 William P. Flavin1,2
William P. Flavin1,2 Helia Hosseini3Jeffrey W. Ruberti4
Helia Hosseini3Jeffrey W. Ruberti4 H. Pirouz Kavehpour3,5
H. Pirouz Kavehpour3,5 Christopher C. Giza2,3,6
Christopher C. Giza2,3,6 Mayumi L. Prins2,3,6*
Mayumi L. Prins2,3,6*- 1Department of Neurology, David Geffen School of Medicine, UCLA, Los Angeles, CA, United States
- 2Steve Tisch BrainSPORT Program, Department of Pediatrics and Neurosurgery, David Geffen School of Medicine, UCLA, Los Angeles, CA, United States
- 3Department of Bioengineering, UCLA, Los Angeles, CA, United States
- 4Department of Bioengineering, Northeastern University, Boston, MA, United States
- 5Department of Mechanical and Aerospace Engineering, UCLA, Los Angeles, CA, United States
- 6Department of Neurosurgery, Brain Injury Research Center, David Geffen School of Medicine, UCLA, Los Angeles, CA, United States
Tau is a protein that has received national mainstream recognition for its potential negative impact to the brain. This review succinctly provides information on the structure of tau and its normal physiological functions, including in hibernation and changes throughout the estrus cycle. There are many pathways involved in phosphorylating tau including diabetes, stroke, Alzheimer’s disease (AD), brain injury, aging, and drug use. The common mechanisms for these processes are put into context with changes observed in mild and repetitive mild traumatic brain injury (TBI). The phosphorylation of tau is a part of the progression to pathology, but the ability for tau to aggregate and propagate is also addressed. Summarizing both the functional and dysfunctional roles of tau can help advance our understanding of this complex protein, improve our care for individuals with a history of TBI, and lead to development of therapeutic interventions to prevent or reverse tau-mediated neurodegeneration.
Introduction
In 1975, while studying the assembly of tubulin into microtubules, the Kirschner laboratory discovered a protein they named “tau,” short for tubulin-associated unit, for its ability to induce tubule formation (1). Then, in 1986, Grundke-Iqbal showed that paired helical filaments, which make up the characteristic neurofibrillary tangles (NFTs) seen in post-mortem brain tissue from Alzheimer’s disease (AD) patients, were composed of tau protein in a hyperphosphorylated state (2, 3). Later, in 1991, Braak and Braak found a characteristic distribution pattern of tau-containing neurofibrillary changes in AD brains (4), thus creating an eponymous staging system which led to an understanding of tau spatiotemporal spread as a causal factor underlying disease progression and severity.
In the past decades since the discovery of a linkage between tau and neurodegenerative disease, there has been intense focus on understanding the mechanisms and risk factors underlying aggregation and propagation of tau pathology in numerous neurodegenerative tauopathies such as AD, chronic traumatic encephalopathy (CTE), frontotemporal dementia (FTD), and progressive supranuclear palsy (PSP), among others. The role of genetic mutations as well as post-translational modifications of the tau protein in the development of neurodegeneration are beginning to be understood, but numerous questions remain unanswered and the goal of therapeutic intervention to prevent aggregation, promote degradation, or halt disease progression remains elusive. One of the most established risk factors for tauopathy is traumatic brain injury (TBI), though despite the link between a history of TBI and the later development of AD or CTE, central questions regarding the mechanistic links are yet unanswered.
This review provides information on the structure of tau, from the gene encoding its six isoforms to the spectrum of possible post-translational modifications, as well as the physiological functions of this protein. In the second part of this review, tau pathology is discussed. We first focus on tau phosphorylation and several pathways that are known to increase this modified form of cellular tau, from diabetes to AD to inflammation and repetitive mild TBI (rmTBI). We then review current knowledge regarding tau pathology following mild and repetitive TBI, including efforts to measure tau as a biomarker for TBI and to quantify tau aggregation in vivo using neuroimaging. We discuss pre-clinical research models of TBI-induced tau aggregation and highlight both the benefits and limitations of current models. Finally, we consider tau as a prion-like self-aggregating protein and summarize mechanisms known to underlie spatiotemporal spreading between cells and between more distant brain regions, ultimately leading to disease progression. We have aimed to advance the understanding of the molecular mechanisms involved in tau aggregation and propagation for all neurodegenerative tauopathies, but also to consider TBI as a complex environmental risk factor that can predispose to and drive pathology in unique ways, thus requiring future research to achieve a deeper understanding of this process.
The tau protein
Tau gene and protein
Tau is a cellular protein that belongs to the microtubule-associated protein (MAP) family and is predominantly found in neurons, though also present in glial cells. The tau gene is located on the long arm of chromosome 17, with 16 exons spanning over 100 kb (5). In humans, exons 4A, 6, and 8 are not present in mRNA, so the alternative splicing of the remaining exons contributes to the 6 protein isoforms that are generated (6). The proteins range from 352 to 441 amino acids with distinct regions (Figure 1). At the C-terminus, the isoforms differ by the presence of 3–4 repeat regions (3R or 4R) which define a microtubule binding domain. This repeat region is involved in axonal transport, development, and the phosphorylation state of tau. Adjacent to these repeats is a basic proline-rich segment which contributes to microtubule binding. At the N-terminus, there are 0–2 inserts of acidic regions (0 N, 1 N, or 2 N) involved in interaction with other cytoskeletal elements, cytoplasmic organelles, and plasma membranes.
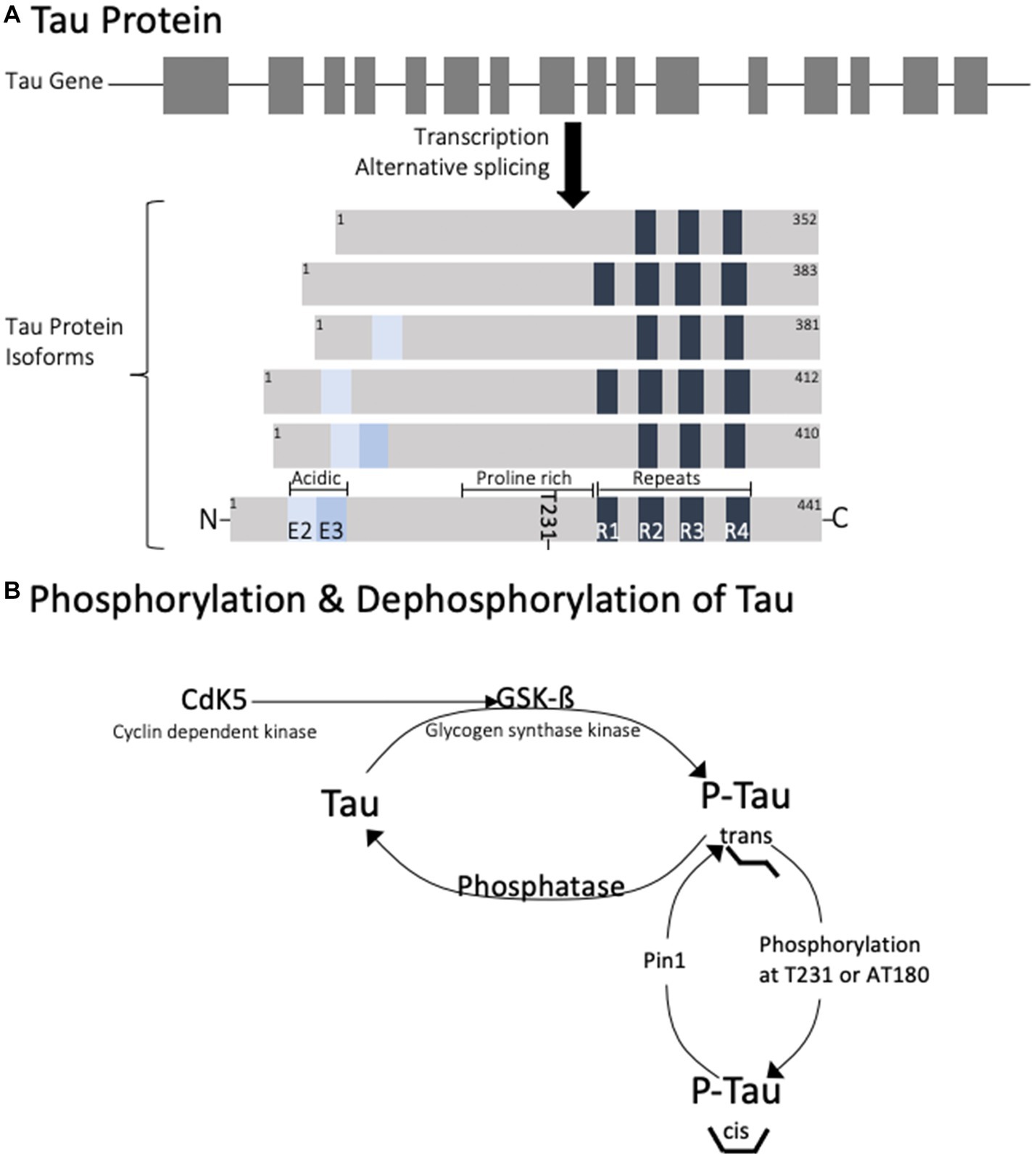
Figure 1. Diagram of the genetic splicing that produces Tau isoforms (A) and the enzymes involved in physiological regulation of tau phosphorylation (B). Note that phosphorylation of tau at T231 is responsible for trans to cis conformational change.
The normal cellular distribution of tau in the brain shows that it is predominantly found in neurons and to a lesser extent in human astrocytes (7). In neurons, tau is present at higher levels in axons than in dendrites (8). This distribution pattern reflects its role in axonal process extension and retraction as well as dendritic spine elongation. Under normal physiologic states the intracellular concentration of tau in a neuron is estimated between 2 and 4 μM (9), (10) with over 99% bound to microtubules (11). Estimations of extracellular concentrations of tau have been obtained in human cerebrospinal fluid (CSF) showing tau around 200 ng/mL (12), (13) and phosphorylated tau at 37.4 ng/mL (13).
Posttranslational modification
There are numerous posttranslational modifications of tau that affect its function. Details of the functional consequences of tau modifications are presented in several reviews (5, 6, 14). The 40 lysine residues can undergo acetylation, ubiquitylation and SUMO (Small Ubiquitin-like Modifier)-ylation. Acetylation of lysine decreases tau binding to microtubules and leads to its aggregation (15). Lysine ubiquitylation increases tau turnover and degradation (16), while SUMOylation can promote phosphorylation of tau and its aggregation (17). In contrast, methylation competes for lysine residues and decreases tau aggregation (18). N-glycosylation, the addition of a saccharide to the N-terminal asparagine, occurs on hyperphosphorylated tau and contributes to its aggregation (19). O-glycosylation, the addition of a saccharide to a hydroxyl oxygen, is instead thought to maintain tau association with microtubules thereby decreasing its aggregation (20). Nitration of a tyrosine residue can decrease the binding strength of tau to microtubules, increasing potential for tau aggregation (10). Glycation occurs in pathological tau when a carbohydrate is added to lysine, generating advanced glycation end products (AGEs), which further contribute to tau aggregation (21).
Phosphorylation is the most extensively studied post translational modification of tau, playing a role in both normal physiologic functions and pathophysiology. Phosphorylation is an important determinant of tau intracellular localization including the axon, somato-dendritic compartment, plasma membrane, and nucleolar organizing regions (22, 23). Axonal tau plays a phosphorylation-dependent role in axonal process extension and retraction, mediated by various kinases acting on serine, threonine, proline and tyrosine. Tau is most abundant in the cytoplasm and a significant proportion of tau has been found in the membrane in an unphosphorylated state. One study using mice showed that inhibition of casein kinase 1 (CK1) or glycogen synthase kinase 3 (GSK3) leads to a decrease in tau phosphorylation and an increase in its membrane fraction. However, the localization of tau to the membrane where it can interact with membrane associated proteins is dependent on Fyn kinase activation (24). Fyn-tau interaction regulates the NMDA receptor signaling via binding to the NR2B subunit involved in the post synaptic dendrite signaling (5). Tau also has a role in maintenance and protection of DNA and RNA in the nucleus (5). In dendrites, tau is responsible for dendritic spine development, elongation and maturation (25). While glycogen synthase kinase (GSK) is responsible for neurite retraction, cyclin dependent kinase (CDK5) is an important regulator in neuronal positioning during development and presumably in synaptogenesis and neurotransmission.
The various cellular functions of tau are regulated through phosphorylation. In the dephosphorylated state tau aggregation is minimal (Figure 2). Phosphorylation of tau is required for physiological functions, but it is tightly regulated. Hyperphosphorylation of tau has been associated with protein aggregation. Normally, 90% of tau exists in the trans conformational state which promotes microtubule stability (26). Phosphorylation at Thr231 causes a conformational shift towards the cis isomeric orientation, which destabilizes microtubule binding and promotes tau aggregation. Cis phosphorylation of tau has been proposed to be a pathological state of the tau protein (27, 28). Pin1 is a prolyl isomerase that dephosphorylates T231 and re-establishes microtubule binding (29). It is currently considered a therapeutic target for diseases caused by tau hyperphosphorylation.
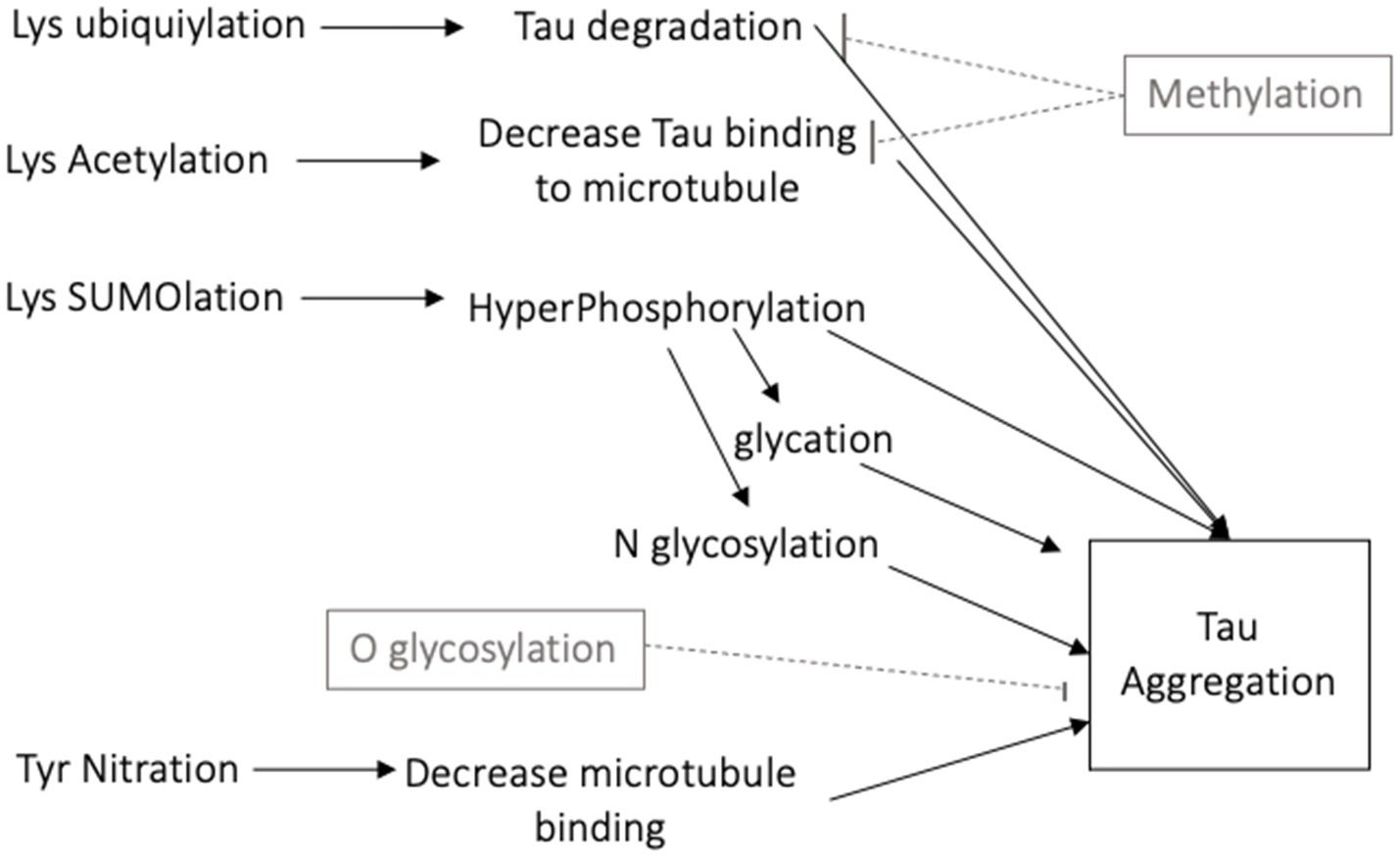
Figure 2. Schematic diagram of the many posttranslational modifications of tau that increase or decrease tau aggregation.
Tau turnover
A major determinant of a protein’s half-life in the cell is the interplay of its degradation and stabilization signals. The 3R and 4R isoforms of tau have different turnover rates, with 4R having a faster rate relative to 3R isoforms. In human induced pluripotent stem cells (iPSCs), the half-life of tau protein is between 6 to 7 days, and in mature CNS this period is estimated to be 23–24 days (30). The half-life of the protein is determined by the rate of degradation, which occurs through the ubiquitin/protease system (UPS) or the autophagy lysosome pathway (ALP). In the UPS system, tau protein is ubiquitylated and degraded in the lysosome or proteasome (5). The 20S core of proteasomes can be blocked with lactacystin in vitro, resulting in decreased degradation of tau (31). Decreased proteasome function may contribute to tau accumulation in both physiologic and pathologic conditions. Other modifications are effective in altering the half-life and function of tau; acetylation by histone acetylases (HATs) increases the stability of tau by acetylating the lysine residues (17). Furthermore, SUMOylation of the lysine 340 residue prevents its ubiquitylation as well as promotes its phosphorylation (17). The mechanisms underpinning targeting of tau to ALP-mediated degradation and its relationship to proteasomal degradation remains incompletely understood.
Physiologic tau regulation
Tau phosphorylation has been linked to normal physiological conditions including states of synaptic plasticity. One well-characterized and predictable model of plasticity that has been used to examine changes in inter-neuronal connectivity is hibernation. Hibernation is an adaptive behavioral strategy to reduce energy expenditure when environmental conditions are unfavorable. The cyclical changes in metabolism, heart and respiratory rate, and neuronal functions are predictable (32). During torpor, neuronal hippocampal synaptic connections decrease, a process that requires activation of cellular microtubule regulation. One cellular mechanism that can regulate these processes is the phosphorylation of tau. In fact, increased phosphorylation of tau is observed during torpor in cortical brains of arctic ground squirrels (33). Upon arousal from hibernation, tau phosphorylation decreases, synaptic connections are reinnervated and metabolism returns to normal. In the ground squirrel, tau hyperphosphorylation was observed at six sites (S199, T205, S214, S262, S396, and S404), but only three of these are dephosphorylated upon arousal (S199, S262, and S404) (34). This pattern of reversible tau phosphorylation has also been observed in the black bear and Syrian hamster (33), but may differ between cerebral regions and phosphorylation sites.
Evidence from hibernation studies has illuminated the relationship between metabolic substrates and the phosphorylation of tau (35). During torpor, cerebral metabolism switches from glucose to ketone use, a fuel change which is thought to induce tau phosphorylation and morphological change. Injection of non-metabolizable 2-deoxy-d-glucose (2DG) into the rat cortex will effectively starve cells metabolically, forcing them to shift towards other oxidizable energy sources. During this time, there was an increase in ß-APP expression and tau phosphorylation measured by the phospho-tau (Ser202 and Thr205) monoclonal antibody AT8 at 7 h post injection, specifically with an increase in cis conformation (36). Reversible tau hyperphosphorylation, primarily in the hippocampus, has also been observed in mice when hypoglycemia was induced by 1–3 days of food deprivation (37). It has been proposed that the decrease in body temperature associated with induced hypoglycemia directly affects kinase and phosphatase activities leading to increased tau phosphorylation (38).
Another physiologic condition associated with significant neuronal plasticity occurs in females during different phases of hormonal changes. In younger pregnant females, there are significant cerebral synaptic changes occurring that require changes in cellular microtubules. Studies have shown that total tau content in the hippocampus decreases during pregnancy, but the proportion of phosphorylated tau increases in rats (39, 40). Female estrogen activates Akt, which inhibits GSK3 thereby inhibiting phosphorylation of tau (41). Later during menopause, decreases in estrogen can reverse the balance between kinases and phosphatases resulting in greater production of p-tau (39). The influence of the estrus cycle and of lifetime hormonal changes on tau regulation and pathology remains incompletely understood.
Pathways to tau phosphorylation
There are many pathways leading to pathological increases in tau phosphorylation, including diabetes, stroke, Alzheimer’s disease (AD), brain injury, aging, and drug use. Examination of the mechanisms described by preclinical research models of these diseases reveals common targets for hyperphosphorylation of tau: activation of kinases or inhibition of protein phosphatases. Evidence from various disease models that utilize these pathways will be briefly reviewed (Figure 3). There are several kinases that have been shown to phosphorylate tau at different sites, including glycogen synthase kinase (GSK3ß), cyclin dependent kinase (CDK5), and c-Jun N terminal kinase (JNK) (42). GSK3ß is so far the most commonly activated kinase in pathological tau hyperphosphorylation. This kinase phosphorylates the greatest number of sites on tau. Normally it is involved in cellular proliferation, migration, glucose regulation and cell death. CDK5 activity is highest in neurons, and with the regulator p35 it is involved in brain development (43). When CDK5 complexes with p35 it can phosphorylate tau. JNK is a mitogen activated protein kinase that is involved in cell proliferation, differentiation, development, inflammation and cell death.
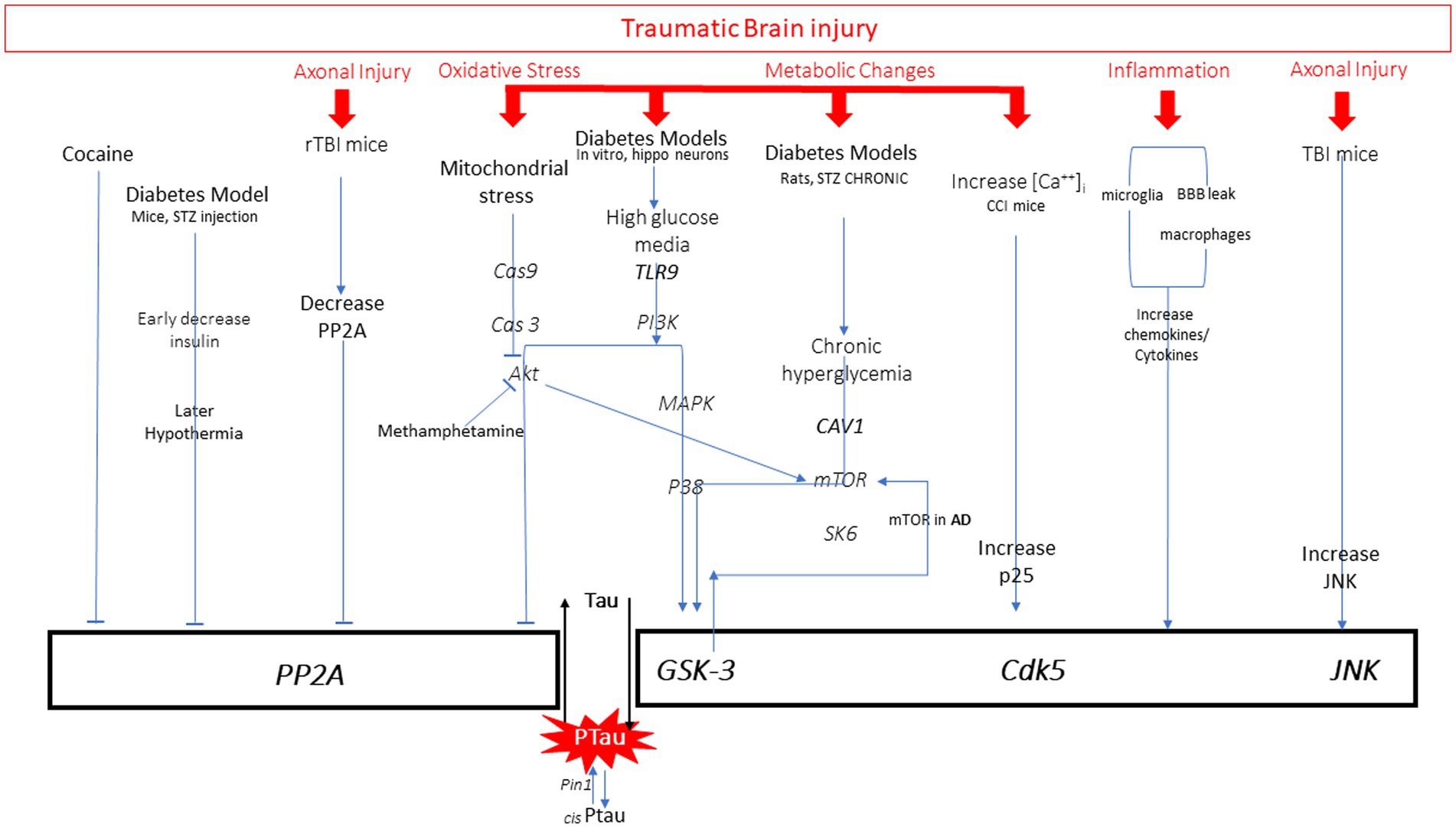
Figure 3. All pathways to tau phosphorylation from numerous diseases and exposures based on literature. It is important to note that those in red are all pathway elements initiated by traumatic brain injury.
Diabetes/Alzheimer’s disease
Activation of one or more kinases is a common mechanism to increase tau phosphorylation. Preclinical models of diabetes have examined the relationship between diabetes-induced hyperglycemia, hypothermia, and insulin dysfunction on p-tau. Tau hyperphosphorylation can be achieved through several mechanisms that activate kinases GSK3 and CDK5. Cultured hippocampal neurons have been used to study effects of high glucose on p-tau in vitro. Sun et al. (44) found increased TLR9 expression in neurons and an increase in tau hyperphosphorylation in the presence of high glucose media. Inhibition of P38/MAPK reversed tau phosphorylation. The GSK3 kinase has also been reported to be activated by hyperglycemia through the decrease in Caveolin-1 (Cav-1), which activates mTOR/SK6 pathway to activate GSK3, thereby increasing tau phosphorylation (45). Many of these pathways involved in diabetes are common to those seen activated in AD. Activation of mTOR has played a prominent role in Aß production and tau phosphorylation through GSK3 activation, which can serve as a feedback loop activating mTOR (46).
Another mechanism that can contribute to hyperphosphorylation of tau is the inhibition of protein phosphatases. This mechanism has been observed under diabetic conditions. Animal models of diabetes can be induced by streptozotocin (STZ) injections which result in decreased circulating insulin and hypothermia. Using this model in mice, it was demonstrated that both decreased insulin and hypothermia can each independently increase phosphorylation of tau (38). Examination of the role of multiple kinases in this process revealed that kinase overactivation is not the primary mechanism, but rather increases in the C-subunit of the PP2A phosphatase enzyme inhibits its function and thereby contributes to the net increase in tau phosphorylation.
Stress, drugs
Kinases are also influenced by stress, inflammation and drug use to mediate tau phosphorylation. Through corticotropin releasing factor receptor (CRFR) pathways, 30 min of restraint stress in mice can also activate several of the kinases that increase tau phosphorylation (47). Mitochondrial stress with release of cytochrome c can initiate Cas9/3 activation which inhibits Akt and thereby increases activation of GSK3ß (48). This mechanism is also associated with increased p-tau in cell cultures exposed to methamphetamine (49). Rats exposed to cocaine do not show changes in CDK5, but do show inhibition of protein phosphatases, which results in less dephosphorylation of tau and results in p-tau accumulation (50). It is important to note that p-tau is elevated in individuals with drug addictions (heroin), showing greater numbers of regions affected and higher p-tau load than age-matched controls (51). Examination of different drug mechanisms is important given this growing body of evidence.
Inflammation
Most disease processes initiate pro-inflammatory responses including release of cytokines. These are elevated in diabetes, AD, TBI and other neurodegenerative disorders. Physiological stress, oxidative stress and pro-inflammatory cytokines can activate the JNK pathway (52). Inhibition of JNK in rat neuronal cultures reduced p-tau, suggesting a role for JNK in tau hyperphosphorylation (53). The role of activated microglia has been studied by injecting IL-1ß into the ventricular space in human alpha-1-antichymotrypsin and tau transgene mice (54). In both mouse models, IL-1ß induced phosphorylation of tau that correlated with JNK activity. Induction of the inflammatory response by injection of lipopolysaccharide (LPS) also induces p-tau through the same pathway (55). IL-1ß is the most studied pro-inflammatory molecule in the tau pathway. However, there is mixed evidence that TNFalpha and IL-6 also can play a role through CDK5, but more studies are required. The type of kinase activated has been shown to be dependent on the duration of the immune stimuli and the age of the subject. Chronic LPS delivery to young transgenic AD mice activates CDK5, whereas GSK3ß is the preferred kinase activated in the aged mice (55). It also has been suggested that the activation of the neuroinflammatory responses through LPS may be sufficient to induce changes, but not to sustain persistent tau phosphorylation. This is an important area of research as the exact mechanism of immune-mediated tau phosphorylation is incompletely understood.
Mild traumatic brain injury and repeat TBI
TBI occurs when the brain moves rapidly inside the skull. This rapid movement causes mechanical deformation of the brain initiating many pathways simultaneously. Initially, the brain movement causes indiscriminate release of neurotransmitters from neurons, which then activate postsynaptic channels allowing large concentrations of ions to move down their concentration gradient. This occurs immediately upon impact, causing an increase in demand for cellular energy as the cells struggle to re-establish ionic equilibrium. Glucose uptake increases early after injury as it is rapidly processed to ATP to fuel the Na+/K+-ATPase. The phase of hyperglycolysis lasts approximately 6 h post injury, and is followed by a more prolonged state of decreased cerebral glucose metabolism that can last for days-weeks in animal models depending on injury severity and age. Depressed glucose metabolism is often accompanied by decreases in cerebral blood flow, and deficits in motor, cognitive and behavioral modalities have been observed during this time (56). Given the relationship between diabetes, AD-related glucose changes, and tau phosphorylation, it is possible that this represents one mechanism through which rmTBI may also contribute to p-tau accumulation. There are currently no studies examining rmTBI metabolism and tau hyperphosphorylation.
Following mTBI there is an increase in both extracellular K+ and intracellular Ca++ caused by intense glutamate release. The accumulation of 45Ca++ after lateral fluid percussion injury is dependent on age and time after injury (57, 58). Immediately after injury, 45Ca++ accumulated in the cortex in P17, P28, and adult rats and resolved within 4 days. However, in P28 and adult rats there was a second increase at 2–4 days in the thalamus that lasted until 14 days post-injury. The presence of increased calcium can activate calcium-dependent cysteine protease pathways that can contribute to axonal cytoskeletal disruption. Transient increases of intracellular Ca++, over 2–5 min, can also lead to an increase in tau phosphorylation in a “dose-dependent” manner, directly correlating with increases in intracellular Ca++. This phosphorylation is achieved by GSK3ß activation (59, 60). Increases in intracellular calcium have also been associated with increased tau phosphorylation 24 h after CCI injury in mice. The post-TBI increase in calcium increased conversion of p35 to p25 producing elevated levels of CDK5/p25 complex that are involved in tau phosphorylation (61).
Single and repetitive mild TBI have been shown to produce varying degrees of axonal injury in preclinical models (62, 63) and human TBI (64). Mechanical disruption of the axons can physically disrupt axonal architecture, and increases in calcium can activate calpain-mediated cytoskeletal degradation as well. These processes can disrupt axonal transport and release tau from microtubules. While the exact mechanism remains unknown, one study demonstrated that three fluid percussion impacts in adult rats results in decreased PP2A, which thereby increases tau phosphorylation. Administration of sodium selenate to the animals elevated PP2A and reversed the p-tau accumulation (65). While TBI can induce tau phosphorylation through decreasing phosphatases, other studies have shown activation of kinases as the mechanism. In one study, a single CCI injury in TgAD mice increased tau phosphorylation through JNK but did not change PKA, CDK5 or GSK3ß activity (66). This contrasts with the Yousuf 2016 study, where increases in intracellular calcium were shown to activate CDK5/p25 complex (61). It is unclear why CCI injuries in separate studies show differential kinase activation, but it is clear more research is required to clarify these differences.
TBI also influences tau phosphorylation through the subsequent inflammation occurring after injury. The temporal profile of the inflammatory response after injury is dependent on age, sex, and injury severity. Acute increases in cytokines have been documented after mTBI in human subjects over 1–3 months (67) and in mice over 7–14 days (68). A preclinical study examining mild TBI with weight drop in adult mice showed evidence of blood brain barrier disruption and increases in serum cytokines such as IL-6 at 90 min post injury (69). There are fewer studies addressing the mechanistic link between rTBI, cytokine profiles, and their consequent effect on tau. Madathil and colleagues (70) examined the effects of single and repetitive mild TBI with a closed head concussive injury in adult rats on microglial morphology over time. After a single mTBI, microglia were found presenting both M1 (pro-inflammatory) and M2 (anti-inflammatory) markers at 6 h. While the pro-inflammatory markers returned to baseline 72 h after a single mTBI, the M1 phenotype remained elevated in the rTBI group at this time point, suggesting a persistent activation of microglia with increasing TBI exposure. No studies have addressed the influence of injury interval on the magnitude and duration of pro-inflammatory responses in the setting of repeat TBI.
Mechanical stretching
TBI is initiated by a mechanical event and initiates numerous biological cascades that compound the physical movement. Biomolecules in solutions exposed to strain can aggregate and change conformation. Recently, tau was studied under extensional flow to determine if strain alone can induce tau aggregation (71). Tau 3R0N (36.7 kDa) was divided into 10 μL droplets and subjected to extensional strain in a modified tensiometer. The diameter of the filament that formed during extension was tracked as a function of time and analyzed for signs of aggregation using tau and two molecules of similar or greater size. Results demonstrated that at high extensional strains, tau protein solution 36.7 kDa and the polyethylene oxide 35 kDa formed aggregates. This is the first study to demonstrate the effects of strain alone on tau protein can cause aggregation. Future studies will need to address how phosphorylated tau behaves and whether specific neuronal microenvironments may create unique strains during injury that contribute to aggregation foci.
Pathological tau changes: evidence of tau after mild and repetitive traumatic brain injury
Clinical mild traumatic brain injury and tauopathy observations
Tau has many different but incompletely understood roles in the setting of traumatic brain injury (TBI), particularly mild TBI (mTBI). The largest body of literature comprises neuropathology cases showing evidence of p-tau in the brains of individuals with a history of repetitive head impacts (predominantly American football players, but also other contact sport athletes and military servicemembers). Cases with these autopsy findings have been diagnosed with chronic traumatic encephalopathy (CTE), and consensus neuropathological criteria have been published and updated (72). Research criteria for diagnosis of CTE in living patients have also been published (73), but difficulty remains due to lack of specificity of common symptoms and the potential contribution of ongoing comorbid conditions on neurobehavioral dysfunction in many patients (74). In these settings, most subjects had a prior history of neurological and behavioral changes that preceded death, and an association between repetitive head impacts and neurocognitive impairment or even neurodegeneration has been reported. Tau has also been examined as a fluid biomarker in CSF and blood after mTBI, and newer molecular ligands are permitting in vivo PET imaging of potential cerebral tau accumulation in case series (75). However, the challenge remains that cerebral tau neuropathology is also reported in individuals without a history of TBI, and the associations reported in case series are not able to determine whether a causal relationship exists between repeated head impacts and the pathological features described.
Tau pathology associated with repeated mTBI, but also those with no TBI
Many case studies have demonstrated hyperphosphorylated tau in brain at autopsy in individuals with a history of repeated head impacts, concussions or mTBI (76, 77). CTE has been defined by the pattern of neuropathology showing hyperphosphorylated tau in a perivascular distribution at the depths of cortical sulci (72). These cases are predominantly in professional American football players, but cases have also been reported in other sports (78) and in military service personnel (79). Other case series looking for CTE neuropathology in military personnel found no evidence of neurodegeneration in a cross-sectional study of 18 military service members (80). While cross-sectional studies have proposed a classification system of increasing severity for CTE across stages, neurodegenerative progression has not clearly been demonstrated in prospective longitudinal studies.
Confounding the causal interpretation of repeated head impacts leading to CTE, neuropathology consistent with CTE has also been described in individuals with no prior history of TBI, including epilepsy (81), substance abuse (82), ALS (83), multiple system atrophy (84) and other neurodegenerative diseases (85). These conflicting data raise the possibility that hyperphosphorylated tau CTE neuropathology may not necessarily be progressive nor be specifically associated with repetitive head impacts (86, 87).
Tau as a biomarker of TBI, mTBI and head impacts
While tau protein may serve as a surrogate measure for axonal injury acutely after TBI, the ability to measure it in vivo is limited. In patients with severe TBI, higher levels of tau in the extracellular fluid, measured using microdialysis, correlated with worse clinical outcomes 6 months post-injury (88). Other studies in severe TBI showed acute elevations of total tau in CSF and associations with long-term outcomes (89, 90). However, total or p-tau did not show CSF elevations chronically after more severe TBI (91). In amateur boxers, total tau was elevated in CSF via lumbar puncture days after a fight, and normalized within 1–3 months (92). Interestingly, a study of TBI biomarkers (including tau) in amateur soccer players found no significant elevations in CSF sampled within a week of a heading training session (93). Some studies of concussion reported elevations of serum tau compared to non-concussed controls, but methodological differences in assays may confound interpretation (89, 94). Ultrasensitive detection of total tau (t-tau) using the Simoa assay kits (Quanterix) has shown increased plasma t-tau levels 1 h after concussion in professional ice hockey players, compared to pre-season levels, and these levels were predictive of time to return-to-play (89). Other studies of mTBI suggest that cleaved tau is a poor predictor of long-term outcome or development of post-concussion syndrome following mTBI (95, 96). In 105 emergency department patients, t-tau levels drawn within 24 h of mTBI were correlated with ordinal outcome Glasgow Outcome Score-Extended (GOSE) but not Rivermead Post-Concussion Symptom Questionnaire (RPCSQ) scores (97). In military personnel undergoing training to breach or force entry into dangerous environments, tau levels in blood 1 h after exposure to blast overpressure did not significantly differ from pre-exposure levels (98). Other chronic military studies showed no significant differences in serum tau between controls and those with a history of prior TBI, but did report significant correlations between tau and symptoms (99). The association of CSF and serum tau levels with mTBI and their relationship to long-term outcomes remains unclear.
Tau neuroimaging in those with a history of repeated mTBI
More recently, studies have begun using radioisotope ligands to attempt to demonstrate tau deposition in living patients (100–102). There have been reports of clinico-pathological correlates between antemortem tau-PET and subsequent CTE tau neuropathology on autopsy (103, 104). Using post-mortem brain tissue samples of pathologically confirmed CTE, autoradiography with tau PET tracer AV-1451 (flortaucipir) did not detect robust binding. This suggests that tau PET tracers may have different binding patterns for different tau conformations in different tauopathies (105). Better characterizing these methods and further longitudinal studies in patients with suspected CTE will be important to more clearly determine progression over time.
In summary, tau has been implicated in both the acute response to repetitive mTBI (rmTBI) and in the setting of chronic neurobehavioral impairment seen in some patients with a history of prior repeat head impact exposure and concussions. While neuropathological consensus criteria for a diagnosis of CTE have been proposed, clinical criteria for this diagnosis proposed for research purposes are nonspecific and overlap with many treatable comorbidities (73, 106), limiting the present utility of these criteria for patients. Further work is necessary to distinguish neuropathology associated with CTE from other conditions that have shown similar findings. Lastly, better prospective longitudinal delineation of neurobehavioral impairments in conjunction with worsening p-tau neuropathology is needed to confirm whether CTE is a progressive illness; advances in in vivo detection using fluid and imaging molecular biomarkers show promise in these critical investigations.
Preclinical mild TBI and tauopathy research
This section will review the existing literature specifically addressing mTBI and rmTBI in animal models with tau outcomes. Many rmTBI models have recently emerged in an effort to address current sports injury concerns. However, in modeling a concussive injury several important criteria must be met. First, the model must demonstrate that the single impact is histologically and behaviorally consistent with clinical mild concussive injuries, for example not causing bleeding, skull fracture, transient behavioral deficits, or any gross pathology. Second, the model must involve a rmTBI protocol that is relevant to the clinical scenario. Lastly, appropriate control groups must be included and relevant time points after injury must be selected to examine acute versus chronic effects. These experimental specifics have been summarized in Table 1. Previous research has established that age at injury influences outcome (57, 107–109) therefore the studies will be grouped into adolescent/young adult, adults, and aged rodents.
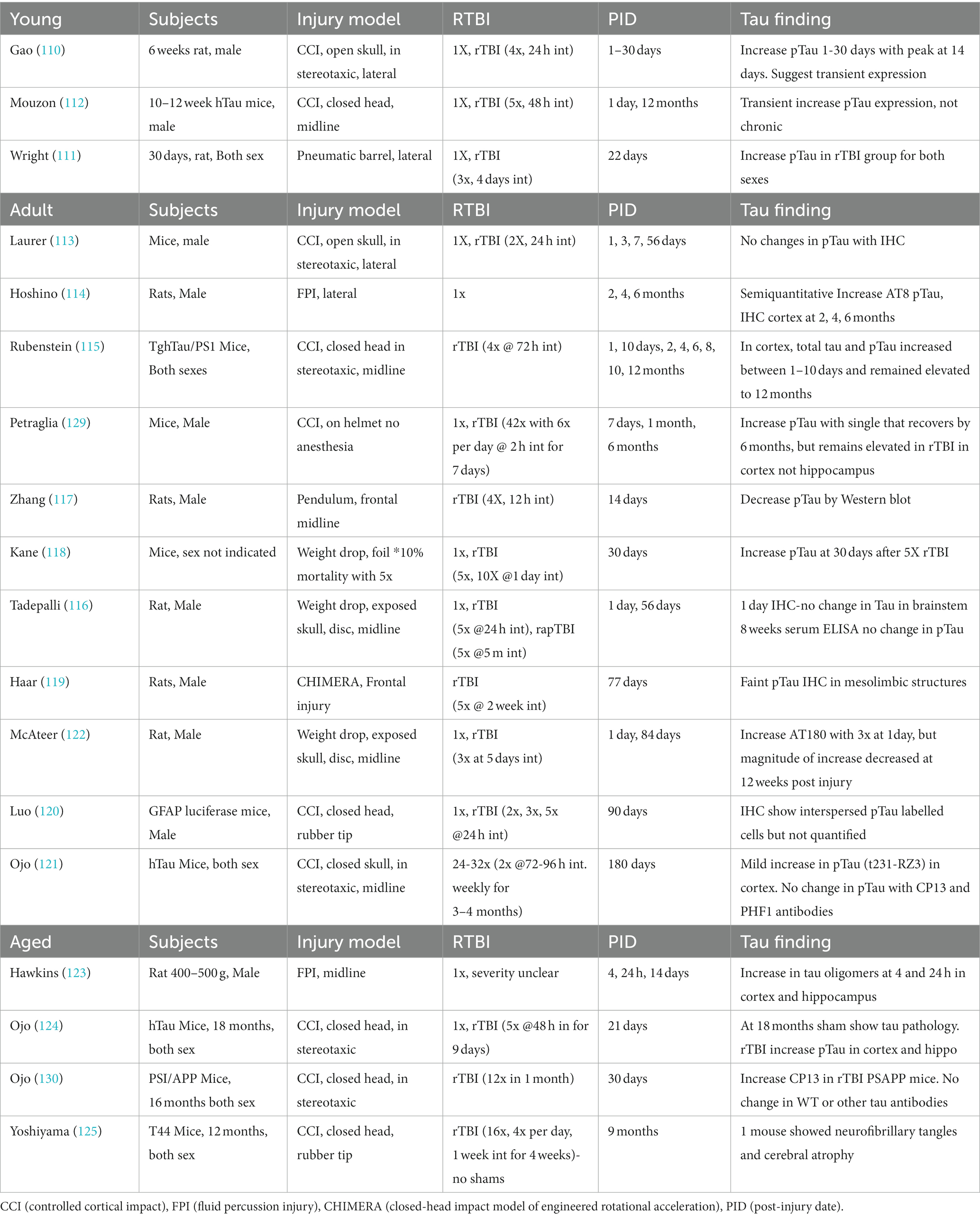
Table 1. Experimental Models of Repeat Mild TBI and Tau.
Consistent with TBI research as a whole, few studies have addressed changes in tau phosphorylation after rmTBI in the adolescent age group. Gao and colleagues examined p-tau within the first month after single and repetitive mTBI and found that p-tau increases during the time period, peaking around 14 days post injury (110). This was observed in both males and females (111, 112). In a rmTBI model using human tau mice, Mouzon and colleagues observed increased p-tau expression at 1d post injury, but not at the 12 months chronic time point (112). Despite differences in the locations and injury specifics, all the studies suggest transient increase in p-tau expression when rmTBI occurs during adolescence.
While a greater number of studies have addressed changes in p-tau after rmTBI in the adult animal models, the findings are varied. There are a number of variables that contribute to the lack of consensus, including wild type versus transgenic animals as well as different injury models, number of injuries, injury intervals, time after injury, and types of p-tau analysis and antibodies used. Among the studies utilizing multiple time points, one study found no changes in tau using immunohistochemistry (113) and three studies demonstrated initial increase in total phosphorylated tau within the first 7–10 days that endured at 6–12 months after rmTBI (114, 115). It is important to note the differences in subjects, injury model, and number of injuries and intervals between these studies. Those studies examining a single acute (< 30 days) timepoint also showed variability; a weight drop injury showed no tau histology changes in brainstem at 1d (116), a frontal pendulum rmTBI showed decreased pTau at 14 days (117), and a weight drop in mice showed increased p-tau at 30 days (118). The remaining studies examining a single chronic time point showed some increase in p-tau but they were not robust (119–121), and one study showed p-tau even decreased compared to 1 day (122). There is a growing need to examine the time course of tau phosphorylation after injury to determine how number of impacts, impact interval, and time contribute to eventual pathology and dysfunction. A single injury may induce more transient plasticity-related changes in tau, whereas repeated injuries with short intervals may result in pathological tau accumulation. Achieving consensus using wild type or transgenic models remains difficult to achieve with so many variations in experimental designs and multiple regional analyses.
Only a handful of studies have addressed TBI-induced tau phosphorylation in the aged animal, and three of the four were conducted in transgenic animals. Increased tau oligomers were observed at 4 and 24 h post injury in the cortex and hippocampus in aged male rats following fluid percussion injury (121, 123). Tau pathology was quite variable in transgenic mice with increases observed with specific antibodies (121, 124) at 21–30 days post injury. RmTBI in a TgT44 mouse model only found 1 animal with neurofibrillary tangles 9 months after injury (125). Quantification of tau pathology in the aged rodent presents several difficulties, as aged sham animals will also develop significant pathology, especially in transgenic animals, and show age related decline in behaviors. Regardless of age, these rmTBI animal models examining tau phosphorylation reveal the difficulties of establishing consensus without investing in a more comprehensive study that takes sex, age, time course and tau quantification into account.
Aggregation and propagation of p-tau
Tau as a prion-like amyloid protein
Misfolding of tau leads to its pathological aggregation from a natively unstructured soluble monomer to a higher-order insoluble hyperphosphorylated multimer in a beta-sheet fibrillar conformation (126, 127). It is this aberrant aggregation and accumulation of tau as seen in neuropil threads (NTs) or neurofibrillary tangles (NFTs) that are the pathological hallmark and defining feature of diverse tauopathies such as Alzheimer’s disease (AD), chronic traumatic encephalopathy (CTE), Pick’s disease (PiD), progressive supranuclear palsy (PSP), argyrophilic grain disease, and corticobasal degeneration (CBD), among others (127). Filaments composed of all 6 tau isoforms make up NFTs in AD and CTE, whereas tangles in PiD include predominantly 3R isoforms and tangles in PSP, AGD, and CBD are composed of mostly 4R isoforms (128).
Though the mechanism by which native tau becomes misfolded and begins to aggregate remains uncertain, missense mutations (P301S and P301L) in the microtubule-binding domain of tau that cause familial frontotemporal dementia have implicated destabilization of microtubule binding in promoting aggregation (131, 132). Physiological phosphorylation of tau occurs as part of its cellular function as discussed previously, but NFTs are known to be hyperphosphorylated compared with normal tau, and phosphorylation has been shown to decrease microtubule binding affinity and promote fibrillization (127, 131). This phosphorylation-driven fibrillization may occur through a liquid–liquid phase separation of tau microtubule-binding repeats that induces aggregation through molecular crowding and electrostatic forces in liquid droplets (133, 134). Binding of tau to microtubules is also thought to be disrupted due to axonal injury during TBI, where microtubule disassembly causes tau dissociation and predisposes it to aggregation, a process hypothesized to underlie CTE pathogenesis (135). We have shown that mechanical stress in the form of extensional strain causes tau to aggregate in solution (71), and others have observed phosphorylated tauopathy induced by concussive or severe TBI in mouse models (79, 136–138). Though the example of familial FTD shows tau aggregation alone is sufficient to cause neurodegeneration, other disease-related amyloid proteins are known to interact with tau and promote its aggregation; interactions with amyloid-beta (Aβ) (139–141) or alpha-synuclein (α-syn) (142, 143) are best studied. Finally, tau aggregation has been triggered by inflammatory stimuli such as inflammasome activation in microglial cells (144, 145), though involvement of additional glial cell populations such as astrocytes or oligodendrocytes may also contribute (146, 147). Once aggregated, since the microtubule-binding region comprises the core of tau filaments, tau aggregation has been shown to be both a toxic gain-and loss-of-function (148–150), as the pathological assembly and physiological function of tau appear to be mutually exclusive.
New insight into the etiology and progression of tauopathies, and numerous other neurodegenerative proteinopathies, stems from the 1982 discovery by Prusiner that a protein alone is the etiological agent of transmissible spongiform encephalopathies such as Creutzfeldt-Jacob disease (CJD). He termed these proteins prions, for proteinaceous infectious particles (151), and in the years since this discovery numerous other proteins, with tau among them, have been shown to behave in a similar fashion to CJD prions at the cellular and biochemical level (128, 135, 152), with the notable exception being that no other disease-related amyloid protein has been shown to be infectious or transmissible between hosts as in CJD (153). In addition to the aforementioned conversion from a physiological, soluble form to a disease-associated, insoluble amyloid form, a hallmark of prions and “prion-like” proteins such as tau is that they amplify their pathological misfolded state through templated recruitment of additional soluble counterparts, triggering further misfolding when naïve monomers contact the misfolded “seed” (Figure 4) (154, 155). Numerous cell culture studies and both transgenic and wild-type animal models have demonstrated the ability of tau fibrils to induce this seed-driven templated amplification of further aggregation, recapitulating Prusiner’s protein-only hypothesis by the induction of NFT insoluble aggregates following cellular uptake of tau fibrils (156–158). Seeding of further aggregation can be inhibited by suppressing the expression of soluble tau, in which case tau aggregates can then be cleared by the autophagy-lysosome pathway and the ubiquitin-proteasome system, though these systems are unable to clear large aggregates (159). Intracellular tau inclusions are known to be dynamic, with fibrillar components undergoing frequent fragmentation and growth facilitating pathological propagation by additional seeding events (159). Fluorescence resonance energy transfer (FRET)-based methods of detecting tau seeds have shown that seeds precede development of NFTs and conventional phosphorylated tau histopathology (160, 161). There is considerable debate regarding which specific composition of tau multimer comprises the minimum seed-competent species, with data from tau mutant transgenic mice suggesting small filamentous species > 10 monomeric components (162), whereas other data demonstrating even tau monomer, derived from sonication of larger tau fibrils, encodes structural information to induce seeding (163).
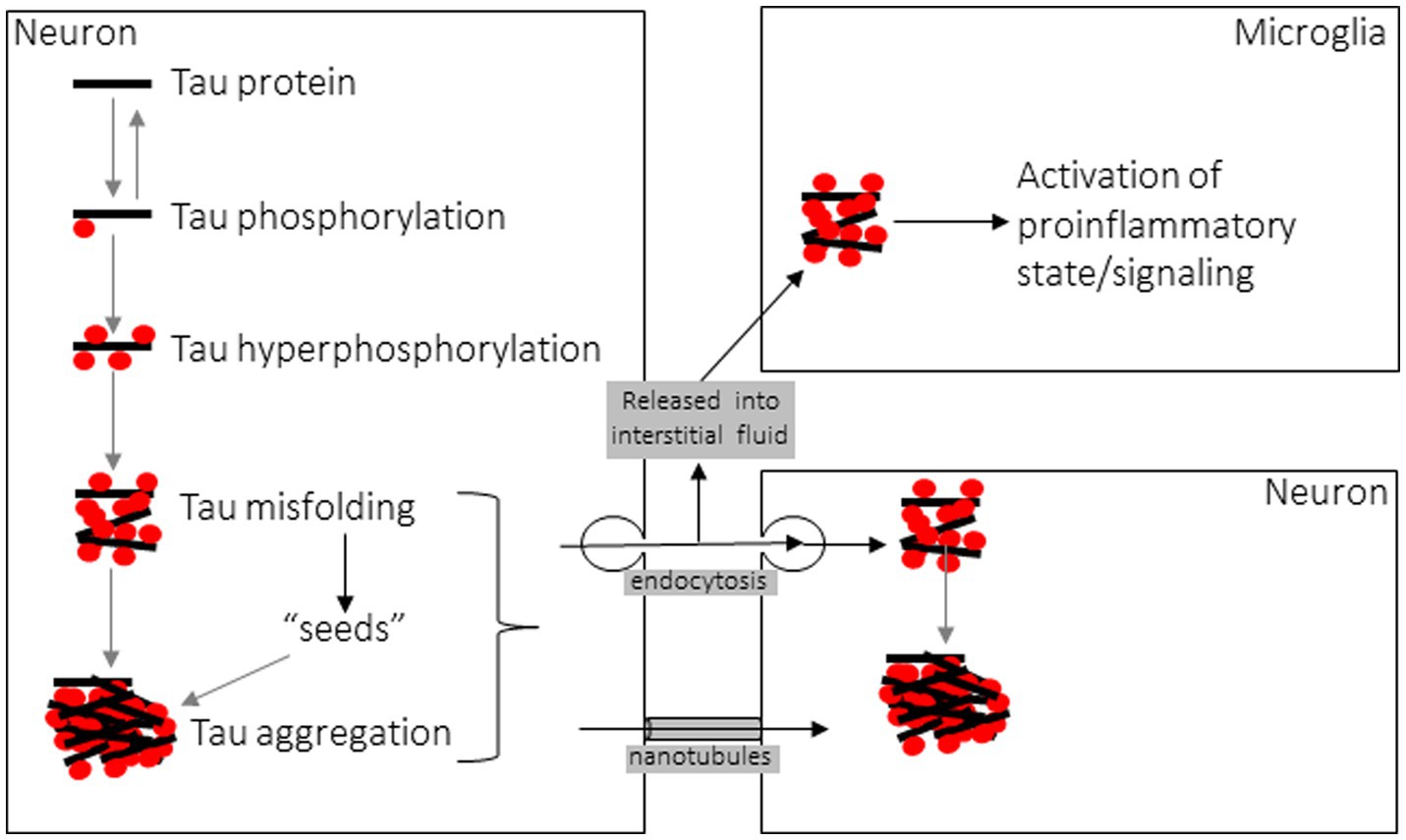
Figure 4. Diagram of Tau aggregation and propagation between cells.
Another hallmark of prions is structural heterogeneity, or misfolding into multiple self-replicating conformations that define unique “strains,” where each conformer produces distinct disease phenotypes that underlie the clinical diversity of prion diseases caused by aggregates of the same protein. Tau aggregation also exhibits this structural polymorphism, resulting in unique strain conformations specified by the templating process and influenced by numerous factors such as isoform composition and post-translational modifications (114, 164). Indeed, seed-competent tau monomers are capable of reconstituting multiple distinct strains owing to different stable monomeric conformations (165). When tau fibrils from different tauopathies are isolated from human brain, the structural polymorphism that defines unique strains has been identified using cryo-electron microscopy, with novel folds observed in AD, CTE, PiD, and CBD (166, 167). Strain conformation is consequential because it imparts unique properties to individual polymorphs. Tau strains exhibit varying levels of potency in their seeding of further aggregation, as strains purified from AD brain exhibited increased seeding potency compared with synthetic tau fibrils (168, 169). Furthermore, strain conformation imparts a unique neuroanatomical and cellular tropism resulting in distinct patterns of neurotoxicity in different tauopathies (170, 171). A specific strain conformation and its associated pathological phenotype of affected cell types and brain regions can be faithfully reproduced when tau strains are re-introduced into naïve cells or successively inoculated into mice (155, 172). These findings highlight how templated amplification of protein misfolding and strain-specific differences in clinicopathologic phenotype define tau as a prion-like amyloid protein, and have given rise to a deeper understanding of the mechanisms underpinning disease progression in tauopathies. While these prion-like mechanisms have been better studied in the context of AD and other primary tauopathies, the overlap with these mechanisms and CTE pathogenesis remains incompletely understood. While McKee and others have hypothesized a similar prion-like behavior of tau in CTE as in other tauopathies (135), a deeper mechanistic understanding is still needed to elucidate whether tau can adopt these prion-like roles following the complex environmental insult of TBI.
Propagation of tau between brain regions
Another prion-like feature of tau pathology is the transmission of aggregated tau from cell-to-cell and between brain regions. Braak described a tau staging system in AD with six stages that distinguish a progressive stereotypical distribution pattern for tau pathology that correlates well with disease severity (4, 173). This characteristic sequential progression of pathological tau in AD is thought to begin in the locus ceruleus and transentorhinal cortex and then spread spatiotemporally through olfactory bulb, hippocampus, and entorhinal regions, and finally neocortical regions such as basal temporal, temporal, insular, and frontal cortices. A similar 4-stage system has been used to describe the ordered and predictable progression of tau pathology in CTE, which co-occurs with axonal disruption and death (72). More recent positron emission tomography (PET) studies using tau-specific ligands in AD and CTE have supported the respective tau staging systems previously established by histological examination of post-mortem brain tissue (174, 175). Though differing tauopathies affect unique circuits, progression staging schemes have also been described for PSP (176), AGD (177), and PiD (178). These findings support the idea that the expansion of misfolded tau pathology through cell-to-cell propagation contributes to differing clinical phenotypes and may be a driver of clinical disease progression.
Intercellular propagation of assembled tau has been seen in numerous experimental models, where expressed cellular tau adopts characteristics of the pathological exogenous tau seeds and this protein misfolding cascade spreads to neighboring cells and interconnected brain regions. Various forms of aggregated tau have been used as seeds for intracerebral injection in animal models, including pre-formed fibrils (PFFs) derived from in vitro assembly of mutant or WT human or mouse tau, brain extracts from transgenic mutant P301S or P301L human tau overexpressing mice, or pathologic tau purified from brains of patients with AD, CBD, AGD, or PSP. Additionally, tau pathology has been initiated via regional overexpression in transgenic animals or transduction of specific brain regions with lentiviral vectors in non-transgenic animals (179, 180). In keeping with the behavior of distinct strains, when seeds derived from tauopathy patients are injected into brains of transgenic mutant or WT tau overexpressing mice or non-transgenic mice, they caused templated amplification of misfolding and cell-to-cell propagation of endogenous tau aggregates with a cell-type and spatiotemporal phenotype that recapitulated the hallmark disease lesions of the extract source (169, 171, 172). Cell-type specificity in tau aggregate transmission is perhaps best illustrated in the case of tau extracts derived from postmortem AD, PSP, and CBD brains injected into non-transgenic mice, where all extracts induced neuronal tau inclusions but only PSP and CBD tau strains were capable of inducing astroglial and oligodendroglial inclusions, a pattern which recapitulates authentic human neuropathology (170). Interestingly, and as further evidence that structural differences endow strains with a unique bioactivity, oligodendroglial tau aggregates induced by PSP or CBD tau strains can propagate to downstream cells along white-matter tracts even when neuronal tau is knocked down, whereas astrocytic tau aggregates do not have this ability (147). Within affected circuits, whether tau aggregates were injected or triggered by local overexpression, synaptic connections appear to enhance tau propagation (179, 180), although transmission can also occur via non-synaptic mechanisms such as along glymphatic or CSF pathways (135). Strain-specific differences in spreading are also observed, with various tau strains exhibiting different rates of propagation (181, 182). This prion-like intercellular spreading of misfolded tau represents a mechanism for the amplification and dissemination of tau pathology throughout the brain, and thus understanding the mechanisms underlying cellular uptake and release of tau aggregates are of utmost importance.
Propagation of tau between cells
In the intercellular propagation life cycle of fibrillar tau, many similarities to prions are again noted when examining mechanisms of cellular entry and egress (Figure 4). Fibrillar assemblies of aggregated tau are predominantly taken up into recipient cells via clathrin-mediated or fluid-phase bulk endocytosis (158, 183), occurring after interaction at the plasma membrane between tau aggregates and heparan sulfate proteoglycans (HSPGs) (184). Recent work has also established a role for low-density lipoprotein receptor-related protein 1 (LRP1) in facilitating tau uptake via endocytosis and subsequent propagation to downstream neurons (185). Expectedly, both aggregate conformation and size dictate entry via endocytosis, as low-molecular weight aggregates or short fibrils, but not monomers, long fibrils, or long filaments, were capable of cellular entry via this mechanism (186). Once contained within the endocytic vesicle, fibrillar tau assemblies are known to induce vesicle rupture as a mechanism of escape from the vesicular compartment to access the cytosol, where templated amplification of further tau aggregation can precede (187). Exogenous tau seeds come into direct contact with intracellular expressed tau as seen by FRET following endocytosis, triggering misfolding (154). In response to this damaging mechanism of cellular invasion, danger receptors such as galectins 3 and 8 detect damaged vesicles and activate autophagy by recruitment of cargo receptors to degrade the burden of amyloid proteins and vesicular debris (187). When seeds are taken up into microglial cells they activate the NLRP3 inflammasome, triggering pro-inflammatory caspase-1 activity and IL-1beta release as well as exacerbating intracellular tau seeding of further aggregation (145, 188). Considering that increasing the degradative burden on proteostatic pathways may predispose these signaling processes and trafficking events to become dysfunctional or overwhelmed, the intracellular consequences of tau aggregate cellular entry may promote further propagation by increasing cellular stress, since lysosomal or autophagic dysfunction are causes of cellular stress known to trigger tau secretion from affected cells (189, 190).
Regarding the process by which tau is released from cells, monomeric tau is constitutively secreted, even from healthy cells, into the interstitial fluid (ISF) and cerebrospinal fluid (CSF) for poorly understood reasons (191, 192), although neuronal activity and calcium influx stimulate this release and blocking pre-synaptic vesicle release inhibits it (193). Secretion of tau occurs through a non-classical pathway that is prevented by low temperature but not blocked by inhibitors of the conventional secretory pathway (194). Because of this secretory mechanism, the resulting extracellular tau is in a membrane-free form, illustrating how antibodies directed against tau can reduce insoluble tau aggregates and inhibit seeding and propagation (154). Tau is detected in CSF and the level correlates with the burden of hyperphosphorylated tau and NFT pathology (195), though ISF levels of monomeric tau are higher and reciprocally decrease as spontaneous or seed-induced aggregation consumes soluble tau into insoluble inclusions (191). Tau is also known to be released from affected cells in a membrane-enclosed form, either in ectosomes or microvesicles formed from outward budding of the plasma membrane or in exosomes released when endosome-derived multivesicular bodies that contain internal vesicles fuse with the plasma membrane (180, 190). Lastly, tau aggregates have been shown to be transmitted between cells via tunneling nanotubes (TNTs), transient filamentous-actin-containing membranous conduits between the cytoplasm of interconnected cells, in a manner similar to prions (196, 197). This mode of intercellular propagation does not require an extracellular phase. Interestingly, exogenous tau is also known to promote the formation of TNT connections that can then facilitate its transmission (196, 197). Understanding the molecular mechanisms involved in the cell-to-cell life cycle of fibrillar tau will allow future work to explore novel therapeutic interventions to counteract or prevent these pathologic processes.
Conclusion
Significant progress has been made to understand the role of tau in neurodegenerative proteinopathies, though tremendous opportunities remain to uncover unique molecular mechanisms that drive individual diseases, and how cellular, genetic, and environmental factors interface to produce distinct pathology in these conditions. Disease-modifying therapeutic approaches that leverage these discoveries would have the potential to benefit millions of patients in the future as our population ages. While the bulk of current research on tau is focused on toxicity questions such as elucidating the causes of de novo tau aggregation, as well as understanding mechanisms of seeding further aggregation, release and uptake in neuronal cells, studying the fibrillar structure of tau aggregates isolated from post-mortem brain may also allow insight into differences between individual tauopathies and facilitate improved diagnostics such fluid biomarkers or as tracer ligands for neuroimaging. Additional study is greatly needed to understand the influence of TBI on the development of tau aggregation, with consideration for the degree of TBI severity as well as if TBI occurs in a repetitive manner over a longer period of exposure. We are now beginning to appreciate the wide degree of structural heterogeneity that exists for fibrillar tau in distinct tauopathies, as well as the numerous cellular factors that may influence the adoption of unique misfolded states. This complexity highlights the need for rigorous investigation into strong risk factors such as TBI to determine how this environmental insult to the brain can predispose to chronic neurodegenerative sequelae. Although central questions still remain open, advances in research techniques and model systems along with an increasing focus on patients with a history of prior TBI will allow the research community to meet the challenge of developing future treatments to halt or even prevent tau-related neurodegeneration.
Author contributions
WPF contributed to writing the aggregation and propagation of tau section, and formulated the final draft. HH contributed to the writing of the original first draft. JWR contributed to the mechanical stretching & tau regulation section along with HPK and CCG contributed to the repeat TBI and clinical history of mild TBI and tau sections. MLP initiated this paper, wrote significant portions and designed the images. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
UCLA Brain Injury Research Center, UCLA Steve Tisch BrainSPORT Program, NS110757, NS104311. WPF was supported by NINDS 5R25NS065723.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Weingarten, MD, Lockwood, AH, Hwo, SY, and Kirschner, MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci. (1975) 72:1858–62. doi: 10.1073/pnas.72.5.1858
2. Grundke-Iqbal, I, Iqbal, K, Tung, YC, Quinlan, M, Wisniewski, HM, and Binder, LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci. (1986) 83:4913–7. doi: 10.1073/pnas.83.13.4913
3. Grundke-Iqbal, I, Iqbal, K, Quinlan, M, Tung, YC, Zaidi, MS, Wisniewski, HM, et al. A component of Alzheimer paired helical filaments. J Biol Chem. (1986) 261:6084–9. doi: 10.1016/s0021-9258(17)38495-8
4. Braak, H, and Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. (1991) 82:239–59. doi: 10.1007/bf00308809
5. Tapia-Rojas, C, Cabezas-Opazo, F, Deaton, CA, Vergara, EH, Johnson, GVW, and Quintanilla, RA. It’s all about tau. Prog Neurobiol. (2019) 175:76. doi: 10.1016/j.pneurobio.2018.12.005
6. Buée, L, Bussière, T, Buée-Scherrer, V, Delacourte, A, and Hof, PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders 11These authors contributed equally to this work. Brain Res Rev. (2000) 33:95–130. doi: 10.1016/s0165-0173(00)00019-9
7. Shin, R-W, Kitamoto, T, and Tateishi, J. Modified tau is present in younger nondemented persons: a study of subcortical nuclei in Alzheimer’s disease and progressive supranuclear palsy. Acta Neuropathol. (1991) 81:517–23. doi: 10.1007/bf00310132
8. Ittner, LM, and Götz, J. Amyloid-β and tau—a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci. (2011) 12:6. doi: 10.1038/nrn2967
9. Gamblin, TC, Chen, F, Zambrano, A, Abraha, A, Lagalwar, S, Guillozet, AL, et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc Natl Acad Sci. (2003) 100:10032–7. doi: 10.1073/pnas.1630428100
10. Reynolds, MR, Berry, RW, and Binder, LI. Site-specific nitration differentially influences τ assembly in vitro †. Biochemistry. (2005) 44:13997–4009. doi: 10.1021/bi051028w
11. King, ME, Kan, H-M, Baas, PW, Erisir, A, Glabe, CG, and Bloom, GS. Tau-dependent microtubule disassembly initiated by prefibrillar β-amyloid. J Cell Biol. (2006) 175:541–6. doi: 10.1083/jcb.200605187
12. Molina, L, Touchon, J, Herpé, M, Lefranc, D, Duplan, L, Cristol, JP, et al. Tau and apo E in CSF. Neuroreport. (1999) 10:3491–5. doi: 10.1097/00001756-199911260-00005
13. Chiaravalloti, A, Barbagallo, G, Ricci, M, Martorana, A, Ursini, F, Sannino, P, et al. Brain metabolic correlates of CSF tau protein in a large cohort of Alzheimer’s disease patients: A CSF and FDG PET study. Brain Res. (2018) 1678:116–22. doi: 10.1016/j.brainres.2017.10.016
14. Alquezar, C, Arya, S, and Kao, AW. Tau post-translational modifications: dynamic transformers of tau function, degradation, and aggregation. Front Neurol. (2021) 11:595532. doi: 10.3389/fneur.2020.595532
15. Cohen, TJ, Guo, JL, Hurtado, DE, Kwong, LK, Mills, IP, Trojanowski, JQ, et al. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. (2011) 2:252. doi: 10.1038/ncomms1255
16. Dickey, CA, Yue, M, Lin, W-L, Dickson, DW, Dunmore, JH, Lee, WC, et al. Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous Phospho-and Caspase-3-cleaved tau species. J Neurosci. (2006) 26:6985–96. doi: 10.1523/jneurosci.0746-06.2006
17. Dorval, V, and Fraser, PE. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J Biol Chem. (2006) 281:9924. doi: 10.1074/jbc.m510127200
18. Funk, KE, Thomas, SN, Schafer, KN, Cooper, GL, Liao, Z, Clark, DJ, et al. Lysine methylation is an endogenous post-translational modification of tau protein in human brain and a modulator of aggregation propensity. Biochem J. (2014) 462:77–88. doi: 10.1042/bj20140372
19. Liu, F, Zaidi, T, Iqbal, K, Grundke-Iqbal, I, Merkle, RK, and Gong, C-X. Role of glycosylation in hyperphosphorylation of tau in Alzheimer’s disease. FEBS Lett. (2002) 512:101–6. doi: 10.1016/s0014-5793(02)02228-7
20. Marcus, JN, and Schachter, J. Targeting post-translational modifications on tau as a therapeutic strategy for Alzheimer’s disease. J Neurogenet. (2011) 25:127–33. doi: 10.3109/01677063.2011.626471
21. Kuret, J, Chirita, CN, Congdon, EE, Kannanayakal, T, Li, G, Necula, M, et al. Pathways of tau fibrillization. Biochimica Et Biophysica Acta Bba-Mol Basis Dis. (2005) 1739:178. doi: 10.1016/j.bbadis.2004.06.016
22. Khatoon, S, Grundke-Iqbal, I, and Iqbal, K. Brain levels of microtubule-associated protein tau are elevated in Alzheimer’s disease: a radioimmuno-slot-blot assay for nanograms of the protein. J Neurochem. (1992) 59:753. doi: 10.1111/j.1471-4159.1992.tb09432.x
23. Papasozomenos, SC. The heat shock-induced hyperphosphorylation of tau is estrogen-independent and prevented by androgens: implications for Alzheimer disease. Proc Natl Acad Sci. (1997) 94:6617. doi: 10.1073/pnas.94.13.6612
24. Pooler, AM, Usardi, A, Evans, CJ, Philpott, KL, Noble, W, and Hanger, DP. Dynamic association of tau with neuronal membranes is regulated by phosphorylation. Neurobiol Aging. (2012) 33:431.e27 38. doi: 10.1016/j.neurobiolaging.2011.01.005
25. Zempel, H, Dennissen, FJA, Kumar, Y, Luedtke, J, Biernat, J, Mandelkow, E-M, et al. Axodendritic sorting and pathological missorting of tau are isoform-specific and determined by axon initial segment architecture. J Biol Chem. (2017) 292:12207. doi: 10.1074/jbc.m117.784702
26. Lim, J, Balastik, M, Lee, TH, Nakamura, K, Liou, Y-C, Sun, A, et al. Pin 1 has opposite effects on wild-type and P301L tau stability and tauopathy. J Clin Invest. (2008) 118:1877–89. doi: 10.1172/jci34308
27. Albayram, O, Angeli, P, Bernstein, E, Baxley, S, Gao, Z, Lu, KP, et al. Targeting prion-like Cis phosphorylated tau pathology in neurodegenerative diseases. J Alzheimers Dis Park. (2018) 8:443. doi: 10.4172/2161-0460.1000443
28. Nakamura, K, Greenwood, A, Binder, L, Bigio, EH, Denial, S, Nicholson, L, et al. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer’s disease. Cells. (2012) 149:232–44. doi: 10.1016/j.cell.2012.02.016
29. Galas, M-C, Dourlen, P, Bégard, S, Ando, K, Blum, D, Hamdane, M, et al. The Peptidylprolyl cis/trans-isomerase Pin1 modulates stress-induced Dephosphorylation of tau in neurons implication in a pathological mechanism related to Alzheimer disease. J Biol Chem. (2006) 281:19296–304. doi: 10.1074/jbc.m601849200
30. Sato, C, Barthélemy, NR, Mawuenyega, KG, Patterson, BW, Gordon, BA, Jockel-Balsarotti, J, et al. Tau kinetics in neurons and the human central nervous system. Neuron. (2018) 98:864. doi: 10.1016/j.neuron.2018.04.035
31. David, DC, Layfield, R, Serpell, L, Narain, Y, Goedert, M, and Spillantini, MG. Proteasomal degradation of tau protein. J Neurochem. (2002) 83:176–85. doi: 10.1046/j.1471-4159.2002.01137.x
32. Arendt, T. Neurodegeneration and plasticity. Int J Dev Neurosci. (2004) 22:514. doi: 10.1016/j.ijdevneu.2004.07.007
33. Stieler, JT, Bullmann, T, Kohl, F, Tøien, Ø, Brückner, MK, Härtig, W, et al. The physiological link between metabolic rate depression and tau phosphorylation in mammalian hibernation. PLoS One. (2011) 6:e14530. doi: 10.1371/journal.pone.0014530
34. Su, B, Wang, X, Drew, KL, Perry, G, Smith, MA, and Zhu, X. Physiological regulation of tau phosphorylation during hibernation. J Neurochem. (2008) 105:2108. doi: 10.1111/j.1471-4159.2008.05294.x
35. Melvin, RG, and Andrews, MT. Torpor induction in mammals: recent discoveries fueling new ideas. Trends Endocrinol Metab. (2009) 20:490–8. doi: 10.1016/j.tem.2009.09.005
36. Grieb, P, Gordon-Krajcer, W, Frontczak-Baniewicz, M, Walski, M, Ryba, MS, Kryczka, T, et al. 2-deoxyglucose induces beta-APP overexpression, tau hyperphosphorylation and expansion of the trans-part of the Golgi complex in rat cerebral cortex. Acta Neurobiol Exp. (2004) 64:491–502.
37. Yanagisawa, M, Planel, E, Ishiguro, K, and Fujita, SC. Starvation induces tau hyperphosphorylation in mouse brain: implications for Alzheimer’s disease. FEBS Lett. (1999) 461:329–33. doi: 10.1016/s0014-5793(99)01480-5
38. Planel, E, Miyasaka, T, Launey, T, Chui, D-H, Tanemura, K, Sato, S, et al. Alterations in glucose metabolism induce hypothermia leading to tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: implications for Alzheimer’s disease. J Neurosci. (2004) 24:2401–11. doi: 10.1523/jneurosci.5561-03.2004
39. Muñoz-Mayorga, D, Guerra-Araiza, C, Torner, L, and Morales, T. Tau phosphorylation in female neurodegeneration: role of estrogens, progesterone, and prolactin. Front Endocrinol. (2018) 9:133. doi: 10.3389/fendo.2018.00133
40. González-Arenas, A, Piña-Medina, AG, González-Flores, O, Gómora-Arrati, P, Carrillo-Martínez, GE, Balandrán-Ruíz, MA, et al. Expression pattern of tau in the rat brain during pregnancy and the beginning of lactation. Brain Res Bull. (2012) 89:108–14. doi: 10.1016/j.brainresbull.2012.07.011
41. Goodenough, S, Schleusner, D, Pietrzik, C, Skutella, T, and Behl, C. Glycogen synthase kinase 3β links neuroprotection by 17β-estradiol to key Alzheimer processes. Neuroscience. (2005) 132:581–9. doi: 10.1016/j.neuroscience.2004.12.029
42. Cavallini, A, Brewerton, S, Bell, A, Sargent, S, Glover, S, Hardy, C, et al. An unbiased approach to identifying tau kinases that phosphorylate tau at sites associated with Alzheimer disease. J Biol Chem. (2013) 288:23331–47. doi: 10.1074/jbc.m113.463984
43. Kimura, T, Ishiguro, K, and Hisanaga, S-I. Physiological and pathological phosphorylation of tau by Cdk 5. Front Mol Neurosci. (2014) 7:65. doi: 10.3389/fnmol.2014.00065
44. Sun, Y, Xiao, Q, Luo, C, Zhao, Y, Pu, D, Zhao, K, et al. High-glucose induces tau hyperphosphorylation through activation of TLR9-P38MAPK pathway. Exp Cell Res. (2017) 359:312–8. doi: 10.1016/j.yexcr.2017.07.032
45. Sun, J, Zhang, X, Wang, C, Teng, Z, and Li, Y. Curcumin decreases hyperphosphorylation of tau by Down-regulating Caveolin-1/GSK-3β in N2a/APP695swe cells and APP/PS1 double transgenic Alzheimer’s disease mice. Am J Chin Med. (2017) 45:1667–82. doi: 10.1142/s0192415x17500902
46. Caccamo, A, Magrì, A, Medina, DX, Wisely, EV, López-Aranda, MF, Silva, AJ, et al. mTOR regulates tau phosphorylation and degradation: implications for Alzheimer’s disease and other tauopathies. Aging Cell. (2013) 12:370–80. doi: 10.1111/acel.12057
47. Rissman, RA, Lee, K-F, Vale, W, and Sawchenko, PE. Corticotropin-releasing factor receptors differentially regulate stress-induced tau phosphorylation. J Neurosci. (2007) 27:6552–62. doi: 10.1523/jneurosci.5173-06.2007
48. Mondragón-Rodríguez, S, Perry, G, Zhu, X, Moreira, PI, Acevedo-Aquino, MC, and Williams, S. Phosphorylation of tau protein as the link between oxidative stress, mitochondrial dysfunction, and connectivity failure: implications for Alzheimer’s disease. Oxidative Med Cell Longev. (2013) 2013:1–6. doi: 10.1155/2013/940603
49. Xu, H, Chen, X, Wang, J, Yang, T, Liu, N, Cheng, J, et al. Involvement of insulin signalling pathway in methamphetamine-induced hyperphosphorylation of tau. Toxicology. (2018) 408:88–94. doi: 10.1016/j.tox.2018.07.002
50. Liu, S-J, Fang, Z-Y, Yang, Y, Deng, H-M, and Wang, J-Z. Alzheimer-like phosphorylation of tau and neurofilament induced by cocaine in vivo. Acta Pharmacol Sin. (2003) 24:512–8.
51. Kovacs, GG, Horvath, MC, Majtenyi, K, Lutz, MI, Hurd, YL, and Keller, E. Heroin abuse exaggerates age-related deposition of hyperphosphorylated tau and p 62-positive inclusions. Neurobiol Aging. (2015) 36:3107. doi: 10.1016/j.neurobiolaging.2015.07.018
52. Metcalfe, MJ, and Figueiredo-Pereira, ME. Relationship between tau pathology and Neuroinflammation in Alzheimer’s disease. Mt Sinai J Medicine J Transl Personalized Med. (2010) 77:50–8. doi: 10.1002/msj.20163
53. Ploia, C, Antoniou, X, Sclip, A, Grande, V, Cardinetti, D, Colombo, A, et al. JNK plays a key role in tau hyperphosphorylation in Alzheimer’s disease models. J Alzheimers Dis. (2011) 26:315–29. doi: 10.3233/jad-2011-110320
54. Tyagi, E, Fiorelli, T, Norden, M, and Padmanabhan, J. Alpha 1-Antichymotrypsin, an inflammatory protein overexpressed in the brains of patients with Alzheimer’s disease, induces tau hyperphosphorylation through c-Jun N-terminal kinase activation. Int J Alzheimers Dis. (2013) 2013:1–11. doi: 10.1155/2013/606083
55. Collins-Praino, LE, and Corrigan, F. Does neuroinflammation drive the relationship between tau hyperphosphorylation and dementia development following traumatic brain injury? Brain Behav Immun. (2017) 60:369–82. doi: 10.1016/j.bbi.2016.09.027
56. Giza, CC, and Hovda, DA. The new neurometabolic cascade of concussion. Neurosurgery. (2014) 75:S24 33. doi: 10.1227/neu.0000000000000505
57. Osteen, CL, Moore, AH, Prins, ML, and Hovda, DA. Age-dependency of 45calcium accumulation following lateral fluid percussion: acute and delayed patterns. J Neurotraum. (2001) 18:162. doi: 10.1089/08977150150502587
58. Fineman, I, Hovda, DA, Smith, M, Yoshino, A, and Becker, DP. Concussive brain injury is associated with a prolonged accumulation of calcium: a45Ca autoradiographic study. Brain Res. (1993) 624:94–102. doi: 10.1016/0006-8993(93)90064-t
59. Hartigan, JA, and Johnson, GVW. Transient increases in intracellular calcium result in prolonged site-selective increases in tau phosphorylation through a glycogen synthase kinase 3β-dependent pathway*. J Biol Chem. (1999) 274:21395–401. doi: 10.1074/jbc.274.30.21395
60. Mattson, MP, Engle, MG, and Rychlik, B. Effects of elevated intracellular calcium levels on the cytoskeleton and tau in cultured human cortical neurons. Mol Chem Neuropathol. (1991) 15:117–42. doi: 10.1007/bf03159951
61. Yousuf, MA, Tan, C, Torres-Altoro, MI, Lu, F-M, Plautz, E, Zhang, S, et al. Involvement of aberrant cyclin-dependent kinase 5/p 25 activity in experimental traumatic brain injury. J Neurochem. (2016) 138:317–27. doi: 10.1111/jnc.13620
62. Prins, ML, Hales, A, Reger, M, Giza, CC, and Hovda, DA. Repeat traumatic brain injury in the juvenile rat is associated with increased axonal injury and cognitive impairments. Dev Neurosci. (2010) 32:518. doi: 10.1159/000316800
63. Xu, X, Cowan, M, Beraldo, F, Schranz, A, McCunn, P, Geremia, N, et al. Repetitive mild traumatic brain injury in mice triggers a slowly developing cascade of long-term and persistent behavioral deficits and pathological changes. Acta Neuropathologica Commun. (2021) 9:60. doi: 10.1186/s40478-021-01161-2
64. Bazarian, JJ, Zhong, J, Blyth, B, Zhu, T, Kavcic, V, and Peterson, D. Diffusion tensor imaging detects clinically important axonal damage after mild traumatic brain injury: a pilot study. J Neurotraum. (2007) 24:1459. doi: 10.1089/neu.2007.0241
65. Tan, XL, Wright, DK, Liu, S, Hovens, C, O’Brien, TJ, and Shultz, SR. Sodium selenate, a protein phosphatase 2A activator, mitigates hyperphosphorylated tau and improves repeated mild traumatic brain injury outcomes. Neuropharmacology. (2016) 108:382–93. doi: 10.1016/j.neuropharm.2016.05.001
66. Tran, HT, Sanchez, L, and Brody, DL. Inhibition of JNK by a peptide inhibitor reduces traumatic brain injury–induced tauopathy in transgenic mice. J Neuropathol Exp Neurol. (2012) 71:116–29. doi: 10.1097/nen.0b013e3182456aed
67. Sun, Y, Bai, L, Niu, X, Wang, Z, Yin, B, Bai, G, et al. Elevated serum levels of inflammation-related cytokines in mild traumatic brain injury are associated with cognitive performance. Front Neurol. (2019) 10:1120. doi: 10.3389/fneur.2019.01120
68. Saber, M, Giordano, KR, Hur, Y, Ortiz, JB, Morrison, H, Godbout, JP, et al. Acute peripheral inflammation and post-traumatic sleep differ between sexes after experimental diffuse brain injury. Eur J Neurosci. (2020) 52:2791–2814. doi: 10.1111/ejn.14611
69. Yang, SH, Gustafson, J, Gangidine, M, Stepien, D, Schuster, R, Pritts, TA, et al. A murine model of mild traumatic brain injury exhibiting cognitive and motor deficits. J Surg Res. (2013) 184:988. doi: 10.1016/j.jss.2013.03.075
70. Madathil, SK, Wilfred, BS, Urankar, SE, Yang, W, Leung, LY, Gilsdorf, JS, et al. Early microglial activation following closed-head concussive injury is dominated by pro-inflammatory M-1 type. Front Neurol. (2018) 9:964. doi: 10.3389/fneur.2018.00964
71. Hosseini, H, Rangchian, A, Prins, M, Giza, C, Ruberti, J, and Kavehpour, HP. Probing flow-induced biomolecular interactions with Micro-extensional rheology: tau protein aggregation. J Biomech Eng. (2020) 142:034501. doi: 10.1115/1.4046330
72. Bieniek, KF, Cairns, NJ, Crary, JF, Dickson, DW, Folkerth, RD, Keene, CD, et al. The second NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. J Neuropathol Exp Neurol. (2021) 80:210–9. doi: 10.1093/jnen/nlab001
73. Katz, DI, Bernick, C, Dodick, DW, Mez, J, Mariani, ML, Adler, CH, et al. National Institute of Neurological Disorders and Stroke consensus diagnostic criteria for traumatic encephalopathy syndrome. Neurology. (2021) 96:848–63. doi: 10.1212/wnl.0000000000011850
74. Asken, BM, Sullan, MJ, DeKosky, ST, Jaffee, MS, and Bauer, RM. Research gaps and controversies in chronic traumatic encephalopathy: A review. JAMA Neurol. (2017) 74:1255. doi: 10.1001/jamaneurol.2017.2396
75. Leuzy, A, Chiotis, K, Lemoine, L, Gillberg, P-G, Almkvist, O, Rodriguez-Vieitez, E, et al. Tau PET imaging in neurodegenerative tauopathies—still a challenge. Mol Psychiatry. (2019) 24:1112–34. doi: 10.1038/s41380-018-0342-8
76. Omalu, BI, DeKosky, ST, Hamilton, RL, Minster, RL, Kamboh, MI, Shakir, AM, et al. Chronic traumatic encephalopathy in a national football league player: part II. Neurosurgery. (2006) 59:1086. doi: 10.1227/01.neu.0000245601.69451.27
77. McKee, AC, Daneshvar, DH, Alvarez, VE, and Stein, TD. The neuropathology of sport. Acta Neuropathol. (2013) 127:29–51. doi: 10.1007/s00401-013-1230-6
78. Lee, EB, Kinch, K, Johnson, VE, Trojanowski, JQ, Smith, DH, and Stewart, W. Chronic traumatic encephalopathy is a common co-morbidity, but less frequent primary dementia in former soccer and rugby players. Acta Neuropathol. (2019) 138:389–99. doi: 10.1007/s00401-019-02030-y
79. Goldstein, LE, Fisher, AM, Tagge, CA, Zhang, XL, Velisek, L, Sullivan, JA, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast Neurotrauma mouse model. Sci Transl Med. (2012) 4:134ra60. doi: 10.1126/scitranslmed.3003716
80. Tripathy, A, Shade, A, Erskine, B, Bailey, K, Grande, A, de Long, JJ, et al. No evidence of increased chronic traumatic encephalopathy pathology or neurodegenerative proteinopathy in former military service members: a preliminary study. J Alzheimers Dis. (2019) 67:1277–89. doi: 10.3233/jad-181039
81. Puvenna, V, Engeler, M, Banjara, M, Brennan, C, Schreiber, P, Dadas, A, et al. Is phosphorylated tau unique to chronic traumatic encephalopathy? Phosphorylated tau in epileptic brain and chronic traumatic encephalopathy. Brain Res. (2016) 1630:240. doi: 10.1016/j.brainres.2015.11.007
82. Noy, S, Krawitz, S, and Bigio, MRD. Chronic traumatic encephalopathy-like abnormalities in a routine neuropathology service. J Neuropathol Exp Neurol. (2016) 75:1154. doi: 10.1093/jnen/nlw092
83. Gao, F, Almeida, S, and Lopez-Gonzalez, R. Dysregulated molecular pathways in amyotrophic lateral sclerosis–frontotemporal dementia spectrum disorder. EMBO J. (2017) 36:2931–50. doi: 10.15252/embj.201797568
84. Koga, S, Sekiya, H, Kondru, N, Ross, OA, and Dickson, DW. Neuropathology and molecular diagnosis of Synucleinopathies. Mol Neurodegener. (2021) 16:83. doi: 10.1186/s13024-021-00501-z
85. Ling, H, Hardy, J, and Zetterberg, H. Neurological consequences of traumatic brain injuries in sports. Mol Cell Neurosci. (2015) 66:114–22. doi: 10.1016/j.mcn.2015.03.012
86. Iverson, GL, Gardner, AJ, Shultz, SR, Solomon, GS, McCrory, P, Zafonte, R, et al. Chronic traumatic encephalopathy neuropathology might not be inexorably progressive or unique to repetitive neurotrauma. Brain. (2019) 142:3672–93. doi: 10.1093/brain/awz286
87. Asken, BM, Brooke, ZS, Stevens, TC, Silvestri, PG, Graham, MJ, Jaffee, MS, et al. Drill-specific head impacts in collegiate football practice: implications for reducing “friendly fire” exposure. Ann Biomed Eng. (2019) 47:2094–108. doi: 10.1007/s10439-018-2088-5
88. Magnoni, S, Esparza, TJ, Conte, V, Carbonara, M, Carrabba, G, Holtzman, DM, et al. Tau elevations in the brain extracellular space correlate with reduced amyloid-β levels and predict adverse clinical outcomes after severe traumatic brain injury. Brain. (2012) 135:1268–80. doi: 10.1093/brain/awr286
89. Shahim, P, Tegner, Y, Gustafsson, B, Gren, M, Ärlig, J, Olsson, M, et al. Neurochemical aftermath of repetitive mild traumatic brain injury. JAMA Neurol. (2016) 73:1308. doi: 10.1001/jamaneurol.2016.2038
90. Öst, M, Nylén, K, Csajbok, L, Öhrfelt, AO, Tullberg, M, Wikkelsö, C, et al. Initial CSF total tau correlates with 1-year outcome in patients with traumatic brain injury. Neurology. (2006) 67:1600–4. doi: 10.1212/01.wnl.0000242732.06714.0f
91. Bagnato, S, Andriolo, M, Boccagni, C, Sant’Angelo, A, ME, DI, and Galardi, G. Dissociation of cerebrospinal fluid amyloid-β and tau levels in patients with prolonged posttraumatic disorders of consciousness. Brain Inj. (2018) 32:1–5. doi: 10.1080/02699052.2018.1479042
92. Zetterberg, H, Smith, DH, and Blennow, K. Biomarkers of mild traumatic brain injury in cerebrospinal fluid and blood. Nat Rev Neurol. (2013) 9:201–10. doi: 10.1038/nrneurol.2013.9
93. Zetterberg, H, Pedersen, M, Lind, K, Svensson, M, Rolstad, S, Eckerström, C, et al. Intra-individual stability of CSF biomarkers for Alzheimer’s disease over two years. J Alzheimers Dis. (2007) 12:255–60. doi: 10.3233/jad-2007-12307
94. Guzel, A, Karasalihoglu, S, Aylanç, H, Temizöz, O, and Hiçdönmez, T. Validity of serum tau protein levels in pediatric patients with minor head trauma. Am J Emerg Med. (2010) 28:399–403. doi: 10.1016/j.ajem.2008.12.025
95. Bazarian, JJ, Zemlan, FP, Mookerjee, S, and Stigbrand, T. Serum S-100B and cleaved-tau are poor predictors of long-term outcome after mild traumatic brain injury. Brain Inj. (2006) 20:765. doi: 10.1080/02699050500488207
96. Ma, M, Lindsell, CJ, Rosenberry, CM, Shaw, GJ, and Zemlan, FP. Serum cleaved tau does not predict postconcussion syndrome after mild traumatic brain injury. Am J Emerg Med. (2008) 26:768. doi: 10.1016/j.ajem.2007.10.029
97. Hossain, I, Mohammadian, M, Takala, RSK, Tenovuo, O, Gil, LA, and Frantzén, J, Gils M van, Hutchinson, PJ, Katila, AJ, Maanpää, H-R, et al. Admission levels of Total tau and β-amyloid isoforms 1–40 and 1–42 in predicting the outcome of mild traumatic brain injury. Front Neurol (2020) 11:325. doi: 10.3389/fneur.2020.00325
98. Edwards, KA, Greer, K, Leete, J, Lai, C, Devoto, C, Qu, B-X, et al. Neuronally-derived tau is increased in experienced breachers and is associated with neurobehavioral symptoms. Sci Rep. (2021) 11:19527. doi: 10.1038/s41598-021-97913-0
99. Pattinson, CL, Gill, JM, Lippa, SM, Brickell, TA, French, LM, and Lange, RT. Concurrent mild traumatic brain injury and posttraumatic stress disorder is associated with elevated tau concentrations in peripheral blood plasma. J Trauma Stress. (2019) 32:546–54. doi: 10.1002/jts.22418
100. Small, GW, Kepe, V, Siddarth, P, Ercoli, LM, Merrill, DA, Donoghue, N, et al. PET scanning of brain tau in retired National Football League Players: preliminary findings. Am J Geriatric Psychiatry. (2013) 21:138–44. doi: 10.1016/j.jagp.2012.11.019
101. Mitsis, EM, Riggio, S, Kostakoglu, L, Dickstein, DL, Machac, J, Delman, B, et al. Tauopathy PET and amyloid PET in the diagnosis of chronic traumatic encephalopathies: studies of a retired NFL player and of a man with FTD and a severe head injury. Transl Psychiatry. (2014) 4:e441. doi: 10.1038/tp.2014.91
102. Stern, RA, Adler, CH, Chen, K, Navitsky, M, Luo, J, Dodick, DW, et al. Tau positron-emission tomography in former National Football League Players. New Engl J Med. (2019) 380:NEJMoa1900757. doi: 10.1056/nejmoa1900757
103. Omalu, B, Small, GW, Bailes, J, Ercoli, LM, Merrill, DA, Wong, K-P, et al. Postmortem autopsy-confirmation of Antemortem [F-18] FDDNP-PET scans in a football player with chronic traumatic encephalopathy. Neurosurgery. (2017) 82:237–46. doi: 10.1093/neuros/nyx536
104. Alosco, ML, Su, Y, Stein, TD, Protas, H, Cherry, JD, Adler, CH, et al. Associations between near end-of-life flortaucipir PET and postmortem CTE-related tau neuropathology in six former American football players. Eur J Nucl Med Mol Imaging. (2023) 50:435–52. doi: 10.1007/s00259-022-05963-x
105. Marquié, M, Agüero, C, Amaral, AC, Villarejo-Galende, A, Ramanan, P, Chong, MST, et al. [18F]-AV-1451 binding profile in chronic traumatic encephalopathy: a postmortem case series. Acta Neuropathologica Commun. (2019) 7:164. doi: 10.1186/s40478-019-0808-1
106. Montenigro, PH, Baugh, CM, Daneshvar, DH, Mez, J, Budson, AE, Au, R, et al. Clinical subtypes of chronic traumatic encephalopathy: literature review and proposed research diagnostic criteria for traumatic encephalopathy syndrome. Alzheimers Res Ther. (2014) 6:68. doi: 10.1186/s13195-014-0068-z
107. Prins, ML, and Hovda, DA. Traumatic brain injury in the developing rat: effects of maturation on Morris water maze acquisition. J Neurotraum. (1998) 15:811. doi: 10.1089/neu.1998.15.799
108. Prins, ML, and Hovda, DA. The effects of age and ketogenic diet on local cerebral metabolic rates of glucose after controlled cortical impact injury in rats. J Neurotraum. (2009) 26:1093. doi: 10.1089/neu.2008-0769
109. Serpa, RO, Ferguson, L, Larson, C, Bailard, J, Cooke, S, Greco, T, et al. Pathophysiology of pediatric traumatic brain injury. Front Neurol. (2021) 12:696510. doi: 10.3389/fneur.2021.696510
110. Gao, H, Han, Z, Bai, R, Huang, S, Ge, X, Chen, F, et al. The accumulation of brain injury leads to severe neuropathological and neurobehavioral changes after repetitive mild traumatic brain injury. Brain Res. (2017) 1657:1–8. doi: 10.1016/j.brainres.2016.11.028
111. Wright, DK, O’Brien, TJ, Shultz, SR, and Mychasiuk, R. Sex matters: repetitive mild traumatic brain injury in adolescent rats. Ann Clin Transl Neur. (2017) 4:654. doi: 10.1002/acn3.441
112. Mouzon, B, Bachmeier, C, Ojo, J, Acker, C, Ferguson, S, Crynen, G, et al. Chronic White matter degeneration, but no tau pathology at one-year post-repetitive mild traumatic brain injury in a tau transgenic model. J Neurotraum. (2019) 36:576–588. doi: 10.1089/neu.2018.5720
113. Laurer, HL, Bareyre, FM, Lee, VM, Trojanowski, JQ, Longhi, L, Hoover, R, et al. Mild head injury increasing the brain’s vulnerability to a second concussive impact. J Neurosurg. (2001) 95:870. doi: 10.3171/jns.2001.95.5.0859
114. Hoshino, S, Tamaoka, A, Takahashi, M, Kobayashi, S, Furukawa, T, Oaki, Y, et al. Emergence of immunoreactivities for phosphorylated tau and amyloid-β protein in chronic stage of fluid percussion injury in rat brain. Neuroreport. (1998) 9:1879–83. doi: 10.1097/00001756-199806010-00039
115. Rubenstein, R, Sharma, DR, Chang, B, Oumata, N, Cam, M, Vaucelle, L, et al. Novel mouse Tauopathy model for repetitive mild traumatic brain injury: evaluation of Long-term effects on cognition and biomarker levels after therapeutic inhibition of tau phosphorylation. Front Neurol. (2019) 10:124. doi: 10.3389/fneur.2019.00124
116. Tadepalli, SA, Bali, ZK, Bruszt, N, Nagy, LV, Amrein, K, Fazekas, B, et al. Long-term cognitive impairment without diffuse axonal injury following repetitive mild traumatic brain injury in rats. Behav Brain Res. (2020) 378:112268. doi: 10.1016/j.bbr.2019.112268
117. Zhang, H, Zhang, Z, Wang, Z, Zhen, Y, Yu, J, and Song, H. Research on the changes in balance motion behavior and learning, as well as memory abilities of rats with multiple cerebral concussion-induced chronic traumatic encephalopathy and the underlying mechanism. Exp Ther Med. (2018) 16:2295–302. doi: 10.3892/etm.2018.6474
118. Kane, MJ, Angoa-Pérez, M, Briggs, DI, Viano, DC, Kreipke, CW, and Kuhn, DM. A mouse model of human repetitive mild traumatic brain injury. J Neurosci Meth. (2012) 203:49. doi: 10.1016/j.jneumeth.2011.09.003
119. Haar, CV, Martens, KM, Bashir, A, McInnes, KA, Cheng, WH, Cheung, H, et al. Repetitive closed-head impact model of engineered rotational acceleration (CHIMERA) injury in rats increases impulsivity, decreases dopaminergic innervation in the olfactory tubercle and generates white matter inflammation, tau phosphorylation and degeneration. Exp Neurol. (2019) 317:99. doi: 10.1016/j.expneurol.2019.02.012
120. Luo, J, Nguyen, A, Villeda, S, Zhang, H, Ding, Z, Lindsey, D, et al. Long-term cognitive impairments and pathological alterations in a mouse model of repetitive mild traumatic brain injury. Front Neurol. (2014) 5:12. doi: 10.3389/fneur.2014.00012
121. Ojo, JO, Mouzon, B, Algamal, M, Leary, P, Lynch, C, Abdullah, L, et al. Chronic Repetitive Mild Traumatic Brain Injury Results in Reduced Cerebral Blood Flow, Axonal Injury, Gliosis, and Increased T-Tau and Tau Oligomers. J Neuropathol Exp Neurol. (2016) 75:636–55. doi: 10.1093/jnen/nlw035
122. McAteer, KM, Corrigan, F, Thornton, E, Turner, RJ, and Vink, R. Short and Long term behavioral and pathological changes in a novel rodent model of repetitive mild traumatic brain injury. PLoS One. (2016) 11:e0160220. doi: 10.1371/journal.pone.0160220
123. Hawkins, BE, Krishnamurthy, S, Castillo-Carranza, DL, Sengupta, U, Prough, DS, Jackson, GR, et al. Rapid accumulation of endogenous tau oligomers in a rat model of traumatic brain injury: possible link between traumatic brain injury and sporadic tauopathies. J Biol Chem. (2013) 288:17050. doi: 10.1074/jbc.m113.472746
124. Ojo, J-O, Mouzon, B, Greenberg, MB, Bachmeier, C, Mullan, M, and Crawford, F. Repetitive mild traumatic brain injury augments tau pathology and glial activation in aged hTau mice. J Neuropathol Exp Neurol. (2013) 72:151. doi: 10.1097/nen.0b013e3182814cdf
125. Yoshiyama, Y, Uryu, K, Higuchi, M, Longhi, L, Hoover, R, Fujimoto, S, et al. Enhanced neurofibrillary tangle formation, cerebral atrophy, and cognitive deficits induced by repetitive mild brain injury in a transgenic tauopathy mouse model. J Neurotraum. (2005) 22:1141. doi: 10.1089/neu.2005.22.1134
126. Goedert, M, Wischik, CM, Crowther, RA, Walker, JE, and Klug, A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc National Acad Sci. (1988) 85:4051–5. doi: 10.1073/pnas.85.11.4051
127. Lee, VM-Y, Balin, BJ, and Trojanowski, JQ. A68: a major subunit of paired helical filaments and Derivatized forms of Normal tau. Science. (1991) 251:675–8. doi: 10.1126/science.1899488
128. Uemura, N, Uemura, MT, Luk, KC, Lee, VM-Y, and Trojanowski, JQ. Cell-to-cell transmission of tau and α-Synuclein. Trends Mol Med. (2020) 26:936–52. doi: 10.1016/j.molmed.2020.03.012
129. Petraglia, AL, Plog, BA, Dayawansa, S, Dashnaw, ML, Czerniecka, K, Walker, CT, et al. The pathophysiology underlying repetitive mild traumatic brain injury in a novel mouse model of chronic traumatic encephalopathy. Surg Neurol Int. (2014) 5:184.
130. Ojo, JO, Leary, P, Lungmus, C, Algamal, M, Mouzon, B, Bachmeier, C, et al. Subchronic Pathobiological Response Following Chronic Repetitive Mild Traumatic Brain Injury in an Aged Preclinical Model of Amyloid Pathogenesis. J Neuropathol Exp Neurol. (2018) 77:1144–1162. doi: 10.1093/jnen/nly101
131. Cowan, CM, Shepherd, D, and Mudher, A. Insights from Drosophila models of Alzheimer’s disease. Biochem Soc T. (2010) 38:988–92. doi: 10.1042/bst0380988
132. Hong, M, Zhukareva, V, Vogelsberg-Ragaglia, V, Wszolek, Z, Reed, L, Miller, BI, et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. (1998) 282:1914–7. doi: 10.1126/science.282.5395.1914
133. Ambadipudi, S, Biernat, J, Riedel, D, Mandelkow, E, and Zweckstetter, M. Liquid–liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein tau. Nat Commun. (2017) 8:275. doi: 10.1038/s41467-017-00480-0
134. Wegmann, S, Eftekharzadeh, B, Tepper, K, Zoltowska, KM, Bennett, RE, Dujardin, S, et al. Tau protein liquid–liquid phase separation can initiate tau aggregation. EMBO J. (2018) 37:e98049. doi: 10.15252/embj.201798049
135. Kriegel, J, Papadopoulos, Z, and McKee, AC. Chronic traumatic encephalopathy: is latency in symptom onset explained by tau propagation? Csh Perspect Med. (2018) 8:a024059. doi: 10.1101/cshperspect.a024059
136. Zanier, ER, Bertani, I, Sammali, E, Pischiutta, F, Chiaravalloti, MA, Vegliante, G, et al. Induction of a transmissible tau pathology by traumatic brain injury. Brain. (2018) 141:2699. doi: 10.1093/brain/awy193
137. Tran, HT, Sanchez, L, Esparza, TJ, and Brody, DL. Distinct temporal and anatomical distributions of amyloid-β and tau abnormalities following controlled cortical impact in transgenic mice. PLoS One. (2011) 6:e25475. doi: 10.1371/journal.pone.0025475
138. Kondo, A, Shahpasand, K, Mannix, R, Qiu, J, Moncaster, J, Chen, C-H, et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature. (2015) 523:431–6. doi: 10.1038/nature14658
139. Bennett, RE, DeVos, SL, Dujardin, S, Corjuc, B, Gor, R, Gonzalez, J, et al. Enhanced tau aggregation in the presence of amyloid β. Am J Pathol. (2017) 187:1601–12. doi: 10.1016/j.ajpath.2017.03.011
140. Gomes, LA, Hipp, SA, Upadhaya, AR, Balakrishnan, K, Ospitalieri, S, Koper, MJ, et al. Aβ-induced acceleration of Alzheimer-related τ-pathology spreading and its association with prion protein. Acta Neuropathol. (2019) 138:913–41. doi: 10.1007/s00401-019-02053-5
141. Vasconcelos, B, Stancu, I-C, Buist, A, Bird, M, Wang, P, Vanoosthuyse, A, et al. Heterotypic seeding of tau fibrillization by pre-aggregated Abeta provides potent seeds for prion-like seeding and propagation of tau-pathology in vivo. Acta Neuropathol. (2016) 131:549–69. doi: 10.1007/s00401-015-1525-x
142. Clinton, LK, Blurton-Jones, M, Myczek, K, Trojanowski, JQ, and LaFerla, FM. Synergistic interactions between A, tau, and -Synuclein: acceleration of neuropathology and cognitive decline. J Neurosci. (2010) 30:7281–9. doi: 10.1523/jneurosci.0490-10.2010
143. Oikawa, T, Nonaka, T, Terada, M, Tamaoka, A, Hisanaga, S, and Hasegawa, M. α-Synuclein fibrils exhibit gain of toxic function, promoting tau aggregation and inhibiting Microtubule assembly*. J Biol Chem. (2016) 291:15046–56. doi: 10.1074/jbc.m116.736355
144. Cherry, JD, Tripodis, Y, Alvarez, VE, Huber, B, Kiernan, PT, Daneshvar, DH, et al. Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol Commun. (2016) 4:112. doi: 10.1186/s40478-016-0382-8
145. Ising, C, Venegas, C, Zhang, S, Scheiblich, H, Schmidt, SV, Vieira-Saecker, A, et al. NLRP3 inflammasome activation drives tau pathology. Nature. (2019) 575:669–73. doi: 10.1038/s41586-019-1769-z
146. Kovacs, GG. Astroglia and tau: new perspectives. Front Aging Neurosci. (2020) 12:96. doi: 10.3389/fnagi.2020.00096
147. Narasimhan, S, Changolkar, L, Riddle, DM, Kats, A, Stieber, A, Weitzman, SA, et al. Human tau pathology transmits glial tau aggregates in the absence of neuronal tau. J Exp Med. (2019) 217:e20190783. doi: 10.1084/jem.20190783
148. Lasagna-Reeves, CA, Castillo-Carranza, DL, Jackson, GR, and Kayed, R. Tau oligomers as potential targets for immunotherapy for Alzheimer’s disease and tauopathies. Curr Alzheimer Res. (2011) 8:659–65. doi: 10.2174/156720511796717177
149. Usenovic, M, Niroomand, S, Drolet, RE, Yao, L, Gaspar, RC, Hatcher, NG, et al. Internalized tau oligomers cause neurodegeneration by inducing accumulation of pathogenic tau in human neurons derived from induced pluripotent stem cells. J Neurosci. (2015) 35:14234–50. doi: 10.1523/jneurosci.1523-15.2015
150. Lee, VM-Y, Brunden, KR, Hutton, M, and Trojanowski, JQ. Developing therapeutic approaches to tau, selected kinases, and related neuronal protein targets. Csh Perspect Med. (2011) 1:a006437. doi: 10.1101/cshperspect.a006437
151. Prusiner, SB. Novel proteinaceous infectious particles cause scrapie. Science. (1982) 216:136–44. doi: 10.1126/science.6801762
152. Peng, C, Trojanowski, JQ, and Lee, VM-Y. Protein transmission in neurodegenerative disease. Nat Rev Neurol. (2020) 16:199–212. doi: 10.1038/s41582-020-0333-7
153. Irwin, DJ, Lee, VM-Y, and Trojanowski, JQ. Parkinson’s disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies. Nat Rev Neurosci. (2013) 14:626–36. doi: 10.1038/nrn3549
154. Kfoury, N, Holmes, BB, Jiang, H, Holtzman, DM, and Diamond, MI. Trans-cellular propagation of tau aggregation by Fibrillar species*. J Biol Chem. (2012) 287:19440–51. doi: 10.1074/jbc.m112.346072
155. Sanders, DW, Kaufman, SK, DeVos, SL, Sharma, AM, Mirbaha, H, Li, A, et al. Distinct tau prion strains propagate in cells and mice and define different Tauopathies. Neuron. (2014) 82:1271–88. doi: 10.1016/j.neuron.2014.04.047
156. Gibbons, GS, Banks, RA, Kim, B, Xu, H, Changolkar, L, Leight, SN, et al. GFP-mutant human tau transgenic mice develop Tauopathy following CNS injections of Alzheimer’s brain-derived pathological tau or synthetic mutant human tau fibrils. J Neurosci. (2017) 37:11485–94. doi: 10.1523/jneurosci.2393-17.2017
157. Woerman, AL, Aoyagi, A, Patel, S, Kazmi, SA, Lobach, I, Grinberg, LT, et al. Tau prions from Alzheimer’s disease and chronic traumatic encephalopathy patients propagate in cultured cells. Proc Natl Acad Sci. (2016) 113:E8187–E 8196. doi: 10.1073/pnas.1616344113
158. Guo, JL, and Lee, VM-Y. Seeding of Normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles*. J Biol Chem. (2011) 286:15317–31. doi: 10.1074/jbc.m110.209296
159. Guo, JL, Buist, A, Soares, A, Callaerts, K, Calafate, S, Stevenaert, F, et al. The dynamics and turnover of tau aggregates in cultured cells INSIGHTS INTO THERAPIES FOR TAUOPATHIES*. J Biol Chem. (2016) 291:13175–93. doi: 10.1074/jbc.m115.712083
160. Furman, JL, Vaquer-Alicea, J, White, CL, Cairns, NJ, Nelson, PT, and Diamond, MI. Widespread tau seeding activity at early Braak stages. Acta Neuropathol. (2017) 133:91–100. doi: 10.1007/s00401-016-1644-z
161. Kaufman, SK, Tredici, KD, Thomas, TL, Braak, H, and Diamond, MI. Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer’s disease and PART. Acta Neuropathol. (2018) 136:57–67. doi: 10.1007/s00401-018-1855-6
162. Jackson, SJ, Kerridge, C, Cooper, J, Cavallini, A, Falcon, B, Cella, CV, et al. Short fibrils constitute the major species of seed-competent tau in the brains of mice transgenic for human P301S tau. J Neurosci. (2016) 36:762–72. doi: 10.1523/jneurosci.3542-15.2016
163. Mirbaha, H, Chen, D, Morazova, OA, Ruff, KM, Sharma, AM, Liu, X, et al. Inert and seed-competent tau monomers suggest structural origins of aggregation. elife. (2018) 7:e36584. doi: 10.7554/elife.36584
164. Arakhamia, T, Lee, CE, Carlomagno, Y, Kumar, M, Duong, DM, Wesseling, H, et al. Posttranslational modifications mediate the structural diversity of Tauopathy strains. Cells. (2020) 180:633–644.e12. doi: 10.1016/j.cell.2020.01.027
165. Sharma, AM, Thomas, TL, Woodard, DR, Kashmer, OM, and Diamond, MI. Tau monomer encodes strains. elife. (2018) 7:e37813. doi: 10.7554/elife.37813
166. Falcon, B, Zivanov, J, Zhang, W, Murzin, AG, Garringer, HJ, Vidal, R, et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature. (2019) 568:420–3. doi: 10.1038/s41586-019-1026-5
167. Shi, Y, Zhang, W, Yang, Y, Murzin, AG, Falcon, B, Kotecha, A, et al. Structure-based classification of tauopathies. Nature. (2021) 598:359–63. doi: 10.1038/s41586-021-03911-7
168. Falcon, B, Cavallini, A, Angers, R, Glover, S, Murray, TK, Barnham, L, et al. Conformation determines the seeding potencies of native and recombinant tau aggregates. J Biol Chem. (2015) 290:1049–65. doi: 10.1074/jbc.m114.589309
169. Guo, JL, Narasimhan, S, Changolkar, L, He, Z, Stieber, A, Zhang, B, et al. Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J Exp Med. (2016) 213:2635–54. doi: 10.1084/jem.20160833
170. Narasimhan, S, and Lee, VMY. Chapter 15 the use of mouse models to study cell-to-cell transmission of pathological tau. Methods Cell Biol. (2017) 141:287–305. doi: 10.1016/bs.mcb.2017.06.009
171. Boluda, S, Iba, M, Zhang, B, Raible, KM, Lee, VM-Y, and Trojanowski, JQ. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol. (2015) 129:221–37. doi: 10.1007/s00401-014-1373-0
172. Clavaguera, F, Lavenir, I, Falcon, B, Frank, S, Goedert, M, and Tolnay, M. “Prion-Like” Templated misfolding in tauopathies. Brain Pathol. (2013) 23:342–9. doi: 10.1111/bpa.12044
173. Braak, H, Alafuzoff, I, Arzberger, T, Kretzschmar, H, and Tredici, KD. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. (2006) 112:389–404. doi: 10.1007/s00401-006-0127-z
174. Mantyh, WG, Spina, S, Lee, A, Iaccarino, L, Soleimani-Meigooni, D, Tsoy, E, et al. Tau positron emission tomographic findings in a former US football player with pathologically confirmed chronic traumatic encephalopathy. JAMA Neurol. (2020) 77:517–21. doi: 10.1001/jamaneurol.2019.4509
175. Schwarz, AJ, Yu, P, Miller, BB, Shcherbinin, S, Dickson, J, Navitsky, M, et al. Regional profiles of the candidate tau PET ligand 18 F-AV-1451 recapitulate key features of Braak histopathological stages. Brain. (2016) 139:1539–50. doi: 10.1093/brain/aww023
176. Williams, DR, Pittman, AM, Revesz, T, Lees, AJ, and de Silva, R. Genetic variation at the tau locus and clinical syndromes associated with progressive supranuclear palsy. Movement Disord. (2007) 22:895–7. doi: 10.1002/mds.21393
177. Saito, Y, Ruberu, NN, Sawabe, M, Arai, T, Tanaka, N, Kakuta, Y, et al. Staging of Argyrophilic grains: an age-associated Tauopathy. J Neuropathol Exp Neurol. (2004) 63:911–8. doi: 10.1093/jnen/63.9.911
178. Irwin, DJ, Byrne, MD, McMillan, CT, Cooper, F, Arnold, SE, Lee, EB, et al. Semi-automated digital image analysis of Pick’s disease and TDP-43 Proteinopathy. J Histochem Cytochem. (2015) 64:54–66. doi: 10.1369/0022155415614303
179. de Calignon, A, Polydoro, M, Suárez-Calvet, M, William, C, Adamowicz, DH, Kopeikina, KJ, et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron. (2012) 73:685–97. doi: 10.1016/j.neuron.2011.11.033
180. Dujardin, S, Bégard, S, Caillierez, R, Lachaud, C, Delattre, L, Carrier, S, et al. Ectosomes: A new mechanism for non-Exosomal secretion of tau protein. PLoS One. (2014) 9:e100760. doi: 10.1371/journal.pone.0100760
181. Dujardin, S, Bégard, S, Caillierez, R, Lachaud, C, Carrier, S, Lieger, S, et al. Different tau species lead to heterogeneous tau pathology propagation and misfolding. Acta Neuropathol Commun. (2018) 6:132. doi: 10.1186/s40478-018-0637-7
182. Kaufman, SK, Sanders, DW, Thomas, TL, Ruchinskas, AJ, Vaquer-Alicea, J, Sharma, AM, et al. Tau prion strains dictate patterns of cell pathology, progression rate, and regional vulnerability in vivo. Neuron. (2016) 92:796–812. doi: 10.1016/j.neuron.2016.09.055
183. Calafate, S, Flavin, W, Verstreken, P, and Moechars, D. Loss of bin 1 promotes the propagation of tau pathology. Cell Rep. (2016) 17:931–40. doi: 10.1016/j.celrep.2016.09.063
184. Holmes, BB, DeVos, SL, Kfoury, N, Li, M, Jacks, R, Yanamandra, K, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci. (2013) 110:E3138–47. doi: 10.1073/pnas.1301440110
185. Rauch, JN, Luna, G, Guzman, E, Audouard, M, Challis, C, Sibih, YE, et al. LRP1 is a master regulator of tau uptake and spread. Nature. (2020) 580:381–5. doi: 10.1038/s41586-020-2156-5
186. Wu, JW, Herman, M, Liu, L, Simoes, S, Acker, CM, Figueroa, H, et al. Small misfolded tau species are internalized via bulk endocytosis and Anterogradely and Retrogradely transported in neurons*. J Biol Chem. (2013) 288:1856–70. doi: 10.1074/jbc.m112.394528
187. Flavin, WP, Bousset, L, Green, ZC, Chu, Y, Skarpathiotis, S, Chaney, MJ, et al. Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol. (2017) 134:629–53. doi: 10.1007/s00401-017-1722-x
188. Stancu, I-C, Cremers, N, Vanrusselt, H, Couturier, J, Vanoosthuyse, A, Kessels, S, et al. Aggregated tau activates NLRP3–ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded tau pathology in vivo. Acta Neuropathol. (2019) 137:599–617. doi: 10.1007/s00401-018-01957-y
189. Mohamed, N-V, Plouffe, V, Rémillard-Labrosse, G, Planel, E, and Leclerc, N. Starvation and inhibition of lysosomal function increased tau secretion by primary cortical neurons. Sci Rep. (2014) 4:5715. doi: 10.1038/srep05715
190. Saman, S, Lee, NCY, Inoyo, I, Jin, J, Li, Z, Doyle, T, et al. Proteins recruited to exosomes by tau overexpression implicate novel cellular mechanisms linking tau secretion with Alzheimer’s disease. J Alzheimers Dis. (2014) 40:S47–70. doi: 10.3233/jad-132135
191. Yamada, K, Cirrito, JR, Stewart, FR, Jiang, H, Finn, MB, Holmes, BB, et al. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci. (2011) 31:13110–7. doi: 10.1523/jneurosci.2569-11.2011
192. Kanmert, D, Cantlon, A, Muratore, CR, Jin, M, O’Malley, TT, Lee, G, et al. C-terminally truncated forms of tau, but not full-length tau or its C-terminal fragments, are released from neurons independently of cell death. J Neurosci. (2015) 35:10851–65. doi: 10.1523/jneurosci.0387-15.2015
193. Wu, JW, Hussaini, SA, Bastille, IM, Rodriguez, GA, Mrejeru, A, Rilett, K, et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci. (2016) 19:1085–92. doi: 10.1038/nn.4328
194. Katsinelos, T, Zeitler, M, Dimou, E, Karakatsani, A, Müller, H-M, Nachman, E, et al. Unconventional secretion mediates the trans-cellular spreading of tau. Cell Rep. (2018) 23:2039–55. doi: 10.1016/j.celrep.2018.04.056
195. Tapiola, T, Alafuzoff, I, Herukka, S-K, Parkkinen, L, Hartikainen, P, Soininen, H, et al. Cerebrospinal fluid β-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch Neurol. (2009) 66:382–9. doi: 10.1001/archneurol.2008.596
196. Tardivel, M, Bégard, S, Bousset, L, Dujardin, S, Coens, A, Melki, R, et al. Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological tau protein assemblies. Acta Neuropathol Commun. (2016) 4:117. doi: 10.1186/s40478-016-0386-4
Keywords: tau, traumatic brain injury, neurodegeneration, brain pathology, repetitive head injury
Citation: Flavin WP, Hosseini H, Ruberti JW, Kavehpour HP, Giza CC and Prins ML (2023) Traumatic brain injury and the pathways to cerebral tau accumulation. Front. Neurol. 14:1239653. doi: 10.3389/fneur.2023.1239653
Edited by:
Ramesh Raghupathi, Drexel University, United StatesReviewed by:
Emanuela Piermarini, Drexel University, United StatesBertrand Russell Huber, Boston University, United States
Copyright © 2023 Flavin, Hosseini, Ruberti, Kavehpour, Giza and Prins. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mayumi L. Prins, bXByaW5zQG1lZG5ldC51Y2xhLmVkdQ==