 Viktor H. Koelzer1,2*
Viktor H. Koelzer1,2* Karl Steuer3Ulrike Camenisch Gross4
Karl Steuer3Ulrike Camenisch Gross4 Dieter Zimmermann4Aino Paasinen-Sohns1Kirsten D. Mertz1
Dieter Zimmermann4Aino Paasinen-Sohns1Kirsten D. Mertz1 Gieri Cathomas1*
Gieri Cathomas1*
- 1Cantonal Hospital Baselland, Institute of Pathology, Liestal, Switzerland
- 2Translational Research Unit (TRU), Institute of Pathology, University of Bern, Bern, Switzerland
- 3Radio Onkologie Allschwil, Allschwil, Switzerland
- 4Division of Diagnostic Molecular Pathology, University Hospital Zürich, Zürich, Switzerland
Background: Personalized therapy of colorectal cancer is influenced by morphological, molecular, and host-related factors. Here, we report the comprehensive clinicopathological and molecular analysis of an extra-gestational colorectal choriocarcinoma in a patient with probable Lynch syndrome.
Case presentation: A 61-year-old female with history of gastric cancer at age 36 presented with a transmurally invasive tumor of the right hemicolon and liver metastasis. A right hemicolectomy was performed. Histopathological analysis showed a mixed trophoblastic and syncytiotrophoblastic differentiation, consistent with choriocarcinoma. Disease progression was rapid under oxaliplatin, capecitabine, irinotecan, and bevacizumab. Molecular phenotyping identified loss of mismatch-repair protein immunostaining for PMS2, microsatellite instability, a lack of MLH1 promoter methylation, and lack of BRAF mutation suggestive of Lynch syndrome. Targeted next-generation sequencing revealed an ataxia telangiectasia mutated (p.P604S) missense mutation. A bleomycin, etoposide, and cisplatin treatment protocol targeting germ cell neoplasia lead to disease remission and prolonged survival of 34 months.
Conclusion: Comprehensive immunohistochemical and genetic testing is essential to identify uncommon cancers possibly related to Lynch syndrome. For rare tumors, personalized therapeutic approaches should take both molecular and morphological information into account.
Background
Colorectal cancer (CRC) ranks among the three most commonly diagnosed malignant tumors worldwide according to the World Health Organization (WHO) (1). Importantly, CRC is a heterogeneous disease with different molecular subtypes, variable clinical course, and prognosis. Optimal treatment requires comprehensive characterization of these features. Research to further develop the histopathological, molecular, and genetic characterization of CRC therefore builds essential knowledge to improve cancer therapy and survival.
The current gold standard to define CRC prognostic groups is the tumor node metastasis (TNM) classification published by the International Union for Cancer Control (UICC) (2). Extensive efforts have been made to aid the projection of post-operative survival and treatment response based on molecular alterations (3). Adverse molecular markers include BRAF mutations found in approximately 10% of patients and the CpG-island methylator phenotype (4, 5). KRAS and NRAS mutations correlate with poor response to drugs inhibiting the epidermal growth factor receptor (EGFR) (6). Microsatellite instability (MSI) is identified in 15–20% of CRC cases and is associated with favorable prognosis due to an increased anti-tumoral host immune response (7). Approximately 80% of MSI CRC arise in the sporadic setting due to hypermethylation of the MLH1 gene, while 20% are associated with germline mutations of the mismatch-repair (MMR) genes, such as MLH1, PMS2, MSH6, or MSH2, in Lynch syndrome. Patients without an identified germline defect in a DNA MMR gene but with MSI and loss of MMR protein expression are likely to have Lynch syndrome if other causes of MSI, such as methylation of the MLH1 promoter, are excluded (8). The suggested WHO terminology for these cases is “probable Lynch syndrome” (8). However, recent studies indicate that pathogenic somatic mutations in MMR genes can also underlie the development of MSI in a subset of cases of early onset CRC (9–11). This group of cases has been termed “Lynch-like” to underline the similar clinicopathological presentation with Lynch syndrome in absence of a proven germline-cause for MMR deficiency (12).
While the clinicopathological and molecular features of sporadic and hereditary CRC have been extensively studied, little is known about the occurrence of rare histopathological variants such as colorectal choriocarcinoma. Choriocarcinoma is a highly malignant neoplasm with trophoblastic differentiation. Gestational choriocarinoma most frequently occurs as a result of a molar pregnancy in pre-menopausal women and represents the vast majority of cases (13). Non-gestational choriocarcinoma can present as a component of ovarian and testicular germ cell tumors, while non-gestational, extra-gonadal choriocarcinomas are exceedingly rare (14). Discrimination of these forms of trophoblastic malignancy is of central importance as significant differences in the genetic origin, oncogenic driver mutations, immunogenicity, and sensitivity to chemotherapy exist (15). In particular, frequent targetable molecular alterations have been described in gestational disease, including an activation of the mitogen-activated kinase pathway (MAPK) through mutations in KRAS and BRAF oncogenes, overexpression of c-MYC, EGFR mutations, and activation of the mammalian target of rapamycin (mTOR) signaling network (16).
In adenocarcinomas of the gastrointestinal tract, choriocarcinomatous differentiation can take several forms. This ranges from the presence of individual beta human gonadotropin (β-HCG) positive malignant syncytiotrophoblastic giant cells in poorly differentiated adenocarcinoma over mixed tumors to pure choriocarcinoma (14). Previous genetic analyses have highlighted a superimposed pattern of genetic changes in adeno- and choriocarcinoma components of mixed tumors suggesting a common stem cell origin (17). However, the molecular pathogenesis and presence of targetable mutations in extra-gestational disease has remained obscure. MMR deficiency and MSI in colorectal choriocarcinoma have not been previously identified. Here, we report the comprehensive morphological, immunohistochemical, and molecular analysis of a colorectal choriocarcinoma in a patient with probable Lynch syndrome.
Case Presentation
A 61-year-old Caucasian post-menopausal female with a history of stage I signet ring cell carcinoma of the stomach at age 36 was evaluated for a change in bowel habits and abdominal discomfort. There was a known family history of breast cancer in a second degree relative (aunt). Physical examination discovered a palpable resistance in the lower right quadrant. Colonoscopy identified a large stenosing mass in the cecum. An endoscopic biopsy was performed showing a poorly differentiated carcinoma with extensive necrosis. Computed tomography (CT) imaging confirmed the diagnosis of a 9.6-cm cecal mass (Figure 1A) and identified four hepatic metastases with a maximum diameter of 2.6 cm (Figure 1B). There was no evidence of a lesion involving the ovaries, the uterus, or a locoregional recurrence of gastric carcinoma. A right hemicolectomy with primary end-to-end anastomosis was performed. A diagnosis of primary colorectal choriocarcinoma in clinical stage IV was made. Pathological tumor stage according to the UICC TNM classification, 7th edition (2) was pT4a, pN0 (0/29), cM1 (HEPAR), L0, V1, Pn0, G3, and R0. Laboratory examinations showed significantly elevated serum β-HCG levels of 70.173 IU/ml (internal reference <2 IU/ml), a normal level of carcinoembryonic antigen (CEA; 1.9 μg/l; internal reference <5.0 μg/l), and CA19.9 (6.7 kU/l; internal reference <35 kU/l). The patient was first treated with oxaliplatin, capecitabine, irinotecan, and bevacizumab (XELOXIRI/bevacizumab) for two cycles according to the standard protocols (18). Rapid radiographic progression of disease and increasing serum levels of β-HCG were recorded under treatment. The therapeutic strategy was therefore adapted toward bleomycin/etoposide/cisplatin (BEP) which has shown efficacy in high-risk gestational and extra-gestational trophoblastic neoplasia (15). A partial remission [RECIST 1.1 (19)] with radiographic evidence of a decrease in the sum of the diameters of the hepatic lesions of over 50% and a decrease in serum β-HCG levels to 8 IU/ml (internal reference range <2 IU/ml) was documented. Close follow-up was maintained.
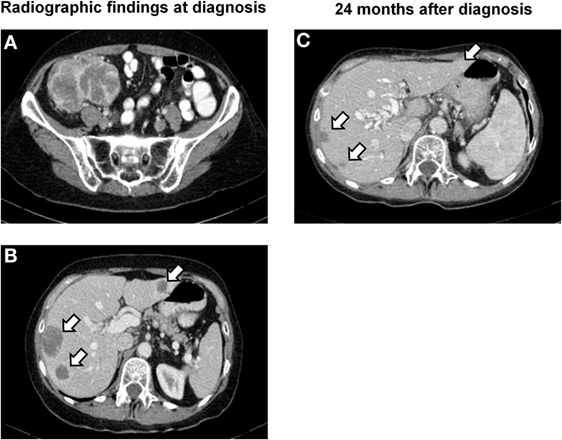
Figure 1. Radiographic findings. (A) CT image of the lower abdomen showing a poorly delineated, partially necrotic tumor in the ileocecal region with a maximum diameter of 9.6 cm. (B) CT scan of the upper abdomen demonstrating hepatic metastases in segments II, VII, and VIII with a maximum diameter of 2.6 cm (segment VI not visible in this image plane). (C) Follow-up examination 24 months later showing subtotal regression of the metastases in segments VII and VIII and total regression of the lesion in segment II.
Fifteen months later, laboratory and radiographic evidence of intra-abdominal tumor progression with new peritoneal lesions was recorded. Third line therapy with five cycles of carboplatin and bleomycin was initiated, leading to stabilization of disease (Figure 1C). New peritoneal, splenic, uterine, ovarian, and rectal lesions were noted on follow-up studies 6 weeks later. Salvage therapy with ifosfamide and vinblastine was started leading to a partial remission [RECIST 1.1 (19)]. The patient recovered well and surgical tumor debulking was performed 2 months later with resection of peritoneal lesions, the rectosigmoid, spleen, and uterus. Following recovery, two cycles of paclitaxel monotherapy were administered. Further treatment was delayed as a result of an intravascular catheter infection. There was rapid progression of hepatic disease with hilar compression. New bone metastases were detected in the spinal column. Due to rapid deterioration, the patient was entered into best supportive care and deceased 34 months after diagnosis. An autopsy was not performed.
Gross Pathology, Histology, and Immunohistochemistry
Gross examination of the resection specimen showed a poorly marginated tumor localized to the cecum and terminal ileum with serosal perforation (Figure 2A). Histopathological analysis revealed a solid carcinoma consisting of a dimorphic population of syncytiotrophoblastic giant cells and mononucleate trophoblastic cells arranged in a plexiform pattern with transmural infiltration of the colonic wall (Figure 2B) and multifocal venous invasion. Immunohistochemical analysis demonstrated strong, diffuse cytoplasmic reactivity for pan-cytokeratin (pan-CK) and β-HCG, while stains for the intestinal transcription factor CDX2 were negative (Figure 3, top). Additional analyses of oncogenes associated with choriocarcinomatous differentiation revealed an overexpression of p53, the p53-associated protein MDM2 (not shown), c-MYC, and p16. Immunohistochemical analysis of the MMR proteins MLH1, PMS2, MSH6, and MSH2, showed an isolated loss of PMS2 protein (Figure 3, bottom right). Antibodies used for immunohistochemical analysis and staining protocols are provided in Table 1.
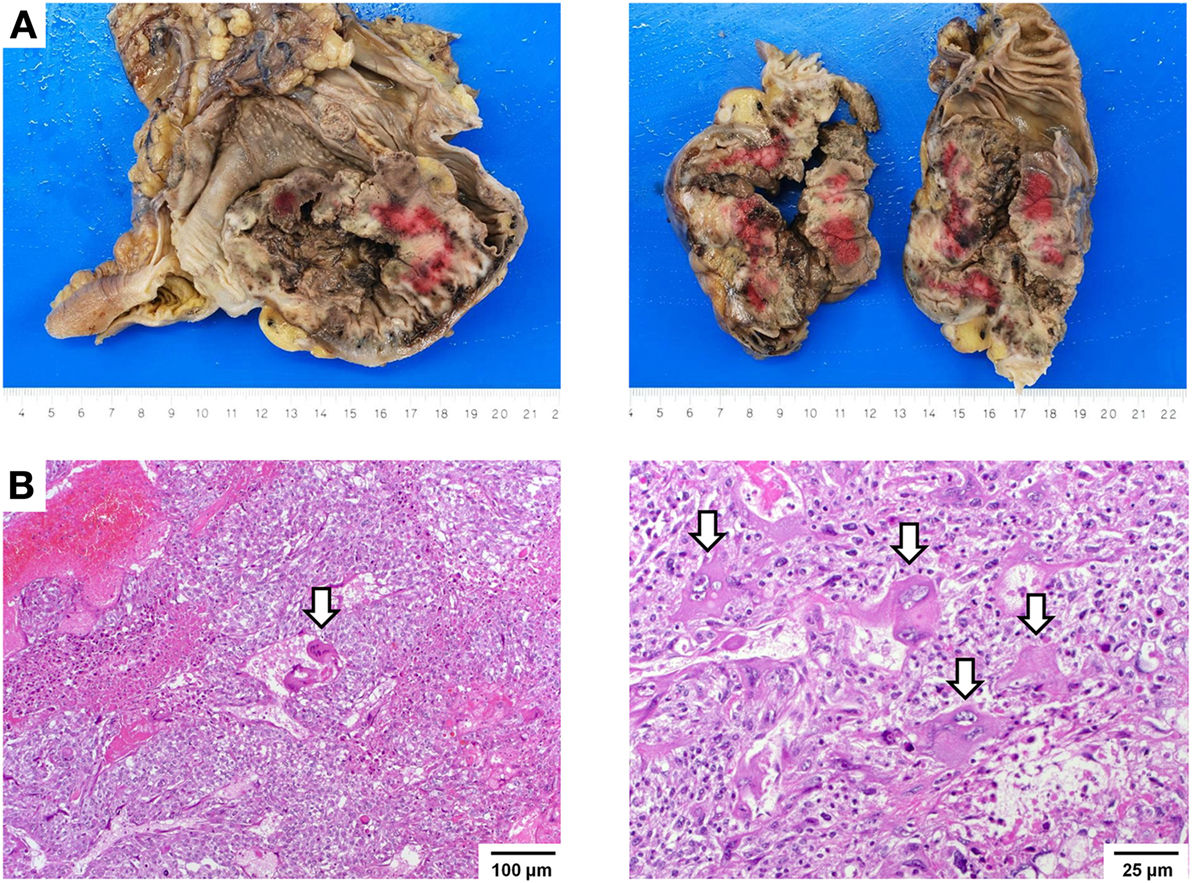
Figure 2. Macroscopic and histopathological images. (A) Gross images showing a hemorrhagic and partially necrotic tumor with transmural infiltration of the colonic wall. (B) Histologic images showing a dimorphic population of syncytiotrophoblastic giant cells (arrows) and mononucleate trophoblastic cells arranged in a plexiform pattern invading the colonic wall with extensive hemorrhagic necrosis (left, 100×). Trophoblastic cells have ample eosinophilic cytoplasm and show markedly atypical nuclei with irregular nuclear membranes, coarse chromatin, and a single, prominent nucleolus. Syncytiotrophoblastic cells (arrows) have multiple nuclei with variable size and abundant eosinophilic to amphiphilic cytoplasm (right, 250×). Cytoplasmic vacuolization and lacunae are frequently observed.
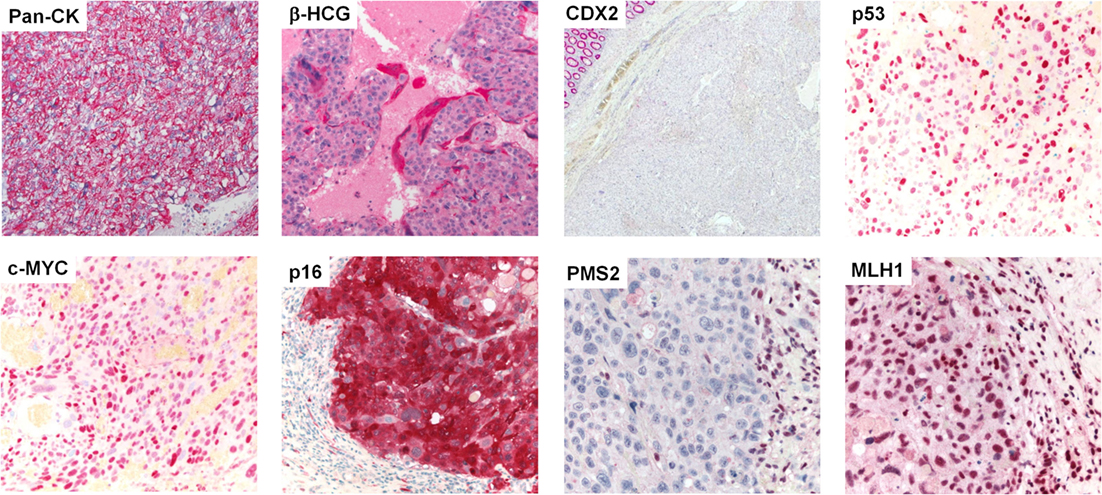
Figure 3. Protein expression profiling by immunohistochemistry. Immunohistochemical stains (from left to right, top to bottom) showing diffuse, strong cytoplasmic reactivity of the neoplastic cell population for pan-CK. β-HCG stain highlights syncytiotrophoblastic giant cells (all 400×). No expression of CDX2 was observed in the tumor cells; note the normal colonic epithelium in the upper left corner (40×). Analysis of postulated oncogenes revealed strong overexpression of p53, c-MYC, and p16. The neoplastic cell population showed an isolated loss of PMS2, while MLH1 was maintained; note the positively stained tumor infiltrating lymphocytes (all 400×).
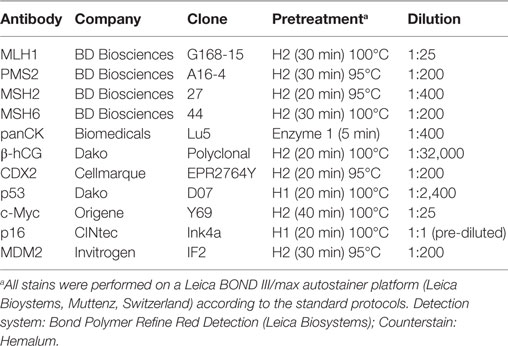
Table 1. Antibodies used for immunohistochemical analysis and staining protocols.
Microsatellite Analysis and Next-Generation Sequencing
Based on a previous history of gastric cancer at age 36 and an isolated loss of PMS2 protein expression by immunohistochemistry, the suspicion of Lynch syndrome was raised. This was further investigated by molecular studies of both tumors (Figure 4A). MSI analysis of the colorectal tumor showed shifted alleles in seven out of the nine microsatellite markers by multiplex PCR assay and high-resolution gel electrophoresis (instable: Bat-25, Bat-26, D2S123, D17S250, Bat-40, D13S153, D18S58; stable: D5S346, D10S197) (Figure 4B). This finding was consistent with an MSI-high genotype. The tumor MLH1 promoter region was un-methylated as revealed by bisulfite sequencing analysis (Figure 4C). Sequencing of BRAF hotspot regions in codons 600 and 601 showed no mutations. Using archival material of the gastric primary from 1985, analysis of six microsatellite markers (Bat-25, Bat-26, D2S123, D5S346, D10S197, and D18S5) was successful. All markers were stable, and there was no evidence of MSI. In consistency with these findings, we found maintained expression of MLH1, PMS2, MSH6, and MSH2 in the gastric carcinoma by immunohistochemistry. No BRAF mutations were identified. MLH1 promoter methylation analysis was not performed. Germline mutation analysis of the PMS2 gene was not possible due to applicable legal regulations.
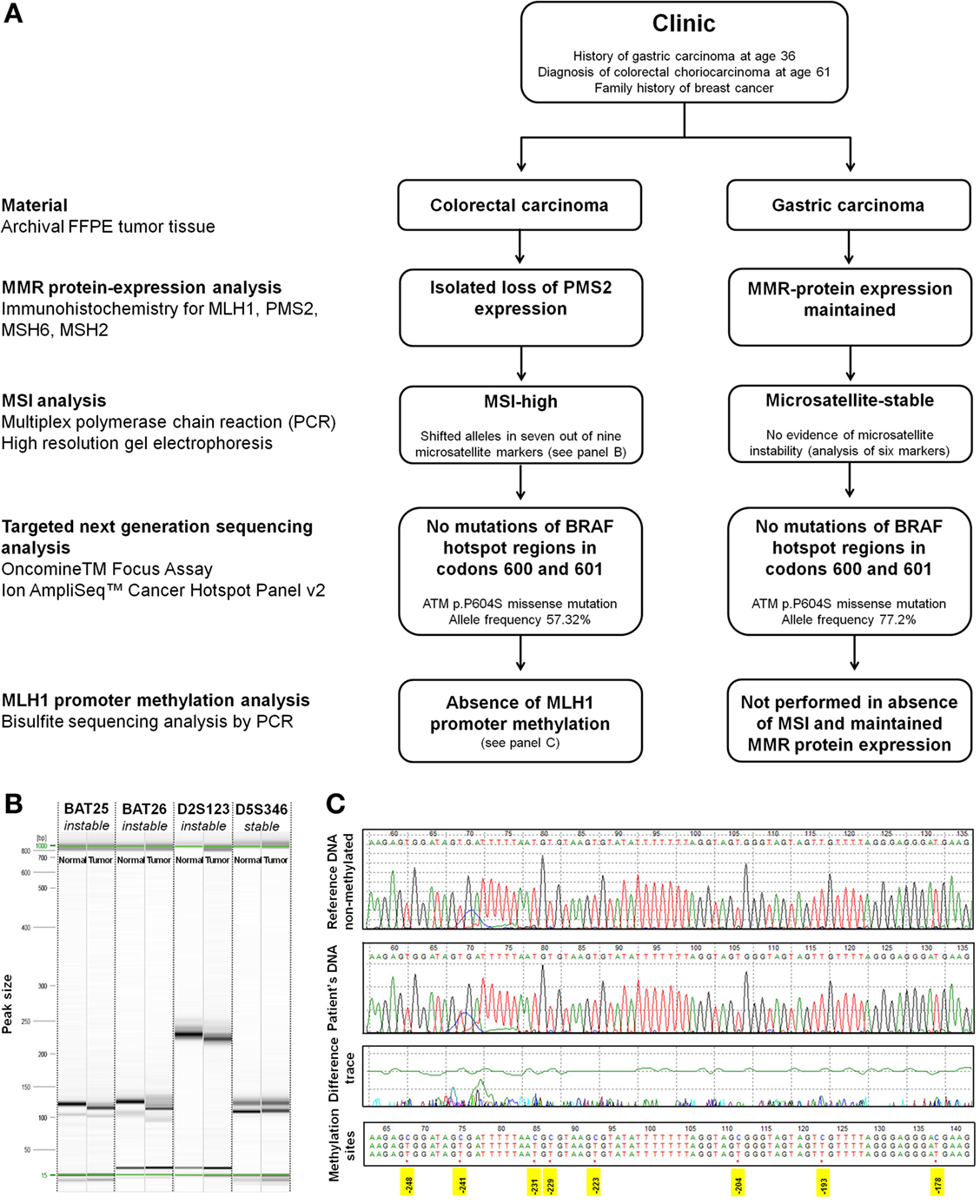
Figure 4. Molecular analysis. (A) Flowchart of molecular analysis. (B) Representative image of high-resolution capillary gel electrophoresis (QIAxcel high-resolution cartridge, QIAGEN AG, Hombrechtikon, Switzerland) of microsatellite markers. One sample of normal tissue and one tumor sample was analyzed for each marker. Electropherograms showed shifted alleles in seven out of the nine analyzed markers in the choriocarcinoma tissue [Bat-25 (shown), Bat-26 (shown), D2S123 (shown), D17S250, Bat-40, D13S153, and D18S58] indicating high-grade microsatellite instability. Two out of the nine markers showed an identical fragment length between normal and tumor tissue [D5S346 (shown) and D10S197]. (C) Bisulfite sequencing. Sequence of the patient DNA sample in comparison to a reference non-methylated DNA (tonsil). Treatment of DNA with bisulfite converts cytosine residues to uracil. Methylated cytosine bases remain unaffected. The tumor MLH1 promoter region was un-methylated as is shown by comparison of the patients DNA with the reference sequence. Positions of cytosins best correlating with MLH1 expression are highlighted in yellow [numbering according to Deng et al. (20)].
To further expand the molecular characterization of colorectal choriocarcinoma, two independent targeted next-generation sequencing (NGS) assays (Oncomine™ Focus Assay and Ion AmpliSeq™ Cancer Hotspot Panel v2, both from Thermo Fischer Scientific, Waltham, MA, USA) were performed according to the manufacturer’s instructions. The Oncomine™ Focus Assay allows the concurrent analysis of DNA and RNA for detection of mutations, copy number variations, and gene fusions in a comprehensive panel of clinically actionable oncogenic driver genes (21). The Ion AmpliSeq™ Cancer Hotspot Panel allows the identification of somatic mutations in a 50 gene panel (22). For NGS analysis, all diagnostic slides of a given case were re-reviewed and representative blocks of formalin-fixed paraffin-embedded material with a tumor cell content exceeding 80% were selected. Areas of necrosis were excluded. Microdissection was not performed. Identical analyses were carried out using archival material of the gastric primary from 1985. While no relevant molecular alterations were identified by the Oncomine™ Focus Assay in the colorectal choriocarcinoma, targeted NGS using the Ion AmpliSeq™ Cancer Hotspot Panel revealed an ataxia telangiectasia mutated (ATM p.P604S) mutation both in the primary tumor and metastatic lesions at an allele frequency of 51.48 and 57.32%, respectively. An identical ATM p.P604S mutation was identified in the gastric primary from 1985 with an allele frequency of 77.2%.
Discussion
Clinical and Therapeutic Aspects
The present study underlines the importance of an integrative assessment of tumor morphology and molecular factors for optimal management of rare tumors. Little is known about the natural history of colorectal choriocarcinoma as only 20 cases have been previously described with variable therapeutic interventions (14, 23, 24). Disease progression is generally rapid with early and aggressive metastasis. Response to adjuvant chemotherapy following surgical resection is poor, and disease-specific survival is commonly less than 1 year after diagnosis (14). A previous study identified responsiveness to oxaliplatin/fluorouracil/folinic acid (mFOLFOX6) and bevacizumab using an in vitro drug sensitivity test; however, therapeutic benefit was limited (14). In the present case, rapid progression of disease was observed under XELOXIRI/bevacizumab leading us to change the treatment strategy toward the BEP regimen for germ cell neoplasia. We observed a rapid clinical response with a drop of serum β-HCG levels from 70.173 to 8 IU/ml and a partial remission of metastatic lesions. However, other authors report limited efficacy of BEP in cases of colorectal choriocarcinoma (14, 24). A possible explanation for this differential therapeutic response may be due to different variants of colorectal choriocarcinoma ranging from mixed tumors with a component of conventional intestinal adenocarcinoma to pure trophoblastic neoplasia (14, 24). Interestingly, a combined treatment regimen targeting both components has previously shown efficacy in a case of mixed colorectal choriocarcinoma leading to long-term disease-free survival of 60 months (25). This suggests that histological tumor composition could be a potential indicator of treatment response.
Remarkably, review of clinicopathological data shows that 9 of the 21 previously reported cases of colorectal choriocarcinoma have arisen in patients under 50 years of age (23, 24). This precedes the median age of CRC diagnosis in western countries by about two decades (26). These data may suggest a possible association of colorectal choriocarcinoma with hereditary cancer syndromes. Indeed, one case of colorectal choriocarcinoma has been previously identified in a patient with juvenile polyposis (23). No specific investigations were reported in the remaining cases, leading to a possible underidentification of patients with hereditary disease. The unusual presentation of colorectal choriocarcinoma at a young age and the possible contribution of background genetic factors therefore require further investigation. In particular, Lynch syndrome may be easily missed if screening for MMR protein expression or MSI testing is not performed (27). Importantly, MSI is also a strong favorable prognostic indicator in CRC patients (28). Although the prognostic impact of MSI in colorectal choriocarcinoma is presently unknown, this factor may have favorably influenced the prognosis in the present case.
Molecular Pathology
DNA mismatch repair is initiated by formation of the DNA-repair complex involving both MSH and MLH heterodimers (29). In sporadic MSI CRC, loss of MLH1 expression is caused by promoter methylation of the MLH1 gene (30). As MLH1 protein is essential for stabilization of PMS2, MLH1 deficiency in sporadic MSI CRC is usually associated with loss of PMS2 expression (31). A further common genetic hallmark of sporadic MSI CRC is the presence of BRAF V600E mutation (32). In contrast, an isolated loss of PMS2 expression is a strong indicator of germline mutations in PMS2 or MLH1 in Lynch syndrome patients (33). Although we were unable to provide the ultimate proof of an MMR germline mutation in the present case due to legal restrictions, the classification as a probable Lynch syndrome is strongly supported by a lack of MLH1 promoter methylation and lack of BRAF mutation as well as isolated loss of PMS2 protein expression (8, 33, 34). PMS2 mutations are associated with a lower penetrance and variable clinical phenotypes ranging from early- or late-onset to apparently sporadic CRC (8, 35). The cumulative CRC risk of female PMS2 mutation carriers by age 70 has been estimated as low as 11% for CRC in a recent large European cohort study of 8 PMS2 families that included a total of 2,548 family members and 377 proven mutation carriers (36). Consequently, only 65.5% of patients with monoallelic PMS2 germline mutations may meet the revised Bethesda guidelines (37). The abovementioned study has also suggested an increased incidence of breast cancer in patients with PMS2 germline mutations [standardized incidence ratio 3.8 (95% CI, 1.9–6.8)], and there is a positive family history for this tumor in our patient (36). Still, the possibility of a Lynch-like syndrome with a somatic PMS2 gene mutation underlying the development of MSI cannot be excluded as somatic mutation analysis of MMR genes was not performed (12). However, somatic MMR gene mutations are most frequently described in MLH1 and MSH2, while somatic PMS2 mutations are uncommon in CRC (9–11).
We performed a comprehensive screen for clinically actionable mutations using two targeted NGS assays to analyze both the present colorectal tumor and the previously diagnosed gastric carcinoma from 1985 (21, 22). This approach identified an identical ATM p.P604S missense mutation with an allele frequency of up to 57.32% in the colorectal primary and 77.2% in the gastric tumor. ATM p.P604S falls into the N-terminal domain of the ATM gene leading to a C > T amino acid substitution at c.1810. On a population level, ATM p.P604S has been described as an infrequent polymorphism present in about 0.5% of the population with conflicting interpretations of pathogenicity (38). While the determination of the impact of ATM p.P604S is beyond the scope of the present study, the assignment of this molecular alteration to a germline polymorphism seems likely considering the identification at high allele frequency in all analyzed samples.
Other studies have identified ATM p.P604S as a somatic mutation in lymphoid neoplasms (39–41), but not in solid tumors. ATM p.P604S has been suggested by other authors to create a new glycosylation site which might interfere with ATM function and protein–protein interaction (42). In particular, ATM is an important stabilizer of the MMR protein complex during DNA repair (43) and induces p53 expression in response to DNA damage (44). Concomitant pathogenic ATM mutations and MMR deficiency in the setting of Lynch syndrome may therefore have further detrimental effects on genomic stability. Selective inhibitors of ATM are currently in preclinical development and may represent future options to treat tumors with defects in the DNA-damage response (45).
In consistence with previously published data on trophoblastic neoplasia, we identify a strong overexpression of p53 and c-MYC in tumor cells (16). As no molecular alterations in p53 and c-MYC genes were identified, these changes may be secondary to signaling alterations during neoplastic progression. Previously described molecular alterations in trophoblastic neoplasia such as EGFR, BRAF, or KRAS mutations, upregulation of BCL-2 (46), loss of p16, and downregulation of E-cadherin (47) were not observed. Interestingly, MSI has not been reported in trophoblastic neoplasia but is a frequent molecular event in primary CRC. The detection of MSI in colorectal choriocarcinoma could thus provide a further argument for an origin of colorectal choriocarcinoma from colonic stem cells.
Conclusion
Colorectal choriocarcinoma is a rare and aggressive cancer which is disproportionately common in young patients. The identification of this subtype may aid the selection of an appropriate therapy and support the use of β-HCG as a clinically actionable biomarker. In addition, an increased suspicion for the detection of hereditary cancer syndromes may be warranted.
Ethics Approval and Consent
Publication of this study was evaluated by the local ethics committee (Ethikkommission Nordwest- und Zentralschweiz) on April 11, 2016. According to the guidelines of the committee, a designated ethics vote and consent is not required for an anonymized case report.
Author Contributions
VK and GC designed and coordinated the study, drafted the manuscript, performed the histopathological and immunohistochemical analysis, and critically discussed molecular data with KM, UG, and DZ. KM helped with rendering the diagnosis, performed and interpreted the molecular analysis, and critically reviewed the manuscript. KS treated the patient at Radio Onkologie Allschwil, helped with rendering the diagnosis, and critically reviewed the manuscript. UG and DZ performed MLH1 promoter methylation analysis and critically reviewed the manuscript. AS performed next-generation sequencing analysis. All the authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
β-HCG, beta human gonadotropin; AFP, alpha-fetoprotein; BEP, bleomycin, etoposide, and cisplatin; CD, cluster of differentiation; CIMP, CpG-island methylator phenotype; CK, cytokeratin; CRC, colorectal cancer; CT, computed tomography; DFS, disease-free survival; EGFR, epidermal growth factor receptor; NGS, next-generation sequencing; OS, overall survival; PLAP, placental alkaline phosphatase; UICC, International Union for Cancer Control; WHO, World Health Organization; XELOXIRI, oxaliplatin, capecitabine, irinotecan.
References
1. Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet (2014) 383:1490–502. doi:10.1016/S0140-6736(13)61649-9
2. Sobin LH, Gospodarowicz MK, Wittekind C; International Union against Cancer. TNM Classification of Malignant Tumours. Chichester, West Sussex; Hoboken, NJ: Wiley-Blackwell (2010).
3. Compton C, Tanabe K, Savarese D. Pathology and Prognostic Determinants of Colorectal Cancer. Waltham, MA: UptoDate (2015).
4. Gallois C, Laurent-Puig P, Taieb J. Methylator phenotype in colorectal cancer: a prognostic factor or not? Crit Rev Oncol Hematol (2016) 99:74–80. doi:10.1016/j.critrevonc.2015.11.001
5. Juo YY, Johnston FM, Zhang DY, Juo HH, Wang H, Pappou EP, et al. Prognostic value of CpG island methylator phenotype among colorectal cancer patients: a systematic review and meta-analysis. Ann Oncol (2014) 25:2314–27. doi:10.1093/annonc/mdu149
6. Allegra CJ, Rumble RB, Hamilton SR, Mangu PB, Roach N, Hantel A, et al. Extended RAS gene mutation testing in metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy: American Society of Clinical Oncology Provisional Clinical Opinion update 2015. J Clin Oncol (2016) 34:179–85. doi:10.1200/JCO.2015.63.9674
7. Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med (2015) 21:1350–6. doi:10.1038/nm.3967
8. Bosman FT; World Health Organization, International Agency for Research on Cancer. WHO Classification of Tumours of the Digestive System. Lyon: International Agency for Research on Cancer (2010).
9. Mensenkamp AR, Vogelaar IP, van Zelst-Stams WA, Goossens M, Ouchene H, Hendriks-Cornelissen SJ, et al. Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in Lynch syndrome-like tumors. Gastroenterology (2014) 146:643–6.e8. doi:10.1053/j.gastro.2013.12.002
10. Geurts-Giele WR, Leenen CH, Dubbink HJ, Meijssen IC, Post E, Sleddens HF, et al. Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers. J pathol (2014) 234:548–59. doi:10.1002/path.4419
11. Haraldsdottir S, Hampel H, Tomsic J, Frankel WL, Pearlman R, de la Chapelle A, et al. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology (2014) 147:1308–1316e1. doi:10.1053/j.gastro.2014.08.041
12. Carethers JM. Differentiating Lynch-like from Lynch syndrome. Gastroenterology (2014) 146:602–4. doi:10.1053/j.gastro.2014.01.041
13. Lurain JR. Gestational trophoblastic disease I: epidemiology, pathology, clinical presentation and diagnosis of gestational trophoblastic disease, and management of hydatidiform mole. Am J Obstet Gynecol (2010) 203:531–9. doi:10.1016/j.ajog.2010.06.073
14. Maehira H, Shimizu T, Sonoda H, Mekata E, Yamaguchi T, Miyake T, et al. A rare case of primary choriocarcinoma in the sigmoid colon. World J Gastroenterol (2013) 19:6683–8. doi:10.3748/wjg.v19.i39.6683
15. May T, Goldstein DP, Berkowitz RS. Current chemotherapeutic management of patients with gestational trophoblastic neoplasia. Chemother Res Pract (2011) 2011:806256. doi:10.1155/2011/806256
16. Shih IeM. Gestational trophoblastic neoplasia – pathogenesis and potential therapeutic targets. Lancet Oncol (2007) 8:642–50. doi:10.1016/S1470-2045(07)70204-8
17. Verbeek W, Schulten HJ, Sperling M, Tiesmeier J, Stoop H, Dinjens W, et al. Rectal adenocarcinoma with choriocarcinomatous differentiation: clinical and genetic aspects. Hum Pathol (2004) 35:1427–30. doi:10.1016/j.humpath.2004.06.005
18. Vasile E, Masi G, Fornaro L, Cupini S, Loupakis F, Bursi S, et al. A multicenter phase II study of the combination of oxaliplatin, irinotecan and capecitabine in the first-line treatment of metastatic colorectal cancer. Br J Cancer (2009) 100:1720–4. doi:10.1038/sj.bjc.6605075
19. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer (2009) 45:228–47. doi:10.1016/j.ejca.2008.10.026
20. Deng G, Chen A, Hong J, Chae HS, Kim YS. Methylation of CpG in a small region of the hMLH1 promoter invariably correlates with the absence of gene expression. Cancer Res (1999) 59:2029–33.
21. Fang P, Yan Z, Labrousse P, Liu W, Biroschak J, Wright J, et al. Oncomine focus assay: simultaneous detection of clinically relevant hotspot mutations, CNVs and gene fusions in 52 oncogenes relevant to solid tumors. Cancer Res (2016) 76(14 Suppl):1397.
22. Tsongalis GJ, Peterson JD, de Abreu FB, Tunkey CD, Gallagher TL, Strausbaugh LD, et al. Routine use of the Ion Torrent AmpliSeq Cancer Hotspot Panel for identification of clinically actionable somatic mutations. Clin Chem Lab Med (2014) 52:707–14. doi:10.1515/cclm-2013-0883
23. Parra-Medina R, Correa PL, Moreno JJ, Lucero PM, Yaspe E, Polo F. Carcinosarcoma with choriocarcinomatous and osteosarcomatous differentiation in a patient with juvenile polyposis syndrome. Rare Tumors (2015) 7:5778. doi:10.4081/rt.2015.5778
24. Oh SK, Kim HW, Kang DH, Choi CW, Choi YY, Lim HK, et al. Primary adenocarcinoma with focal choriocarcinomatous differentiation in the sigmoid colon. Korean J Gastroenterol (2015) 66:291–6. doi:10.4166/kjg.2015.66.5.291
25. Harada M, Inoue T, Hamano K. Choriocarcinoma of the sigmoid colon: report of a case. Surg Today (2012) 42:93–6. doi:10.1007/s00595-011-0026-3
26. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin (2015) 65:87–108. doi:10.3322/caac.21262
27. Shia J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J Mol Diagn (2008) 10:293–300. doi:10.2353/jmoldx.2008.080031
28. Guastadisegni C, Colafranceschi M, Ottini L, Dogliotti E. Microsatellite instability as a marker of prognosis and response to therapy: a meta-analysis of colorectal cancer survival data. Eur J Cancer (2010) 46:2788–98. doi:10.1016/j.ejca.2010.05.009
29. Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res (2008) 18:85–98. doi:10.1038/cr.2007.115
30. Bettstetter M, Dechant S, Ruemmele P, Grabowski M, Keller G, Holinski-Feder E, et al. Distinction of hereditary nonpolyposis colorectal cancer and sporadic microsatellite-unstable colorectal cancer through quantification of MLH1 methylation by real-time PCR. Clin Cancer Res (2007) 13:3221–8. doi:10.1158/1078-0432.CCR-06-3064
31. Mohd AB, Palama B, Nelson SE, Tomer G, Nguyen M, Huo X, et al. Truncation of the C-terminus of human MLH1 blocks intracellular stabilization of PMS2 and disrupts DNA mismatch repair. DNA Repair (Amst) (2006) 5:347–61. doi:10.1016/j.dnarep.2005.11.001
32. Schafroth C, Galvan JA, Centeno I, Koelzer VH, Dawson HE, Sokol L, et al. VE1 immunohistochemistry predicts BRAF V600E mutation status and clinical outcome in colorectal cancer. Oncotarget (2015) 6:41453–63. doi:10.18632/oncotarget.6162
33. Dudley B, Brand RE, Thull D, Bahary N, Nikiforova MN, Pai RK. Germline MLH1 mutations are frequently identified in Lynch syndrome patients with colorectal and endometrial carcinoma demonstrating isolated loss of PMS2 immunohistochemical expression. Am J Surg Pathol (2015) 39:1114–20. doi:10.1097/PAS.0000000000000425
34. Perez-Carbonell L, Alenda C, Paya A, Castillejo A, Barbera VM, Guillen C, et al. Methylation analysis of MLH1 improves the selection of patients for genetic testing in Lynch syndrome. J Mol Diagn (2010) 12:498–504. doi:10.2353/jmoldx.2010.090212
35. Goodenberger ML, Thomas BC, Riegert-Johnson D, Boland CR, Plon SE, Clendenning M, et al. PMS2 monoallelic mutation carriers: the known unknown. Genet Med (2016) 18:13–9. doi:10.1038/gim.2015.27
36. ten Broeke SW, Brohet RM, Tops CM, van der Klift HM, Velthuizen ME, Bernstein I, et al. Lynch syndrome caused by germline PMS2 mutations: delineating the cancer risk. J Clin Oncol (2015) 33:319–25. doi:10.1200/JCO.2014.57.8088
37. Senter L, Clendenning M, Sotamaa K, Hampel H, Green J, Potter JD, et al. The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology (2008) 135:419–28. doi:10.1053/j.gastro.2008.04.026
38. Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res (2016) 44:D862–8. doi:10.1093/nar/gkv1222
39. Gronbaek K, Worm J, Ralfkiaer E, Ahrenkiel V, Hokland P, Guldberg P. ATM mutations are associated with inactivation of the ARF-TP53 tumor suppressor pathway in diffuse large B-cell lymphoma. Blood (2002) 100:1430–7. doi:10.1182/blood-2002-02-0382
40. Fang NY, Greiner TC, Weisenburger DD, Chan WC, Vose JM, Smith LM, et al. Oligonucleotide microarrays demonstrate the highest frequency of ATM mutations in the mantle cell subtype of lymphoma. Proc Natl Acad Sci U S A (2003) 100:5372–7. doi:10.1073/pnas.0831102100
41. Greiner TC, Dasgupta C, Ho VV, Weisenburger DD, Smith LM, Lynch JC, et al. Mutation and genomic deletion status of ataxia telangiectasia mutated (ATM) and p53 confer specific gene expression profiles in mantle cell lymphoma. Proc Natl Acad Sci U S A (2006) 103:2352–7. doi:10.1073/pnas.0510441103
42. Offit K, Gilad S, Paglin S, Kolachana P, Roisman LC, Nafa K, et al. Rare variants of ATM and risk for Hodgkin’s disease and radiation-associated breast cancers. Clin Cancer Res (2002) 8:3813–9.
43. Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev (2000) 14:927–39. doi:10.1101/gad.14.8.927
44. Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science (1998) 281:1674–7. doi:10.1126/science.281.5383.1674
45. Weber AM, Ryan AJ. ATM and ATR as therapeutic targets in cancer. Pharmacol Ther (2015) 149:124–38. doi:10.1016/j.pharmthera.2014.12.001
46. Candelier JJ, Frappart L, Yadaden T, Poaty H, Picard JY, Prevot S, et al. Altered p16 and Bcl-2 expression reflects pathologic development in hydatidiform moles and choriocarcinoma. Pathol Oncol Res (2013) 19:217–27. doi:10.1007/s12253-012-9572-2
Keywords: colorectal cancer, choriocarcinoma, Lynch syndrome, microsatellite instability, ataxia telangiectasia mutated, molecular pathology, next-generation sequencing, personalized medicine
Citation: Koelzer VH, Steuer K, Gross UC, Zimmermann D, Paasinen-Sohns A, Mertz KD and Cathomas G (2016) Colorectal Choriocarcinoma in a Patient with Probable Lynch Syndrome. Front. Oncol. 6:252. doi: 10.3389/fonc.2016.00252
Received: 30 July 2016; Accepted: 11 November 2016;
Published: 29 November 2016
Edited by:
Tiziana Venesio, Istituto per la Ricerca e la Cura del Cancro (IRCC), ItalyReviewed by:
Gisela Keller, Technische Universität München, GermanyDaniela Furlan, University of Insubria, Italy
Copyright: © 2016 Koelzer, Steuer, Gross, Zimmermann, Paasinen-Sohns, Mertz and Cathomas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Viktor H. Koelzer, dmtvZWx6ZXJAZ21haWwuY29t;
Gieri Cathomas, Z2llcmkuY2F0aG9tYXNAa3NibC5jaA==