 Pareena Chaitanuwong
Pareena Chaitanuwong Heather E. Moss
Heather E. Moss- 1Ophthalmology Department, Rajavithi Hospital, Ministry of Public Health, Bangkok, Thailand
- 2Department of Ophthalmology, College of Medicine, Rangsit University, Bangkok, Thailand
- 3Department of Ophthalmology, Stanford University, Palo Alto, CA, United States
- 4Department of Neurology & Neurological Sciences, Stanford University, Palo Alto, CA, United States
Background: Optic neuritis (ON) is an inflammatory condition of the optic nerve that can lead to significant visual impairment. It is often associated with multiple sclerosis (MS) but can also occur in other demyelinating diseases, such as neuromyelitis optica spectrum disorder (NMOSD) and myelin oligodendrocyte glycoprotein antibody disease (MOGAD). Understanding the current therapeutic approaches and emerging treatment strategies is critical for optimizing patient outcomes.
Objective: This review provides a focused overview of current therapies for demyelinating optic neuritis associated with MS, NMOSD, and MOGAD. Less common autoimmune optic neuropathies, non-autoimmune causes (e.g., infections) and pediatric optic neuritis are not covered.
Methods: A review of the literature was conducted, including clinical trials, observational studies, and expert recommendations on the treatment and management of demyelinating ON. The efficacy, safety, and limitations of various therapeutic modalities were assessed.
Results: High-dose intravenous corticosteroids remain the mainstay of acute demyelinating ON treatment, accelerating visual recovery but not altering long-term visual outcomes. Immunomodulatory therapies, such as disease-modifying treatments for MS, play a crucial role in preventing recurrent episodes in demyelinating diseases. Emerging therapies, including re-myelination agents, neuroprotective strategies, and novel immunotherapies, show promise in improving visual prognosis and reducing long-term disability.
Conclusion: While corticosteroids remain the primary treatment for acute demyelinating ON, ongoing research into neuroprotective and re-myelinating therapies offers hope for better visual recovery and long-term management. Future studies should focus on optimizing treatment strategies and exploring novel therapeutics to enhance patient outcomes.
1 Introduction
Optic neuritis (ON) is a demyelinating disorder of the optic nerve that can lead to sudden vision loss in one or both eyes. Optic neuritis can be caused by both infectious and non-infectious factors. The most common non-infectious cause of optic neuritis worldwide is multiple sclerosis (MS) (1), but it can also occur in association with other demyelinating diseases, such as neuromyelitis optica spectrum disorder (NMOSD) and myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD), or without an identifiable cause. The relative incidence of different causes varies according to geographic location.
The standard treatment for optic neuritis is intravenous methylprednisolone (IVMP) (2), which has been shown to hasten the recovery of vision but not impact long-term visual outcome of MS and idiopathic forms. In recent years, there has been a growing interest in the use of other treatments for optic neuritis, such as oral steroids, plasma exchange, and other immunomodulatory drugs.
This review focuses on current treatment strategies for demyelinating ON in the context of MS, NMOSD, and MOGAD. Rare autoimmune [e.g., Chronic Relapsing Inflammatory Optic Neuritis (CRION), Autoimmune glial fibrillary acid protein (GFAP), autoimmune collapsin response-mediator protein-5 (CRMP5)], non-autoimmune causes (e.g., infectious ON) and pediatric optic neuritis are beyond the scope of this article. Furthermore, we will discuss the potential future directions of research in the field of optic neuritis treatment.
2 Overview of autoimmune optic neuritis
Autoimmune ON is primarily categorized into two types: typical ON (idiopathic or MS-associated) and atypical ON. The common atypical ON cases, characterized by biomarkers such as aquaporin-4 (AQP4) and myelin oligodendrocyte glycoprotein (MOG) antibodies, represents distinct CNS demyelinating diseases. Less common autoimmune optic neuropathies such as CRION, GFAP-associated astrocytopathy, and paraneoplastic CRMP5 optic neuropathy, are differentiated by clinical presentation and testing. Differentiating between typical and atypical ON is essential for understanding prognosis and treatment variations. While the Optic Neuritis Treatment Trial (ONTT) (3) remains foundational in guiding the acute treatment of typical optic neuritis, several limitations must be acknowledged in light of evolving understanding. Notably, the study was conducted prior to the identification of biomarkers for NMOSD and MOGAD. Furthermore, patients with bilateral, recurrent or severe ON phenotypes were excluded. As a result, its findings are not generalizable to NMOSD or MOGAD-associated optic neuritis. In addition, the ONTT was predominantly composed of young white females from North America, limiting its applicability to more diverse populations. The emergence of serological testing and advanced MRI biomarkers has significantly refined ON diagnostic and prognostic stratification, necessitating an updated framework for interpreting ONTT results in today's clinical practice (3).
2.1 Typical optic neuritis (MS-associated, idiopathic)
Typical ON usually presents with unilateral vision loss in young adults and tends to recover spontaneously. While most patients regain good visual acuity, MRI and CSF analysis are important for assessing MS risk. Recent diagnostic criteria updates now allow earlier MS diagnosis in ON patients, which may influence early therapeutic decisions (4–6).
2.2 Atypical optic neuritis (NMO, MOG)
Atypical ON is often bilateral, more severe, and recurrent, with poorer visual prognosis. NMOSD-associated ON is linked with AQP4-IgG and often involves the optic chiasm (7). MOGAD-associated ON is typically seen in younger patients and presents with marked disc edema and good recovery, though relapses are not common (8, 9). Accurate diagnosis of both conditions via antibody testing and/or clinical criteria is essential, as treatment differs from typical ON (10, 11).
As our understanding of optic neuritis expands, especially with the identification of atypical forms such as NMOSD and MOGAD, accurate classification has become increasingly important for guiding management. These subtypes differ markedly in clinical presentation, diagnostic pathways, and therapeutic approaches. Due to overlapping features and evolving diagnostic criteria, clinicians may face challenges in early differentiation. To support clinical decision-making, Table 1 provides a visual summary that contrasts typical and atypical ON, highlighting key diagnostic clues and outlining distinct treatment strategies for demyelinating ON. Management of optic neuritis can be challenging due to decisions for treatment being required prior to availability of supporting evidence (MRI, NMO serology, MOG serology) to triage as to the underlying cause. In these situations clinical features can assist in preliminary triage as MS/idiopathic, NMO-SD (severe vision loss, bilateral involvement) and MOG (severe disc edema). It is particularly important to be alert for features of NMO-SD so that therapy can be escalated accordingly.
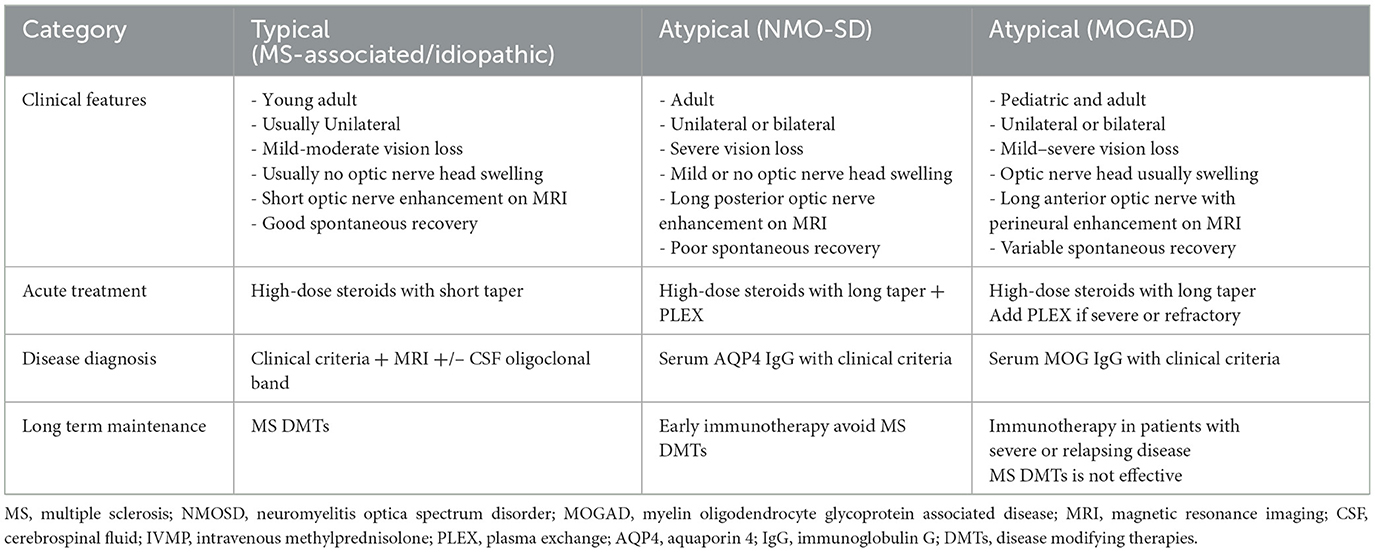
Table 1. Simplified clinical and therapeutic approach to typical vs. atypical optic neuritis.
3 Treatment approaches for optic neuritis and review of clinical studies
3.1 Acute treatment
High-dose corticosteroids, either oral or intravenous (IV), are the primary treatments for acute autoimmune ON. The largest trial, ONTT, found that high dose intravenous corticosteroids (1 g/day for 3 consecutive days) expedite initial visual recovery within the first 2 weeks, by about 1–2 lines of Snellen visual acuity. Interestingly, low-dose oral prednisone (1 mg/kg) was associated with increased risk of ON relapse within the initial 2 years, discouraging its use. Recent studies have shown that high-dose oral corticosteroids may be non-inferior to high-dose intravenous steroid treatments for treating MS relapses (12). Additionally, high-dose oral methylprednisolone may be a cost-effective alternative with comparable patient satisfaction to traditional intravenous administration (13).
Notably, ONTT results may not be broadly generalizable, especially for atypical ON variants like NMOSD-ON. Acute flares of NMOSD-ON, including optic neuritis, should be treated with IVMP (1 g/day for 3–5 consecutive days with or without a PO prednisone taper). Early treatment has been shown to correlate with preservation of peripapillary retinal nerve fiber layer. Retrospective studies suggest that IV corticosteroids alone might be suboptimal for visual recovery in NMOSD-ON, and that concurrent or sequential plasma exchange (PLEX) may improve outcomes. PLEX can be initiated in IVMP-refractory disease, with one series reporting average final visual acuity of 20/50 in NMOSD-ON patients receiving sequential IVMP and PLEX compared to 20/400 in those receiving IVMP alone. In two non-randomized studies of acute NMOSD (including some ON cases), 40%−50% of attacks treated with PLEX within 2 days of symptom onset experienced complete recovery, and 0%−5% recovering fully with PLEX initiation after 20 days (14, 15). Although high-quality randomized controlled trial data specifically addressing the impact of PLEX in NMOSD are currently lacking, retrospective studies suggest potential benefits from early PLEX therapy when combined with high-dose corticosteroids.
In cases of MOGAD, much like with other ON, the standard approach to acute treatment typically involves IVMP, which tends to yield rapid responses in most patients (16, 17). One retrospective study encompassing both AQP4-IgG+NMOSD and MOGAD cases found a potential benefit from initiating treatment at an earlier stage (18). For individuals experiencing severe attacks with significant disability at the peak of the attack, it is advisable to consider the early implementation of a combined therapy involving intravenous corticosteroids and plasma exchange (PLEX) (19).
Several studies have evaluated non-steroid treatments for acute optic neuritis, but none have shown a clear benefit (Table 2). Some of the failure to demonstrate benefit might relate to trial design. Future trials should prioritize earlier administration post-injury, focus on well-characterized patient subgroups with features likely to benefit from the intervention (e.g., active demyelination but limited neurodegeneration in the case of re-myelinating agents), and adopt more sensitive and specific outcome measures, such as functional imaging and electrophysiological assessments targeting affected white matter tracts. Due to varying recovery patterns in different ON subtypes (NMOSD, MOG, etc.), expert-opinion-based treatment approaches are being proposed.
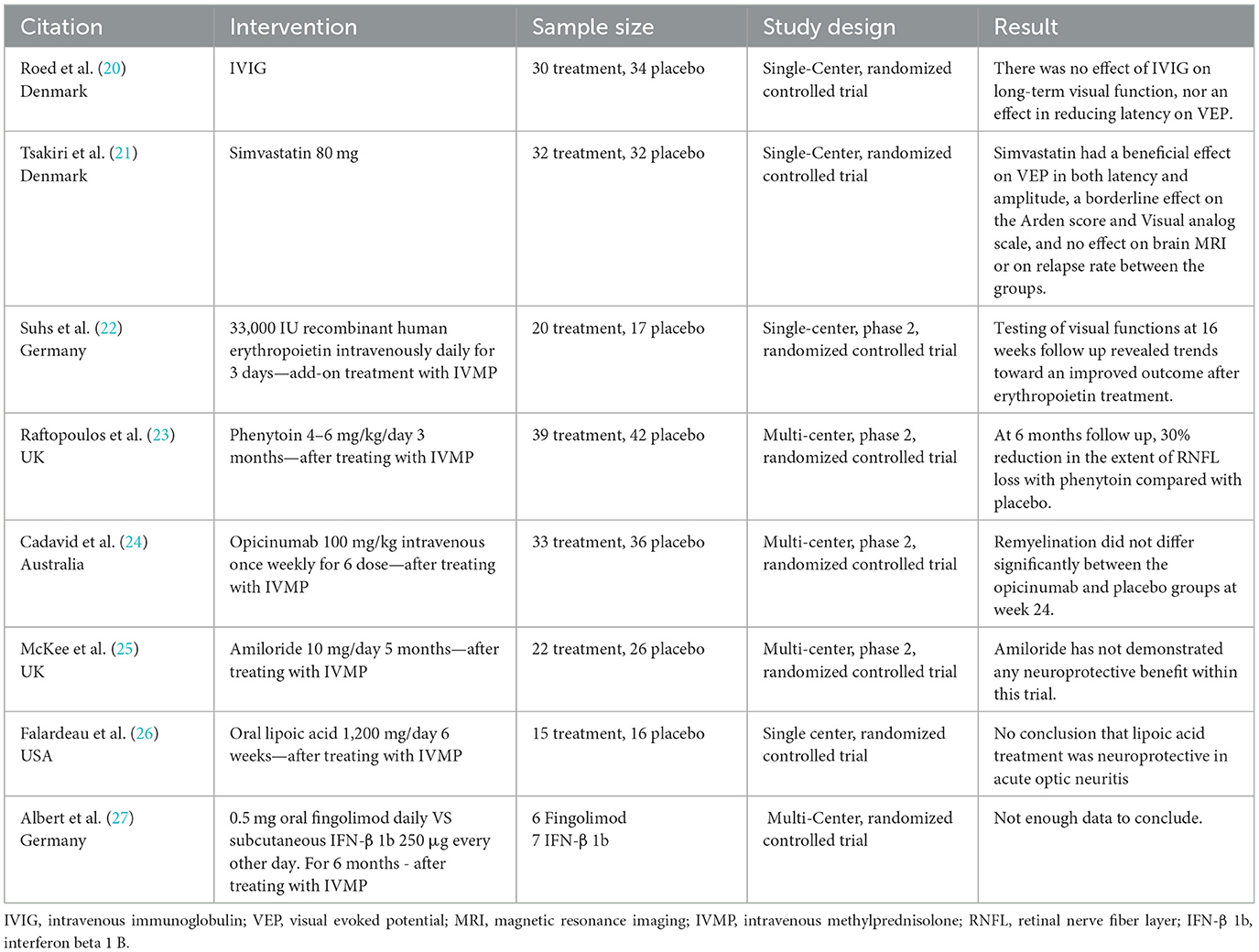
Table 2. Research on non-steroid treatments for acute optic neuritis.
3.2 Long term management
Prevention of recurrence and preservation of long-term visual function are the primary objectives of long-term treatment following acute optic neuritis. The prognosis following the initial attack, the likelihood of relapses and the appropriate treatment are contingent upon the underlying inflammatory condition (i.e., MS, NMO or MOG). When a diagnosis of MS, NMO or MOG cannot be established it is important to reassess the patient on a regular basis to determine if diagnostic criteria have been met so that appropriate treatment can be initiated.
Optic neuritis associated with NMOSD typically exhibits the poorest visual prognosis from the outset, and subsequent relapses can worsen this. In NMOSD, extended courses of oral prednisone are typically given following acute therapy [intravenous methylprednisolone (IVMP) with or without PLEX], with consideration for early initiation of immunosuppressive disease modifying therapy to reduce risk of future episodes of optic neuritis.
Similarly, MOGAD presents a less favorable visual prognosis compared to multiple sclerosis (MS) optic neuritis. After the administration of IVMP, patients are often started on an oral prednisone tapering regimen, which typically extends beyond the two-week duration recommended by the ONTT, and consideration of PLEX for severe cases with poor recovery. Disease modifying therapy initiation in MOGAD is based on relapses and persistence of serum MOG antibodies.
Multiple sclerosis (MS) treatment encompasses a comprehensive array of strategies aimed at managing the disease's complex facets. Many FDA-approved medications have demonstrated efficacy in managing MS over the long term. Disease-modifying therapy (DMT) forms the cornerstone of management of MS by reducing relapse frequency and neurological disability. These therapies have evolved over time, with newer options replacing earlier, less targeted immunosuppressive treatments. Treatment strategies vary, with escalation from low efficacy to high efficacy medications. However, a newer approach involves initiating higher-efficacy treatments at diagnosis to achieve better relapse control.
3.2.1 Multiple sclerosis DMT
MS disease modifying therapies act by modulating the immune system through diverse mechanisms, including sequestration of lymphocytes, alteration of cytokine secretion patterns, and immune cell depletion and vary by efficacy, adverse effects and route of administration (Table 3). Newly developed DMTs, including monoclonal antibodies exhibit heightened efficacy compared to traditional oral and injectable options. While infusion reactions and autoimmune side effects remain potential concerns, their overall benefits are significant.
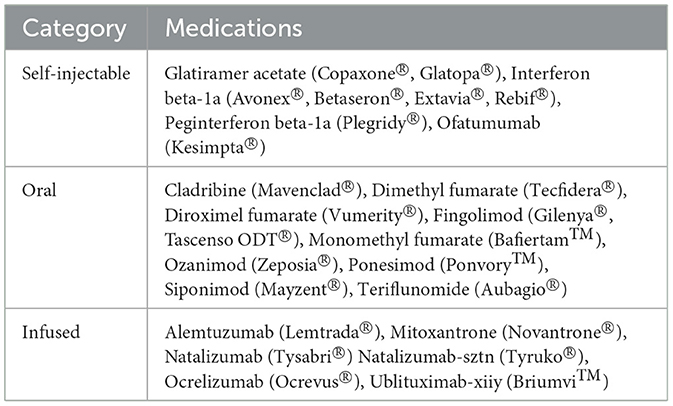
Table 3. The table categorizes the FDA-approved disease-modifying therapies (DMTs) for multiple sclerosis (MS) by their method of administration (29).
Selection of specific MS DMT are tailored to individual patient's needs. Although DMT treatment is typically long-term, people with stable disease while on certain DMTs may choose to de-escalate therapy. The reader is referred to the excellent review by McGinley et al. for more information (28).
3.2.2 NMOSD
Certain drugs that are approved for multiple sclerosis (MS) treatment should not be used in patients with neuromyelitis optica spectrum disorder (NMOSD) (30, 31), especially in those who are AQP4-IgG-positive. Some of these drugs, such as glatiramer acetate, are simply ineffective in preventing NMOSD attacks (32). Others, such as interferon beta (31), natalizumab (33), fingolimod, alemtuzumab (34), and dimethyl fumarate (35), have been reported to trigger severe NMOSD attacks.
Mitoxantrone may have some effect on reducing the frequency of NMOSD attacks, but it should no longer be used due to its unfavorable safety profile and the limited duration of treatment (36). Cyclophosphamide is another drug that has been tried in NMOSD, but the results have been conflicting. It is not recommended for use in NMOSD due to the limited total dose allowance and potentially severe side effects (37).
Many studies have been conducted in the last few years to develop medications to treat NMOSD. These new drugs have shown significant promise in clinical trials, reducing the frequency of relapses and improving neurological function in NMOSD patients. As a result, four new drugs have been approved in the U.S. for the treatment of NMOSD: eculizumab, inebilizumab, satralizumab, and ravulizumab- cwvz.
• Eculizumab is a monoclonal antibody that blocks the complement protein C5. It is the only drug approved for both AQP4-IgG seropositive and AQP4-IgG seronegative NMOSD (38).
• Inebilizumab is a monoclonal antibody that targets CD19-positive B cells. It is approved for the treatment of AQP4-IgG seropositive NMOSD (39).
• Satralizumab is a monoclonal antibody that targets the interleukin-6 receptor. It is approved for the treatment of AQP4-IgG seropositive NMOSD as an adjunct therapy to other immunosuppressants or as monotherapy (40, 41).
• Ravulizumab-cwvz is a long-acting C5 complement inhibitor. It is approved for the treatment of adult patients with AQP4-IgG seropositive NMOSD (42).
In addition to these new drugs, other immunosuppressants, such as azathioprine, mycophenolate mofetil, and rituximab, are also commonly used to treat NMOSD based on historical experience. However, these drugs are not FDA-approved for NMOSD. The choice of treatment for NMOSD should be individualized based on the patient's clinical presentation, serologic status, and response to previous treatments. Additional information can be found in the excellent review by Gospe et al. (43).
3.2.3 MOGAD
Long-term management of myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) focuses on preventing relapses and limiting cumulative neurological damage. The risk of relapse is influenced by several factors, including persistent seropositivity for MOG-IgG, especially when present at high titers, and a history of multiple clinical events. Conversely, pediatric patients (44), male patients presenting with spinal cord involvement and those who receive corticosteroid therapy for at least 1 month during the first attack are more likely to experience a monophasic disease course and a longer time to first relapse (45). Additionally, seroconversion to negative MOG-IgG has been associated with a reduced risk of recurrence (45). About half of people with MOGAD only have one relapse, and those who do have relapses often make a good recovery after the first one (46). Some experts advise that patients with MOGAD who make a full recovery after the first attack do not need to take long-term immunosuppressive drugs until they are proven to have relapsing disease by having another relapse (47).
Currently, there are no FDA approved treatments for MOGAD. Disease-modifying MS medications, such as interferon-β and glatiramer acetate have been shown to be ineffective in MOGAD, while data on natalizumab are ambiguous (25). In contrast, immunosuppressants such as mycophenolate mofetil (MMF), azathioprine, intravenous immunoglobulin (IVIG), and rituximab have all been associated with reductions in the annualized relapse rate (ARR) (48, 49). A recent multi-center retrospective study found that IVIG was associated with the lowest relapse rate among a variety of immunosuppressants (50). Furthermore, a recent meta-analysis of 41 primarily retrospective observational studies demonstrated that azathioprine, MMF, rituximab, IVIG, and tocilizumab are effective in reducing relapse risk in both pediatric and adult patients with MOGAD (51). Despite these promising findings, it is important to note that, to date, no randomized controlled trials have been conducted for MOGAD, underscoring the urgent need for high-quality prospective studies to guide evidence-based treatment strategies.
4 Future directions in optic neuritis treatment
Current treatments for optic neuritis primarily aim to reduce acute inflammation and prevent long-term relapses, but they are not always effective. There is an urgent need for new and more effective treatments for optic neuritis. Some of the most promising future directions in optic neuritis treatment include:
• Neuroprotective treatments that aim to prevent damage to the optic nerve during inflammatory attacks. Promising agents include memantine, erythropoietin, interferon-beta, phenytoin, and clemastine. However, recent literature reviews have not yet demonstrated significant clinical differences, and further studies are needed in the future (52).
• Remyelination therapies that promote the repair of damaged myelin in the optic nerve. Notable remyelinating agents under clinical investigation for treating optic neuritis in multiple sclerosis (MS) include Ibudilast and Mesenchymal Stem Cells (MSC) (53).
Among remyelination-promoting therapies, opicinumab, a monoclonal antibody targeting LINGO1, a CNS protein that inhibits remyelination, was developed based on preclinical studies showing promotion of oligodendrocyte survival, axonal regeneration, and remyelination. Phase I trials confirmed its safety, leading to three Phase II studies: RENEW (optic neuritis) (24), SYNERGY (MS) (54), and AFFINITY (MS). While RENEW showed delayed improvement in visual evoked potentials, none of the trials met their primary endpoints, and development was discontinued following the failure of AFFINITY (55). Despite promising results in animal models, the human clinical translation of opicinumab has been limited by challenges such as blood-brain barrier penetration, identification of reliable biomarkers, and determining the optimal treatment window (56). Early intervention, ideally within 25 days of symptom onset, and careful patient selection appear critical, with better responses seen in older patients with optic neuritis and younger MS patients with preserved CNS structure (24, 54, 55).
• Gene therapy is a rapidly advancing field with the potential to develop regenerative treatments for optic neuritis and other optic neuropathy. One promising approach involves delivering neuroprotective gene-encoding proteins directly into the optic nerve. This strategy is currently under preclinical investigation and may also hold therapeutic potential for other neurological disorders in the future (51). Another emerging direction is gene therapy aimed at enabling or enhancing retinal ganglion cell (RGC) regeneration and functional integration (57). While this remains in the experimental stage, such therapies could ultimately transform care for patients with irreversible vision loss. Large-scale collaborative efforts, such as the RGC Repopulation, Stem Cell Transplantation, and Optic Nerve Regeneration (RReSTORe) Consortium, are essential to overcoming current challenges and translating these advances into clinical practice (58). For more information, the author suggests an excellent review by Esposito et al. on this topic (59).
Further research is needed to elucidate the underlying pathophysiological mechanisms of MS-, NMOSD- and MOGAD-associated optic neuritis. A better understanding of these mechanisms, may facilitate the development of newer, more precise and targeted therapies aimed at preserving vision and improving quality of life for the affected individuals.
5 Conclusion
In conclusion, optic neuritis is a complex condition with various underlying causes. It presents with sudden and often painful vision loss and can be associated with demyelinating diseases like multiple sclerosis (MS), neuromyelitis optica spectrum disorder (NMOSD), and myelin oligodendrocyte glycoprotein antibody disease (MOGAD). While intravenous methylprednisolone (IVMP) remains the standard treatment for acute optic neuritis of these varieties, its impact on long-term visual outcomes is limited. Therefore, exploring alternative treatments has become an area of growing interest.
Typical optic neuritis, often associated with MS, tends to have a favorable prognosis, with most patients experiencing significant visual recovery. However, monitoring and early diagnosis are crucial for identifying those at risk of developing MS. Treatment strategies for MS involve disease-modifying therapies (DMTs), which aim to reduce relapse frequency and disability.
Atypical optic neuritis, especially in the context of NMOSD and MOGAD, presents unique challenges. Differentiating between typical and atypical forms is essential, as their prognoses and treatment approaches differ significantly. NMOSD, characterized by the presence of AQP4-IgG antibodies, benefits from PLEX in the acute setting and immunosuppressive therapies like eculizumab, inebilizumab, and satralizumab in the long term. MOGAD ON can have excellent recovery and minimal risk of relapse, but some cases have poor recovery and relapsing disease. In these more severe cases PLEX and chronic treatments like mycophenolate mofetil (MMF), azathioprine, intravenous immunoglobulin (IVIG), and rituximab show promise in reducing relapse rates.
Ongoing research in neuroprotective therapies, remyelination strategies, gene therapy and disease-specific pathophysiology holds promise for the development of targeted and effective treatments for optic neuritis.
Author contributions
PC: Conceptualization, Data curation, Methodology, Writing – original draft, Writing – review & editing. HM: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. P30 026877 (NIH), unrestricted grant from Research to Prevent Blindness.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Toosy AT, Mason DF, Miller DH. Optic neuritis. Lancet Neurol. (2014) 13:83–99. doi: 10.1016/S1474-4422(13)70259-X
2. Beck RW, Gal RL. Treatment of acute optic neuritis: a summary of findings from the optic neuritis treatment trial. Arch Ophthalmol. (2008) 126:994–5. doi: 10.1001/archopht.126.7.994
3. Beck RW. The optic neuritis treatment trial. Arch Ophthalmol. (1988) 106:1051–3 doi: 10.1001/archopht.1988.01060140207023
4. Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. (2018) 17:162–73 doi: 10.1016/S1474-4422(17)30470-2
5. Montalban. Revisions of the McDonald Criteria. Scientific Session 1: New diagnostic Criteria. Denmark: ECTRIMS Congress Copenhagen (2024).
6. Huang J, Khademi M, Fugger L, Lindhe Ö, Novakova L, Axelsson M, et al. Inflammation-related plasma and CSF biomarkers for multiple sclerosis. Proc Natl Acad Sci U S A. (2020) 117:12952–60. doi: 10.1073/pnas.1912839117
7. Winter A, Chwalisz B. MRI characteristics of NMO, MOG and MS related optic neuritis. Semin Ophthalmol. (2020) 35:333–42. doi: 10.1080/08820538.2020.1866027
8. Jurynczyk M, Messina S, Woodhall MR, Raza N, Everett R, Roca-Fernandez A, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. (2017) 140:3128–38. doi: 10.1093/brain/awx276
9. Cobo-Calvo A, Ruiz A, Maillart E, Audoin B, Zephir H, Bourre B, et al. Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults: the MOGADOR study. Neurology. (2018) 90:e1858–e69. doi: 10.1212/WNL.0000000000005560
10. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. (2015) 85:177–89. doi: 10.1212/WNL.0000000000001729
11. Banwell B, Bennett JL, Marignier R, Kim HJ, Brilot F, Flanagan EP, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: international MOGAD panel proposed criteria. Lancet Neurol. (2023) 22:268–82. doi: 10.1016/S1474-4422(22)00431-8
12. Burton JM, O'Connor PW, Hohol M, Beyene J. Oral versus intravenous steroids for treatment of relapses in multiple sclerosis. Cochrane Database Syst Rev. (2012) 12:Cd006921. doi: 10.1002/14651858.CD006921.pub3
13. Horta-Hernández AM, Esaclera-Izquierdo B, Yusta-Izquierdo A, Martín-Alcalde E, Blanco-Crespo M, Álvarez-Nonay A, et al. High-dose oral methylprednisolone for the treatment of multiple sclerosis relapses: cost-minimisation analysis and patient's satisfaction. Eur J Hosp Pharm. (2019) 26:280–4. doi: 10.1136/ejhpharm-2018-001499
14. Kleiter I, Gahlen A, Borisow N, Fischer K, Wernecke KD, Hellwig K, et al. Apheresis therapies for NMOSD attacks: a retrospective study of 207 therapeutic interventions. Neurol Neuroimmunol Neuroinflamm. (2018) 5:e504. doi: 10.1212/NXI.0000000000000504
15. Bonnan M, Valentino R, Debeugny S, Merle H, Fergé JL, Mehdaoui H, et al. Short delay to initiate plasma exchange is the strongest predictor of outcome in severe attacks of NMO spectrum disorders. J Neurol Neurosurg Psychiatry. (2018) 89:346–51. doi: 10.1136/jnnp-2017-316286
16. Marignier R, Hacohen Y, Cobo-Calvo A, Pröbstel AK, Aktas O, Alexopoulos H, et al. Myelin-oligodendrocyte glycoprotein antibody-associated disease. Lancet Neurol. (2021) 20:762–72. doi: 10.1016/S1474-4422(21)00218-0
17. Ramanathan S, Mohammad S, Tantsis E, Nguyen TK, Merheb V, Fung VSC, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry. (2018) 89:127–37. doi: 10.1136/jnnp-2017-316880
18. Stiebel-Kalish H, Hellmann MA, Mimouni M, Paul F, Bialer O, Bach M, et al. Does time equal vision in the acute treatment of a cohort of AQP4 and MOG optic neuritis? Neurology Neuroimmunol Neuroinflamm. (2019) 6:e572. doi: 10.1212/NXI.0000000000000572
19. Weinshenker BG, O'Brien PC, Petterson TM, Noseworthy JH, Lucchinetti CF, Dodick DW, et al. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann Neurol. (1999) 46:878–86. doi: 10.1002/1531-8249(199912)46:6<878::aid-ana10>3.0.co;2-q
20. Roed H, Langkilde A, Sellebjerg F, Lauritzen M, Bang P, Mørup A, et al. A double-blind, randomized trial of IV immunoglobulin treatment in acute optic neuritis. Neurology. (2005) 64:804–10. doi: 10.1212/01.WNL.0000152873.82631.B3
21. Tsakiri A, Kallenbach K, Fuglø D, Wanscher B, Larsson H, Frederiksen J. Simvastatin improves final visual outcome in acute optic neuritis: a randomized study. Mult Scler J. (2012) 18:72–81. doi: 10.1177/1352458511415452
22. Sühs KW, Hein K, Sättler MB, Görlitz A, Ciupka C, Scholz K, et al. A randomized, double-blind, phase 2 study of erythropoietin in optic neuritis. Ann Neurol. (2012) 72:199–210. doi: 10.1002/ana.23573
23. Raftopoulos R, Hickman SJ, Toosy A, Sharrack B, Mallik S, Paling D, et al. Phenytoin for neuroprotection in patients with acute optic neuritis: a randomised, placebo-controlled, phase 2 trial. Lancet Neurol. (2016) 15:259–69. doi: 10.1016/S1474-4422(16)00004-1
24. Cadavid D, Balcer L, Galetta S, Aktas O, Ziemssen T, Vanopdenbosch L, et al. Safety and efficacy of opicinumab in acute optic neuritis (RENEW): a randomised, placebo-controlled, phase 2 trial. Lancet Neurol. (2017) 16:189–99. doi: 10.1016/S1474-4422(16)30377-5
25. McKee JB, Cottriall CL, Elston J, Epps S, Evangelou N, Gerry S, et al. Amiloride does not protect retinal nerve fibre layer thickness in optic neuritis in a phase 2 randomised controlled trial. Mult Scler J. (2019) 25:246–55. doi: 10.1177/1352458517742979
26. Falardeau J, Fryman A, Wanchu R, Marracci GH, Mass M, Wooliscroft L, et al. Oral lipoic acid as a treatment for acute optic neuritis: a blinded, placebo controlled randomized trial. Mult Scler J Exp Transl Clin. (2019) 5:2055217319850193. doi: 10.1177/2055217319850193
27. Albert C, Mikolajczak J, Liekfeld A, Piper SK, Scheel M, Zimmermann HG, et al. Fingolimod after a first unilateral episode of acute optic neuritis (MOVING) – preliminary results from a randomized, rater-blind, active-controlled, phase 2 trial. BMC Neurol. (2020) 20:1–12. doi: 10.1186/s12883-020-01645-z
28. McGinley MP, Goldschmidt CH, Rae-Grant AD. Diagnosis and treatment of multiple sclerosis: a review. JAMA. (2021) 325:765–79. doi: 10.1001/jama.2020.26858
29. Wingerchuk DM, Carter JL. Multiple sclerosis: current and emerging disease-modifying therapies and treatment strategies. Mayo Clinic Proc. (2014) 89:225–40. doi: 10.1016/j.mayocp.2013.11.002
30. Barnett MH, Sutton I. Neuromyelitis optica: not a multiple sclerosis variant. Curr Opin Neurol. (2012) 25:215–20. doi: 10.1097/WCO.0b013e3283533a3f
31. Shimizu J, Hatanaka Y, Hasegawa M, Iwata A, Sugimoto I, Date H, et al. IFNβ-1b may severely exacerbate Japanese optic-spinal MS in neuromyelitis optica spectrum. Neurology. (2010) 75:1423–7. doi: 10.1212/WNL.0b013e3181f8832e
32. Ayzenberg I, Schöllhammer J, Hoepner R, Hellwig K, Ringelstein M, Aktas O, et al. Efficacy of glatiramer acetate in neuromyelitis optica spectrum disorder: a multicenter retrospective study. J Neurol. (2016) 263. doi: 10.1007/s00415-015-7991-1
33. Kornberg MD, Newsome SD. Unmasking and provoking severe disease activity in a patient with NMO spectrum disorder. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e66. doi: 10.1212/NXI.0000000000000066
34. Kowarik MC, Hoshi M, Hemmer B, Berthele A. Failure of alemtuzumab as a rescue in a NMOSD patient treated with rituximab. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e208. doi: 10.1212/NXI.0000000000000208
35. Javadian N, Magrouni H, Ghaffarpour M, Ranji-Burachaloo S. Severe relapses of neuromyelitis optica spectrum disorder during treatment with dimethyl fumarate. Clin Neuropharmacol. (2021) 44:21–2. doi: 10.1097/WNF.0000000000000430
36. Enriquez CAG, Espiritu AI, Pasco PMD. Efficacy and tolerability of mitoxantrone for neuromyelitis optica spectrum disorder: a systematic review. J Neuroimmunol. (2019) 332:126–34. doi: 10.1016/j.jneuroim.2019.04.007
37. Kageyama T, Miyamoto K, Ozaki A, Komori M, Okunomiya T, Kambe D, et al. Long-term effect of cyclosporine A in the prophylaxis of neuromyelitis optica spectrum disorders (P2.160). Neurology. (2016) 86(16 Suppl):P2.160. doi: 10.1212/WNL.86.16_supplement.P2.160
38. Pittock SJ, Berthele A, Fujihara K, Kim HJ, Levy M, Palace J, et al. Eculizumab in aquaporin-4–positive neuromyelitis optica spectrum disorder. N Engl J Med. (2019) 381:614–25. doi: 10.1056/NEJMoa1900866
39. Cree BA, Bennett JL, Kim HJ, Weinshenker BG, Pittock SJ, Wingerchuk DM, et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet. (2019) 394:1352–63. doi: 10.1016/S0140-6736(19)31817-3
40. Traboulsee A, Greenberg BM, Bennett JL, Szczechowski L, Fox E, Shkrobot S, et al. Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: a randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol. (2020) 19:402–12. doi: 10.1016/S1474-4422(20)30078-8
41. Yamamura T, Kleiter I, Fujihara K, Palace J, Greenberg B, Zakrzewska-Pniewska B, et al. Trial of satralizumab in neuromyelitis optica spectrum disorder. N Engl J Med. (2019) 381:2114–24. doi: 10.1056/NEJMoa1901747
42. Pittock SJ, Barnett M, Bennett JL, Berthele A, de Sèze J, Levy M, et al. Ravulizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. Ann Neurol. (2023) 93:1053–68. doi: 10.1002/ana.26626
43. Gospe SM, Chen JJ, Bhatti MT. Neuromyelitis optica spectrum disorder and myelin oligodendrocyte glycoprotein associated disorder-optic neuritis: a comprehensive review of diagnosis and treatment. Eye. (2021) 35:753–68. doi: 10.1038/s41433-020-01334-8
44. Bruijstens AL, Breu M, Wendel E-M, Wassmer E, Lim M, Neuteboom RF, et al. E.U. paediatric MOG consortium consensus: part 4 – outcome of paediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur J Paediatr Neurol. (2020) 29:32–40. doi: 10.1016/j.ejpn.2020.10.007
45. Huda S, Whittam D, Jackson R, Karthikeayan V, Kelly P, Linaker S, et al. Predictors of relapse in MOG antibody associated disease: a cohort study. BMJ Open. (2021) 11:e055392. doi: 10.1136/bmjopen-2021-055392
46. Han JY, Kim SY, Kim W, Kim H, Cho A, Choi J, et al. Phenotype of relapsing myelin oligodendrocyte glycoprotein antibody-associated disease in Children. J Clin Neurol. (2025) 21:65–73. doi: 10.3988/jcn.2024.0276
47. Matteo G, Thomas F, Giacomo G, Silvia S, Eleonora R, Stefano M, et al. Prognostic relevance of quantitative and longitudinal MOG antibody testing in patients with MOGAD: a multicentre retrospective study. J Neurol Neurosurg Psychiatry. (2023) 94:201. doi: 10.1136/jnnp-2022-330237
48. Chen JJ, Flanagan EP, Bhatti MT, Jitprapaikulsan J, Dubey D, Lopez Chiriboga AS, et al. Steroid-sparing maintenance immunotherapy for MOG-IgG associated disorder. Neurology. (2020) 95:e111–e20. doi: 10.1212/WNL.0000000000009758
49. Li S, Ren H, Xu Y, Xu T, Zhang Y, Yin H, et al. Long-term efficacy of mycophenolate mofetil in myelin oligodendrocyte glycoprotein antibody-associated disorders. Neurology. (2020) 7:e705. doi: 10.1212/NXI.0000000000000705
50. Whittam DH, Karthikeayan V, Gibbons E, Kneen R, Chandratre S, Ciccarelli O, et al. Treatment of MOG antibody associated disorders: results of an international survey. J Neurol. (2020) 267:3565–77. doi: 10.1007/s00415-020-10026-y
51. Chang X, Zhang J, Li S, Wu P, Wang R, Zhang C, et al. Meta-analysis of the effectiveness of relapse prevention therapy for myelin-oligodendrocyte glycoprotein antibody-associated disease. Mult Scler Relat Disord. (2023) 72:104571. doi: 10.1016/j.msard.2023.104571
52. Tsai TH, Lin CW, Chan LW, Tew TB, Chen TC. Neuroprotective effects of novel treatments on acute optic neuritis–a meta-analysis. Biomedicines. (2022) 10:192. doi: 10.3390/biomedicines10010192
53. Aneesh A, Liu A, Moss HE, Feinstein D, Ravindran S, Mathew B, et al. Emerging concepts in the treatment of optic neuritis: mesenchymal stem cell-derived extracellular vesicles. Stem Cell Res Ther. (2021) 12:594. doi: 10.1186/s13287-021-02645-7
54. Cadavid D, Mellion M, Hupperts R, Edwards KR, Calabresi PA, Drulović J, et al. Safety and efficacy of opicinumab in patients with relapsing multiple sclerosis (SYNERGY): a randomised, placebo-controlled, phase 2 trial. Lancet Neurol. (2019) 18:845–56. doi: 10.1016/S1474-4422(19)30137-1
56. Neumann B, Foerster S, Zhao C, Bodini B, Reich DS, Bergles DE, et al. Problems and pitfalls of identifying remyelination in multiple sclerosis. Cell Stem Cell. (2020) 26:617–9. doi: 10.1016/j.stem.2020.03.017
57. Harvey AR. Gene therapy and the regeneration of retinal ganglion cell axons. Neural Regen Res. (2014) 9:232–3. doi: 10.4103/1673-5374.128213
58. Soucy JR, Aguzzi EA, Cho J, Gilhooley MJ, Keuthan C, Luo Z, et al. Retinal ganglion cell repopulation for vision restoration in optic neuropathy: a roadmap from the RReSTORe consortium. Mol neurodegener. (2023) 18:64. doi: 10.1186/s13024-023-00655-y
Keywords: optic neuritis, multiple sclerosis, corticosteroids, treatment in optic neuritis, emerging therapies
Citation: Chaitanuwong P and Moss HE (2025) Optic neuritis: a comprehensive review of current therapies and emerging treatment strategies. Front. Neurol. 16:1605075. doi: 10.3389/fneur.2025.1605075
Received: 02 April 2025; Accepted: 02 June 2025;
Published: 18 June 2025.
Edited by:
Sachin Kedar, Emory University, United StatesReviewed by:
Essam Mohamed Elmatbouly Saber, Benha University, EgyptXiangxiang Liu, Capital Medical University, China
Ana Banc, University of Medicine and Pharmacy Iuliu Hatieganu, Romania
Copyright © 2025 Chaitanuwong and Moss. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heather E. Moss, aGVtb3NzQHN0YW5mb3JkLmVkdQ==