 Emilie Faivre1
Emilie Faivre1 Joana E. Coelho2
Joana E. Coelho2 Katja Zornbach3
Katja Zornbach3 Enas Malik4
Enas Malik4 Younis Baqi4,5
Younis Baqi4,5 Marion Schneider4
Marion Schneider4 Lucrezia Cellai1
Lucrezia Cellai1 Kevin Carvalho1
Kevin Carvalho1 Shéhérazade Sebda6
Shéhérazade Sebda6 Martin Figeac6Sabiha Eddarkaoui1Raphaëlle Caillierez1
Martin Figeac6Sabiha Eddarkaoui1Raphaëlle Caillierez1 Yijuang Chern7Michael Heneka8,9
Yijuang Chern7Michael Heneka8,9 Nicolas Sergeant1
Nicolas Sergeant1 Christa E. Müller4
Christa E. Müller4 Annett Halle3,8
Annett Halle3,8 Luc Buée1
Luc Buée1 Luisa V. Lopes2
Luisa V. Lopes2 David Blum1*
David Blum1*- 1Université de Lille, Inserm, CHU-Lille, LabEx DISTALZ, Jean-Pierre Aubert Research Centre UMR-S1172, Alzheimer & Tauopathies, Lille, France
- 2Instituto de Medicina Molecular, Faculdade de Medicina de Lisboa, Universidade de Lisboa, Lisbon, Portugal
- 3Center of Advanced European Studies and Research, Bonn, Germany
- 4PharmaCenter Bonn, Pharmaceutical Institute, Pharmaceutical Chemistry I, University of Bonn, Bonn, Germany
- 5Department of Chemistry, Faculty of Science, Sultan Qaboos University, Muscat, Oman
- 6Plateau de Génomique Fonctionnelle et Structurale, CHU Lille, University of Lille, Lille, France
- 7Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan
- 8German Center for Neurodegenerative Diseases (DZNE), Bonn, Germany
- 9Department of Neurodegenerative Diseases and Geropsychiatry/Neurology, University of Bonn Medical Center, Bonn, Germany
Consumption of caffeine, a non-selective adenosine A2A receptor (A2AR) antagonist, reduces the risk of developing Alzheimer’s disease (AD) and mitigates both amyloid and Tau lesions in transgenic mouse models of the disease. While short-term treatment with A2AR antagonists have been shown to alleviate cognitive deficits in mouse models of amyloidogenesis, impact of a chronic and long-term treatment on the development of amyloid burden, associated neuroinflammation and memory deficits has never been assessed. In the present study, we have evaluated the effect of a 6-month treatment of APPsw/PS1dE9 mice with the potent and selective A2AR antagonist MSX-3 from 3 to 9-10 months of age. At completion of the treatment, we found that the MSX-3 treatment prevented the development of memory deficits in APP/PS1dE9 mice, without significantly altering hippocampal and cortical gene expressions. Interestingly, MSX-3 treatment led to a significant decrease of Aβ1-42 levels in the cortex of APP/PS1dE9 animals, while Aβ1-40 increased, thereby strongly affecting the Aβ1-42/Aβ1-40 ratio. Together, these data support the idea that A2AR blockade is of therapeutic value for AD.
Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disorder in the elderly. AD is characterized by a progressive cognitive decline and neuropathologically defined by two hallmarks: extracellular deposits constisting of aggregated β-amyloid (Aβ) peptides and intraneuronal fibrillar aggregates of hyper- and abnormally phosphorylated Tau proteins (Masters et al., 1985; Duyckaerts et al., 2015). AD is dependent on various genetic and environmental factors (Reitz et al., 2011; Cuyvers and Sleegers, 2016). Among protective factors, several epidemiological studies have reported an inverse relation between caffeine intake, age-related cognitive impairments and the risk to develop AD later in life (for reviews see Flaten et al., 2014; Cunha, 2016). In accordance, we and others have shown that caffeine is beneficial towards memory impairments and pathology in transgenic mouse models of AD (Arendash et al., 2006, 2009; Cao et al., 2009; Laurent et al., 2014).
Beneficial effects of caffeine have been ascribed to its ability to block adenosine A2A receptors (A2ARs), a G protein-coupled receptor whose endogenous ligand is adenosine (Cunha, 2016). In line with a role of A2ARs in AD, an association between a polymorphism of the ADORA2A gene with hippocampal volume in mild cognitive impairment and AD has been recently reported (Horgusluoglu-Moloch et al., 2017). Like caffeine, pharmacological and genetic A2AR blockade was found to reduce hippocampal pathology, neuroinflammation and memory deficits in a model of AD-like Tau pathology (Laurent et al., 2016). A2AR blockade or deletion was also found to counteract synaptotoxicity and memory deficits acutely induced by Aβ peptides (Dall’lgna et al., 2003, 2007; Canas et al., 2009). In line, recent data emphasize in transgenic models of amyloidogenesis (APP/PS1dE9 and hAPP-J20), that short-term treatments (from 1 to 3 weeks) with selective A2AR antagonists (SCH58260 i.p. or KW6002 p.o.) revert memory alterations (Viana da Silva et al., 2016; Orr et al., 2018; Silva et al., 2018). However, the impact of a long-term and chronic A2AR blockade on the development of amyloid pathology and associated memory impairment has not been investigated yet.
In the present study, we explored the outcomes of a chronic pharmacological blockade of A2AR in the APPswe/PS1dE9 transgenic mouse model, using the selective water-soluble antagonist MSX-3. Our data demonstrate that chronic MSX-3 treatment delivered from 3 to 9-10 months of age in APPsw/PS1dE9 mice improves spatial memory deficits and moderately reduces cortical amyloid load. These data support the notion that targeting A2ARs is of therapeutic interest for AD.
Materials and Methods
Animals
In this study, we used heterozygous male APPswe/PS1dE9 (herein referred to as APP/PS1, C57Bl6/J background; Jankowsky et al., 2001) and littermates controls. All animals were maintained in standard cages under conventional laboratory conditions (12 h/12 h light/dark cycle, 22°C), with ad libitum access to food and water. Animals were maintained 5–6 per cage with genotype segregated with enrichment as the form of small cylinder “cocoon,” which offer the animal the possibility to fulfill their natural nesting instinct. The animals were used in compliance with European standards for the care and use of laboratory animals and experimental protocols approved by the local Animal Ethical Committee (agreement APAFIS#2264-2015101320441671 from CEEA75, Lille, France).
MSX-3 Treatment
MSX-3 is a water-soluble prodrug of the potent and highly selective A2AR antagonist MSX-2 (Sauer et al., 2000), that crosses the blood-brain barrier (Collins et al., 2010). The drug was administered through drinking water at 0.3 g/L, a dose previously shown to provide benefit in Tau transgenic mice (Laurent et al., 2016). Chronic delivery at this dose achieved, in 10-month-old C57Bl6/J mice, plasma and brain concentrations of MSX-2 of about 13 nM and 50 nM, respectively (Supplementary Figure S1), compatible with A2AR blockade (MSX-2 Ki = ca. 8 nM). Animals randomized according to their body weight, were assigned to the four following experimental groups: WT/H2O, WT/MSX-3, APP/PS1/H2O and APP/PS1/MSX-3. The MSX-3 solution was kept in bottles protected from light and changed weekly. Treatment started at 3 months of age, when amyloid pathology begins and before memory impairments in APP/PS1 mice, and continued until 9–10 months of age, when mice exhibit cortical and hippocampal amyloid pathology and memory deficits. MSX-3 consumption was assessed throughout treatment for each experimental cage. In average, mice consumed 6.6 ± 0.6 ml of the MSX-3 solution per day, corresponding to an average daily intake of 2 mg of the antagonist.
Anxiety Assessment Using Elevated Plus Maze
The elevated plus maze was used to investigate anxiety-related behavior. The apparatus consisted of a plus-shaped maze with two closed and two open arms (30 cm long × 6.5 cm wide). Mice were placed at the center of the maze with their face in the direction of a closed arm and were allowed to explore freely for 5 min. Distance moved, velocity and time spent in arms were recorded using the Ethovision XT tracking system (Noldus).
Spatial Memory Assessment Using the Morris Water Maze Task
Spatial memory abilities were evaluated in the standard hidden platform (PF) acquisition and retention version of the Morris Water Maze as previously described (Laurent et al., 2016). A 100-cm circular pool was filled with water, opacified with non-toxic white paint and kept at 21°C. A 10-cm round PF was hidden 1 cm beneath the surface of the water at a fixed position. Four positions around the edge of the tank were arbitrarily designated 1, 2, 3 and 4, thus dividing the tank into four quadrants (clockwise): target (hidden-PF contained), adjacent 1, opposite and adjacent 2. During the learning procedure, each mouse was given four swimming trials per day (15 min inter-trial interval) for five consecutive days. The start position (1, 2, 3, or 4) was pseudo-randomized across trials. A trial consisted of placing the mouse into the water facing the outer edge of the pool in one of the virtual quadrants and allowing it to escape to the hidden PF. A trial terminated when the animal reached the PF where it was allowed to remain for 15 s. If the animal failed to find the target before 120 s, it was manually guided to the PF where it was allowed to stay for 15 s. After completion of a trial, mice were removed from the pool and placed back to their home cages. Distance traveled to locate the hidden escape PF (path length) and swimming speed (i.e., velocity, as a measure of possible motor defects that could interfere with their ability to perform in this task) were recorded using the Ethovision XT tracking system (Noldus). Seventy-two hours following the acquisition phase, a probe trial was conducted. During this probe trial (60 s), the PF was removed and search pattern of the mice was tracked again. Proportion of time spent in the target quadrant (T) vs. averaged non-target quadrants (O) was determined.
Sacrifice and Brain Tissue Preparation
Sacrifice of animals took place in the afternoon. Mice were deeply anesthetized with pentobarbital sodium (50 mg/kg, i.p.), then transcardially perfused with cold NaCl (0.9%). Brains were removed and one half of the hemisphere were post-fixed in 4% paraformaldehyde fixative in PBS (pH 7.4) for a week at 4°C and transferred to 30% sucrose solution overnight before being frozen. Coronal brains sections (35 μm) were obtained using a Leica cryostat. Free-floating sections were selected according the stereological rules, with the first section taken at random and every ninth sections afterwards and were stored in PBS-azide (0.2%) at 4°C. Cortex and hippocampus of the other hemisphere were dissected using a coronal acrylic slicer (Delta Microscopies) at 4°C and stored at −80°C until use.
Immunohistochemistry and Image Analysis
Antibodies used in this study are listed in Table 1. For Aβ immunohistochemistry (IHC), sections were pretreated with 80% formic acid for 3 min and were permeabilized with 0.2% Triton X-100/sodium phosphate buffer. Sections were then blocked with 10% “Mouse On Mouse” Kit serum (Vector Laboratories) for 1 h before incubation with mouse biotinylated anti-Aβ antibody (6E10) at 4°C overnight. After washing in PBS, the sections were incubated with the ABC kit (Vector Laboratories) for 2 h and developed using DAB (Sigma). Images were acquired using Leica ICC50 HD microscope. Quantification of the 6E10 staining intensity was performed using Mercator software (Explora Nova, Mountain View, CA, USA). The number of plaques, the average plaque size and the plaque burden, expressed as percentage of analyzed area, were calculated in the cortex and hippocampus of the APP/PS1 mice. For immunofluorescence studies, coronal brain sections were washed with sodium phosphate buffer and permeabilized with 0.2% Triton X-100/sodium phosphate buffer. Sections where blocked with normal goat serum (1/100; Vector Laboratories) before incubated with an anti-GFAP antibody (Table 1) at 4°C overnight. Primary antibody was detected with Alexa Fluor 488 or 633 goat anti-rabbit IgG (1/500, Thermo Fisher). After washes, sections were blocked with donkey serum (1/100; Sigma) and were incubated for 48 h at 4°C with an anti-A2AR antibody followed by an incubation with Alexa Fluor 595 donkey anti-guinea pig IgG (1/500; Jackson) antibody for 2 h at RT. To visualize amyloid plaques, sections were pretreated with 80% formic acid for 3 min, blocked with 10% of Mouse On Mouse Kit serum (Vector Laboratories) for 1 h before incubated in mouse biotinylated anti-Aβ antibody at 4°C overnight. Sections were then incubated with streptavidin Alexa Fluo 488 conjugate (1/1000; Thermo Fisher Scientific). Sections were finally incubated with DAPI (1/5000; Sigma-Aldrich) for 10 min and treated for 10 min in 0.3% Suden Black (Sigma-Aldrich) with 70% ethanol to block autofluorescence. Images were acquired using a Zeiss LSM 710 confocal laser-scanning microscope to define co-localization of A2AR with glial markers and amyloid plaques. 3D reconstruction of 2D confocal z stacks was performed using Imaris software (Bitplane, South Windsor, CT, USA).
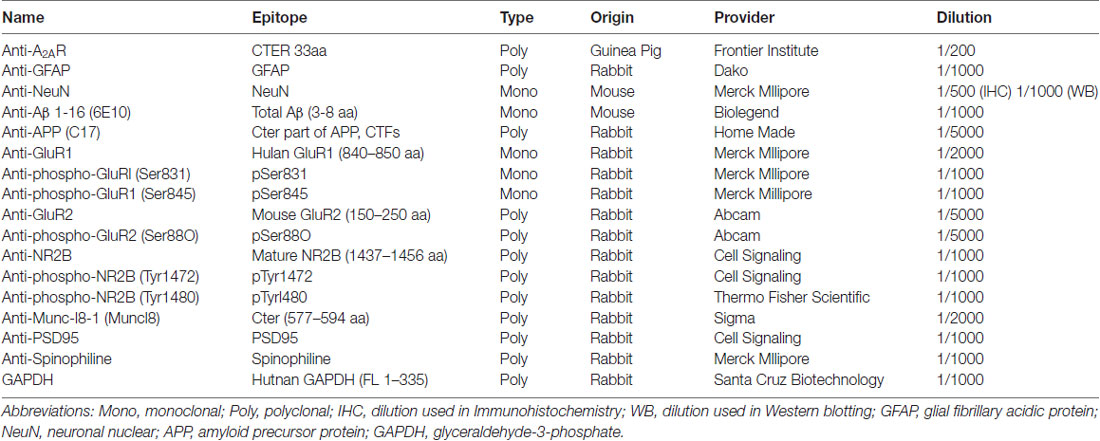
Table 1. Antibodies used in this study.
Western Blots
For all biochemical experiments, tissue was homogenized in 200 μL Tris buffer (pH 7.4) containing 10% sucrose and protease inhibitors (Complete; Roche Diagnostics GmbH), sonicated, and kept at −80°C until use. Protein amounts were evaluated using the BCA assay (Pierce). Protein amounts were evaluated using the BCA assay (Pierce), subsequently diluted with LDS 2X supplemented with reducing agents (Invitrogen) and then separated on 4%–12% NuPage Novex gels (Invitrogen). Proteins were transferred to nitrocellulose membranes, saturated (5% non-fat dry milk or 5% BSA) in TNT (Tris 15 mM pH 8, NaCl 140 mM, 0.05% Tween) and incubated with primary (see Table 1) overnight and then corresponding secondary antibodies (peroxidase labeled horse anti-rabbit 1/5000 or anti-mouse 1/50,000, Vector Laboratories). Immunoreactivity was visualized using chemiluminescence kits (ECLTM, Amersham Bioscience) and a LAS3000 imaging system (Fujifilm). Results were normalized to GAPDH and quantifications were performed using ImageJ software (Scion Software).
mRNA Extraction and Quantitative Real-Time RT-PCR Analysis
Total RNA was extracted from hippocampi and cortex, and purified using the RNeasy Lipid Tissue Mini Kit (Qiagen, France). One microgram of total RNA was reverse-transcribed using the Applied Biosystems High-Capacity cDNA reverse transcription kit. Quantitative real-time RT-PCR analysis was performed on an Applied Biosystems Prism 7900 System using Power SYBR Green PCR Master Mix. The thermal cycler conditions were as follows: hold for 10 min at 95°C, followed by 45 cycles of a two-step PCR consisting of a 95°C step for 15 s followed by a 60°C step for 25 s. Sequences of primers used are given in Table 2. Cyclophilin A was used as internal control. Amplifications were carried out in triplicate and the relative expression of target genes was determined by the ΔΔCT method.

Table 2. Primer sequences used in this study.
ELISA Measurements
Brain levels of human Aβ1-40 and Aβ1-42 were measured using ELISA kits (Invitrogen, Carlsbad, CA, USA; IBL-International, Hamburg, Germany) following manufactured’ instructions. Briefly, for hippocampal and cortical samples, 20 μg of protein were diluted in Guanidine/Tris buffer (Guanidine HCl 5 M and Tris 50 mM pH 8), sonicated and incubated for 1 h at 4°C under agitation. Samples were then diluted in a BSAT-DPBS solution (KCl, KH2PO4, NaCl, Na2HPO4, BSA 5%, Tween-20 0.03% pH 7.4). The homogenates were centrifuged at 12,000 g for 15 min at 4°C. Supernatants were collected for the analysis of Aβ1-40 and Aβ1-42 by colorimetric immunoassays. Absorbance was measured by Multiskan Ascent counter (ThermoLab Systems). The normalized amounts of Aβ were expressed as pg/mL.
Evaluation of Microglial Phagocytosis
The effect of acute A2AR blockade on microglial phagocytosis was quantified in an in situ live cerebral slices assay similar to what has been described (Krabbe et al., 2013; Savage et al., 2015). APP/PS1 mice were crossbred with Csfr1r-EGFP mice (Sasmono et al., 2003) to readily visualize microglia. Coronal acute cerebral slices with a thickness of 130 μm were prepared from 12 month-old APP/PS1-Csf1r-EGFP mice and Csf1r-EGFP wildtype littermates using a vibratome (Leica VT1200 S). Acute slices were pre-incubated with the indicated concentration of MSX-3 (10–5000 nM) or vehicle for 60 min in artificial cerebrospinal fluid (aCSF) under constant carbogen saturation (three acute cerebral slices per condition). Live acute slices were then incubated with FCS-coated fluorescent microspheres (2 μm diameter, flash red, Bang Laboratories Inc.) at a concentration of 1.1 × 107 microspheres/mL for 60 min at 37°C in HBSS together with the indicated concentration of MSX-3 or vehicle. Slices were washed and fixed in 4% PFA. Aβ dense-core plaques were stained with 0.001% thiazine red (Sigma Aldrich) in PBS. Five corresponding regions of interest (ROIs) were recorded in the isocortex of each cerebral slice using a Nikon eclipse Ti-E confocal microscope (60× objective, 20 μm z stack, 1 μm z slice interval) and the phagocytic index, i.e., quotient between number of fluorescent microspheres internalized by GFP-positive plaque-associated microglia and the total number of plaque-associated microglia per ROI was quantified by an investigator blinded to the treatment condition.
Transcriptomic Analysis (Agilent Microarray)
Total RNA yield and quality were assessed on the Agilent 2100 bioanalyzer (Agilent Technologies, Massy, France). One color whole Mouse (074809_D_F_20150624 slides) 60-mer oligonucleotides 8× 60k microarrays (Agilent Technologies) were used to analyze gene expression. Six biological replicates for each condition were prepared for a total of 48 samples (Cortex or Hippocampus of WT-H2O/WT-MSX-3/APP- H2O/APP-MSX-3). cRNA labeling, hybridization and detection were carried out according to supplier’s instructions (Agilent Technologies). For each microarray, Cyanine 3-labeled cRNA were synthesized with the low input QuickAmp labeling kit from 50 ng of total RNA. RNA Spike-In were added to all tubes and used as positive controls of labeling and amplification steps. The labeled cRNA were purified and 600 ng of each cRNA were then hybridized and washed following manufacturer’s instructions. Microarrays were scanned on an Agilent G2505C scanner and data extracted using Agilent Feature Extraction Software© (FE version 10.7.3.1). Microarray data are available through the GEO depository from NCBI (accession no. GSE113141).
The statistical analyses were performed with Genespring® software version GX13.0 (Agilent Technologies). Microarrays have been normalized to the 75th percentile. Probes below background under all conditions were removed from the analysis. Differentially expressed genes were identified by a moderated t-test with correction of multiple tests by the Benjamini Hochberg (BH) method. We selected significantly deregulated probes with a corrected p-value of less than 0.05 and with an expression differential of at least 1.5× (FC1.5). Differentially expressed probes were further analyzed in term of molecular function and biological process using the Ingenuity Pathways Analysis (Qiagen Inc.) software.
Statistics
Results are expressed as means ± SEM. Differences between mean values were determined using the Student’s t-test, Two Way-analysis of variance (ANOVA) or One Way-ANOVA followed by a post hoc Fisher’s LSD test using Graphpad Prism Software. P values < 0.05 were considered significant.
Results
A2AR Is Overexpressed by Astrocytes in APP/PS1 Transgenic Mice
We first examined A2AR expression levels in APP/PS1 mice using IHC and confocal microscopy. Similar to what has been previously described in AD patients and in different APP models (Orr et al., 2015, 2018; Lee et al., 2018), we observed that APP/PS1 mice exhibit a progressive hippocampal upsurge of A2AR immunoreactivity (Figures 1G–I), as compared to WT littermates (Figures 1A–C). Importantly, A2AR immunopositive signal observed in the striatum and hippocampus of APP/PS1 mice was found abolished in APP/PS1 animals with genetically deleted A2AR (Supplementary Figure S2) supporting the specificity of the A2AR immunostaining provided in Figure 1. In APP/PS1 mice, increased A2AR expression was observed in astrocytes in the hippocampus, starting at 3 months of age, and the cortex, starting at 9 months of age (Figures 1D–F,J–N). Astrocytic A2AR overexpression was not found exclusively in the vicinity of 6E10-positive plaques but also in reactive astrocytes located at larger distance to plaques (Figure 1O). These data therefore indicate that the development of amyloid pathology elicits astrocytic A2AR upsurge in APP/PS1 mice.
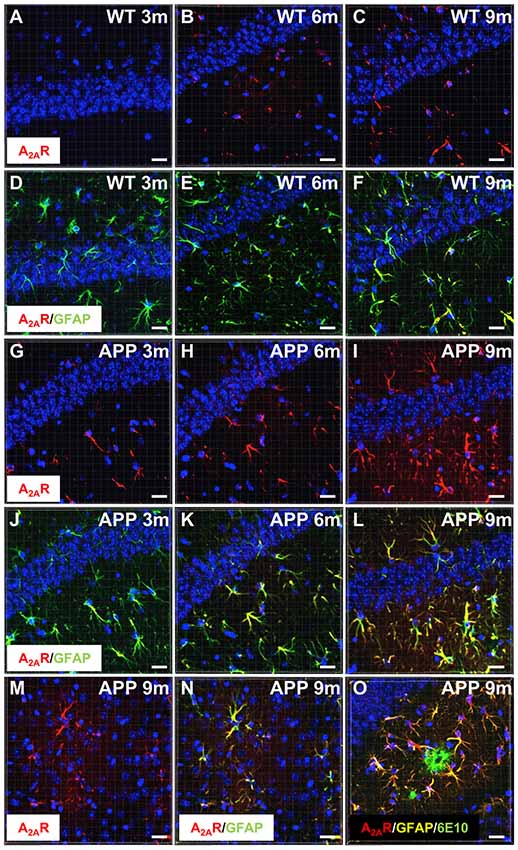
Figure 1. Astrocytic upsurge of A2ARs in APP/PS1 mice. Representative photomicrographs of hippocampal immunostaining for the A2A receptor (A2AR; red) (A–C,G–I) and merged with the astrocyte marker GFAP (green) (D–F,J–L) from WT mice (A–F) and the APP/PS1 mice (G–L), at different ages: 3 months (A,D,G,J), 6 months (B,E,H,K), and 9–10 months (C,F,I,L). Representative photomicrographs of A2AR expression (red) (M) and the merged with GFAP marker (green; N) in the cortex of 9-month-old APP/PS1 mice. Representative photomicrograph of A2AR expression (red) GFAP (yellow) and 6E10-positive amyloid plaque marker (green) in the hippocampus of 9 month-old APP/PS1 mice (O). Cell nuclei were labeled with DAPI (blue). Scale bar = 20 μm.
A2AR Pharmacological Blockade Prevents Spatial Memory Impairments in APP/PS1 Transgenic Mice
In the present experimental paradigm, APP/PS1 mice and littermate controls were treated with MSX-3 in drinking water at 0.3 g/L, using a treatment paradigm we previously described to promote a significant benefit in a model of AD-like Tau pathology (Laurent et al., 2016). Mice were treated from 3 to 9-10 months of age, i.e along the development of amyloid pathology and cognitive deficits in this transgenic strain. At completion of the treatment, we evaluated the impact of the chronic A2AR blockade on anxiety and spatial learning and memory using, respectively, the Elevated Plus Maze and the Morris Water Maze. We analyzed the impact of MSX-3 upon anxiety using the Elevated Plus Maze task. As shown in Supplementary Figure S3, we did not find any significant impact of MSX-3 upon velocity, distance moved and percentage of time spent in both closed and open arms (p > 0.05), suggesting that the treatment did not significantly impact anxiety behavior in both WT and APP/PS1 mice. Regarding Morris Water Maze, during the training phase, all groups showed a decrease in path length across trials (p < 0.05, two-way ANOVA). APP/PS1 animals demonstrated a slight learning deficit at day 2 (p = 0.0002, Two-Way ANOVA followed by Fisher LSD post hoc test) with no impact of the treatment (p > 0.05; Figure 2A). Neither APP/PS1 genotype nor the MSX-3 treatment influenced mouse velocity (p > 0.05; Figure 2B). Seventy-two hours following acquisition, a probe trial was performed to evaluate spatial memory. Regardless of treatment (H2O or MSX-3), WT littermates exhibited a significant preference for the target quadrant (Figure 2C; WT H2O: p = 0.0003; WT MSX3: p = 0.039 vs. O quadrants using One-way ANOVA followed by LSD Fisher post hoc test). As expected at this age, APP/PS1 H2O mice showed spatial memory deficits, evidenced by the absence of preference for the target vs. the non-target (O) quadrants (Figure 2C; p > 0.05 vs. O quadrants using One-way ANOVA followed by LSD Fisher post hoc test). ANOVA analysis also indicated that the percentage of time spent in target quadrant for APP/PS1 H2O mice was significantly reduced as compared to WT H2O animals (p = 0.037). In sharp contrast, the blockade of A2AR by MSX-3 significantly alleviated spatial memory impairment in APP/PS1 mice, as demonstrated by a significant preference of MSX-3-treated APP/PS1 mice for the target quadrant (Figure 2C, p = 0.0002 vs. O quadrants using One-way ANOVA followed by LSD Fisher post hoc test). ANOVA analysis also indicated that APP/PS1 mice treated with MSX-3 spent a higher percentage of time in the target quadrant as compared to water-treated APP/PS1 animals (p = 0.027; Figure 2C). Altogether, these data indicated that early onset chronic A2AR blockade prevents the development of spatial memory alterations in APP/PS1 mice.
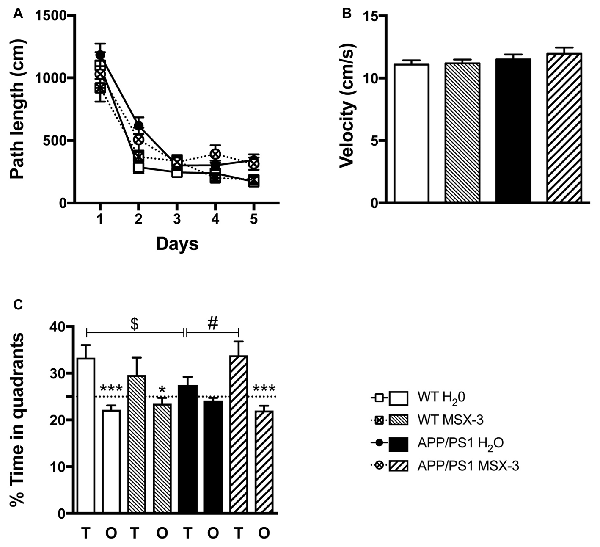
Figure 2. A2AR blockade prevents spatial memory impairments in APP/PS1 transgenic mice. Effect of MSX-3 treatment on spatial learning and memory using the Morris water-maze task. (A) Learning, as indicated by the equivalent path length needed to find the hidden platform (PF). At day 2, APP/PS1 mice exhibited a slight but significant higher path length as compared to WT animals (***p < 0.001 vs. WT H2O using One-way analysis of variance (ANOVA) followed by LSD Fisher post hoc test). (B) All genotypes exhibited a comparable velocity in the maze, suggesting no motor deficits. (C) Spatial memory was assessed 72 h after the last day of learning. Results represent the percentage of time spent in the target (T) vs. non-target (O) quadrants. WT mice (both treated with water or MSX-3) spent significantly more time in the T quadrant, indicative of a preserved spatial memory. While APP/PS1 mice exhibited spatial memory deficits as underlined by their lack of preference for the T quadrant, APP/PS1-MSX-3 treated mice behaved as WT mice, suggesting a rescue of memory impairment. *p < 0.05 ***p < 0.001 T vs. O; $p < 0.05 WT vs. APP/PS1; #p < 0.05 APP/PS1-H2O vs. APP/PS1 MSX-3 using One-way ANOVA followed by LSD Fisher post hoc test; N = 12–16 per group; Results are expressed as mean ± SEM.
Effect of Chronic A2AR Blockade on Hippocampal Synaptic Markers in APP/PS1 Transgenic Mice
To evaluate whether the beneficial effects of A2AR blockade on spatial memory in the APP/PS1 mice could be ascribed to changes in the expression of hippocampal synaptic markers, we performed western blot evaluation of neuronal, pre/post-synaptic proteins as well as glutamatergic receptors. While MSX-3 did not modulate the expression of the studied neuronal and postsynaptic markers (NeuN, spinophiline and PSD95; p > 0.05, vs. APP/PS1 H2O mice using Student’s t-test; Supplementary Figure S4A) nor the expression and phosphorylation of AMPA and NMDA receptor subunits (p > 0.05, vs. APP/PS1 H2O mice using Student’s t-test; Supplementary Figure S4B), we found that MSX-3 treatment significantly increased the hippocampal expression of the presynaptic marker Munc-18 in the APP/PS1 mice treated with MSX-3 (p = 0.037 vs. APP/PS1 H2O using Student’s t-test; Supplementary Figure S4A). Notably, none of the markers studied was found modified in the cortex of APP/PS1 MSX-3 animals (not shown).
Effect of Chronic A2AR Blockade on Cortical and Hippocampal Transcriptome of APP/PS1 Transgenic Mice
In order to provide potential molecular insights on how chronic A2AR blockade by MSX-3 improves memory of APP/PS1 mice, we evaluated gene expression changes in the cortex and hippocampus in the different groups of mice using Agilent technology. First, we determined gene expression changes between APP/PS1 H2O vs. WT H2O. In the cortex, 519 probes were significantly changed (BH adjusted p-values < 0.05) with a major effect (FC > 10) for Clec7a, Itgax, Cst, Ccl3, Ccl4 and Cxcl10 (Supplementary Table S1). In the hippocampus, 125 probes were significantly altered (BH adjusted p-values < 0.05; Supplementary Table S3) and we found the same highly modified genes, indicating highly concordant results between structures. The study of molecular functions by Ingenuity Pathway Analysis highlighted a significant number of deregulated genes in pathways related to immune functions (Supplementary Tables S2, S4), in accordance with the known link between amyloid load and the development of parenchymal neuroinflammation (Heneka et al., 2015 for review), which is known to favor cognitive deficits (Marciniak et al., 2015; Laurent et al., 2018 and references herein). Using quantitative PCR analysis, we validated several neuroinflammatory markers (GFAP, CD68, TLR2, CCL5 and CCL3) in both the cerebral cortex and the hippocampus of APP/PS1 H2O mice as compared to WT H2O animals (p < 0.001 vs. WT using One Way ANOVA followed by LSD Fisher post hoc text; Supplementary Figures S5A,B). We were then interested in comparing gene expression changes in the mouse groups treated with MSX-3. Notably, A2ARs were shown to modulate the function and activation of brain innate immune cells (i.e., microglia and astrocytes; Cunha, 2016 for review) and blockade of these receptors may resolve brain neuroinflammation (Rebola et al., 2011; Laurent et al., 2016). Surprisingly, no significant effect of MSX-3, particularly for markers associated to immune functions (see also Supplementary Figures S5A,B), was observed in the cortex or hippocampus of WT or APP/PS1. Altogether, these data suggest that memory improvement in APP/PS1 treated with MSX-3 is associated with a weak transcriptional effect and a notable absence of impact on neuroinflammation.
Effect of Chronic A2AR Blockade on Amyloid Burden in APP/PS1 Transgenic Mice
Age-dependent spatial memory impairment in APP/PS1 has been shown to correlate with increased brain amyloid burden (Savonenko et al., 2005; Garcia-Alloza et al., 2006; Zhang et al., 2011). Previous studies demonstrated that long-term caffeine treatment mitigates memory defects in APPsw mice, while reducing brain Aβ production (Arendash et al., 2006, 2009). While this is still controversial (Lu et al., 2016), other data indicated that A2AR might impact on the production of Aβ in vitro (Nagpure and Bian, 2014). Altogether, these data supported a possible involvement of A2ARs in Aβ production and/or accumulation and, therefore upon memory. Using 6E10 IHC, we first performed the analysis of cortical and hippocampal Aβ plaque load in APP/PS1 mice treated with water or MSX-3 (Figures 3A,B). We found that the treatment with MSX-3 significantly reduced, in the cortex (Figure 3D) but not in the hippocampus (Figure 3C), the density of plaques of lower size (between 50–150 μm2) as compared with APP/PS1 H2O animals (50–150 μm2, p < 0.001; 100–150 μm2, p < 0.05 vs. APP/PS1 H2O using Two-Way ANOVA followed by LSD Fisher post hoc test). Microglia surrounding amyloid plaques have an impaired phagocytic capacity (Krabbe et al., 2013; Savage et al., 2015). Since adenosine signaling and A2AR activation has been shown to impair microglial phagocytosis in cell culture and in situ (Orr et al., 2009; Bulavina et al., 2013), we tested the possibility that MSX-3 could enhance the phagocytic capacity of microglia, thereby potentially explaining the moderate plaque reduction seen in the cortex of chronically MSX-3-treated APP/PS1 animals. To test whether A2AR blockade has an immediate effect on microglial phagocytosis as has been shown for other microglial functions (Gyoneva et al., 2014, 2016), we used an in situ live acute slice assay and evaluated whether acute A2AR blockade with MSX-3 in live cerebral slices could enhance the phagocytic capacity of microglia surrounding amyloid plaques. However, although our results confirmed that 12-month-old APP/PS1 mice exhibit a reduced phagocytic activity of microglia as compared with littermate controls, acute treatment of acute slices from APP/PS1 mice with MSX-3 of up to 5 μM failed to normalize phagocytosis of plaque-associated microglia (Figure 3E), suggesting that acute A2AR blockade does not significantly affect microglial phagocytosis.
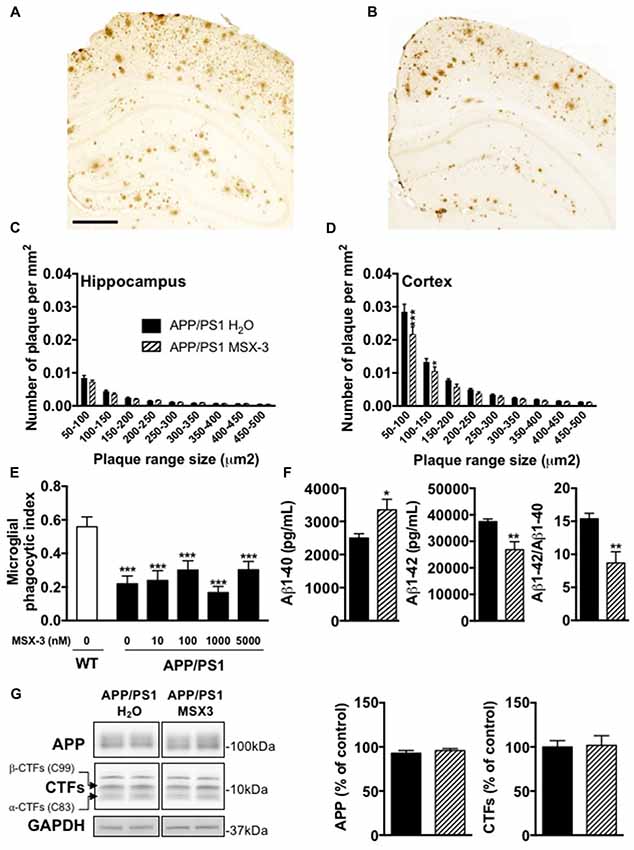
Figure 3. Impact of MSX-3 treatment on amyloid load, Aβ levels and phagocytic capacity of microglia in APP/PS1 mice. Representative images of 6E10 staining in the brains of 10 months old APP/PS1 treated with water (A) or MSX-3 (B). Scale bar = 500 μm. Distribution of amyloid plaques size were examined in the hippocampus (C) and cortex (D) of APP/PS1 mice. We found that the treatment with MSX-3 significantly reduced, in the cortex (D) but not in the hippocampus (C), the density of plaques of lower size (between 50–150 μm2) as compared with APP/PS1 H2O animals (50–150 μm2, p < 0.001; 100–150 μm2, p < 0.05 vs. APP/PS1 H2O using Two-Way ANOVA followed by LSD Fisher post hoc test; N = 7–9/group). (E) Effect of acute MSX-3 treatment on microglial phagocytic index determined in an in situ live cerebral slice assay of 12-month-old APP/PS1-Csf1r-EGFP mice and Csf1r-EGFP wildtype littermates after 60 min pre-incubation with the indicated concentration of MSX-3. Datasets were tested for significance with the One-way ANOVA and represent data from three independent experiments. ***p < 0.001 vs. WT (F) As measured by ELISA, MSX-3 treatment decreased Aβ1–42 levels in the cortex of APP/PS1 mice while Aβ1–40 levels was found increased. Overall, the Aβ1–42/Aβ1–40 ratio was found significantly reduced by the A2A antagonist treatment (*p < 0.05, **p < 0.01 vs. APP/PS1 H2O using Student’s t-test; N = 7–11/group). (G) Western blot analysis performed in cortex of water and MSX-3 treated APP/PS1dE9 mice did not revealed any change in APP and Carboxyterminal fragments (CTFs) expression (N = 6/group). Results are expressed as mean ± SEM.
Next, we measured the concentration of Aβ1-42 and Aβ1-40 in the cortex and the hippocampus. The cortex of MSX-3-treated APP/PS1 mice exhibited a significant decrease of Aβ1-42 levels (p = 0.0011 vs. APP/PS1 H2O using Student’s t-test; Figure 3F) while Aβ1-40 levels were increased (p = 0.012 vs. APP/PS1 H2O using Student’s t-test; Figure 3F). Overall, in the cortex, the Aβ42/Aβ40 ratio was found to be significantly reduced in APP/PS1 MSX-3 mice as compared to APP/PS1 H2O animals (p = 0.0015 vs. APP/PS1 H2O using Student’s t-test; Figure 3F). These cortical changes were not accompanied by a modification of APP expression or C-terminal fragments (CTF) of APP (Figure 3G). Also, none of the parameters studied was found changed in the hippocampus of APP/PS1 MSX-3 animals (p > 0.05, Student’s t-test; not shown). Altogether, these data indicated that early and chronic A2AR blockade reduces the development of cortical amyloid burden in APP/PS1 mice.
Discussion
The present study demonstrates that an early, chronic and long-term treatment with a specific A2AR antagonist, starting from an asymptomatic stage, prevents spatial memory impairments and reduces, at least in part, the development of amyloidogenesis in APPswe/PS1dE9 mice. These data extend previous findings showing that short-term treatment with different antagonists (KW6002 p.o. and SCH58261 i.p.; Viana da Silva et al., 2016; Orr et al., 2018; Silva et al., 2018) improves memory of three different models of amyloidogenesis (APP/PS1 and hAPP-J20). Here, we also provide the first experimental evidence that, in vivo, early blockade of A2ARs can reduce brain amyloid levels. Taken together with previous studies demonstrating the ability of A2AR blockade to reduce Tau hyperphosphorylation and associated cognitive decline (Laurent et al., 2016; Zhao et al., 2017), these data support that A2AR signaling is a target of interest in AD.
We found a progressive age-dependent astrocytic A2AR upsurge in the hippocampus of APP/PS1dE9 mice, in accordance with recent studies demonstrating similar receptor dysregulation in several mouse models of cerebral amyloidosis (hAPP-J20, APP KI; Orr et al., 2015, 2018; Lee et al., 2018). A2AR dysregulation likely contributes to memory deficits since conditional astrocytic-specific A2AR deletion (Orr et al., 2015) or pharmacological blockade (Orr et al., 2018; the present data) of the receptors improve memory performance in APP mice. The idea that astrocytic upregulation of A2AR might be detrimental for memory in AD models is in line with its physiological ability to control both glutamate and GABA uptake by these glial cells (Nishizaki et al., 2002; Matos et al., 2012; Cristóvão-Ferreira et al., 2013). In support of this, we recently demonstrated that A2AR deletion improves memory while normalizing glutamate/GABA balance in the hippocampus of Tau transgenic mice (Laurent et al., 2016). Interestingly, recent data demonstrated that specific deletion of astrocytic A2ARs leads to neuronal adaptative changes in glutamatergic synapses, characterized by increased evoked release of glutamate from nerve terminals or enhanced density of NR2B (Matos et al., 2015). Further, it remains also possible that modulation of astrocytic A2ARs impacts the function of synaptic A2ARs. Indeed, deletion of A2AR in astrocytes has been shown to enhance the ability of A2AR agonist CGS2160 to promote glutamate release by synaptosomes (Matos et al., 2015). Therefore, astrocytic A2AR upsurge is likely prone to favor synaptic dysfunctions leading to memory deficits in APP/PS1 mice. Further, we cannot rule out that the benefit afforded by the pharmacological A2AR blockade in APP/PS1 mice also result from the specific blockade of A2AR on other receptor subpopulations, notably at the neuronal level. Indeed, neuronal upsurge of A2AR have been observed in the brain of AD patients (Temido-Ferreira et al., 2018) and recent work demonstrated that APP/PS1 mice exhibit a significant increase of A2AR binding on synaptic hippocampal membranes (Viana da Silva et al., 2016; Silva et al., 2018). It is thus highly conceivable that APP/PS1 mice exhibit both astrocytic and synaptic A2AR upsurge, the latter being probably difficult to capture using classical immunohistofluorescence. In this regard, neuronal/synaptic modulation of A2ARs could also play an instrumental role in synaptic and memory deficits of APP mice. Indeed, mimicking neuronal A2AR upsurge using conditional transgenic models or optogenetically enhancing intracellular signaling of the receptor was sufficient to promote plasticity and memory deficits (Giménez-Llort et al., 2007; Li et al., 2015; Batalha et al., 2016). Thus, it is likely that the pharmacological blockade of A2ARs also acts at the neuronal level to normalize memory deficits. It is also important to emphasize that the link between synaptic A2AR upregulation and memory deficits goes far beyond the AD context, as demonstrated by the laboratories of Rodrigo Cunha and Luisa Lopes. For instance, synaptic A2AR have been found dysregulated in models of aging, stress or depression with behavioral manifestations normalized by A2AR receptor antagonists (Lopes et al., 1999; Rebola et al., 2003; Batalha et al., 2013; Kaster et al., 2015; Machado et al., 2017). Interestingly, in regard to presumable neuronal-based mechanisms, we observed, among the synaptic markers studied, an enhanced level of Munc-18 protein as seen by Western blot. Munc-18 is a neuronal (presynaptic) protein required for synaptic vesicles exocytosis (Carr and Rizo, 2010) and is considered as an important regulator of synaptic transmission and presynaptic strength (Toonen and Verhage, 2007; Genc et al., 2014), which is a crucial process in neuronal information processing and memory formation (Abbott and Regehr, 2004). Thus, enhancement of hippocampal Munc18 expression could also therefore contribute to memory improvement promoted by MSX-3. Besides memory impairments, it is important to mention that AD is also associated with neuropsychiatric symptoms. Recently, a study emphasized that long-term oral treatment with caffeine, a non-selective antagonist of A2ARs, exacerbate some behavioral symptoms in the triple Tg AD model (Baeta-Corral et al., 2018). While we only addressed the impact of MSX-3 on anxiety in our study behavior, which appears not affected by the antagonist, it will be important, in the future and from a therapeutic perspective, to evaluate more closely the impact of long-term A2AR blockade on AD-related behavioral symptoms.
Our microarray data indicate that both cortex and hippocampus of 9–10-month-old APP/PS1 mice exhibit gene deregulation as compared to littermate controls. As expected, the number of deregulated genes was higher in the cortex, which harbors a more advanced pathology compared to the hippocampus in this mouse model (Kim et al., 2012). Several pathways were strongly dysregulated by the development of amyloid pathology (Suh and Checler, 2002), among them processes related to immune functions and neuroinflammation, which we confirmed by qPCR analysis. Interestingly and surprisingly, none of these pathways were impacted by MSX-3 treatment. Furthermore, chronic MSX-3 treatment of WT mice did not have a significant impact on the transcriptome either. These data indicate that, overall, chronic A2AR inhibition is associated with very weak transcriptional effects. Actually, to the best of our knowledge, only one study reported an evaluation of transcriptomic changes following A2AR blockade. This study reported that constitutive deletion of A2AR in the striatum of knock-out mice leads to a significant deregulation of 152 genes compared to WT littermates (Yu et al., 2009). These data are difficult to compare to our experiments since the striatum is highly enriched in A2AR as compared to cortex and hippocampus, with a major post-synaptic localization (Blum et al., 2003a,b; Cunha, 2016). The observation that MSX-3 improves memory of APP/PS1 mice is however in accordance with its acknowledged ability to fine tune synaptic plasticity in the hippocampus (Cunha, 2016). Recent data notably emphasized that A2AR blockade can lead within minutes to an improvement of plasticity deficits in APP/PS1 mice (Viana da Silva et al., 2016).
However, improved memory in APP/SP1 MSX-3 mice in our data was found to be associated with reduced amyloid load. In APP/PS1dE9 mice, amyloid burden, and particularly hippocampal and cortical Aβ42 levels, strongly correlate with spatial memory deficits (Puoliväli et al., 2002; Garcia-Alloza et al., 2006; Sipos et al., 2007; Zhang et al., 2011). Our results demonstrate that MSX-3 treatment promotes a moderate but significant reduction of amyloid plaques in the cortex of APP/PS1 mice. Importantly, a significant reduction of the Aβ1-42/Aβ1-40 ratio was also observed in the cortex of APP/PS1 mice following A2AR blockade. To our knowledge, this is the first report demonstrating an effect of an A2AR ligand on amyloid pathology in vivo, in agreement with previous studies showing that chronic caffeine treatment reduces brain soluble Aβ in APPsw mice (Arendash et al., 2006, 2009). The decrease of amyloid burden following A2AR blockade found in the cortex of APP/PS1 mice, a brain region involved in spatial navigation, could therefore explain, at least in part, the beneficial effect of MSX-3 on spatial memory. Reasons explaining a specific effect of MSX-3 on cortical amyloid pathology vs. hippocampus remain unclear. Similarly, the mechanisms underlying the changes of Aβ1-42/Aβ1-40 ratio and 6E10 immunoreactivity warrant further evaluations. However, the reduction of the Aβ1-42/Aβ1-40 is consistent with the reduction of amyloid burden, especially of small amyloid plaques. In fact, in vitro data indicated that Aβ42 facilitates amyloid nucleation while Aβ40 allows for Aβ elongation (Snyder et al., 1994). In the present work, small plaques, reflecting amyloid nucleation, are decreased presumably due to the reduced amount of Aβ42 peptides. It cannot also be excluded that MSX-2 might bind to Aβ and act as a direct amyloid aggregation inhibitor, which warrants further exploration in the future. Interestingly, caffeine was shown, in vivo and in vitro to reduce both Aβ1-42 and Aβ1-40 levels (Arendash et al., 2006). This contrast with our data which indicate that MSX-3 modulates Aβ1-42/Aβ1-40 ratio, reducing Aβ1-42 and enhancing Aβ1-40 levels. Aβ length is under the control of the carboxypeptidase activity of the gamma-secretase which cleaves the Aβ peptide from its carboxy-terminal region. Thus, lack of PS1 exon 9 and exon 10 generates longer Aβ peptides (Le Guennec et al., 2017). Although speculative, MSX3 could promote the carboxypeptidase activity of the gamma-secretase and thus modify the ratio of Aβ towards the production of shorter species of Aβ peptides. In line with this hypothesis, A2AR and gamma-secretase have been co-localized to endosomes and A2ARs have been suggested to modulate the gamma-secretase activity (Lu et al., 2016). Although controversial, our results also support a regulatory activity of A2AR towards the gamma-secretase in APP/PS1 mouse. However, the underlying mechanism remains to be elucidated.
In conclusion, we have shown for the first time that a chronic and long-lasting treatment with an A2AR antagonist reduces amyloid pathology and improves memory in a model of AD. Considering previous converging studies in different models of AD (Orr et al., 2015, 2018; Laurent et al., 2016; Viana da Silva et al., 2016; Silva et al., 2018), the present findings further highlight A2AR as a promising therapeutic target in AD.
Author Contributions
EF, JC, KZ, EM, YB, MS, LC, KC, SS, SE and RC performed experiments, analyzed data and corrected the manuscript. MF, YC, MH, NS, CM, AH, LB, LL and DB supervised the work, analyzed data and wrote the manuscript.
Funding
This work was supported by a cross-border grant from LECMA/Alzheimer Forschung Initiative/Vaincre Alzheimer (to DB and CM). We hereby thank Frédéric Leprêtre for submitting transcriptomics data to GEO. EF and KC are supported by Université de Lille, LC was supported by Italian Society of Pharmacology and LabEx DISTALZ.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Bio Imaging Center Lille Nord de France (Campus Hospito-universitaire) as well as A. Bongiovanni and M. Tardivel for access microscopes and imagery analysis platform. We thank the animal core facility (animal facilities of Université de Lille) of Plateformes en Biologie Santé de Lille as well as M. Besegher-Dumoulin, C. Declerck, J. Devassine, Y. Lepage, C. Meunier, C. Degraeve and D. Taillieu for transgenic mouse production and care. Our laboratory is also supported by the France Alzheimer, FHU VasCog research network (Lille, France), and programs d’investissements d’avenir LabEx (excellence laboratory) Development of Innovative Strategies for a Transdisciplinary approach to ALZheimer’s disease (DISTALZ), ANR (ADORATAU, SPREADTAU, GRAND), Fondation pour la Recherche Médicale, Fondation Plan Alzheimer, Inserm, CNRS, Université de Lille, Lille Métropole Communauté Urbaine, Région Hauts-de-France (COGNADORA), DN2M. AH is member of the DFG-funded Cluster of Excellence ImmunoSensation (EXC 1023).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2018.00235/full#supplementary-material
References
Abbott, L. F., and Regehr, W. G. (2004). Synaptic computation. Nature 431, 796–803. doi: 10.1038/nature03010
Arendash, G. W., Mori, T., Cao, C., Mamcarz, M., Runfeldt, M., Dickson, A., et al. (2009). Caffeine reverses cognitive impairment and decreases brain amyloid-β levels in aged Alzheimer’s disease mice. J. Alzheimers Dis. 17, 661–680. doi: 10.3233/JAD-2009-1087
Arendash, G. W., Schleif, W., Rezai-Zadeh, K., Jackson, E. K., Zacharia, L. C., Cracchiolo, J. R., et al. (2006). Caffeine protects Alzheimer’s mice against cognitive impairment and reduces brain-amyloid production. Neuroscience 142, 941–952. doi: 10.1016/j.neuroscience.2006.07.021
Baeta-Corral, R., Johansson, B., and Giménez-Llort, L. (2018). Long-term treatment with low-dose caffeine worsens BPSD-like profile in 3xTg-AD mice model of Alzheimer’s disease and affects mice with normal aging. Front. Pharmacol. 9:79. doi: 10.3389/fphar.2018.00079
Batalha, V. L., Ferreira, D. G., Coelho, J. E., Valadas, J. S., Gomes, R., Temido-Ferreira, M., et al. (2016). The caffeine-binding adenosine A2A receptor induces age-like HPA-axis dysfunction by targeting glucocorticoid receptor function. Sci. Rep. 6:31493. doi: 10.1038/srep31493
Batalha, V. L., Pego, J. M., Fontinha, B. M., Costenla, A. R., Valadas, J. S., Baqi, Y., et al. (2013). Adenosine A2A receptor blockade reverts hippocampal stress-induced deficits and restores corticosterone circadian oscillation. Mol. Psychiatry 18, 320–331. doi: 10.1038/mp.2012.8
Blum, D., Galas, M.-C., Pintor, A., Brouillet, E., Ledent, C., Muller, C. E., et al. (2003a). A dual role of adenosine A2A receptors in 3-nitropropionic acid-induced striatal lesions: implications for the neuroprotective potential of A2A antagonists. J. Neurosci. 23, 5361–5369. doi: 10.1523/JNEUROSCI.23-12-05361.2003
Blum, D., Hourez, R., Galas, M.-C., Popoli, P., and Schiffmann, S. N. (2003b). Adenosine receptors and Huntington’s disease: implications for pathogenesis and therapeutics. Lancet Neurol. 2, 366–374. doi: 10.1016/s1474-4422(03)00411-3
Bulavina, L., Szulzewsky, F., Rocha, A., Krabbe, G., Robson, S. C., Matyash, V., et al. (2013). NTPDase1 activity attenuates microglial phagocytosis. Purinergic Signal. 9, 199–205. doi: 10.1007/s11302-012-9339-y
Canas, P. M., Porciuncula, L. O., Cunha, G. M. A., Silva, C. G., Machado, N. J., Oliveira, J. M. A., et al. (2009). Adenosine A2A receptor blockade prevents synaptotoxicity and memory dysfunction caused by β-amyloid peptides via p38 mitogen-activated protein kinase pathway. J. Neurosci. 29, 14741–14751. doi: 10.1523/JNEUROSCI.3728-09.2009
Cao, C., Cirrito, J. R., Lin, X., Wang, L., Verges, D. K., Dickson, A., et al. (2009). Caffeine suppresses amyloid-β levels in plasma and brain of Alzheimer’s disease transgenic mice. J. Alzheimers Dis. 17, 681–697. doi: 10.3233/JAD-2009-1071
Carr, C. M., and Rizo, J. (2010). At the junction of SNARE and SM protein function. Curr. Opin. Cell Biol. 22, 519–527. doi: 10.1016/j.ceb.2010.04.006
Collins, L. E., Galtieri, D. J., Brennum, L. T., Sager, T. N., Hockemeyer, J., Müller, C. E., et al. (2010). Oral tremor induced by the muscarinic agonist pilocarpine is suppressed by the adenosine A2A antagonists MSX-3 and SCH58261, but not the adenosine A1 antagonist DPCPX. Pharmacol. Biochem. Behav. 94, 561–569. doi: 10.1016/j.pbb.2009.11.011
Cristóvão-Ferreira, S., Navarro, G., Brugarolas, M., Pérez-Capote, K., Vaz, S. H., Fattorini, G., et al. (2013). A1R-A2A R heteromers coupled to Gsand Gi/0proteins modulate GABA transport into astrocytes. Purinergic Signal. 9, 433–449. doi: 10.1007/s11302-013-9364-5
Cunha, R. A. (2016). How does adenosine control neuronal dysfunction and neurodegeneration? J. Neurochem. 139, 1019–1055. doi: 10.1111/jnc.13724
Cuyvers, E., and Sleegers, K. (2016). Genetic variations underlying Alzheimer’s disease: evidence from genome-wide association studies and beyond. Lancet Neurol. 15, 857–868. doi: 10.1016/S1474-4422(16)00127-7
Dall’lgna, O. P., Fett, P., Gomes, M. W., Souza, D. O., Cunha, R. A., and Lara, D. R. (2007). Caffeine and adenosine A2A receptor antagonists prevent β-amyloid (25–35)-induced cognitive deficits in mice. Exp. Neurol. 203, 241–245. doi: 10.1016/j.expneurol.2006.08.008
Dall’lgna, O. P., Porciúncula, L. O., Souza, D. O., Cunha, R. A., and Lara, D. R. (2003). Neuroprotection by caffeine and adenosine A2A receptor blockade of β -amyloid neurotoxicity. Br. J. Pharmacol. 138, 1207–1209. doi: 10.1038/sj.bjp.0705185
Duyckaerts, C., Braak, H., Brion, J., Buée, L., Del Tredici, K., Goedert, M., et al. (2015). PART is part of Alzheimer disease. Acta Neuropathol. 129, 749–756. doi: 10.1007/s00401-015-1390-7
Flaten, V., Laurent, C., Coelho, J. E., Sandau, U., Batalha, V. L., Burnouf, S., et al. (2014). From epidemiology to pathophysiology: what about caffeine in Alzheimer’s disease? Biochem. Soc. Trans. 42, 587–592. doi: 10.1042/BST20130229
Garcia-Alloza, M., Robbins, E. M., Zhang-Nunes, S. X., Purcell, S. M., Betensky, R. A., Raju, S., et al. (2006). Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol. Dis. 24, 516–524. doi: 10.1016/j.nbd.2006.08.017
Genc, O., Kochubey, O., Toonen, R. F., Verhage, M., and Schneggenburger, R. (2014). Munc18–1 is a dynamically regulated PKC target during short-term enhancement of transmitter release. Elife 3:e01715. doi: 10.7554/eLife.01715
Giménez-Llort, L., Schiffmann, S. N., Shmidt, T., Canela, L., Camón, L., Wassholm, M., et al. (2007). Working memory deficits in transgenic rats overexpressing human adenosine A2A receptors in the brain. Neurobiol. Learn. Mem. 87, 42–56. doi: 10.1016/j.nlm.2006.05.004
Gyoneva, S., Shapiro, L., Lazo, C., Garnier-Amblard, E., Smith, Y., Miller, G. W., et al. (2014). Adenosine A2A receptor antagonism reverses inflammation-induced impairment of microglial process extension in a model of Parkinson’s disease. Neurobiol. Dis. 67, 191–202. doi: 10.1016/j.nbd.2014.03.004
Gyoneva, S., Swanger, S. A., Zhang, J., Weinshenker, D., and Traynelis, S. F. (2016). Altered motility of plaque-associated microglia in a model of Alzheimer’s disease. Neuroscience 330, 410–420. doi: 10.1016/j.neuroscience.2016.05.061
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405. doi: 10.1016/S1474-4422(15)70016-5
Horgusluoglu-Moloch, E., Nho, K., Risacher, S. L., Kim, S., Foroud, T., Shaw, L. M., et al. (2017). Targeted neurogenesis pathway-based gene analysis identifies ADORA2A associated with hippocampal volume in mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 60, 92–103. doi: 10.1016/j.neurobiolaging.2017.08.010
Jankowsky, J. L., Slunt, H. H., Ratovitski, T., Jenkins, N. A., Copeland, N. G., and Borchelt, D. R. (2001). Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol. Eng. 17, 157–165. doi: 10.1016/s1389-0344(01)00067-3
Kaster, M. P., Machado, N. J., Silva, H. B., Nunes, A., Ardais, A. P., Santana, M., et al. (2015). Caffeine acts through neuronal adenosine A2A receptors to prevent mood and memory dysfunction triggered by chronic stress. Proc. Natl. Acad. Sci. U S A 112, 7833–7838. doi: 10.1073/pnas.1423088112
Kim, T.-K., Lee, J.-E., Park, S.-K., Lee, K.-W., Seo, J.-S., Im, J.-Y., et al. (2012). Analysis of differential plaque depositions in the brains of Tg2576 and Tg-APPswe/PS1dE9 transgenic mouse models of Alzheimer disease. Exp. Mol. Med. 44, 492–502. doi: 10.3858/emm.2012.44.8.056
Krabbe, G., Halle, A., Matyash, V., Rinnenthal, J. L., Eom, G. D., Bernhardt, U., et al. (2013). Functional impairment of microglia coincides with beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS One 8:e60921. doi: 10.1371/journal.pone.0060921
Laurent, C., Buée, L., and Blum, D. (2018). Tau and neuroinflammation: what impact for Alzheimer’s disease and tauopathies? Biomed. J. 41, 21–33. doi: 10.1016/j.bj.2018.01.003
Laurent, C., Burnouf, S., Ferry, B., Batalha, V. L., Coelho, J. E., Baqi, Y., et al. (2016). A2A adenosine receptor deletion is protective in a mouse model of Tauopathy. Mol. Psychiatry 21, 97–107. doi: 10.1038/mp.2014.151
Laurent, C., Eddarkaoui, S., Derisbourg, M., Leboucher, A., Demeyer, D., Carrier, S., et al. (2014). Beneficial effects of caffeine in a transgenic model of Alzheimer’s disease-like tau pathology. Neurobiol. Aging 35, 2079–2090. doi: 10.1016/j.neurobiolaging.2014.03.027
Le Guennec, K., Veugelen, S., Quenez, O., Szaruga, M., Rousseau, S., Nicolas, G., et al. (2017). Deletion of exons 9 and 10 of the Presenilin 1 gene in a patient with Early-onset alzheimer disease generates longer amyloid seeds. Neurobiol. Dis. 104, 97–103. doi: 10.1016/j.nbd.2017.04.020
Lee, C.-C., Chang, C.-P., Lin, C.-J., Lai, H.-L., Kao, Y.-H., Cheng, S.-J., et al. (2018). Adenosine augmentation evoked by an ENT1 inhibitor improves memory impairment and neuronal plasticity in the APP/PS1 mouse model of Alzheimer’s disease. Mol. Neurobiol. doi: 10.1007/s12035-018-1030-z [Epub ahead of print].
Li, P., Rial, D., Canas, P. M., Yoo, J.-H., Li, W., Zhou, X., et al. (2015). Optogenetic activation of intracellular adenosine A2A receptor signaling in the hippocampus is sufficient to trigger CREB phosphorylation and impair memory. Mol. Psychiatry 20, 1339–1349. doi: 10.1038/mp.2014.182
Lopes, L. V., Cunha, R. A., and Ribeiro, J. A. (1999). Increase in the number, G protein coupling and efficiency of facilitatory adenosine A2A receptors in the limbic cortex, but not striatum, of aged rats. J. Neurochem. 73, 1733–1738. doi: 10.1046/j.1471-4159.1999.731733.x
Lu, J., Cui, J., Li, X., Wang, X., Zhou, Y., Yang, W., et al. (2016). An Anti-Parkinson’s disease drug via targeting adenosine A2A receptor enhances amyloid-β generation and γ-secretase activity. PLoS One 11:e0166415. doi: 10.1371/journal.pone.0166415
Machado, N. J., Simões, A. P., Silva, H. B., Ardais, A. P., Kaster, M. P., Garção, P., et al. (2017). Caffeine reverts memory but not mood impairment in a depression-prone mouse strain with up-regulated adenosine A2A receptor in hippocampal glutamate synapses. Mol. Neurobiol. 54, 1552–1563. doi: 10.1007/s12035-016-9774-9
Marciniak, E., Faivre, E., Dutar, P., Alves Pires, C., Demeyer, D., Caillierez, R., et al. (2015). The Chemokine MIP-1α/CCL3 impairs mouse hippocampal synaptic transmission, plasticity and memory. Sci. Rep. 5:15862. doi: 10.1038/srep15862
Masters, C. L., Simms, G., Weinman, N. A., Multhaup, G., McDonald, B. L., and Beyreuther, K. (1985). Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U S A 82, 4245–4249. doi: 10.1073/pnas.82.12.4245
Matos, M., Augusto, E., Santos-Rodrigues, A. D., Schwarzschild, M. A., Chen, J.-F., Cunha, R. A., et al. (2012). Adenosine A2A receptors modulate glutamate uptake in cultured astrocytes and gliosomes. Glia 60, 702–716. doi: 10.1002/glia.22290
Matos, M., Shen, H. Y., Augusto, E., Wang, Y., Wei, C. J., Wang, Y. T., et al. (2015). Deletion of adenosine A2A receptors from astrocytes disrupts glutamate homeostasis leading to psychomotor and cognitive impairment: relevance to schizophrenia. Biol. Psychiatry 78, 763–774. doi: 10.1016/j.biopsych.2015.02.026
Nagpure, B. V., and Bian, J. S. (2014). Hydrogen sulfide inhibits A2A adenosine receptor agonist induced β-amyloid production in SH-SY5Y neuroblastoma cells via a cAMP dependent pathway. PLoS One 9:e88508. doi: 10.1371/journal.pone.0088508
Nishizaki, T., Nagai, K., Nomura, T., Tada, H., Kanno, T., Tozaki, H., et al. (2002). A new neuromodulatory pathway with a glial contribution mediated via A2A adenosine receptors. Glia 39, 133–147. doi: 10.1002/glia.10100
Orr, A. G., Orr, A. L.,Li, X. J., Gross, R. E., and Traynelis, S. F. (2009). Adenosine A2A receptor mediates microglial process retraction. Nat. Neurosci. 12, 872–878. doi: 10.1038/nn.2341
Orr, A. G., Hsiao, E. C., Wang, M. M., Ho, K., Kim, D. H., Wang, X., et al. (2015). Astrocytic adenosine receptor A2A and Gs-coupled signaling regulate memory. Nat. Neurosci. 18, 423–434. doi: 10.1038/nn.3930
Orr, A. G., Lo, I., Schumacher, H., Ho, K., Gill, M., Guo, W., et al. (2018). Istradefylline reduces memory deficits in aging mice with amyloid pathology. Neurobiol. Dis. 110, 29–36. doi: 10.1016/j.nbd.2017.10.014
Puoliväli, J., Wang, J., Heikkinen, T., Heikkilä, M., Tapiola, T., van Groen, T., et al. (2002). Hippocampal Abeta42 levels correlate with spatial memory deficit in APP and PS1 double transgenic mice. Neurobiol. Dis. 9, 339–347. doi: 10.1006/nbdi.2002.0481
Rebola, N., Sebastião, A. M., de Mendonca, A., Oliveira, C. R., Ribeiro, J. A., and Cunha, R. A. (2003). Enhanced adenosine A2A receptor facilitation of synaptic transmission in the hippocampus of aged rats. J. Neurophysiol. 90, 1295–1303. doi: 10.1152/jn.00896.2002
Rebola, N., Simões, A. P., Canas, P. M., Tomé, A. R., Andrade, G. M., Barry, C. E., et al. (2011). Adenosine A2A receptors control neuroinflammation and consequent hippocampal neuronal dysfunction. J. Neurochem. 117, 100–111. doi: 10.1111/j.1471-4159.2011.07178.x
Reitz, C., Brayne, C., and Mayeux, R. (2011). Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 7, 137–152. doi: 10.1038/nrneurol.2011.2
Sasmono, R. T., Oceandy, D., Pollard, J. W., Tong, W., Pavli, P., Wainwright, B. J., et al. (2003). A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood 101, 1155–1163. doi: 10.1182/blood-2002-02-0569
Sauer, R., Maurinsh, J., Reith, U., Fülle, F., Klotz, K. N., and Müller, C. E. (2000). Water-soluble phosphate prodrugs of 1-propargyl-8-styrylxanthine derivatives, A2A-selective adenosine receptor antagonists. J. Med. Chem. 43, 440–448. doi: 10.1021/jm9911480
Savage, J. C., Jay, T., Goduni, E., Quigley, C., Mariani, M. M., Malm, T., et al. (2015). Nuclear receptors license phagocytosis by trem2+ myeloid cells in mouse models of Alzheimer’s disease. J. Neurosci. 35, 6532–6543. doi: 10.1523/JNEUROSCI.4586-14.2015
Savonenko, A., Xu, G. M., Melnikova, T., Morton, J. L., Gonzales, V., Wong, M. P. F., et al. (2005). Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer’s disease: relationships to β-amyloid deposition and neurotransmitter abnormalities. Neurobiol. Dis. 18, 602–617. doi: 10.1016/j.nbd.2004.10.022
Silva, A. C., Lemos, C., Gonçalves, F. Q., Pliássova, A. V., Machado, N. J., Silva, H. B., et al. (2018). Blockade of adenosine A2A receptors recovers early deficits of memory and plasticity in the triple transgenic mouse model of Alzheimer’s disease. Neurobiol Dis. 31, 72–81. doi: 10.1016/j.nbd.2018.05.024
Sipos, E., Kurunczi, A., Kasza, Á., Horváth, J., Felszeghy, K., Laroche, S., et al. (2007). β-Amyloid pathology in the entorhinal cortex of rats induces memory deficits: implications for Alzheimer’s disease. Neuroscience 147, 28–36. doi: 10.1016/j.neuroscience.2007.04.011
Snyder, S. W., Ladror, U. S., Wade, W. S., Wang, G. T., Barrett, L. W., Matayoshi, E. D., et al. (1994). Amyloid-beta aggregation: selective inhibition of aggregation in mixtures of amyloid with different chain lengths. Biophys. J. 67, 1216–1228. doi: 10.1016/s0006-3495(94)80591-0
Suh, Y.-H., and Checler, F. (2002). Amyloid precursor protein, presenilins, and alpha synuclein: molecular pathogenesis and pharmacological applications in Alzheimer’s disease. Pharmacol. Rev. 54, 469–525. doi: 10.1124/pr.54.3.469
Temido-Ferreira, M., Ferreira, D. G., Batalha, V. L., Marques-Morgado, I., Coelho, J. E., Pereira, P., et al. (2018). Age-related shift in LTD is dependent on neuronal adenosine A2A receptors interplay with mGluR5 and NMDA receptors. Mol. Psychiatry doi: 10.1038/s41380-018-0110-9 [Epub ahead of print].
Toonen, R. F. G., and Verhage, M. (2007). Munc18–1 in secretion: lonely Munc joins SNARE team and takes control. Trends Neurosci. 30, 564–572. doi: 10.1016/j.tins.2007.08.008
Viana da Silva, S., Haberl, M. G., Zhang, P., Bethge, P., Lemos, C., Gonçalves, N., et al. (2016). Early synaptic deficits in the APP/PS1 mouse model of Alzheimer’s disease involve neuronal adenosine A2A receptors. Nat. Commun. 7:11915. doi: 10.1038/ncomms11915
Yu, L., Coelho, J. E., Zhang, X., Fu, Y., Tillman, A., Karaoz, U., et al. (2009). Uncovering multiple molecular targets for caffeine using a drug target validation strategy combining A2A receptor knockout mice with microarray profiling. Physiol. Genomics 37, 199–210. doi: 10.1152/physiolgenomics.90353.2008
Zhang, W., Hao, J., Liu, R., Zhang, Z., Lei, G., Su, C., et al. (2011). Soluble Aβ levels correlate with cognitive deficits in the 12-month-old APPswe/PS1dE9 mouse model of Alzheimer’s disease. Behav. Brain Res. 222, 342–350. doi: 10.1016/j.bbr.2011.03.072
Keywords: Alzheimer’s disease, amyloid, adenosine receptor, A2A, memory
Citation: Faivre E, Coelho JE, Zornbach K, Malik E, Baqi Y, Schneider M, Cellai L, Carvalho K, Sebda S, Figeac M, Eddarkaoui S, Caillierez R, Chern Y, Heneka M, Sergeant N, Müller CE, Halle A, Buée L, Lopes LV and Blum D (2018) Beneficial Effect of a Selective Adenosine A2A Receptor Antagonist in the APPswe/PS1dE9 Mouse Model of Alzheimer’s Disease. Front. Mol. Neurosci. 11:235. doi: 10.3389/fnmol.2018.00235
Received: 25 April 2018; Accepted: 15 June 2018;
Published: 12 July 2018.
Edited by:
Detlev Boison, Legacy Health, United StatesReviewed by:
Annakaisa Haapasalo, A.I. Virtanen Institute for Molecular Sciences, Univeristy of Eastern Finland, FinlandRodrigo A. Cunha, Faculdade de Medicina, Universidade de Coimbra, Portugal
Copyright © 2018 Faivre, Coelho, Zornbach, Malik, Baqi, Schneider, Cellai, Carvalho, Sebda, Figeac, Eddarkaoui, Caillierez, Chern, Heneka, Sergeant, Müller, Halle, Buée, Lopes and Blum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Blum, ZGF2aWQuYmx1bUBpbnNlcm0uZnI=