 Xinran Ma
Xinran Ma Xiaoxuan Liu
Xiaoxuan Liu Xiaohui Duan
Xiaohui Duan Dongsheng Fan
Dongsheng Fan- 1Department of Neurology, Peking University Third Hospital, Beijing, China
- 2Key Laboratory for Neuroscience, National Health Commission/Ministry of Education, Peking University, Beijing, China
- 3Beijing Key Laboratory of Biomarker and Translational Research in Neurodegenerative Diseases, Beijing, China
- 4Department of Neurology, China-Japan Friendship Hospital, Beijing, China
Background: Periaxins (encoded by PRX) play an important role in the stabilization of peripheral nerve myelin. Mutations in PRX can lead to Charcot-Marie-Tooth disease type 4F (CMT4F).
Methods: In this study, we screened for PRX mutations using next-generation sequencing and whole-exome sequencing in a large Chinese CMT cohort consisting of 465 unrelated index patients and 650 healthy controls. Sanger sequencing was used for the validation of all identified variants. We also reviewed all previously reported PRX-related CMT cases and summarized the clinical manifestations and genetic features of PRX-related CMTs.
Results: The hit rate for biallelic PRX variants in our cohort of Chinese CMT patients was 0.43% (2/465). One patient carried a previously unreported splice-site mutation (c.25_27 + 9del) compound heterozygous with a known nonsense variant. Compiling data on CMT4F cases and PRX variants from the medical literature confirmed that early-onset (95.2%), distal amyotrophy or weakness (94.0%), feet deformity (75.0%), sensory impairment or sensory ataxia (65.5%), delayed motor milestones (60.7%), and spinal deformity (59.5%) are typical features for CMT4F. Less frequent features were auditory impairments, respiratory symptoms, late onset, dysarthria or hoarseness, ophthalmic problems, and central nervous system involvement. The two cases with biallelic missense mutations have later onset age than those with nonsense or frameshift mutations. We did not note clear correlations between the type and site of mutations and clinical severity or distinct constellations of symptoms.
Conclusion: Consistent with observations in other countries and ethnic groups, PRX-related CMT is rare in China. The clinical spectrum is wider than previously anticipated.
1. Introduction
Charcot-Marie-Tooth disease (CMT) is the most common form of inherited peripheral neuropathy (1) with a large degree of clinical and genetic heterogeneity. According to the results of electrophysiological studies especially the upper limb motor nerve conduction velocity (MNCV), CMT is classified into demyelinating forms, including CMT1 (autosomal dominant) and CMT4 (autosomal recessive); an axonal form, CMT2; and an intermediate form (2). Over 100 genes have been found to be associated with CMTs (https://neuromuscular.wustl.edu/).
CMT4F is a globally rare form of autosomal recessive inherited demyelinating CMT that is characterized by early-onset and slow progression (3). PRX, the causative gene for CMT4F (4, 5), was reported to be mutated in 1.2% (1/82) of a Chinese CMT cohort (6). PRX encodes two proteins, L-periaxin and S-periaxin, through alternative splicing (7). Periaxin is involved in the maintenance and stabilization of myelin (8). To date, over 60 pathogenic PRX mutations have been identified (Human Gene Mutation Database, Professional Version), most of which are nonsense and frameshift mutations (9). Splice-site mutations of PRX are rare. Due to the small number of reported CMT patients with PRX mutations, knowledge of PRX and CMT4F is still limited.
In this study, we screened for PRX mutations in a large Chinese CMT cohort and identified three additional CMT4F patients and a previously unreported splice-site mutation of PRX. We expanded the genetic and clinical spectrum of PRX-related CMT. We also reviewed previously reported CMT4F cases to provide a comprehensive overview of this rare form of CMT.
2. Methods
2.1. Materials and participants
Between January 2007 and February 2022, 465 unrelated index patients with CMT were enrolled in the CMT centers at Peking University Third Hospital and China-Japan Friendship Hospital. The family history, age of onset, clinical features, CMT neuropathy score, and electrophysiological features were collected in detail for each patient. Based on the electrophysiological criteria, 276 families were diagnosed with demyelinating CMT, 137 families were diagnosed with axonal CMT, and 52 families were diagnosed with intermediate CMT. Among the 465 index patients, 163 patients were diagnosed with CMT1A via multiplex ligation-dependent probe amplification (MLPA) and 88 patients underwent direct sequencing only for PMP22, MPZ, GJB1, and MFN2. The remaining 214 index patients and their relatives underwent whole-exome sequencing (WES) or next-generation panel testing screening. The present study was approved by the Ethics Committee of Peking University Third Hospital (IRB 00006761). Informed consent forms were signed by all the participants or their parents.
2.2. Genetic tests and bioinformatics analysis
2.2.1. DNA extraction and gene sequencing
Peripheral blood samples were collected from all participants. Genomic DNA was extracted by a DNA isolation kit (Bioteke, AU1802). Concentration assays and quality control were performed using standard methods. Next-generation sequencing (NGS) targeting CMT-related genes including PRX (NM_181882.2) or WES was conducted in 214 index patients. All variants identified were then validated by Sanger sequencing for segregation analysis. For NGS, the sample preparation and sequencing procedure was performed following the Illumina instructions. The KAPA library preparation kit (Kapa Biosystems, KR0453) was used to prepare the DNA libraries. Pooled libraries were hybridized with capture probes on the Agilent SureSelect XT2 target enrichment system. Then sequencing targeting CMT-related genes was conducted on a HiSeq2500 platform. For WES, an Agilent Human All Exon V6 kit and HiSeq2500 platform were used. The direct Sanger sequencing was conducted on an ABI 3730XL DNA analyzer (Applied Biosystems, Waltham, MA, USA).
2.2.2. Classification and assessment of mutations
The pathogenicity of the variants was assessed according to guidelines proposed by the American College of Medical Genetics and Genomics (ACMG) (10). Population databases, including dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP), 1000 Genomes (http://www.1000genomes.org/), GnomAD (http://gnomad.broadinstitute.org/), and Exome Sequencing Project (http://evs.gs.washington.edu/), and 650 healthy Chinese individuals were used as controls to exclude polymorphisms and benign variants. We used prediction software including Human Splicing Finder version 3.1 (http://www.umd.be/HSF/) for in silico analysis of variants.
2.3. Literature review
We reviewed all previously reported PRX-related CMT4F cases in PubMed. The Human Gene Mutation Database, Professional Version (HGMD pro), and ClinicalVar database were searched for all reported pathogenic mutations. Mutations identified as pathogenic and likely to be pathogenic were included, while those with unknown significance or with a diagnosis other than CMT were excluded.
3. Results
3.1. Clinical manifestation
In total, 465 unrelated patients diagnosed with CMT were enrolled in this study. Pathogenic mutations of PRX were detected in two patients, including one isolated case and another individual who had an affected sibling. Their detailed clinical data are shown in Table 1.
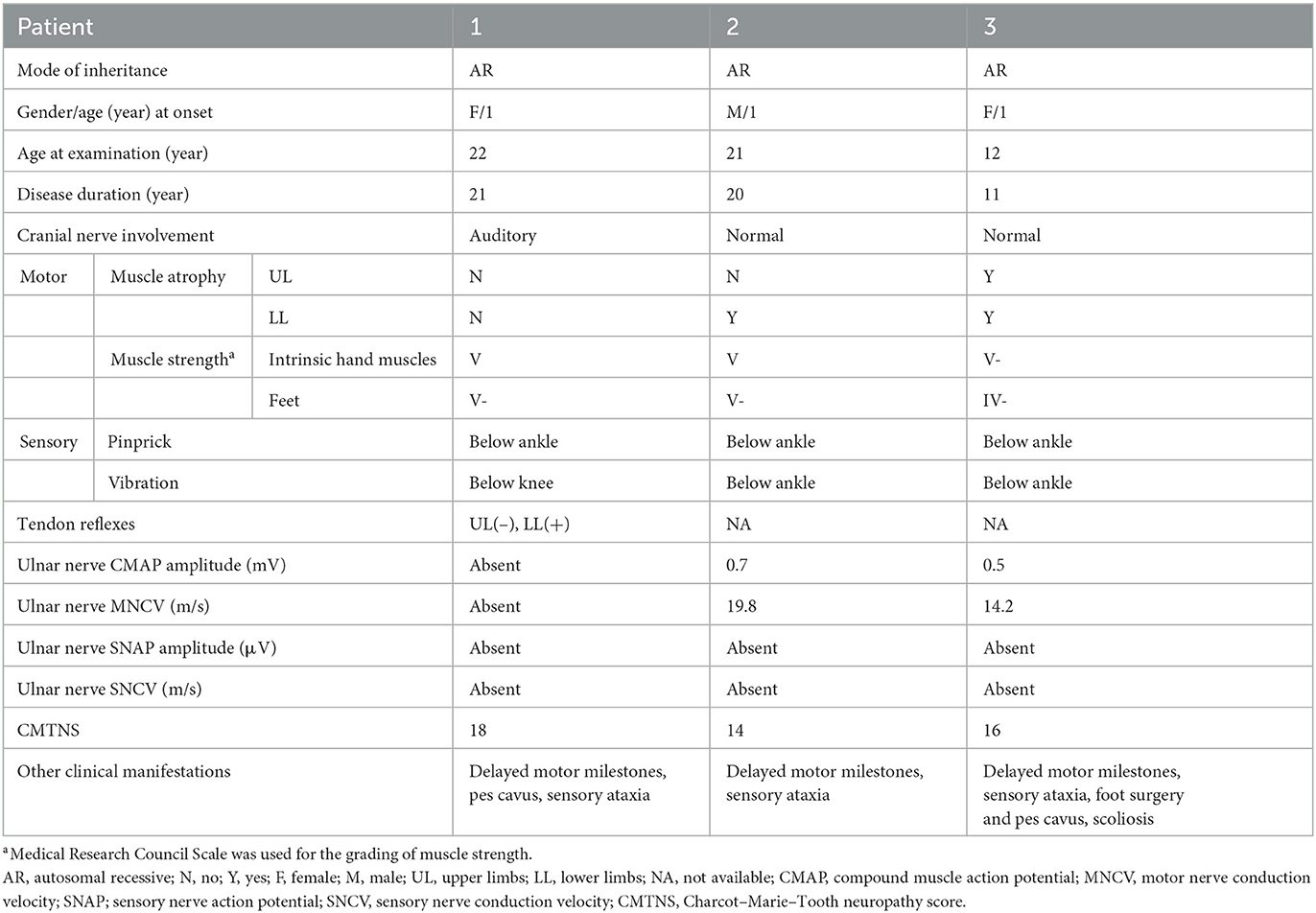
Table 1. Clinical features of the three CMT4F patients identified in this study.
Patient 1 was a 28-year-old woman who was the only child of healthy, non-consanguineous parents and had no affected relatives (Figure 1). Her mother took cold medicine during pregnancy. Patient 1 presented with delayed motor milestones and subsequently presented with motor deficits and unsteady gait. No intellectual disability or vision problems were reported. Her symptoms progressed slowly in the following 28 years. On her visit at the age of 22 years, no abnormality of the cranial nerve was found on physical examination. Severe sensory ataxia was observed. She also had pes cavus but no scoliosis. Pinprick sensitivity was decreased in the distal lower limbs. Vibration sensation was absent below the knees. Mild weakness was present in the feet only, with no severe muscle atrophy. Tendon reflexes were absent in the bilateral upper limbs and decreased in the lower limbs. No obvious abnormalities were found on the brain and whole-spine magnetic resonance imaging except for cervical intervertebral disk herniation. She underwent electrophysiological examinations at the age of 22 years, and the compound muscle action potential (CMAP) and sensory nerve action potential (SNAP) of the bilateral ulnar and median were undetectable in nerve conduction studies. The distal latency of CMAP was prolonged in the bilateral facial nerves, indicating demyelinating neuropathies. She also presented with prolonged latency in the waves III and V of the right side during an brainstem auditory evoked potential (BAEP) test. No biopsy was conducted. She complained of tinnitus starting at the age of 27 years. At age 28, the hearing test was conducted, and bilateral impairment of high-frequency sound was detected.
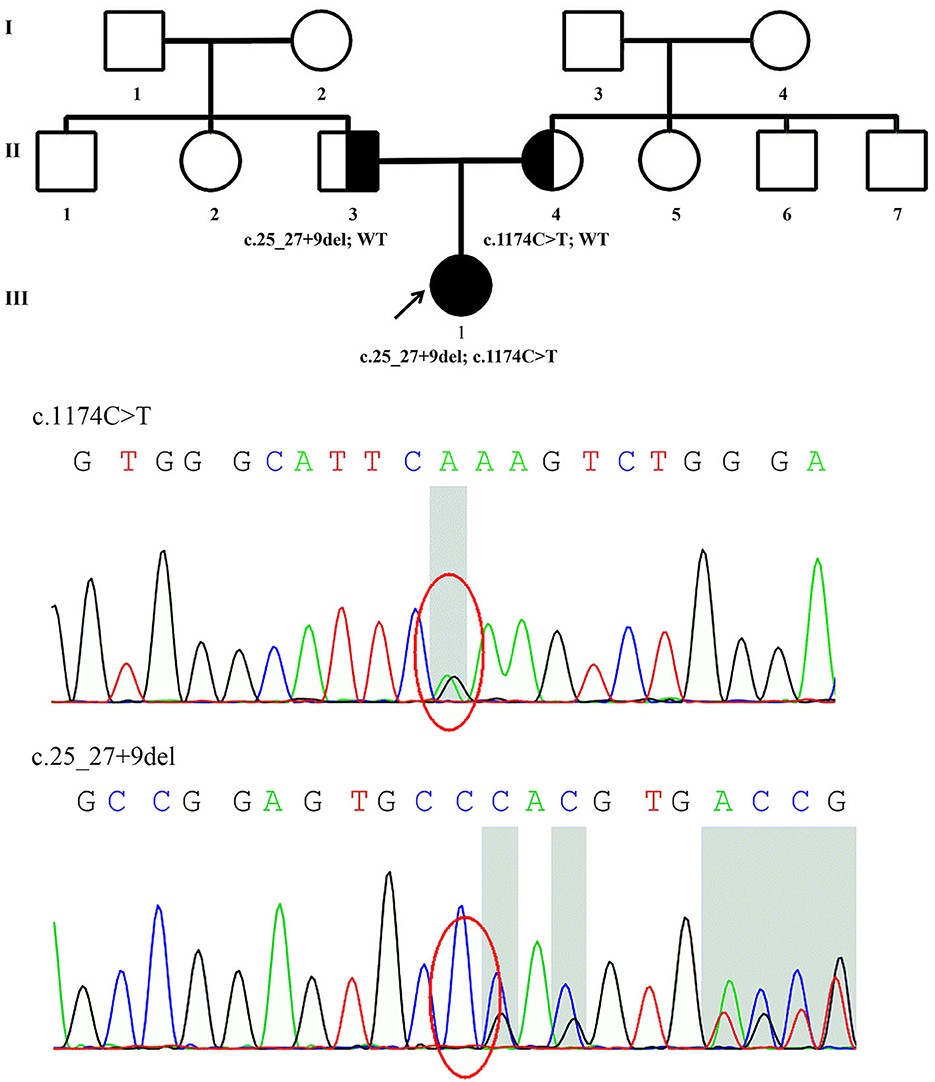
Figure 1. Family pedigree of Patient 1 and genomic sequencing electropherograms. A novel PRX splice-site mutation was detected. The reverse strand is shown for the c.1174C>T variant. WT, wild type.
Patient 2 was a 21-year-old man who had a history of delayed motor milestones, with independent walking delayed until 3 years old, balance problems, and numbness starting at 16 years of age. He was the first child of healthy, non-consanguineous parents from the same region. He had a younger affected sister and three healthy siblings. Physical examination showed severe sensory ataxia and sensory loss in the distal lower limbs. Weakness was mild. Nerve conduction studies indicated severe demyelination and axonal neuropathy.
Patient 3 was a 12-year-old girl who was the younger sister of Patient 2. She had delayed motor milestones, with independent walking delayed until 26 months of age. During childhood, she developed gait difficulties and balance problems. The symptoms progressed moderately. On examination at age 12, she had scoliosis and bilateral pes cavus. She had moderate to severe weakness and atrophy in the distal part of her lower limbs. Her Romberg sign was positive. Vibration sensation was absent in her lower limbs. Electrophysiological studies showed demyelinating neuropathy with mildly reduced CMAP amplitude.
3.2. Genetic results
The details of all identified pathogenic or likely pathogenic mutations are shown in Table 2. Compound heterozygosity for PRX variants was detected in Patient 1. The mutation p.R392X is a previously reported pathogenic mutation (11), while c.25_27 + 9del is a novel splice-site mutation of PRX. This novel deletion mutation alters the exon 4-intron 4 junction. In silico analysis using Human Splicing Finder showed this mutation will inevitably destroy the intron four donor splice site. According to the ACMG guidelines, this novel mutation has very strong evidence for pathogenicity. Segregation studies showed that the mutations were located on different parental chromosomes (Figure 1). A homozygous mutation p.R953X (4) was found in the two siblings (Patients 2 and 3). This mutation was segregating in their family and their parents were heterozygous carriers.
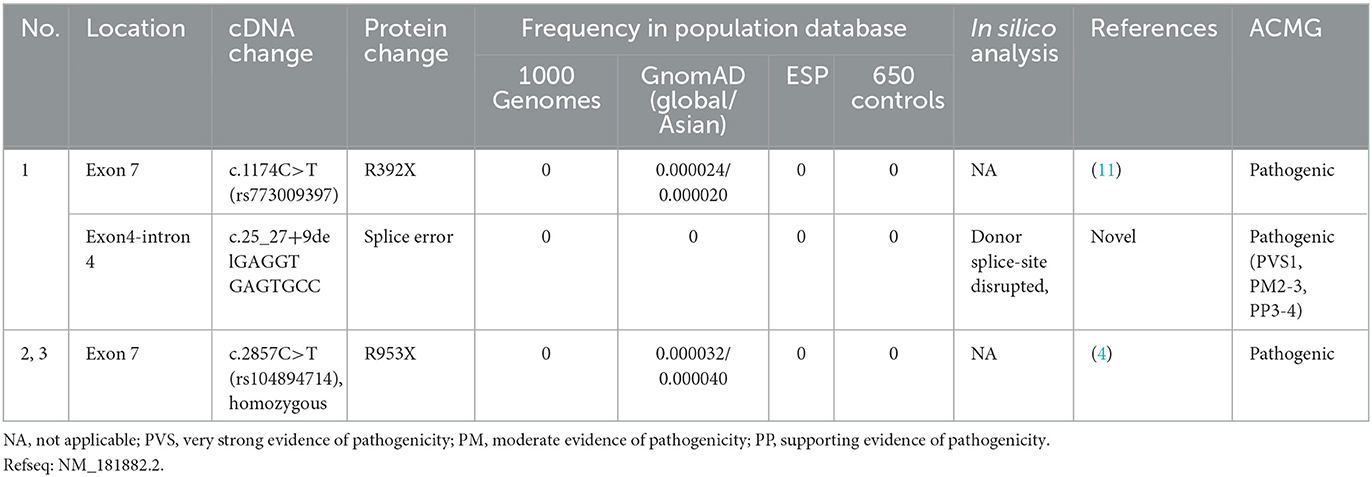
Table 2. Genotypes of the three CMT4F patients.
3.3. Literature review of PRX-related CMTs
Currently, over 60 PRX mutations that are either pathogenic or likely pathogenic (Figure 2; those with a diagnosis other than autosomal recessive CMT were excluded) have been derived from nearly 70 CMT4F pedigrees (Supplementary Table).
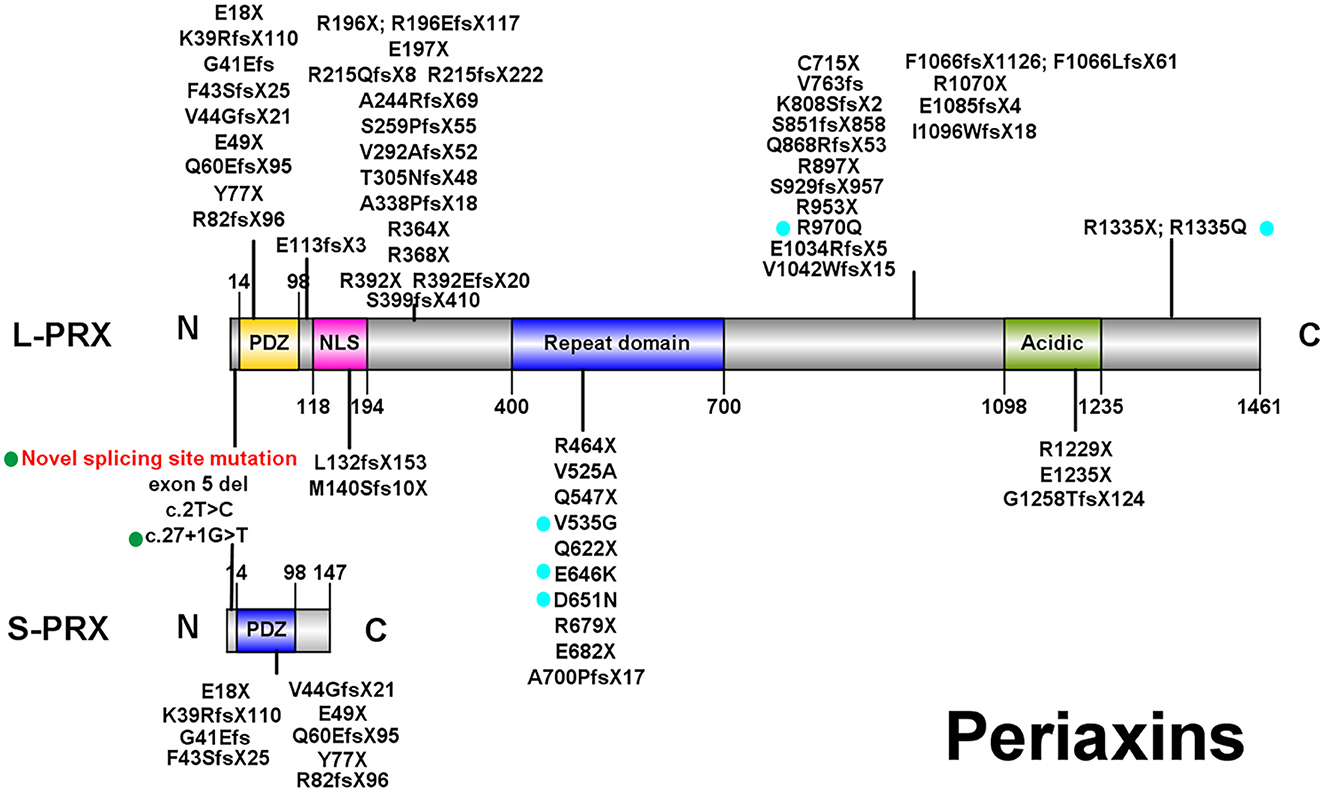
Figure 2. Illustrations of PRX. All pathogenic or likely pathogenic mutations of PRX from previously reported CMT4F cases and this study are shown. Green points: splice-site mutations. Blue points: nonsense and frameshift mutations. PDZ, PSD-95/Discs Large/ZO-1 domain; NLS, nuclear localization signal domain.
Eighty-four patients have available clinical information, and their clinical features are summarized in Table 3. The symptoms usually start in the first decade of life, and only four patients (4.8%) have late onset (after 20 years old). The most frequent symptoms are distal weakness or muscle atrophy (94%), feet deformity (75%), sensory impairment or sensory ataxia (65.5%), delayed motor milestones (60.7%), and spinal deformity (59.5%). Auditory impairments, ophthalmic symptoms, and respiratory symptoms are reported in 16.7%, 16.7%, and 8.3% of patients, respectively. CNS involvement is recorded in 16 patients (19.0%), including epilepsy in eight patients (9.5%), structural abnormality in six patients (7.1%), intellectual disability in four patients (4.8%), and pyramidal and cerebellar signs in two and one patients, respectively. Dysarthria or hoarseness is recorded in three CMT4F patients. Symptoms occasionally recorded in CMT4F patients include diabetes (12), congenital loss of nails (13), tongue fasciculation (14, 15), asymmetrical involvement (16), proximal weakness (15), and trigeminal neuralgia (17).
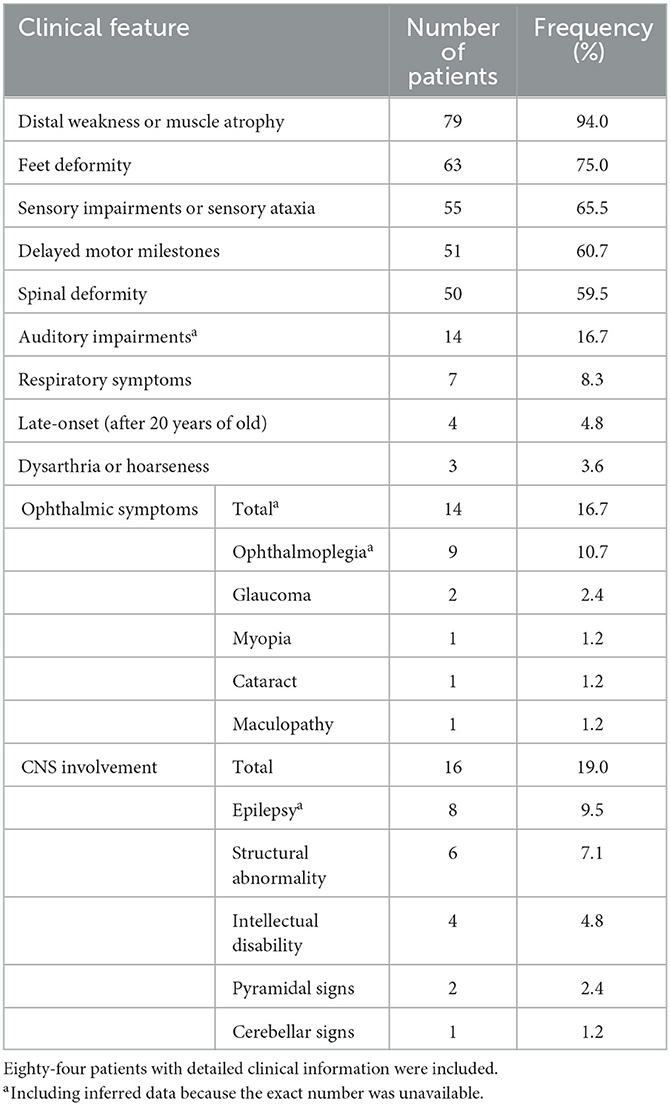
Table 3. Clinical features of all previously reported CMT4F cases.
Nerve conduction studies usually show a dramatic decrease in MNCV and undetectable SNAP (18). Elevated protein levels in cerebrospinal fluid have been reported (19). In one autopsy case, enlarged spinal nerve roots and demyelination of the phrenic, recurrent laryngeal, and oculomotor nerves were found (20, 21). Biopsy usually shows demyelination, classic onion bulbs, or sometimes the formation of basal lamina onion bulbs, tomacula, and myelin folding (18).
Currently, two cases with biallelic missense PRX mutations have been reported (22, 23). Their symptoms started at the age of 18 and 28, respectively, which appeared later than other patients. However, this needs more confirmation. No other obvious correlations between the type and site of mutations and clinical severity or distinct constellations of symptoms were found.
4. Discussion
We screened for PRX mutations in a large Chinese CMT cohort via NGS and WES. Two index patients with PRX mutations were found in the 465 CMT patient cohort. They were diagnosed with CMT4F. To the best of our knowledge, this is the largest Chinese CMT cohort to date and the first PRX screening study in China, which helps to clarify the distribution and characterization of PRX-related CMT in China. A splice-site mutation of PRX was identified, which is very rare in PRX-related CMT. Our study expanded the genetic spectrum of PRX-related CMT.
In our study, PRX mutations contributed to 0.43% of CMT cases (CMT1A included). In a previous study in the Chinese population, 1.2% (1/82) (6) of CMT cases were linked to PRX mutations. The discrepancy may be due to differing sample sizes, sequencing methods, or geographic distribution of patients. The frequency we reported is similar to previous studies from other countries, such as 0.4% in Japan (24), 1.6% in Spain (17), 0.35% in Canada (25) (2% of positive results), and 0.3% in a cross-country study (26).
PRX, located in chromosome 19 and containing seven exons (4), encodes two isoforms of periaxin through alternative splicing (7). L-periaxin (exon 4, 5, 6, and 7) and S-periaxin (exon 4, 5, and 6 and intron 6) share the same PSD-95/Discs Large/ZO-1 (PDZ) domain, which mediates the interaction between periaxin and other proteins (4, 7). L-periaxin also contains a nuclear localization signal (NLS) domain, a repeat domain, and an acidic domain (4) (Figure 2). L-periaxin links the cytoskeleton of Schwann cells to the basal lamina via its interactions with other proteins. The basic domains in the N-terminal of L-periaxin, which also function as NLS domains, bind with Drp2 and form the Prx/Drp2/Dag complex (27). The C-terminal of L-periaxin influences the function of this complex (28) and binds with β4 integrins (29). L-periaxin also binds with ezirin, which links the membrane and cytoskeleton, through both N- and C-terminals (30). The function of S-periaxin is not fully understood. Currently, it is believed to regulate the function of L-periaxin (31, 32). The loss of periaxin leads to instability of the peripheral nerve myelin and demyelinating neuropathy in murine models (8). Periaxin has recently been found to be expressed in the endothelium of small vessels in the human brain and influences the function of the blood–brain barrier (33).
Considering both the available literature and our study, the typical PRX-related CMT phenotype is characterized by early onset and slow progression of the disease, predominant sensory involvement, sensory ataxia, and distal amyotrophy or weakness (12, 18). Scoliosis is usually present in PRX-related CMT patients over the age of 12 years (12) but was only found in one of our patients.
As more cases have been reported, PRX-related CMT has shown a certain variability in clinical manifestations. A few patients may have late onset (22) or rapid onset (14). Newly recognized manifestations include auditory problems (12, 14, 15), ophthalmic problems (12), dysarthria (12, 23), hoarseness (22), vocal cord palsy (20), respiratory problems (12, 20), and involvement of the CNS (16, 23, 34–37). Abnormal BAEPs are common in CMT cases. The previously reported PRX-mutated CMT cases showed wave I delay (14, 15), which indicated auditory nerve lesions. In our study, Patient 1 presented with delayed waves III and V in BAEP, which is common in demyelinating CMT (38). Prolonged latencies in vestibular-evoked myogenic potentials were also reported in other CMT4F cases (39). These findings indicate that auditory and vestibular pathways may also be impaired in CMT4F. Periaxin is also expressed in lens fibers and is involved in the maintenance and development of the lens (40). Heterozygous mutations in PRX have been detected in pedigrees with congenital cataracts (41, 42). This may partially explain the ophthalmic symptoms, such as myopia and cataracts, observed in CMT4F. Cranial nerve demyelination was confirmed by pathology (20). The involvement of the phrenic and recurrent laryngeal nerves in CMT4F needs more attention because respiratory problems are sometimes fatal (20). Manifestations of CNS involvement include epilepsy (37, 43), delayed development of intelligence and cognitive deficits (36, 37), pyramidal signs (16), cerebellar signs (23), and structural abnormalities (34, 35, 37). In one case, this may be unrelated to the PRX mutation (37). The role of periaxin in the blood–brain barrier may partially explain these phenotypes (33). More research is needed to be carried out to clarify the function of periaxin in the CNS. However, the rare symptoms are not commonly observed and might be coincidental rather than part of the phenotype.
Consistent with a previous study (11), no obvious phenotype–genotype correlations were found in CMT4F in our study except that the onset age seems to be later in those with biallelic missense mutations. Over 60 pathogenic PRX mutations were detected. Most of them are nonsense or frameshift mutations, suggesting that a loss of function is the pathogenic mechanism for PRX-related neuropathies. Most mutations are located in exon 7, which is the largest exon encoding 90% of the protein (44). Currently, there are a few known pathogenic mutations for CMT which affect both L- and S-periaxin, including our novel splice-site mutation c.25_27 + 9del. Similar to our family with the splice-site variant, most patients with mutations affecting L- and S-periaxin did not develop symptoms distinct from those observed in patients with mutations in exon 7. However, a report on a single family with a homozygous p.Y77X mutation suggested that mutations affecting both types of periaxin could lead to a more severe phenotype (43).
5. Conclusion
We have elucidated the clinical and genetic features of PRX-related CMTs and reported a novel splice-site mutation, c.25_27 + 9del. PRX-related CMT accounted for 0.43% of the whole CMT cohort (CMT1A included) in China. The review of the literature showed that most CMT4F cases presented with early-onset and slow-progression demyelinating CMT. The discovery of rare symptoms indicates that the clinical spectrum is wider than previously anticipated.
Data availability statement
The datasets presented in this study can be found in online repositories. The data presented in the study are deposited in the GenBank, accession number PRJNA946142. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by Ethics Committee of Peking University Third Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
XL conceived the study, established the cohort, and revised the manuscript. XM analyzed the data and wrote the manuscript. XD and DF participated in the establishment of the cohort and provided technical support. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the National Natural Science Foundation of China (81873784 and 82071426), Clinical Cohort Construction Program of Peking University Third Hospital, Grant/Award Number: BYSYDL2019002 and BYSYDL2021007, and Clinical Medicine Plus X-Youth Scholars Project, Peking University, the Fundamental Research Funds for the Central Universities (PKU2021LCXQ019).
Acknowledgments
The authors would like to thank the patients and their families for their participation in this project.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2023.1148044/full#supplementary-material
References
1. Martyn CN, Hughes RA. Epidemiology of peripheral neuropathy. J Neurol Neurosurg Psychiatry. (1997) 62:310–8. doi: 10.1136/jnnp.62.4.310
2. Pareyson D, Marchesi C. Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol. (2009) 8:654–67. doi: 10.1016/S1474-4422(09)70110-3
3. Kijima K, Numakura C, Shirahata E, Sawaishi Y, Shimohata M, Igarashi S, et al. Periaxin mutation causes early-onset but slow-progressive Charcot-Marie-Tooth disease. J Hum Genet. (2004) 49:376–9. doi: 10.1007/s10038-004-0162-3
4. Boerkoel CF, Takashima H, Stankiewicz P, Garcia CA, Leber SM, Rhee-Morris L, et al. Periaxin mutations cause recessive Dejerine-Sottas neuropathy. Am J Hum Genet. (2001) 68:325–33. doi: 10.1086/318208
5. Guilbot A, Williams A, Ravise N, Verny C, Brice A, Sherman DL, et al. A mutation in periaxin is responsible for CMT4F, an autosomal recessive form of Charcot-Marie-Tooth disease. Hum Mol Genet. (2001) 10:415–21. doi: 10.1093/hmg/10.4.415
6. Sun B, Chen Z, Ling L, Yang F, Huang X. Clinical and genetic spectra of Charcot-Marie-Tooth disease in Chinese Han patients. J Peripher Nerv Syst. (2017) 22:13–8. doi: 10.1111/jns.12195
7. Dytrych L, Sherman DL, Gillespie CS, Brophy PJ. Two PDZ domain proteins encoded by the murine periaxin gene are the result of alternative intron retention and are differentially targeted in Schwann cells. J Biol Chem. (1998) 273:5794–800. doi: 10.1074/jbc.273.10.5794
8. Gillespie CS, Sherman DL, Fleetwood-Walker SM, Cottrell DF, Tait S, Garry EM, et al. Peripheral demyelination and neuropathic pain behavior in periaxin-deficient mice. Neuron. (2000) 26:523–31. doi: 10.1016/S0896-6273(00)81184-8
9. DiVincenzo C, Elzinga CD, Medeiros AC, Karbassi I, Jones JR, Evans MC, et al. The allelic spectrum of Charcot-Marie-Tooth disease in over 17,000 individuals with neuropathy. Mol Genet Genomic Med. (2014) 2:522–9. doi: 10.1002/mgg3.106
10. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
11. Choi YJ, Hyun YS, Nam SH, Koo H, Hong YB, Chung KW, et al. Novel compound heterozygous nonsense PRX mutations in a Korean Dejerine-Sottas neuropathy family. J Clin Neurol. (2015) 11:92–6. doi: 10.3988/jcn.2015.11.1.92
12. Renouil M, Stojkovic T, Jacquemont ML, Lauret K, Boue P, Fourmaintraux A, et al. [Charcot-Marie-Tooth disease associated with periaxin mutations (CMT4F): clinical, electrophysiological and genetic analysis of 24 patients]. Rev Neurol. (2013) 169:603–12. doi: 10.1016/j.neurol.2013.07.004
13. Auer-Grumbach M, Fischer C, Papic L, John E, Plecko B, Bittner RE, et al. Two novel mutations in the GDAP1 and PRX genes in early onset Charcot-Marie-Tooth syndrome. Neuropediatrics. (2008) 39:33–8. doi: 10.1055/s-2008-1077085
14. Takashima H, Boerkoel CF, De Jonghe P, Ceuterick C, Martin JJ, Voit T, et al. Periaxin mutations cause a broad spectrum of demyelinating neuropathies. Ann Neurol. (2002) 51:709–15. doi: 10.1002/ana.10213
15. Baets J, Deconinck T, De Vriendt E, Zimon M, Yperzeele L, Van Hoorenbeeck K, et al. Genetic spectrum of hereditary neuropathies with onset in the first year of life. Brain. (2011) 134(Pt 9):2664–76. doi: 10.1093/brain/awr184
16. Crimi M, Tarawneh A. The genetic counseling in a patient affected by congenital polyneuropathy after a “diagnostic odyssey” recently solved with WES approach. Eur J Transl Myol. (2022) 32:10361. doi: 10.4081/ejtm.2022.10361
17. Sivera R, Sevilla T, Vilchez JJ, Martinez-Rubio D, Chumillas MJ, Vazquez JF, et al. Charcot-Marie-Tooth disease: genetic and clinical spectrum in a Spanish clinical series. Neurology. (2013) 81:1617–25. doi: 10.1212/WNL.0b013e3182a9f56a
18. Marchesi C, Milani M, Morbin M, Cesani M, Lauria G, Scaioli V, et al. Four novel cases of periaxin-related neuropathy and review of the literature. Neurology. (2010) 75:1830–8. doi: 10.1212/WNL.0b013e3181fd6314
19. Chen YH, Zhang H, Meng LB, Tang XY, Gong T, Yin J. Novel mutation in the periaxin gene causal to Charcot-Marie-Tooth disease type 4F. J Int Med Res. (2020) 48:300060519862064. doi: 10.1177/0300060519862064
20. Maeda K, Yamamoto Y, Ohuchi M, Sakashita T, Shiohara M, Namura T, et al. Pathological evidence of demyelination in the recurrent laryngeal, phrenic, and oculomotor nerves in Charcot-Marie-Tooth disease 4F. eNeurologicalSci. (2021) 25:100358. doi: 10.1016/j.ensci.2021.100358
21. Shintaku M, Maeda K, Shiohara M, Namura T, Kushima R. Neuropathology of the spinal nerve roots, spinal cord, and brain in the first autopsied case of Charcot-Marie-Tooth disease 4F with a D651N mutation in the periaxin gene. Neuropathology. (2021) 41:281–7. doi: 10.1111/neup.12731
22. Tokunaga S, Hashiguchi A, Yoshimura A, Maeda K, Suzuki T, Haruki H, et al. Late-onset Charcot-Marie-Tooth disease 4F caused by periaxin gene mutation. Neurogenetics. (2012) 13:359–65. doi: 10.1007/s10048-012-0338-5
23. Schabhuttl M, Wieland T, Senderek J, Baets J, Timmerman V, De Jonghe P, et al. Whole-exome sequencing in patients with inherited neuropathies: outcome and challenges. J Neurol. (2014) 261:970–82. doi: 10.1007/s00415-014-7289-8
24. Yoshimura A, Yuan JH, Hashiguchi A, Ando M, Higuchi Y, Nakamura T, et al. Genetic profile and onset features of 1005 patients with Charcot-Marie-Tooth disease in Japan. J Neurol Neurosurg Psychiatry. (2019) 90:195–202. doi: 10.1136/jnnp-2018-318839
25. Volodarsky M, Kerkhof J, Stuart A, Levy M, Brady LI, Tarnopolsky M, et al. Comprehensive genetic sequence and copy number analysis for Charcot-Marie-Tooth disease in a Canadian cohort of 2517 patients. J Med Genet. (2021) 58:284–8. doi: 10.1136/jmedgenet-2019-106641
26. Fridman V, Bundy B, Reilly MM, Pareyson D, Bacon C, Burns J, et al. CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis. J Neurol Neurosurg Psychiatry. (2015) 86:873–8. doi: 10.1136/jnnp-2014-308826
27. Sherman DL, Fabrizi C, Gillespie CS, Brophy PJ. Specific disruption of a schwann cell dystrophin-related protein complex in a demyelinating neuropathy. Neuron. (2001) 30:677–87. doi: 10.1016/S0896-6273(01)00327-0
28. Sherman DL, Brophy PJ. A murine model of Charcot-Marie-Tooth disease 4F reveals a role for the C-terminus of periaxin in the formation and stabilization of Cajal bands. Wellcome Open Res. (2018) 3:20. doi: 10.12688/wellcomeopenres.13673.1
29. Raasakka A, Linxweiler H, Brophy PJ, Sherman DL, Kursula P. Direct binding of the flexible C-terminal segment of periaxin to beta4 integrin suggests a molecular basis for CMT4F. Front Mol Neurosci. (2019) 12:84. doi: 10.3389/fnmol.2019.00084
30. Guo T, Zhang L, Xiao H, Yang Y, Shi Y. Ezrin interacts with L-periaxin by the “head to head and tail to tail” mode and influences the location of L-periaxin in Schwann cell RSC96. Biochim Biophys Acta Gen Subj. (2020) 1864:129520. doi: 10.1016/j.bbagen.2020.129520
31. Yang Y, Shi Y. L-periaxin interacts with S-periaxin through its PDZ domain. Neurosci Lett. (2015) 609:23–9. doi: 10.1016/j.neulet.2015.10.020
32. de Morree A, Droog M, Grand Moursel L, Bisschop IJ, Impagliazzo A, Frants RR, et al. Self-regulated alternative splicing at the AHNAK locus. FASEB J. (2012) 26:93–103. doi: 10.1096/fj.11-187971
33. Wang MM, Zhang X, Lee SJ, Maripudi S, Keep RF, Johnson AM, et al. Expression of periaxin (PRX) specifically in the human cerebrovascular system: PDZ domain-mediated strengthening of endothelial barrier function. Sci Rep. (2018) 8:10042. doi: 10.1038/s41598-018-28190-7
34. Kuperberg M, Lev D, Blumkin L, Zerem A, Ginsberg M, Linder I, et al. Utility of whole exome sequencing for genetic diagnosis of previously undiagnosed pediatric neurology patients. J Child Neurol. (2016) 31:1534–9. doi: 10.1177/0883073816664836
35. Delague V, Bareil C, Tuffery S, Bouvagnet P, Chouery E, Koussa S, et al. Mapping of a new locus for autosomal recessive demyelinating Charcot-Marie-Tooth disease to 19q131-133 in a large consanguineous Lebanese family: exclusion of MAG as a candidate gene. Am J Hum Genet. (2000) 67:236–43. doi: 10.1086/302980
36. Karakaya M, Storbeck M, Strathmann EA, Delle Vedove A, Holker I, Altmueller J, et al. Targeted sequencing with expanded gene profile enables high diagnostic yield in non-5q-spinal muscular atrophies. Hum Mutat. (2018) 39:1284–98. doi: 10.1002/humu.23560
37. Makrythanasis P, Nelis M, Santoni FA, Guipponi M, Vannier A, Bena F, et al. Diagnostic exome sequencing to elucidate the genetic basis of likely recessive disorders in consanguineous families. Hum Mutat. (2014) 35:1203–10. doi: 10.1002/humu.22617
38. Rance G, Ryan MM, Bayliss K, Gill K, O'Sullivan C, Whitechurch M. Auditory function in children with Charcot-Marie-Tooth disease. Brain. (2012) 135(Pt 5):1412–22. doi: 10.1093/brain/aws085
39. Poovaiah P, Rajasekaran AK, Yuvraj P, Belur YK, Atchayaram N. Audiovestibular dysfunction in siblings with Charcot-Marie-Tooth disease 4F: a case series. J Am Acad Audiol. (2021) 32:616–24. doi: 10.1055/s-0042-1744105
40. Maddala R, Skiba NP, Lalane R, Sherman DL, Brophy PJ, Rao PV. Periaxin is required for hexagonal geometry and membrane organization of mature lens fibers. Dev Biol. (2011) 357:179–90. doi: 10.1016/j.ydbio.2011.06.036
41. Yuan L, Yi J, Lin Q, Xu H, Deng X, Xiong W, et al. Identification of a PRX variant in a Chinese family with congenital cataract by exome sequencing. QJM. (2016) 109:731–5. doi: 10.1093/qjmed/hcw058
42. Jones JL, McComish BJ, Staffieri SE, Souzeau E, Kearns LS, Elder JE, et al. Pathogenic genetic variants identified in Australian families with paediatric cataract. BMJ Open Ophthalmol. (2022) 7:e001064. doi: 10.1136/bmjophth-2022-001064
43. Zaman Q, Khan MA, Sahar K, Rehman G, Khan H, Rehman M, et al. Novel Variants in MPV17, PRX, GJB1, and SACS cause charcot-marie-tooth and spastic ataxia of charlevoix-saguenay type diseases. Genes. (2023) 14:328. doi: 10.3390/genes14020328
Keywords: Charcot-Marie-Tooth disease, periaxin, mutation, whole-exome sequencing, next-generation sequencing
Citation: Ma X, Liu X, Duan X and Fan D (2023) Screening for PRX mutations in a large Chinese Charcot-Marie-Tooth disease cohort and literature review. Front. Neurol. 14:1148044. doi: 10.3389/fneur.2023.1148044
Received: 19 January 2023; Accepted: 06 June 2023;
Published: 04 July 2023.
Edited by:
Christiane Schneider-Gold, St. Josef-Hospital, GermanyReviewed by:
Kazumasa Saigoh, Kindai University Hospital, JapanNobuyuki OKA, Konoe Rehabilitation Hospital, Japan
Jan Senderek, Ludwig Maximilian University of Munich, Germany
Copyright © 2023 Ma, Liu, Duan and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoxuan Liu, bHVjeWFuX2xpdUBiam11LmVkdS5jbg==