. Therefore, strategies that target noncoding RNA may be used to stabilize the microenvironment during the early stages of injury.
Circular RNAs (circRNAs) are non-coding RNAs found in the cytoplasm that regulate transcriptional and posttranscriptional gene expression. While there is little evidence available that documents the role of circRNA in the PNS, recent evidence suggests that they may be functionally associated with processes that regulate autophagy. Zhou et al. (13) reported significant discrepancies in the expression of circRNAs immediately after PNI; the elimination of circRNA.2837 resulted in the amplification of autophagy in primary spinal nerves. Likewise, the downregulation of circRNA in vitro could block static nerve injury by promoting autophagy via targeting miR-34a (13).
Resveratrol
Resveratrol activates autophagy and thus has an impact on many signals that promote or alleviate pathology, including those contributing to nerve injury and CNS/PNS diseases. Although its mechanism of action remains unclear, RSV is believed to promote autophagy via its interactions with signaling pathways associated with mTOR, AMPK, SIRT1, PI3K/Akt, and MAPK. RSV also suppresses the TLR4/NF-κB signaling to activate autophagy, thereby accelerating clearance and promoting regeneration (14). Administration of RSV may thus be an effective strategy for the treatment of PNS and CNS injury.
Rapamycin
In mammals, mTOR is a downstream factor of PI3K/Akt signaling (195) and regulates the formation of myelin and the growth of axons. Results from previous studies revealed that mTOR inhibition activates autophagy and thus promotes the clearance of damaged cellular components and preserves homeostasis (196–198). A study of PNI in rats reported that treatment with the mTOR inhibitor, rapamycin, promotes peripheral nerve regeneration and functional recovery after injury (15). While this may appear to be paradoxical given the role of mTOR in promoting negative regulation of myelin formation, autophagy detected in SCs under these circumstances may be the result of an mTOR-independent pathway. We suspect that this is likely given the timing of intervention after injury; in other words, timely intervention with mTOR inhibitors might enhance myelin clearance.
Epothilone B
While EropB was initially used to treat cancer, additional studies revealed that this agent could promote recovery from CNS diseases (199, 200). The results of recent research suggest that EropB can aid with the structural and functional repair of damaged peripheral nerves (16). In contrast to the aforementioned strategy, EropB enhances autophagy in the PNS and promotes the migration of SCs. This process can be suppressed in the presence of the autophagy inhibitor, 3-MA. Additional research suggests that EropB may provide crucial regulation of this response by suppressing the PI3K/Akt pathway.
Metformin
Metformin is a first-line anti-hyperglycemic agent used to treat type II diabetes mellitus. Metformin has also been used to relieve neurological problems, including neuropathic pain in the spine (201), cognitive decline, and memory loss (202). The results of a recent study reveal that metformin can induce autophagy in the early stages after injury and enhance the number of autophagosomes. This facilitates recovery by increasing the rate and extent of autophagy which results in more effective removal of myelin fragments (17).
Inhibition of β-Site amyloid precursor protein cleaving enzyme 1
β-Site amyloid precursor protein cleaving enzyme 1 is an aspartyl protease known for its role in producing amyloid-β peptides in association with Alzheimer's disease. In the PNS, BACE1 cleaves neuregulin 1 type III (72) and, indirectly, APP (203). Reductions in BACE1 activity in gene-deleted rats enhance the clearance of myelin debris and promote axon regeneration compared to the wild-type (204, 205). By contrast, BACE1 over-expression leads to significant reductions in the length of the regenerated axons as well as the number of neuromuscular junctions (19). Treatment with a BACE1 inhibitor results in improved nerve regrowth and clearance of debris 7 days after a crushing injury (206). This rapid clearance is facilitated by the earlier influx of macrophages that are capable of more effective phagocytosis (79). The mechanisms underlying this response involve the upregulation of both tumor necrosis factor receptor 1 (TNFR1) and its downstream transcription factor, NF-κB, at the distal site of the injury.
E6020
E6020 is a lipid A mimetic and TLR4 agonist that promotes macrophage-mediated phagocytosis of accumulated debris. E6020 also induced cytokine production, activation of intracellular NF-κB signaling, and an overall “activated” macrophage morphology in experiments performed in vitro. Treatment with E6020 resulted in a larger macrophage infiltrate. Although these results cannot be paralleled with LPS, E6020-induced clearance might be controlled in the appropriate range and thus used to limit over-clearance (20).
Inhibition of signal regulatory proteinα
A SIRPα-dependent mechanism suppressed macrophage activation and thus impeded myelin clearance in vivo. Earlier studies revealed that CD47 associated with myelin interacted with SIRPα on macrophages to downregulate phagocytosis in the CNS (207). This interaction, combined with IL-10, constraints inflammation-induced macrophage phagocytosis and thus may protect healthy cells (208). Recent studies have characterized this interaction in the PNS. Injured rats undergoing SIRPα inhibition respond with rapid clearance of myelin, axon regeneration, and repair of the affected nerve. This effect results from blocking the binding between SIRPα and CD47 and removing CD47 from myelin fragments (21).
Conclusion
Regeneration after PNI is influenced by many factors and molecular mechanisms. The extent of the injury and the ability to establish and provide ongoing support for a microenvironment that supports regeneration are among these critical features. Previous studies and clinical strategies have focused primarily on interventions that might promote axonal and myelin regeneration after injury; there has been little to no attention paid to the process of myelin clearance. Here, we discuss several strategies that might be used to promote myelin clearance. Collectively, current evidence suggests that clearance of myelin debris is beneficial for the process of regeneration during the early stages yet harmful if the process has been delayed. An improved understanding of mechanisms associated with myelin clearance after a PNI might contribute to new and effective therapeutic strategies to promote regeneration after injury. These findings will also provide essential contributions to our understanding of demyelinating neural disorders, degenerative diseases, and diseases of the CNS.
There are still many issues that need to be investigated, including the key pathways and the potential range of intervention strategies, as well as the aforementioned paradoxes and negative influences, among others. For example, one study features a nerve regeneration strategy involving partial enzymatic digestion of myelin debris. This mechanism facilitates the rapid removal of debris and creates a favorable microenvironment that has promoted the regeneration of damaged nerves in experimental rat models. However, as reported by Tsuang et al. (108), repair exhibited by the high-dose group was significantly less than that observed among those treated with lower doses. These results also suggest that treatment should be decided on a per-case basis and might focus on the rapid removal of cellular debris. Additional experiments will be needed to re-evaluate and optimize these strategies.
Statements
Author contributions
Conceptualization: YY and YW. Investigation: YY and SW. Writing—original draft preparation: YY and MZ. Writing—review and editing, project administration, funding acquisition, and Supervision: YW. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (Grant: 81973926), the Provincial Natural Science Foundation (Grant: LH2021H090), and the Outstanding Innovative Talents Support Program of Heilongjiang University of Chinese Medicine (2018RCL10).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Huebner EA Strittmatter SM . Axon regeneration in the peripheral and central nervous systems. Results Probl Cell Differ. (2009) 48:339–51. 10.1007/400_2009_19
2.
Morton PD Johnstone JT Ramos AY Liebl DJ Bunge MB Bethea JR . Nuclear factor-κB activation in Schwann cells regulates regeneration and remyelination. Glia. (2012) 60:639–50. 10.1002/glia.22297
3.
Morton PD Dellarole A Theus MH Walters WM Berge SS Bethea JR . Activation of NF-κB in Schwann cells is dispensable for myelination in vivo. J Neurosci. (2013) 33:9932–6. 10.1523/JNEUROSCI.2483-12.2013
4.
Qu W-R Zhu Z Liu J Song D-B Tian H Chen B-P et al . Interaction between Schwann cells and other cells during repair of peripheral nerve injury. Neural Regen Res. (2021) 16:93–8. 10.4103/1673-5374.286956
5.
Makoukji J Belle M Meffre D Stassart R Grenier J Shackleford G et al . Lithium enhances remyelination of peripheral nerves. Proc Natl Acad Sci U S A. (2012) 109:3973–8. 10.1073/pnas.1121367109
6.
Chen Y Weng J Han D Chen B Ma M Yu Y et al . GSK3β inhibition accelerates axon debris clearance and new axon remyelination. Am J Transl Res. (2016) 8:5410–20.
7.
Makoukji J Shackleford G Meffre D Grenier J Liere P Lobaccaro J-MA et al . Interplay between LXR and Wnt/β-catenin signaling in the negative regulation of peripheral myelin genes by oxysterols. J Neurosci. (2011) 31:9620–9. 10.1523/JNEUROSCI.0761-11.2011
8.
Wu H Lu Y Shen C Patel N Gan L Xiong WC et al . Distinct roles of muscle and motoneuron LRP4 in neuromuscular junction formation. Neuron. (2012) 75:94–107. 10.1016/j.neuron.2012.04.033
9.
Hui T-K Lai X-S Dong X Jing H Liu Z Fei E et al . Ablation of Lrp4 in Schwann cells promotes peripheral nerve regeneration in mice. Biology (Basel). (2021) 10:452. 10.3390/biology10060452
10.
Shen Y-AA Chen Y Dao DQ Mayoral SR Wu L Meijer D et al . Phosphorylation of LKB1/Par-4 establishes Schwann cell polarity to initiate and control myelin extent. Nat Commun. (2014) 5:4991. 10.1038/ncomms5991
11.
Brügger V Duman M Bochud M Münger E Heller M Ruff S et al . Delaying histone deacetylase response to injury accelerates conversion into repair Schwann cells and nerve regeneration. Nat Commun. (2017) 8:14272. 10.1038/ncomms14272
12.
Müller C Hochhaus NM Fontana X Luhmann HJ White R . SncRNA715 inhibits schwann cell myelin basic protein synthesis. PLoS ONE. (2015) 10:e0136900. 10.1371/journal.pone.0136900
13.
Zhou Z-B Niu Y-L Huang G-X Lu J-J Chen A Zhu L . Silencing of circRNA.2837 plays a protective role in sciatic nerve injury by sponging the miR-34 family via regulating neuronal autophagy. Mol Ther Nucleic Acids. (2018) 12:718–29. 10.1016/j.omtn.2018.07.011
14.
Zhang J Ren J Liu Y Huang D Lu L . Resveratrol regulates the recovery of rat sciatic nerve crush injury by promoting the autophagy of Schwann cells. Life Sci. (2020) 256:117959. 10.1016/j.lfs.2020.117959
15.
Huang H-C Chen L Zhang H-X Li S-F Liu P Zhao T-Y et al . Autophagy promotes peripheral nerve regeneration and motor recovery following sciatic nerve crush injury in rats. J Mol Neurosci. (2016) 58:416–23. 10.1007/s12031-015-0672-9
16.
Zhou J Li S Gao J Hu Y Chen S Luo X et al . Epothilone B facilitates peripheral nerve regeneration by promoting autophagy and migration in schwann cells. Front Cell Neurosci. (2020) 14:143. 10.3389/fncel.2020.00143
17.
Liu L Tian D Liu C Yu K Bai J . Metformin enhances functional recovery of peripheral nerve in rats with sciatic nerve crush injury. Med Sci Monit. (2019) 25:10067–76. 10.12659/MSM.918277
18.
Liu L Fissel JA Tasnim A Borzan J Gocke A Calabresi PA et al . Increased TNFR1 expression and signaling in injured peripheral nerves of mice with reduced BACE1 activity. Neurobiol Dis. (2016) 93:21–7. 10.1016/j.nbd.2016.04.002
19.
Tallon C Rockenstein E Masliah E Farah MH . Increased BACE1 activity inhibits peripheral nerve regeneration after injury. Neurobiol Dis. (2017) 106:147–57. 10.1016/j.nbd.2017.07.003
20.
Church JS Milich LM Lerch JK Popovich PG McTigue DM . E6020, a synthetic TLR4 agonist, accelerates myelin debris clearance, Schwann cell infiltration, and remyelination in the rat spinal cord. Glia. (2017) 65:883–99. 10.1002/glia.23132
21.
Elberg G Liraz-Zaltsman S Reichert F Matozaki T Tal M Rotshenker S . Deletion of SIRPα (signal regulatory protein-α) promotes phagocytic clearance of myelin debris in Wallerian degeneration, axon regeneration, and recovery from nerve injury. J Neuroinflammation. (2019) 16:277. 10.1186/s12974-019-1679-x
22.
Stoll G Griffin JW Li CY Trapp BD . Wallerian degeneration in the peripheral nervous system: participation of both Schwann cells and macrophages in myelin degradation. J Neurocytol. (1989) 18:671–83. 10.1007/BF01187086
23.
Shen ZL Lassner F Bader A Becker M Walter GF Berger A . Cellular activity of resident macrophages during Wallerian degeneration. Microsurgery. (2000) 20:255–61. 10.1002/1098-2752(2000)20:5<255::aid-micr6>3.0.co;2-a
24.
Rao SNR Pearse DD . Regulating axonal responses to injury: the intersection between signaling pathways involved in axon myelination and the inhibition of axon regeneration. Front Mol Neurosci. (2016) 9:33. 10.3389/fnmol.2016.00033
25.
Pereira JA Lebrun-Julien F Suter U . Molecular mechanisms regulating myelination in the peripheral nervous system. Trends Neurosci. (2012) 35:123–34. 10.1016/j.tins.2011.11.006
26.
Jessen KR Mirsky R Lloyd AC . Schwann cells: development and role in nerve repair. Cold Spring Harb Perspect Biol. (2015) 7:a020487. 10.1101/cshperspect.a020487
27.
Brosius Lutz A Barres BA . Contrasting the glial response to axon injury in the central and peripheral nervous systems. Dev Cell. (2014) 28:7–17. 10.1016/j.devcel.2013.12.002
28.
Chen Z-L Yu W-M Strickland S . Peripheral regeneration. Annu Rev Neurosci. (2007) 30:209–33. 10.1146/annurev.neuro.30.051606.094337
29.
Goethals S Ydens E Timmerman V Janssens S . Toll-like receptor expression in the peripheral nerve. Glia. (2010) 58:1701–9. 10.1002/glia.21041
30.
Namgung U . The role of Schwann cell-axon interaction in peripheral nerve regeneration. Cells Tissues Organs. (2014) 200:6–12. 10.1159/000370324
31.
Schwab ME . Nogo and axon regeneration. Curr Opin Neurobiol. (2004) 14:118–24. 10.1016/j.conb.2004.01.004
32.
Vargas ME Barres BA . Why is Wallerian degeneration in the CNS so slow?Annu Rev Neurosci. (2007) 30:153–79. 10.1146/annurev.neuro.30.051606.094354
33.
Fujita Y Endo S Takai T Yamashita T . Myelin suppresses axon regeneration by PIR-B/SHP-mediated inhibition of Trk activity. EMBO J. (2011) 30:1389–401. 10.1038/emboj.2011.55
34.
Mueller M Wacker K Ringelstein EB Hickey WF Imai Y Kiefer R . Rapid response of identified resident endoneurial macrophages to nerve injury. Am J Pathol. (2001) 159:2187–97. 10.1016/S0002-9440(10)63070-2
35.
Lindborg JA Mack M Zigmond RE . Neutrophils are critical for myelin removal in a peripheral nerve injury model of wallerian degeneration. J Neurosci. (2017) 37:10258–77. 10.1523/JNEUROSCI.2085-17.2017
36.
Zigmond RE Echevarria FD . Macrophage biology in the peripheral nervous system after injury. Prog Neurobiol. (2019) 173:102–21. 10.1016/j.pneurobio.2018.12.001
37.
Balakrishnan A Belfiore L Chu T-H Fleming T Midha R Biernaskie J et al . Insights into the role and potential of schwann cells for peripheral nerve repair from studies of development and injury. Front Mol Neurosci. (2020) 13:608442. 10.3389/fnmol.2020.608442
38.
Nave K-A . Myelination and support of axonal integrity by glia. Nature. (2010) 468:244–52. 10.1038/nature09614
39.
Feltri ML Poitelon Y Previtali SC . How Schwann cells sort axons: new concepts. Neuroscientist. (2016) 22:252–65. 10.1177/1073858415572361
40.
Jessen KR Mirsky R . The success and failure of the schwann cell response to nerve injury. Front Cell Neurosci. (2019) 13:33. 10.3389/fncel.2019.00033
41.
Liu B Xin W Tan J-R Zhu R-P Li T Wang D et al . Myelin sheath structure and regeneration in peripheral nerve injury repair. Proc Natl Acad Sci U S A. (2019) 116:22347–52. 10.1073/pnas.1910292116
42.
Svaren J Meijer D . The molecular machinery of myelin gene transcription in Schwann cells. Glia. (2008) 56:1541–51. 10.1002/glia.20767
43.
Chernousov MA Yu W-M Chen Z-L Carey DJ Strickland S . Regulation of Schwann cell function by the extracellular matrix. Glia. (2008) 56:1498–507. 10.1002/glia.20740
44.
Monk KR Oshima K Jörs S Heller S Talbot WS . Gpr126 is essential for peripheral nerve development and myelination in mammals. Development. (2011) 138:2673–80. 10.1242/dev.062224
45.
Glenn TD Talbot WS . Signals regulating myelination in peripheral nerves and the Schwann cell response to injury. Curr Opin Neurobiol. (2013) 23:1041–8. 10.1016/j.conb.2013.06.010
46.
Heller BA Ghidinelli M Voelkl J Einheber S Smith R Grund E et al . Functionally distinct PI 3-kinase pathways regulate myelination in the peripheral nervous system. J Cell Biol. (2014) 204:1219–36. 10.1083/jcb.201307057
47.
Newbern J Birchmeier C . Nrg1/ErbB signaling networks in Schwann cell development and myelination. Semin Cell Dev Biol. (2010) 21:922–8. 10.1016/j.semcdb.2010.08.008
48.
Tawk M Makoukji J Belle M Fonte C Trousson A Hawkins T et al . Wnt/beta-catenin signaling is an essential and direct driver of myelin gene expression and myelinogenesis. J Neurosci. (2011) 31:3729–42. 10.1523/JNEUROSCI.4270-10.2011
49.
Jessen KR Mirsky R . The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci. (2005) 6:671–82. 10.1038/nrn1746
50.
Arthur-Farraj P Wanek K Hantke J Davis CM Jayakar A Parkinson DB et al . Mouse schwann cells need both NRG1 and cyclic AMP to myelinate. Glia. (2011) 59:720–33. 10.1002/glia.21144
51.
Beard JL Wiesinger JA Connor JR . Pre- and postweaning iron deficiency alters myelination in Sprague-Dawley rats. Dev Neurosci. (2003) 25:308–15. 10.1159/000073507
52.
Cheli VT Santiago González DA Marziali LN Zamora NN Guitart ME Spreuer V et al . The divalent metal transporter 1 (DMT1) Is required for iron uptake and normal development of oligodendrocyte progenitor cells. J Neurosci. (2018) 38:9142–59. 10.1523/JNEUROSCI.1447-18.2018
53.
Santiago González DA Cheli VT Wan R Paez PM . Iron metabolism in the peripheral nervous system: the role of DMT1, ferritin, and transferrin receptor in schwann cell maturation and myelination. J Neurosci. (2019) 39:9940–53. 10.1523/JNEUROSCI.1409-19.2019
54.
Santiago González DA Cheli VT Rosenblum SL Denaroso G Paez PM . Ceruloplasmin deletion in myelinating glial cells induces myelin disruption and oxidative stress in the central and peripheral nervous systems. Redox Biol. (2021) 46:102118. 10.1016/j.redox.2021.102118
55.
Jessen KR Mirsky R . Negative regulation of myelination: relevance for development, injury, and demyelinating disease. Glia. (2008) 56:1552–65. 10.1002/glia.20761
56.
Arthur-Farraj PJ Latouche M Wilton DK Quintes S Chabrol E Banerjee A et al . c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron. (2012) 75:633–47. 10.1016/j.neuron.2012.06.021
57.
Kim M Wende H Walcher J Kühnemund J Cheret C Kempa S et al . Maf links Neuregulin1 signaling to cholesterol synthesis in myelinating Schwann cells. Genes Dev. (2018) 32:645–57. 10.1101/gad.310490.117
58.
Liu X Peng S Zhao Y Zhao T Wang M Luo L et al . AMPK negatively regulates peripheral myelination via activation of c-Jun. Mol Neurobiol. (2017) 54:3554–64. 10.1007/s12035-016-9913-3
59.
Fontana X Hristova M Da Costa C Patodia S Thei L Makwana M et al . c-Jun in Schwann cells promotes axonal regeneration and motoneuron survival via paracrine signaling. J Cell Biol. (2012) 198:127–41. 10.1083/jcb.201205025
60.
Norrmén C Figlia G Pfistner P Pereira JA Bachofner S Suter U . mTORC1 is transiently reactivated in injured nerves to promote c-Jun elevation and Schwann cell dedifferentiation. J Neurosci. (2018) 38:4811–28. 10.1523/JNEUROSCI.3619-17.2018
61.
Dennis EA . The growing phospholipase A2 superfamily of signal transduction enzymes. Trends Biochem Sci. (1997) 22:1–2. 10.1016/s0968-0004(96)20031-3
62.
Florio F Ferri C Scapin C Feltri ML Wrabetz L D'Antonio M . Sustained expression of negative regulators of myelination protects schwann cells from dysmyelination in a charcot-marie-tooth 1B mouse model. J Neurosci. (2018) 38:4275–87. 10.1523/JNEUROSCI.0201-18.2018
63.
Kioussi C Gross MK Gruss P . Pax3: a paired domain gene as a regulator in PNS myelination. Neuron. (1995) 15:553–62. 10.1016/0896-6273(95)90144-2
64.
Blanchard AD Sinanan A Parmantier E Zwart R Broos L Meijer D et al . Oct-6 (SCIP/Tst-1) is expressed in Schwann cell precursors, embryonic Schwann cells, and postnatal myelinating Schwann cells: comparison with Oct-1, Krox-20, and Pax-3. J Neurosci Res. (1996) 46:630–40. 10.1002/(SICI)1097-4547(19961201)46:5<630::AID-JNR11>3.0.CO;2-0
65.
Doddrell RDS Dun X-P Moate RM Jessen KR Mirsky R Parkinson DB . Regulation of Schwann cell differentiation and proliferation by the Pax-3 transcription factor. Glia. (2012) 60:1269–78. 10.1002/glia.22346
66.
Sheu JY Kulhanek DJ Eckenstein FP . Differential patterns of ERK and STAT3 phosphorylation after sciatic nerve transection in the rat. Exp Neurol. (2000) 166:392–402. 10.1006/exnr.2000.7508
67.
Harrisingh MC Perez-Nadales E Parkinson DB Malcolm DS Mudge AW Lloyd AC . The Ras/Raf/ERK signalling pathway drives Schwann cell dedifferentiation. EMBO J. (2004) 23:3061–71. 10.1038/sj.emboj.7600309
68.
Morgan L Jessen KR Mirsky R . The effects of cAMP on differentiation of cultured Schwann cells: progression from an early phenotype (04+) to a myelin phenotype (P0+, GFAP-, N-CAM-, NGF-receptor-) depends on growth inhibition. J Cell Biol. (1991) 112:457–67. 10.1083/jcb.112.3.457
69.
Napoli I Noon LA Ribeiro S Kerai AP Parrinello S Rosenberg LH et al . A central role for the ERK-signaling pathway in controlling Schwann cell plasticity and peripheral nerve regeneration in vivo. Neuron. (2012) 73:729–42. 10.1016/j.neuron.2011.11.031
70.
Cervellini I Galino J Zhu N Allen S Birchmeier C Bennett DL . Sustained MAPK/ERK activation in adult schwann cells impairs nerve repair. J Neurosci. (2018) 38:679–90. 10.1523/JNEUROSCI.2255-17.2017
71.
Michailov GV Sereda MW Brinkmann BG Fischer TM Haug B Birchmeier C et al . Axonal neuregulin-1 regulates myelin sheath thickness. Science. (2004) 304:700–3. 10.1126/science.1095862
72.
Taveggia C Zanazzi G Petrylak A Yano H Rosenbluth J Einheber S et al . Neuregulin-1 type III determines the ensheathment fate of axons. Neuron. (2005) 47:681–94. 10.1016/j.neuron.2005.08.017
73.
Fricker FR Bennett DL . The role of neuregulin-1 in the response to nerve injury. Future Neurol. (2011) 6:809–22. 10.2217/fnl.11.45
74.
Jung J Cai W Lee HK Pellegatta M Shin YK Jang SY et al . Actin polymerization is essential for myelin sheath fragmentation during Wallerian degeneration. J Neurosci. (2011) 31:2009–15. 10.1523/JNEUROSCI.4537-10.2011
75.
Shin YK Jang SY Park JY Park SY Lee HJ Suh DJ et al . The Neuregulin-Rac-MKK7 pathway regulates antagonistic c-jun/Krox20 expression in Schwann cell dedifferentiation. Glia. (2013) 61:892–904. 10.1002/glia.22482
76.
Parkinson DB Bhaskaran A Arthur-Farraj P Noon LA Woodhoo A Lloyd AC et al . c-Jun is a negative regulator of myelination. J Cell Biol. (2008) 181:625–37. 10.1083/jcb.200803013
77.
Yang DP Kim J Syed N Tung Y-J Bhaskaran A Mindos T et al . p38 MAPK activation promotes denervated Schwann cell phenotype and functions as a negative regulator of Schwann cell differentiation and myelination. J Neurosci. (2012) 32:7158–68. 10.1523/JNEUROSCI.5812-11.2012
78.
Dummula K Vinukonda G Chu P Xing Y Hu F Mailk S et al . Bone morphogenetic protein inhibition promotes neurological recovery after intraventricular hemorrhage. J Neurosci. (2011) 31:12068–82. 10.1523/JNEUROSCI.0013-11.2011
79.
Liu X Zhao Y Peng S Zhang S Wang M Chen Y et al . BMP7 retards peripheral myelination by activating p38 MAPK in Schwann cells. Sci Rep. (2016) 6:31049. 10.1038/srep31049
80.
Kuroda M Muramatsu R Maedera N Koyama Y Hamaguchi M Fujimura H et al . Peripherally derived FGF21 promotes remyelination in the central nervous system. J Clin Invest. (2017) 127:3496–509. 10.1172/JCI94337
81.
Zhang Y Jiang K Xie G Ding J Peng S Liu X et al . FGF21 impedes peripheral myelin development by stimulating p38 MAPK/c-Jun axis. J Cell Physiol. (2021) 236:1345–61. 10.1002/jcp.29942
82.
Wang J Ren K-Y Wang Y-H Kou Y-H Zhang P-X Peng J-P et al . Effect of active Notch signaling system on the early repair of rat sciatic nerve injury. Artif Cells Nanomed Biotechnol. (2015) 43:383–9. 10.3109/21691401.2014.896372
83.
Park HT Feltri ML . Rac1 GTPase controls myelination and demyelination. Bioarchitecture. (2011) 1:110–3. 10.4161/bioa.1.3.16985
84.
Tricaud N Park HT . Wallerian demyelination: chronicle of a cellular cataclysm. Cell Mol Life Sci. (2017) 74:4049–57. 10.1007/s00018-017-2565-2
85.
Fannon AM Sherman DL Ilyina-Gragerova G Brophy PJ Friedrich VL Colman DR . Novel E-cadherin-mediated adhesion in peripheral nerve: Schwann cell architecture is stabilized by autotypic adherens junctions. J Cell Biol. (1995) 129:189–202. 10.1083/jcb.129.1.189
86.
Woodhoo A Alonso MBD Droggiti A Turmaine M D'Antonio M Parkinson DB et al . Notch controls embryonic Schwann cell differentiation, postnatal myelination and adult plasticity. Nat Neurosci. (2009) 12:839–47. 10.1038/nn.2323
87.
Jessen KR Mirsky R . Control of Schwann cell myelination. F1000 Biol Rep. (2010) 2:19. 10.3410/B2-19
88.
Boerboom A Dion V Chariot A Franzen R . Molecular mechanisms involved in schwann cell plasticity. Front Mol Neurosci. (2017) 10:38. 10.3389/fnmol.2017.00038
89.
Brinkmann BG Quintes S . Zeb2: Inhibiting the inhibitors in Schwann cells. Neurogenesis (Austin). (2017) 4:e1271495. 10.1080/23262133.2016.1271495
90.
Zhu X Ding S Qiu C Shi Y Song L Wang Y et al . SUMOylation negatively regulates angiogenesis by targeting endothelial NOTCH signaling. Circ Res. (2017) 121:636–49. 10.1161/CIRCRESAHA.117.310696
91.
Fergani IF Frick LR . Wrestling and wrapping: a perspective on SUMO proteins in schwann cells. Biomolecules. (2021) 11:1055. 10.3390/biom11071055
92.
Taveggia C . Schwann cells-axon interaction in myelination. Curr Opin Neurobiol. (2016) 39:24–9. 10.1016/j.conb.2016.03.006
93.
Figlia G Gerber D Suter U . Myelination and mTOR. Glia. (2018) 66:693–707. 10.1002/glia.23273
94.
Figlia G Norrmén C Pereira JA Gerber D Suter U . Dual function of the PI3K-Akt-mTORC1 axis in myelination of the peripheral nervous system. Elife. (2017) 6:e29241. 10.7554/eLife.29241
95.
Saher G Quintes S Möbius W Wehr MC Krämer-Albers E-M Brügger B et al . Cholesterol regulates the endoplasmic reticulum exit of the major membrane protein P0 required for peripheral myelin compaction. J Neurosci. (2009) 29:6094–104. 10.1523/JNEUROSCI.0686-09.2009
96.
El Soury M Fornasari BE Morano M Grazio E Ronchi G Incarnato D et al . Soluble neuregulin1 down-regulates myelination genes in Schwann cells. Front Mol Neurosci. (2018) 11:157. 10.3389/fnmol.2018.00157
97.
Chen Z-L Strickland S . Laminin gamma1 is critical for Schwann cell differentiation, axon myelination, and regeneration in the peripheral nerve. J Cell Biol. (2003) 163:889–99. 10.1083/jcb.200307068
98.
Yu W-M Chen Z-L North AJ Strickland S . Laminin is required for Schwann cell morphogenesis. J Cell Sci. (2009) 122:929–36. 10.1242/jcs.033928
99.
Ghidinelli M Poitelon Y Shin YK Ameroso D Williamson C Ferri C et al . Laminin 211 inhibits protein kinase A in Schwann cells to modulate neuregulin 1 type III-driven myelination. PLoS Biol. (2017) 15:e2001408. 10.1371/journal.pbio.2001408
100.
La Marca R Cerri F Horiuchi K Bachi A Feltri ML Wrabetz L et al . TACE (ADAM17) inhibits Schwann cell myelination. Nat Neurosci. (2011) 14:857–65. 10.1038/nn.2849
101.
Linneberg C Harboe M Laursen LS . Axo-glia interaction preceding CNS myelination is regulated by bidirectional eph-ephrin signaling. ASN Neuro. (2015) 7:1759091415602859. 10.1177/1759091415602859
102.
Iwasato T Katoh H Nishimaru H Ishikawa Y Inoue H Saito YM et al . Rac-GAP alpha-chimerin regulates motor-circuit formation as a key mediator of EphrinB3/EphA4 forward signaling. Cell. (2007) 130:742–53. 10.1016/j.cell.2007.07.022
103.
Harboe M Torvund-Jensen J Kjaer-Sorensen K Laursen LS . Ephrin-A1-EphA4 signaling negatively regulates myelination in the central nervous system. Glia. (2018) 66:934–50. 10.1002/glia.23293
104.
Chen R Yang X Zhang B Wang S Bao S Gu Y et al . EphA4 negatively regulates myelination by inhibiting schwann cell differentiation in the peripheral nervous system. Front Neurosci. (2019) 13:1191. 10.3389/fnins.2019.01191
105.
Salzer JL . Schwann cell myelination. Cold Spring Harb Perspect Biol. (2015) 7:a020529. 10.1101/cshperspect.a020529
106.
Feng X Takayama Y Ohno N Kanda H Dai Y Sokabe T et al . Increased TRPV4 expression in non-myelinating Schwann cells is associated with demyelination after sciatic nerve injury. Commun Biol. (2020) 3:716. 10.1038/s42003-020-01444-9
107.
Miller RH . Calcium control of myelin sheath growth. Nat Neurosci. (2018) 21:2–3. 10.1038/s41593-017-0043-7
108.
Tsuang F-Y Chen M-H Lin F-H Yang M-C Liao C-J Chang W-H et al . Partial enzyme digestion facilitates regeneration of crushed nerve in rat. Transl Neurosci. (2020) 11:251–63. 10.1515/tnsci-2020-0112
109.
Mizushima N Levine B Cuervo AM Klionsky DJ . Autophagy fights disease through cellular self-digestion. Nature. (2008) 451:1069–75. 10.1038/nature06639
110.
Dikic I Elazar Z . Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. (2018) 19:349–64. 10.1038/s41580-018-0003-4
111.
Jang SY Shin YK Park SY Park JY Rha S-H Kim JK et al . Autophagy is involved in the reduction of myelinating Schwann cell cytoplasm during myelin maturation of the peripheral nerve. PLoS ONE. (2015) 10:e0116624. 10.1371/journal.pone.0116624
112.
Ko P-Y Yang C-C Kuo Y-L Su F-C Hsu T-I Tu Y-K et al . Schwann-cell autophagy, functional recovery, and scar reduction after peripheral nerve repair. J Mol Neurosci. (2018) 64:601–10. 10.1007/s12031-018-1056-8
113.
Belgrad J De Pace R Fields RD . Autophagy in myelinating glia. J Neurosci. (2020) 40:256–66. 10.1523/JNEUROSCI.1066-19.2019
114.
Gomez-Sanchez JA Carty L Iruarrizaga-Lejarreta M Palomo-Irigoyen M Varela-Rey M Griffith M et al . Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J Cell Biol. (2015) 210:153–68. 10.1083/jcb.201503019
115.
Marinelli S Nazio F Tinari A Ciarlo L D'Amelio M Pieroni L et al . Schwann cell autophagy counteracts the onset and chronification of neuropathic pain. Pain. (2014) 155:93–107. 10.1016/j.pain.2013.09.013
116.
Chrast R Saher G Nave K-A Verheijen MHG . Lipid metabolism in myelinating glial cells: lessons from human inherited disorders and mouse models. J Lipid Res. (2011) 52:419–34. 10.1194/jlr.R009761
117.
Singh R Kaushik S Wang Y Xiang Y Novak I Komatsu M et al . Autophagy regulates lipid metabolism. Nature. (2009) 458:1131–5. 10.1038/nature07976
118.
Lüningschrör P Slotta C Heimann P Briese M Weikert UM Massih B et al . Absence of Plekhg5 results in myelin infoldings corresponding to an impaired schwann cell autophagy, and a reduced t-cell infiltration into peripheral nerves. Front Cell Neurosci. (2020) 14:185. 10.3389/fncel.2020.00185
119.
Li R Li D Wu C Ye L Wu Y Yuan Y et al . Nerve growth factor activates autophagy in Schwann cells to enhance myelin debris clearance and to expedite nerve regeneration. Theranostics. (2020) 10:1649–77. 10.7150/thno.40919
120.
Lee IH Cao L Mostoslavsky R Lombard DB Liu J Bruns NE et al . A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. (2008) 105:3374–9. 10.1073/pnas.0712145105
121.
Romeo-Guitart D Leiva-Rodriguez T Forés J Casas C . Improved motor nerve regeneration by SIRT1/Hif1a-mediated autophagy. Cells. (2019) 8:E1354. 10.3390/cells8111354
122.
Hubbard BP Sinclair DA . Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol Sci. (2014) 35:146–54. 10.1016/j.tips.2013.12.004
123.
Carbajal KS Schaumburg C Strieter R Kane J Lane TE . Migration of engrafted neural stem cells is mediated by CXCL12 signaling through CXCR4 in a viral model of multiple sclerosis. Proc Natl Acad Sci U S A. (2010) 107:11068–73. 10.1073/pnas.1006375107
124.
Ardelt AA Bhattacharyya BJ Belmadani A Ren D Miller RJ . Stromal derived growth factor-1 (CXCL12) modulates synaptic transmission to immature neurons during post-ischemic cerebral repair. Exp Neurol. (2013) 248:246–53. 10.1016/j.expneurol.2013.06.017
125.
Zendedel A Johann S Mehrabi S Joghataei M-T Hassanzadeh G Kipp M et al . Activation and regulation of NLRP3 inflammasome by intrathecal application of sdf-1a in a spinal cord injury model. Mol Neurobiol. (2016) 53:3063–75. 10.1007/s12035-015-9203-5
126.
Gao D Tang T Zhu J Tang Y Sun H Li S . CXCL12 has therapeutic value in facial nerve injury and promotes Schwann cells autophagy and migration via PI3K-AKT-mTOR signal pathway. Int J Biol Macromol. (2019) 124:460–8. 10.1016/j.ijbiomac.2018.10.212
127.
Medina DL Di Paola S Peluso I Armani A De Stefani D Venditti R et al . Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. (2015) 17:288–99. 10.1038/ncb3114
128.
Tong Y Song F . Intracellular calcium signaling regulates autophagy via calcineurin-mediated TFEB dephosphorylation. Autophagy. (2015) 11:1192–5. 10.1080/15548627.2015.1054594
129.
Rusmini P Cortese K Crippa V Cristofani R Cicardi ME Ferrari V et al . Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy. (2019) 15:631–51. 10.1080/15548627.2018.1535292
130.
Reed CB Frick LR Weaver A Sidoli M Schlant E Feltri ML et al . Deletion of calcineurin in schwann cells does not affect developmental myelination, but reduces autophagy and delays myelin clearance after peripheral nerve injury. J Neurosci. (2020) 40:6165–76. 10.1523/JNEUROSCI.0951-20.2020
131.
Logan AM Mammel AE Robinson DC Chin AL Condon AF Robinson FL . Schwann cell-specific deletion of the endosomal PI 3-kinase Vps34 leads to delayed radial sorting of axons, arrested myelination, and abnormal ErbB2-ErbB3 tyrosine kinase signaling. Glia. (2017) 65:1452–70. 10.1002/glia.23173
132.
Wang H Wang X Zhang K Wang Q Cao X Wang Z et al . Rapid depletion of ESCRT protein Vps4 underlies injury-induced autophagic impediment and Wallerian degeneration. Sci Adv. (2019) 5:eaav4971. 10.1126/sciadv.aav4971
133.
Pollak DD Minh BQ Cicvaric A Monje FJ . A novel fibroblast growth factor receptor family member promotes neuronal outgrowth and synaptic plasticity in aplysia. Amino Acids. (2014) 46:2477–88. 10.1007/s00726-014-1803-2
134.
Moncion A Lin M O'Neill EG Franceschi RT Kripfgans OD Putnam AJ et al . Controlled release of basic fibroblast growth factor for angiogenesis using acoustically-responsive scaffolds. Biomaterials. (2017) 140:26–36. 10.1016/j.biomaterials.2017.06.012
135.
Jiang Y Liang J Li R Peng Y Huang J Huang L . Basic fibroblast growth factor accelerates myelin debris clearance through activating autophagy to facilitate early peripheral nerve regeneration. J Cell Mol Med. (2021) 25:2596–608. 10.1111/jcmm.16274
136.
Li R Li D-H Zhang H-Y Wang J Li X-K Xiao J . Growth factors-based therapeutic strategies and their underlying signaling mechanisms for peripheral nerve regeneration. Acta Pharmacol Sin. (2020) 41:1289–300. 10.1038/s41401-019-0338-1
137.
Ravikumar B Sarkar S Davies JE Futter M Garcia-Arencibia M Green-Thompson ZW et al . Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. (2010) 90:1383–435. 10.1152/physrev.00030.2009
138.
Brosius Lutz A Chung W-S Sloan SA Carson GA Zhou L Lovelett E et al . Schwann cells use TAM receptor-mediated phagocytosis in addition to autophagy to clear myelin in a mouse model of nerve injury. Proc Natl Acad Sci U S A. (2017) 114:E8072–80. 10.1073/pnas.1710566114
139.
Bhattacharya A Muller FL Liu Y Sabia M Liang H Song W et al . Denervation induces cytosolic phospholipase A2-mediated fatty acid hydroperoxide generation by muscle mitochondria. J Biol Chem. (2009) 284:46–55. 10.1074/jbc.M806311200
140.
Tan ECTH Bahrami S Kozlov AV Kurvers HAJM Ter Laak HJ Nohl H et al . The oxidative response in the chronic constriction injury model of neuropathic pain. J Surg Res. (2009) 152:84–8. 10.1016/j.jss.2008.03.035
141.
Abruzzo PM di Tullio S Marchionni C Belia S Fanó G Zampieri S et al . Oxidative stress in the denervated muscle. Free Radic Res. (2010) 44:563–76. 10.3109/10715761003692487
142.
Kensler TW Wakabayashi N Biswal S . Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. (2007) 47:89–116. 10.1146/annurev.pharmtox.46.120604.141046
143.
Zhang L Johnson D Johnson JA . Deletion of Nrf2 impairs functional recovery, reduces clearance of myelin debris and decreases axonal remyelination after peripheral nerve injury. Neurobiol Dis. (2013) 54:329–38. 10.1016/j.nbd.2013.01.003
144.
Griffin JW George R Ho T . Macrophage systems in peripheral nerves. A review. J Neuropathol Exp Neurol. (1993) 52:553–60. 10.1097/00005072-199311000-00001
145.
Müller M Stenner M Wacker K Ringelstein EB Hickey WF Kiefer R . Contribution of resident endoneurial macrophages to the local cellular response in experimental autoimmune neuritis. J Neuropathol Exp Neurol. (2006) 65:499–507. 10.1097/01.jnen.0000229239.43866.d1
146.
Ydens E Amann L Asselbergh B Scott CL Martens L Sichien D et al . Profiling peripheral nerve macrophages reveals two macrophage subsets with distinct localization, transcriptome and response to injury. Nat Neurosci. (2020) 23:676–89. 10.1038/s41593-020-0618-6
147.
Gaudet AD Popovich PG Ramer MS . Wallerian degeneration: gaining perspective on inflammatory events after peripheral nerve injury. J Neuroinflammation. (2011) 8:110. 10.1186/1742-2094-8-110
148.
Narciso MS Mietto B de S Marques SA Soares CP Mermelstein C et al . Sciatic nerve regeneration is accelerated in galectin-3 knockout mice. Exp Neurol. (2009) 217:7–15. 10.1016/j.expneurol.2009.01.008
149.
Ramaglia V King RHM Nourallah M Wolterman R de Jonge R Ramkema M et al . The membrane attack complex of the complement system is essential for rapid Wallerian degeneration. J Neurosci. (2007) 27:7663–72. 10.1523/JNEUROSCI.5623-06.2007
150.
Ramaglia V Wolterman R de Kok M Vigar MA Wagenaar-Bos I King RHM et al . Soluble complement receptor 1 protects the peripheral nerve from early axon loss after injury. Am J Pathol. (2008) 172:1043–52. 10.2353/ajpath.2008.070660
151.
Brück W . The role of macrophages in Wallerian degeneration. Brain Pathol. (1997) 7:741–52. 10.1111/j.1750-3639.1997.tb01060.x
152.
Dailey AT Avellino AM Benthem L Silver J Kliot M . Complement depletion reduces macrophage infiltration and activation during Wallerian degeneration and axonal regeneration. J Neurosci. (1998) 18:6713–22.
153.
Liu L Lioudyno M Tao R Eriksson P Svensson M Aldskogius H . Hereditary absence of complement C5 in adult mice influences Wallerian degeneration, but not retrograde responses, following injury to peripheral nerve. J Peripher Nerv Syst. (1999) 4:123–33.
154.
Chernov AV Dolkas J Hoang K Angert M Srikrishna G Vogl T et al . The calcium-binding proteins S100A8 and S100A9 initiate the early inflammatory program in injured peripheral nerves. J Biol Chem. (2015) 290:11771–84. 10.1074/jbc.M114.622316
155.
Schiopu A Cotoi OS . S100A8 and S100A9: DAMPs at the crossroads between innate immunity, traditional risk factors, and cardiovascular disease. Mediators Inflamm. (2013) 2013:828354. 10.1155/2013/828354
156.
Boivin A Pineau I Barrette B Filali M Vallières N Rivest S et al . Toll-like receptor signaling is critical for Wallerian degeneration and functional recovery after peripheral nerve injury. J Neurosci. (2007) 27:12565–76. 10.1523/JNEUROSCI.3027-07.2007
157.
Vallieres N Berard JL David S Lacroix S . Systemic injections of Blipopolysaccharide accelerates myelin phagocytosis during Wallerian degeneration in the injured mouse spinal cord. Glia. (2006) 53:103–13. 10.1002/glia.20266
158.
Nguyen MD Julien J-P Rivest S . Innate immunity: the missing link in neuroprotection and neurodegeneration?Nat Rev Neurosci. (2002) 3:216–27. 10.1038/nrn752
159.
Takeda K Kaisho T Akira S . Toll-like receptors. Annu Rev Immunol. (2003) 21:335–76. 10.1146/annurev.immunol.21.120601.141126
160.
Willis D Li KW Zheng J-Q Chang JH Smit AB Smit A et al . Differential transport and local translation of cytoskeletal, injury-response, and neurodegeneration protein mRNAs in axons. J Neurosci. (2005) 25:778–91. 10.1523/JNEUROSCI.4235-04.2005
161.
Langenhan T Piao X Monk KR . Adhesion G protein-coupled receptors in nervous system development and disease. Nat Rev Neurosci. (2016) 17:550–61. 10.1038/nrn.2016.86
162.
Mogha A Benesh AE Patra C Engel FB Schöneberg T Liebscher I et al . Gpr126 functions in Schwann cells to control differentiation and myelination via G-protein activation. J Neurosci. (2013) 33:17976–85. 10.1523/JNEUROSCI.1809-13.2013
163.
Mogha A Harty BL Carlin D Joseph J Sanchez NE Suter U et al . Gpr126/Adgrg6 has schwann cell autonomous and nonautonomous functions in peripheral nerve injury and repair. J Neurosci. (2016) 36:12351–67. 10.1523/JNEUROSCI.3854-15.2016
164.
Martini R Fischer S López-Vales R David S . Interactions between Schwann cells and macrophages in injury and inherited demyelinating disease. Glia. (2008) 56:1566–77. 10.1002/glia.20766
165.
Upragarin N Landman WJM Gaastra W Gruys E . Extrahepatic production of acute phase serum amyloid A. Histol Histopathol. (2005) 20:1295–307. 10.14670/HH-20.1295
166.
Jang SY Shin YK Lee HY Park JY Suh DJ Kim JK et al . Local production of serum amyloid a is implicated in the induction of macrophage chemoattractants in Schwann cells during wallerian degeneration of peripheral nerves. Glia. (2012) 60:1619–28. 10.1002/glia.22382
167.
Spreyer P Schaal H Kuhn G Rothe T Unterbeck A Olek K et al . Regeneration-associated high level expression of apolipoprotein D mRNA in endoneurial fibroblasts of peripheral nerve. EMBO J. (1990) 9:2479–84
168.
Ganfornina MD Do Carmo S Martínez E Tolivia J Navarro A Rassart E et al . ApoD, a glia-derived apolipoprotein, is required for peripheral nerve functional integrity and a timely response to injury. Glia. (2010) 58:1320–34. 10.1002/glia.21010
169.
García-Mateo N Ganfornina MD Montero O Gijón MA Murphy RC Sanchez D . Schwann cell-derived Apolipoprotein D controls the dynamics of post-injury myelin recognition and degradation. Front Cell Neurosci. (2014) 8:374. 10.3389/fncel.2014.00374
170.
Roberts SL Dun X Doddrell RDS Mindos T Drake LK Onaitis MW et al . Sox2 expression in Schwann cells inhibits myelination in vivo and induces influx of macrophages to the nerve. Development. (2017) 144:3114–25. 10.1242/dev.150656
171.
Parrinello S Napoli I Ribeiro S Wingfield Digby P Fedorova M Parkinson DB et al . EphB signaling directs peripheral nerve regeneration through Sox2-dependent Schwann cell sorting. Cell. (2010) 143:145–55. 10.1016/j.cell.2010.08.039
172.
Deng B Lv W Duan W Liu Y Li Z Song X et al . FGF9 modulates Schwann cell myelination in developing nerves and induces a pro-inflammatory environment during injury. J Cell Biochem. (2018) 119:8643–58. 10.1002/jcb.27105
173.
Brown WJ Chambers K Doody A . Phospholipase A2 (PLA2) enzymes in membrane trafficking: mediators of membrane shape and function. Traffic. (2003) 4:214–21. 10.1034/j.1600-0854.2003.00078.x
174.
Yaksh TL Kokotos G Svensson CI Stephens D Kokotos CG Fitzsimmons B et al . Systemic and intrathecal effects of a novel series of phospholipase A2 inhibitors on hyperalgesia and spinal prostaglandin E2 release. J Pharmacol Exp Ther. (2006) 316:466–75. 10.1124/jpet.105.091686
175.
Lopez-Vales R Navarro X Shimizu T Baskakis C Kokotos G Constantinou-Kokotou V et al . Intracellular phospholipase A(2) group IVA and group VIA play important roles in Wallerian degeneration and axon regeneration after peripheral nerve injury. Brain. (2008) 131:2620–31. 10.1093/brain/awn188
176.
Yang J Gu Y Huang X Shen A Cheng C . Dynamic changes of ICAM-1 expression in peripheral nervous system following sciatic nerve injury. Neurol Res. (2011) 33:75–83. 10.1179/016164110X12714125204353
177.
Vougioukas VI Roeske S Michel U Brück W . Wallerian degeneration in ICAM-1-deficient mice. Am J Pathol. (1998) 152:241–9.
178.
Avellino AM Dailey AT Harlan JM Sharar SR Winn RK McNutt LD et al . Blocking of up-regulated ICAM-1 does not prevent macrophage infiltration during Wallerian degeneration of peripheral nerve. Exp Neurol. (2004) 187:430–44. 10.1016/j.expneurol.2004.02.004
179.
Liou J-T Lee C-M Lin Y-C Chen C-Y Liao C-C Lee H-C et al . P-selectin is required for neutrophils and macrophage infiltration into injured site and contributes to generation of behavioral hypersensitivity following peripheral nerve injury in mice. Pain. (2013) 154:2150–9. 10.1016/j.pain.2013.06.042
180.
Zhou T Zheng Y Sun L Badea SR Jin Y Liu Y et al . Microvascular endothelial cells engulf myelin debris and promote macrophage recruitment and fibrosis after neural injury. Nat Neurosci. (2019) 22:421–35. 10.1038/s41593-018-0324-9
181.
Hall SE Savill JS Henson PM Haslett C . Apoptotic neutrophils are phagocytosed by fibroblasts with participation of the fibroblast vitronectin receptor and involvement of a mannose/fucose-specific lectin. J Immunol. (1994) 153:3218–27.
182.
Yoon C Korade Z Carter BD . Protein kinase A-induced phosphorylation of the p65 subunit of nuclear factor-kappaB promotes Schwann cell differentiation into a myelinating phenotype. J Neurosci. (2008) 28:3738–46. 10.1523/JNEUROSCI.4439-07.2008
183.
Chen Y Wang H Yoon SO Xu X Hottiger MO Svaren J et al . HDAC-mediated deacetylation of NF-κB is critical for Schwann cell myelination. Nat Neurosci. (2011) 14:437–41. 10.1038/nn.2780
184.
Li L Li Y Fan Z Wang X Li Z Wen J et al . Ascorbic acid facilitates neural regeneration after sciatic nerve crush injury. Front Cell Neurosci. (2019) 13:108. 10.3389/fncel.2019.00108
185.
Huff TC Sant DW Camarena V Van Booven D Andrade NS Mustafi S et al . Vitamin C regulates Schwann cell myelination by promoting DNA demethylation of pro-myelinating genes. J Neurochem. (2021) 157:1759–73. 10.1111/jnc.15015
186.
Lütjohann D Papassotiropoulos A Björkhem I Locatelli S Bagli M Oehring RD et al . Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J Lipid Res. (2000) 41:195–8.
187.
Leoni V Masterman T Diczfalusy U De Luca G Hillert J Björkhem I . Changes in human plasma levels of the brain specific oxysterol 24S-hydroxycholesterol during progression of multiple sclerosis. Neurosci Lett. (2002) 331:163–6. 10.1016/s0304-3940(02)00887-x
188.
Teunissen CE Dijkstra CD Polman CH Hoogervorst ELJ von Bergmann K Lütjohann D . Decreased levels of the brain specific 24S-hydroxycholesterol and cholesterol precursors in serum of multiple sclerosis patients. Neurosci Lett. (2003) 347:159–62. 10.1016/s0304-3940(03)00667-0
189.
Sundaram VK Massaad C Grenier J . Liver X receptors and their implications in the physiology and pathology of the peripheral nervous system. Int J Mol Sci. (2019) 20:E4192. 10.3390/ijms20174192
190.
Gribble KD Walker LJ Saint-Amant L Kuwada JY Granato M . The synaptic receptor Lrp4 promotes peripheral nerve regeneration. Nat Commun. (2018) 9:2389. 10.1038/s41467-018-04806-4
191.
Pooya S Liu X Kumar VBS Anderson J Imai F Zhang W et al . The tumour suppressor LKB1 regulates myelination through mitochondrial metabolism. Nat Commun. (2014) 5:4993. 10.1038/ncomms5993
192.
Beirowski B Babetto E Golden JP Chen Y-J Yang K Gross RW et al . Metabolic regulator LKB1 is crucial for Schwann cell-mediated axon maintenance. Nat Neurosci. (2014) 17:1351–61. 10.1038/nn.3809
193.
He X Zhang L Queme LF Liu X Lu A Waclaw RR et al . A histone deacetylase 3-dependent pathway delimits peripheral myelin growth and functional regeneration. Nat Med. (2018) 24:338–51. 10.1038/nm.4483
194.
Bauer NM Moos C van Horssen J Witte M van der Valk P Altenhein B et al . Myelin basic protein synthesis is regulated by small non-coding RNA 715. EMBO Rep. (2012) 13:827–34. 10.1038/embor.2012.97
195.
Sherman DL Krols M Wu L-MN Grove M Nave K-A Gangloff Y-G et al . Arrest of myelination and reduced axon growth when Schwann cells lack mTOR. J Neurosci. (2012) 32:1817–25. 10.1523/JNEUROSCI.4814-11.2012
196.
Xue Z Zhang S Huang L He Y Fang R Fang Y . Increased expression of Beclin-1-dependent autophagy protects against beta-amyloid-induced cell injury in PC12 cells [corrected]. J Mol Neurosci. (2013) 51:180–6. 10.1007/s12031-013-9974-y
197.
Dunlop EA Tee AR . mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol. (2014) 36:121–9. 10.1016/j.semcdb.2014.08.006
198.
Carroll B Dunlop EA . The lysosome: a crucial hub for AMPK and mTORC1 signalling. Biochem J. (2017) 474:1453–66. 10.1042/BCJ20160780
199.
Ballatore C Brunden KR Huryn DM Trojanowski JQ Lee VM-Y Smith AB . Microtubule stabilizing agents as potential treatment for Alzheimer's disease and related neurodegenerative tauopathies. J Med Chem. (2012) 55:8979–96. 10.1021/jm301079z
200.
Ruschel J Hellal F Flynn KC Dupraz S Elliott DA Tedeschi A et al . Axonal regeneration. Systemic administration of epothilone B promotes axon regeneration after spinal cord injury. Science. (2015) 348:347–52. 10.1126/science.aaa2958
201.
Weng W Yao C Poonit K Zhou X Sun C Zhang F et al . Metformin relieves neuropathic pain after spinal nerve ligation via autophagy flux stimulation. J Cell Mol Med. (2019) 23:1313–24. 10.1111/jcmm.14033
202.
Rotermund C Machetanz G Fitzgerald JC . The therapeutic potential of metformin in neurodegenerative diseases. Front Endocrinol (Lausanne). (2018) 9:400. 10.3389/fendo.2018.00400
203.
Nikolaev A McLaughlin T O'Leary DDM Tessier-Lavigne M . N-APP binds DR6 to cause axon pruning and neuron death via distinct caspases. Nature. (2009) 457:981–9. 10.1038/nature07767
204.
Farah MH Pan BH Hoffman PN Ferraris D Tsukamoto T Nguyen T et al . Reduced BACE1 activity enhances clearance of myelin debris and regeneration of axons in the injured peripheral nervous system. J Neurosci. (2011) 31:5744–54. 10.1523/JNEUROSCI.6810-10.2011
205.
Farah MH . BACE1 influences debris clearance and axonal regeneration in injured peripheral nerve. J Peripher Nerv Syst. (2012) 17:30–3. 10.1111/j.1529-8027.2012.00428.x
206.
Tallon C Farah MH . Beta secretase activity in peripheral nerve regeneration. Neural Regen Res. (2017) 12:1565–74. 10.4103/1673-5374.217319
207.
Gitik M Liraz-Zaltsman S Oldenborg P-A Reichert F Rotshenker S . Myelin down-regulates myelin phagocytosis by microglia and macrophages through interactions between CD47 on myelin and SIRPα (signal regulatory protein-α) on phagocytes. J Neuroinflammation. (2011) 8:24. 10.1186/1742-2094-8-24
208.
Bian Z Shi L Guo Y-L Lv Z Tang C Niu S et al . Cd47-Sirpα interaction and IL-10 constrain inflammation-induced macrophage phagocytosis of healthy self-cells. Proc Natl Acad Sci U S A. (2016) 113:E5434–5443. 10.1073/pnas.1521069113
Summary
Keywords
Schwann cell, myelin clearance, peripheral nerve injury, demyelination, autophagy, macrophages
Citation
Yuan Y, Wang Y, Wu S and Zhao MY (2022) Review: Myelin clearance is critical for regeneration after peripheral nerve injury. Front. Neurol. 13:908148. doi: 10.3389/fneur.2022.908148
Received
26 April 2022
Accepted
17 November 2022
Published
16 December 2022
Volume
13 - 2022
Edited by
Melissa R. Andrews, University of Southampton, United Kingdom
Reviewed by
Assunta Virtuoso, University of Campania Luigi Vanvitelli, Italy; Nathalie Sol-Foulon, INSERM U1127 Institut du Cerveau et de la Moelle épinière (ICM), France
Updates
Copyright
© 2022 Yuan, Wang, Wu and Zhao.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Wang swallow-1113@163.com
This article was submitted to Neurotrauma, a section of the journal Frontiers in Neurology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.