 Michelle L. Boland
Michelle L. Boland Aparajita H. Chourasia
Aparajita H. Chourasia Kay F. Macleod
Kay F. Macleod- 1The Ben May Department for Cancer Research, The University of Chicago, Chicago, IL, USA
- 2Committee on Molecular Metabolism and Nutrition, The University of Chicago, Chicago, IL, USA
- 3Committee on Cancer Biology, The University of Chicago, Chicago, IL, USA
A mechanistic understanding of how mitochondrial dysfunction contributes to cell growth and tumorigenesis is emerging beyond Warburg as an area of research that is under-explored in terms of its significance for clinical management of cancer. Work discussed in this review focuses less on the Warburg effect and more on mitochondria and how dysfunctional mitochondria modulate cell cycle, gene expression, metabolism, cell viability, and other established aspects of cell growth and stress responses. There is increasing evidence that key oncogenes and tumor suppressors modulate mitochondrial dynamics through important signaling pathways and that mitochondrial mass and function vary between tumors and individuals but the significance of these events for cancer are not fully appreciated. We explore the interplay between key molecules involved in mitochondrial fission and fusion and in apoptosis, as well as in mitophagy, biogenesis, and spatial dynamics of mitochondria and consider how these distinct mechanisms are coordinated in response to physiological stresses such as hypoxia and nutrient deprivation. Importantly, we examine how deregulation of these processes in cancer has knock on effects for cell proliferation and growth. We define major forms of mitochondrial dysfunction and address the extent to which the functional consequences of such dysfunction can be determined and exploited for cancer diagnosis and treatment.
Introduction
Mitochondrial integrity is central to efficient cellular energy production and cell survival in the face of environmental stresses, such as nutrient deprivation and ischemia, but also in response to genotoxic agents, such as those used in cancer therapies (1–4). How changes in mitochondrial mass and function affects the basic biology of cancer or determine the clinical outcome for cancer patients stands out as having unexplored diagnostic and therapeutic potential.
Mitochondria and the Warburg Effect
Defective mitochondria was proposed by Otto Warburg to explain his observation that tumor cells undergo increased aerobic glycolysis (the so-called “Warburg effect”) compared to normal cells (5). While mutations in key Krebs cycle enzymes support the notion that mitochondrial metabolism is inherently defective in at least a few human cancers (6), evidence that dysfunctional mitochondria are the major cause of the Warburg effect is limited (5). Instead, accumulating evidence supports altered expression and activity of key glycolytic enzymes in tumor cells. For example, altered expression of phosphoglycerate dehydrogenase, phosphoglycerate mutase 1, and pyruvate kinase M2 has been shown to reduce the rate of glycolytic flux to pyruvate and increase flux to biosynthetic pathways (7), such as serine biosynthesis (8, 9) and the pentose phosphate pathway (10). Interestingly, increased glucose metabolism and the Warburg effect also promote tumor cell survival through redox regulation of cytochrome c and inhibition of apoptosis (11). This may explain the Warburg effect and tumor growth without necessarily invoking defective mitochondria, as discussed elegantly in recent reviews (5, 12–18).
Mitochondrial Genome Mutations in Cancer
While mitochondrial dysfunction does not necessarily explain the Warburg effect, there is significant evidence that tumors do indeed accumulate defective mitochondria (19–21). Homoplasmic mutations in the mitochondrial genome have been found in primary tumors (22) and linked to both increased primary tumor growth (23) and metastasis (24). The tumor-promoting effects of mitochondrial genome mutation, such as in the genes encoding subunit 1 of cytochrome oxidase (CO) or various subunits of NADH dehydrogenase (ND) is due in part to increased levels of cytosolic and mitochondrial reactive oxygen species (ROS) resulting from electron escape from the respiratory chain when these genes products have reduced function (19–21, 23, 24). There is also evidence that more efficient electron chain activity and complex I activity in particular limits breast tumor growth and metastasis in part by maintaining high NAD+/NADH levels (25). However, as recently discussed (26), more research is required to establish the extent to which mitochondrial genome mutations actually drive tumor growth and progression, as opposed to being a marker or readout of mitochondrial dysfunction itself.
Mitochondrial ROS in Cancer
Increased ROS levels primarily emanating from the mitochondria are a noted feature of transformed cells that are variously attributed to inefficiencies in electron transport at the respiratory chain, increased metabolic demand, reduced ROS scavenging, oncogene-induced replicative stress, and altered mitochondrial dynamics (27–30). Oncogene-induced ROS promotes tumorigenesis in numerous ways, including stabilization of hypoxia-inducible factor (HIF)-α, induction of oxidative base damage to DNA, increased calcium flux, inactivation of key phosphatases, such as Pten and activation of both the NRF2 and NF-κB transcription factors (27, 29). Elevated ROS levels in tumor cells compared to normal cells has been exploited experimentally to kill cancer cells specifically by chemically pushing ROS levels over a critical homeostatic threshold that is incompatible with either growth or survival of tumor cells but tolerable by normal cells (31–33). Indeed, many current genotoxic agents used in the clinic, such as cisplatin and certain alkaloids, rely on ROS production for their efficacy (29, 34, 35). However, one major side-effect of increasing ROS systemically in a clinical setting is the damaging effect of elevated ROS on normal tissues that can lead to disrupted differentiation and defective immune cell function, particularly in cell types, such as macrophages and neutrophils, where ROS is inherently elevated to perform critical signaling roles (29). Also, not all tumor cell types are equally sensitive to ROS induction with differences in the sensitivity of epithelial cells (resistant) compared to cells of mesenchymal origin (sensitive) (32). While appreciating the important signaling role mitochondrial ROS plays in cancer etiology and treatment response, this topic has recently been thoroughly reviewed elsewhere (29) and is not the focus of this review.
Mitochondria are Highly Dynamic
Mitochondria are highly dynamic organelles responding to cellular stress through changes in overall mass, interconnectedness, and sub-cellular localization (1–3)(Figure 1). Change in overall mitochondrial mass reflects an altered balance between mitochondrial biogenesis (increased mitochondrial genome duplication combined with increased protein mass added to mitochondria) and rates of mitophagy (degradation of mitochondria at the autophagosome) (36–38). In addition, the extent to which mitochondria are interconnected to each other as a single continuous mitochondrial reticulum is determined by the extent of mitochondrial fusion while conversely, mitochondrial fission results in fragmented mitochondria of smaller overall dimensions (2, 39, 40). Mitochondria are also dynamic in terms of where they are located in the cell with increased perinuclear localization under hypoxia compared to normoxia (41), at axonal termini in neurons (42), and movement along microtubules toward lysosomes under conditions that promote mitophagy (43). The extent to which control of mitochondrial dynamics, not only rates of fission versus fusion, but also changes in mitochondrial mass and sub-cellular spatial organization (Figure 1), is deregulated in cancer has been less frequently reviewed than Warburg (5), ROS (29), and mitochondrial genome DNA (mtDNA) mutations (26) and is the focus of this review. We shall also assess whether a rational basis exists to target key aspects of mitochondrial dynamics to treat cancer.
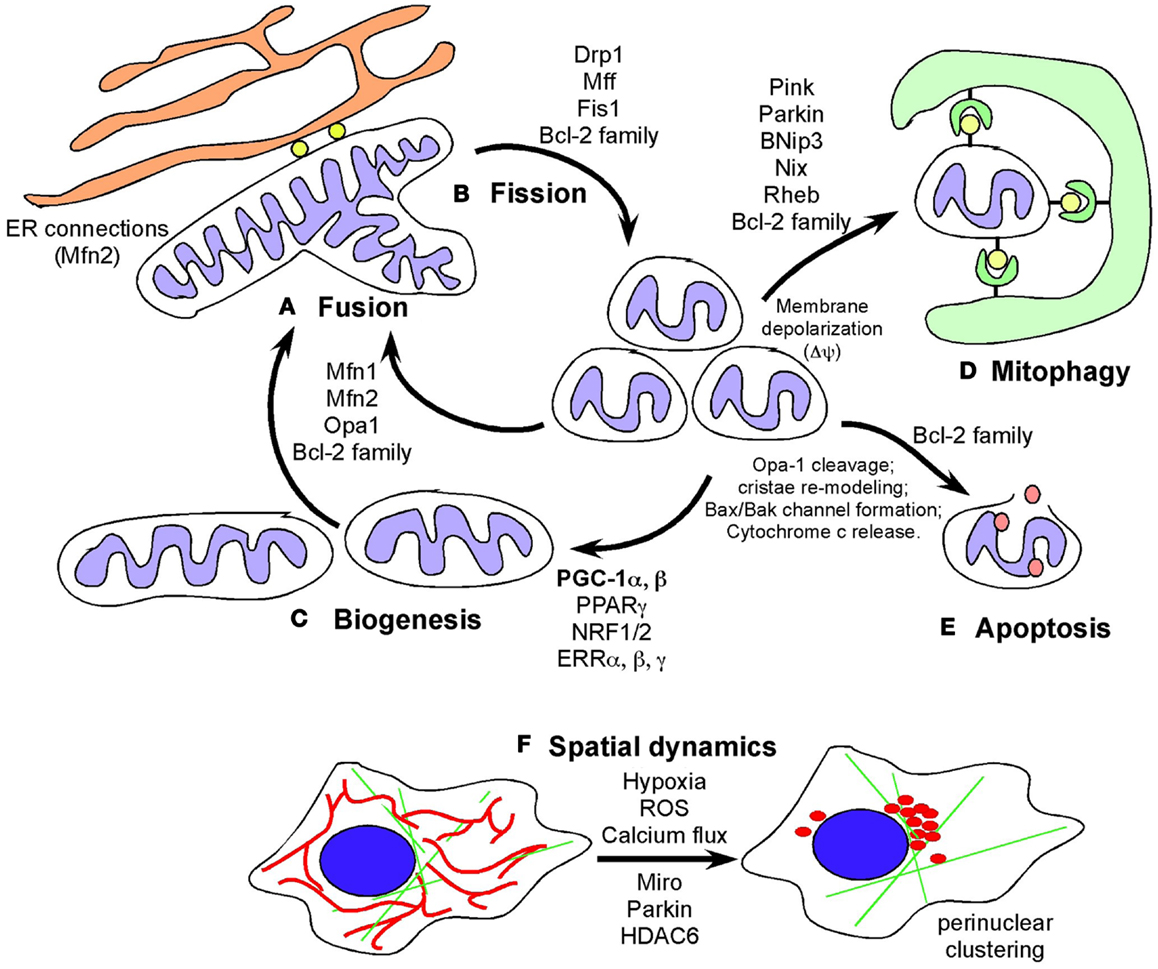
Figure 1. Defining mitochondrial dynamics. (A) Mitochondrial fusion requires the action of fusion proteins, OPA-1 at the IMM and Mitofusin 1 and Mitofusin 2 at the OMM promoting the fusion of membranes of juxtaposed mitochondria. Mitochondrial fusion is selective for polarized mitochondria and promoted by growth on oxidative carbon sources, such as galactose, that also induces respiratory chain protein expression, increased cristae density and formation of respiratory chain supercomplexes and increased OXPHOS. Fusion likely also contributes to increased respiration and mitochondrial metabolism by promoting increased diffusion of intermediate metabolites and reducing agents. Fusion also limits mitophagy and apoptosis. (B) Mitochondrial fission is promoted by the GTPase activity of the dynamin-related protein (DRP1) that is recruited to mitochondria in response to stresses, such as hypoxia, where DRP1 interacts with its mitochondrial receptors (Mff1, Fis1 and others) to pinch off mitochondria into smaller units. Mitochondrial fission depolarizes mitochondria but mitochondria generally recover. Failure to restore membrane potential is thought to target mitochondria for degradation by autophagy or depending on other stresses, result in apoptosis. Cleavage of OPA-1 promotes apoptosis. (C) Biogenesis is induced by nutrient deprivation and in response to oxidative stress and requires the coordinated expression of nuclear and mitochondrial encoded genes that are co-regulated by transcription factors NRF1/2, PPARγ, ERRα, β, γ, and the key transcriptional co-factor, PGC-1α. Mitochondrial biogenesis is required for cell growth to produce increased metabolites and energy and defects in biogenesis are frequently lethal to cells and organisms. (D) Mitophagy is a specialized form of autophagy in which mitochondria are targeted to nascent phagophores and engulfed by autophagosomes that fuse with lysosomes to degrade the encapsulated mitochondria. Mitochondrial fragmentation is required for mitophagy and induction of fusion protects mitochondria from degradation under starvation conditions. Mitophagy is promoted by a number of different mechanisms including Pink1/Parkin-mediated pathways and also the BNIP3/NIX pathway. (E) Apoptosis is a terminal event that is promoted by the activity of BH3-only members of the Bcl-2 superfamily of cell death regulators. When apoptosis is inhibited, novel functions for BAK/BAX and other Bcl-2 family members has been revealed. (F) Mitochondrial spatial dynamics is relatively under-studied but mitochondria respond to key stresses, including hypoxia and calcium flux in the cell, by changing their sub-cellular localization, including coalescing around the nucleus and changing their proximity to the ER. Mitochondrial migration in cells is modulated by Miro, a Ca2+ dependent small G protein as well as by poorly understood effects of Parkin and HDAC6.
Fission Versus Fusion in Cancer
Re-modeling of the mitochondrial network in cells is mechanically regulated by key dynamin-related fission and fusion gene products and takes place in response to hypoxia, cell cycle cues, changing energy demands, and other cellular stresses (1, 2, 39, 40, 44–46).
The Mechanics of Fission and Fusion
Mitochondrial fusion is promoted by homotypic/heterotypic interactions of the Mitofusin 1 and Mitofusin 2 dynamin-related GTPases at the outer mitochondrial membrane (OMM) of adjacent mitochondria and by Opa-1, also a dynamin-related GTPase, at the inner mitochondrial membrane (IMM) (1, 39, 40, 45). Inhibition or loss of any one of these proteins impedes mitochondrial fusion leading to increased mitochondrial fragmentation and is associated with clinical neuropathy in Charcot–Marie–Tooth disease and Autosomal Dominant Optic Atrophy, highlighting the critical role played by mitochondrial fusion in cellular homeostasis, particularly in the nervous system (39, 42).
Mitochondrial fission requires the recruitment of a different dynamin-related GTPase, Drp1 to the OMM where it forms ring-like oligomers that pinch off mitochondria into smaller fragmented mitochondria (39). Fission is important ahead of mitosis to ensure even distribution of mitochondria to daughter cells (47–49) but also occurs when cells undergo mitophagy or apoptosis (2, 40, 44, 50, 51). Recruitment of cytosolic Drp1 to the mitochondria during fission is a regulated process involving post-translational modification of Drp1 (49, 52–54) and its interaction with putative receptors at the OMM such as Mitochondrial Fission Factor (Mff), Fis1, MiD49, MiD51, and possibly other proteins with which Drp1 interacts (45, 55–59). Recent work has also highlighted a role for the endoplasmic reticulum (ER) that is intimately associated with mitochondria, in determining the sites at which fission will occur (60). The constriction of mitochondria at points of contact with the ER are set up prior to recruitment of Drp1 to mitochondria and independent of Mff. Intriguingly, the mitochondrial fusion protein Mfn2 also plays a role in tethering mitochondria to the ER, in a manner required for proper calcium uptake by the mitochondria from the ER (61). Screens in yeast have identified additional putative molecular regulators of mitochondrial tethering to the cell cortex and the ER in ways that regulate mitochondrial positioning in cells and inheritance by daughter cells (62) but whether similar mechanisms operate in mammalian cells is unclear.
Stress-Induced Changes in Rates of Fission and Fusion
Beyond the actual mechanics of mitochondrial fission and fusion, the signaling pathways that regulate these processes are only just coming to the fore as mechanisms that may be disrupted in cancer. These signaling pathways respond to specific stresses and serve to coordinate mitochondrial dynamics with other aspects of cellular physiology. Drugs that inhibit protein synthesis, including mTOR inhibitors, as well as other stresses such as ultra-violet (UV) light have been shown to promote so-called “stress-induced mitochondrial hyperfusion” that relies upon canonical fusion proteins (Opa-1, Mfn1) (46), although, how these stress signaling pathways activate the fusion machinery is not clear. Stress-induced hyperfusion of this kind promotes ATP production through more efficient oxidative phosphorylation (OXPHOS) (63), and also inhibits mitophagy and prevents apoptosis (46, 64, 65).
The functional consequences of altered rates of fission or fusion for cellular metabolism, cell cycle kinetics and cell viability are a “work in progress” relying on numerous systems and technical approaches. In Drosophila, Yorkie-mediated up-regulation of Opa-1 and mitochondrial fusion was required for Yorkie/YAP-dependent proliferation and tumorigenesis (66). In mammalian systems, glucose deprivation and use of galactose as an alternative source of carbon, resulted in cells switching from glycolysis to OXPHOS, as expected, but significantly caused a marked increase in mitochondrial fusion and an accompanying increase in respiratory chain protein expression and cristae density, without any increase in mitochondrial mass (67).
Consistent with a critical role for mitochondrial fusion in regulating metabolism, inactivation of Mitofusin 1, Mitofusin 2, or Opa-1 inhibits oxidative metabolism and cell growth (68, 69). If fusion promotes mitochondrial ATP production (63), it is reasonable to postulate that this is achieved by increasing the efficiency of the electron transport chain (ETC) that is key to OXPHOS. This may be achieved by increasing the numbers/density of ETC complexes through increased expression of key components, as has been reported (67), or by altering the composition of specific respiratory chain components into different supercomplexes to maximize utilization of specific substrates, such as NADH in the presence of glucose versus FAD in the presence of fatty acids (70). Recent data has shown that assembly of respiratory chain complexes (RCC) into supercomplexes and increased respiratory efficiency is promoted by tighter cristae formation that is dependent on the fusion protein, Opa-1 (71). Conversely, disruption of cristae formation by knockout of Opa-1 in mice promoted mitochondrial fragmentation, cristae dissolution, and reduced RCC formation and respiration (71). Thus fusion may promote respiration through Opa-1 dependent effects on cristae density and formation of respiratory chain supercomplexes. Additionally, fusion may induce mitochondrial membrane potential changes that promote uptake of pyruvate or other substrates that fuel OXPHOS. Clearly, a more continuous mitochondrial lumen achieved by increased fusion is likely to promote more rapid diffusion of ADP, NADH, FADH2, and other matrix metabolites required to drive more efficient OXPHOS. In this way, fusion would also likely promote increased carbon flux through the Krebs’ cycle, more efficient rates of fatty acid oxidation and possibly increased activity of other metabolic pathways located at the mitochondria.
It has been suggested that fusion may promote respiratory efficiency by promoting complementation of mtDNA mutations (51). While this may be the case in some instances, the failure to detect homoplasmic mutations in mtDNA encoded subunits of CO, ND, ATP synthase, or cytochrome b in primary cells, or indeed more widely in tumor cells suggests that complementation of mtDNA mutations is not the key role of mitochondrial fusion in metabolism. Indeed, increased fusion has been shown to promote OXPHOS in very short time frames (67), indicating that the effects of fusion on oxidative metabolism are post-translational and not primarily dependent on gene complementation between mitochondrial genomes.
The effects of mitochondrial fusion in promoting oxidative metabolism at the mitochondria imply a contrasting role for mitochondrial fission in inhibiting oxidative metabolism, perhaps by decreasing substrate uptake, disrupting cristae, and respiratory complex formation and/or limiting diffusion of reducing equivalents. Oxygen is the major electron acceptor from complex IV of the respiratory chain, and thus it benefits the cell to decrease OXPHOS under limiting oxygen conditions (hypoxia) both to maximize efficient use of the smaller amounts of oxygen available but also to prevent generation of damaging ROS. Hypoxia limits OXPHOS in a number of ways but promoting mitochondrial fission may be one of the key mechanisms. Hypoxia promotes mitochondrial fission by modulating Drp1 activity and interaction with Fis1 (72). Hypoxia-induced expression of Siah2, an E3 ubiquitin ligase (73) targets the mitochondrial scaffold protein, anchoring protein 121 (AKAP121) for degradation (72) thereby preventing protein kinase A (PKA) dependent inhibition of Drp1. These observations, amongst others, are consistent with an inhibitory role for mitochondrial fission in OXPHOS. Increased fission linked to deregulated expression of Drp1 (increased) and Mfn2 (decreased) has been observed in tumor cells (74) but to what extent this contributes to the Warburg effect or other aspects of tumor growth remains to be determined.
Coordination of Rates of Fusion/Fission with Cell Cycle
Several reports indicate that increased mitochondrial fusion is required not only for efficient oxidative metabolism (68, 69) but is necessary for proliferation and entry into S-phase (63). In this latter study, mitochondrial membrane polarization and hyperfusion of mitochondria occurring at the G1/S transition was required for cyclin E (CCNE) expression and S-phase entry (63). Additional studies have also shown that mitochondrial bioenergetics are linked to cell cycle progression (75). However, artificially inducing hyperfusion, either through treatment with mdivi (a drug that inhibits Drp1) (63) or expression of dominant negative Drp1 (76), resulted in untimely induction of hyperfusion and sustained cyclin E over-expression at inappropriate phases of cell cycle, such as G2/M (76). This in turn was accompanied by replication stress, DNA damage, centrosomal amplification, and chromosomal instability (63, 76), all known features of cells over-expressing cyclin E (77). Interestingly, while cyclin E over-expression was required for proliferation and genome instability arising from hyperfusion or dysfunctional fission (63, 76), the specific factors produced by hyperfusion that resulted in cyclin E up-regulation have not been identified. Increased ROS has been previously reported to modulate levels of key cell cycle proteins, such as Emi-1 (78) but neither increased mitochondrial ROS nor altered ATP production (76) explain increased proliferation induced by mitochondrial hyperfusion (although this arguably requires further validation), leaving us with the unanswered question of what drives cyclin E expression in these circumstances.
Glycolysis is also cell cycle regulated and increases at the G1/S-phase boundary due in part to stabilization of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase, isoform 3 (Pfkfb3) that is normally turned over by APC/Cdh1 in early stages of G1 phase of cell cycle (79). Interestingly, Drp1 is also turned over by APC/Cdh1 in G1 phase of cell cycle in a CDK-dependent manner (47, 49) suggesting a possible mechanism by which up-regulation of glycolysis may be coordinated with mitochondrial dynamics. Evidence that glutaminolysis is cell cycle regulated is sparse but since c-Myc and RB/E2F both modulate glutamine uptake (80–83), it would not be surprising if glutaminolysis, like glycolysis, was up-regulated as cells enter S-phase.
Dual Role of Bcl-2 Family Members in Mitochondrial Dynamics and Apoptosis
In addition to effects on cellular metabolism and cell cycle, mitochondrial fusion can delay cytochrome c release and apoptosis (46, 84), with Opa-1 oligomerization inhibiting pro-apoptotic cristae remodeling independent of its role in fusion (85) (Figure 2). Disruption of Opa-1 oligomers in a Bax/Bak-dependent manner by the pro-apoptotic BID protein was independent of mitochondrial membrane permeabilization (86) but was associated with pro-apoptotic cristae remodeling (a requirement for cytochrome c release) and Opa-1 deficient cells are more susceptible to apoptosis (87, 88). Interestingly, prohibitins promote cellular proliferation and resistance to apoptosis by inhibiting OPA-1 cleavage at the IMM thereby promoting fusion and normal cristae morphology (89).
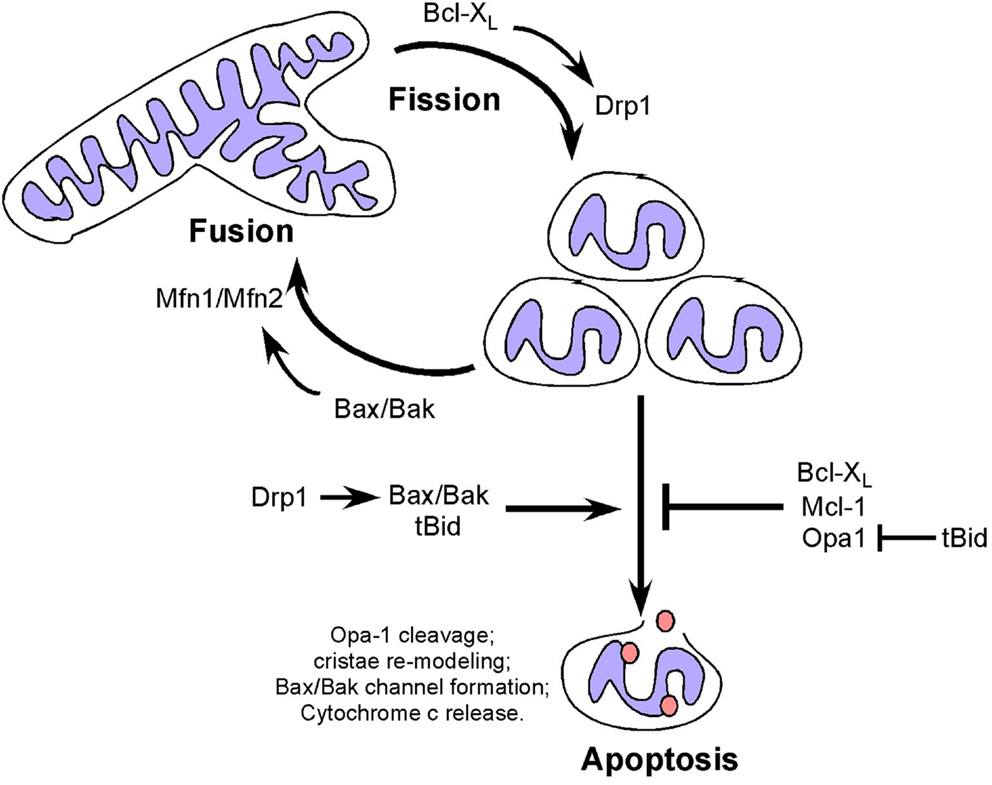
Figure 2. Dual and apparently opposing roles of Bcl-2 family members and fission/fusion proteins in apoptosis and mitochondrial dynamics. Mitochondrial fission and fusion proteins appear to modulate apoptosis through activities that are distinct from their roles in mitochondrial dynamics but which involve members of the Bcl-2 family. Conversely, Bcl-2 family members modulate mitochondrial fission and fusion in a manner that appears to be independent of their functions in apoptosis.
In contrast to mitochondrial fusion, mitochondrial fission promotes mitochondrial membrane depolarization, cytochrome c release and apoptosis (90), with Drp1 promoting Bax oligomerization through mechanisms independent of its GTPase activity (91), possibly explaining how fragmented mitochondria are more amenable to Bax/Bak channel formation. Thus both mitochondrial fission and fusion proteins appear to modulate apoptosis through activities that are distinct from their roles in mitochondrial dynamics but which involve members of the Bcl-2 family (Figure 2).
Members of the Bcl-2 superfamily of cell death regulators are extensively characterized for their key role in regulating apoptosis (2, 44) but they also have an emerging role in mitochondrial dynamics (44, 92, 93). Bak and Bax are essential for apoptosis, such that Bax/Bak double knockout cells are resistant to apoptosis (94, 95). When Bak and Bax are activated by pro-apoptotic signals, they undergo oligomerization to form a channel in the OMM through which cytochrome c is released resulting in the formation of the apoptosome and activation of caspases (96). Apoptosis-resistant Bax/Bak null cells exhibit extensive mitochondrial fragmentation that is rescued by over-expression of Bak in the absence of apoptotic signaling suggesting that Bak and Bax can promote mitochondrial fusion (92). Indeed, the soluble form of Bax interacts directly with Mfn2 to promote its GTPase activity while both Bak and Bax interact with Mfn1 (90, 92, 97). Conversely, the anti-apoptotic Bcl-XL has been shown to promote mitochondrial fission in neurons through interactions with Drp1 that promote its GTPase activity (98). More recently, Mcl-1 has been implicated in modulating mitochondrial dynamics through an amino-terminal truncated form that localizes to the mitochondrial matrix, in contrast to full-length Mcl-1 at the OMM (99). Truncated Mcl-1 in the matrix is required for mitochondrial fusion and assembly of the F0F1-ATP synthase and for efficient respiration (99). This novel function for Mcl-1, distinct from its anti-apoptotic function may explain the observed heart failure in Mcl-1 null mice, in which cardiomyocytes exhibited aberrant mitochondrial structure and defects in respiration that were not rescued by Bax/Bak deletion (100), although the defects were partially rescued by deletion of cyclophilin D, a key regulator of the mitochondrial permeability transition pore (101). Thus there is regulation of the apoptotic activity of Bcl-2 related proteins by fusion/fission proteins and conversely regulation of fission/fusion protein activity by Bcl-2 related proteins. What is not clear is whether the activities of Bcl-2 proteins, and Bax/Bak in particular, in apoptosis and fission/fusion are exclusive. The increase in mitochondrial fragmentation taking place during apoptosis suggests that the pro-fusion activity of Bak/Bax is suppressed by their pro-apoptotic functions but formal experimental evidence supporting this is lacking. Similarly, it is not known whether reduced OXPHOS resulting from increased fission may contribute to cellular susceptibility to apoptosis. Finally, it is not clear whether altered mitochondrial dynamics contributes to the oncogenic activity of key Bcl-2 family members, such as Bcl-2 or Bcl-XL that are both over-expressed in certain human malignancies (102). For example, increased Bcl-XL expression in tumors may promote mitochondrial fission, as seen in neurons (98), that in turn would be predicted to limit mitochondrial OXPHOS thereby promoting the Warburg effect.
Altered Mitochondrial Mass in Cancer – Biogenesis Versus Mitophagy
Mitochondrial mass in cells is regulated by both changes in mitochondrial biogenesis and mitophagy, two processes that are tightly regulated in response to cellular stress, including nutrient availability, oxidative damage, and redox state. Mitochondrial biogenesis and mitophagy are both influenced by the activity of key oncogenes and tumor suppressors, and much has recently been discovered about how these processes are coordinated in cultured cells and in mouse models. However, surprisingly little evidence is available that examines changes in mitochondrial mass in primary tumors in vivo. Based on what is known about regulation of mitochondrial mass in tumor cell lines, one might expect that changes in cancers might be linked to specific oncogenic lesions, for example c-Myc amplification, or to localized regional effects of hypoxia/ischemia, or indeed be reflective of or contribute to evolving tumor heterogeneity. Our data identifies significant variation in mitochondrial mass between tumors in different individuals (Figure 3) but whether this relates to tumor grade, molecular sub-type, therapy response, and/or recurrence-free survival is not clear. However, this would be clinically relevant if it allowed improved stratification of cancer patients for treatment purposes.
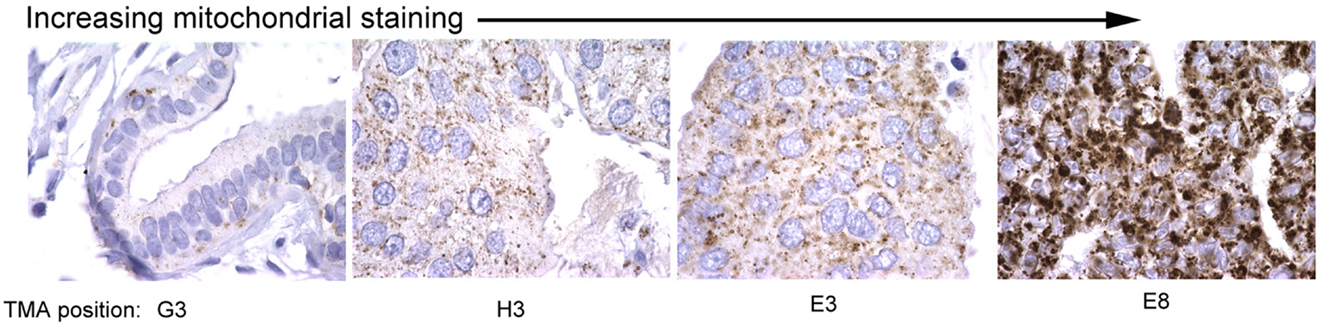
Figure 3. Variation in mitochondrial staining in human breast cancers. Immunohistochemical staining for mitochondrial 60 kDa antigen (Biogenex clone 113-1) reveals marked variations in mitochondrial staining between different primary human breast cancers with some tumors showing very low staining (left) and others very high staining (right). Differences in mitochondrial mass between different primary tumors examined in this study was greater than intra-tumor heterogeneity in mitochondrial mass. The significance of these differences in mitochondrial mass for tumor growth, progression, and therapy response is unknown.
Mitochondrial Biogenesis in Cancer
Mitochondrial biogenesis involves replication of the mitochondrial genome and coordinated expression of both nuclear and mitochondrial encoded gene products (3, 103, 104). Critical nuclear-encoded mitochondrial proteins, such as Tfam, Tfb2, and the mitochondrial RNA polymerase (Polrmt) are translated in the cytosol and encode a mitochondrial targeting sequence allowing their regulated import into the mitochondrial network where they are sorted for function (105). Key ETC proteins (subunits of ND and cytochrome b, for example) are encoded by the mitochondrial genome and since these proteins are translated in the mitochondrial matrix, coordinated induction of mitochondrial encoded tRNAs and rRNA expression is also required (104). The entire process is highly responsive to redox stress, nutrient availability, and mitochondrial function (104, 106) and defective mitochondrial biogenesis results in embryonic lethality and disease (3, 107). Mitochondrial mass increases in proportion to cell size (108) although opinions differ on whether mitochondrial biogenesis is cell cycle regulated (109, 110).
Mitochondrial biogenesis depends upon the activity of a hierarchy of nuclear transcription factors, that includes peroxisome-proliferator activator receptor-alpha (PPARα), PPAR-γ, nuclear respiratory factor 1 (NRF1), nuclear respiratory factor 2 (NRF2), and estrogen related receptors (ERR) α, β, γ (36, 103). NRF1 and NRF2 both modulate expression of respiratory chain components, such as cytochrome c and CO subunits in addition to anti-oxidant genes, while ERR factors regulate expression of genes involved in fatty acid oxidation, Krebs cycle, and OXPHOS (103). All of these transcription factors are critically dependent for their activity on PPAR-γ co-activator 1-alpha (PGC-1α) that was first identified as a co-activator of PPARγ in brown adipocytes. PGC-1α is now recognized as the major integrator of transcriptional responses to nutrient stress (Figure 4) that together with structurally and functionally related proteins, PGC-1β and PRC (PGC-related co-activator) promotes mitochondrial biogenesis, cellular metabolism, and anti-oxidant responses through coordinated activation of the afore-mentioned transcription factors, including NRF2, ERRα, and PPARγ (103, 106).
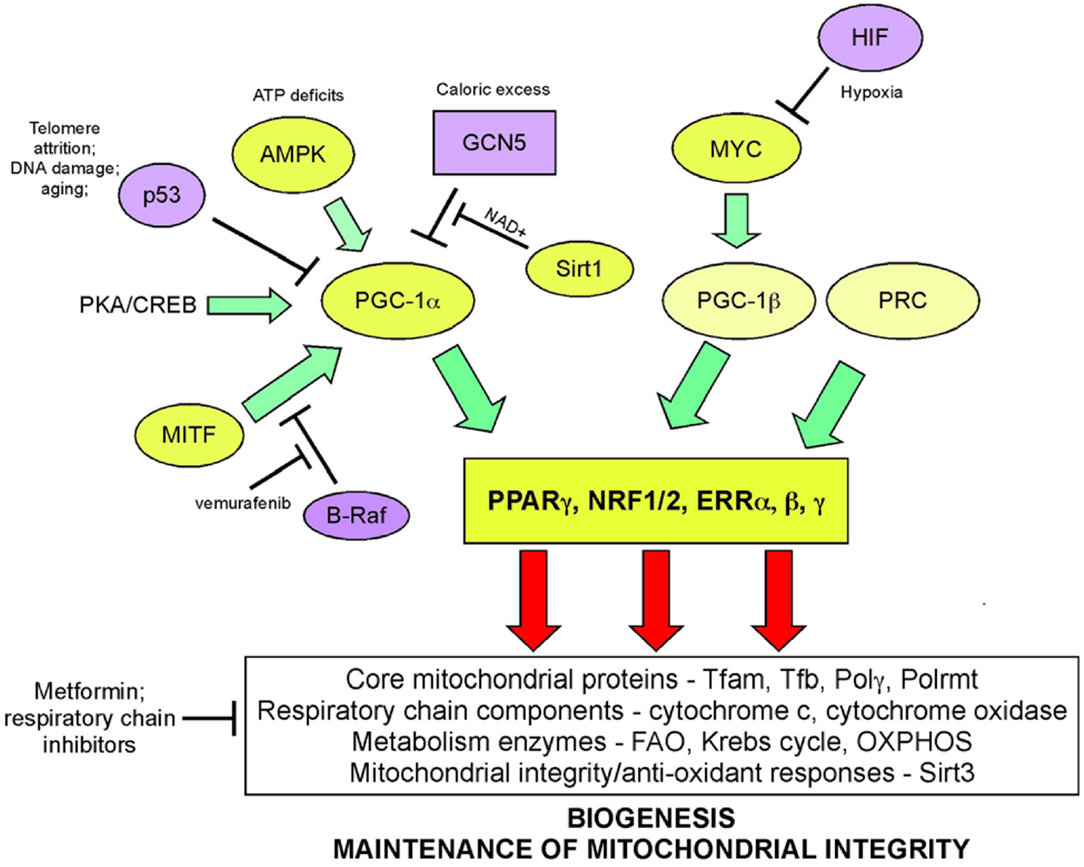
Figure 4. Signaling pathways regulating biogenesis in response to stress. Mitochondrial mass is increased in response to nutrient stress through increased mitochondrial biogenesis. The peroxisome-proliferator activator receptor-gamma co-activator 1-alpha (PGC-1α) is key to coordinating responses to nutrient availability and induction of biogenesis through its co-activation of NRF1/2, ERRα, β, γ, and PPARγ. These transcription factors induce expression of key nuclear-encoded genes, such as Tfam and mitochondrial polymerases, but also induce expression of metabolic enzymes active at the mitochondria and other proteins that are imported into the mitochondria. Mitochondrial encoded proteins required for respiratory chain function are also induced secondary to the actions of PGC-1α and its related proteins, PGC-1β, and PRC. Both p53 (through inhibition of PGC-1α) and c-MYC (through activation of PGC-1β) modulate biogenesis. Recent data highlights a role for MITF-induced PGC-1α activity and mitochondrial metabolism in a subset of human melanomas, that is sensitive to B-Raf inhibitors (since B-Raf blocks the action of MITF on PGC-1α).
Nutrient supply and energy balance in the cell modulates PGC-1α activity at both the transcriptional and post-translational level (106) (Figure 4). Cells respond to mitochondrial damage by increasing mitochondrial biogenesis and this is also dependent on up-regulated PGC-1α expression (111, 112). PGC-1α is transcriptionally activated by PPARs, mTOR (acting on YY1), and CREB (downstream of PKA signaling) leading to increased mitochondrial biogenesis (106). At the post-translational level, PGC-1α is regulated by both phosphorylation and acetylation events. Phosphorylation by the energy sensor AMP-dependent kinase (AMPK) activates PGC-1α while GCN5-mediated acetylation inhibits PGC-1α activity (106). Deacetylation of PGC-1α by NAD+ dependent SIRT1 promotes mitochondrial biogenesis and ensures that the activity of PGC-1α is sensitive to both the energy and the redox balance in the cell (113). PGC-1α co-activation of ERRα in turn promotes expression of mitochondrial SIRT3 that ensures effective scavenging of ROS at the mitochondria through activation of mitochondrial superoxide dismutase, amongst other mitochondrial sirtuin targets (114).
Key tumor suppressors and oncogenes regulate mitochondrial biogenesis. The c-Myc oncogene stimulates mitochondrial biogenesis through induction of PGC-1β expression (115) leading to increased expression of key mitochondrial proteins, including Tfam, Polγ, and NRF1 (116, 117). Regulation by c-Myc may explain how certain aspects of mitochondrial biogenesis, such as mtDNA replication appears to be cell cycle regulated, at least in some systems (109). HIF-1 by contrast inhibits biogenesis by promoting c-Myc degradation and by activating Mxi-1, a repressor of c-Myc transcriptional activity (115). Together with the role of HIF-1 in promoting mitophagy (as will be discussed later), HIF-1 mediated repression of mitochondrial biogenesis explains in part how mitochondrial mass is reduced in response to hypoxia. HIF-1α protein is stabilized by loss of SIRT3, a downstream target of PGC-1α (118), suggesting that signals that promote biogenesis, such as starvation-induced PGC-1α activity act in part to block ROS-induced HIF-1 stabilization and its inhibitory effect on biogenesis.
Increased PGC-1α expression has recently been implicated in the etiology of a subset of human melanomas as a result of its induction by the melanocyte-specific transcription factor, MITF (119, 120) (Figure 4). PGC-1α expressing melanomas exhibited high expression of mitochondrial proteins and a dependence on oxidative metabolism, that contrasted with PGC-1α low-expressing melanomas that were more glycolytic (119, 120). PGC-1α expressing melanomas were highly dependent on PGC-1α for growth and progression, possibly to protect against ROS-induced apoptosis (120). Intriguingly, the key melanoma oncogene, B-Raf was shown to suppress oxidative metabolism by inhibiting MITF-induced induction of PGC-1α and melanomas treated with B-Raf inhibitors, such as vemurafenib, were critically dependent on oxidative metabolism for survival suggesting that inhibitors of mitochondrial metabolism may synergize with B-Raf inhibitors in melanoma therapy (119). An obvious choice of such a drug is Metformin/Phenformin that inhibits complex I of the respiratory chain and is already approved for the treatment of type II diabetes.
PGC-1α has also been shown to drive HIF-independent expression of VEGF through co-activation of ERR-α and thus promote angiogenesis, although the relevance of these findings for cancer has not been directly tested (121).
Recent work demonstrated that PGC-1α is transcriptionally repressed by p53 in response to telomere dysfunction (122), possibly explaining reduced mitochondrial biogenesis during the aging process and significantly, mitochondrial dysfunction and elevated ROS in cancer when p53 is mutated (123, 124). When telomerase was inactivated in Atm null T cell lymphomas, tumors initially grew more slowly but over time, more aggressive tumors emerged that had activated the alternative lengthening of telomeres (ALT) pathway (125). Intriguingly, PGC-1β showed consistent copy-number alteration in ALT+ tumors and increased expression of PGC-1β and its targets, NRF2, TFAM, SOD2, and catalase were also detected (125) suggesting that there was a selective advantage to emerging tumors of increasing both mitochondrial biogenesis and ROS scavenging. ALT+ tumors showed increased mitochondrial dysfunction and ROS, possibly as a result of transcriptional inhibition of PGC-1α by p53 earlier in the pathogenesis of these tumors (122).
In summary, evidence suggests that mitochondrial biogenesis is tumor promoting by increasing metabolite and energy generation, and indeed biogenesis is positively regulated by the c-Myc oncogene and repressed by the p53 tumor suppressor. However, a different argument suggests that the production of new healthy mitochondria may be tumor suppressive by promoting oxidative metabolism, limiting ROS and HIF-α stabilization. Thus, whether mitochondrial biogenesis promotes or limits cancer may depend on context, such as tissue type, stage of tumor progression or on specific exogenous stresses present in the microenvironment.
Mitophagy in Cancer
Macro-autophagy is a catabolic process that plays a housekeeping role in eliminating protein aggregates and malfunctioning organelles, such as mitochondria, and is also activated in response to nutrient deprivation to provide energy (126–130). The specialized form of autophagy in which mitochondria are specifically targeted for degradation at the autophagolysosome is known as mitophagy. Numerous studies indicate that mitochondrial fragmentation and mitochondrial membrane depolarization precede mitophagy (64, 65, 131) and it has been suggested that mitophagy and fusion are opposing fates of dysfunctional mitochondria (43). Mitochondrial fission is a major source of depolarized mitochondria and conversely fusion is selective for polarized and respiring mitochondria (131, 132). In response to nutrient deprivation, healthy mitochondria are protected from mitophagy by mitochondrial fusion resulting from PKA induced down-regulation of Drp1 activity (64, 65) while dysfunctional/depolarized mitochondria appear to be specifically targeted for degradation possibly due to selective depletion of the fusion protein Opa-1, that is proteolytically cleaved in response to depolarization (133, 134).
Following on from fragmentation and depolarization, mitochondria are targeted to phagophore membranes through a growing number of mechanisms (Figure 5) including the Parkin/Pink1 gene products that are mutated in human Parkinson’s Disease (38) as well as the BNIP3/NIX proteins that are hypoxia-inducible and regulated by key tumor suppressors such as RB and p53 (135).
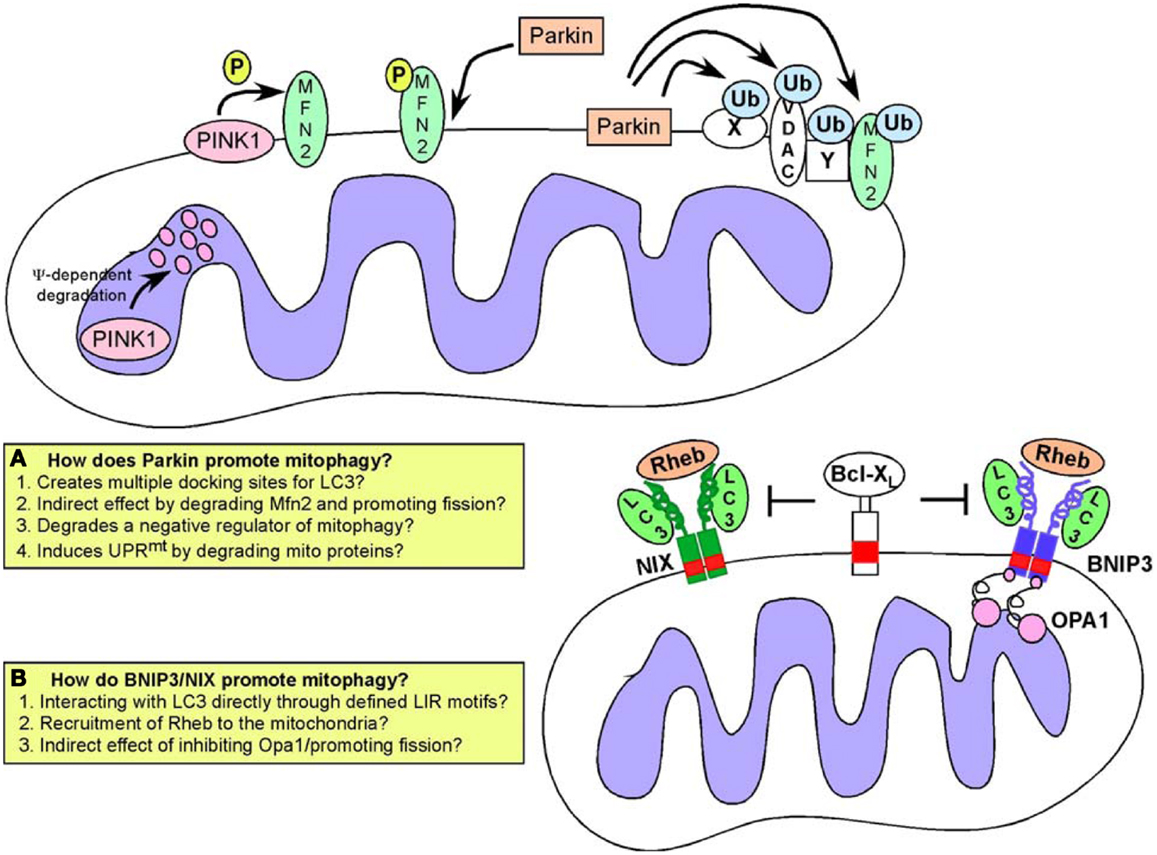
Figure 5. Mitophagy pathways. Turnover of mitochondria at the autophagosome (mitophagy) is required to maintain a healthy pool of mitochondria. Defects in mitophagy result in accumulation of dysfunctional mitochondria. Two major pathways have been identified that regulate targeting of mitochondria to the autophagosome: (A) the Pink1/Parkin pathway in which activation of Pink1 kinase following mitochondrial depolarization leads to phosphorylation of Mitofusin by Pink1, that then acts as a receptor for Parkin. Recruitment of Parkin, a E3 ubiquitin ligase, results in ubiquitination of multiple mitochondrial substrates but how this leads to mitophagy is still a matter for debate, and possible explanations for how Parkin functions are highlighted in the inset box; (B) An alternative pathway involves the activity of BNIP3 and NIX, both of which are hypoxia inducible but also regulated by FoxOs, E2Fs, and p53. BNIP3 and NIX have both been shown to interact directly with processed LC3 through a conserved LIR motif in their amino terminal ends. This interaction has been proposed to explain how BNIP3 and NIX target mitochondria to the autophagosome. Both BNIP3 and NIX are also known to interact with Rheb, and with Bcl-2/Bcl-XL but the significance of these interactions for mitophagy are not clear.
Parkin and PINK1 in Mitophagy
PINK1 (Pten-induced putative kinase-1) is a serine/threonine kinase that undergoes voltage-dependent proteolysis at the IMM in healthy mitochondria but accumulates at the OMM in response to depolarization (136, 137). PINK1 phosphorylates the fusion protein Mfn2 at the OMM and phosphorylated Mfn2 then acts as a receptor for the E3 ubiquitin ligase Parkin (138) selectively recruiting Parkin to damaged mitochondria from the cytosol (139, 140). Parkin has a number of substrates at the OMM including its own receptor, Mfn2 but also Mfn1, voltage-dependent anion channel (VDAC), and Miro (38, 141, 142). These proteins are ubiquitinated by Parkin and it was originally proposed that Parkin-mediated ubiquitination of such proteins created a docking site for the LC3-interacting protein p62/SQSTM1 (143–145) thereby linking Parkin activity to mitochondrial degradation at the autophagosome. However, it is now clear that a number of Parkin substrates are targeted for degradation by the ubiquitin-proteasome system independent of autophagy-mediated degradation (141). Furthermore, no single Parkin substrate has been shown to be required for mitophagy (146) leading to the suggestion that Parkin promotes mitophagy indirectly by either promoting fragmentation (through degradation of Mfn1/Mfn2) or by removing a negative regulator of mitophagy from the surface of the mitochondria (38, 141) (Figure 5). Alternatively, by promoting degradation of mitochondrial proteins, Parkin may be inducing an imbalance in mitochondrial versus nuclear-encoded proteins (particularly proteins involved in respiration) that has been shown to induce the mitochondrial unfolded protein response (UPRmt). This in turn activates adaptive mitochondrial stress signaling that is reported to improve “fitness” and promote longevity (147), so-called “mitohormesis” (123, 148, 149). Mitophagy along with mitochondrial biogenesis may be part of such compensatory mechanisms needed to restore mitochondrial function, although this has not been properly examined.
In addition to promoting mitochondrial turnover through increasing fragmentation, Parkin also modulates transport of mitochondria along microtubules to a perinuclear region where autophagosomes are concentrated (140, 146). This may be due to Parkin-mediated turnover of Miro, a protein required to tether kinesin motor protein complexes to the OMM (142). Additionally, HDAC6, a ubiquitin-binding protein deacetylase is recruited to mitochondria by Parkin activity where it promotes autophagosome-lysosome fusion and trafficking of mitochondria along microtubules (144, 150). Interestingly, a more recently discovered Parkin substrate termed PARIS represses mitochondrial biogenesis through transcriptional inhibition of PGC-1α expression (151), consistent with multiple roles for Parkin in mitochondrial homeostasis. Clearly, there is still much to be understood about the significance of these interlinking functions of Parkin/Pink1 in mitochondrial dynamics, mitophagy, and biogenesis for cellular physiology.
Human Parkin has been mapped to a common fragile site at chromosome 6q25–q26 that is found deleted in ovarian, lung, and breast cancer (152) and Parkin mutant mice are susceptible to spontaneous liver tumors (153). Parkin has also been shown to promote lipid uptake by hepatocytes, by modulating turnover of the fatty acid binding protein CD36. Further, Parkin null mice are resistant to weight gain and insulin resistance induced by feeding a high-fat diet (154). However, it is not clear whether this function in lipid metabolism in the liver relates to the function of Parkin in preventing liver tumors in mice. Parkin has also been identified as a p53 target gene and reported to prevent the Warburg effect and promote oxidative metabolism, likely through effects on mitochondrial integrity (155) providing another mechanism to explain how Parkin functions as a tumor suppressor. However, the role of Parkin as a tumor suppressor is at odds with data suggesting that mitophagy is tumor-promoting and required to maintain a healthy pool of mitochondria that are functional for TCA cycle and other aspects of metabolism upon which tumor cells depend for growth (156, 157). However, these latter studies inhibited macro-autophagy generically, not just mitophagy, and while the presence of abnormal mitochondria and defective mitochondrial metabolism suggested that defective mitophagy played a part in the observed retardation of tumor growth, the contribution of defects in turnover of ER, peroxisomes, or protein aggregates to the tumor phenotype was not examined.
BNIP3 and NIX in Mitophagy and Mitochondrial Integrity
The hypoxia-inducible genes BNIP3 and NIX (also known as BNIP3L) are also implicated in promoting mitophagy (135, 145, 158) (Figure 5). BNIP3 and NIX function as redox-resistant homo-dimers at the OMM and their integration into the OMM is dependent on dimerization. Both BNIP3 and NIX were originally thought to function as BH3-only proteins to promote cell death (135) but more recent work indicates that the BH3 domain in these proteins is weakly conserved and redundant for function (159). Additionally, several normal tissues express these proteins at high levels in the absence of cell death (160, 161) and it is now likely that additional signals/stresses are required for cell death associated with over-expression of either BNIP3 or NIX (162, 163). More consistent with a role on the adaptive response to hypoxia (164), both BNIP3 and NIX have been shown to promote mitophagy through interactions with LC3-related molecules and to possess an LC3-interacting region (LIR) at their unstructured amino-termini (165, 166). Thus, it has been proposed that, similar to ATG32 in yeast (167, 168), BNIP3 and NIX act as molecular adaptors targeting mitochondria directly to the autophagosome for degradation. NIX has been shown to be required for mitochondrial clearance from maturing reticulocytes (169, 170), while BNIP3 is involved in modulating mitochondrial integrity through mitophagy in skeletal muscle and liver (161, 171).
Interestingly, both BNIP3 and NIX interact with Bcl-2 and Bcl-XL through their amino terminal 49 amino acids (172) suggesting that binding of BNIP3/NIX to Bcl-2 or Bcl-XL may interfere with binding to processed LC3 that is dependent on an overlapping LIR motif. While reducing mitochondrial mass in cells in response to hypoxia seems likely to benefit the cell in terms of preventing excess ROS generation when oxygen is limiting (164), it is not clear whether the interaction of mitochondrial BNIP3 and/or NIX with processed LC3 at the autophagosome is a regulated interaction since not all mitochondria are turned over by mitophagy under hypoxic conditions (173). It remains to be tested whether additional events at the mitochondria, such as elevated ROS, membrane depolarization, or indeed altered electron flux at the respiratory chain modulates BNIP3/NIX structure to induce interactions with LC3 or other proteins involved in mitophagy. While not yet fully elucidated, it appears that in addition to its role in regulating mitophagy through interactions with LC3-related proteins, that BNIP3 may modulate OXPHOS and lipid metabolism in additional ways that are relevant to understanding tumor metabolism and disease (161, 174, Chourasia et al., under review).
As is observed for mitophagy involving Parkin activity, BNIP3 associated mitophagy is preceded by mitochondrial fragmentation and perinuclear clustering of mitochondria under hypoxic conditions (41, 173). Over-expression of exogenous BNIP3 promotes mitochondrial fragmentation without necessarily inducing mitophagy. This has been attributed to the inhibitory interaction of BNIP3 with Opa-1 resulting in disruption of Opa-1 complexes and cristae remodeling (175, 176). BNIP3 has also been reported to induce translocation of Drp1 to mitochondria and over-expression of either Mfn1 or dominant negative Drp1 inhibited both mitochondrial fragmentation and mitophagy induced by BNIP3 (56). This work also reported that BNIP3 induced Parkin translocation to the mitochondria in a Drp1-dependent manner. Similar to BNIP3, NIX has also been shown to promote Parkin recruitment to mitochondria (177). Parkin recruitment in this manner may reflect indirect effects of BNIP3/NIX on mitochondrial membrane potential and it remains to be determined to what extent BNIP3 or NIX depends upon Parkin to promote mitophagy.
BNIP3 has also been reported to interact with Rheb, a small GTPase that acts positively upstream of mTOR to promote cell growth (178). Similar to the interaction of BNIP3 with Bcl-2 and Bcl-XL (172), Rheb was reported to interact with BNIP3 in a manner dependent on the transmembrane domain of BNIP3 consistent with Rheb only interacting with BNIP3 dimers at the OMM. Rheb binding also required the 30 amino terminal residues of BNIP3 (178) suggesting that Bcl-2 and Bcl-XL may modulate the BNIP3-Rheb interaction although this has not been examined. It was suggested that BNIP3 repressed Rheb activity resulting in reduced mTOR activity and slower cell growth (178) but it is not clear if this is consistent with the interaction of BNIP3 with Rheb taking place exclusively at the OMM.
Interestingly, Rheb has recently been implicated in modulating mitophagy independent of mTOR in response to the altered metabolic state of the cell (179). Growth of cells under conditions that promote high levels of OXPHOS recruited Rheb to the OMM where it was shown to interact directly with NIX and with processed LC3. Furthermore, over-expression of Rheb in cells promoted LC3 processing and increased mitophagy (179). Intriguingly, this function of Rheb appeared to be independent of mTOR activity but dependent on NIX expression (179). This work suggested that NIX plays a role in recruiting Rheb to mitochondria under conditions of high OXPHOS leading to increased mitophagy required to maintain a healthy pool of mitochondria under high rates of oxidative metabolism. This contrasts with the previous study showing a role for BNIP3 in repressing Rheb activity (178) and while it is possible that NIX functions distinctly from BNIP3 with regards to regulation of Rheb, further work is needed to reconcile these disparate findings.
Expression of both BNIP3 and NIX is tightly regulated. Both are hypoxia-inducible HIF target genes (180, 181) although BNIP3 is more readily induced by relatively small decreases in oxygen compared to NIX that is only induced at much lower oxygen levels; an observation that is attributed to the differential dependence of BNIP3 and NIX expression on the two different transactivation domains of HIF-1α (182–184). They both also show markedly different tissue-specific patterns of expression with BNIP3 most strongly expressed in heart, liver and muscle while NIX is expressed strongly in hematopoietic tissues and in testes (160, 161). In addition to transcriptional regulation by HIFs, BNIP3 is regulated by RB/E2Fs (173), NF-κB (185), FoxO3 (171), oncogenic Ras (186, 187), and p53 (188), while NIX is also regulated by p53 (189).
Both BNIP3 and NIX are deregulated in human cancer with elevated expression of both genes detected at pre-malignant stages of several different tumor types, but they appear to be down-regulated upon progression to invasive and malignant cancers (190–193). Epigenetic silencing of the BNIP3 promoter appears to be the most common mechanism explaining down-regulation of BNIP3 during malignant progression in lung, colorectal, hematologic, liver, and pancreatic cancers (194–200) although genomic deletion (201) and repression by specific microRNAs (202) has also been reported. Knockdown of BNIP3 in the 4T07 orthotopic mammary tumor model promoted tumor growth and metastasis (203) while genetic targeting of BNip3 accelerated the growth and metastasis of mammary tumors in the MMTV-PyVT mouse model of breast cancer (Chourasia et al., under review), both results supporting a tumor suppressor/metastasis suppressor function for BNIP3. Thus, similar to effects of Parkin deletion, loss of BNIP3 appears to promote tumorigenesis in mouse models consistent with a tumor suppressor function for mitophagy.
Additional Links between Mitochondria and Autophagy
Mitochondria contribute OMM lipids to nascent autophagosomes (204) and autophagosomes form at the junction of mitochondria with the ER (205). Given that ER associated with mitochondria also regulates sites of mitochondrial fission (60), we postulate that connections between the ER and the mitochondria may be important in coordinating mitochondrial fragmentation with autophagosome formation during mitophagy. Both BNIP3 and NIX have been reported to localize to the ER as well as the mitochondria (165, 206) but whether they are involved in such coordination is not yet clear.
Inhibition of autophagy can to lead to apoptosis in part due to accumulation of defective mitochondria that release cytochrome c and activate the apoptosome (207). Autophagy and apoptosis are coordinately regulated in part through modulation of Beclin-1 activity by Bcl-2/Bcl-XL at the ER with Bcl-2/Bcl-XL inhibiting autophagy by binding directly to Beclin-1 (208, 209). Conversely, autophagy proteins have also been shown to function in apoptosis with Atg12 acting as a BH3-only protein to inhibit anti-apoptotic Bcl-2 (210) while calpain cleavage of Atg5 induces a truncated form of Atg5 that can bind to and inhibit Bcl-XL (211). While cleaved Atg5 promotes cytochrome c release and apoptosis (211), full-length Atg7 binds to p53 to prevent p53-dependent cell cycle and cell death (212). Various signaling pathways modulate this balance between autophagy and apoptosis. For example, starvation – induced Jnk1 signaling phosphorylates Bcl-2 disrupting its interaction with Beclin-1 (209). Under conditions of oxidative stress, nuclear HMGB1 is released to interact with Beclin-1 displacing Bcl-2 to promote autophagy (213). Conversely, apoptosis is promoted at the expense of autophagy as a result of calpain-mediated cleavage of key autophagy regulators, Atg5 and Beclin-1 (211, 214) and caspase-3 cleavage of Beclin-1, Atg4D, and GABARAPL1 (215, 216).
Unanswered Questions about Role of Mitophagy in Cellular Metabolism and Cancer
Currently it is not clear to what extent the two major known mechanisms regulating mitophagy in mammalian cells (PINK1/PARKIN and BNIP3/NIX) (Figure 5) are dependent on each other or function independently. Interestingly, the mitophagy defect observed in Nix null erythroblasts can be rescued by mitochondrial depolarization with CCCP (169) suggesting that different mitophagy mechanisms may be somewhat redundant and explaining the lack of more severe phenotypes in mice genetically deleted for Parkin, BNIP3, or NIX (154, 160, 217). Recent work also identified a novel mechanism by which hypoxia promotes mitophagy, through dephosphorylation of the FUNDC1 protein at the OMM (218). De-phosphorylated FUNDC1 interacted with LC3 through a conserved LIR motif in FUNDC1 (218). This indicates that additional mechanisms regulating mitophagy may yet be discovered and suggest the existence of multiple redundant pathways modulating mitochondrial turnover.
How much mitochondrial damage or dysfunction can be tolerated by cells, and for how long, without loss of viability, is an additional unknown. Nor is it clear how rapidly cells accumulate damaged mitochondria once mitophagy is inhibited. The kinetics of mitochondrial damage accumulation will likely vary from cell type to cell type and in proportion to how much oxidative or metabolic stress is imposed. Studies examining a specific defect in mitophagy in adult liver in BNip3 null mice indicated that accumulation of defective mitochondria increased over time explaining increasing defects in cellular metabolism and lipid metabolism as the mice aged (161). Initially, there was accumulation of lipid and defective mitochondria in young mice, but over time increased hepatocyte cell death was observed and the mice developed steatohepatitis (161). Mitochondrial defects due to inactivation of mtDNA polymerase activity have been linked previously to aging (219, 220) with mtDNA mutations early in development causing respiration defects particularly in aging neural and hematopoietic progenitor cells (221, 222). Some of the aging effects are likely due to accumulation of ROS-induced mtDNA mutations in line with the “free radical theory of aging” since mouse life span can be increased and age-related phenotypes can be ameliorated through over-expression of mitochondrial catalase (223, 224). However, it remains to be determined to what extent defective mitophagy affects aging and which specific tissues are more susceptible to aging due to defective mitophagy. Clearly, since cancer is a disease of old age, defective mitophagy may contribute to tumorigenesis in an age-dependent manner. However, this remains to be formally tested.
Significance of Mitophagy for Cancer Treatment?
The duality of macro-autophagy function in cancer (both pro- and anti-tumorigenic, likely as a function of tumor stage, driving oncogene, and/or tissue type) makes it clinically questionable to generically target the entire autophagy process. However, a more effective therapeutic response in terms of long-term cancer patient survival may be possible by specifically targeting mitophagy. Inhibition of mitophagy increases ROS production at the mitochondria that may promote cell killing for at least a subset of tumor cells. Because mitochondria in normal cells are less likely to be dysfunctional and therefore less sensitive to mitophagy inhibition, by inducing ROS indirectly, we may avoid potentially harmful effects of supra-physiological ROS levels on normal cells. Furthermore, inhibition of mitophagy may disrupt fatty acid oxidation and Krebs cycle at the mitochondria and preferentially disrupt tumor cell growth that is also more dependent on mitochondrial citrate production for lipid synthesis than are normal cells. The combined effect of increased ROS and reduced mitochondrial metabolism arising from inhibition of mitophagy may be synergistic and promote efficient tumor cell killing. An alternative approach may be to combine mitophagy inhibition with drugs that induce other forms of mitochondrial stress signaling, such as Metformin that inhibits respiratory chain complex I or with antibiotics, such as tetracycline/doxcycline that inhibit mitochondrial protein translation, thereby inducing a “mitonuclear” protein imbalance and a mitochondrial unfolded protein response (UPRmt) (147, 148) that might be predicted to depend on mitophagy to clear damaged/dysfunctional mitochondria.
Retrograde Signaling from the Mitochondria to the Nucleus in Cancer
While most mitochondrial proteins are encoded by the nuclear genome and control of nuclear gene expression is key to mitochondrial function, it is also clear that mitochondria signal to the nucleus and such “retrograde” signaling is an area of increasingly important investigation (26). Mitochondrial dysfunction (Figure 6), defined as loss of membrane potential, defective respiration, defects in synthesis of iron-sulfur clusters, and/or the mitochondrial unfolded protein response (UPRmt), has been shown to alter nuclear gene expression through a variety of different mechanisms. For example, mitochondrial dysfunction can induce genome instability due to defective iron-sulfur complex synthesis in the mitochondrial matrix (225). Clearly, release of cytochrome c signals mitochondrial dysfunction and leads to apoptosis that is tumor suppressive, but short of inducing apoptosis, there are several other mechanisms of mitochondrial stress signaling that affect tumor cell growth. Altered metabolite levels, increased calcium (Ca2+) release from the mitochondria, elevated ROS production, reduced production of ATP or NADH arising from altered metabolism, changes in activity of mitochondrial kinases or other cellular enzymes/proteins dependent on redox state, Ca2+ levels or Fe/S complexes are among several major mechanisms put forward to explain how mitochondrial stress signaling affects nuclear gene expression (Figure 7), as discussed below.
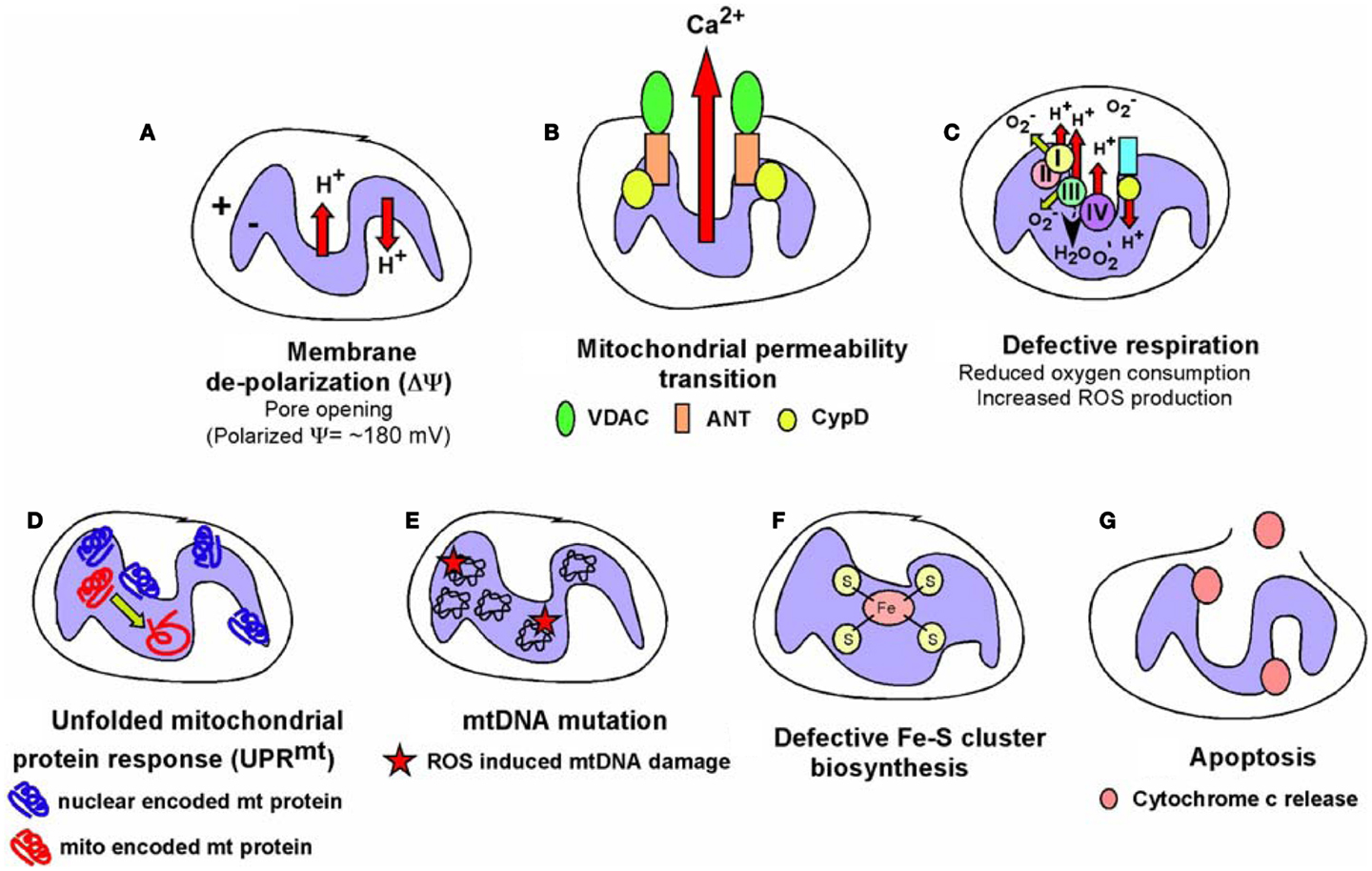
Figure 6. Types of mitochondrial dysfunction. We have attempted to define “mitochondrial dysfunction” in this review and the figure summarizes the major types of mitochondrial dysfunction that are known. (A) Mitochondrial inner membrane de-polarization (ΔΨ) during which there is loss of membrane potential; (B) mitochondrial membrane permeability transition (MPT) during which opening of the permeability transition pore (consisting of VDAC, ANT and usually association of Cyclophilin D) can lead to non-apoptotic cell death; (C) defective respiration/oxygen consumption due to altered expression of respiratory chain components, poisoning with respiratory complex inhibitors or many other stresses; (D) the Unfolded Mitochondrial Protein Response (UPRmt) can arise when there is an imbalance in expression of mitochondrial encoded mitochondrial proteins relative to nuclear encoded mitochondrial proteins, resulting in dysfunctional mitochondria; (E) damage to the mitochondrial genome most commonly reported as a result of oxidative damage to bases arising from respiratory chain defects; (F) defects in the production of iron-sulfur complexes in the mitochondrial matrix leading to defects not just in respiratory chain components but also other cellular enzymes; (G) release of cytochrome c anchored at the inner mitochondrial membrane via cardiolipin can result in formation of the apoptosome and activation of caspases leading to apoptosis. Some of these aberrant mitochondrial behaviors are inter-dependent, for example, membrane depolarization is a factor in mitochondrial permeability transition, defective respiration, and apoptosis amongst other consequences, but frequently can stand alone as a signal, for example to promote mitophagy. The consequences for the cell of these different forms of mitochondrial dysfunction are described in the text and below in Figure 7.
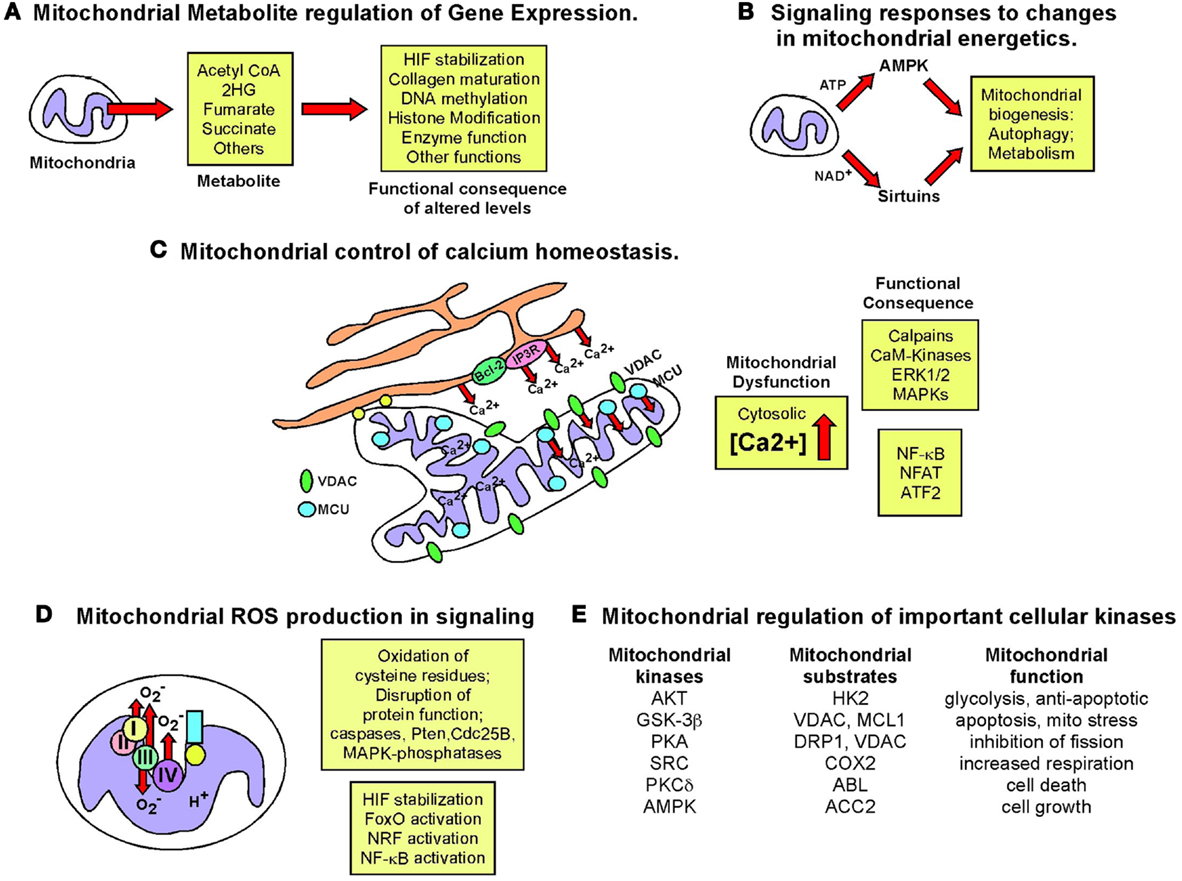
Figure 7. Retrograde signaling from mitochondria to nucleus. The role of the nucleus in regulating mitochondrial function has been examined extensively but the importance of mitochondrial events in signaling to the nucleus and to other cell growth mechanisms has been relatively under-studied. The figure summarizes some key signaling consequences of dysfunctional mitochondria. (A) Mitochondrial control of nuclear gene expression through effects of altered production of certain metabolites, such as α-ketoglutarate and succinate, on epigenetic modification of histones, stabilization of key transcription factors, such as HIF, in addition to effects on other enzymes and proteins. (B) Altered production of NAD+, ATP, and other changes in mitochondrial metabolism can modulate key signaling molecules in the cell, such as AMPK and the Sirtuins. (C) Mitochondria play a key role in buffering against Ca2+ flux into the cytosol from the extra-cellular environment or following release from the ER and failure of the mitochondria to execute this key function can result in altered Ca2+ signaling in the cell. (D) Mitochondrial ROS production has been one of the most extensively studied mediators of mitochondrial dysfunction and activity that elicits its effects on transcription factor activity as well as activity of key enzymes in the cell, such as caspases and phosphatases. (E) Important cellular kinases are known to localize to the mitochondria and altered mitochondrial dynamics and function may modulate the activity of these kinases not just at the mitochondria but at other sub-cellular locations if released from the mitochondria.
Mitochondrial Control of Epigenetics
Epigenetic control of nuclear gene expression is highly sensitive to mitochondrial function (226, 227). For example, levels of histone acetylation are regulated by availability of acetyl CoA produced from citrate exported from the mitochondria (228). In addition, fumarate, succinate, and other TCA cycle intermediates produced at the mitochondria regulate nuclear gene expression through effects on histone modifying enzymes, DNA demethylases, prolyl-hydroxylases, and other cellular dioxygenase enzymes (6, 13, 18, 226, 227, 229). Recent research progress in this area has been fueled by evidence showing that human cancer development is linked to mutations in genes encoding enzymes in the TCA cycle, such as isocitrate dehydrogenase-1 (IDH1) and -2 (IDH2) in glioblastoma and AML (230–232), as well as in fumarate hydratase (FH) and succinate dehydrogenase (SDH) in other rarer malignancies (6). Mutation of these genes in cancer leads to a buildup of their substrates; fumarate and succinate in the case of FH and SDH mutations (6, 226), or conversion of their regular substrate to a new “oncometabolite” in the case of mutant IDH1/IDH2 converting isocitrate to 2-hydroxyglutarate (229, 231). Inhibition of respiration through binding of the mitochondrial chaperone TRAP1 to SDH also resulted in elevated succinate levels and promoted tumor growth (233).
The key mechanism of action of these “oncometabolites” is their ability to compete with the structurally related α-ketoglutarate, a required co-factor for the afore-mentioned cellular dioxygenase enzymes (234–237) resulting in reduced activity of these key cellular enzymes. Amongst these enzymes are the prolyl-hydroxylases that promote turnover of HIF-α subunits, the TET2 DNA demethylase and the JmjC histone lysine demethylases (227, 229). Thus, the downstream consequences of increased fumarate, succinate, or 2HG include the accumulation of HIF-α and increased HIF target gene expression (235, 238), defects in collagen maturation (239), as well as hypermethylation and altered histone code due to disruption of epigenetic control of gene expression (236, 240, 241). This in turn was linked to altered gene expression patterns, defects in cellular differentiation and accumulation of immature stem cells and progenitors in affected tissues that can lead to cancer (240–244).
In addition to a role in inhibiting dioxygenases by competing with α-ketoglutarate for reversible binding, fumarate and succinate can also modulate cell growth by covalently modifying key signaling molecules. For example, elevated fumarate levels have been shown to stabilize HIF by covalently reacting with glutathione to produce an alternative substrate for glutathione reductase resulting in increased ROS production, as well as decreased NADPH (245). Succinylation of cysteine residues in KEAP1 in the cytosol results in activation of the NRF2 anti-oxidant pathway and up-regulated expression of stress response genes, such as heme oxygenase (246, 247). Similarly, succinylation of mitochondrial aconitase causes defects in iron-sulfur cluster binding (248). Tumor cells can tolerate such defects in the TCA cycle brought about by IDH1, FH or SDH mutation by redirecting the use of metabolites. In particular, glutamine can be used to generate citrate by reductive carboxylation (249, 250) or diverted to heme biosynthesis and degradation, thereby partially restoring NADH production and limiting accumulation of fumarate and succinate (251). This latter pathway is up-regulated in FH mutant tumor cells and inhibition of heme oxygenase specifically killed FH mutant tumor cells suggesting a novel therapeutic approach to treating cancers with FH mutation (251). Finally, the identification of novel protein modifications, including malonylation, succinylation, and butyrylation suggest the existence of novel regulatory pathways that may be sensitive to mitochondrial metabolites (252). Clearly, the more we understand about how metabolic pathways at the mitochondria are deregulated in cancer, the more likely it seems that we will identify novel signaling pathways that are aberrantly activated by accumulation or alternate fates of specific metabolites.
Effects of Mitochondrial Dysfunction on Calcium Homeostasis
Mitochondria play a critical role in buffering intracellular calcium levels in part due to their localization close to calcium channels in the ER, such as the inositol-1,4,5-triphosphate-sensitive channels [Ins(1,4,5)P3R] that release Ca2+ from the ER in response to inositol-1,4,5-triphosphate (253). Mitochondria located at such microdomains of high Ca2+ concentrations rapidly take up the released divalent cation through the VDAC at the OMM and the mitochondrial calcium uniporter (MCU) at the IMM (253). While Ca2+-binding proteins can buffer cytosolic Ca2+ to some extent, the quantity of Ca2+ that the mitochondria can take up and buffer against is significantly larger. While VDACs are readily permeable to Ca2+ at the OMM and interact with Ins(1,4,5)P3Rs at the ER (254, 255), it has been suggested that levels and activity of VDAC may regulate the amount of Ca2+ that crosses the OMM (256). VDACs are subject to multiple levels of regulation including expression levels, post-translational modification, and protein–protein interactions all of which can limit Ca2+ uptake. Mitochondrial dysfunction results in increased cytosolic Ca2+, since only energized mitochondria can take up Ca2+ (253). Interestingly, members of the Bcl-2 family localized at the ER can modulate activity of the Ins(1,4,5)P3R and thereby regulate Ca2+ release and uptake by the mitochondria, with attendant effects on mitochondrial function and apoptosis (257).
The failure of mitochondria to take up Ca2+ effectively in response to its release from the ER or influx through the plasma membrane, directly affects mitochondrial activity. For example, key mitochondrial enzymes, including several TCA cycle enzymes are Ca2+ modulated (254, 258). Also, changes in mitochondrial matrix volume induced by altered Ca2+ uptake impact the activity of the ETC (258) and altered cytosolic Ca2+ concentration affects mitochondrial localization in the cell (259). Calcium inhibits mitochondrial movement in the cell through regulation of Miro (mitochondrial Rho GTPase), a Ca2+ binding Ras-like small G protein at the OMM that controls the interaction and movement of mitochondria along microtubules (259).
Failure of mitochondria to take up Ca2+ also results in aberrant activation of cytosolic enzymes such as calpain proteases (260) and Ca2+/calmodulin-dependent kinases (261) that can in turn alter cellular signaling cascades with dramatic effects on cell growth and viability leading to cancer (262). For example, mitochondrial stress induced in cultured cells through depletion of mtDNA (following growth in ethidium bromide) resulted in loss of mitochondrial membrane potential and elevated cytosolic Ca2+ levels that in turn led to increased glycolysis, increased ERK1/ERK2 and PKC activity (these enzymes are Ca2+ dependent), and increased tumor cell invasion associated with increased expression of cathepsin L and TGF-β (263, 264). Significantly, activation of calcineurin protein phosphatase by increased cytosolic Ca2+ in this system lead to dephosphorylation of IκBβ, and activation of NF-κB, as well as ATF2 and NFAT (265, 266). The pro-tumorigenic activities of NF-κB are well documented and include promoting resistance to apoptosis in addition to effects on cell migration and cell metabolism, through effects on HIF-1α (267–269). In summary, deregulated calcium homeostasis is one of the major consequences for the cell of dysfunctional mitochondria (Figure 7) that can result in dramatic changes in gene expression.
Mitochondrial Reactive Oxygen Signaling Modulates Cell Growth and Differentiation
As already mentioned above, mitochondria are the major source of cellular ROS and the contribution of ROS to mitochondrial stress signaling in cell growth, cellular senescence, and differentiation is significant (27, 29, 78, 270, 271). For example, ROS is required for KRas driven tumorigenesis (272) and anti-oxidants that quench ROS are anti-tumorigenic in certain systems (273, 274).
There are numerous mechanisms by which mitochondrial ROS can alter cell signaling but one of the major consequences of increased ROS and altered cellular redox state is the oxidation of thiol groups in cysteine residues in relevant proteins (27, 275). For example, the cysteine at the active site of caspases is inhibited by ROS production (276) as is the cysteine at the active site of many cellular phosphatases, including the Pten tumor suppressor (277), the CDC25B oncogene (278), and MAPK phosphatases (279).
The other key mechanism by which mitochondrial ROS is known to modulate cell signaling is through stabilization of HIF-1 subunits (280–284), as a result of prolyl hydroxylase inhibition (29). Increased HIF levels feed back to modulate mitochondrial respiration through induction of target genes such pyruvate dehydrogenase kinase-1 (PDK1) that inhibits conversion of pyruvate to acetyl CoA to feed the Krebs cycle and provide reducing agents for OXPHOS (285, 286), as well as the LON protease that degrades the regular COX4-1 subunit, and induction of COX4-2, an alternative isoform of COX4, that allows more efficient oxygen utilization and respiration under limiting oxygen conditions (287). NDUFA4L2, an inhibitory subunit of ND/complex I, is also induced by HIF-1α and limits respiration and ROS production (288). HIF-1α also protects cells from apoptosis associated with increased ROS, for example through induction of molecules such as ATIA that maintains mitochondrial thioredoxin 2 in a reduced form required for its anti-oxidant activity (289). Significantly, ATIA is up-regulated in human glioblastoma (289). Mitochondrial ROS-induced stabilization of HIF-α also explains in large part the pro-tumorigenic effect of deleting key regulatory molecules, including SirT3 (118), REDD1 (290), and BNIP3 (Chourasia et al., under review). Stabilization of HIF either through effects on accumulation of TCA cycle intermediates, as discussed above, or due to elevated mitochondrial ROS production, leads to increased angiogenesis, EMT, a switch to glycolytic metabolism, and priming of the pre-metastatic niche amongst many of the known tumorigenic effects of HIF activity (291–296).
FoxO transcription factors are another key signaling component in the response to elevated ROS levels and their induction activates not only anti-oxidant responses (increased expression of catalase and SOD2) but also cell cycle arrest and/or cell death (297, 298). For example, mitochondrial ROS in Drosophila as a result of inhibition of mitochondrial respiration lead to activation of a G1 cell cycle arrest in part due to activation of FoxO transcription factors (299). Interestingly, by antagonizing c-Myc, FoxO3a has also been shown to limit nuclear-encoded mitochondrial gene expression thereby limiting mitochondrial biogenesis under conditions of oxidative stress (300–302).
Finally, the KEAP1-NRF2 anti-oxidant signaling axis is activated by increased ROS due to the redox sensitivity of KEAP1 (303, 304). KEAP1 normally binds to NRF2 in the cytosol and promotes its degradation at the proteasome. ROS-induced dissociation of KEAP1 stabilizes NRF2 allowing it to translocate to the nucleus where it induces genes involved in quenching ROS (303). NRF2 is also stabilized by accumulation of p62/Sqstm1 that is often linked to defects in autophagy (305, 306). NRF2 stabilization promotes metabolic reprograming toward anabolic pathways, such as nucleotide biosynthesis thereby promoting tumor cell growth (307). NRF2 also promotes tumor cell survival by limiting levels of damaging ROS and constitutive activation of the KEAP1-NRF2 pathway has been detected in human cancers, either through activating mutations in NRF2 or through inactivating mutations in KEAP1 (308–310) and activation of NRF2 is associated with poor prognosis and therapy resistance (311). As a key regulator of mitochondrial biogenesis, as well as responses to ROS and autophagy defects, NRF2 activity is thus intimately linked to determining how the cell responds to mitochondrial dysfunction in terms of cell growth and tumorigenesis.
It is important to consider however that ROS has a relatively short diffusion distance in solution and thus mitochondrial ROS signaling may rely on proximity of ROS-producing mitochondria to their sites of action/targets. Intriguingly, perinuclear clustering of mitochondria induced by hypoxia was associated with increased nuclear ROS and was required for maximal HIF-1α DNA binding and target gene (VEGF) expression (41). These observations suggest that mitochondrial movement may play a role in allowing mitochondrial ROS to signal more directly to the nucleus. Perinuclear mitochondrial hubs that form in response to hypoxia and ROS (41) may also act to limit mitochondrial uptake of Ca2+ from the ER or the plasma membrane, thereby spatially regulating the effects of Ca2+ signaling in the cell.
Altered Mitochondrial Metabolism Signaling via AMPK and Sirtuins
Defective oxidative metabolism and reduced ATP levels in cells activate AMPK (312) and certain drugs are known to induce AMPK as a result of inhibiting mitochondrial respiration, such as Metformin that inhibits complex I of the ETC and resveratrol that inhibits the F0F1 ATPase (313). AMPK plays a key role in mitochondrial homeostasis and while activated by mitochondrial dysfunction, feeds back to promote both mitochondrial biogenesis through activation of PGC-1α (314, 315) and mitophagy by activating ULK1 and inhibiting mTOR (316, 317), thereby improving the overall “health” of the mitochondrial pool in cells.
In addition to AMPK, the sirtuins serve as metabolic sensors of mitochondrial well-being due to their function as NAD+ dependent deacetylases (318). In particular, the mitochondrial sirtuins (SirT3, SirT4, and SirT5) are sensitive to the mitochondrial pool of NAD+ that is in turn determined by metabolic activity at the mitochondrion, with NAD+ levels increased by OXPHOS and reduced by fatty acid oxidation. The best characterized mitochondrial sirtuin, SirT3 deacetylates a number of critical enzymes involved in fatty acid metabolism (LCAD), the TCA cycle (IDH2), and OXPHOS (SDHB, complex I, II, V) in addition to cyclophilin D and UCP2 that modulate mitochondrial permeability and electron flow respectively (318, 319). The cytosolic and mitochondrial pools of NAD+/NADH are separate but can equilibrate through transfer via the malate-aspartate shuttle and thus mitochondrial metabolism may also influence nuclear and cytosolic sirtuins. Interestingly, nuclear SirT1 promotes mitochondrial biogenesis in response to nutrient deprivation through deacetylation and activation of PGC-1α (320), as discussed above, and similar to AMPK, SirtT1 may also promote mitophagy in response to nutrient deprivation through deacetylation of key autophagy genes, including Atg5, Atg7, and Atg8 (321).
Mitochondrial Localization of Kinases Involved in Stress Response Signaling
Kinases known to play key roles in cellular stress responses have been detected at the mitochondria, including AKT, GSK-3β, PKA, ABL, PKC, AMPK, SRC, ATM, and others (313, 322–328). While substrates for some of these kinases at the mitochondria have been identified, the significance of localization of some of the other kinases is less clear.
AKT is a major growth promoting kinase that acts by inhibiting apoptosis in the presence of glucose and by activating mTOR (329). AKT also promotes glycolysis by phosphorylating hexokinase II (HKII) and promoting its interaction with VDAC at the mitochondria (328). HKII is required for tumor initiation and maintenance in mouse models (330). Failure of HKII to interact at the mitochondria with VDAC results in apoptosis (328), and thus, AKT plays a role in coupling mitochondrial metabolism with cell viability.
AKT also phosphorylates and inactivates GSK-3β a cellular kinase that localizes to the mitochondria under certain circumstances. Mitochondrial GSK-3β phosphorylates MCL-1 and VDAC amongst other mitochondrial targets (325, 331, 332). GSK-3β mediated phosphorylation of MCL-1 promoted its degradation and increased apoptosis (331), while phosphorylation of VDAC by GSK-3β resulted in increased mitochondrial membrane permeability, again predisposing to apoptosis (325, 333). Interestingly, GSK-3β also phosphorylates Drp1 resulting in elongated mitochondrial morphology that mitigates against cell death (334). This suggests that GSK-3β activity (either pro- or anti-apoptotic) is modulated by mitochondrial stress, although the precise mechanism of such a regulatory switch at the mitochondria is unclear. Obviously, GSK-3β is also known to phosphorylate and promote the proteasomal degradation of c-Myc, cyclin D1, and β-catenin (335–338) and thus one may postulate that activities of GSK-3β at the mitochondria influence nuclear oncogene activity. This would represent a novel perspective on retrograde signaling from the mitochondria to the nucleus.
Other kinases located at the mitochondria include PKA that associates with the mitochondria via adaptor molecules such as Rab32 and other A-kinase AKAPs (322, 339, 340) where it has been shown to phosphorylate VDAC (323), Drp1 (72), and other mitochondrial proteins. Localization of PKA to the mitochondria via AKAPs is subject to regulation by hypoxia and other physiological stresses (72, 341). For example, hypoxia destabilizes AKAP121 through induction of SIAH2, a mitochondrial ubiquitin ligase, thereby limiting oxidative capacity under conditions of low oxygen. Interestingly, AKAP121 also appears to promote mitochondrial localization of SRC-tyrosine kinase (342) where SRC appears to regulate CO activity and respiratory activity (342, 343), and other mitochondrial substrates for SRC family kinases are likely (344).
Association of protein kinase C-delta (PKCδ) with the mitochondria is induced by increased ROS (327) and this is turn recruits other signaling molecules, including the ABL tyrosine kinase that is associated with loss of membrane potential and non-apoptotic cell death (326). Again, whether these important kinases also play a role in mitochondrial function and signaling independent of cell death is not clear.
The localization of AMPK to the mitochondria is likely linked to its ability to modulate mitochondrial metabolism. Acetyl CoA carboxylase-2 (ACC2) is a well-established AMPK target that localizes to the OMM where it regulates lipid metabolism by controlling production of malonyl CoA (313). Inhibition of ACC2 (and ACC1) by AMPK boosts NADPH homeostasis under energy crisis and promotes tumor cell survival, anchorage independent growth, and tumor formation in vivo (345).
Finally, mitochondrial uncoupling activates ATM kinase, a fraction of which was shown to be located at the mitochondria (346). Loss of Atm in genetically engineered mouse models led to mitochondrial dysfunction suggesting the presence of a feedback loop, although the key substrates of ATM kinase in modulating mitochondrial homeostasis are not known (346). Of note, it has been reported that p53 tumor suppressor expression is sensitive to inhibition of mitochondrial respiration by unknown mechanisms (347) but whether p53 is the key substrate of mitochondrial ATM in mitochondrial stress signaling has not been examined.
In summary, there are numerous ways in which the mitochondria signals to the nucleus (Figure 7) and the consequences of mitochondrial dysfunction can therefore impact cell growth significantly.
Oncogenic Control of Mitochondrial Function
A growing number of tumor suppressor genes and oncogenes are being investigated for their ability to regulate mitochondrial function either through effects on the expression and/or activity of components of the ETC or other key metabolic enzymes at the mitochondria or through effects on mitochondrial biogenesis and mitophagy. Some more recent findings in this area of seminal interest about two key tumor suppressors (p53 and RB) and two key oncogenes (Myc and KRas) and how they modulate mitochondrial function and metabolism are discussed here.
The p53 Tumor Suppressor Regulates Mitochondrial Function at Multiple Levels
The p53 tumor suppressor gene is the most commonly mutated gene in human cancer with inactivating mutations found in its DNA binding domain that result in loss of its normal transcriptional properties with gain of dominant negative or novel functions frequently the result (348, 349). Many of the tumor suppressor functions of p53 are attributed to its role as a transcriptional regulator of nuclear-encoded genes in response to stresses such as DNA damage, nutrient deprivation, and aberrant oncogene activity (350). The outcome of activating normal p53 in response to these stresses is induction of downstream target genes, such as p21Waf1 that induces a G1 cell cycle arrest or induction of pro-apoptotic genes like Puma and Bax (350, 351). While p21Waf1 is key to the ability of p53 to induce growth arrest (352), there are numerous downstream effectors of p53-induced apoptosis (350), including p53 itself (353, 354).
In recent years, the ability of p53 to regulate cell growth processes, other than proliferation or apoptosis, has emerged, including roles for p53 in modulating expression of genes involved in mitochondrial biogenesis (through repression of PGC-1α), autophagy (355, 356), and mitochondrial metabolism (356, 357) (Figure 8). P53 indirectly affects mitophagy and mitochondrial quality control through induction of genes that regulate autophagy, such as Dram and Atg7 (355, 356). Recent work has identified a role for p53 in limiting the accumulation of damaged mitochondria in cancer by enforcing a growth arrest. Specifically, loss of Atg7 in KRas driven lung cancers caused accumulation of damaged mitochondria, defective fatty acid oxidation, and a growth arrest that retarded tumor growth (157). Inactivation of p53 alleviated growth arrest to some extent, although autophagy deficient tumor cells were unable to mobilize lipid stores and tumors continued to grow more slowly than control tumors that were functional for autophagy (157). Nevertheless, these results are consistent with a role for p53 in sensing defects in autophagy and/or mitochondrial function. Indeed, p53 may act more directly to modulate autophagy and responses to defects in autophagy (358) by interacting with Atg7 (212).
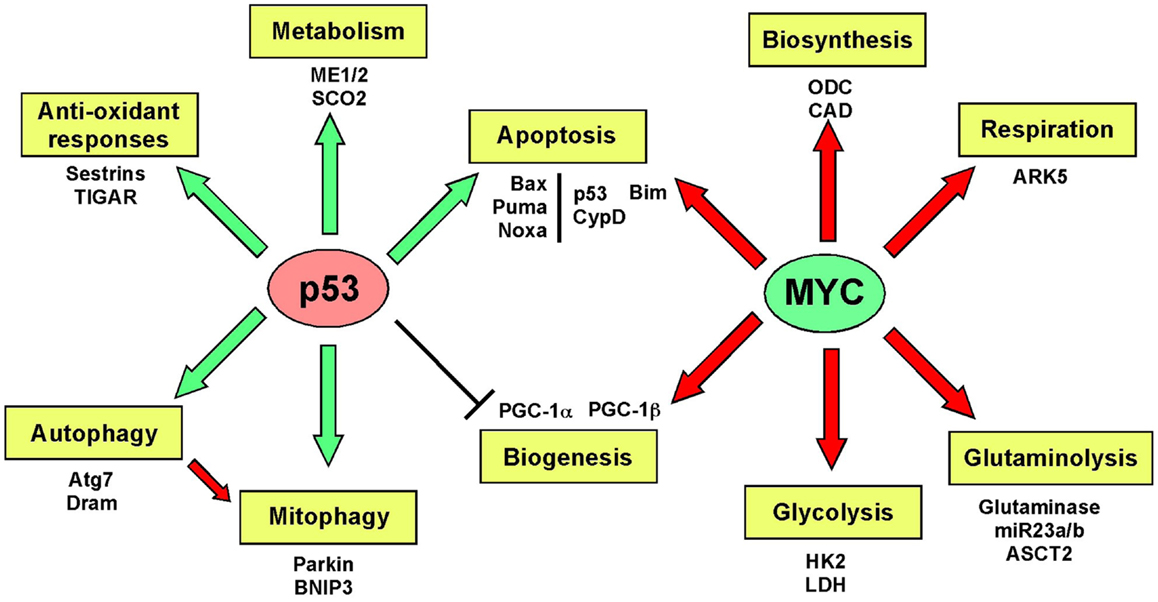
Figure 8. Oncogene and tumor suppressor gene regulation of mitochondria. The key activities of major oncogenes and tumor suppressors are increasingly being linked to effects on mitochondrial function. In particular, both p53 and Myc, inarguably two of the most significant tumor related genes in the human genome, have been shown to modulate several different aspects of mitochondrial function and in some instances, this has been shown to be key to their role in cancer, as discussed in greater length in the text.
p53 also regulates expression of genes encoding regulators of the ETC [such as CO/Sco2 (359)], TCA cycle enzymes [such as malic enzymes ME1/ME2 (360)] in addition to modulators of glucose metabolism [such as HKII and specific glucose transporters (357)], and the pentose phosphate pathway [such as TIGAR (361)] in the cytosol. This transcriptional activity of p53 in metabolism has major significance for cell growth. For example, suppression of malic enzymes by p53 results in markedly lower NADPH production required for lipid synthesis and glutaminolysis (360).
p53 has been reported to be induced by AMPK through phosphorylation on serine 15 in response to nutrient stress resulting in a starvation-induced growth arrest (362) although it has been pointed out that the AMPK phosphorylation site in p53 (serine 15) is a weak AMPK consensus site (313) and thus perhaps that effects of AMPK activation on p53 are indirect. Given that serine 15 phosphorylation modulates the interaction of p53 with MDM2 to promote p53 degradation, reduced activity of AKT under nutrient deprivation resulting in lower nuclear MDM2 levels [AKT promotes nuclear localization of MDM2 and inhibits its interaction with p19/ARF (363)] may be a more likely explanation for increased levels of p53 in response to starvation. Nevertheless, p53 is induced by nutrient deprivation and its activity limits glycolysis and promotes OXPHOS, and indeed p53 null mice exhibit deficiencies in respiration and exercise performance (359).
In addition to these important functions as a transcriptional activator of genes that modulate mitochondrial turnover and metabolism at the mitochondria, p53 also plays a direct non-transcriptional role at the mitochondria. P53 has been detected at the mitochondria itself where it has been reported to promote cytochrome c release and apoptosis (353, 354). Specifically, mitochondrial p53 can function as a BH3-only protein that interacts with anti-apoptotic Bcl-2 and Bcl-XL potentiating the pro-apoptotic activity of Puma, a target of nuclear p53 (364). More recently, mitochondrial p53 has been shown under conditions of oxidative stress to interact directly with cyclophilin D in the mitochondrial matrix. P53 uptake by mitochondria was dependent on mitochondrial membrane potential and interaction with mitochondrial chaperones (365). This resulted in opening of the mitochondrial permeability transition pore and induction of necrosis (365) that was not mitigated by nuclear functions of p53 in anti-oxidant gene expression induction (sestrins, glutathione peroxidase) but was blocked by cyclosporine-A, an inhibitor of cyclophilin D. Thus, p53 functions not just as a “guardian of the genome,” but also as a guardian of mitochondrial integrity and function.
Modulation of Mitochondrial Metabolism and Cell Viability by the RB Tumor Suppressor
The RB tumor suppressor gene is commonly deleted in human retinoblastoma, osteosarcoma, and small cell lung carcinoma, but other genes upstream in the RB pathway, including p16/INK4A (CDKN2A) and Cyclin D1 (CCND1) are more commonly deregulated in human cancer than the RB tumor suppressor gene itself (366, 367). Indeed some cancers maintain functional RB at late stages in disease progression (368, 369) and it is not yet clear what the selective pressures are to maintain functional pRB at late stages of some tumor types but not others.
The RB tumor suppressor is considered primarily as a regulator of cell cycle checkpoints and induces a G1 arrest through repression of E2F transcription factors in response to numerous stresses, including growth factor deprivation, DNA damage, and hypoxia (370, 371). The role of pRB in establishing a cell cycle arrest has also been key to our understanding of how pRB promotes both cellular senescence (372, 373) and terminal differentiation (374–379). pRB/E2F complexes have also been shown to regulate genes involved in programed cell death, such as caspase-3, p73, and Apaf1 (380).
In addition to regulating genes involved in cell cycle control and cell death however, there has been over the past decade a growing appreciation that pRB and E2Fs together can modulate genes involved in metabolism and mitochondrial homeostasis (381), including BNIP3 (173), pyruvate dehydrogenase kinase 4 (382), as well as other mitochondrial genes (383). Recently, a role for RB in inhibiting glutaminolysis has emerged from studies in mammalian cells (83) and from metabolomic analyses in Drosophila (82). pRB was shown to repress expression of the glutamine transporter, and glutaminase-1 (83) and loss of RBF1 in flies led to metabolic reprograming such that glutamine flux to glutathione was increased and RBF1 deficient flies were more sensitive to oxidative stress (82). Thus RB/E2Fs are now considered to be significant modulators of cellular metabolism, although the relevance of these functions to the role of RB as a tumor suppressor in vivo remains to be determined.
Given the canonical role of RB/E2Fs as transcriptional regulators, it is not surprising that until recently attention was focused on effects of RB loss on expression of E2F regulated metabolism and mitochondrial genes (383, 384). However, like p53, pRB has also now been detected at the mitochondria (385, 386). pRB was shown to interact with and conformationally activate Bax to induce apoptosis (386), a function consistent with its role as a tumor suppressor. However, it remains to be seen whether other functions for pRB at the mitochondria can be determined that may explain the unexplained dependence on functional pRB at late stages of certain cancers (368, 369).
Over-Expression of c-MYC in Cancer Promotes Dependence on Functional Mitochondria
The c-Myc oncogene is over-expressed in over 70% of all human cancers where it functions as a transcriptional regulator of genes involved in cell cycle (p21Waf1, cdc25A, Cdk4, cyclin D2), cell death (Bim, p53), replicative senescence (Tert, Bmi1) (387), genome stability [BRCA1/2, MUTS (388, 389), protein translation (ribosomal RNAs) (390)], cell adhesion (collagen, fibronectin, integrins) (391), angiogenesis (thrombospondin) (392), the tumor microenvironment (393), mitochondrial biogenesis (PGC-1β) (115), mitochondrial function (VDAC), and metabolism (glutaminase, lactate dehydrogenase-A/LDHA) (81, 394).
Myc-driven tumors regress rapidly when Myc is inhibited/turned off, as demonstrated in elegant switchable mouse models (395) and it has also been recently reported that KRas driven lung tumors are also dependent on Myc activity (396), highlighting the importance of c-Myc as a driving oncogene (81) and emphasizing the importance of how tumors become “addicted” to Myc. Induction of glutaminolysis at the mitochondria is a key factor explaining how tumors become “addicted” to Myc (80). Myc induces expression of key enzymes in glutaminolysis such as glutaminase (through repression of miR23a/b) (397) and ACST2 (the glutamine transporter) (80). Thus, Myc expressing tumors are dependent on glutamine as an anapleurotic source of carbon for the TCA cycle, as a source of nitrogen for nucleotide biosynthesis, to produce ATP and to generate lipids via reductive carboxylation at the mitochondria (249, 250). Withdrawal of glutamine causes Myc-driven tumor cells to apoptose (398, 399) and this is now being exploited for therapeutic purposes, as recently reported for N-Myc-driven neuroblastomas (400). A synthetic lethal screen identified Myc regulated molecules required to support glutaminolysis in c-Myc-driven tumors (401). Specifically, Myc was shown to induce expression of AMPK-related kinase 5 (ARK5) thereby promoting increased mitochondrial respiratory chain capacity required for glutaminolysis. Significantly, inhibition of ARK5 led to apoptosis of Myc-driven tumor cells (401), again with important therapeutic implications.
In addition to glutaminolysis, Myc regulates glucose metabolism by inducing expression of key glycolytic enzymes, including LDHA, HKII. Glycolysis is important in tumors not just as a low level source of ATP but also to provide precursors for biosynthetic pathways, including serine and nucleotide biosynthesis, and Myc promotes biosynthetic processes through induction of carbamoyl phosphate synthase, aspartate transcarbamylase, dihydroorotase (CAD), and ornithine decarboxylase (ODC) among other genes (81). Like Myc, HIF-1α also promotes glycolysis by inducing expression of glycolytic enzymes and when Myc expression is deregulated in cancers due to translocation or amplification, Myc, and HIF-1α cooperate to regulate glucose metabolism. However, hypoxia induces a growth arrest in normal cells and when Myc is expressed at normal levels in cells (not amplified or translocated), HIF-1α antagonizes Myc by displacing it from complexes with Max, by inducing Mxi-1, a repressive binding partner of Myc and by promoting Myc protein degradation at the proteasome (115, 296, 402). In this way, HIF-1α uncouples glycolysis from biosynthesis under hypoxic conditions and promotes mitophagy at the expense of biogenesis. By contrast, HIF-2α synergizes with Myc to stabilize Myc-Max dimers and to promote Myc target gene expression, cell growth, and genome stability (296, 388), although HIF-2α expression is more tissue restricted.
In summary, by increasing mitochondrial mass through induction of mitochondrial biogenesis (117) and promoting glutaminolysis at the mitochondria (80) (Figure 8), Myc oncogenes make tumors more dependent on mitochondrial function, not less. This may suggest that Myc-dependent tumors would be more susceptible to defects in mitophagy, mitochondrial fusion, or other key processes required for mitochondrial quality control.
Activation of KRas
Activated KRas is one of the most prevalent oncogenic events in cancer of the pancreas, lung, and small intestine (403). In pancreatic cancer, activated KRas is linked to reprograming of tumor metabolism both through increased glycolytic flux to lactate, hexosamine biosynthesis, and non-oxidative pentose phosphate pathway (404). Also, KRas induces up-regulation of an alternative glutaminolysis pathway that converts glutamine-derived aspartate to oxaloacetate in the cytosol allowing pancreatic tumor cells to buffer against ROS through increased glutathione production (405). These results indicate that KRas driven tumors have evolved to be independent of mitochondrial metabolism (since both glycolysis and the alternative use of glutamine take place in the cytosol). However, other work points to a critical role for autophagy in KRas driven tumorigenesis by promoting mitochondrial metabolism (156, 157) suggesting that mitochondrial function is required for KRas tumorigenesis. It was suggested that KRas activation causes mitochondrial dysfunction, including increased ROS and reduced OXPHOS (406) but these studies were performed using a doxycycline inducible system that by itself induces mitochondrial dysfunction due to inhibition of mitochondrial protein synthesis and the UPRmt. Cells expressing activated KRas do exhibit reduced respiration associated with decreased expression of components of complex I of the respiratory chain (407) and activation of KRas does lead to increased c-Myc protein stability (408) suggesting that some alterations in mitochondrial function associated with KRas activation are in fact driven by increased levels of c-Myc.
Conclusion
As the major energy and metabolite source in the cell, it stands to reason that mitochondrial function is deregulated in cancer and there is growing interest in understanding how altered mitochondrial function may be targeted to inhibit tumor growth. Emerging data identifies key oncogenes and tumor suppressors as modulators of different aspects of mitochondrial metabolism and dynamics. Interestingly, different tumor types may be more or less sensitive to modulation of mitochondrial function depending on which oncogenic lesions drive that tumor type. This is a new and exciting avenue in the continued “war on cancer.”
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to members of the MacLeod lab for critical reading of the manuscript and for financial support from the National Institutes of Health (T32-DK780073 to Michelle L. Boland, T32 CA009594 to Aparajita H. Chourasia, and RO1 CA131188 to Kay F. Macleod).
References
1. Chan DC. Mitochondria: dynamic organelles in disease, aging and development. Cell (2006) 125:1241–52. doi: 10.1016/j.cell.2006.06.010
2. Martinou JC, Youle RJ. Mitochondria in apoptosis:Bcl-2 family members and mitochondrial dynamics. Dev Cell (2011) 21:92–101. doi:10.1016/j.devcel.2011.06.017
3. Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell (2012) 148:1145–59. doi:10.1016/j.cell.2012.02.035
4. Wallace DC, Fan WW, Procaccio V. Mitochondrial energetics and therapeutics. Annu Rev Pathol (2010) 5:297–348. doi:10.1146/annurev.pathol.4.110807.092314
5. Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer (2011) 11:325–37. doi:10.1038/nrc3038
6. Gottlieb E, Tomlinson IPM. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer (2005) 5:857–66. doi:10.1038/nrc1737
7. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: the metabolic requirements of cell proliferation. Science (2009) 324:1029–33. doi:10.1126/science.1160809
8. Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet (2011) 43:869–74. doi:10.1038/ng.890
9. Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature (2011) 476:346–50. doi:10.1038/nature10350
10. Hitosugi T, Zhou L, Elf S, Fan J, Kang HB, Seo JH, et al. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell (2012) 22:585–600. doi:10.1016/j.ccr.2012.09.020
11. Vaughn AE, Deshmukh M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat Cell Biol (2008) 10:1477–83. doi:10.1038/ncb1807
12. Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer Discov (2012) 2:881–98. doi:10.1158/2159-8290.CD-12-0345
13. Deberardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic programming fuels cell growth and proliferation. Cell Metab (2008) 7:11–20. doi:10.1016/j.cmet.2007.10.002
14. Deberardinis RJ, Thompson CB. Cellular metabolism and disease: what do metabolic outliers teach us? Cell (2012) 148:1132–44. doi:10.1016/j.cell.2012.02.032
15. Locasale JW, Cantley LC. Metabolic flux and the regulation of mammalian cell growth. Cell Metab (2011) 14:443–51. doi:10.1016/j.cmet.2011.07.014
16. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol (2011) 27:441–64. doi:10.1146/annurev-cellbio-092910-154237
17. Metallo CM, Vander Heiden MG. Understanding metabolic regulation and its influence on cell physiology. Mol Cell (2013) 49:388–98. doi:10.1016/j.molcel.2013.01.018
18. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell (2012) 21:297–308. doi:10.1016/j.ccr.2012.02.014
19. Chatterjee A, Dasgupta SK, Sidransky D. Mitochondrial subversion in cancer. Cancer Prev Res (2011) 4:638–54. doi:10.1158/1940-6207.CAPR-10-0326
20. Galluzzi L, Morselli E, Kepp O, Vitale I, Rigoni A, Vacchelli E, et al. Mitochondrial gateways to cancer. Mol Aspects Med (2010) 31:1–20. doi:10.1016/j.mam.2009.08.002
21. Ralph SJ, Rodríguez-Enríquez S, Neuzil J, Saavedra E, Moreno-Sánchez R. The causes of cancer revisited: “mitochondrial malignancy” and ROS-induced oncogenic transformation – why mitochondria are targets for cancer therapy. Mol Aspects Med (2010) 31:145–70. doi:10.1016/j.mam.2010.02.008
22. Polyak K, Li Y, Zhu H, Lengauer C, Willson JK, Markowitz SD, et al. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat Genet (1998) 20:291–3. doi:10.1038/3108
23. Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S A (2005) 102:719–24. doi:10.1073/pnas.0408894102
24. Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yaaguchi A, Imanishi H, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science (2008) 320:661–4. doi:10.1126/science.1156906
25. Santidrian AF, Matsuno-Yagi A, Ritland M, Seo BB, Leboeuf SE, Gay LJ, et al. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J Clin Invest (2013) 123:1068–81. doi:10.1172/JCI64264
27. Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci (2010) 35:505–13. doi:10.1016/j.tibs.2010.04.002
28. Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell (2006) 10:175–6. doi:10.1016/j.ccr.2006.08.015
29. Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell (2012) 48:158–67. doi:10.1016/j.molcel.2012.09.025
30. Wheaton WW, Chandel NS. Hypoxia. 2. Hypoxia regulates cellular metabolism. Am J Physiol Cell Physiol (2011) 300:C385–93. doi:10.1152/ajpcell.00485.2010
31. Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature (2011) 475:231–4. doi:10.1038/nature10167
32. Shaw AT, Winslow MR, Magensantz M, Ouyang C, Dowdle J, Subramanian A, et al. Selective killing of K-ras mutant cancer cells by small molecule inhibitors of oxidative stress. Proc Natl Acad Sci U S A (2011) 108:8773–8. doi:10.1073/pnas.1105941108
33. Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen ZY, Pelicano H, et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by b-phenylethyl isothiocynate. Cancer Cell (2006) 10:241–52. doi:10.1016/j.ccr.2006.08.009
34. Liu H, Knabb JR, Spike BT, MacLeod KF. Elevated Parp activity sensitizes Rb deficient cells to DNA damage-induced necrosis. Mol Cancer Res (2009) 7:1099–109. doi:10.1158/1541-7786.MCR-08-0439
35. Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach. Nat Rev Drug Discov (2009) 8:579–91. doi:10.1038/nrd2803
36. Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev (2004) 18:357–68.
37. Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta (2008) 1777:1092–7. doi:10.1016/j.bbabio.2008.05.001
38. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Biol (2011) 12:9–14. doi:10.1038/nrm3028
39. Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Biol (2007) 8:870–9. doi:10.1038/nrm2275
40. Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev (2008) 22:1577–90. doi:10.1101/gad.1658508
41. Al-Mehdi AB, Pastukh VM, Swiger BM, Reed DJ, Patel MR, Bardwell GC, et al. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci Signal (2012) 5:ra47. doi:10.1126/scisignal.2002712
42. Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci (2008) 9:505–18. doi:10.1038/nrn2417
43. Twig G, Shirihai OS. The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal (2011) 14:1939–51. doi:10.1089/ars.2010.3779
44. Autret A, Martin SJ. Emerging role for members of the Bl-2 family in mitochondrial morphogenesis. Mol Cell (2009) 36:355–63. doi:10.1016/j.molcel.2009.10.011
45. Otera H, Mihara K. Molecular mechanisms and physiologic functions of mitochondrial dynamics. J Biochem (2011) 149:241–51. doi:10.1093/jb/mvr002
46. Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J (2009) 28:1589–600. doi:10.1038/emboj.2009.89
47. Horn SR, Thomenius MJ, Johnson ES, Freel CD, Wu JQ, Coloff JL, et al. Regulation of mitochondrial morphology by APC/CCdh1-mediated control of Drp1 stability. Mol Biol Cell (2011) 22:1207–16. doi:10.1091/mbc.E10-07-0567
48. Kashatus DF, Lim KH, Brady DC, Pershing NL, Cox AD, Counter CM. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat Cell Biol (2011) 13:1108–15. doi:10.1038/ncb2310
49. Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem (2007) 282:11521–9. doi:10.1074/jbc.M607279200
50. Youle RJ, Karbowski M. Mitochondrial fission in apoptosis. Nat Rev Mol Biol (2005) 6:657–63. doi:10.1038/nrm1697
51. Youle RJ, van der Bliek AM. Mitochondrial fission, fusion and stress. Science (2012) 337:1062–5. doi:10.1126/science.1219855
52. Cereghetti GM, Stangherlin A, de Brito OM, Chang CR, Blackstone C, Bernardi P, et al. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci U S A (2008) 105:15803–8. doi:10.1073/pnas.0808249105
53. Chang CR, Blackstone C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann N Y Acad Sci (2010) 1201:34–9. doi:10.1111/j.1749-6632.2010.05629.x
54. Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol (2007) 177:439–50. doi:10.1083/jcb.200610042
55. Ishihara N, Otera H, Oka T, Mihara K. Regulation and physiologic functions of GTPases in mitochondrial fusion and fission in mammals. Antioxid Redox Signal (2012) 19:389–99. doi:10.1089/ars.2012.4830
56. Lee YK, Lee HY, Hanna RA, Gustafsson AB. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol (2011) 301:H1924–31. doi:10.1152/ajpheart.00368.2011
57. Losón OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49 and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell (2013) 24:659–67. doi:10.1091/mbc.E12-10-0721
58. Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, et al. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol (2010) 191:1141–58. doi:10.1083/jcb.201007152
59. Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep (2011) 12:565–73. doi:10.1038/embor.2011.54
60. Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science (2011) 334:358–62. doi:10.1126/science.1207385
61. de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature (2008) 456:605–10. doi:10.1038/nature07534
62. Lackner LL, Ping H, Graef M, Murley A, Nunnari J. Endoplasmic reticulum-associated mitochondria-cortex tether functions in the distribution and inheritance of mitochondria. Proc Natl Acad Sci U S A (2013) 110:E458–67. doi:10.1073/pnas.1215232110
63. Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci U S A (2009) 106:11960–5. doi:10.1073/pnas.0904875106
64. Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol (2011) 13:589–98. doi:10.1038/ncb2220
65. Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondrial from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A (2011) 108:10190–5. doi:10.1073/pnas.1107402108
66. Nagaraj R, Gururaja-Rao S, Jones KT, Slattery M, Negre N, Braas D, et al. Control of mitochondrial structure and function by the Yorkie/YAP oncogenic pathway. Genes Dev (2012) 26:2027–37. doi:10.1101/gad.183061.111
67. Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, Capaldi RA. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res (2004) 64:985–93. doi:10.1158/0008-5472.CAN-03-1101
68. Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem (2003) 278:17190–7. doi:10.1074/jbc.M212754200
69. Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem (2005) 280:26185–92. doi:10.1074/jbc.M503062200
70. Lapuente-Brun E, Moreno-Loshuertos R, Acín-Pérez R, Latorre-Pellicer A, Colás C, Balsa E, et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science (2013) 340:1567–70. doi:10.1126/science.1230381
71. Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell (2013) 155:160–71. doi:10.1016/j.cell.2013.08.032
72. Kim H, Scimia MC, Wilkinson D, Trelles RD, Wood MR, Bowtell D, et al. Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol Cell (2011) 44:532–44. doi:10.1016/j.molcel.2011.08.045
73. Nakayama K, Frew IJ, Hagensen M, Skals M, Habelhah H, Bhoumik A, et al. Siah2 regulates stability of prolyl-hydroxylases, controls HIF1alpha abundance, and modulates physiological responses to hypoxia. Cell (2004) 117:941–52. doi:10.1016/j.cell.2004.06.001
74. Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, et al. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J (2012) 26:2175–86. doi:10.1096/fj.11-196543
75. Schieke S, McCoy JP Jr, Finkel T. Coordination of mitochondrial bioenergetics with G1 phase cell cycle progression. Cell Cycle (2008) 7:1782–7. doi:10.4161/cc.7.12.6067
76. Qian W, Choi SE, Gibson GA, Watkins SC, Bakkenist CJ, Van Houten B. Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress. J Cell Sci (2012) 125:5745–57. doi:10.1242/jcs.109769
77. Spruck CH, Won KA, Reed SI. Deregulated cyclin E induces chromosome instability. Nature (1999) 401:297–300. doi:10.1038/45836
78. Havens CG, Ho H, Yoshioka N, Dowdy SF. Regulation of late G1/S phase transition and APCCdh1 by reactive oxygen. Mol Cell Biol (2006) 26:4701–11. doi:10.1128/MCB.00303-06
79. Almeida A, Bolaños JP, Moncada S. E3 ubiquitin ligase APC/C-Cdh1 accounts for the Warburg effect by linking glycolysis to cell proliferation. Proc Natl Acad Sci U S A (2010) 107:738–41. doi:10.1073/pnas.0913668107
80. Dang CV. Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer Res (2010) 70:859–62. doi:10.1158/0008-5472.CAN-09-3556
82. Nicolay BN, Gameiro PA, Tschöp K, Korenjak M, Heilmann AM, Asara JM, et al. Loss of RBF1 changes glutamine catabolism. Genes Dev (2013) 27:182–96. doi:10.1101/gad.206227.112
83. Reynolds MR, Lane AN, Robertson B, Kemp S, Liu Y, Hill BG, et al. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene (2013) 1:1–13. doi:10.1038/onc.2012.635
84. Jahani-Asl A, Cheung EC, Neuspie LM, MacLaurin JG, Fortin A, Park DS, et al. Mitofusin 2 protects cerebellar granule neurons against injury-induced cell death. J Biol Chem (2007) 282:23788–98. doi:10.1074/jbc.M703812200
85. Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell (2006) 126:177–89. doi:10.1016/j.cell.2006.06.025
86. Yamaguchi R, Lartigue L, Perkins G, Scott RT, Dixit A, Kushnareva Y, et al. Opa1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, and independent of Bak oligomerization. Mol Cell (2008) 31:557–69. doi:10.1016/j.molcel.2008.07.010
87. Arnoult D, Grodet A, Lee YJ, Estaquier J, Blackstone C. Release of OPA1 during apoptosis participates in the rapid and complete release of cytochrome c and subsequent mitochondrial fragmentation. J Biol Chem (2005) 280:35742–50. doi:10.1074/jbc.M505970200
88. Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell (2004) 15:5001–11. doi:10.1091/mbc.E04-04-0294
89. Merkwirth C, Dargazanli S, Tatsuta T, Geimer S, Löwer B, Wunderlich FT, et al. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev (2008) 22:476–88. doi:10.1101/gad.460708
90. Brooks C, Wei Q, Feng L, Dong G, Tao Y, Mei L, et al. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci U S A (2007) 104:11649–54. doi:10.1073/pnas.0703976104
91. Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R, et al. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell (2010) 142:889–901. doi:10.1016/j.cell.2010.08.017
92. Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature (2006) 443:658–62. doi:10.1038/nature05111
93. Karbowski M, Youle RJ. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ (2003) 10:870–80. doi:10.1038/sj.cdd.4401260
94. Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, et al. The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol Cell (2000) 6:1389–99. doi:10.1016/S1097-2765(00)00136-2
95. Wei MC, Zong W, Cheng E, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science (2001) 292:727–30. doi:10.1126/science.1059108
96. Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science (2004) 305:626–9. doi:10.1126/science.1099320
97. Hoppins S, Edlich F, Cleland MM, Banerjee S, McCaffery JM, Youle RJ, et al. The soluble form of Bax regulates mitochondrial fusion via MFN2 homotypic complexes. Mol Cell (2011) 41:150–60. doi:10.1016/j.molcel.2010.11.030
98. Li H, Chen Y, Jones AF, Sanger RH, Collis LP, Flannery R, et al. Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci U S A (2008) 105:2169–74. doi:10.1073/pnas.0711647105
99. Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol (2012) 14:575–83. doi:10.1038/ncb2488
100. Wang X, Bathina M, Lynch J, Koss B, Calabrese C, Frase S, et al. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev (2013) 27:1351–64. doi:10.1101/gad.215855.113
101. Thomas RL, Roberts DJ, Kubli DA, Lee YJ, Quinsay MN, Owens JB, et al. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev (2013) 27:1365–77. doi:10.1101/gad.215871.113
102. Kelly PN, Strasser A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ (2011) 18:1414–24. doi:10.1038/cdd.2011.17
103. Scarpulla RC, Vega RB, Kelly DP. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab (2012) 23:459–66. doi:10.1016/j.tem.2012.06.006
104. Wenz T. Regulation of mitochondrial biogenesis and PGC-1a under cellular stress. Mitochondrion (2013) 13:134–42. doi:10.1016/j.mito.2013.01.006
105. Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell (2009) 138:628–44. doi:10.1016/j.cell.2009.08.005
106. Dominy JE, Lee YS, Gerhart-Hines Z, Puigserver P. Nutrient-dependent regulation of PGC-1a’s acetylation state and metabolic function through the enzymatic activities of Sirt1/GCN5. Biochim Biophys Acta (2010) 1804:1676–83. doi:10.1016/j.bbapap.2009.11.023
107. Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, et al. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet (1998) 18:231–6. doi:10.1038/ng0398-231
108. Kitami T, Logan DJ, Negri J, Hasaka T, Tolliday NJ, Carpenter AE, et al. A chemical screen probing the relationship between mitochondrial content and cell size. PLoS One (2012) 7:e33755. doi:10.1371/journal.pone.0033755
109. Martínez-Diez M, Santamaría G, Ortega AD, Cuezva JM. Biogenesis and dynamics of mitochondria during the cell cycle: significance of 3′UTRs. PLoS One (2006) 1:e107. doi:10.1371/journal.pone.0000107
110. Posakony JW, England JM, Attardi G. Mitochondrial growth and division during the cell cycle in HeLa cells. J Cell Biol (1977) 74:468–91. doi:10.1083/jcb.74.2.468
111. Rasbach KA, Schnellmann RG. Signaling of mitochondrial biogenesis following oxidant injury. J Biol Chem (2007) 282:2355–62. doi:10.1074/jbc.M608009200
112. Rohas LM, St-Pierre J, Uldry M, Jager S, Handschin C, Spiegelman BM. A fundamental system of cellular energy homeostasis regulated by PGC-1a. Proc Natl Acad Sci U S A (2007) 104:7933–8. doi:10.1073/pnas.0702683104
113. Canto C, Auwerx J. NAD+ as a signaling molecule modulating metabolism. Cold Spring Harb Symp Quant Biol (2011) 76:291–8. doi:10.1101/sqb.2012.76.010439
114. Bell EL, Guarente L. The SirT3 divining rod points to oxidative stress. Mol Cell (2011) 42:561–8. doi:10.1016/j.molcel.2011.05.008
115. Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of c-Myc activity. Cancer Cell (2007) 11:407–20. doi:10.1016/j.ccr.2007.04.001
116. Kim J, Lee JH, Iyer VR. Global identification of MYC target genes reveals its direct role in mitochondrial biogenesis and its E-box usage in vivo. PLoS One (2008) 3:e1798. doi:10.1371/journal.pone.0001798
117. Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O’Donnell KA, et al. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol (2007) 25:6225–34. doi:10.1128/MCB.25.14.6225-6234.2005
118. Finley LW, Carracedo A, Lee JH, Souza AL, Egia A, Zhang J, et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Cancer Cell (2011) 19:416–28. doi:10.1016/j.ccr.2011.02.014
119. Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1a and MITF. Cancer Cell (2013) 23:303–15. doi:10.1016/j.ccr.2013.02.003
120. Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, et al. PGC1a expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell (2013) 23:287–301. doi:10.1016/j.ccr.2012.11.020
121. Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature (2008) 451:1008–12. doi:10.1038/nature06613
122. Sahin E, Colla S, Liesa M, Moslehi J, Müller FL, Guo M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature (2012) 470:359–65. doi:10.1038/nature09787
123. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell (2013) 153:1194–217. doi:10.1016/j.cell.2013.05.039
124. Sahin E, dePinho RA. Axis of ageing: telomeres, p53 and mitochondria. Nat Rev Cancer (2012) 13:397–404. doi:10.1038/nrm3352
125. Hu J, Hwang SS, Liesa M, Gan B, Sahin E, Jaskelioff M, et al. Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell (2012) 148:651–63. doi:10.1016/j.cell.2011.12.028
126. Glick D, Barth S, MacLeod KF. Autophagy: cellular and molecular mechanisms. J Pathol (2010) 221:3–12. doi:10.1002/path.2697
127. Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell (2010) 40:280–93. doi:10.1016/j.molcel.2010.09.023
128. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell (2008) 132:27–42. doi:10.1016/j.cell.2007.12.018
129. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell (2011) 147:728–41. doi:10.1016/j.cell.2011.10.026
130. Rabinowitz JD, White E. Autophagy and metabolism. Science (2010) 330:1344–8. doi:10.1126/science.1193497
131. Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J (2008) 27:433–46. doi:10.1038/sj.emboj.7601963
132. Graef M, Nunnari J. Mitochondria regulate autophagy by conserved signaling pathways. EMBO J (2011) 30:2101–14. doi:10.1038/emboj.2011.104
133. Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, et al. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol (2009) 187:1023–36. doi:10.1083/jcb.200906084
134. Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol (2009) 187:959–66. doi:10.1083/jcb.200906083
135. Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy and mitophagy. Cell Death Differ (2009) 16:939–46. doi:10.1038/cdd.2009.16
136. Narendra D, Walker JE, Youle R. Mitochondrial quality control mediated by PINK1 and Parkin: links to parkinsonism. Cold Spring Harb Perspect Biol (2012) 4:a011338. doi:10.1101/cshperspect.a011338
137. Narendra DP, Jin SM, Tanaka AJ, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol (2010) 8:e1000298. doi:10.1371/journal.pbio.1000298
138. Chen Y, Dorn GW. PINK1-phosphorylated Mitofusin-2 is a Parkin receptor for culling damaged mitochondria. Science (2013) 340:471–5. doi:10.1126/science.1231031
139. Narendra D, Tanaka AJ, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol (2008) 83:795–803. doi:10.1083/jcb.200809125
140. Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A (2010) 107:378–83. doi:10.1073/pnas.0911187107
141. Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, et al. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet (2011) 20:1726–37. doi:10.1093/hmg/ddr048
142. Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell (2011) 147:893–906. doi:10.1016/j.cell.2011.10.018
143. Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol (2010) 12:119–31. doi:10.1038/ncb2012
144. Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP. Disease-causing mutations in Parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J Cell Biol (2010) 189:671–9. doi:10.1083/jcb.201001039
145. Novak I. Mitophagy: a complex mechanism of mitochondrial removal. Antioxid Redox Signal (2012) 17:794–802. doi:10.1089/ars.2011.4407
146. Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy (2010) 6:1090–106. doi:10.4161/auto.6.8.13426
147. Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature (2013) 497:451–7. doi:10.1038/nature12188
148. Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell (2011) 144:79–91. doi:10.1016/j.cell.2010.12.016
149. Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab (2007) 6:280–93. doi:10.1016/j.cmet.2007.08.011
150. Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J (2010) 29:969–80. doi:10.1038/emboj.2009.405
151. Shin JH, Ko HS, Kang HS, Lee YK, Lee YI, Pletinkova O, et al. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell (2011) 144:689–702. doi:10.1016/j.cell.2011.02.010
152. Cesari R, Martin ES, Calin GA, Pentimalli F, Bichi R, McAdams H, et al. Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25-q27. Proc Natl Acad Sci U S A (2003) 100:5956–61. doi:10.1073/pnas.0931262100
153. Fujiwara M, Marusawa H, Wang HQ, Iwai A, Ikeuchi K, Imai Y, et al. Parkin as a tumor suppressor gene for hepatocellular carcinoma. Oncogene (2008) 27:6002–11. doi:10.1038/onc.2008.199
154. Kim KY, Stevens MV, Akter MH, Rusk SE, Huang RJ, Cohen A, et al. Parkin is a lipid-responsive regulator of fat uptake in mice and mutant human cells. J Clin Invest (2011) 121:3701–12. doi:10.1172/JCI44736
155. Zhang C, Lin M, Wu R, Wang X, Yang BG, Levine AJ, et al. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc Natl Acad Sci U S A (2011) 108:16259–64. doi:10.1073/pnas.1113884108
156. Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzumbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev (2011) 25:460–70. doi:10.1101/gad.2016311
157. Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev (2013) 27:1447–61. doi:10.1101/gad.219642.113
158. Chinnadurai G, Vijayalingam S, Gibson SB. BNIP3 subfamily BH3-only proteins: mitochondrial stress sensors in normal and pathological functions. Oncogene (2009) 27:S114–27. doi:10.1038/onc.2009.49
159. Hardwick JM, Youle RJ. Snapshot: Bcl2 proteins. Cell (2009) 138:404.e1–404.e2. doi:10.1016/j.cell.2009.07.003
160. Diwan A, Koesters AG, Odley AM, Pushkaran S, Baines CP, Spike BT, et al. Unrestrained erythroblast development in Nix−/− mice reveals a mechanism for apoptotic modulation of erythropoiesis. Proc Natl Acad Sci USA (2007) 104:6794–9. doi:10.1073/pnas.0610666104
161. Glick D, Zhang W, Beaton M, Marsboom G, Gruber M, Simon MC, et al. BNip3 regulates mitochondrial function and lipid metabolism in the liver. Mol Cell Biol (2012) 32:2570–84. doi:10.1128/MCB.00167-12
162. Graham RM, Frazier DP, Thompson JW, Haliko S, Li H, Wasserlauf BJ, et al. A unique pathway of cardiac myocyte death caused by hypoxia-acidosis. J Exp Biol (2004) 207:3189–200. doi:10.1242/jeb.01109
163. Tracy K, MacLeod KF. Regulation of mitochondrial integrity, autophagy and cell survival by BNIP3. Autophagy (2007) 3:616–9.
164. Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is a HIF-1 dependent adaptive metabolic response to hypoxia. J Biol Chem (2008) 283:10892–903. doi:10.1074/jbc.M800102200
165. Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with BNip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem (2012) 287:19094–104. doi:10.1074/jbc.M111.322933
166. Schwarten M, Mohrluder J, Ma P, Stoldt M, Thielman Y, Stangler T, et al. Nix binds to GABARAP: a possible crosstalk between apoptosis and autophagy. Autophagy (2009) 5:690–8. doi:10.4161/auto.5.5.8494
167. Kanki T, Wang KKW, Caio Y, Baba M, Klionsky DJ. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell (2009) 17:98–109. doi:10.1016/j.devcel.2009.06.014
168. Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondrial-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell (2009) 17:87–97. doi:10.1016/j.devcel.2009.06.013
169. Sandoval H, Thiagarajan P, Dasgupta SK, Scumacker A, Prchal JT, Chen M, et al. Essential role for Nix in autophagic maturation of red cells. Nature (2008) 454:232–5. doi:10.1038/nature07006
170. Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA (2007) 104:19500–5. doi:10.1073/pnas.0708818104
171. Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab (2007) 6:458–71. doi:10.1016/j.cmet.2007.11.001
172. Ray R, Chen G, Vande Velde C, Cizeau J, Park JH, Reed JC, et al. BNIP3 heterodimerizes with Bcl-2/Bcl-XL and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J Biol Chem (2000) 275:1439–48. doi:10.1074/jbc.275.2.1439
173. Tracy K, Dibling BC, Spike BT, Knabb JR, Schumacker P, MacLeod KF. BNIP3 is a RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol (2007) 27:6229–42. doi:10.1128/MCB.02246-06
174. Chaanine AH, Gordon RE, Kohlbrenner E, Benard L, Jeong D, Hajjar RJ. Potential role of BNIP3 in cardiac re-modeling, myocardial stiffness and endoplasmic-reticulum-mitochondrial calcium homeostasis in diastolic and systolic heart failure. Circ Heart Fail (2013) 6:572–83. doi:10.1161/CIRCHEARTFAILURE.112.000200
175. Landes T, Emorine LJ, Courilleau D, Rojo M, Belenguer P, Arnaune-Pelloquin L. The BH3-only BNip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. EMBO Rep (2010) 11:459–65. doi:10.1038/embor.2010.50
176. Quinsay MN, Lee YS, Rikka S, Sayen MR, Molkentin JD, Gottlieb RA, et al. BNip3 mediates permeabilization of mitochondria and release of cytochrome c via a novel mechanism. J Mol Cell Cardiol (2009) 48:1146–56. doi:10.1016/j.yjmcc.2009.12.004
177. Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, et al. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem (2010) 285:27879–90. doi:10.1074/jbc.M110.119537
178. Li Y, Wang Y, Kim E, Beemiller P, Wang CY, Swanson J, et al. Bnip3 mediates the hypoxia-induced inhibition on mTOR by interacting with Rheb. J Biol Chem (2007) 282:35803–13. doi:10.1074/jbc.M705231200
179. Melser S, Chatelain EH, Lavie J, Mahfouf W, Jose C, Obre E, et al. Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab (2013) 17:719–30. doi:10.1016/j.cmet.2013.03.014
180. Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci U S A (2000) 97:9082–7. doi:10.1073/pnas.97.16.9082
181. Guo K, Searfoss G, Krolikowski D, Pagnoni M, Franks C, Clark K, et al. Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ (2001) 8:367–76. doi:10.1038/sj.cdd.4400810
182. Dayan F, Roux D, Brahimi-Horn C, Pouyssegur J, Mazure NM. The oxygen sensing factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bi-functional transcriptional character of hypoxia-inducible factor-1a. Cancer Res (2006) 66:3688–98. doi:10.1158/0008-5472.CAN-05-4564
183. Kasper LH, Boussouar F, Boyd K, Xu W, Biesen M, Rehg J, et al. Two transcriptional mechanisms cooperate for the bulk of HIF-1-responsive gene expression. EMBO J (2005) 24:3846–58. doi:10.1038/sj.emboj.7600846
184. Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature (2006) 441:437–43. doi:10.1038/nature04871
185. Shaw J, Yurkova N, Zhang T, Gang H, Aguilar F, Weidman D, et al. Antagonism of E2F-1 regulated Bnip3 transcription by NF-kappaB is essential for basal cell survival. Proc Natl Acad Sci U S A (2008) 105:20734–9. doi:10.1073/pnas.0807735105
186. Kalas W, Swiderek E, Rapak A, Kopij M, Rak JW, Strzadala L. H-ras up-regulates expression of BNIP3. Anticancer Res (2011) 31:2869–75.
187. Wu SY, Lan SH, Cheng DE, Chen WK, Shen CH, Lee YR, et al. Ras-related tumorigenesis is suppressed by BNIP3-mediated autophagy through inhibition of cell proliferation. Neoplasia (2011) 13:1171–82.
188. Feng X, Liu X, Zhang W, Xiao W. p53 directly suppresses BNIP3 expression to protect against hypoxia-induced cell death. EMBO J (2011) 30:3397–415. doi:10.1038/emboj.2011.248
189. Fei P, Wang W, Kim SH, Wang S, Burns TF, Sax JK, et al. Bnip3L is induced by p53 under hypoxia, and its knockdown promotes tumor growth. Cancer Cell (2005) 6:597–609. doi:10.1016/j.ccr.2004.10.012
190. Koop EA, van Laar T, van Wichen DF, de Weger RA, van der Wall E, van Diest PJ. Expression of BNIP3 in invasive breast cancer: correlations with the hypoxic response and clinicopathological features. BMC Cancer (2009) 9:175. doi:10.1186/1471-2407-9-175
191. Okami J, Simeone DM, Logsdon CD. Silencing of the hypoxia-inducible cell death protein BNIP3 in pancreatic cancer. Cancer Res (2004) 64:5338–46. doi:10.1158/0008-5472.CAN-04-0089
192. Sowter HM, Ferguson M, Pym C, Watson P, Fox SB, Han C, et al. Expression of the cell death genes BNip3 and Nix in ductal carcinoma in situ of the breast; correlation of BNip3 levels with necrosis and grade. J Path (2003) 201:573–80. doi:10.1002/path.1486
193. Tan EY, Campo L, Han C, Turley H, Pezzella F, Gatter KC, et al. BNIP3 as a progression marker in primary human breast cancer; opposing functions in in situ versus invasive cancer. Clin Cancer Res (2007) 13:467–74. doi:10.1158/1078-0432.CCR-06-1466
194. Abe T, Toyota M, Suzuki H, Murai M, Akino K, Ueno M, et al. Upregulation of BNIP3 by 5-aza-2’-deoxycytidine sensitizes pancreatic cancer cells to hypoxia-mediated cell death. J Gastroenterol (2005) 40:504–10. doi:10.1007/s00535-005-1576-1
195. Bacon AL, Fox SB, Turley H, Harris AL. Selective silencing of the hypoxia-inducible factor 1 target gene BNIP3 by histone deacetylation and methylation in colorectal cancer. Oncogene (2006) 26:132–41. doi:10.1038/sj.onc.1209761
196. Calvisi DF, Ladu S, Gorden A, Farina M, Lee JS, Conner E, et al. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J Clin Invest (2007) 117:2713–22. doi:10.1172/JCI31457
197. Castro M, Grau L, Puerta P, Gimenez L, Venditti J, Quadrelli S, et al. Multiplexed methylation profiles of tumor suppressor genes and clinical outcome in lung cancer. J Transl Med (2010) 8:1–11. doi:10.1186/1479-5876-8-86
198. Erkan M, Kleef J, Esposito I, Giese T, Ketterer K, Buchler MW, et al. Loss of BNIP3 expression is a late event in pancreatic cancer contributing to chemoresistance and worsened prognosis. Oncogene (2005) 24:4421–32. doi:10.1038/sj.onc.1208642
199. Murai M, Toyota M, Satoh A, Suzuki H, Akino K, Mita H, et al. Aberrant methylation associated with silencing BNIP3 expression in haematopoietic tumours. Br J Cancer (2005) 92:1165–72. doi:10.1038/sj.bjc.6602422
200. Murai M, Toyota M, Suzuki H, Satoh A, Sasaki Y, Akino K, et al. Aberrant methylation and silencing of the BNIP3 gene in colorectal and gastric cancer. Clin Cancer Res (2005) 11:1021–7.
201. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature (2010) 463:899–905. doi:10.1038/nature08822
202. Chen X, Gong J, Zeng H, Chen N, Huang R, Huang Y, et al. MicroRNA145 targets BNIP3 and suppresses prostate cancer progression. Cancer Res (2010) 70:2728–38. doi:10.1158/0008-5472.CAN-09-3718
203. Manka D, Spicer Z, Millhorn DE. Bcl-2/Adenovirus E1B 19 kDa interacting protein-3 knockdown enables growth of breast cancer metastases in the lung, liver and bone. Cancer Res (2005) 65:11689–93. doi:10.1158/0008-5472.CAN-05-3091
204. Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell (2010) 141:656–67. doi:10.1016/j.cell.2010.04.009
205. Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, et al. Autophagosomes form at ER-mitochondria contact sites. Nature (2013) 495:389–93. doi:10.1038/nature11910
206. Diwan A, Matkovich SJ, Yuan Q, Zhao W, Yatani A, Brown JH, et al. Endoplasmic reticulum-mitochondria crosstalk in NIX-mediated murine cell death. J Clin Invest (2009) 119:203–12. doi:10.1172/JCI36445
207. Boya P, González-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol (2005) 25:1025–40. doi:10.1128/MCB.25.3.1025-1040.2005
208. Pattingre S, Tassa A, Qu X, Garuti R, Linag XH, Mizushima N, et al. Bcl-2 anti-apoptotic proteins inhibit Beclin 1-dependent autophagy. Cell (2005) 122:927–39. doi:10.1016/j.cell.2005.07.002
209. Wei G, Pattingre S, Sinha S, Bassik MC, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell (2008) 30:678–88. doi:10.1016/j.molcel.2008.06.001
210. Rubinstein AD, Eisenstein M, Ber Y, Bialik S, Kimchi A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol Cell (2011) 44:698–709. doi:10.1016/j.molcel.2011.10.014
211. Yousefi S, Perozzo R, Schmid I, Zieiecki A, Schaffner T, Scapozza L, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol (2006) 8:1124–32. doi:10.1038/ncb1482
212. Lee IH, Kawai Y, Fergusson MM, Rovira II, Bishop AJ, Motoyama N, et al. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science (2012) 336:225–8. doi:10.1126/science.1218395
213. Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol (2010) 190:881–92. doi:10.1083/jcb.200911078
214. Russo R, Berliocchi L, Adornetto A, Varano GP, Cavaliere F, Nucci C, et al. Calpain-mediated cleavage of Beclin-1 and autophagy deregulation following retinal ischemic injury in vivo. Cell Death Dis (2011) 2:e144. doi:10.1038/cddis.2011.29
215. Betin VM, Lane JD. Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J Cell Sci (2009) 122:2554–66. doi:10.1242/jcs.046250
216. Wirawan E, Vande Walle L, Kersse K, Cornelis S, Claerhout S, Vanoverberghe I, et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis (2010) 1:e18. doi:10.1038/cddis.2009.16
217. Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, et al. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of BNip3 restrains postinfarction remodeling in mice. J Clin Invest (2007) 117:2825–33. doi:10.1172/JCI32490
218. Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol (2012) 14:177–85. doi:10.1038/ncb2422
219. Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science (2005) 309:481–4. doi:10.1126/science.1112125
220. Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature (2004) 429:417–23. doi:10.1038/nature02517
221. Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA, et al. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat Genet (2008) 40:392–4. doi:10.1038/ng.95
222. Ahlqvist KJ, Hämäläinen RH, Yatsuga S, Uutela M, Terzioglu M, Götz A, et al. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab (2012) 15:100–9. doi:10.1016/j.cmet.2011.11.012
223. Lee HY, Choi CS, Birkenfeld AL, Alves TC, Jornayvaz FR, Jurczak MJ, et al. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab (2010) 12:668–74. doi:10.1016/j.cmet.2010.11.004
224. Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science (2005) 308:1909–11. doi:10.1126/science.1106653
225. Veatch JR, McMurray MA, Nelson ZW, Gottschling DE. Mitochondrial dysfunction leads to nuclear genome instability via an iron-sulfur cluster defect. Cell (2009) 137:1247–58. doi:10.1016/j.cell.2009.04.014
226. Kaelin WGJ, McKnight SL. Influence of metabolism on epigenetics and disease. Cell (2013) 153:56–69. doi:10.1016/j.cell.2013.03.004
227. Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell Metab (2012) 16:9–17. doi:10.1016/j.cmet.2012.06.001
228. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science (2009) 324:1076–80. doi:10.1126/science.1164097
229. Losman JA, Kaelin WGJ. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev (2013) 27:836–52. doi:10.1101/gad.217406.113
230. Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med (2009) 361:1058–66. doi:10.1056/NEJMoa0903840
231. Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell (2010) 17:225–34. doi:10.1016/j.ccr.2010.01.020
232. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med (2009) 360:765–73. doi:10.1056/NEJMoa0808710
233. Sciacovelli M, Guzzo G, Morello V, Frezza C, Zheng L, Nannini N, et al. The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab (2013) 17:988–99. doi:10.1016/j.cmet.2013.04.019
234. Koivunen P, Hirsilä M, Remes AM, Hassinen IE, Kivirikko KI, Myllyharju J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J Biol Chem (2007) 282:4524–32. doi:10.1074/jbc.M610415200
235. Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell (2005) 7:77–85. doi:10.1016/j.ccr.2004.11.022
236. Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev (2012) 26:1326–38. doi:10.1101/gad.191056.112
237. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell (2011) 19:17–30. doi:10.1016/j.ccr.2010.12.014
238. Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science (2009) 324:261–5. doi:10.1126/science.1170944
239. Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio II, et al. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev (2012) 26:2038–49. doi:10.1101/gad.198200.112
240. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell (2010) 18:553–67. doi:10.1016/j.ccr.2010.11.015
241. Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature (2012) 483:479–83. doi:10.1038/nature10866
242. Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brüstle A, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature (2012) 488:656–9. doi:10.1038/nature11323
243. Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature (2012) 483:474–8. doi:10.1038/nature10860
244. Losman JA, Looper RE, Koivunen P, Lee SH, Schneider RK, McMahon C, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science (2013) 339:1621–5. doi:10.1126/science.1231677
245. Sullivan LB, Garcia-Martinez E, Nguyen H, Mullen AR, Dufour E, Sudarshan S, et al. The Proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell (2013) 51:236–48. doi:10.1016/j.molcel.2013.05.003
246. Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell (2011) 20:524–37. doi:10.1016/j.ccr.2011.09.006
247. Ooi A, Wong JC, Petillo D, Roossien D, Perrier-Trudova V, Whitten D, et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell (2011) 20:511–23. doi:10.1016/j.ccr.2011.08.024
248. Ternette N, Yang M, Laroyia M, Kitagawa M, O’Flaherty L, Wolhulter K, et al. Inhibition of mitochondrial aconitase by succination in fumarate hydratase deficiency. Cell Rep (2013) 3:689–700. doi:10.1016/j.celrep.2013.02.013
249. Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature (2011) 481:380–4. doi:10.1038/nature10602
250. Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature (2011) 481:385–8. doi:10.1038/nature10642
251. Frezza C, Zheng L, Folger O, Rajagopalan KN, MacKenzie ED, Jerby L, et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature (2011) 477:225–8. doi:10.1038/nature10363
252. Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell (2011) 146:1016–28. doi:10.1016/j.cell.2011.08.008
253. Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signaling. Nat Rev Mol Cell Biol (2012) 13:566–78. doi:10.1038/nrm3412
254. Rowland AA, Voeltz GK. Endoplasmic reticulum mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol (2012) 13:607–15. doi:10.1038/nrm3440
255. Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski MR, Cavagna D, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol (2006) 175:901–11. doi:10.1083/jcb.200608073
256. Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, et al. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J Cell Biol (2002) 159:613–24. doi:10.1083/jcb.200205091
257. White C, Li CY, Yang J, Petrenko NB, Madesh M, Thompson CB, et al. The endoplasmic reticulum gateway to apoptosis by Bcl-XL modulation of the InsP3R. Nat Cell Biol (2005) 7:1021–8. doi:10.1038/ncb1302
258. McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev (1990) 70:391–425.
259. Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, et al. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A (2008) 105:20728–33. doi:10.1073/pnas.0808953105
260. Storr SJ, Carragher NO, Frame MC, Parr T, Martin SG. The calpain system and cancer. Nat Rev Cancer (2011) 11:364–74. doi:10.1038/nrc3050
261. Racioppi L, Means AR. Calcium/calmodulin-dependent protein kinase kinase 2: roles in signaling and pathophysiology. J Biol Chem (2012) 287:31658–65. doi:10.1074/jbc.R112.356485
262. Prevarskaya N, Skryma R, Shuba Y. Calcium in tumour metastasis: new roles for known actors. Nat Rev Cancer (2011) 11:609–18. doi:10.1038/nrc3105
263. Amuthan G, Biswas G, Zhang SY, Klein-Szanto A, Vijayasarathy C, Avadhani NG. Mitochondria-to-nucleus stress signaling induces phenotypic changes, tumor progression and cell invasion. EMBO J (2001) 20:1910–20. doi:10.1093/emboj/20.8.1910
264. Amuthan G, Biswas G, Ananadatheerthavarada HK, Vijayasarathy C, Shephard HM, Avadhani NG. Mitochondrial stress-induced calcium signaling, phenotypic changes and invasive behavior in human lung carcinoma A549 cells. Oncogene (2002) 21:7839–49. doi:10.1038/sj.onc.1205983
265. Biswas G, Anandatheerthavarada HK, Zaidi M, Avadhani NG. Mitochondria to nucleus stress signaling: a distinctive mechanism of NFkappaB/Rel activation through calcineurin-mediated inactivation of IkappaBbeta. J Cell Biol (2003) 161:507–19. doi:10.1083/jcb.200211104
266. Biswas G, Tang W, Sondheimer N, Guha M, Bansal S, Avadhani NG. A distinctive physiological role for IkappaBbeta in the propagation of mitochondrial respiratory stress signaling. J Biol Chem (2008) 283:12586–94. doi:10.1074/jbc.M710481200
267. Karin M, Greten FR. NF-kB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol (2005) 5:749–59. doi:10.1038/nri1703
268. Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol (2009) 1:1–14. doi:10.1101/cshperspect.a000034
269. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. NF-kB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1a. Nature (2008) 453:807–11. doi:10.1038/nature06905
270. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants and aging. Cell (2005) 120:483–95. doi:10.1016/j.cell.2005.02.001
271. Tormos KV, Anso E, Hamanaka RB, Eisenbart J, Joseph J, Kalyanaraman B, et al. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab (2011) 14:537–44. doi:10.1016/j.cmet.2011.08.007
272. Weinberg F, Hamanaka RB, Wheaton WW, Weinberg S, Joseph JW, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A (2010) 107:8788–93. doi:10.1073/pnas.1003428107
273. Gao P, Zhang H, Dinavahi R, Li F, Xiang Y, Raman V, et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell (2007) 12:230–8. doi:10.1016/j.ccr.2007.08.004
274. Ma Q, Cavallin LE, Yan B, Zhu S, Duran EM, Wang H, et al. Antitumorigenesis of antioxidants in a transgenic Rac1 model of Kaposi’s sarcoma. Proc Natl Acad Sci U S A (2009) 106:8683–8. doi:10.1073/pnas.0812688106
275. Tonks NK. Redox redux: revisiting PTPs and the control of cell signaling. Cell (2005) 121:667–70. doi:10.1016/j.cell.2005.05.016
276. Hampton MB, Fadeel B, Orrenius S. Redox regulation of the caspases during apoptosis. Ann N Y Acad Sci (1998) 854:328–35. doi:10.1111/j.1749-6632.1998.tb09913.x
277. Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem (2002) 277:20336–42. doi:10.1074/jbc.M111899200
278. Buhrman G, Parker B, Sohn J, Rudolph J, Mattos C. Structural mechanism of oxidative regulation of the phosphatase Cdc25B via an intramolecular disulfide bond. Biochemistry (2005) 44:5307–16. doi:10.1021/bi058011y
279. Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell (2005) 120:649–61. doi:10.1016/j.cell.2004.12.041
280. Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, et al. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab (2005) 1:409–14. doi:10.1016/j.cmet.2005.05.002
281. Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem (2000) 275:25130–8. doi:10.1074/jbc.M001914200
282. Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab (2005) 1:401–8. doi:10.1016/j.cmet.2005.05.001
283. Guzy RD, Schumacker PT. Oxygen sensing by mitochondria at Complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol (2006) 91:807–19.
284. Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT, et al. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab (2005) 1:393–9. doi:10.1016/j.cmet.2005.05.003
285. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1 mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab (2006) 3:177–85. doi:10.1016/j.cmet.2006.02.002
286. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab (2006) 3:187–97. doi:10.1016/j.cmet.2006.01.012
287. Fukuda R, Zhang H, Kim J, Shimoda LA, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell (2007) 129:111–22. doi:10.1016/j.cell.2007.01.047
288. Tello D, Balsa E, Acosta-Iborra B, Fuertes-Yebra E, Elorza A, Ordóñez Á, et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1α decreases oxygen consumption by inhibiting Complex I activity. Cell Metab (2011) 14:768–79. doi:10.1016/j.cmet.2011.10.008
289. Choksi S, Lin YS, Pobezinskaya Y, Chen L, Park C, Morgan M, et al. A HIF-1 target, ATIA, protects cells from apoptosis by modulating the mitochondrial thioredoxin, TRX1. Mol Cell (2011) 42:597–609. doi:10.1016/j.molcel.2011.03.030
290. Horak P, Crawford AR, Vadysirisack DD, Nash ZM, DEeYoung MP, Sgroi D, et al. Negative feedback control of HIF-1 through REDD1-regulated ROS suppresses tumorigenesis. Proc Natl Acad Sci U S A (2010) 107:4675–80. doi:10.1073/pnas.0907705107
291. Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer (2003) 3:401–10. doi:10.1038/nrc1093
292. Erler JT, Bennewith KL, Nicolau M, Dornhofer N, Kong C, Le QT, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature (2006) 440:1222–6. doi:10.1038/nature04695
293. Yang MH, Wu KJ. TWIST activation by hypoxia inducible factor-1 (HIF-1). Cell Cycle (2008) 7:2090–6. doi:10.4161/cc.7.14.6324
294. Dewhirst MW, Cao Y, Moeller BJ. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer (2008) 8:425–37. doi:10.1038/nrc2397
295. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell (2012) 148:399–408. doi:10.1016/j.cell.2012.01.021
296. Keith B, Johnson RS, Simon MC. HIF-1a and HIF-2a: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer (2012) 12:9–22. doi:10.1038/nrc3183
297. Fu Z, Tindall DJ. FOXOs, cancer and regulation of apoptosis. Oncogene (2008) 27:2312–9. doi:10.1038/onc.2008.24
298. Kops GJPL, Dansen TB, Polderman E, Saarloos I, Wirtz KWA, Coffer PJ, et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature (2002) 419:316–21. doi:10.1038/nature01036
299. Owusu-Ansah E, Yavari A, Mandal S, Banerjee U. Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nat Genet (2008) 40:356–61. doi:10.1038/ng.2007.50
300. Ferber EC, Peck B, Delpuech O, Bell GP, East P, Schulze A. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ (2012) 19:968–79. doi:10.1038/cdd.2011.179
301. Jensen KS, Binderup T, Jensen KT, Therkelsen I, Borup R, Nilsson E, et al. FoxO3A promotes metabolic adaptation to hypoxia by antagonizing Myc function. EMBO J (2011) 30:4554–70. doi:10.1038/emboj.2011.323
302. Peck B, Ferber EC, Schulze A. Antagonism between FOXO and MYC regulates cellular powerhouse. Front Oncol (2013) 3:96. doi:10.3389/fonc.2013.00096
303. Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci (2009) 34:176–88. doi:10.1016/j.tibs.2008.12.008
304. Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun (1997) 236:313–22. doi:10.1006/bbrc.1997.6943
305. Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol (2010) 12:213–23. doi:10.1038/ncb2021
306. Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, et al. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol (2010) 30:3275–85. doi:10.1128/MCB.00248-10
307. Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell (2012) 22:66–79. doi:10.1016/j.ccr.2012.05.016
308. DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature (2011) 475:106–9. doi:10.1038/nature10189
309. Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci U S A (2008) 105:13568–73. doi:10.1073/pnas.0806268105
310. Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med (2006) 3:e420. doi:10.1371/journal.pmed.0030420
311. Solis LM, Behrens C, Dong W, Suraokar M, Ozburn NC, Moran CA, et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin Cancer Res (2010) 16:3743–53. doi:10.1158/1078-0432.CCR-09-3352
312. Hardie DG. AMP-activated protein kinase – an energy sensor that regulates all aspects of cell function. Genes Dev (2011) 25:1895–908. doi:10.1101/gad.17420111
313. Mihaylova MM, Shaw RJ. The AMPK signaling pathway coordinates cell growth, autophagy and metabolism. Nat Rev Cell Biol (2011) 13:1016–23. doi:10.1038/ncb2329
314. Jäger S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A (2007) 104:12017–22. doi:10.1073/pnas.0705070104
315. Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, et al. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A (2002) 99:15983–7. doi:10.1073/pnas.252625599
316. Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science (2011) 331:456–61. doi:10.1126/science.1196371
317. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol (2011) 13:132–41. doi:10.1038/ncb2152
318. Sack MN, Finkel T. Mitochondrial metabolism, sirtuins and aging. Cold Spring Harb Perspect Biol (2012) 4:1–10. doi:10.1101/cshperspect.a013102
319. Verdin E, Hirschey MD, Finley LWS, Haigis MC. Sirtuin regulation of mitochondria – energy production, apoptosis, and signaling. Trends Biochem Sci (2010) 35:669–75. doi:10.1016/j.tibs.2010.07.003
320. Canto C, Auwerx J. Interference between PARPs and SIRT1: a novel approach to healthy ageing? Aging (2011) 3:543–7.
321. Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A (2008) 105:3374–9. doi:10.1073/pnas.0712145105
322. Alto NM, Soderling J, Scott JD. Rab32 is an A-kinase anchoring protein and participates in mitochondrial dynamics. J Cell Sci (2002) 158:659–68. doi:10.1083/jcb.200204081
323. Bera AK, Ghosh S, Das S. Mitochondrial VDAC can be phosphorylated by cyclic AMP-dependent protein kinase. Biochem Biophys Res Commun (1995) 209:213–7. doi:10.1006/bbrc.1995.1491
324. Das S, Wong R, Rajapakse N, Murphy E, Steenbergen C. Glycogen synthase kinase 3 inhibition slows mitochondrial adenine nucleotide transport and regulates voltage-dependent anion channel phosphorylation. Circ Res (2008) 103:983–91. doi:10.1161/CIRCRESAHA.108.178970
325. Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, et al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest (2004) 113:1535–49. doi:10.1172/JCI200419906
326. Kumar S, Bharti A, Mishra NC, Raina D, Kharbanda S, Saxena S, et al. Targeting of the c-Abl tyrosine kinase to mitochondria in the necrotic cell death response to oxidative stress. J Biol Chem (2001) 276:17281–5. doi:10.1074/jbc.M101414200
327. Majumder PK, Mishra NC, Sun X, Bharti A, Kharbanda S, Saxena S, et al. Targeting of protein kinase C delta to mitochondria in the oxidative stress response. Cell Growth Differ (2001) 12:465–70.
328. Robey RB, Hay N. Is Akt the “Warburg kinase”? – Akt energy metabolism interactions and oncogenesis. Semin Cancer Biol (2009) 19:25–31. doi:10.1016/j.semcancer.2008.11.010
329. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell (2007) 129:1261–74. doi:10.1016/j.cell.2007.06.009
330. Patra KC, Wang QJ, Bhaskar PT, Miller L, Wang Z, Chandel NS, et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell (2013) 24:213–28. doi:10.1016/j.ccr.2013.06.014
331. Maurer U, Chavret C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell (2006) 21:749–60. doi:10.1016/j.molcel.2006.02.009
332. Sheldon KL, Maldonado EN, Lemasters JJ, Rostovtseva TK, Bezrukov SM. Phosphorylation of voltage-dependent anion channel by serine/threonine kinases governs its interaction with tubulin. PLoS One (2011) 6:e25539. doi:10.1371/journal.pone.0025539
333. Martel C, Allouche M, Esposti DD, Fanelli E, Boursier C, Henry C, et al. Glycogen synthase kinase 3-mediated voltage-dependent anion channel phosphorylation controls outer mitochondrial membrane permeability during lipid accumulation. Hepatology (2013) 57:93–102. doi:10.1002/hep.25967
334. Chou CH, Lin CC, Yang MC, Wei CC, Liao HD, Lin RC, et al. GSK3beta-mediated Drp1 phosphorylation induced elongated mitochondrial morphology against oxidative stress. PLoS One (2012) 7:e49112. doi:10.1371/journal.pone.0049112
335. Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3 beta regulates cyclin D1 proteolysis and subcellular localisation. Genes Dev (1998) 12:3499–511.
336. Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3B to the APC-b-catenin complex and regulation of complex assembly. Science (1996) 272:1023–6. doi:10.1126/science.272.5264.1023
337. Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev (2000) 14:1–14.
338. Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol (2004) 6:308–18. doi:10.1038/ncb1110
339. Ilouz R, Bubis J, Wu J, Yim YY, Deal MS, Kornev AP, et al. Localization and quaternary structure of the PKA RIβ holoenzyme. Proc Natl Acad Sci U S A (2012) 109:12443–8. doi:10.1073/pnas.1209538109
340. Means CK, Lygren B, Langeberg LK, Jain A, Dixon RE, Vega AL, et al. An entirely specific type I A-kinase anchoring protein that can sequester two molecules of protein kinase A at mitochondria. Proc Natl Acad Sci U S A (2011) 108:1227–35. doi:10.1073/pnas.1107182108
341. Carlucci A, Adornetto A, Scorziello A, Viggiano D, Foca M, Cuomo O, et al. Proteolysis of AKAP121 regulates mitochondrial activity during cellular hypoxia and brain ischaemia. EMBO J (2008) 27:1073–84. doi:10.1038/emboj.2008.33
342. Livigni A, Scorziello A, Agnese S, Adornetto A, Carlucci A, Garbi C, et al. Mitochondrial AKAP121 links cAMP and src signaling to oxidative metabolism. Mol Biol Cell (2006) 17:263–71. doi:10.1091/mbc.E05-09-0827
343. Miyazaki T, Neff L, Tanaka S, Horne WC, Baron R. Regulation of cytochrome c oxidase activity by c-Src in osteoclasts. J Cell Biol (2003) 160:709–18. doi:10.1083/jcb.200209098
344. Tibaldi E, Brunati AM, Massimino ML, Stringaro A, Colone M, Agostinelli E, et al. Src-Tyrosine kinases are major agents in mitochondrial tyrosine phosphorylation. J Cell Biochem (2008) 104:840–9. doi:10.1002/jcb.21670
345. Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature (2012) 485:661–5. doi:10.1038/nature11066
346. Valentin-Vega YA, MacLean KH, Tait-Mulder J, Milasta S, Steeves M, Dorsey FC, et al. Mitochondrial dysfunction in ataxia-telangiectasia. Blood (2012) 119:1490–500. doi:10.1182/blood-2011-08-373639
347. Compton S, Kim C, Griner NB, Potluri P, Scheffler IE, Sen S, et al. Mitochondrial dysfunction impairs tumor suppressor p53 expression/function. J Biol Chem (2011) 286:20297–312. doi:10.1074/jbc.M110.163063
348. Muller PAJ, Vousden KH. p53 mutations in cancer. Nat Cell Biol (2013) 15:2–8. doi:10.1038/ncb2641
349. Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell (2009) 137:413–31. doi:10.1016/j.cell.2009.04.037
350. Bieging KT, Attardi LD. Deconstructing p53 transcriptional networks in tumor suppression. Trends Cell Biol (2012) 22:97–106. doi:10.1016/j.tcb.2011.10.006
351. Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer (2002) 2:594–604. doi:10.1038/nrc864
352. Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature (1995) 377:552–7. doi:10.1038/377552a0
353. Marchenko ND, Zaika A, Moll UM. Death signal-induced localization of p53 protein to mitochondria. J Biol Chem (2000) 275:16202–12. doi:10.1074/jbc.275.21.16202
354. Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell (2003) 11:577–90. doi:10.1016/S1097-2765(03)00050-9
355. Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell (2006) 126:121–34. doi:10.1016/j.cell.2006.05.034
356. Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev (2013) 27:1016–31. doi:10.1101/gad.212282.112
358. Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol (2008) 10:676–87. doi:10.1038/ncb1730
359. Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, et al. p53 regulates mitochondrial respiration. Science (2006) 312:1650–3. doi:10.1126/science.1126863
360. Jiang P, Du W, Mancuso A, Wellen KE, Yang X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature (2013) 493:689–93. doi:10.1038/nature11776
361. Bensaad K, Tsuruta A, Selak MA, Vidal MNC, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell (2006) 126:107–20. doi:10.1016/j.cell.2006.05.036
362. Jones RG, Plas D, Kubek S, Buzzai M, Mu J, Xu Y, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell (2005) 18:283–93. doi:10.1016/j.molcel.2005.03.027
363. Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol (2001) 3:973–82. doi:10.1038/35060032
364. Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD, Green DR. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science (2005) 309:1732–5. doi:10.1126/science.1114297
365. Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell (2012) 149:1536–48. doi:10.1016/j.cell.2012.05.014
366. Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell (2002) 2:103–12. doi:10.1016/S1535-6108(02)00102-2
367. Weinberg RA. The retinoblastoma protein and cell cycle control. Cell (1995) 81:323–30. doi:10.1016/0092-8674(95)90385-2
368. MacLeod KF. The RB tumor suppressor: a gatekeeper to hormone independence in prostate cancer? J Clin Invest (2010) 120:4179–82. doi:10.1172/JCI45406
369. Sharma A, Yeow WS, Ertel A, Coleman I, Thangavel C, Cornstock CES, et al. Novel functions of the retinoblastoma tumor suppressor in controlling androgen signaling and prostate cancer progression. J Clin Invest (2010) 120:4478–92. doi:10.1172/JCI44239
370. Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer (2008) 8:1–12. doi:10.1038/nrc2399
371. MacLeod KF. The role of the RB tumor suppressor pathway in oxidative stress responses in the hematopoietic system. Nat Rev Cancer (2008) 8:769–81. doi:10.1038/nrc2504
372. Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell (2003) 113:703–16. doi:10.1016/S0092-8674(03)00401-X
373. Sage J, Miller AL, Perez-Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature (2003) 424:223–8. doi:10.1038/nature01764
374. Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer (2009) 9:785–97. doi:10.1038/nrc2696
375. Chong JL, Wenzel PL, Saenz-Robles MT, Nair V, Ferrey A, Hagan JP, et al. E2f1-3 switch from activators in progenitor cells to repressors in differentiating cells. Nature (2009) 462:930–4. doi:10.1038/nature08677
376. Dirlam A, Spike BT, MacLeod KF. De-regulated E2f-2 activity underlies cell cycle and maturation defects in Rb null erythroblasts. Mol Cell Biol (2007) 27:8713–28. doi:10.1128/MCB.01118-07
377. Fajas L, Egler V, Reiter R, Miard S, Lefebvre AM, Auwerx J. PPARg controls cell proliferation and apoptosis in an RB-dependent manner. Onocgene (2003) 22:4186–93. doi:10.1038/sj.onc.1206530
378. Fajas L, Landsberg RL, Huss-Garcia Y, Sardet C, Lees JA, Auwerx J. E2Fs regulate adipocyte differentiation. Dev Cell (2002) 3:39–49. doi:10.1016/S1534-5807(02)00190-9
379. Sankaran VG, Orkin SH, Walkley CR. Rb intrinsically promotes erythropoiesis by coupling cell cycle exit with mitochondrial biogenesis. Genes Dev (2008) 22:463–75. doi:10.1101/gad.1627208
380. Chau BN, Wang JYJ. Coordinated regulation of life and death by RB. Nat Rev Cancer (2003) 3:130–8. doi:10.1038/nrc993
381. Aguilar V, Fajas L. Cycling through metabolism. EMBO Mol Med (2010) 2:338–48. doi:10.1002/emmm.201000089
382. Hsieh MCF, Das D, Sambandam N, Zhang MQ, Nahle Z. Regulation of the PDK4 isozyme by the RB-E2F1 complex. J Biol Chem (2008) 283:27410–7. doi:10.1074/jbc.M802418200
383. Lopez-Bigas N, Kisiel TA, DeWaal DC, Holmes KB, Volkert TL, Gupta S, et al. Genome-wide analysis of the H3K4 histone demethylase RBP2 reveals a transcriptional program controlling differentiation. Mol Cell (2008) 31:520–30. doi:10.1016/j.molcel.2008.08.004
384. Blanchet E, Annicotte JS, Lagarrigue S, Aguilar V, Clapé C, Chavey C, et al. E2F transcription factor-1 regulates oxidative metabolism. Nat Cell Biol (2011) 13:1146–52. doi:10.1038/ncb2309
385. Ferecatu I, Le Floch N, Bergeaud M, Rodriguez-Enfedaque A, Rincheval V, Oliver L, et al. Evidence for a mitochondrial localization of the retinoblastoma protein. BMC Cell Biol (2009) 10:50. doi:10.1186/1471-2121-10-50
386. Hilgendorf KI, Leshchiner ES, Nedelcu S, Maynard MA, Calo E, Ianari A, et al. The retinoblastoma protein induces apoptosis directly at the mitochondria. Genes Dev (2013) 27:1–13. doi:10.1101/gad.211326.112
387. Wang J, Xie LY, Allan S, Beach D, Hannon GJ. Myc activates telomerase. Genes Dev (1998) 12:1769–74.
388. Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2a promotes hypoxic cell proliferation by enhancing c-Myc transcriptional activity. Cancer Cell (2007) 11:335–47. doi:10.1016/j.ccr.2007.02.006
389. Koshiji M, To KKW, Hammer S, Kumamoto K, Harris AL, Modrich P, et al. HIF-1a induces genetic instability by transcriptionally downregulating Mutsa expression. Mol Cell (2005) 17:793–803. doi:10.1016/j.molcel.2005.02.015
390. Johnson SA, Dubeau L, Johnson DL. Enhanced RNA polymerase III-dependent transcription is required for oncogenic transformation. J Biol Chem (2008) 283:19184–91. doi:10.1074/jbc.M802872200
391. Liu H, Radisky DC, Yang D, Xu R, Radisky ES, Bissell MJ, et al. MYC suppresses cancer metastasis by direct transcriptional silencing of αv and β3 integrin subunits. Nat Cell Biol (2012) 14:567–74. doi:10.1038/ncb2491
392. Baudino TA, McKay C, Pendeville-Samain H, Nilsson JA, MacLean KH, White EL, et al. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev (2002) 16:2530–43.
393. Sodir NM, Swigart LB, Karnezis AN, Hanahan D, Evan GI, Soucek L. Endogenous Myc maintains the tumor microenvironment. Genes Dev (2011) 25:907–16. doi:10.1101/gad.2038411
394. Shim H, Chun YS, Lewis BC, Dang CV. A unique glucose-dependent apoptotic pathway induced by c-Myc. Proc Natl Acad Sci U S A (1998) 95:1511–6. doi:10.1073/pnas.95.4.1511
395. Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, et al. Modelling Myc inhibition as a cancer therapy. Nature (2008) 455:679–83. doi:10.1038/nature07260
396. Soucek L, Whitfield JR, Sodir NM, Massó-Vallés D, Serrano E, Karnezis AN, et al. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev (2013) 27:504–12. doi:10.1101/gad.205542.112
397. Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature (2009) 458:762–5. doi:10.1038/nature07823
398. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A (2008) 105:18782–7. doi:10.1073/pnas.0810199105
399. Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol (2007) 178:93–105. doi:10.1083/jcb.200703099
400. Qing G, Li B, Vu A, Skuli N, Walton ZE, Liu X, et al. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell (2012) 22:631–44. doi:10.1016/j.ccr.2012.09.021
401. Liu L, Ulbrich J, Müller J, Wüstefeld T, Aeberhard L, Kress TR, et al. Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature (2012) 483:608–12. doi:10.1038/nature10927
402. Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE. HIF-1a induces cell cycle arrest by functionally counteracting Myc. EMBO J (2004) 23:1949–56. doi:10.1038/sj.emboj.7600196
403. American CS. Cancer Facts & Figures 2011. (2011). Available at: http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/
documents/document/acspc-029771.pdf
404. Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell (2012) 149:656–70. doi:10.1016/j.cell.2012.01.058
405. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature (2013) 496:101–5. doi:10.1038/nature12040
406. Hu S, Balakrishnan A, Bok RA, Anderton B, Larson PEZ, Nelson SJ, et al. 13C-pyruvate imaging reveals alterations in glycolysis that precede cMyc-induced tumor formation and regression. Cell Metab (2011) 14:131–42. doi:10.1016/j.cmet.2011.04.012
407. Baracca A, Chiaradonna F, Sgarbi G, Solaini G, Alberghina L, Lenaz G. Mitochondrial complex I decrease is responsible for bioenergetic dysfunction in K-ras transformed cells. Biochim Biophys Acta (2010) 1797:314–23. doi:10.1016/j.bbabio.2009.11.006
Keywords: mitochondria, oxidative phosphorylation, mitophagy, mitochondrial biogenesis, mitochondrial fusion and fission, cancer metabolism, cell cycle control
Citation: Boland ML, Chourasia AH and Macleod KF (2013) Mitochondrial dysfunction in cancer. Front. Oncol. 3:292. doi: 10.3389/fonc.2013.00292
Received: 10 October 2013; Accepted: 17 November 2013;
Published online: 02 December 2013.
Edited by:
Lorenzo Galluzzi, Institut National de la Santé et de la Recherche Medicale, FranceReviewed by:
Michelangelo Campanella, Royal Veterinary College and University College London, UKNaoufal Zamzami, Institut National de la Santé et de la Recherche Médicale, France
Vladimir Gogvadze, Karolinska Institutet, Sweden
Copyright: © 2013 Boland, Chourasia and Macleod. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kay F. Macleod, The Ben May Department for Cancer Research, The University of Chicago Comprehensive Cancer Center, The Gordon Center for Integrative Sciences, W338, 929 East 57th Street, Chicago, IL 60637, USA e-mail: kmacleod@uchicago.edu