 Alexander C. Ervin
Alexander C. Ervin James D. Blakemore*
James D. Blakemore*- Department of Chemistry, University of Kansas, Lawrence, KS, United States
The redox properties of actinide-containing species strongly influence their reactivity, speciation, and interfacial behavior, but the experimental quantification of the electrochemical characteristics of molecular actinide complexes in nonaqueous media has not received the attention it deserves. Here, results from hydrodynamic methods and electrochemical simulations of U(VI)/U(V) redox are reported, including quantification of heterogeneous electron-transfer kinetics and estimation of chemical reversibility of U(VI)/U(V) interconversion at electrodes in acetonitrile-based electrolyte. The complexes investigated are recently reported U(VI) and U(V) complexes in which the uranyl ion (UO2n+) is encapsulated in a macrocyclic 18-crown-6-like moiety templated by a Pt(II) center. These complexes feature the most positive value UVI/UV reduction potential yet reported and are thus particularly relevant to study of facile U(V) generation from U(VI) precursors as well as uranium electroanalysis. Rotating disk electrode (RDE) studies have been used to quantify the diffusion coefficients of the U(VI) and U(V) complexes, and standard heterogeneous electron transfer rate constants (k0) for the redox have been determined using a conventional Koutecký-Levich analysis. Rotating ring-disk electrode (RRDE) studies have been used to directly interrogate the chemical reversibility of U(VI)-U(V) interconversion, confirming that reduction of the U(VI) complex at an Au disk is associated with formation of the U(V) analogue that can be readily re-oxidized at a Pt ring under hydrodynamic (rotating) conditions. Because measurements of the type reported here are generally associated with current flows that are larger than those found in corresponding quiescent (unstirred) conditions, our findings suggest that hydrodynamic methods could be advantageous for design of electroanalytical approaches to detection of actinide species and study of their redox properties.
1 Introduction
Non-destructive assay techniques that can quantify the elemental (actinide) content of nuclear materials are critical for the work of the nuclear safeguards community. This is because analytical tools are needed to analyze a variety of sample types and to generate evidence of the purposes (declared or otherwise) of regulated nuclear materials. The actinide elements uranium and plutonium are particularly relevant to the work in this area, given their possible uses as nuclear fuels and possible targets of diversion. Of course, significant historical and contemporary work has examined a variety of analytical approaches to safeguarding the chemistry of these elements, including a wide range of techniques of varying sensitivities and detection modalities.
Electrochemical methods are particularly useful for characterization of systems containing the early actinides. This is attributable, in part, to the distinct reduction potentials displayed by species containing uranium (U), neptunium (Np), and plutonium (Pu). The long-standing tabulated reduction potentials for the An(VI)/An(V) couples of U, Np, and Pu show that U(V) is particularly difficult to access (E°(UVI/UV) = +0.16 V vs. the standard hydrogen electrode, abbreviated SHE) in comparison to both Np(V) (E°(NpVI/NpV) = +1.24 V vs. SHE) and Pu(V) (E°(PuVI/PuV) = +1.02 V vs. SHE) (Bard et al., 1985). In light of the unique thermodynamic potentials associated with each of the actinyl cations in the series of U, Np, and Pu, one could envision development of ion-selective electrode technologies for assaying their presence in solutions of interest. However, a prerequisite for the development of such technologies would involve addressing issues arising from both electrochemical irreversibility and chemical reactivity, since such effects can complicate or even mask expected behavior at electrodes.
A portion of our interest in recent years has been drawn to studies of the chemistry and electrochemistry of well-defined molecular coordination complexes featuring actinyl cations (see Figure 1). Our work is motivated by the importance of redox processes in governing the reactivity and speciation of actinide complexes, as well as the importance of developing approaches to detecting and identifying particular actinide-containing molecular species in solution. Prior work in this area has been particularly informative, including the classic work of Wester and Sullivan on the voltammetric properties of uranyl carbonate species like [UO2(CO3)3]4– (Wester and Sullivan, 1980). In this work, it was found that virtually colorless U(V) could be produced from U(VI) in solutions of pH 11-12 with high total carbonate concentrations. Coulometry and spectrochemical experiments provided ample evidence of the generation of U(V) under these conditions, affording insights into the rather elusive properties of U(V) in aqueous media. Electrolysis studies have also been shown to afford access to Np(V) and Pu(V) species in aqueous carbonate solutions, including elegant Raman characterization (Madic et al., 1983). Voltammetry and digital simulations have examined the role of coordination environment in the electrochemical characteristics of various species containing UO22+ as well (Morris, 2002).
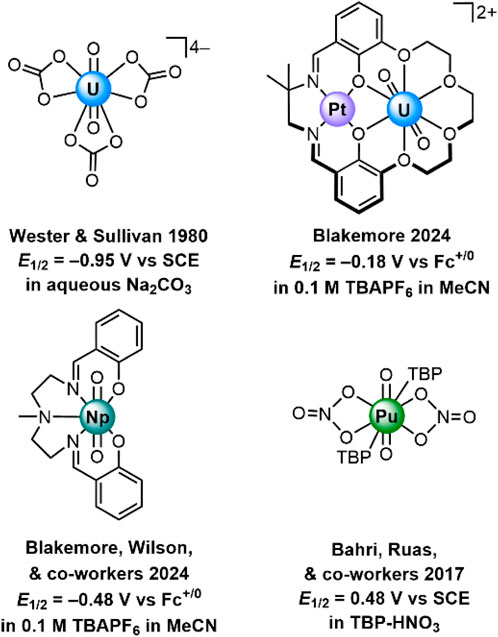
Figure 1. Examples of electrochemically characterized An(VI)/An(V) redox couples for complexes containing actinyl moieties. TBP is tributylphosphate.
Voltammetry is useful for studies of non-aqueous redox chemistry as well. Of note, both the Pu(VI)/Pu(V) and Pu(IV)/Pu(III) couples have been recently measured in tributylphosphate/nitric acid (Bahri et al., 2017). In this work, both couples were found to be electrochemically quasi-reversible and thus associated with relatively fast heterogeneous electron-transfer kinetics. This finding is in accord with prior studies of aqueous plutonium systems showing that, for example, Pu(VI)/Pu(V) conversion is quite fast but that other interconversions like Pu(V)/Pu(IV) tend to be slow (Newton, 1975). In collaboration with Richard Wilson at Argonne National Laboratory, our own group has recently investigated the electrochemical properties of neptunium coordination complexes, finding that a workhorse chelating pentadentate ligand affords significant stabilization of otherwise rather oxidizing [NpVIO2]2+ (Mikeska et al., 2024). Considering all this, electrochemistry appears quite promising as a technique for distinguishing the redox properties of various actinide species and affording insights into the chemical reactivity involved in interconversions between oxidation states. And, as we have shown recently, electrochemical methods are also useful for electrogeneration of reactive actinide species and tracking the products of reaction with exogeneous reagents (Golwankar et al., 2024b).
Recently, we reported the synthesis and isolation of [UVIO2] and [UVO2], complexes in which the uranium is encapsulated in a macrocyclic 18-crown-6-like moiety templated by a Pt(II) center (Golwankar et al., 2024a). These complexes (see Figure 2) are unique from a number of perspectives, including both their coordination environment and redox properties. From the perspective of coordination environment, [UVIO2] and [UVO2] represent unusual examples of crown-encapsulated uranyl species that are stable in solution, as demonstrated by nuclear magnetic resonance and electronic spectroscopies. [UVO2] is also the first reported crown-ligated U(V) species. From the perspective of electrochemistry, [UVIO2] and [UVO2] are unique in that the reduction potential associated with their interconversion (E1/2 = −0.18 V vs. ferrocenium/ferrocene, denoted hereafter as Fc+/0) is the most positive yet reported for any non-oxo-functionalized uranyl complex. These unique features of our crown-encapsulated uranyl complexes suggest a number of exciting opportunities for further development, including electroanalytical work. This is because, in principle, the dramatic shift in reduction potential away from the rather negative values associated with conventional UO22+-containing species could be rather useful. Very negative reduction potentials are typically associated with unselective reactions, particularly under aqueous conditions where electrode-catalyzed proton reduction to form H2 could outcompete desirable reactions such as uranium reduction. In other words, the properties of [UVIO2] and [UVO2] afford the opportunity to study uranium redox under only mildly reducing conditions. Indeed, we have already shown that conversion of [UVIO2] to [UVO2] can be accomplished with only mildly reducing decamethylferrocene (Cp*2Fe; E1/2(FeIII/FeII) = −0.48 V vs. Fc+/0) (Connelly and Geiger, 1996).
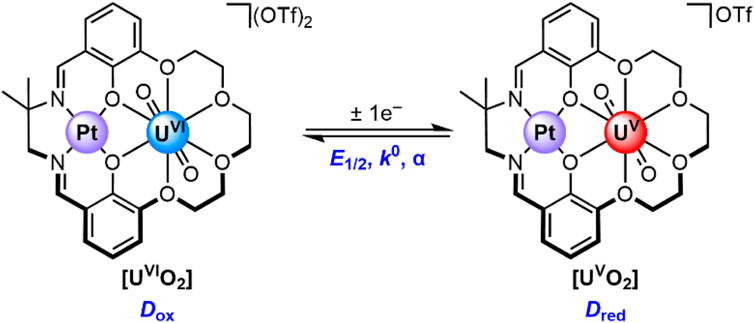
Figure 2. The U(VI) and U(V) species in this study. The parameters describing this redox couple include the U(VI)/U(V) half-wave potential (E1/2), the standard heterogeneous electron-transfer rate constant (k0), and the symmetry factor (α). The diffusion coefficient for the U(VI) and U(V) species are denoted as Dox and Dred, respectively.
However, although the most common electrochemical techniques of cyclic voltammetry and bulk electrolysis have been extensively applied throughout the various realms of actinide chemistry and nuclear science, hydrodynamic methods have received far less attention in the field. Two reports concern electrodeposition—one regarding generation of plutonium metal under stirred conditions (Becerril et al., 1993) and one regarding generation of uranium metal from a molten salt eutectic (Rappleye and Simpson, 2017). The hydrodynamic current response of [UCl6]2– in two ionic liquids has also been studied with a rotating disk electrode (Nikitenko et al., 2005). Hydrodynamic methods in electrochemistry are appealing as they involve convective (rather than purely diffusion-limited) mass transport and therefore, can be used to measure species at lower concentrations and with improved figures of merit compared to more routine techniques involving quiescent (unstirred) solutions of analyte(s) (Bard et al., 2022; Pleskov and Filinovskii, 1976). The most well-known hydrodynamic methods feature rotating electrodes and feature configurations in which laminar flow is achieved at the working electrode. In rotating disk electrochemistry (RDE), convection is used to constantly draw analyte to the surface of a disk working electrode, and in rotating ring-disk electrochemistry (RRDE), convection is also used for active transport of electrogenerated species from the disk (generator) electrode to the ring (collector) electrode for potential-dependent characterization (Sherman et al., 2016). RDE studies, particularly Koutecký-Levich analysis, can be used to experimentally quantify heterogeneous electron-transfer kinetics while generator-collector RRDE studies can be used to understand the chemical reversibility of individual redox waves in voltammetry. Considering all this, we anticipated that hydrodynamic electrochemical study of our recently isolated crown-encapsulated complexes [UVIO2] and [UVO2] could be attractive for demonstrating electroanalytical tools related to nuclear forensics and safeguards as well as for providing insights into the electrochemical redox displayed by [UVIO2] and [UVO2]. Both of these aspects represent opportunities to expand knowledge of approaches to study of actinide redox chemistry.
Here, we report electroanalytical studies of the interconversion of the crown-encapsulated complexes [UVIO2] and [UVO2]. Digital simulations of conventional cyclic voltammetry data collected with quiescent solutions with either gold (Au) or platinum (Pt) electrodes were used to extract the key parameters governing the electrode behavior of the system, including heterogeneous electron-transfer rate constant (k0), transfer coefficient (α), and the diffusion coefficients of both [UVIO2] (Dox) and [UVO2] (Dred). Comparison of these data to those obtained by direct RDE experimentation and Koutecký-Levich analysis suggests a faster rate of electron transfer can be achieved under hydrodynamic conditions. RRDE experiments were used to provide a quantitative measure of the chemical reversibility of interconversion of [UVIO2] and [UVO2] as well, providing a complement to estimates of chemical reversibility that can be obtained from cyclic voltammetry studies. Taken together, these results expand the toolbox of electrochemical approaches suitable for study of actinide redox chemistry and suggest further opportunities for deployment of hydrodynamic methods in the fields of nuclear forensics.
2 Materials and methods
2.1 General consideration
All manipulations were carried out in dry N2-filled gloveboxes (Vacuum Atmospheres Co., Hawthorne, CA, USA) or under N2/Ar atmosphere using standard Schlenk techniques unless otherwise noted. All solvents were of commercial grade and dried over activated alumina using a PPT Glass Contour (Nashua, NH) solvent purification system prior to use, and were stored over molecular sieves. All chemicals were obtained from major commercial suppliers and used as received or after extensive drying. The complexes in this study, [UVIO2] and [UVO2], were synthesized according to literature procedures (Golwankar et al., 2024a).
Caution! Depleted uranium is a weak alpha-particle emitter; all manipulations of U-containing materials should be carried out in a laboratory equipped with appropriate radiation safety protocols.
2.2 Electrochemical methods
Quiescent voltammetry experiments were carried out in a N2-filled glovebox in dry, degassed acetonitrile (MeCN). 0.10 M tetra(n-butylammonium) hexafluorophosphate ([nBu4N]+[PF6]–); Sigma-Aldrich, electrochemical grade) served as the solvent and supporting electrolyte. Measurements were carried out with a Gamry Reference 600+ Potentiostat/Galvanostat (Gamry Instruments, Warminster, PA, USA), using a standard three-electrode configuration. The working electrode was either a Au or Pt button electrode (CH Instruments, Austin, TX, USA; surface area: 0.0314 cm2), the counter electrode was a platinum wire (Kurt J. Lesker, Jefferson Hills, PA; 99.99%, 0.5 mm diameter), and a silver wire immersed in electrolyte served as a quasireference electrode (CH Instruments). The reference was separated from the working solution by a Vycor frit (Bioanalytical Systems, Inc., West Lafayette, IN, USA). Ferrocene (Sigma Aldrich, St. Louis, MO, USA; twice sublimed) was added to the electrolyte solution prior to the beginning of each experiment; the midpoint potential of the ferrocenium/ferrocene couple (denoted as Fc+/0) served as an external standard for comparison of the recorded potentials. Concentrations of analytes for cyclic voltammetry were typically 1 mM unless otherwise noted.
As described above, experimentation conducted in order to prepare this report utilized [nBu4N]+[PF6]– as the supporting electrolyte, a Ag+/0 quasireference electrode, and ferrocene as the external standard for potential measurements. These materials were chosen because we anticipated that they represent the most commonly utilized conditions for nonaqueous (organic) electrochemical studies of inorganic/coordination complexes. Similarly, acetonitrile was used as the solvent for our work because it is among the most commonly encountered solvents in studies of inorganic and organometallic redox chemistry. The compounds of interest in this study are also quite soluble in acetonitrile, motivating its use in this particular case as well. It is our hope that use of commonly available reagents and conditions will broaden interest in studies of nonaqueous actinide redox chemistry.
Simulations of the quiescent voltammetry data were carried out using DigiElch8.FD.
Rotating disk (RDE) and ring-disk electrode (RRDE) electrochemical experiments were carried out outside the glovebox and on the benchtop either under ambient conditions or under a positive pressure of argon gas. Measurements were carried out with a CH Instruments 7104E bipotentiostat, using standard three electrode (for RDE) or four-electrode (for RRDE) configurations, and the rotation rate was set using an ASR speed controller (Pine Research Instrumentation, Inc.). The working electrode for the RDE measurements was a gold disk electrode (Pine Research Instrumentation, Inc., Durham, NC; surface area: 0.196 cm2), while the RRDE measurements utilized a Au disk electrode (Pine Research Instrumentation, Inc., Durham, NC; surface area: 0.126 cm2) as the first working electrode and a Pt ring electrode (Pine Research Instrumentation, Inc.; thickness: 0.5 mm) as the second working electrode. The disk and ring electrodes were components of a Pine ChangeDisk tip system mated to a custom ASR-rotator-compatible shaft.
3 Results
3.1 Cyclic voltammetry studies of [UVIO2] and [UVO2]
Recently, we reported the synthesis, isolation, and redox properties of [UVIO2] and [UVO2] (Golwankar et al., 2024a). Cyclic voltammetry (CV) on highly-oriented pyrolytic graphite (HOPG) electrodes and related chemical redox experiments were used to demonstrate that the conversion of [UVIO2] to [UVO2] can be achieved at an exceptionally positive reduction potential of E1/2 = −0.18 V vs. Fc+/0. This represents a potential that is ca. 1.3 V more positive than U(VI)/U(V) redox cycling with a conventional dianionic ligand (Golwankar et al., 2024b). The results of synthetic studies using either decamethylferrocene (Cp*2Fe) or cobaltocene (Cp2Co) suggested that conversion of [UVIO2] to [UVO2] can be driven with high yields (97% with both reductants). Accordingly, CV data collected under conventional quiescent conditions showed electrochemically quasi-reversible U(VI)/U(V) redox in solutions prepared with [UVIO2]. However, in this work, apart from noting qualitatively similar anodic and cathodic peak currents, no measure was made of chemical reversibility in the electrochemical work. And, similarly, no quantification of the heterogeneous electron-transfer kinetics was attempted, apart from noting a peak-to-peak separation (ΔEp) of 98 mV at a scan rate of 100 mV/s. (This value suggests quasi-reversible behavior, given the established theoretical value of ΔEp of 57 mV for electron transfer at the fast limit.)
To provide the data needed for electrochemical simulations, we measured CV data utilizing a Au working electrode under inert atmospheric conditions for [UVIO2], which showed a U(VI)/U(V) couple at E1/2 = −0.18 V vs. Fc+/0, which is in good agreement with the voltametric response that was observed on HOPG working electrodes in our previous report (vide supra). In the new studies reported here, the couple again appeared to be quasi-reversible in nature based on a ΔEp of 85 mV at 100 mV/s. Extensive voltammetric cycling was also conducted in which ten cycles were scanned sequentially (see Supplementary Figure S1) on [UVIO2] to demonstrate that this redox couple is stable and the oxidation/reduced species persists in solution. These conclusions were confirmed by observing only minor deviations of the raw peak currents across the ten voltammetric cycles. Specifically, we found the peak cathodic current (ip,c) = −5.26 ± 0.02 μA and peak anodic current (ip,a) = 4.32 ± 0.02 μA, suggesting the Au working surface was not fouled to any significant degree upon reduction/oxidation of [UVIO2] (see Supplementary Figure S2). Voltammetry data at various scan rates were collected to interrogate the diffusional nature of the compounds involved in this redox couple (see Figure 3A). The peak currents extracted from the chosen scan rates for both cathodic and anodic peaks indeed obey the Randles-Ševčík equation, as for a diffusional redox couple both peak anodic and cathodic currents should trend linearly with respect to the square root of scan rate (see Figure 3B). The average ratio of the peak anodic to cathodic current across the scan rates chosen in this study was determined to be 0.96 ± 0.03 (see Supplementary Figure S3), in good agreement with the ideal value of 1 for an electrochemically reversible couple.
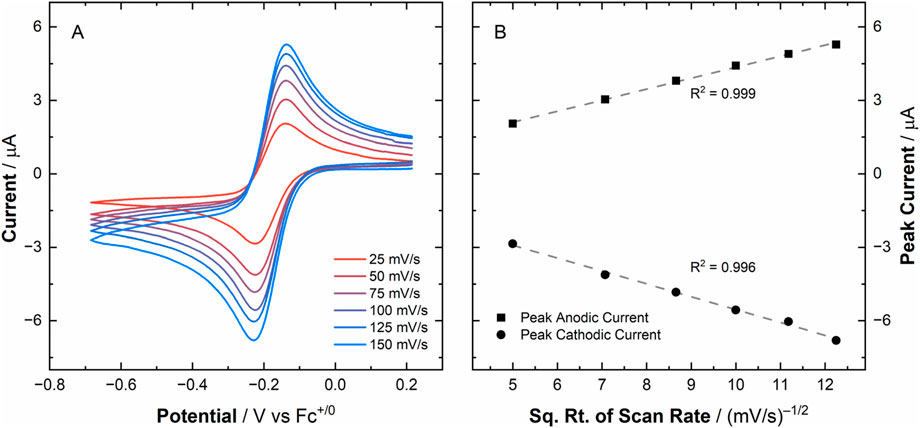
Figure 3. Panel (A) Scan-rate-dependent cyclic voltammetry data for [UVIO2] in MeCN electrolyte (0.1 M tetra(n-butylammonium) hexafluorophosphate, referred to hereafter as TBAPF6). Panel (B) Plot of raw peak currents vs. square root of scan rate, demonstrating the diffusional nature of both the oxidized and reduced species involved in this redox process.
In an effort to better understand the observed redox profile of [UVIO2], we chemically synthesized the reduced form of the complex, [UVO2]. Following the procedure reported in our previous study (Golwanker et al., 2024a), a slight excess (1.05 equivalents with respect to [UVIO2]) of Cp*2Fe was added dropwise to a solution of [UVIO2] in MeCN and was allowed to stir for 30 min. The solvent was removed in vacuo, and the resulting green powder was washed with diethyl ether (3 × 10 mL) to remove any remaining unreacted Cp*2Fe. The remaining powder, a mixture of [UVO2] and the co-generated [Cp*2Fe]+[OTf]– salt, was washed with 1,2-dichloroethane (3 × 5 mL) to remove the co-generated ferrocenium salt, resulting in a pale-yellow powder of what has previously been shown by our group to be analytically pure [UVO2].
Using the chemically prepared [UVO2], we thus conducted analogous CV measurements to that of [UVIO2]. The voltammetric response obtained for [UVO2] was virtually identical to that of [UVIO2], with E1/2 = −0.18 V vs. Fc+/0 and ΔEp = 100 mV. Multi-cycling experiments also displayed results akin to those observed for [UVIO2], ip,c = −1.71 ± 0.01 μA and ip,a = 2.44 ± 0.03 μA (see Supplementary Figures S4, S5). Peak currents extracted from the scan-rate-dependent data across two orders of magnitude demonstrated linear behavior as a function of the square root of scan rate, as well as an average peak anodic to cathodic current ratio of 1.01 ± 0.04 (see Supplementary Figures S6, S7). Although the conclusions that can be drawn from these data with regards to the U(VI)/U(V) redox chemistry of this system are essentially indistinguishable to those obtained from experimentation with [UVIO2], lower signal-to-noise was observed in all the voltammograms associated with these measurements, presumably due to the lower concentration of analyte; 0.4 mM of [UVO2] was utilized compared to 1 mM of [UVIO2]. This was necessary as [UVO2] was found to be less soluble in MeCN compared to [UVIO2].
Building upon the studies of [UVIO2] and [UVO2] on Au electrodes, we also interrogated the redox behavior of [UVIO2] and [UVO2] on Pt working electrodes. The results obtained from the CV experiments for [UVIO2] showed results similar to that on Au with an E1/2 = −0.18 V vs. Fc+/0 (ΔEp = 90 mV), and E1/2 = −0.18 V vs. Fc+/0 (ΔEp = 85 mV) for [UVO2]. However, over ten cycles, a diminished current response for [UVIO2] was observed of both the peak cathodic and anodic currents (ip,c = −4.95 ± 0.08 μA, ip,a = 4.64 ± 0.04 μA) compared to the current response on Au electrodes for the same cycling regime (ip,c = −5.26 ± 0.02 μA and ip,a = 4.32 ± 0.02 μA). This could be an indication of a non-Faradaic process altering the surface of the electrode over the time course of the experiment (see Supplementary Figures S8, S9). [UVO2] displayed a more stable current response over the same cycling regime, ip,c = −1.71 ± 0.02 μA and ip,a = 2.47 ± 0.02 μA (see Supplementary Figures S12, S13), suggesting that the undesirable interaction of analyte with the Pt electrode is confined to the U(VI) form of the complex only. Nevertheless, even though [UVIO2] presents reduced stability on the Pt electrode on the time course of the experiment, the scan-rate-dependent data were in good agreement with the anticipated behavior based on the Randles-Ševčík equation. Both the cathodic and anodic peak currents across the six scan rates for [UVIO2] and [UVO2] demonstrated linear trends with respect to the square root of scan rate (see Supplementary Figures S10, S14). The ratio of the peak anodic to peak cathodic current for the set of experiments for [UVO2] displayed an average value of 1.01 ± 0.04, in line with the behavior observed on Au. However, the corresponding data for [UVIO2] displayed a drastic deviation from unity with an average value of 0.78 ± 0.04 (see Supplementary Figures S11, S15), which could be attributed to instability of [UVIO2] at a Pt surface. Apparently, high concentrations of the U(VI) species cannot be held stably at polycrystalline Pt electrode surfaces.
To understand and quantify the fundamental parameters which describe the electrochemical and chemical reversibility of the [UVIO2]/[UVO2] redox couple, digital simulations were carried out using the collected CV data for [UVIO2] (Britz, 2016). The half-wave potential (E1/2), diffusion coefficients (Dox for [UVIO2] and Dred for [UVO2]), heterogeneous electron-transfer rate constant (k0), symmetry factor (α), and the formal reduction potential (E°′) for the [UVIO2]/[UVO2] redox couple were determined by simulating a range of scan rates which were experimentally measured for [UVIO2] on both Au and Pt working electrodes (see Table 1). Each voltammogram was simulated with a single, chemically reversible, and 1e– system that does not undergo follow-up chemical reactivity. The parameters Dox, Dred, k0, and α were subjected to a sensitivity analysis and validation procedure for each set of simulations by analyzing the arithmetic mean and standard deviation of each parameter across the various scan rates (see Supplementary Figures S16–S26). Generally, the profiles of the simulated voltammograms associated with the experimental data collected with Au working electrodes are in close agreement with the experimental data (see Figure 4), indicating the simulated parameters for [UVIO2] are good descriptors for this redox process. However, the profiles of the simulated voltammograms associated with the experiments that utilized Pt electrodes were found to be in significantly poorer agreement than the Au experiments. This behavior could be quantified by comparing the goodness-of-fit (GOOF) parameters calculated at each scan rate across the two sets of simulations. The GOOF is an output parameter which is calculated with the residual current values associated with ten fixed grid points along the simulation curve, and is determined following a least-squares regression fitting of the simulation output to the experimental data. In this case, GOOF values closer to 0 are indicative of a better fit to experimental data. For the simulations of the experiments utilizing Au working electrodes, the average goodness-of-fit value was calculated to be 0.0262 ± 0.0088, whereas for Pt working electrode simulations, the average value was calculated to be 0.0389 ± 0.0117 (see Supplementary Table S1). The larger goodness-of-fit value for the Pt working electrode simulations is in line with the conventional interpretation of the voltammetric profile and suggests that detrimental interactions can take place between [UVIO2] and the Pt surface. However, at the Au surface, modeling of a single well-behaved redox manifold is sufficient to reproduce the experimental data.
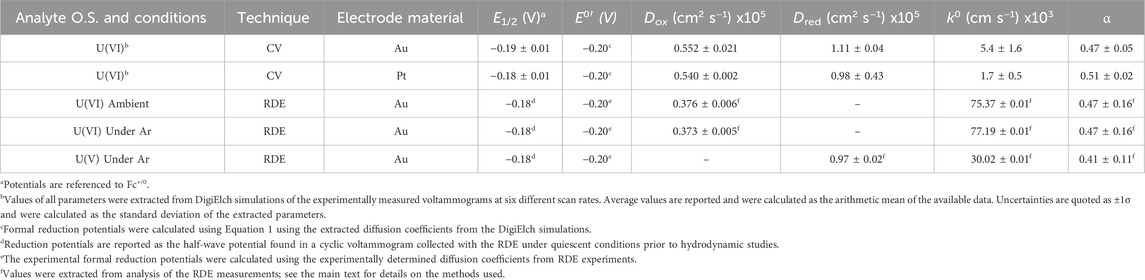
Table 1. Comparison of simulated and experimental parameters for redox reactivity of [UVIO2] and [UVO2].
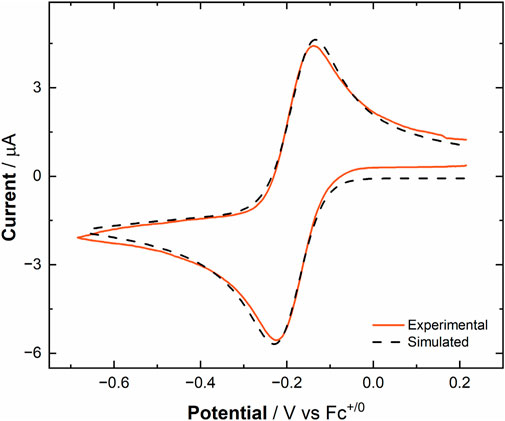
Figure 4. Experimental and simulated cyclic voltammograms of [UVIO2] in MeCN ([U] = 1.0 mM, working electrode: Au button (0.0314 cm2), electrolyte: 0.1 M TBAPF6, scan rate: 100 mV/s).
Examination of the extracted parameters from the simulation results confirms that the simulated half-wave potentials for both sets of simulations align well with the previously observed reduction potentials of [UVIO2], −0.18 V vs. Fc+/0 on both Au and Pt (simulated results: −0.19 V and −0.18 V for Au and Pt, respectively). The diffusion coefficients for [UVIO2] (5.52 × 10−6 cm2/s) and [UVO2] (1.11 × 10−5 cm2/s) extracted from the simulations also fall within a reasonable range when compared to previous electrochemical analyses from both our group (Mikeska et al., 2024) and other groups. For example, these calculated diffusion coefficients compare well to the diffusion coefficients for both ferrocene (Cp2Fe) and ferrocenium (Cp2Fe+) (both FeII and FeIII = 2.6 × 10−5 cm2/s), one of the most common non-aqueous external reference compounds (Bond et al., 2000). Indeed, the extracted diffusion coefficients for [UVIO2] and [UVO2] can be considered reasonable for this study as the macrocyclic and heterobimetallic nature of the complexes would be expected to contribute to larger hydrodynamic radii and, thus, smaller diffusion coefficients than those found in typical small-molecule inorganic complexes studied in non-aqueous media. Importantly, the new electrochemical simulations reported here for the [UVIO2]/[UVO2] redox couple confirms a key finding of our prior synthetic chemical work—the compounds in this study do not display solution speciation beyond the formal oxidation state change generated either at the electrode surface or with a chemical redox reagent.
Using the diffusion coefficients obtained from simulations, the formal reduction potential of the [UVIO2]/[UVIO2] couple under the studied conditions (denoted as E°′) can be calculated using Nernstian behavior from the following equation:
In this equation E is the measured E1/2, E°′ is the formal reduction potential (or diffusion adjusted reduction potential), R is the ideal gas constant (8.314 J mol−1 K−1), T is temperature (in K), n is the number of electrons, F is Faraday’s constant (96,485 C mol−1), Dred is the diffusion coefficient of the reduced complex, and Dox is the diffusion coefficient of the oxidized complex (both in cm2 sec−1). Applying the obtained diffusion coefficients from the simulations to Equation 1, the calculated formal reduction potential for the [UVIO2]/[UVO2] was found to be −0.20 V vs. Fc+/0. This value is in good agreement with the experimentally observed reduction potential (−0.18 V vs. Fc+/0). The larger value of Dred in comparison with Dox is sensible as well, since the reduced form of the complex, [UVO2], is less charge dense and is formally only a monocationic species. [UVIO2] is formally a dicationic species and thus is likely closely-associated or ion-paired in solution with a relevant counteranion such as triflate (available from the synthesized solid) or hexafluorophosphate (available in vast excess from the supporting electrolyte).
Importantly, the digital simulations of the quiescent voltammetry also provided values of k0 and α, with the heterogeneous electron-transfer rate constants obtained from these simulations (0.0054 and 0.0017 cm/s for Au and Pt, respectively) residing in the realm of electrochemically quasi-reversible systems (Matsuda and Ayabe, 1955). This is in line with our prior proposal based solely on the ΔEp values obtained from CV measurements. The obtained k0 values also compare well with values obtained for the redox manifold of other uranyl systems, with k0 values ranging from 0.0028 to 0.0092 cm/s for systems studied under aqueous conditions (Morris, 2002). The obtained symmetry factors (0.47 ± 0.05 and 0.51 ± 0.02 for Au and Pt respectively) are also consistent with the expected value of 0.5 from the Butler-Volmer kinetic model for a typical redox system.
3.2 Rotating disk electrochemical analysis
Having extracted apparent values of the key parameters describing the [UVIO2]/[UVO2] couple with digital simulations of the data collected in quiescent solutions with cyclic voltammetry, we turned to hydrodynamic methods to directly experimentally quantify the parameters that govern this redox pair. As mentioned above, rotating disk electrode (RDE) electrochemistry utilizes convection to pull the bulk analyte to the surface of the electrode, circumventing the limitations of diffusion typically associated with more routine electrochemical measurements. Using a Au disk electrode affixed to the base of a rotating shaft (either under ambient conditions or a positive pressure of argon), the current response of the analyte can be measured at various rotation rates. From these experiments, a Koutecký-Levich analysis was applied to experimentally determine the parameters Dox, Dred, k0, and α (Levich, 1962; Bard et al., 2022).
To provide an initial determination of the validity of pursuing these RDE experiments, a CV was collected of [UVIO2] in the RDE cell (see Figure 5 top panel), which showed a U(VI)/U(V) couple at E1/2 = −0.18 V vs. Fc+/0 with ΔEp = 100 mV, consistent with results from the quiescent electrochemistry carried out in the glovebox. Encouraged by the result of this CV, we conducted linear sweep voltammetry (LSV) measurements, scanning from more oxidizing to more reducing potentials at a scan rate of 50 mV/s, to probe the current response of [UVIO2] under hydrodynamic conditions. The obtained current response from the LSV’s at rotation rates ranging from 500 to 2000 RPM (52.36–209.44 rad/s) displayed an increased current flow at increased rotation rate, of course, as expected as the diffusion limitation decreases upon increasing rotation rate. To estimate the current in the absence of mass transport limitations, six potentials were chosen at values negative of E°′, as estimated from the diffusion coefficients extracted from the CV simulations (vide supra). A Koutecký-Levich plot was constructed from these experiments by plotting the reciprocal of the current at each chosen overpotential against the reciprocal of the square root of angular rotation rate (see Figure 6; Supplementary Figures S31, S32; Supplementary Table S3). The slopes of these lines are expected to be nearly identical, placing the lines parallel with one another on the basis of the Koutecký-Levich (KL) equation, given below in Equation 2.
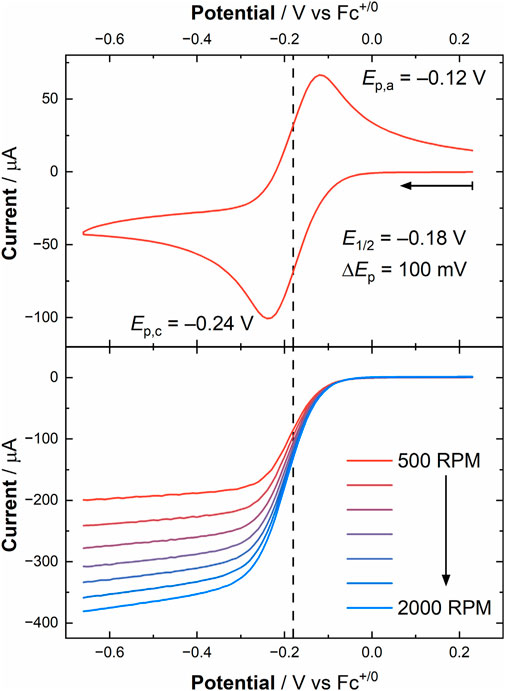
Figure 5. Upper panel: cyclic voltammogram of [UVIO2] obtained from the RDE experiment in MeCN under ambient conditions ([U] = 1.0 mM, working electrode: Au disk (0.196 cm2), electrolyte: 0.1 M TBAPF6, scan rate: 100 mV/s). Lower panel: linear sweep voltammograms at increasing rotation rates in MeCN ([U] = 1.0 mM, working electrode: Au disk (0.196 cm2), electrolyte: 0.1 M TBAPF6, scan rate: 50 mV/s). The vertical dashed line in both panels corresponds to the E1/2 value obtained from the cyclic voltammogram (E1/2 = −0.18 V vs. Fc+/0).
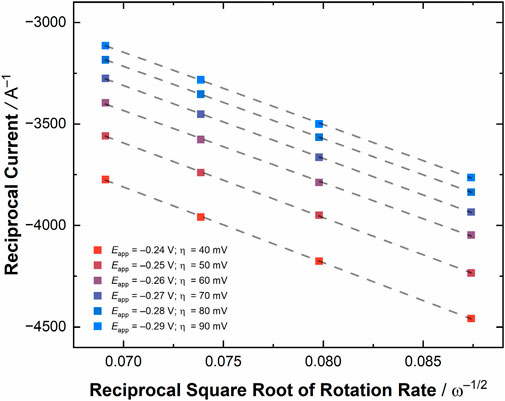
Figure 6. A zoomed-in region of the Koutecký-Levich plot for [UVIO2] at various overpotentials (for the full plot, see Supplementary Figure S32). The overpotential (η) values are given in mV in the figure legend. The linear fit of each line is shown as a dashed gray line visualizing the parallel nature of the lines and expected Koutecký-Levich behavior of the system at the selected overpotentials.
In this expression, iK is the current in the absence of mass transport (Amps), n is the number of electrons, F is Faraday’s constant, A is the area of the electrode (cm2), Cox* is the initial concentration of the bulk solution, Dox is the diffusion coefficient for the oxidized species, ω is the angular rotation rate of the electrode (radians sec−1), and ν is the kinematic viscosity of the solution (cm2 sec−1). As the kinematic viscosity of 0.1 M TBAPF6 in MeCN can be assumed to be nearly indistinguishable from the reported kinematic viscosity for 0.1 M tetraethylammonium perchlorate (TEAP) in MeCN (Tsushima et al., 1994), the only unknown variable in the expression related to the slope of the KL lines is the diffusion coefficient of the oxidized species. Therefore, the average slope of the lines was computed (36,409 ± 609 s1/2 C−1 cm−1/2) and this value used to determine the value of the diffusion coefficient for [UVIO2] (Dox) of 3.76 ± 0.06 × 10−6 cm2/s. This value is in quite reasonable agreement with the value of Dox extracted with the simulations of the quiescent voltammetry (5.52 × 10−6 cm2/s). To confirm the reasonableness of our method and result, we also carried out a determination of the diffusion coefficient of Cp2Fe under similar conditions, and found the value of this parameter to be 2.5 ± 0.06 × 10−6 cm2/s (see Supplementary Figures S27–S30; Supplementary Table S2), quite similar to the literature value (vide supra). Considering all this, the KL method appears reliable for analyzing the diffusional nature of actinide-containing complexes, such as [UVIO2], even in nonaqueous media and on the benchtop.
To determine both k0 and α, further analysis of the constructed KL plot was required. Based on Equation 2, the reciprocal of iK (kinetic current), corresponds to the intercept of these KL lines, which can be interpreted as the expected current at an infinite rotation rate. Using the kinetic currents across multiple overpotenials, the heterogeneous electron-transfer rate constant and symmetry factor can be obtained from the equations given below.
Where iK is the kinetic current, F is Faraday’s constant, A is the area of the electrode, C*ox is the initial concentration of the bulk solution, and kf(E) is the forward rate constant as a function of the applied (over)potential (expressed below in Equation 4).
In this expression, k0 is the heterogeneous electron-transfer rate constant, α is the symmetry factor, n is the number of electrons passed, F is Faraday’s constant, R is the ideal gas constant, T is temperature, E is the applied (over)potential, and E°′ is the formal reduction potential. Combining Equations 3 and 4, the below equation can be obtained, Equation 5.
Equation 5 is a function of overpotential. For this system, the applied potential is always more negative than the formal potential, and thus we note that the absolute value of the difference between the overpotential and formal potential was utilized to avoid an invalid mathematical result. The values of the applied potential were always chosen to be more negative than the formal potential in our study of [UVIO2] because our interest in this work concerned the reduction process to form [UVO2]. With this in mind, the observed kinetic currents can be plotted as a function of overpotential (see Figure 7). From this plot, the intercept can be converted to the experimentally determined k0, as at zero overpotential k0 is equivalent to iK/(FACox*). Upon solving for k0, a heterogeneous electron-transfer rate constant of 0.0754 ± 0.0004 cm/s was obtained for [UVIO2], a value consistent with rather fast electron-transfer kinetics at the surface of the gold electrode. (The uncertainty on the value of k0 comes from the error analysis on the y-intercept of the linear fit of the experimental data to Equation 5.) The symmetry factor, α, could also be solved using Equation 5. The average α value was obtained by solving for α at each overpotential and taking the arithmetic average, determined to be 0.47 ± 0.16 which is in excellent agreement with the simulated value of 0.47 ± 0.05.
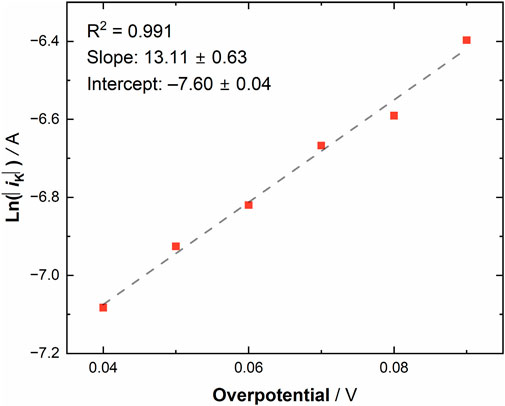
Figure 7. Natural log of the absolute value of the observed kinetic currents for [UVIO2] (in MeCN electrolyte under ambient conditions) vs. overpotential.
In an effort to eliminate potential side reactivity or chemical instability caused by ambient O2 (and, as much as possible, H2O), the RDE measurements were repeated using a positive pressure of argon gas in the electrochemical cell. This set of experiments was conducted using similar potential ranges to the measurements conducted under ambient conditions. The obtained CV under these more inert conditions resulted in a virtually identical voltammogram, with E1/2 = −0.18 V vs. Fc+/0 and ΔEp = 80 mV (see Supplementary Figure S33). These data compare very well with related values from quiescent electrochemistry and from the RDE experiment under ambient conditions. We then conducted LSVs with rotation rates ranging from 500 to 2000 RPM, at a scan rate of 50 mV/s, scanning from more oxidizing to more reducing potentials to obtain the hydrodynamic current response of this system under these more inert conditions. Applying the KL approach to this set of LSVs, and using the same overpotential values from the experiment under ambient conditions, a diffusion coefficient of 3.73 ± 0.05 × 10−6 cm2/s was obtained. This value is indistinguishable, within error, from the obtained diffusion coefficient under ambient conditions. Using the KL method, we then applied Equation 5 to determine k0 and α under these conditions, obtaining a heterogeneous electron-transfer rate constant of 0.0772 ± 0.0001 cm/s, a value slightly greater than that obtained from the ambient atmospheric experiments, and a symmetry factor of 0.47 ± 0.16, identical to that of the prior experiment (see Supplementary Figures S34–S36; Supplementary Table S4).
The foregoing results, taken together, demonstrate that [UVIO2] is stable, at least on the time scale of the electrochemical experiment (∼1 h), to the presence O2. It also demonstrates that it is stable in the presence of ambient humidity (H2O) from the atmosphere of our laboratories, although we note that our laboratories are climate controlled and these experiments were conducted on days that were not particularly humid in Lawrence, Kansas. We would highlight here that, as one peer reviewer remarked upon reading this report, laboratory humidity is often omitted from the description of experimental details in the literature but nonetheless is often a non-negligible factor in conducting otherwise routine experiments. At this stage, we have not conducted RDE work of the type reported here with [UVIO2] on days of widely varying humidity, and thus no data are in hand regarding humidity-dependent effects. However, we actively considered the role of liquid H2O (added to the experimental conditions) and these results are described in Section 4 of this paper. Nonetheless, a role for ambient humidity in influencing the performance of our system is relevant to the nuclear forensics applications of hydrodynamic voltammetry, and thus we highlight it as an area deserving of further attention when/if needed.
With the hydrodynamic voltammetry of [UVIO2] at the RDE completed, we next set out to experimentally determine the diffusion coefficient of the reduced species [UVO2] under similar conditions. At the outset, we anticipated that [UVO2] could be more sensitive to O2 and/or H2O than the U(VI) species, and therefore we only conducted these studies under an Ar atmosphere. Following the procedure applied to [UVIO2], but under Ar, a CV in quiescent solution was obtained using the stationary Au disk working electrode showing values of E1/2 = −0.18 V vs. Fc+/0 and ΔEp = 110 mV (see Supplementary Figure S37). The peak-to-peak separation for [UVO2] was thus a bit greater at 100 mV/s than the value for [UVIO2] of 100 mV. Superficially, we would usually consider this difference marginal under our experimental conditions.
Thus, we followed the initial quiescent CV experiment conducted on the stationary RDE with LSV studies, using the same rotation rates applied in the [UVIO2] experiments. However, in this set of experiments, the LSVs started at more reducing potentials and were swept to more oxidizing potentials, as we were aiming to interrogate the oxidation of [UVO2] to [UVIO2] rather than the previously studied reduction. Applying the KL method to this set of data, a diffusion coefficient of [UVO2] was obtained as Dred = 9.71 ± 0.16 × 10-5 cm2/sec, comparing well with the diffusion coefficient obtained from CV simulations (1.11 ± 0.04 × 10-5 cm2/s). Analogous to the [UVIO2] experiments, the KL method was used to determine k0 (0.0302 ± 0.0001 cm/s) and α (0.41 ± 0.11), (see Supplementary Figures S38–S40; Supplementary Table S5). Although the obtained α value is in reasonable agreement with both simulation and the results from the hydrodynamic analysis of [UVIO2], the observed k0 is 2.5 times less than the k0 value calculated from the results for [UVIO2]. This is in accord with the greater ΔEp observed for [UVO2], however. And, by extension, this result is also in accord with the observation that, on Au working electrodes, [UVIO2] and [UVO2] do not display detectable electrode fouling behavior or cycling/time dependent behaviors.
3.3 Rotating ring-disk electrochemical analysis
With the experimentally determined parameters governing the [UVIO2]/[UVO2] redox pair in hand, we turned to rotating ring-disk electrode (RRDE) electrochemistry in an attempt to directly quantify the chemical reversibility of this redox pair under convective conditions. RRDE electrochemistry is exceptionally well poised to speak to the chemical stability of both [UVIO2] and [UVO2] because the use of two working electrodes provides unique data not available from a simple cyclic voltammogram collected in a quiescent solution. Similar to the RDE measurements, laminar flow is induced by rotating the electrode, but in the RRDE method, the disk and ring electrodes are held co-planar and are separated by an inert spacer material (for this study a RRDE electrode shaft constructed out of PEEK and a Teflon spacer was used). The co-planar electrode configuration in combination with the induction of laminar flow allows for the electrogenerated species at the disk electrode to be detected at the ring electrode, as the flow of the solution is directed outward from the center of the electrode.
Importantly, in RRDE measurements, the ratio of the “generator” current at the disk electrode and the “collector” current at the ring electrode is described mathematically by a fixed collection efficiency that depends on the specific geometric details of the disk, ring, and separator. Depending on these parameters and the rotation rate, the transit time from the disk to the ring is fixed as well. Therefore, for a chemically reversible process in which an electrogenerated species is stable in solution for at least the timescale of transit from disk to ring (ca. 500 ms on average for conventional rotation rates), one can use the ratio of disk and ring currents to provide a quantitation of the degree of chemical reversibility of a redox couple. Such a quantitation provides an alternative to the voltammetric approach of examining the ratio of cathodic and anodic peak currents. Stated another way, the ratio of ring current to disk current should be near unity for a reversible process when adjusted by the theoretical collection efficiency of the electrode configuration being used.
The theoretical collection efficiency (NT) can be determined for a given electrode configuration Equation 6:
In this expression, NT is only dependent on the radius of the disk, r1, the inner diameter of the ring, r2, and the outer diameter of the ring, r3. In the expression, γ is equal to (r2/r1)3–1 and β is equal to [(r33/r13) – (r23/r13)]. Applying Equation 6 to the electrode used in this study, where the disk radius was 2 mm (r1), the ring to disk gap caused by the Teflon spacer was 0.75 mm, and the inner diameter of the ring was 3.25 mm (r2), and the outer diameter of the ring was 3.75 mm (r3), a theoretical collection efficiency of 25.6% was obtained.
Considering all this, an RRDE experiment represents a four-electrode configuration. In the approach taken in our work, the first working electrode was the disk electrode (Au disk, 0.126 cm2) and was used for generating the desired species in solution by scanning across a chosen potential window. The second working electrode was the ring electrode (Pt ring, thickness: 0.5 mm) was used as the collector electrode by holding at a potential sufficiently reducing (or oxidizing) to electrochemically regenerate the starting analyte. To determine the efficiency of our RRDE set-up, the oxidation of Cp2Fe to Cp2Fe+ was used as a test analyte. LSVs were obtained at a scan rate of 50 mV/s at the same set of rotation rates applied to the RDE experiments (see Supplementary Figure S41). However, to experimentally determine the collection efficiency, Ne, the absolute value of the ratio of the disk current and ring current was used, given in Equation 7.
Here, iR is the current measured at the ring electrode and iD is the current measured at the disk electrode. In this case, three disk potentials (0.405 V, 0.505 V, and 0.605 V vs. Fc+/0) were used to quantify both iR and iD, and then the apparent efficiencies were averaged across each rotation rate to yield an experimental collection efficiency of 23.8% ± 0.7% (see Supplementary Table S6). This is in good agreement with the expected/theoretical collection efficiency, NT, of 25.6%. The observation that Ne is lower than NT for our system is reasonable, and suggests influences of imperfect laminar flow and disruption of perfect co-planarity of the two working electrode surfaces in our apparatus.
With the validation from the experiments with Cp2Fe completed, we moved to measuring the RRDE current response with [UVIO2]. Performing LSVs under ambient conditions, the disk electrode was scanned across a potential window from 0.30 V to −0.59 V vs. Fc+/0 (50 mV/s, 500 to 2000 RPM). This series of experiments yielded current responses similar to those observed in the RDE experiments (see Figure 8 lower panel). However, due to utilizing a smaller disk electrode (0.126 cm2 for RRDE experiments vs. 0.196 cm2 for RDE experiments) an overall decrease in current was observed despite the nominally identical solution concentrations between the experiments. As anticipated, the current response corresponding to the oxidation of the electro-generated [UVO2] was measured at the ring electrode (fixed potential: 0.3 V vs. Fc+/0, see Figure 8 upper panel). This observation bolsters the evidence that both [UVIO2] and [UVO2] are stable in MeCN on the time scale of these experiments. However, contrasting with the result from the Cp2Fe experiment, the observed collection efficiency of this experiment was calculated to be 12.6% ± 1.0% (see Supplementary Table S7). This value of Ne is nearly half the theoretical collection efficiency (NT) of 25.6%.
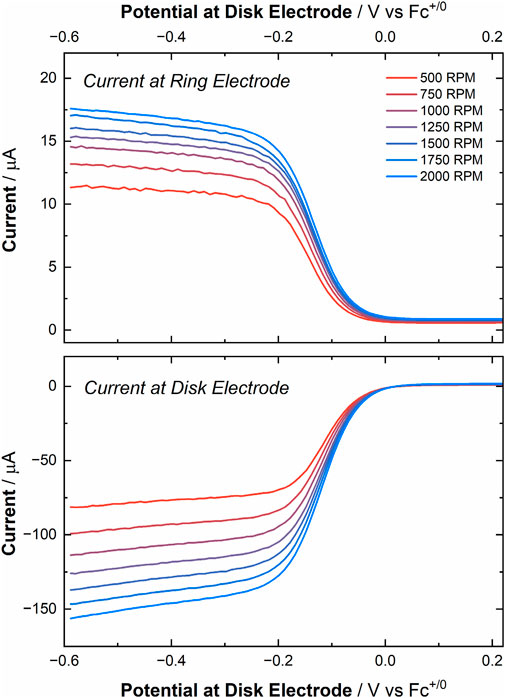
Figure 8. Top panel: Current response at various rotation rates (500–2000 RPM) at the ring working electrode, corresponding to the oxidation of electro-generated [UVO2] to [UVIO2] (fixed potential of 0.3 V vs. Fc+/0). Bottom panel: Linear sweep voltammograms of [UVIO2] at various rotation rates (potential range: 0.3 to −0.59 V vs. Fc+/0).
In other words, the collection efficiency was 49% of the expected value, suggesting a degree of chemical irreversibility in the RRDE experimentation that was not observed in all cyclic voltammetry investigations conducted to date. We theorized, however, that the suboptimal collection efficiency observed for [UVO2] generation under ambient conditions could be attributable to reactivity of the U(V) species with solvated O2 (and/or H2O) in the MeCN electrolyte, and therefore we conducted analogous experiment under a positive pressure of argon in the electrochemical cell on the benchtop. Although this experiment yielded a marginally larger collection efficiency, 13.9% ± 1.1% (see Supplementary Figure S42; Supplementary Table S8), the calculated collection efficiency in this case is still near half (54%) of the expected theoretical value for a perfectly reversible redox process occurring at the disk. Of note, however, the ring electrode for our RRDE set-up is limited by our available equipment to being a polycrystalline Pt electrode. It is thus important to mention that we have observed (vide supra) significantly poorer electrochemical behavior, including poorer electrochemical reversibility, with Pt electrodes than with Au electrodes for experiments involving [UVIO2]. This behavior on Pt as measured in CV and RDE work thus appears in the RRDE results as well.
The hydrodynamic current response of solutions prepared with pure samples of [UVO2] were also examined with RRDE electrochemistry—in this case to identify the production of [UVIO2] at the disk electrode by re-reduction to re-form [UVO2] at the ring. LSVs were thus conducted (with a positive pressure of argon gas flow in the electrochemical cell) by sweeping the disk electrode across a potential window of −0.6 V to –0.2 V vs. Fc+/0 (50 mV/s, 500 to 2000 RPM), which yielded the expected sigmoidal current responses across all RPM rates. However, similar to the case of the RRDE studies with [UVIO2], the experimental collection efficiency for [UVO2] was less than the theoretical collection efficiency, calculated to be 9.8% ± 1.2% (see Supplementary Figure S43; Supplementary Table S9). This corresponds to a collection efficiency of only 38% of the expected value for ideal chemical reversibility with our apparatus. On the basis of the results from RRDE measurements conducted with [UVIO2], the suboptimal collection efficiency was anticipated. However, the further decrease from the 54% of optimal collection achieved with [UVIO2] under Ar flow to 38% of optimal collection with [UVO2] was not anticipated. At this stage, we theorize that the location of our rotatating electrode and bipotentiostat outside of our inert-atmosphere glovebox may be driving these results. This is because the measurements with the RDE and RRDE were collected outside of the glovebox, in glass cells in which adventitious water is certainly present in greater concentrations than typically encountered in the glovebox. The water could make its way into our solutions either from sticking to the glass cells and electrode connections used for the mesasurements, or from the brief handling of solutions under ambient air to load them into the cell. Thus, at this time, we ascribe the unexpected deviations from the theoretical collection efficiency in these measurements both to a possible role of polycrystalline platinum electrodes in driving chemical irreversibility as well as a possible role of adventitious water in driving reactivity with both [UVIO2] and [UVO2].
To wrap up the set of hydrodynamic experiments reported here, we conducted RRDE measurements with cyclic voltammetry under hydrodynamic conditions in order to rigorously assign the relationship between cathodic disk current for reduction of [UVIO2] and anodic ring current for re-oxidation of nascent [UVO2] produced at the disk. Such voltammetry represents both a complementary view of the chemical reversibility of the redox process being interrogated as well as a test of chemical reversibility. With the ring potential set to +0.25 V vs. Fc+/0 (a potential sufficiently positive to oxidize any [UVO2] electro-generated at the disk), the disk electrode was swept cyclically across a potential window of +0.25 V to −0.65 V vs. Fc+/0 (see Figure 9 red trace). The data from this RRDE-CV experiment show that as the disk potential is swept closer to the value (approaching E1/2 or E°′) necessary to reduce [UVIO2], current responses are observed at the disk electrode corresponding to the reduction of [UVIO2] (Figure 9 purple trace) and at the ring electrode corresponding to the oxidation of the electro-generated [UVO2] (Figure 9 blue trace). The reverse behavior can also be observed, in that as the disk potential sweeps back to values that (in a quiescent solution) would be sufficiently positive to oxidize [UVO2] present at the electrode in solution, a decrease in ring current back to the baseline value is observed. Thus, when the disk is no longer generating [UVO2], very little anodic current is observed at the ring. Stated another way, this symmetric current response is indicative of [UVO2] being freely diffusional in solution, and able to make the journey from the disk to the ring intact. The electrochemical process for U(V) production can thus be safely concluded to be selectively turned on and off based on the potential applied to the Au disk working electrode.
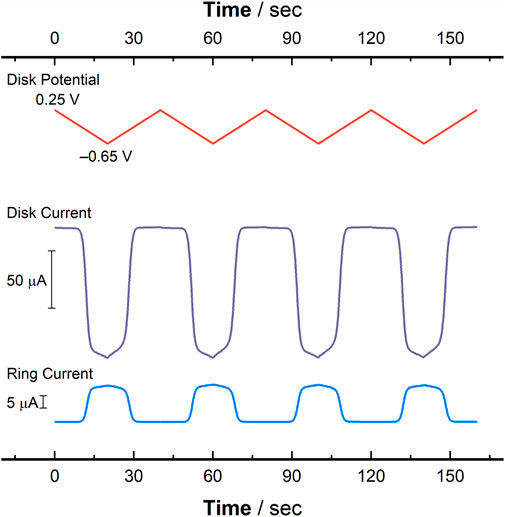
Figure 9. Data from rotating ring-disk cyclic voltammetry (RRDE-CV) studies conducted with a solution containing [UVIO2] plotted as a function of time ([U] = 1.0 mM, working electrode: Au disk (0.126 cm2), electrolyte: 0.1 M TBAPF6, scan rate: 50 mV/s). Red trace: applied disk potential, in V vs. Fc+/0. Purple trace: disk current observed for [UVIO2], corresponding to the current at the reducing electrode. Blue trace: ring current observed for [UVIO2], corresponding to the reoxidation of the electro-generated reduced species (ring potential: +0.3 V vs. Fc+/0).
4 Discussion
This work was targeted at demonstrating the usefulness of hydrodynamic (RDE and RRDE) methods for electroanalysis and quantification of the fundamental electrochemical characteristics of a U(VI)/U(V) redox couple in non-aqueous media. Along this line, we chose to investigate a macrocyclic system recently developed in our laboratory for which both the [UVIO2] and [UVO2] forms can be synthetically isolated. We felt that such an approach would be valuable, in that the ability to chemically isolate both the formal oxidation states of the complex affords the opportunity to separately interrogate the redox couple in both the reductive and oxidative directions.
Through the Koutecký-Levich analysis carried out with the RDE, we measured a k0 value for the target U(VI)/U(V) redox couple under ambient laboratory atmosphere of 0.0754 ± 0.0004 cm s−1, a value that is over an order of magnitude larger than the value of 0.0054 ± 0.0016 cm s−1 that was extracted from cyclic voltammetry data collected in an inert-atmosphere glovebox. In principle, the discrepancy between the experimentally determined and simulated k0 could be ascribable in part to the electrochemically active surface area of the rotating-disk electrode being larger than anticipated, due to imperfections of the electrode surface; a large roughness factor would contribute to higher than anticipated currents. However, we consider this possibility unlikely given the order-of-magnitude increase in apparent electron-transfer rate.
Instead, at this stage, we anticipate that impedance effects from the necessarily circuitous electrical wiring required to interface an electrochemical cell inside the glovebox with our potentiostat outside the glovebox might provide an often-undetected limitation on charge transfer kinetics. In support of this proposal, our group has often quoted a “fast limit” of electron transfer on highly oriented pyrolytic graphite electrodes in our glovebox of 0.010 cm s−1, a value that is well below the apparent k0 in the RDE configuration studied here with a gold electrode of ca. 0.075 cm s−1. But, qualitative evidence against this proposal came from comparing the peak-to-peak separation (ΔEp) of the Cp2Fe+/Cp2Fe couple on polycrystalline Au electrodes measured under both conditions; as expected, more concentrated ferrocene solutions gave greater ΔEp values under both conditions, but the values of ΔEp measured in control runs were within the conventional range in all cases. Detailed investigations would seem to be necessary to investigate the possibility of a substantive influence on the measured k0 values from circuit impedance.
Nonetheless, we are quite satisfied by the finding that quite fast electron transfer is achievable for the [UVIO2]/[UVO2] redox couple under hydrodynamic conditions, as it shows that the Pt(II)-templated macrocycle supporting the complexes can engender rapid charge transfer to/from uranium. This ability complements the shift of the U(VI)/U(V) reduction potential to the most positive value yet measured for a non-oxo-functionalized uranyl system. Together, these characteristics make the Pt(II)-templated macrocycle quite attractive for further development of electrochemical assays for uranium, when solution conditions are maintained that are suitable for binding of appropriate uranyl species.
A distinctive feature of the [UVIO2]/[UVO2] redox couple is excellent performance on gold electrodes. The instantaneous performance of a gold working electrode in a single cycle of voltammetry lasting 16 s was found to be essentially and reliably equivalent to the performance of the same working electrode over the course of a series of RDE experiments used for detailed analysis lasting 15 min. This performance can be ascribed to a virtually total lack of electrode fouling behavior on gold electrodes, both by the U(VI) and U(V) forms of the complex. From quiescent multicycling voltammetry experiments, this behavior could be visualized over ten (or presumably more) cycles. And, from the RDE work, it could be visualized by rotating linear sweep voltammograms that essentially follow “textbook” predictions. From an electroanalysis perspective, the lack of fouling is quite attractive as it suggests that an apparatus assembled for routine quantification of uranium in samples would require little downtime if using a uranium-ligating platform like our Pt(II)-templated macrocycle that inherently avoids undesirable interactions with the electrode surface.
However, we anticipate at this stage that although our Pt(II)-templated macrocycle has several attractive features, it is not ideal in every conceivable dimension. This is because of the specter of environment-dependent reactivity of the [UVIO2] and [UVO2] species studied in this work. On the U(VI) side, evidence from both quiescent voltammetry and rotating measurements has identified a possible reactivity pattern with Pt electrodes. On the U(V) side, there does not appear to be a detrimental reactivity pattern with Pt electrodes, but electrogenerated U(VI) appeared in RRDE experiments to be particularly prone to undesirable behavior at the Pt ring electrode. Taken together, these observations suggest two possibilities. First, the Pt(II)-templated macrocycle in [UVIO2] could be engaging in a “platinaphilic” interaction with the Pt electrode surface. This possibility could be investigated in future work with macrocycles templated by another dication. Second, the well-known ability of platinum electrodes to support surface-associated H-atom or hydride equivalents could be serving to degrade [UVIO2] by H-atom transfer or proton-coupled electron transfer behavior. Such a possibility is sensible given the champion characteristic of platinum electrodes as dihydrogen-evolving catalysts, and is certainly worth investigating given the typically harsh conditions required for conversion of U(VI) materials into lower valent states. An intriguing early report of platinum-catalyzed reduction of Pu(IV) with a mixture of 4% H2/96% Ar suggests that such reactivity might be feasible. (Rainey, 1965).
The possibility of platinum-adsorbed hydrogen/hydride reactivity raises the final point of discussion that we would mention, namely the potential role of adventitious water in affecting the chemical properties of [UVIO2] and [UVO2]. One reviewer of this paper noted that [UVO2] would seem to have a sufficiently negative reduction potential to reduce O2 but presumably not sufficiently negative to drive reduction of adventitious H2O. It struck the same reviewer as unlikely, however, that the compound could drive O2 conversion to O2⋅– via a purely outer-sphere 1e– reduction. The reviewer noted that the coordination sphere of the uranium center (for both the U(VI) and U(V) oxidation states) is fully occupied, meaning that an inner-sphere pathway for electron transfer is presumably blocked. The ligand exchange rate in both oxidation states can be reasonably anticipated to be relatively slow in acetonitrile, meaning that any inner sphere processes with exogeneous reagents like oxygen or water would be retarded from this perspective as well.
In our laboratory, we typically handle coordination complexes under rigorously air- and moisture-free conditions, either in gloveboxes or with Schlenk-type glassware. To execute the hydrodynamic studies reported here, however, we handled the complexes for short periods under ambient conditions. Although there was no obvious reaction of either the darkly red solutions of [UVIO2] or the pale-yellow solutions of [UVO2] with ambient water/oxygen, this cannot be taken as rigorous evidence supporting a lack of reactivity, particularly with H2O. H2O could be responsible for a variety of outcomes, including loss of [UO2] from the macrocyclic ligand, H+/H•/H−reactivity, or disproportionation behaviors. These possibilities merit additional chemical work, particularly for the more reducing [UVO2] system.
As a first qualitative test, however, we added 1000 equivalents of purified H2O (18.2 MΩ from a MilliQ system) via syringe to a working solution of [UVIO2] in acetonitrile-based electrolyte on the benchtop after conclusion of a series of hydrodynamic measurements. The starting dark red solution underwent an immediate color change to bright yellow, suggesting rapid decomplexation of uranyl from the Pt(II)-templated macrocycle. This color change is in accord with the appearance of uranyl triflate, UO2(OTf)2, in water as well as the bright yellow coloration of the monometallic Pt(II)-templated macrocycle in organic solvents like acetonitrile. A great number of exciting experiments wait on the horizon based on this result, including efforts to identify reaction products, the stoichiometry of the water-induced uranyl displacement reaction, the thermodynamics of ligand binding, and the kinetics of these processes.
And finally, we note that the hydrodynamic nature of these studies, in which solutions are continually stirred, could be anticipated to give rise to more significant effects from adventitious water than would be encountered in quiescent experiments. This is both because of stirring solutions under atmospheres that could be “contaminated” with H2O or over glassware and electrode materials with surface-associated H2O. Along this line, the marginally slower k0 measured by RDE (and implied by voltammetric simulations) for oxidation of [UVO2] in comparison to reduction of [UVIO2] might provide evidence of specific interactions or reactivity between [UVO2] and H2O. This possibility and the other experimental opportunities mentioned above are currently under investigation in our laboratory.
5 Conclusion
Taken together, this work demonstrates the high degree of usefulness that hydrodynamic techniques can offer toward electroanalysis and measurement of the fundamental electrochemical characteristics of actinide redox couples. In this study, we focused on a rather unique U(VI)/U(V) redox couple in order to quantify electron-transfer kinetics and chemical reversibility in particular. Moving forward, we anticipate that hydrodynamic measurements could grow to be useful tools for the nuclear forensics and safeguards communities. However, careful preliminary attention to the chemical characteristics of the target element(s) and their oxidation states would seem to be required, as well as attention to possible interactions of the species to be formed as analytes with the relevant working electrode surfaces. An unavoidable limitation of the initial studies reported here would seem to have been reliance on a platinum ring electrode for RRDE studies of a redox couple that appears to intrinsically misbehave on platinum. Nonetheless, such a problem can be overcome by screening and thorough comparison of a variety of electrode materials prior to beginning hydrodynamic measurements. And, as demonstrated by the quite satisfactory agreement in this report between findings from digital simulation of quiescent voltammetry and direct quantification of individual parameters from hydrodynamic measurements, the investigation of electrochemistry in both stirred and unstirred solutions can yield complementary insights into the properties of redox processes of interest.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
AE: Conceptualization, Investigation, Writing – original draft, Writing – review and editing. JB: Conceptualization, Funding acquisition, Investigation, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences through the Early Career Research Program (DE-SC0019169).
Acknowledgments
This paper is dedicated to Prof. George S. Wilson in appreciation of his scholarly contributions to chemistry and his unflagging enthusiasm in support of students, collaborators, and colleagues.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnuen.2025.1639874/full#supplementary-material
References
Bahri, M., Ruas, A., Labbé, E., and Moisy, P. (2017). Electrochemical characterization of plutonium in n-tributyl phosphate. Dalton Trans. 46, 4943–4949. doi:10.1039/C6DT04765C
Bard, A. J., Faulkner, L. R., and White, H. S. (2022). Electrochemical methods: fundamentals and applications, 3rd edn. John Wiley and Sons.
Bard, A. J., Parsons, R., and Jordan, J. (1985). Standard potentials in aqueous solution. Marcel Dekker.
Becerril, V., Meas, V., Tejera, R., and Ozil, P. (1993). An improved rotating disc cathode cell for electrodeposition of actinides. The case of plutonium. Nucl. Instrum. Methods Phys. Res. A 328, 512–516. doi:10.1016/0168-9002(93)90668-8
Bond, A. M., Oldham, K. B., and Snook, G. A. (2000). Use of the ferrocene oxidation process to provide both reference electrode potential calibration and a simple measurement (via semiintegration) of the uncompensated resistance in cyclic voltammetric studies in high-resistance organic solvents. Anal. Chem. 72, 3492–3496. doi:10.1021/ac000020j
Connelly, N. G., and Geiger, W. E. (1996). Chemical redox agents for organometallic chemistry. Chem. Rev. 96, 877–910. doi:10.1021/cr940053x
Golwankar, R. R., Ervin, A. C., Makoś, M. Z., Mikeska, E. R., Glezakou, V.-A., and Blakemore, J. D. (2024a). Synthesis, isolation, and study of heterobimetallic uranyl crown ether complexes. J. Am. Chem. Soc. 146, 9597–9604. doi:10.1021/jacs.3c12075
Golwankar, R. R., Makoś, M. Z., Cajiao, N., Neidig, M. L., Oliver, A. G., Day, C. S., et al. (2024b). Activation and functionalization of the uranyl ion by electrochemical reduction. Inorg. Chem. 63, 24542–24553. doi:10.1021/acs.inorgchem.4c03349
Madic, C., Hobart, D., and Begun, G. (1983). Raman spectrometric studies of actinide (V) and-(VI) complexes in aqueous sodium carbonate solution and of solid sodium actinide (V) carbonate compounds. Inorg. Chem. 22, 1494–1503. doi:10.1021/ic00152a015
Matsuda, H., and Ayabe, Y. (1955). Zur Theorie der Randles-Sevčikschen Kathodenstrahl-Polarographie. Berichte Bunsenges. für Phys. Chem. 59, 494–503. doi:10.1002/bbpc.19550590605
Mikeska, E. R., Curry, T. D., Wilson, R. E., and Blakemore, J. D. (2024). Electroanalytical characterization of Np (VI)/Np (V) redox in a pentadentate ligand environment and stabilization of [NpVO2]+ by hydrogen bonding. Dalton Trans. 53, 19126–19142. doi:10.1039/D4DT02557A
Morris, D. E. (2002). Redox energetics and kinetics of uranyl coordination complexes in aqueous solution. Inorg. Chem. 41, 3542–3547. doi:10.1021/ic0201708
Newton, T. W. (1975). The kinetics of the oxidation-reduction reactions of uranium, neptunium, plutonium, and americium in aqueous solutions. Technical Information Center, Office of Public Affairs, U.S. Oak Ridge, TN: Energy Research and Development Administration.
Nikitenko, S. I., Cannes, C., LE Naour, C., Moisy, P., and Trubert, D. (2005). Spectroscopic and electrochemical studies of U(IV)−Hexachloro complexes in hydrophobic room-temperature ionic liquids [BuMeIm] [Tf2N] and [MeBu3N] [Tf2N]. Inorg. Chem. 44, 9497–9505. doi:10.1021/ic051065b
Pleskov, Y. V., and Filinovskii, V. Y. (1976). The rotating disc electrode. Consultants Bureau, Plenum Publishing.
Rainey, R. H. (1965). Hydrogen reduction of Pu(IV) to Pu(III). Nucl. App. 1, 310–311. doi:10.13182/NT65-A20527
Rappleye, D., and Simpson, M. F. (2017). Application of the rotating cylinder electrode in molten LiCl-KCl eutectic containing uranium (III)-and magnesium (II)-chloride. J. Nucl. Mat. 487, 362–372. doi:10.1016/j.jnucmat.2017.02.037
Sherman, B. D., Sheridan, M. V., Wee, K.-R., Song, N., Dares, C. J., Fang, Z., et al. (2016). Analysis of homogeneous water oxidation catalysis with collector–generator cells. Inorg. Chem. 55, 512–517. doi:10.1021/acs.inorgchem.5b02182
Tsushima, M., Tokuda, K., and Ohsaka, T. (1994). Use of hydrodynamic chronocoulometry for simultaneous determination of diffusion coefficients and concentrations of dioxygen in various media. Anal. Chem. 66, 4551–4556. doi:10.1021/ac00096a024
Keywords: actinides, crown ethers, cyclic voltammetry, rotating electrodes, reversibility
Citation: Ervin AC and Blakemore JD (2025) Hydrodynamic characterization of the redox chemistry of crown-encapsulated uranyl complexes. Front. Nucl. Eng. 4:1639874. doi: 10.3389/fnuen.2025.1639874
Received: 02 June 2025; Accepted: 30 June 2025;
Published: 24 July 2025.
Edited by:
Kattathu Mathew, Oak Ridge National Laboratory (DOE), United StatesReviewed by:
Xian Tang, University of South China, ChinaIsrael Zilbermann, Ben-Gurion University of the Negev, Israel
Copyright © 2025 Ervin and Blakemore. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James D. Blakemore, Ymxha2Vtb3JlQGt1LmVkdQ==