 Ciro Maria Noioso*
Ciro Maria Noioso* Liliana BevilacquaGabriella Maria AcerraPaola Della ValleMarina Serio
Liliana BevilacquaGabriella Maria AcerraPaola Della ValleMarina Serio Claudia Vinciguerra
Claudia Vinciguerra Giuseppe PiscosquitoAntonella TorielloPaolo Barone
Giuseppe PiscosquitoAntonella TorielloPaolo Barone Aniello Iovino
Aniello Iovino- Neurology Unit, University Hospital “San Giovanni di Dio e Ruggi d'Aragona”, University of Salerno, Salerno, Italy
Introduction: Miller Fisher syndrome (MFS) is considered a rare variant of Guillain-Barré syndrome (GBS), a group of acute-onset immune-mediated neuropathies characterized by the classic triad of ataxia, areflexia, and ophthalmoparesis. The present review aimed to provide a detailed and updated profile of all aspects of the syndrome through a collection of published articles on the subject, ranging from the initial description to recent developments related to COVID-19.
Methods: We searched PubMed, Scopus, EMBASE, and Web of Science databases and gray literature, including references from the identified studies, review studies, and conference abstracts on this topic. We used all MeSH terms pertaining to “Miller Fisher syndrome,” “Miller Fisher,” “Fisher syndrome,” and “anti-GQ1b antibody.”
Results: An extensive bibliography was researched and summarized in the review from an initial profile of MFS since its description to the recent accounts of diagnosis in COVID-19 patients. MFS is an immune-mediated disease with onset most frequently following infection. Anti-ganglioside GQ1b antibodies, detected in ~85% of patients, play a role in the pathogenesis of the syndrome. There are usually no abnormalities in MFS through routine neuroimaging. In rare cases, neuroimaging shows nerve root enhancement and signs of the involvement of the central nervous system. The most consistent electrophysiological findings in MFS are reduced sensory nerve action potentials and absent H reflexes. Although MFS is generally self-limited and has excellent prognosis, rare recurrent forms have been documented.
Conclusion: This article gives an updated narrative review of MFS with special emphasis on clinical characteristics, neurophysiology, treatment, and prognosis of MFS patients.
1. Introduction
Miller Fisher syndrome (MFS) is considered a rare variant of Guillain-Barré syndrome (GBS), a group of acute-onset immune-mediated neuropathies characterized by the classic triad of ataxia, areflexia, and ophthalmoparesis. The present review aimed to provide a detailed and updated profile of all aspects of the syndrome through a collection of published articles on the subject ranging from the initial description to recent developments related to COVID-19.
2. Methods
We searched PubMed, Scopus, EMBASE, and Web of Science databases and gray literature, including references from the identified studies, review studies, and conference abstracts on this topic. We used all MeSH terms pertaining to “Miller Fisher syndrome,” “Miller Fisher,” “Fisher syndrome,” and “anti-GQ1b antibody.”
3. Epidemiology
The worldwide annual incidence of GBS is ~1–2/100,000 inhabitants. Of these, MFS represents a small fraction of the total, with the percentage varying according to the area considered. There is a slight male predominance, and MFS can occur in all age groups. The incidence is higher in Asian countries, where it can reach 15–25% of GBS cases: an 11-year retrospective study in Taiwan estimated a relative incidence of ~18%, and other studies found a 9% incidence in Hong Kong and a 7.7% incidence in Thailand (1–3). It is lower in the West, where it accounts for ~1–7% of GBS cases: an Italian study estimated the incidence in Europe to be between 0.04 and 0.18 cases per 1,00,000 inhabitants or ~6.6% of GBS patients; while a 7% incidence was recorded in Spain (4, 5). Such a low incidence justifies the absence of randomized trials on patients. All currently published studies appear to be retrospective, with very limited case studies.
There is no consensus regarding the classification of GBS and its variants. A more inclusive approach, based on clinical features, was proposed by the GBS Classification Group in 2014, placing both GBS and MFS within a continuous spectrum disorder, considering apparent differences in pathogenesis, treatment, and prognosis (6) (Table 1).
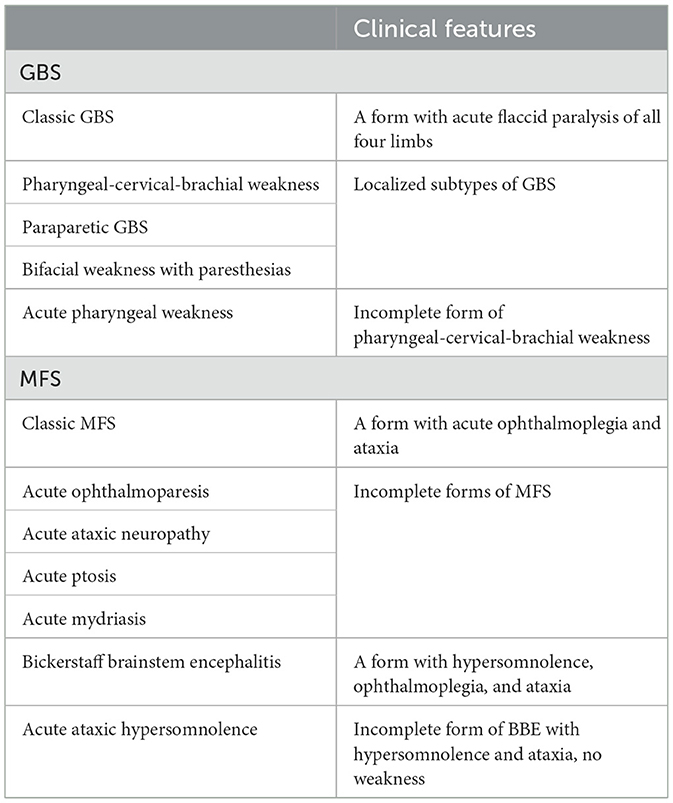
Table 1. GBS, MFS, and their subtypes.
4. Clinical features
MFS classically presents itself with a symptomatological triad characterized by ophthalmoparesis, ataxia, and osteotendinous areflexia, appearing in ~80% of patients (7, 8). Ophthalmoparesis, usually bilateral, progresses to complete external ophthalmoplegia in 1–2 weeks. Ataxia, often very severe, may cause an inability to walk without support despite normal strength. Areflexia, a less specific element of the triad (absent in 18%), may also be limited to an isolated body area (9). The symptomatological triad is also frequently associated with the presence of extra signs such as ptosis (60%), facial nerve palsy (30–50%), sensory deficits (20–50%), and hyposthenia (20–25%) (10–13) (Table 2).

Table 2. Extra signs associated with the symptomatological triad.
Interesting evidence emerges from a retrospective study tracing the 10-year experience of a third-level center. It takes into account 19 cases of MFS documented from 1995 to 2005: epidemiologically, there is a clear male prevalence (M>F 84%), early age onset (36 years on average), and autumn–winter peak (73% of cases), which corresponds with the higher incidence of respiratory infections (URIs). This study showed that the average interval between infection and the onset of neurological symptoms is 7 days (ranging from 1 to 30) and that diplopia is the main onset symptom (63%).
The clinical timing for onset varies according to the symptom considered: ophthalmoparesis appears to be the earliest symptom, presenting itself at ~7 days (variability 1–30 days), followed by ataxia, which on average appears at ~10 days (variability 1–30 days), while areflexia, the symptom with the most variable onset of the three, presents itself on average at 14 days (variability 4–45 days) (14).
A longitudinal assessment of the pathology thus becomes essential for diagnosis.
MFS with atypical manifestations has also been described: possible onset with bilateral internal ophthalmoplegia, unilateral external ophthalmoplegia, bilateral abducent nerve palsy, and isolated bilateral ptosis (15–18).
The literature reports cases of generally bilateral optic neuritis, causing visual impairments with no signs of pain in eye movement, color desaturation, or field deficit. The optic fundi are normal. Orbital magnetic resonance imaging or visual evoked potentials (VEPs) revealed bilateral optic neuropathy (19, 20).
5. Pathogenesis
The origin of MFS has been shown to be immunologic. The infectious antecedent appears to be present in the majority of patients: previous infection of the upper respiratory apparatus is most frequent (56–76% of patients). Gastrointestinal infection (4%), typical of classical GBS, and isolated fever (2%) are rarer. MFS associated with autoimmune or neoplastic disorders is also possible (1, 13, 21–24). Additional factors that increase the risk of the disease comprise the utilization of specific medications (such as heroin, isotretinoin, and streptokinase), implementation of TNF-alpha antagonist treatment, bone marrow transplantation, and surgical procedures (25–27).
In this light, the higher incidence of MFS in the Far East could also be related to the higher rate of infections. Pathogens most frequently implicated are Campylobacter jejuni (21%) and Haemophilus influenzae (8%) (22, 28). In most cases, the pathogen responsible is not known (28).
The infectious etiology had already been suspected by Miller Fisher and then corroborated by Koga (8, 28). The underlying mechanism seems to be that of “molecular mimicry:” The immune system's activation of the lipo-oligosaccharides (LOS) present on the membrane of these pathogens, which are similar in shape to gangliosides (GQ1b, GM1, and GD1a), would lead to the production of autoantibodies. If the antibody produced is GM1 or GD1b, the classic form of GBS is produced, whereas if it is GQ1b, MFS is produced (29).
Antibodies against GQ1b have a sensitivity of 85% and a specificity of 100% (30).
GQ1b is a ganglioside present in the paranodal myelin, mainly at the level of oculomotor nerves (III-IV and VI cranial nerves), dorsal root ganglia (DRG), and fibers of neuromuscular spindles (Figure 1).

Figure 1. The underlying mechanism of MFS seems to be that of “molecular mimicry.” The immune system's activation of the lipo-oligosaccharides (LOS) present on the membrane of some pathogens, most frequently Campylobacter jejuni, which are similar in shape to gangliosides (GQ1b, GM1, and GD1a), would lead to the production of autoantibodies. If the antibody produced is GM1 or GD1b, the classic form of GBS with acute motor axonal neuropathy is produced, whereas if it is GQ1b, MFS is produced (29). GQ1b is a ganglioside present in the paranodal myelin, mainly at the level of oculomotor nerves (III-IV and VI cranial nerves), dorsal root ganglia (DRG), and fibers of neuromuscular spindles. The localization of the ganglioside explains the symptomatological triad of patients.
The localization of the ganglioside explains the symptomatological triad of patients with ophthalmoparesis, ataxia, and areflexia due to the involvement of the muscle spindles (1). The possible involvement of the optic nerve with optic neuritis is explained by the presence of the GQ1b epitope in the optic nerve (31).
The passage of the autoantibodies would take place at the level of the blood–nerve barrier of the neuromuscular junctions (NMJs), an area rich in gangliosides, which are dependent on rapid cell turnover for its function, as is also shown by histological analyses (32).
GQ1b is expressed in the NMJs of the oculomotor nerves and of muscle spindles and serves as a target for autoantibodies producing the characteristic outlook of MFS.
Antibodies against GQ1b ganglioside are present in Bickerstaff brainstem encephalitis (BBE) as well as MFS. BBE is an even rarer related condition in which ataxia and ophthalmoparesis are accompanied by impaired consciousness and hyperreflexia. The presence of these symptoms points to anti-GQ1b antibodies having pathologic effects in the central nervous system as well as in the cranial and peripheral nerves.
At the level of the nodes of Ranvier, three distinct areas can be identified:
• Juxtaparanodal region, where the compacted myelin is tightly attached to the axolemma.
• Paranodal region, where the myelin is attached to the axolemma but not organized as a compact structure.
• The node of Ranvier, in which the axolemma is not lined with myelin and is in direct contact with the extracellular fluid, despite being covered by the microvillous Schwann cells.
Fundamental to longitudinal impulse conduction is the paranodal region. At the level of the paranode, there is, in fact, a formation consisting of Contactin-1 (CNTN1) and CASPR, expressed by neurons, which bind the NF-155 counterpart, and is of glial origin. This paranodal axon-glial formation is responsible for ion channel clustering, propagating the action potential and blocking the lateral diffusion of membrane proteins in myelinated nerve fibers (Figure 2).
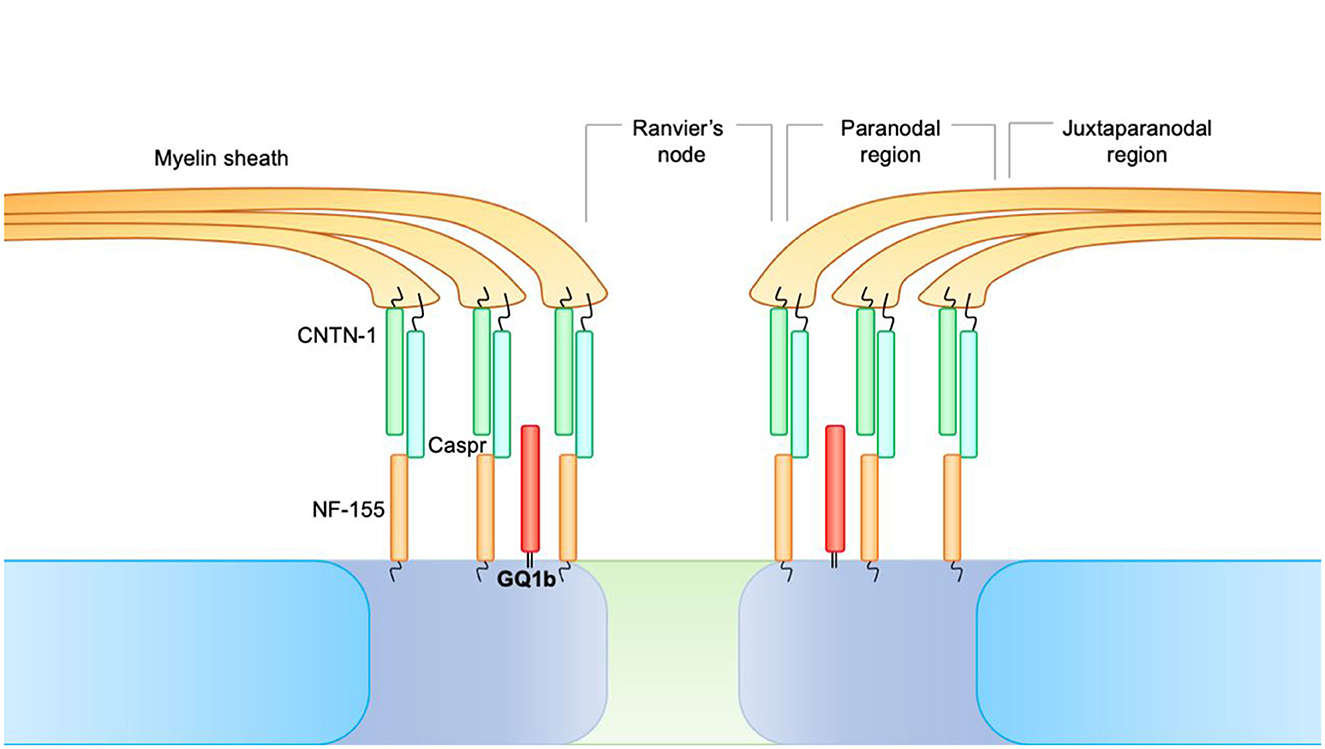
Figure 2. At the level of the nodes of Ranvier, three distinct areas can be identified. 1. Juxtaparanodal region, where the compacted myelin is tightly attached to the axolemma. 2. Paranodal region, where the myelin is attached to the axolemma but not organized as a compact structure. 3. The node of Ranvier, in which the axolemma is not lined with myelin and is in direct contact with the extracellular fluid, despite being covered by the microvillous Schwann cells. At the level of the paranode, there is a formation consisting of Contactin-1 (CNTN1) and CASPR, expressed by neurons, which bind the NF-155 counterpart and are of glial origin. This paranodal axon-glial formation is responsible for ion channel clustering, propagating the action potential, and blocking the lateral diffusion of membrane proteins in myelinated nerve fibers.
GQ1b acts by stabilizing this paranodal formation (33). GQ1b autoantibodies, acting against the paranodal formation, cause acute blockade of anterograde propagation of the electrical impulse, justifying the acute symptomatic onset (29).
Certain cases have been reported of anti-glutamic acid decarboxylase (GAD) seropositive patients, including those with anti-GQ1b seronegative forms (34). Anti-GAD plays an uncertain role in the pathogenesis of MFS. Titers, though inferior to that found in stiff person syndrome, appeared increased in these patients' serum. A progressive decrease in titers is associated with clinical and electrophysiological improvement. The pathogenetic role of these antibodies would involve causing the disruption of GABA synthesis through GAD inhibition in the brainstem and cerebellum (35–38). GAD is the rate-limiting enzyme for the synthesis of the inhibitory gamma-aminobutyric acid (GABA) (38). Supporting evidence of a broader role of anti-GAD antibodies in the pathogenesis of MFS is the case, described by Javaid et al., of a patient presenting seronegative BBE with anti-GAD seropositivity (39).
5.1. Relation to SARS-CoV-2
COVID-19 (coronavirus disease 2019) appears to be a possible infective antecedent to MFS.
COVID-19 is associated with several systemic symptoms, the most frequent being gastrointestinal, cardiovascular, dermatological, and neurological symptoms (40).
A retrospective study on 214 hospitalized patients reported neurological complications in 36% of the cases examined with involvement of the CNS (24.8%) greater than the peripheral nervous system (8.9%) (41). These varied from lighter forms such as headache, dizziness, myalgia, and anosmia to more severe symptoms such as encephalopathy, encephalitis, necrotizing hemorrhagic encephalopathy, stroke, epileptic seizures, and Guillain-Barré syndrome (40).
Infection may be either hematogenous or via retrograde neural propagation along the olfactory pathway in which ACE2 (angiotensin-converting enzyme 2) functional receptor is more present (42).
An Italian study demonstrated how the percentage of GBS patients following SARS-CoV2 infection substantially increased during the pandemic compared to the previous 30 years (43–45).
According to the ALBACOVID registry, GBS patients accounted for 0.5% of hospitalizations due to COVID-19 (46). A systematic review by Aladawj found that, out of 99 COVID-19-related GBS cases described in the literature, nine presented the Miller Fisher variant (47).
Although a rare occurrence, multiple cases of MFS following COVID-19 vaccination have been described across several countries (48–50). Unlike GBS associated with vaccination, it does not appear to be related to the viral vector vaccination (51).
6. Diagnosis
The diagnosis of MFS is based on clinical aspects and may be supported by laboratory investigations. Various “formes frustes” and overlap syndromes have been reported to date, thereby expanding the understanding of this clinical spectrum. In the cerebrospinal fluid (CSF), hyperproteinorrachia, a marker of GBS, may not be an early finding and may not be present in all cases, i.e., it can be found in ~47% of patients. Of these patients, the incidence of hyperproteinorrachia increases progressively over the first 3 weeks from 66% in week 1 to 82% in week 3 (1).
More frequent is the finding in overlap syndromes: In these syndromes, the incidence of hyperproteinorrachia is similar to that of classic GBS (1).
In contrast, anti-GQ1b antibodies are found in the CSF of almost 85% of patients with MFS (1). This percentage is higher in other studies (22). The anti-GQ1b Ab dosage appears to be much more useful than the proteins in the CSF during the 1st week. Ab anti-GD1a (28%) and Ab anti-GM1 (15%), whose pathogenic role has not yet been demonstrated, may also be found (52).
Anti-GQ1b antibodies are also present in the forms of GBS in overlap with ophthalmoparesis in acute isolated ophthalmoplegia and in BBE: these forms constitute the “Anti-GQ1b spectrum” (53).
Although MFS belongs to the group of peripheral neuropathies, the hypothesis of concomitant central involvement has been put forward by different authors (54–56).
Fisher considered ataxia as the manifestation of an unusual peripheral neuron lesion but also reported it as the cerebellar type as it was disproportionate to sensory loss (8).
Some authors reported a cerebellar type of ataxia and supranuclear ophthalmoplegia with Bell's sign positive in MFS and suggested additional involvement of the central nervous system (57).
As proof of this finding, a study was conducted on MFS patients using 18F-fluorodeoxyglucose-positron emission tomography (FDG-PET) to determine the involvement of the central nervous system. Cerebral glucose metabolism in 10 patients with MFS was compared with that of 60 age- and sex-matched normal controls using PET. Group analyses described increased metabolism in the cerebellar vermis, bilateral cerebellar hemispheres, pontine tegmentum, midbrain tectum, right inferior frontal cortex (Brodmann area 47), and left thalamus and decreased metabolism in the bilateral occipital cortices (Brodmann area 18 and 19). The hypermetabolism in those disorders has been attributed to inflammatory changes, and stereotaxic biopsy indeed revealed inflammation with lymphocytic infiltrations and reactive gliosis in one patient (58).
Hypermetabolism of the right inferior frontal cortex would be associated with hyperactivation to adjust locomotion and limb movements, which are impaired by diplopia and ataxia. A similar finding of sustained prefrontal hyperactivity is also observed during ataxic gait in patients with infratentorial stroke, which also suggests a compensatory mechanism for impaired locomotor control (59).
Hypometabolism of the associative visual cortex (Brodmann area 18 and 19) has been interpreted as an adaptive functional suppression of the cortical responses to deranged visual inputs (60).
MFS is not generally associated with abnormalities in brain imaging. Hyperintensity in T2W at the level of the brainstem and the cranial and spinal nerve roots can be found in 2% of cases, providing evidence supporting central involvement (61–63).
Electrophysiologically, the neurophysiological alterations are generally minor when compared to the ones presented in GBS. The hallmarks typical of acquired demyelinating polyneuropathies such as reduced motor conduction velocity (MCV), marked temporal dispersion, and conduction blocks are not present here. Motor and sensory conduction studies are generally within normal limits. Sensory neuropathy characterized by amplitude reduction disproportionate to the slowing of sensory conduction velocities or prolongation of the distal latencies may appear (64, 65).
More indicative is the study of late responses, which allows the evaluation of the most proximal segment of nerves such as plexuses and roots, inaccessible to routine nerve conduction studies (NCSs). The F response, a late motor response, generally appears to be normal; in rare cases, it is possible to find the prolongation or absence of this reflex (65).
A constant abnormality in patients with MFS appears to be the absence of the H reflex (66, 67). The disappearance of this late reflex, the neurophysiological equivalent of the myotatic reflex, appears to be ascribable to the impairment of the fibers of the neuromuscular spindle that present the GQ1b epitope on the surface, which is responsible for ataxia and areflexia. These fibers cannot be investigated with routine NCSs, but only in mixed nerve studies and with the H reflex. Recovery from this neurophysiological alteration is associated with patient recovery (13).
7. Therapy
As MFS is a rare condition, there are no randomized, double-blind, placebo-controlled trials on treatment and retrospective studies are controversial.
A large retrospective study of 92 patients, including 28 treated with intravenous immunoglobulin (IVIG) (0.4 g/kg/day × 5 days), 23 treated with plasma exchange (PLEX; 2–6 cycles, average 4), and 41 untreated, retrospectively assessing the time of recovery from ophthalmoplegia and ataxia, showed that the survival rate was ~100%. The resolution of areflexia could not be retrospectively assessed as it did not alter the patients' day-to-day living. IVIG slightly improved the patient's condition and recovery time by reducing the binding of anti-GQ1b antibodies and limiting the pathologic effects of the antibodies (68). Plasmapheresis would neither improve symptomatology nor recovery time (69).
The natural history of the disease is self-limiting. Therapy would not affect the patient's outcome. The natural course of MFS is characterized by excellent recovery.
Although rare cases of recurrent MFS have also been recorded in the literature, a review by Ishii et al. found that 12% of MFS patients presented a recurrent form (70). Clinical, electrophysiological, and laboratory analyses of these patients presented no apparent differences from that of non-recurrent MFS. Epidemiological analysis revealed a younger median onset age (22 years as opposed to 37 years in non-recurrent forms). Although there is ample variability in the number of recurrences, all cases examined presented similar symptoms to the first occurrence, though less severe (70).
As reported by Chida, the recurrence of MFS in these patients may depend on a gene polymorphism: the presence of HLA-DR2 antigen is significantly higher than that in patients with monophasic MFS and healthy controls (71).
MFS was found to overlap with other disorders within 7 days of onset in ~50% of cases: classic GBS (15%), pharyngeal-cervical-brachial GBS (23%), and BBE (12%), which may place MFS within a continuous spectrum disorder (6, 72, 73). Early recognition and IVIG or PLEX are recommended in these cases (6, 72, 74, 75).
The average resolution time of symptoms varies depending on the symptom considered: recovery from ataxia takes 35 days (10–121), ophthalmoplegia takes 3 months (93 days, 18–244), and areflexia of the 3 symptoms is the most variable (64 days, 10–650). At outpatient follow-up, all patients were free of ataxia, ophthalmoplegia, and areflexia after an average of 6 months, returning to their normal activities with no functional limitations (14).
8. Seronegative forms
Particular attention should be paid to the clinical forms of MFS that show CSF negativity for GQ1b.
As shown in a study published in the Journal of Neurology in 2012, 12% of MFS are seronegative and ~70% of these are positive for GM1. These forms, clinically similar to GQ1b+ forms, differ in that they almost exclusively affect men and for a higher frequency of gastroenteritis as an infectious antecedent (76).
These forms are generally paraneoplastic and are more frequently associated with Burkitt lymphoma, diffuse large B cell lymphoma, chronic lymphocytic leukemia, Hodgkin's disease, and leptomeningeal signet ring cell carcinomatosis of lung cancer (21, 77, 78).
9. Conclusion
This article aimed to provide an updated narrative review of the clinical features, neurophysiology, treatment, and prognosis of patients with MFS.
MFS is a rare, immune-mediated disorder that presents itself with a classical triad of ataxia, areflexia, and ophthalmoplegia. Apart from these classic clinical signs, other signs may be associated with it, such as optic neuritis, facial palsy, limb weakness, and superficial sensory loss.
Anti-ganglioside GQ1b antibodies play a role in the pathogenesis of the syndrome. These antibodies can be found in ~85% of patients, peaking in the 1st week, whereas albuminocytological dissociation in the cerebrospinal fluid (CSF) appears later. Electrophysiologically reduced sensory nerve action potentials and absent H reflexes are also characteristic. The natural course of MFS is characterized by excellent recovery.
Although no abnormalities are usually found in MFS by routine neuroimaging, contrast enhancement in T2-weighted sequences of nerve roots and signs of the involvement of the central nervous system have been described in some cases, supporting the hypothesis of central and peripheral involvement.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was funded by the University of Salerno/Department of Medicine and Surgery, “Fondi di Ateneo per la Ricerca di Base (FARB) 2022”.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Teener JW. Miller Fisher's syndrome. In: Seminars in Neurology. New York, NY: Thieme Medical Publishers (2012). p. 512–6. doi: 10.1055/s-0033-1334470
2. Ma YM, Liu TKT, Wong V. Guillain-Barre syndrome in southern Chinese children: 32 year experience in Hong Kong. Pediatr Int Off J Jpn Pediatr Soc. (2010) 52:13–9. doi: 10.1111/j.1442-200X.2009.02951.x
3. Lolekha P, Phanthumchinda K. Miller-Fisher syndrome at King Chulalongkorn Memorial Hospital. J Med Assoc Thail Chotmaihet Thangphaet. (2009) 92:471–7.
4. Clinical E, Neurology EP. Guillain-Barré syndrome variants in Emilia-Romagna, Italy, 1992-3: incidence, clinical features, and prognosis. Emilia-Romagna Study Group on clinical and epidemiological problems in neurology. J Neurol Neurosurg Psychiatry. (1998) 65:218–24. doi: 10.1136/jnnp.65.2.218
5. Téllez-Zenteno JF, Jacinto-Tinajero JC, Avila-Funes A, García-Ramos G, Negrete-Pulido O, Sentíes-Madrid H. Guillain-Barre syndrome. Experience in a third level hospital. Rev Investig Clin Organo Hosp Enfermedades Nutr. (2001) 53:311–4.
6. Wakerley BR, Uncini A, Yuki N. Guillain-Barré and Miller Fisher syndromes—new diagnostic classification. Nat Rev Neurol. (2014) 10:537–44. doi: 10.1038/nrneurol.2014.138
7. Collier J. Peripheral neuritis. The Morrison Lectures, 1932, delivered before the Royal College of Physicians of Edinburgh, May 1932. Edinb Med J. (1932) 39:601–18.
8. Fisher M. An unusual variant of acute idiopathic polyneuritis (syndrome of ophthalmoplegia, ataxia and areflexia). N Engl J Med. (1956) 255:57–65. doi: 10.1056/NEJM195607122550201
9. Truong J, Conley J, Ashurst J. Miller-Fisher syndrome: a case report and review of the literature. Clin Pract Cases Emerg Med. (2020) 4:653–5. doi: 10.5811/cpcem.2020.7.48507
10. Lyu RK, Tang LM, Cheng SY, Hsu WC, Chen ST. Guillain-Barré syndrome in Taiwan: a clinical study of 167 patients. J Neurol Neurosurg Psychiatry. (1997) 63:494–500. doi: 10.1136/jnnp.63.4.494
11. Mori M, Kuwabara S, Fukutake T, Yuki N, Hattori T. Clinical features and prognosis of Miller Fisher syndrome. Neurology. (2001) 56:1104–6. doi: 10.1212/WNL.56.8.1104
12. Kollár K, Liptai Z, Rosdy B, Móser J. Guillain-Barré syndrome in childhood. Ideggyogyaszati Szle. (2009) 62:399–404.
13. Arányi Z, Kovács T, Sipos I, Bereczki D. Miller Fisher syndrome: brief overview and update with a focus on electrophysiological findings. Eur J Neurol. (2012) 19:15–20.e1–3. doi: 10.1111/j.1468-1331.2011.03445.x
14. San-Juan OD, Martínez-Herrera JF, García JM, Gonzalez-Aragon MF, Del Castillo-Calcáneo JD, Pérez-Neri I. Miller Fisher syndrome: 10 years' experience in a third-level center. Dysphagia. 62:149–54. doi: 10.1159/000226599
15. Bae JS, Kim JK, Kim SH, Kim OK. Bilateral internal ophthalmoplegia as an initial sole manifestation of Miller Fisher syndrome. J Clin Neurosci Off J Neurosurg Soc Australas. (2009) 16:963–4. doi: 10.1016/j.jocn.2008.09.009
16. Smith J, Clarke L, Severn P, Boyce R. Unilateral external ophthalmoplegia in Miller Fisher syndrome: case report. BMC Ophthalmol. (2007) 7:1–3. doi: 10.1186/1471-2415-7-7
17. Wertheim MS, Benzimra JD, Jadresic LP, Ferris JD. Ocular presentation of pediatric Miller-Fisher syndrome. J Pediatr Ophthalmol Strabismus. (2008) 45:245–6. doi: 10.3928/01913913-20080701-16
18. Stalpers XL, Verhagen WIM, Meulstee J. Isolated bilateral ptosis as the only ophthalmologic sign in the Fisher variant of Guillain-Barré syndrome. J Neuro-Ophthalmol Off J North Am Neuro-Ophthalmol Soc. (2009) 29:354–5. doi: 10.1097/WNO.0b013e3181b41445
19. Zeng Q, Li J, Feng SX, Xiao PY, Zheng YF, Zhang C, et al. Anti-GQ1b antibody syndrome with visual impairment: a retrospective case series. J Integr Neurosci. (2022) 21:81. doi: 10.31083/j.jin2103081
20. Lolekha P, Phanthumchinda K. Optic neuritis in a patient with Miller-Fisher syndrome. J Med Assoc Thail Chotmaihet Thangphaet. (2008) 91:1909–13.
21. Gentile S, Messina M, Rainero I, Lo Giudice R, De Martino P, Pinessi L. Miller Fisher syndrome associated with Burkitt's lymphoma. Eur J Neurol. (2006) 13:430. doi: 10.1111/j.1468-1331.2006.01254.x
22. Lo YL. Clinical and immunological spectrum of the Miller Fisher syndrome. Muscle Nerve. (2007) 36:615–27. doi: 10.1002/mus.20835
23. Roquer J, Pou Serradell A. Paraneoplastic Miller-Fisher syndrome? Neurol Barc Spain. (1992) 7:40.
24. Csépány T, Boczán J, Magyar MT, Molnár S, Csiba L, Décsy J, et al. Miller Fisher syndrome–a presenting clinical manifestation of lung cancer in a previously apparently healthy individual. J Neurol. (2004) 251:898–900. doi: 10.1007/s00415-004-0465-5
25. Shin ISJ, Baer AN, Kwon HJ, Papadopoulos EJ, Siegel JN. Guillain-Barré and Miller Fisher syndromes occurring with tumor necrosis factor alpha antagonist therapy. Arthritis Rheum. (2006) 54:1429–34. doi: 10.1002/art.21814
26. Pritchard J, Appleton R, Howard R, Hughes RC. Guillain-Barré syndrome seen in users of isotretinoin. Br Med J. (2004) 328:1537. doi: 10.1136/bmj.328.7455.1537
27. Yu EH, Geraldi-Samara D. Pharyngeal-Cervico-Brachial/Miller Fisher overlap syndrome with infliximab exposure. J Clin Neuromuscul Dis. (2020) 21:157–8. doi: 10.1097/CND.0000000000000274
28. Koga M, Gilbert M, Li J, Koike S, Takahashi M, Furukawa K, et al. Antecedent infections in Fisher syndrome: a common pathogenesis of molecular mimicry. Neurology. (2005) 64:1605–11. doi: 10.1212/01.WNL.0000160399.08456.7C
29. Martín-Aguilar L, Pascual-Goñi E, Querol L. Autoantibodies in immune-mediated inflammatory neuropathies. Med Clin. (2019) 153:360–7. doi: 10.1016/j.medcle.2019.06.015
30. Chiba A, Kusunoki S, Obata H, Machinami R, Kanazawa I. Serum anti-GQ1b IgG antibody is associated with ophthalmoplegia in Miller Fisher syndrome and Guillain-Barré syndrome: clinical and immunohistochemical studies. Neurology. (1993) 43:1911–7. doi: 10.1212/WNL.43.10.1911
31. Yuki N. Fisher syndrome and Bickerstaff brainstem encephalitis (Fisher-Bickerstaff syndrome). J Neuroimmunol. (2009) 215:1–9. doi: 10.1016/j.jneuroim.2009.05.020
32. Willison HJ, O'Hanlon GM. The immunopathogenesis of Miller Fisher syndrome. J Neuroimmunol. (1999) 100:3–12. doi: 10.1016/S0165-5728(99)00213-1
33. Susuki K, Baba H, Tohyama K, Kanai K, Kuwabara S, Hirata K, et al. Gangliosides contribute to stability of paranodal junctions and ion channel clusters in myelinated nerve fibers. Glia. (2007) 55:746–57. doi: 10.1002/glia.20503
34. Dagklis IE, Papagiannopoulos S, Theodoridou V, Kazis D, Argyropoulou O, Bostantjopoulou S. Miller-Fisher syndrome: are anti-GAD antibodies implicated in its pathophysiology? Case Rep Neurol Med. (2016) 2016:3431849. doi: 10.1155/2016/3431849
35. Fouka P, Alexopoulos H, Akrivou S, Trohatou O, Politis PK, Dalakas MC. GAD65 epitope mapping and search for novel autoantibodies in GAD-associated neurological disorders. J Neuroimmunol. (2015) 281:73–7. doi: 10.1016/j.jneuroim.2015.03.009
36. Pietrini V, Pavesi G, Andreetta F. Miller Fisher syndrome with positivity of anti-GAD antibodies. Clin Neurol Neurosurg. (2013) 115:2399–400. doi: 10.1016/j.clineuro.2012.11.008
37. Shoraka AR, Fang X, Hamouda D, Gogia B, Li X. Ataxia and ophthalmoplegia: an atypical case of Miller Fisher syndrome (MFS) with anti-GAD antibody. Int J Neurosci. (2022) 132:994–8. doi: 10.1080/00207454.2020.1859502
38. Dalakas MC. Stiff-person syndrome and GAD antibody-spectrum disorders: GABAergic neuronal excitability, immunopathogenesis and update on antibody therapies. Neurother J Am Soc Exp Neurother. (2022) 19:832–47. doi: 10.1007/s13311-022-01188-w
39. Javaid H, Ejaz U, Slowinski Z. Case of Bickerstaff encephalitis with positive glutamic acid decarboxylase antibodies. Br Med J Case Rep. (2020) 13:e234814. doi: 10.1136/bcr-2020-234814
40. Maury A, Lyoubi A, Peiffer-Smadja N, de Broucker T, Meppiel E. Neurological manifestations associated with SARS-CoV-2 and other coronaviruses: a narrative review for clinicians. Rev Neurol. (2021) 177:51–64. doi: 10.1016/j.neurol.2020.10.001
41. Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. J Am Med Assoc Neurol. (2020) 77:683–90. doi: 10.1001/jamaneurol.2020.1127
42. Conde Cardona G, Quintana Pájaro LD, Quintero Marzola ID, Ramos Villegas Y, Moscote Salazar LR. Neurotropism of SARS-CoV 2: mechanisms and manifestations. J Neurol Sci. (2020) 412:116824. doi: 10.1016/j.jns.2020.116824
43. Gigli GL, Bax F, Marini A, Pellitteri G, Scalise A, Surcinelli A, et al. Guillain-Barré syndrome in the COVID-19 era: just an occasional cluster? J Neurol. (2021) 268:1195–7. doi: 10.1007/s00415-020-09911-3
44. Gutiérrez-Ortiz C, Méndez-Guerrero A, Rodrigo-Rey S, San Pedro-Murillo E, Bermejo-Guerrero L, Gordo-Mañas R, et al. Miller Fisher syndrome and polyneuritis cranialis in COVID-19. Neurology. (2020) 95:e601–5. doi: 10.1212/WNL.0000000000009619
45. Li Z, Li X, Shen J, Chan MTV, Wu WKK. Miller Fisher syndrome associated with COVID-19: an up-to-date systematic review. Environ Sci Pollut Res Int. (2021) 28:20939–44. doi: 10.1007/s11356-021-13233-w
46. Romero-Sánchez CM, Díaz-Maroto I, Fernández-Díaz E, Sánchez-Larsen Á, Layos-Romero A, García-García J, et al. Neurologic manifestations in hospitalized patients with COVID-19: the ALBACOVID registry. Neurology. (2020) 95:e1060–70. doi: 10.1212/WNL.0000000000009937
47. Aladawi M, Elfil M, Abu-Esheh B, Abu Jazar D, Armouti A, Bayoumi A, et al. Guillain Barre syndrome as a complication of COVID-19: a systematic review. Can J Neurol Sci. 49:38–48. doi: 10.1017/cjn.2021.102
48. Dang YL, Bryson A. Miller-Fisher Syndrome and Guillain-Barre Syndrome overlap syndrome in a patient post Oxford-AstraZeneca SARS-CoV-2 vaccination. Br Med J Case Rep. (2021) 14:e246701. doi: 10.1136/bcr-2021-246701
49. Nishiguchi Y, Matsuyama H, Maeda K, Shindo A, Tomimoto H. Miller Fisher syndrome following BNT162b2 mRNA coronavirus 2019 vaccination. BMC Neurol. (2021) 21:452. doi: 10.1186/s12883-021-02489-x
50. Neophytou P, Artemiadis A, Hadjigeorgiou GM, Zis P. Miller Fischer syndrome after COVID-19 infection and vaccine: a systematic review. Acta Neurol Belg. (2023) 23:5. doi: 10.1007/s13760-023-02336-5
51. Liang H, Cao Y, Zhong W, Ma Z, Liu J, Chen H. Miller-Fisher syndrome and Guillain-Barre syndrome overlap syndrome following inactivated COVID-19 vaccine: case report and scope review. Hum Vaccines Immunother. (2022) 18:2125753. doi: 10.1080/21645515.2022.2125753
52. Nishimoto Y, Odaka M, Hirata K, Yuki N. Usefulness of anti-GQ1b IgG antibody testing in Fisher syndrome compared with cerebrospinal fluid examination. J Neuroimmunol. (2004) 148:200–5. doi: 10.1016/j.jneuroim.2003.11.017
53. Shahrizaila N, Yuki N. Bickerstaff brainstem encephalitis and Fisher syndrome: anti-GQ1b antibody syndrome. J Neurol Neurosurg Psychiatry. (2013) 84:576–83. doi: 10.1136/jnnp-2012-302824
54. Yeh JH, Chen WH, Chen JR, Chiu HC. Miller Fisher syndrome with central involvement: successful treatment with plasmapheresis. Ther Apher Off J Int Soc Apher Jpn Soc Apher. (1999) 3:69–71. doi: 10.1046/j.1526-0968.1999.00104.x
55. Berlit P, Rakicky J. The Miller Fisher syndrome. Review of the literature. J Clin Neuroophthalmol. (1992) 12:57–63.
56. Tezer FI, Gurer G, Karatas H, Nurlu G, Saribas O. Involvement of the central nervous system in Miller Fisher syndrome: a case report. Clin Neurol Neurosurg. (2002) 104:377–9. doi: 10.1016/S0303-8467(02)00016-1
57. Keane JR, Finstead BA. Upward gaze paralysis as the initial sign of Fisher's syndrome. Arch Neurol. (1982) 39:781–2. doi: 10.1001/archneur.1982.00510240043012
58. Scheid R, Lincke T, Voltz R, von Cramon DY, Sabri O. Serial 18F-fluoro-2-deoxy-D-glucose positron emission tomography and magnetic resonance imaging of paraneoplastic limbic encephalitis. Arch Neurol. (2004) 61:1785–9. doi: 10.1001/archneur.61.11.1785
59. Kim YK, Kim JS, Jeong SH, Park KS, Kim SE, Park SH. Cerebral glucose metabolism in Fisher syndrome. J Neurol Neurosurg Psychiatry. (2009) 80:512–7. doi: 10.1136/jnnp.2008.154765
60. Mihara M, Miyai I, Hatakenaka M, Kubota K, Sakoda S. Sustained prefrontal activation during ataxic gait: a compensatory mechanism for ataxic stroke? Neuroimage. (2007) 37:1338–45. doi: 10.1016/j.neuroimage.2007.06.014
61. Taphoorn MJ, Uitdehaag BM, Lanting P. A brain-stem lesion in the Miller Fisher syndrome demonstrated by CT and MRI. J Neurol. (1991) 238:243. doi: 10.1007/BF00314791
62. Giroud M, Mousson C, Chalopin JM, Rifle G, Dumas R. Miller-Fisher syndrome and pontine abnormalities on MRI: a case report. J Neurol. (1990) 237:489–90. doi: 10.1007/BF00314769
63. Garcia-Rivera CA, Rozen TD, Zhou D, Allahyari P, Niknam R, Dougherty D, et al. Miller Fisher syndrome: MRI findings. Neurology. (2001) 57:1755. doi: 10.1212/WNL.57.10.1755
64. Sauron B, Bouche P, Cathala HP, Chain F, Castaigne P. Miller Fisher syndrome: clinical and electrophysiologic evidence of peripheral origin in 10 cases. Neurology. (1984) 34:953–6. doi: 10.1212/WNL.34.7.953
65. Fross RD, Daube JR. Neuropathy in the Miller Fisher syndrome: clinical and electrophysiologic findings. Neurology. (1987) 37:1493–8. doi: 10.1212/WNL.37.9.1493
66. Jamal GA, Ballantyne JP. The localization of the lesion in patients with acute ophthalmoplegia, ataxia and areflexia (Miller Fisher syndrome). A serial multimodal neurophysiological study. Brain J Neurol. (1988) 111:95–114. doi: 10.1093/brain/111.1.95
67. Dachy B, Deltenre P, Deconinck N, Dan B. The H reflex as a diagnostic tool for Miller Fisher syndrome in pediatric patients. J Clin Neurosci Off J Neurosurg Soc Australas. (2010) 17:410–1. doi: 10.1016/j.jocn.2009.06.014
68. Jacobs BC, O'Hanlon GM, Bullens RWM, Veitch J, Plomp JJ, Willison HJ. Immunoglobulins inhibit pathophysiological effects of anti-GQ1b-positive sera at motor nerve terminals through inhibition of antibody binding. Brain J Neurol. (2003) 126:2220–34. doi: 10.1093/brain/awg235
69. Mori M, Kuwabara S, Fukutake T, Hattori T. Intravenous immunoglobulin therapy for Miller Fisher syndrome. Neurology. (2007) 68:1144–6. doi: 10.1212/01.wnl.0000258673.31824.61
70. Ishii J, Yuki N, Kawamoto M, Yoshimura H, Kusunoki S, Kohara N. Recurrent Guillain-Barré syndrome, Miller Fisher syndrome and Bickerstaff brainstem encephalitis. J Neurol Sci. (2016) 364:59–64. doi: 10.1016/j.jns.2016.03.008
71. Chida K, Nomura H, Konno H, Takase S, Itoyama Y. Recurrent Miller Fisher syndrome: clinical and laboratory features and HLA antigens. J Neurol Sci. (1999) 165:139–43. doi: 10.1016/S0022-510X(99)00095-7
72. Sekiguchi Y, Mori M, Misawa S, Sawai S, Yuki N, Beppu M, et al. How often and when Fisher syndrome is overlapped by Guillain-Barré syndrome or Bickerstaff brainstem encephalitis? Eur J Neurol. (2016) 23:1058–63. doi: 10.1111/ene.12983
73. Kuwabara S. Is ‘Bickerstaff brainstem encephalitis' really encephalitis? J Neurol Neurosurg Psychiatry. (2012) 2012:304655. doi: 10.1136/jnnp-2012-304655
74. Hughes RC, Wijdicks EFM, Barohn R, Benson E, Cornblath DR, Hahn AF, et al. Practice parameter: immunotherapy for Guillain-Barré syndrome: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. (2003) 61:736–40. doi: 10.1212/WNL.61.6.736
75. Mori M, Kuwabara S. Fisher syndrome. Curr Treat Options Neurol. (2011) 13:71–8. doi: 10.1007/s11940-010-0103-8
76. Koga M, Gilbert M, Takahashi M, Li J, Hirata K, Kanda T, et al. GQ1b-seronegative Fisher syndrome: clinical features and new serological markers. J Neurol. (2012) 259:1366–74. doi: 10.1007/s00415-011-6360-y
77. Nakatsuji Y, Sadahiro S, Watanabe S, Fujimura H, Abe K, Koguchi K, et al. Leptomeningeal signet-ring cell carcinomatosis presenting with ophthalmoplegia, areflexia and ataxia. Clin Neuropathol. (2001) 20:272–5.
Keywords: Miller Fisher, Miller Fisher syndrome, anti-GQ1b antibody, ataxia, ophthalmoparesis
Citation: Noioso CM, Bevilacqua L, Acerra GM, Della Valle P, Serio M, Vinciguerra C, Piscosquito G, Toriello A, Barone P and Iovino A (2023) Miller Fisher syndrome: an updated narrative review. Front. Neurol. 14:1250774. doi: 10.3389/fneur.2023.1250774
Received: 30 June 2023; Accepted: 07 August 2023;
Published: 24 August 2023.
Edited by:
German Moris, SESPA, SpainReviewed by:
Yuji Tomizawa, Juntendo University, JapanCopyright © 2023 Noioso, Bevilacqua, Acerra, Della Valle, Serio, Vinciguerra, Piscosquito, Toriello, Barone and Iovino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ciro Maria Noioso, Yy5ub2lvc29AeWFob28uY29t