 Chengpeng Yu
Chengpeng Yu Zeyang Ding
Zeyang Ding Huifang Liang†
Huifang Liang† Bixiang Zhang
Bixiang Zhang Xiaoping Chen
Xiaoping Chen- Hepatic Surgery Center, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
Transcriptional intermediary factor 1 γ (TIF1γ), also known as TRIM33, RFG7, PTC7, or Ectodermin, is an E3 ubiquitin-ligase family member with a ring-box-coiled-coil region. It can regulate TGF-β/Smad signaling in two different ways in different cellular contexts. On one hand, TIF1γ can monoubiquitinate Smad4 to inhibit the formation of Smad2/3/4 nuclear complexes. On the other hand, TIF1γ can function as a cofactor of phosphorylated (p)-Smad2/3, competing with Smad4 to inhibit the formation of the Smad2/3/4 complex. In addition, TIF1γ has been reported to play a role in transcription elongation, cellular differentiation, embryonic development, and mitosis. As transforming growth factor-β (TGF-β) superfamily signaling plays an important role in the occurrence and development of cancer, and TIF1γ was reported to be involved in the regulation of TGF-β superfamily signaling, studies on TIF1γ during the last decade have focused on its role in the development of cancer. However, TIF1γ can function either as a tumor suppressor or promoter in different cellular contexts, yet there are few reviews focusing on the roles of TIF1γ in cancer. Hence, in this paper we systematically review and discuss the roles of TIF1γ in cancer. Firstly, we review the biological features, the regulatory mechanisms and the related signaling pathways of TIF1γ. Next, we illustrate the roles of TIF1γ in different tumors. We then provide a tentative hypothesis that explains the dual roles of TIF1 γ in cancer. Finally, we provide our viewpoint regarding the future developments of cancer research focusing on TIF1γ, especially in relation to the effects of TIF1γ on tumoral immunity.
Introduction
Transcriptional intermediary factor 1 γ (TIF1γ), synonymous with TRIM33, RFG7, PTC7, or Ectodermin, is an E3 ubiquitin-ligase family member with a ring-box-coiled-coil region (1). It has been reported to play a role in transcription elongation (2, 3), DNA repair (4), differentiation of cells (2, 5, 6), embryonic development (6–9), mitosis (10), and dermatomyositis (11–13). As transforming growth factor-β (TGF-β) superfamily signaling plays an important role in the occurrence and development of cancer (14–16), and TIF1γ was reported to be involved in the regulation of TGF-β superfamily signaling (17, 18), recent studies on TIF1γ have focused on its role in tumorigenesis (10, 19–24).
Perplexingly, TIF1γ can function either as a tumor suppressor or promoter in different cells. In many different tumors, such as non-small-cell lung cancer, breast cancer, glioma, and clear cell renal cell carcinoma (21, 23, 25, 26), TIF1γ acts as a tumor suppressor and its expression is decreased. However, in B lymphoblastic leukemia, pancreatic cancer, and cervical carcinoma (10, 27, 28), TIF1γ functions as a tumor promoter and prevents the apoptosis of tumor cells. However, there are few reviews focusing on the dual and contradictory roles of TIF1γ in cancer. We therefore systematically review and discuss the roles of TIF1γ in cancer in this paper. Firstly, we review the biological features, the regulatory mechanisms and the related signaling pathways of TIF1γ. Next, we illustrate the roles of TIF1γ in different tumors. We then provide a tentative hypothesis that explains the dual roles of TIF1 γ in cancer. Finally, we provide our viewpoint regarding the future developments of cancer research focusing on TIF1γ, especially in relation to the effects of TIF1γ on tumor immunity.
The Biological Functions of TIF1γ
TIF1γ is a 123 kDa protein consisting of 1120 amino acids encoded by the trim33 gene, which is 118,415 bps in length and contains 21 exons and 20 introns, encoded on chromosome 1 in humans (29). The TIF1γ protein consist of several different domains. At the N terminus, there is a ring-box-coiled-coil (RBCC) unit, containing a RING domain, B boxes, and a coiled-coil domain, which is involved in the ubiquitination of Smad4 (30), TGF-beta1 receptor (TβRI) (31), and β-catenin (23), as well as the sumoylation of SnoN1 (32). A PHD domain and a bromodomain at the C terminus can interact with histones 3 and 4 (33, 34). Between these regions, there is a middle linker which can interact with activated Smad2 and Smad3. The middle linker is less well-conserved, which explains why the other members of the TIF1γ family cannot interact with Smad proteins [Figure 1; (33)].
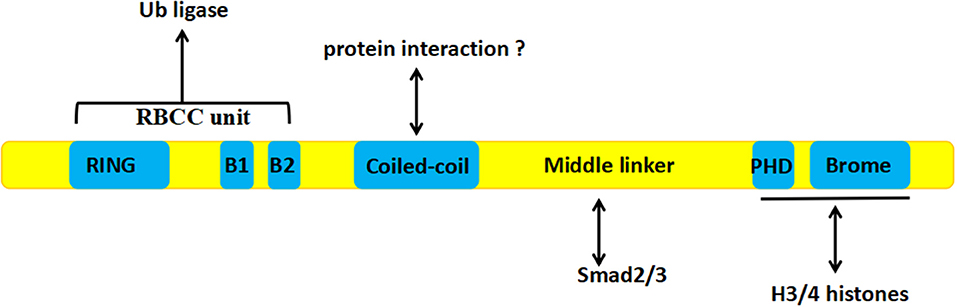
Figure 1. The structure of TIF1γ: The RBCC unit at the N terminus is involved in ubiquitination, A PHD domain and a bromodomain at the C terminus can interact with histones 3 and 4, and the middle linker can interact with activated Smad2 and Smad3.
The Regulatory Mechanisms of TIF1γ
SRY-related HMG-box2 (SOX2) was reported to be involved in the transcriptional regulation of TIF1γ and can bind to the putative SRY-binding sites of the TIF1γ promoter, which represses the expression of TIF1γ at the mRNA and thus protein level (21). Furthermore, Jingushi et al. reported that miR-629 is involved in the post-transcriptional regulation of TIF1γ and can bind to a specific sequence in the 3′-UTR of TIF1γ mRNA and promote the degradation of TIF1γ mRNA (25). At the same time, miR-429/miR-200b-3p was also reported to be involved in the post-transcriptional regulation of TIF1γ and to be able to bind to a specific sequence in the 3′-UTR of TIF1γ mRNA, which promotes its degradation. Additionally, the circular RNA PTK2 can bind directly to miR-429/miR-200b-3p to protect TIF1γ mRNA from targeting by miR-429/miR-200b-3p (35). Moreover, Yuki et al. reported that v-abl Abelson murine leukemia viral oncogene homolog 1(c-Abl) tyrosine kinase takes part in the post-translational regulation of TIF1γ and can regulate its phosphorylation at tyrosines 524, 610, and 1,048, which inhibits the interaction of TIF1γ with Smad2/3 (36). At the same time, the Ad5 E4-ORF3 protein can promote the initial conjugation of SUMO3 to TIF1γ, inducing its sumoylation and proteasomal degradation [Figure 2; (37, 38)].
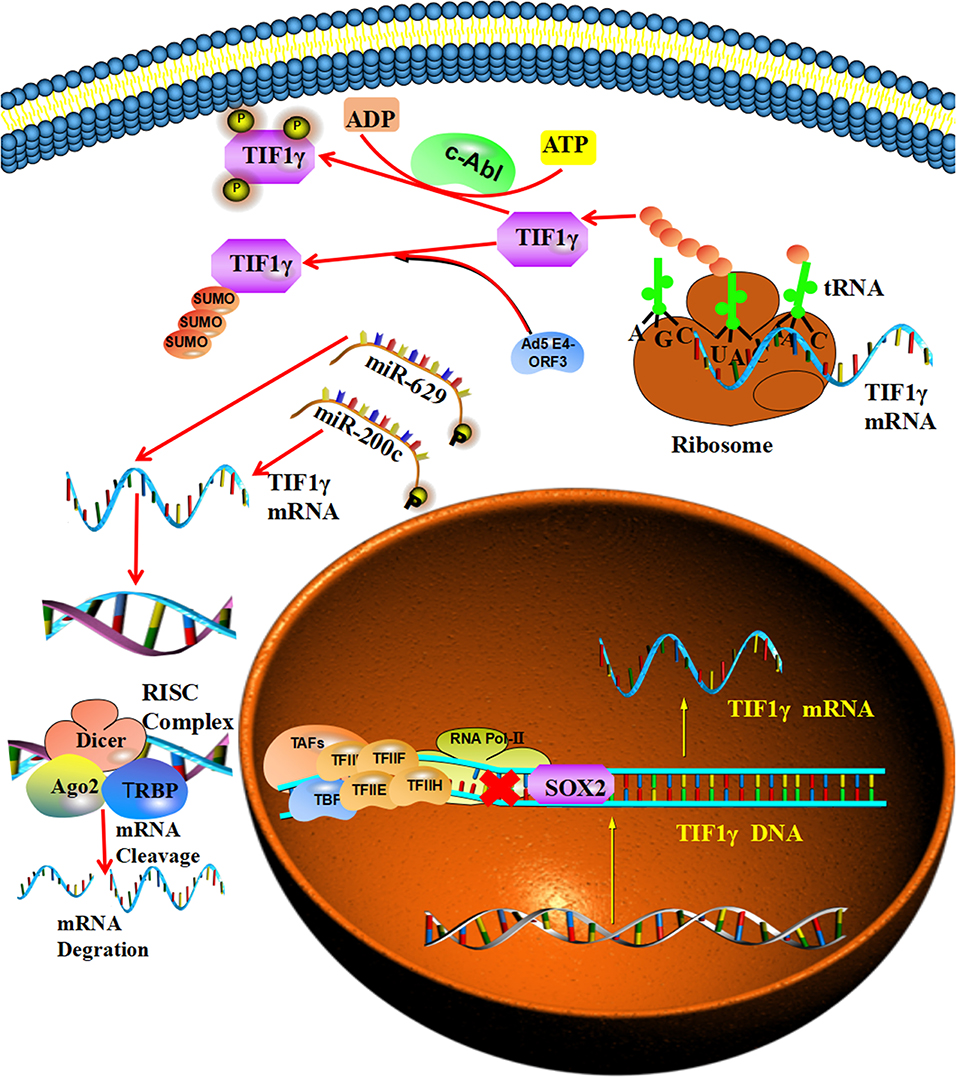
Figure 2. The regulatory mechanisms of TIF1γ: SOX2 can bind to the putative SRY-binding sites of the TIF1γ promoter and repress its expression. MiR-200c and miR-629 can bind to specific sequences in the 3′-UTR of TIF1γ mRNA and promote its degradation. The c-Abl tyrosine kinase can regulate the tyrosine phosphorylation of TIF1γ. In addition, the Ad5 E4-ORF3 protein can promote the initial SUMO3 conjugation to TIF1γ and induce its sumoylation.
The Physiological Roles of TIF1γ
As a transcriptional intermediary factor, TIF1γ takes part in the transcriptional regulation of a number of genes by interacting with other transcriptional factors. For example, TIF1γ can promote the transcription elongation of hematopoietic genes by interacting with FACT, p-TEFb, and the SCL complex, and TIF1γ deficiency reduced the full-length transcript level of these genes (2). At the same time, TIF1γ controls hematopoiesis and the specification of the germ layer and regulates cell growth by antagonizing TGFβ signaling (2, 7). Furthermore, TIF1γ can promote the repair of DNA damage by interacting with Amplified in Liver Cancer 1 (ALC1) in a poly(ADP-ribose) polymerase (PARP)-dependent manner (4).
Signalings Pathways Related to TIF1γ in Cancer
The Inhibitory Effect of TIF1γ on TGF-β/Smad Signaling
TGF-β plays a vital role in the regulation of cellular proliferation, differentiation, apoptosis, motility, invasion, and immune responses (18, 39–41). Smad proteins can be phosphorylated by TGF-β and translocate to the nucleus, which results in the transcriptional activation of downstream target genes (32, 42, 43). Increasing numbers of studies show that TGF-β/Smad signaling is involved in tumor growth, metastasis and the epithelial–mesenchymal transition (EMT) (44–47). Specifically, TGFβ/Smad signaling can function as a tumor suppressor to inhibit tumor growth and metastasis by regulating the downstream genes, such as p21, p53, c-myc, and snail (48, 49). Deletions or mutations of TGFβ/Smad signaling were detected in many cancers (48). For example, mutations of Smad2 were found in cervical cancer, colorectal cancer and hepatocellular carcinoma (50–52). At the same time, mutations of Smad4 are more frequent in some cancers, such as colon cancer, gastric cancer, and pancreatic tumors (53–55). TIF1γ can regulate TGF-β/Smad signaling in two different ways in different cellular contexts. On the one hand, it can monoubiquitinate Smad4 and inhibit the formation of Smad nuclear complexes (7, 30). On the other hand, TIF1γ can function as a cofactor of phosphorylated (p)-Smad2/3, competing with Smad4 to inhibit the formation of the Smad2/3/4 complex (17). Additionally, TIF1γ requires sumoylation mediated by ubiquitin carrier 9 to exert its inhibitory effect on TGFβ/Smad4 signaling (24, 37). Numerous studies have demonstrated that TIF1γ can inhibit tumor growth, TGF-β-induced epithelial mesenchymal transition and metastasis, and that its expression is reduced in non-small-cell lung cancer and breast cancer [Figure 3; (21, 22, 35)]. However, FAM/USP9x can reverse the ubiquitination of Smad4 and counteract the activity of TIF1γ in TGF-β/Smad signaling (30). Moreover, forkhead box M1 (FOXM1) can also counteract the activity of TIF1γ in TGF-β/Smad signaling by interfering with the interaction between TIF1γ and Smad4 (26). In addition, αB-crystallin can interact with TIF1γ and disrupt the monoubiquitination of Smad4, which favors the formation of the Smad2/3/4 complex and enhances TGF-β/Smad signaling (56).
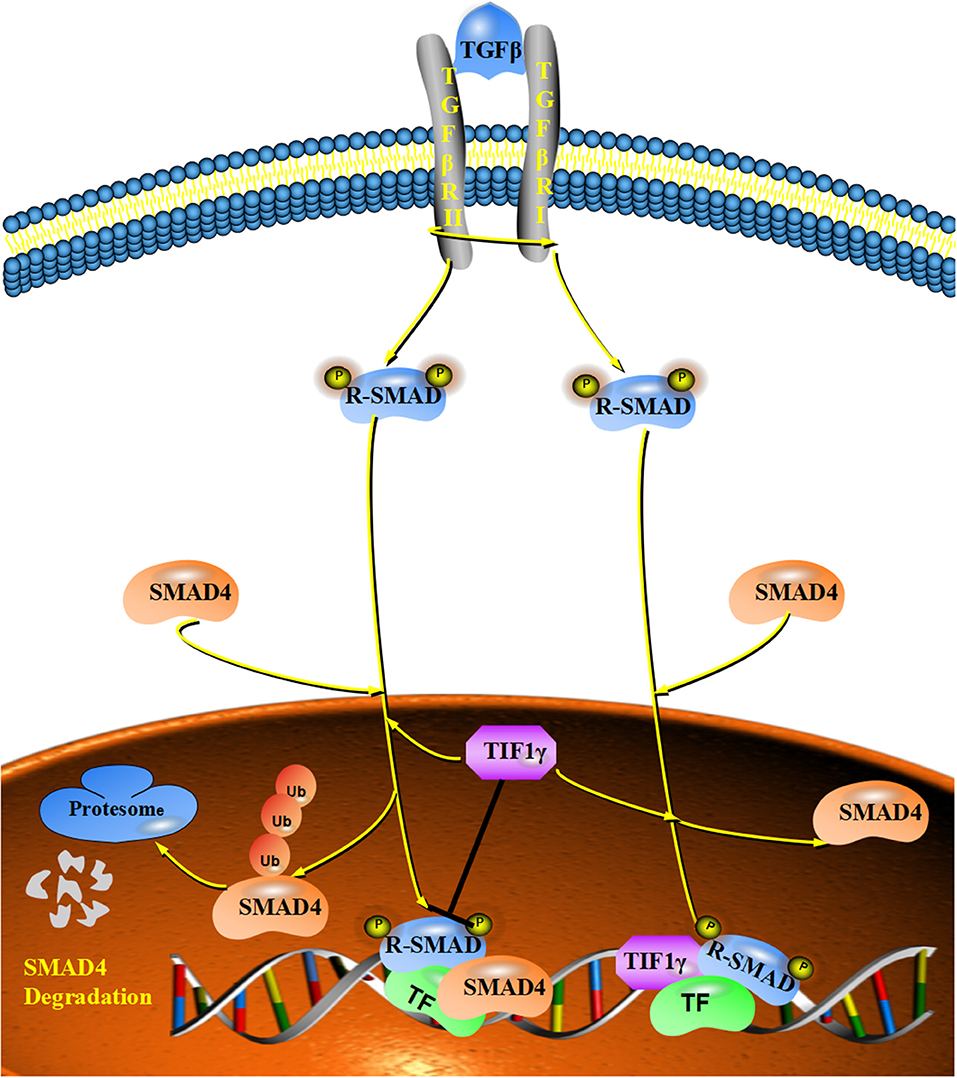
Figure 3. The inhibitory effect of TIF1γ on TGF-β/Smad signaling: TIF1γ can regulate TGF-β/Smad signaling in two different ways. On the one hand, it can monoubiquitinate Smad4 and inhibit the formation of Smad nuclear complexes. On the other hand, TIF1γ can function as a cofactor of phosphorylated (p)-Smad2/3, competing with Smad4 to inhibit the formation of the Smad2/3/4 complex.
The Inhibitory Effect of TIF1γ on Wnt/β-Catenin Signaling
The Wnt signaling pathway exerts an important role in regulating stem cell self-renewal, cell proliferation, differentiation, adhesion, and migration (57–61). Wnt protein can protect β-catenin from being phosphorylated by disrupting the “destruction complex” of β-catenin (62–64), which can enable β-catenin to translocate to the nucleus and form a complex with TCF/LEF (T-cell specific transcription factor/lymphoid enhancer-binding factor), which induces the expression of Wnt-targeted genes (65–67). Wnt/β-catenin signaling can influence tumor growth and metastasis by regulating the expression of the downstream genes, such as c-myc, cyclin D1, and Snail (68). Increasing numbers of studies report the dysregulation of Wnt/β-catenin signaling in many human cancers (68). For example, increased expression of Wnt ligands was detected in colon cancer, breast cancer and lung cancer (53, 69, 70). Furthermore, mutations of β-catenin were founded in colon cancer, gastric cancer, and hepatocellular carcinoma (69, 71, 72). Moreover, TIF1γ was reported to regulate Wnt/β-catenin signaling by interacting with and ubiquitylating nuclear β-catenin with the assistance of protein kinase Cδ, which degrades nuclear β-catenin and inhibits cell proliferation and tumorigenesis in glioblastoma (23). These studies provide new insights into the development of human cancers caused by aberrant activation of β-catenin (Figure 4).
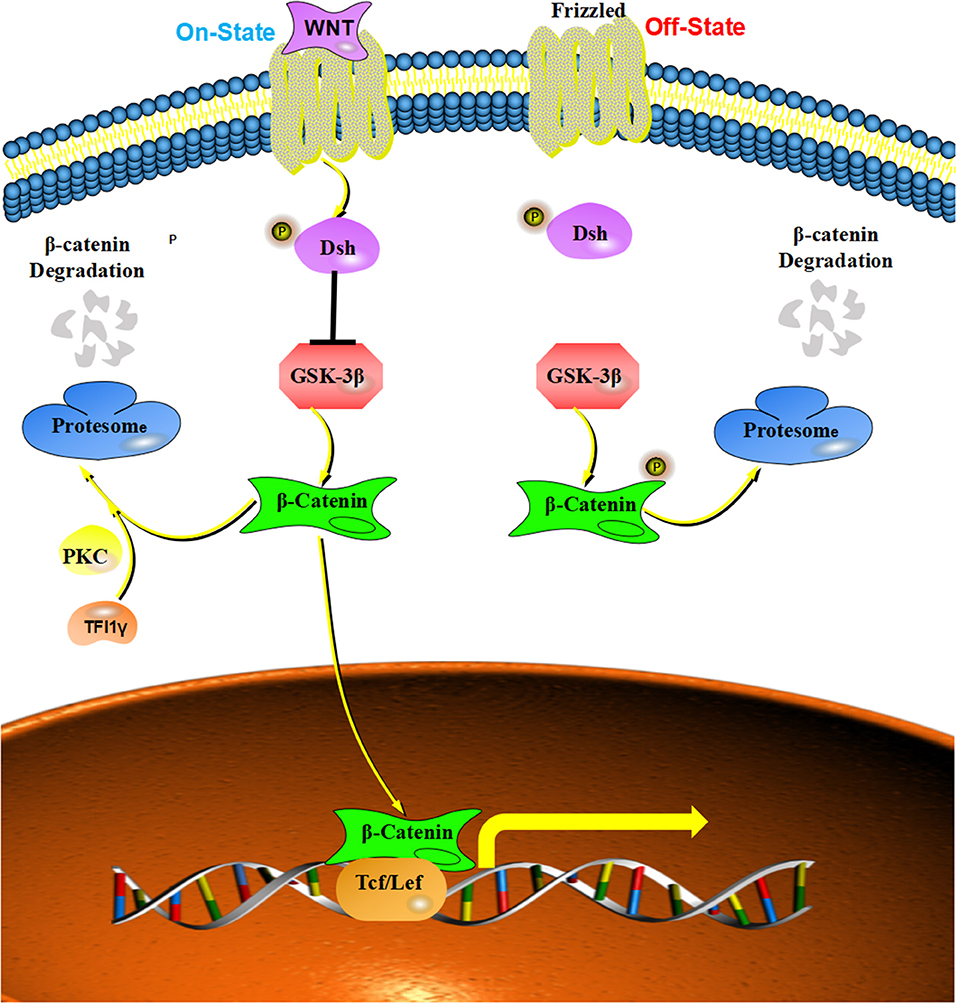
Figure 4. The inhibitory effect of TIF1γ on Wnt/β-catenin signaling: Wnt protein can protected β-catenin from being phosphorylated by disrupting the “destruction complex,” and TIF1γ can ubiquitylate nuclear β-catenin with the assistance of protein kinase Cδ (PKC) to inhibit Wnt/β-catenin signaling.
The Positive Effect of TIF1γ on DHX33-NLRP3 Signaling
Nod-like receptor 3 (NLRP3) is a member of the eponymous receptor family, which can perceive multiple types of stimulatory molecules (73–76), such as ATP, crystalline reagents and the microbial toxin nigericin, and form a macromolecular signaling complex with its adaptor protein ASC and procaspase-1 to induce inflammasome assembly (77–80). DHX33, a member of the DExD/H-box helicase family, is a cytosolic RNA sensor that can bind to and activate NLRP3 to oligomerize and recruit the adaptor protein ASC and cause the cleavage of pro-caspase-1 to the active form of caspase-1 (81, 82). Caspase-1 then transforms pro-IL-1β and pro-interleukin (IL)-18 into their biologically active mature secreted forms to induce inflammation (83, 84). There is increasing evidence that the expression of the NLRP3 inflammasome is dysregulated in many cancers, such as head and neck squamous cell carcinoma, hepatocellular carcinoma, and colorectal cancer (71, 85, 86). Furthermore, the overactivation of NLRP3 was related to poor survival and tumor invasiveness in head and neck squamous cell carcinoma and breast cancer (85, 87). At the same time, NLRP3 inflammasome takes part in the resistance to radiotherapy and chemotherapy in oral squamous cell carcinoma and glioblastoma (88, 89). While the activation of the NLRP3 inflammasome complex needs the assistance of TIF1γ, TIF1γ can bind to and ubiquitinate DHX33 at lysine 218, which helps DHX33 activate NLRP3 under dsRNA stimulation (90). Accordingly, a knockdown of TIF1γ disrupted the dsRNA-induced NLRP3 inflammasome activation in macrophages (90). Furthermore, increasing numbers of studies demonstrate that the NLRP3 inflammasome plays a vital role in the metastasis of tumors (89, 91, 92). Taken together, these results imply that TIF1γ might influence the metastasis of tumors via DHX33-NLRP3 signaling (Figure 5).
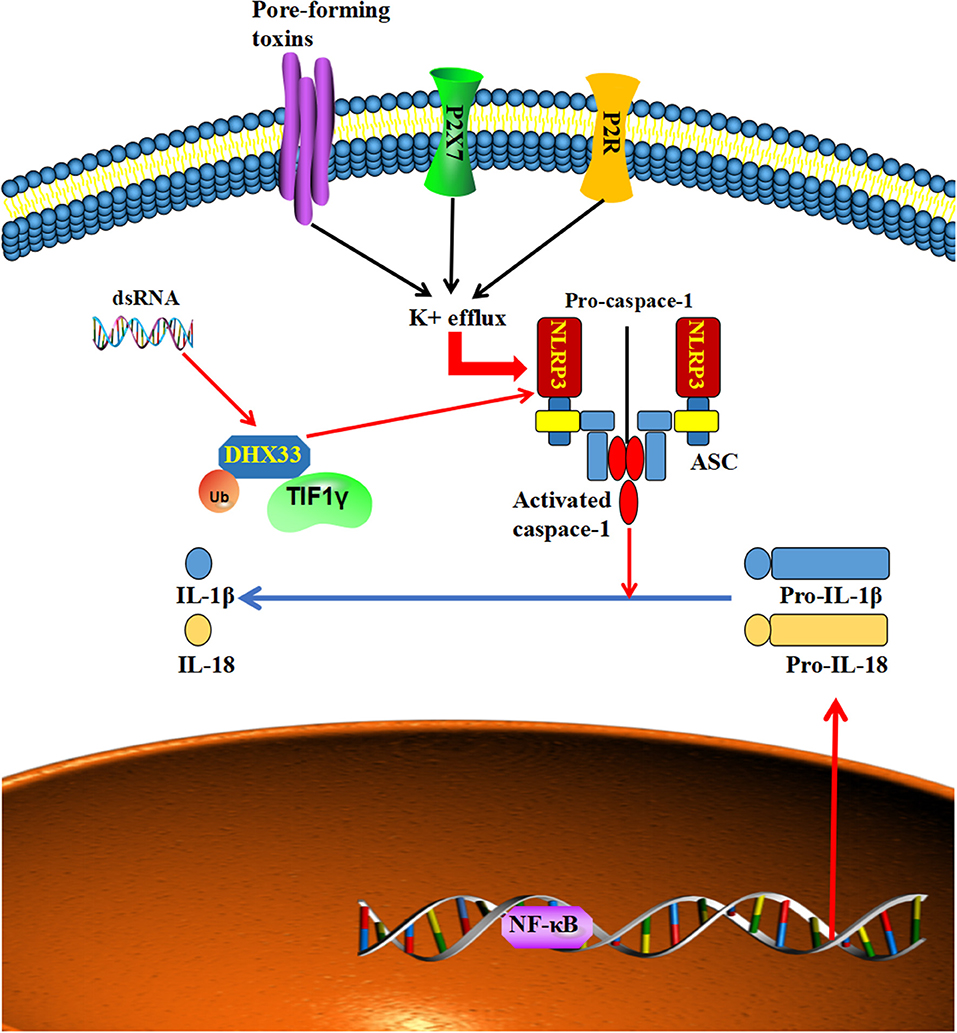
Figure 5. Multiple types of stimulatory signals can activate NLRP3 to form a macromolecular signaling complex with its adaptor protein ASC, which causes the cleavage of pro-caspase-1 to the active form of caspase-1, which in turn transforms pro-IL-1β and pro-interleukin (IL)-18 into their biologically active, mature secreted forms. Furthermore, TIF1γ can ubiquitinate and assist DHX33 to interact with, and activate NLRP3 to form a macromolecular signaling complex to produce mature IL-1β and IL-18.
The Roles of TIF1γ in Different Types of Tumors (Table 1)
The Expression of TIF1γ in Tumors
The expression of TIF1γ varies in different tumors. The expression of TIF1γ is low in most tumors, such as liver cancer, pancreatic cancer, lung cancer, renal carcinoma, and glioblastoma (20, 21, 23, 25, 27). However, its expression is increased in some tumors, such as colorectal cancer and breast cancer (Table 1). At the same time, the roles of TIF1γ also vary in different tumors. In some tumors, TIF1γ functions as a tumor promoter and prevents the apoptosis of tumor cells, but it also acts as a tumor suppressor in other tumors and inhibits the growth of tumor cells.
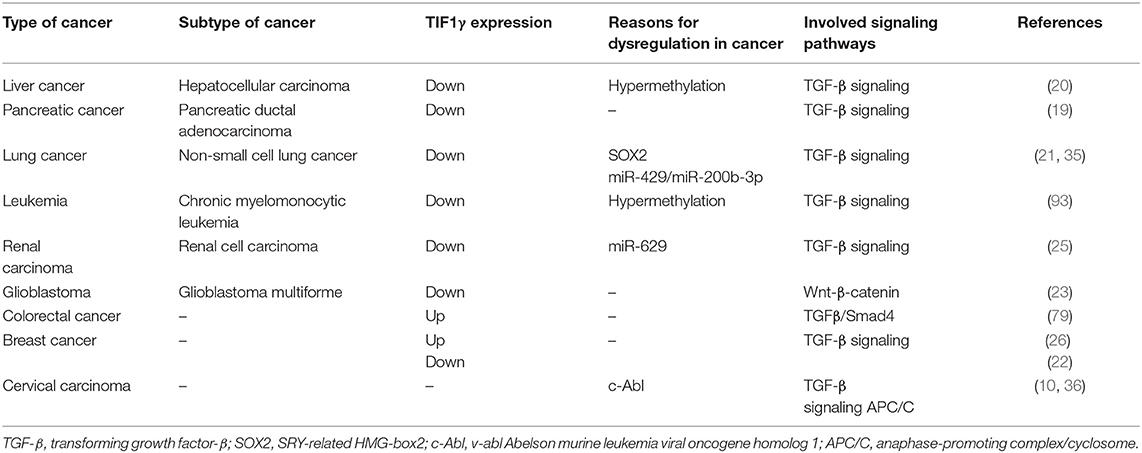
Table 1. The dysregulated expression and target genes of TIF1γ in cancer.
TIF1γ in Liver Cancer
In one of our own earlier studies (20), we found that the CpG islands in the TIF1γ promoter were hypermethylated and the expression of TIF1γ was reduced in hepatocellular carcinoma (HCC), especially in samples from advanced HCC. At the same time, the decreased expression of TIF1γ was an independent and significant risk factor for recurrence and survival after curative resection (20). Furthermore, the combined measurement of TIF1γ and p-Smad2 was found to be a more powerful predictor of poor prognosis in HCC patients. Interestingly, TIF1γ plays a double role in HCC cells. It favors tumor growth in early-, but not in advanced-stage HCC. However, TIF1γ inhibits the invasion and metastasis of both early- and advanced-stage HCC. Mechanistically, TIF1γ can suppress TGF-β/Smad signaling by monoubiquitinating Smad4 and inhibiting the formation of the Smad2/3/4 complex to regulate tumor growth and metastasis. Specifically, TIF1γ can relieve TGFβ-induced growth inhibition and favor tumor growth in early-stage HCC. In advanced-stage HCC, TIF1γ in turn inhibits TGF-β-induced tumor invasion and metastasis. Furthermore, we confirmed that the downstream cascades of TGF-β/Smad signaling, such as c-myc, p21/cip1, p15/ink4b, and protein kinase B–signaling transactivation, are also downregulated by TIF1γ (20). At the same time, another study reported that TIF1γ can interact with TIF1α and TIF1β to form a regulatory complex that suppresses murine hepatocellular carcinoma (94). Furthermore, TIF1γ can also interact with TIF1α to inhibit VL30 retrotransposons, and thus plays an important role in retroviral restriction and antiviral defense, which broadens what is known about the roles of the TRIM family of proteins in the endogenous retrovirus (ERV)-derived oncogenic regulatory network (95).
TIF1γ in Pancreatic Cancer
TIF1γ expression was reported to be decreased in pancreatic cancer tissues (19, 27, 96). At the same time, the expression of TIF1γ was inversely correlated with Smad 4 expression in pancreatic cancer cell lines and the overexpression of TIF1γ suppressed TGFβ signaling to inhibit the growth and invasion of pancreatic cancer cells (27). Furthermore, TIF1γ inactivation was found to cooperate with KrasG12D activation to induce cystic pancreatic tumors that resemble human intraductal papillary mucinous neoplasms (19).
TIF1γ in Colorectal Cancer
TIF1γ was found to be overexpressed in colorectal cancer and its expression levels were found to be associated with advanced tumor stage (7, 79). Furthermore, the expression of TIF1γ attenuated TGF-β-induced growth inhibition (7). At the same time, increased TIF1γ expression was correlated with a loss of Smad4 in colorectal cancer and predicted a poor prognosis for colorectal cancer patients (79). However, another study reported that the knockdown of TIF1γ resulted in genomic instability and cancer progression in colorectal cancer by regulating mitotic checkpoints (28). At the same time, TIF1γ was reported to interact with ALC1 (Amplified in Liver Cancer 1) and is involved in DNA repair in a Poly (ADP-ribose) polymerase 1 (PARP1)-dependent manner (4). Furthermore, Shi et al. reported that the loss of TIF1γ in colorectal cancer cell lines can cause resistance to the bromodomain and extraterminal domain (BET) protein inhibitors via MYC and TGF-β-dependent mechanisms (97). This further implies that TIF1γ also acts as a tumor suppressor in colorectal cancer. However, the underlying mechanisms that can explain the contradictory results require further research.
TIF1γ in Breast Cancer
TIF1γ expression was reported to be slightly reduced in human breast cancer tissues, compared to normal breast tissues. Moreover, the overexpression of FOXM1 in breast cancer can interact with Smad3/Smad4 and inhibit the binding of TIF1γ to Smad4 to prevent its ubiquitination, which can attenuate the inhibitory effects of TIF1γ on TGF-β signaling to promote the metastasis of breast cancer (26). However, another study reported that TIF1γ expression was increased in 35.9 % of breast cancer patients and its expression was related to younger age, estrogen receptor (ER) negativity, and tumors larger than 2 cm. Additionally, TIF1γ overexpression was related to poor prognosis in breast cancer patients (22), but the contradictory results require more thorough investigation. Furthermore, the deletion of TIF1γ was found to enhance TGFβ-induced growth inhibition in breast cell lines via Smad4 in MDA-MB468 signaling (7). In addition, TIF1γ can inhibit the EMT of mammary epithelial cells and terminal differentiation of mammary alveolar epithelial cells by antagonizing Smad4 (9, 47).
TIF1γ in Lung Cancer
TIF1γ was reported to be decreased in non-small cell lung cancer (NSCLC), but the CpG islands in the TIF1γ promoter were not found to be hypermethylated (98). Furthermore, Wang et al. reported that the expression of TIF1γ was downregulated by the overexpression of SOX2 in NSCLC tissues (21), and the reduced expression of TIF1γ was associated with poor survival of the patients (35). Furthermore, knockdown of TIF1γ was found to promote TGF-β-induced EMT and invasion of NSCLC cells in vitro and favor their metastasis. Conversely, the knockdown of SOX2 attenuated TGF-β-induced EMT and invasion of NSCLC cells. At the same time, expression of the circular RNA PTK2 was reported to be decreased in metastatic NSCLC tissues compared to non-metastatic NSCLC tissues, and was found to protect TIF1γ from miR-429/miR-200b-3p-mediated downregulation (35). Finally, the overexpression of circPTK2 was found to promote TIF1γ expression and suppress TGF-β-induced EMT and NSCLC cell invasion (35).
TIF1γ in Chronic Myelomonocytic Leukemia
TIF1γ was reported to be downregulated in a subset of chronic myelomonocytic leukemia (CMLL) patients (93, 99), and tif1gΔ/Δ mice were confirmed to develop a CMML-like myeloproliferative disease with monocytic features. Furthermore, TIF1γ was found to regulate the differentiation of hematopoietic progenitor populations (17) and promote the expansion of the granulomonocytic progenitor compartment. At the same time, the response of hematopoietic cells to TGF-β is suppressed in tif1γΔ/Δ mice (93). Finally, the CpG sequences of TIF1γ were found to be hypermethylated and a demethylating agent recovered the normal epigenetic status of the TIF1γ promoter and the expression of TIF1γ in human cells (93), which implies that TIF1γ is an epigenetically regulated tumor suppressor gene in hematopoietic cells.
TIF1γ in Other Tumors
TIF1γ expression was found to be decreased and associated with pathological stages and grades in clear cell renal cell carcinoma, and the overexpression of TIF1γ inhibited the growth and invasion of its tumor cells (25). At the same time, TIF1γ expression was decreased and inversely correlated with the levels of β-catenin Ser715 phosphorylation in primary glioblastoma multiforme (GBM) specimens, and the overexpression of TIF1γ inhibited the growth of GBM cells by destabilizing β-catenin (23). However, TIF1γ can promote tumor cell survival by being recruited by PU.1 to bind to two lineage-specific enhancers near the Bim gene and antagonizing PU.1 function in B lymphoblastic leukemia (B-ALL) cells (100). At the same time, TIF1γ can also favor the proliferation of tumor cells by binding to the anaphase-promoting complex/cyclosome (APC/C) to promote the mitosis in HeLa cells (10). In addition, the knockdown of TIF1γ can enhance the TGF-β-induced elongation of HeLa cells (36).
A Tentative Hypothesis That Explains the Dual Roles of TIF1γ in Cancer
As presented in this review, TIF1γ can function either as a tumor suppressor or promoter according to the different cellular contexts. For example, TIF1γ acts as a tumor suppressor and inhibits the tumor growth of non-small-cell lung cancer (35). By contrast, TIF1γ act as a tumor promoter in B lymphoblastic leukemia and can prevent the apoptosis of tumor cells (100). The potential underlying mechanism that causes these contrary results remains a mystery. We hypothesized that the different functions of TIF1γ might be attributed to inconsistent experimental results. On the one hand, as an intermediary transcriptional factor, TIF1γ can regulate the transcription of target genes. For example, TIF1γ was found to be recruited by PU.1 to bind to two lineage-specific enhancers near the Bim gene to antagonize PU.1 function and promote the survival B-ALL cells (100, 101). On the other hand, as an E3 ubiquitin-ligase family member, TIF1γ can monoubiquitinate targeted proteins leading to their degradation. For example, TIF1γ can monoubiquitinate Smad4 and suppress TGFβ signaling to inhibit the growth and invasion of pancreatic cancer cells (27). However, the mechanisms underlying the dual roles of TIF1γ in other tumors still require further research.
Prospects and Conclusions
As TIF1γ was reported to regulate the fate and differentiation of hematopoietic cells, increasing studies have reported on the roles of TIF1γ in immunity (17, 102–104). Ferri et al. reported that TIF1γ can be recruited by PU.1 to bind to the Ifnb1 Control Element (ICE) and regulate the chromatin structure of the interferon-β gene (Ifnb1), which suppresses its transcription by preventing the recruitment of CBP/p300 in the late phase of macrophage activation (105). Additionally, TIF1γ was also reported to regulate the production and activation of macrophages (106). At the same time, it was also reported that TIF1γ is involved in and regulates macrophage motility (107). TIF1γ was also reported to regulate the differentiation of granulomonopoiesis in mice (108). Furthermore, it was reported that TIF1γ also controls the lineage expansion of invariant natural killer T (iNKT) cells (109). In addition, TIF1γ is involved in the differentiation and development of T helper 17 (Th17) cells and can decrease the production of IL-10 to regulate the cells' proinflammatory function (110), and numerous studies have demonstrated that Th17 cells, macrophages, and iNKT cells play important roles in antitumor immunity (111–117). Taken together, it can be speculated that TIF1γ might be involved in tumoral immunity and this question certainly merits further investigation in future studies.
The abnormal expression of TIF1γ has been evidenced in many kinds of tumors and plays a vital role in cancer progression and metastasis (19, 21, 25). Furthermore, TIF1γ might become a potential prognostic marker for cancer patients. For example, increased TIF1γ expression predicted a poor prognosis for colorectal cancer patients (20, 79). At the same time, TIF1γ might become a potential therapeutic target for cancer treatment. Abundant evidence demonstrates that TIF1γ is downregulated and plays an important tumor suppressive role in multiple types of cancer (19, 20, 23). Our team also showed that lentivirus-mediated TIF1γ overexpression could inhibit the invasion and metastasis of HCC cells in vivo (20). However, the exact mechanisms underlying the dual roles of TIF1γ in cancer are still unclear. Moreover, the functions of other domains of TIF1γ, such as the B boxes and the coiled coil domain, also remains unclear. Solving these problems will help us better understand the conflicting roles of TIF1γ in cancer.
Author Contributions
XC and BZ provided direction and guidance throughout the preparation of this manuscript. CY, ZD, and HL wrote and edited the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the State Key Project on Infectious Diseases of China (grant no. 2018ZX10723204-003), the National Natural Science Foundation of China (grant no. 81572855 to XC, grant no. 81572427 to BZ, grant nos. 81874065 and 81500565 to ZD), and the Hepato-Biliary-Pancreatic Malignant Tumor Investigation Fund of Chen Xiaoping Foundation for the Development of Science and Technology of Hubei Province (CXPJJH11800001-2018356 to ZD).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
2. Bai X, Kim J, Yang Z, Jurynec MJ, Akie TE, Lee J, et al. TIF1gamma controls erythroid cell fate by regulating transcription elongation. Cell. (2010) 142:133–43. doi: 10.1016/j.cell.2010.05.028
3. Bai X, Trowbridge JJ, Riley E, Lee JA, DiBiase A, Kaartinen VM, et al. TiF1-gamma plays an essential role in murine hematopoiesis and regulates transcriptional elongation of erythroid genes. Dev Biol. (2013) 373:422–30. doi: 10.1016/j.ydbio.2012.10.008
4. Kulkarni A, Oza J, Yao M, Sohail H, Ginjala V, Tomas-Loba A, et al. Tripartite Motif-containing 33 (TRIM33) protein functions in the poly(ADP-ribose) polymerase (PARP)-dependent DNA damage response through interaction with Amplified in Liver Cancer 1 (ALC1) protein. J Biol Chem. (2013) 288:32357–69. doi: 10.1074/jbc.M113.459164
5. Guo J, Qin W, Xing Q, Gao M, Wei F, Song Z, et al. TRIM33 is essential for osteoblast proliferation and differentiation via BMP pathway. J Cell Physiol. (2017) 232:3158–69. doi: 10.1002/jcp.25769
6. Rajderkar S, Panaretos C, Kaartinen V. Trim33 regulates early maturation of mouse embryoid bodies in vitro. Biochem Biophys Rep. (2017) 12:185–92. doi: 10.1016/j.bbrep.2017.10.002
7. Dupont S, Zacchigna L, Cordenonsi M, Soligo S, Adorno M, Rugge M, et al. Germ-layer specification and control of cell growth by Ectodermin, a Smad4 ubiquitin ligase. Cell. (2005) 121:87–99. doi: 10.1016/j.cell.2005.01.033
8. Falk S, Joosten E, Kaartinen V, Sommer L. Smad4 and Trim33/Tif1γ redundantly regulate neural stem cells in the developing cortex. Cerebral Cortex. (2014) 24:2951–63. doi: 10.1093/cercor/bht149
9. Hesling C, Lopez J, Fattet L, Gonzalo P, Treilleux I, Blanchard D, et al. Tif1γ is essential for the terminal differentiation of mammary alveolar epithelial cells and for lactation through SMAD4 inhibition. Development. (2013) 140:167–75. doi: 10.1242/dev.085068
10. Sedgwick GG, Townsend K, Martin A, Shimwell NJ, Grand RJ, Stewart GS, et al. Transcriptional intermediary factor 1γ binds to the anaphase-promoting complex/cyclosome and promotes mitosis. Oncogene. (2013) 32:4622–33. doi: 10.1038/onc.2012.501
11. Zachou A, Zouvelou V, Papadimas GK, Rentzos M, Papadopoulos C. Anti-TIF1-γ autoantibodies-positive dermatomyositis: where is the malignancy? Rheumatology. (2018) 57:756. doi: 10.1093/rheumatology/kex342
12. Bernet LL, Lewis MA, Rieger KE, Casciola-Rosen L, Fiorentino DF. Ovoid palatal patch in dermatomyositis: a novel finding associated with anti-TIF1γ (p155) antibodies. JAMA Dermatol. (2016) 152:1049–51. doi: 10.1001/jamadermatol.2016.1429
13. Muro Y, Ishikawa A, Sugiura K, Akiyama M. Clinical features of anti-TIF1-α antibody-positive dermatomyositis patients are closely associated with coexistent dermatomyositis-specific autoantibodies and anti-TIF1-γ or anti-Mi-2 autoantibodies. Rheumatology. (2012) 51:1508–13. doi: 10.1093/rheumatology/kes073
14. Colak S, Ten Dijke P. Targeting TGF-β signaling in cancer. Trends Cancer. (2017) 3:56–71. doi: 10.1016/j.trecan.2016.11.008
15. Luo J, Chen XQ, Li P. The role of TGF-β and Its receptors in gastrointestinal cancers. Transl Oncol. (2019) 12:475–84. doi: 10.1016/j.tranon.2018.11.010
16. Loffek S. Transforming of the tumor microenvironment: implications for TGF-β inhibition in the context of immune-checkpoint therapy. J Oncol. (2018) 2018:9732939. doi: 10.1155/2018/9732939
17. He W, Dorn DC, Erdjument-Bromage H, Tempst P, Moore MA, Massague J. Hematopoiesis controlled by distinct TIF1gamma and Smad4 branches of the TGFbeta pathway. Cell. (2006) 125:929–41. doi: 10.1016/j.cell.2006.03.045
19. Vincent DF, Yan KP, Treilleux I, Gay F, Arfi V, Kaniewski B, et al. Inactivation of TIF1gamma cooperates with Kras to induce cystic tumors of the pancreas. PLoS Genet. (2009) 5:e1000575. doi: 10.1371/journal.pgen.1000575
20. Ding ZY, Jin GN, Wang W, Chen WX, Wu YH, Ai X, et al. Reduced expression of transcriptional intermediary factor 1 gamma promotes metastasis and indicates poor prognosis of hepatocellular carcinoma. Hepatology. (2014) 60:1620–36. doi: 10.1002/hep.27273
21. Wang L, Yang H, Lei Z, Zhao J, Chen Y, Chen P, et al. Repression of TIF1γ by SOX2 promotes TGF-β-induced epithelial-mesenchymal transition in non-small-cell lung cancer. Oncogene. (2016) 35:867–77. doi: 10.1038/onc.2015.141
22. Kassem L, Deygas M, Fattet L, Lopez J, Goulvent T, Lavergne E, et al. TIF1γ interferes with TGFβ1/SMAD4 signaling to promote poor outcome in operable breast cancer patients. BMC Cancer. (2015) 15:453. doi: 10.1186/s12885-015-1471-y
23. Xue J, Chen Y, Wu Y, Wang Z, Zhou A, Zhang S, et al. Tumour suppressor TRIM33 targets nuclear beta-catenin degradation. Nat Commun. (2015) 6:6156. doi: 10.1038/ncomms7156
24. Yin X, Xu C, Zheng X, Yuan H, Liu M, Qiu Y, et al. SnoN suppresses TGF-β-induced epithelial-mesenchymal transition and invasion of bladder cancer in a TIF1γ-dependent manner. Oncol Rep. (2016) 36:1535–41. doi: 10.3892/or.2016.4939
25. Jingushi K, Ueda Y, Kitae K, Hase H, Egawa H, Ohshio I, et al. miR-629 targets TRIM33 to promote TGFβ/Smad signaling and metastatic phenotypes in ccRCC. Mol Cancer Res. (2015) 13:565–74. doi: 10.1158/1541-7786.MCR-14-0300
26. Xue J, Lin X, Chiu WT, Chen YH, Yu G, Liu M, et al. Sustained activation of SMAD3/SMAD4 by FOXM1 promotes TGF-β-dependent cancer metastasis. J Clin Invest. (2014) 124:564–79. doi: 10.1172/JCI71104
27. Ligr M, Wu X, Daniels G, Zhang D, Wang H, Hajdu C, et al. Imbalanced expression of Tif1γ inhibits pancreatic ductal epithelial cell growth. Am J Cancer Res. (2014) 4:196–210.
28. Pommier RM, Gout J, Vincent DF, Alcaraz LB, Chuvin N, Arfi V, et al. TIF1γ suppresses tumor progression by regulating mitotic checkpoints and chromosomal stability. Cancer Res. (2015) 75:4335–50. doi: 10.1158/0008-5472.CAN-14-3426
29. Venturini L, You J, Stadler M, Galien R, Lallemand V, Koken MH, et al. TIF1γ, a novel member of the transcriptional intermediary factor 1 family. Oncogene. (1999) 18:1209–17. doi: 10.1038/sj.onc.1202655
30. Dupont S, Mamidi A, Cordenonsi M, Montagner M, Zacchigna L, Adorno M, et al. FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell. (2009) 136:123–35. doi: 10.1016/j.cell.2008.10.051
31. Quere R, Saint-Paul L, Carmignac V, Martin RZ, Chretien ML, Largeot A, et al. Tif1γ regulates the TGF-β 1 receptor and promotes physiological aging of hematopoietic stem cells. Proc Natl Acad Sci USA. (2014) 111:10592–7. doi: 10.1073/pnas.1405546111
32. Ikeuchi Y, Dadakhujaev S, Chandhoke AS, Huynh MA, Oldenborg A, Ikeuchi M, et al. TIF1γ protein regulates epithelial-mesenchymal transition by operating as a small ubiquitin-like modifier (SUMO) E3 ligase for the transcriptional regulator SnoN1. J Biol Chem. (2014) 289:25067–78. doi: 10.1074/jbc.M114.575878
33. Heldin CH, Moustakas A. A new twist in Smad signaling. Dev Cell. (2006) 10:685–6. doi: 10.1016/j.devcel.2006.05.006
34. Agricola E, Randall RA, Gaarenstroom T, Dupont S, Hill CS. Recruitment of TIF1γ to chromatin via its PHD finger-bromodomain activates its ubiquitin ligase and transcriptional repressor activities. Mol Cell. (2011) 43:85–96. doi: 10.1016/j.molcel.2011.05.020
35. Wang L, Tong X, Zhou Z, Wang S, Lei Z, Zhang T, et al. Circular RNA hsa_circ_0008305 (circPTK2) inhibits TGF-β-induced epithelial-mesenchymal transition and metastasis by controlling TIF1γ in non-small cell lung cancer. Mol Cancer. (2018) 17:140. doi: 10.1186/s12943-018-0889-7
36. Yuki R, Tatewaki T, Yamaguchi N, Aoyama K, Honda T, Kubota S, et al. Desuppression of TGF-β signaling via nuclear c-Abl-mediated phosphorylation of TIF1γ /TRIM33 at Tyr-524,−610, and−1048. Oncogene. (2018) 38:637–55. doi: 10.1038/s41388-018-0481-z
37. Sohn SY, Hearing P. The adenovirus E4-ORF3 protein functions as a SUMO E3 ligase for TIF-1γ sumoylation and poly-SUMO chain elongation. Proc Natl Acad Sci USA. (2016) 113:6725–30. doi: 10.1073/pnas.1603872113
38. Forrester NA, Patel RN, Speiseder T, Groitl P, Sedgwick GG, Shimwell NJ, et al. Adenovirus E4orf3 targets transcriptional intermediary factor 1γ for proteasome-dependent degradation during infection. J Virol. (2012) 86:3167–79. doi: 10.1128/JVI.06583-11
39. Massague J. TGFβ signalling in context. Nat Rev Mol Cell Biol. (2012) 13:616–30. doi: 10.1038/nrm3434
40. Wu MY, Hill CS. Tgf-beta superfamily signaling in embryonic development and homeostasis. Dev Cell. (2009) 16:329–43. doi: 10.1016/j.devcel.2009.02.012
41. Gratchev A. TGF-β signalling in tumour associated macrophages. Immunobiology. (2017) 222:75–81. doi: 10.1016/j.imbio.2015.11.016
42. Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. (2000) 103:295–309. doi: 10.1016/S0092-8674(00)00121-5
43. Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer. (2010) 10:415–24. doi: 10.1038/nrc2853
44. Centurione L, Aiello FB. DNA repair and cytokines: TGF-β, IL-6, and thrombopoietin as different biomarkers of radioresistance. Front Oncol. (2016) 6:175. doi: 10.3389/fonc.2016.00175
45. Barcellos-Hoff MH, Cucinotta FA. New tricks for an old fox: impact of TGFβ on the DNA damage response and genomic stability. Sci Signal. (2014) 7:re5. doi: 10.1126/scisignal.2005474
46. Derynck R, Muthusamy BP, Saeteurn KY. Signaling pathway cooperation in TGF-β-induced epithelial-mesenchymal transition. Curr Opin Cell Biol. (2014) 31:56–66. doi: 10.1016/j.ceb.2014.09.001
47. Hesling C, Fattet L, Teyre G, Jury D, Gonzalo P, Lopez J, et al. Antagonistic regulation of EMT by TIF1γ and Smad4 in mammary epithelial cells. EMBO Rep. (2011) 12:665–72. doi: 10.1038/embor.2011.78
48. Samanta D, Datta PK. Alterations in the Smad pathway in human cancers. Front Biosci. (2012) 17:1281–93. doi: 10.2741/3986
49. Seoane J, Gomis RR. TGF-β family signaling in tumor suppression and cancer progression. Cold Spring Harb Perspect Biol. (2017) 9:a022277. doi: 10.1101/cshperspect.a022277
50. Fukushima T, Mashiko M, Takita K, Otake T, Endo Y, Sekikawa K, et al. Mutational analysis of TGF-beta type II receptor, Smad2, Smad3, Smad4, Smad6 and Smad7 genes in colorectal cancer. J Exp Clin Cancer Res. (2003) 22:315–20.
51. Maliekal TT, Antony ML, Nair A, Paulmurugan R, Karunagaran D. Loss of expression, and mutations of Smad 2 and Smad 4 in human cervical cancer. Oncogene. (2003) 22:4889–97. doi: 10.1038/sj.onc.1206806
52. Yakicier MC, Irmak MB, Romano A, Kew M, Ozturk M. Smad2 and Smad4 gene mutations in hepatocellular carcinoma. Oncogene. (1999) 18:4879–83. doi: 10.1038/sj.onc.1202866
53. Thiagalingam S, Lengauer C, Leach FS, Schutte M, Hahn SA, Overhauser J, et al. Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat Genet. (1996) 13:343–6. doi: 10.1038/ng0796-343
54. Hahn SA, Seymour AB, Hoque AT, Schutte M, da Costa LT, Redston MS, et al. Allelotype of pancreatic adenocarcinoma using xenograft enrichment. Cancer Res. (1995) 55:4670–5.
55. Levy L, Hill CS. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. (2006) 17:41–58. doi: 10.1016/j.cytogfr.2005.09.009
56. Bellaye PS, Wettstein G, Burgy O, Besnard V, Joannes A, Colas J, et al. The small heat-shock protein αB-crystallin is essential for the nuclear localization of Smad4: impact on pulmonary fibrosis. J Pathol. (2014) 232:458–72. doi: 10.1002/path.4314
57. Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. (2013) 13:11–26. doi: 10.1038/nrc3419
58. Kypta RM, Waxman J. Wnt/β-catenin signalling in prostate cancer. Nat Rev Urol. (2012) 9:418–28. doi: 10.1038/nrurol.2012.116
59. Cui J, Jiang W, Wang S, Wang L, Xie K. Role of Wnt/β-catenin signaling in drug resistance of pancreatic cancer. Curr Pharm Des. (2012) 18:2464–71. doi: 10.2174/13816128112092464
60. Wang B, Tian T, Kalland KH, Ke X, Qu Y. Targeting Wnt/β-catenin signaling for cancer immunotherapy. Trends Pharmacol Sci. (2018) 39:648–58. doi: 10.1016/j.tips.2018.03.008
61. Hu T, Li C. Convergence between Wnt-β-catenin and EGFR signaling in cancer. Mol Cancer. (2010) 9:236. doi: 10.1186/1476-4598-9-236
62. He L, Zhou H, Zeng Z, Yao H, Jiang W, Qu H. Wnt/β-catenin signaling cascade: a promising target for glioma therapy. J Cell Physiol. (2019) 234:2217–28. doi: 10.1002/jcp.27186
63. Ring A, Kim YM, Kahn M. Wnt/catenin signaling in adult stem cell physiology and disease. Stem Cell Rev Rep. (2014) 10:512–25. doi: 10.1007/s12015-014-9515-2
64. Hussain M, Xu C, Lu M, Wu X, Tang L, Wu X. Wnt/β-catenin signaling links embryonic lung development and asthmatic airway remodeling. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:3226–42. doi: 10.1016/j.bbadis.2017.08.031
65. Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. (2012) 149:1192–205. doi: 10.1016/j.cell.2012.05.012
66. Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell. (2017) 169:985–99. doi: 10.1016/j.cell.2017.05.016
67. Harb J, Lin PJ, Hao J. Recent development of Wnt signaling pathway inhibitors for cancer therapeutics. Curr Oncol Rep. (2019) 21:12. doi: 10.1007/s11912-019-0763-9
68. Yao H, Ashihara E, Maekawa T. Targeting the Wnt/β-catenin signaling pathway in human cancers. Expert Opin Ther Targets. (2011) 15:873–87. doi: 10.1517/14728222.2011.577418
69. Akiri G, Cherian MM, Vijayakumar S, Liu G, Bafico A, Aaronson SA. Wnt pathway aberrations including autocrine Wnt activation occur at high frequency in human non-small-cell lung carcinoma. Oncogene. (2009) 28:2163–72. doi: 10.1038/onc.2009.82
70. Holcombe RF, Marsh JL, Waterman ML, Lin F, Milovanovic T, Truong T. Expression of Wnt ligands and frizzled receptors in colonic mucosa and in colon carcinoma. Mol Pathol. (2002) 55:220–6. doi: 10.1136/mp.55.4.220
71. Woo DK, Kim HS, Lee HS, Kang YH, Yang HK, Kim WH. Altered expression and mutation of beta-catenin gene in gastric carcinomas and cell lines. Int J Cancer. (2001) 95:108–13. doi: 10.1002/1097-0215(20010320)95:2<108::AID-IJC1019>3.0.CO;2-%23
72. Kitaeva MN, Grogan L, Williams JP, Dimond E, Nakahara K, Hausner P, et al. Mutations in beta-catenin are uncommon in colorectal cancer occurring in occasional replication error-positive tumors. Cancer Res. (1997) 57:4478–81.
73. Moossavi M, Parsamanesh N, Bahrami A, Atkin SL, Sahebkar A. Role of the NLRP3 inflammasome in cancer. Mol Cancer. (2018) 17:158. doi: 10.1186/s12943-018-0900-3
74. Ahechu P, Zozaya G, Marti P, Hernandez-Lizoain JL, Baixauli J, Unamuno X, et al. NLRP3 inflammasome: a possible link between obesity-associated low-grade chronic inflammation and colorectal cancer development. Front Immunol. (2018) 9:2918. doi: 10.3389/fimmu.2018.02918
75. Tartey S, Kanneganti TD. Differential role of the NLRP3 inflammasome in infection and tumorigenesis. Immunology. (2019) 156:329–38. doi: 10.1111/imm.13046
76. Maruyama K, Nemoto E, Yamada S. Mechanical regulation of macrophage function - cyclic tensile force inhibits NLRP3 inflammasome-dependent IL-1β secretion in murine macrophages. Inflamm Regen. (2019) 39:3. doi: 10.1186/s41232-019-0092-2
77. Weigt SS, Palchevskiy V, Belperio JA. Inflammasomes and IL-1 biology in the pathogenesis of allograft dysfunction. J Clin Invest. (2017) 127:2022–9. doi: 10.1172/JCI93537
78. Choi AJ, Ryter SW. Inflammasomes: molecular regulation and implications for metabolic and cognitive diseases. Mol Cells. (2014) 37:441–8. doi: 10.14348/molcells.2014.0104
79. Jain S, Singhal S, Francis F, Hajdu C, Wang JH, Suriawinata A, et al. Association of overexpression of TIF1γ with colorectal carcinogenesis and advanced colorectal adenocarcinoma. World J Gastroenterol. (2011) 17:3994–4000. doi: 10.3748/wjg.v17.i35.3994
80. Franchi L, Munoz-Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. (2012) 13:325–32. doi: 10.1038/ni.2231
81. Schroder K, Tschopp J. The inflammasomes. Cell. (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
82. Mitoma H, Hanabuchi S, Kim T, Bao M, Zhang Z, Sugimoto N, et al. The DHX33 RNA helicase senses cytosolic RNA and activates the NLRP3 inflammasome. Immunity. (2013) 39:123–35. doi: 10.1016/j.immuni.2013.07.001
83. Afonina IS, Zhong Z, Karin M, Beyaert R. Limiting inflammation-the negative regulation of NF-κB and the NLRP3 inflammasome. Nat Immunol. (2017) 18:861–9. doi: 10.1038/ni.3772
84. Haneklaus M, O'Neill LA. NLRP3 at the interface of metabolism and inflammation. Immunol Rev. (2015) 265:53–62. doi: 10.1111/imr.12285
85. Wei Q, Mu K, Li T, Zhang Y, Yang Z, Jia X, et al. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab Invest. (2014) 94:52–62. doi: 10.1038/labinvest.2013.126
86. Bae JY, Lee SW, Shin YH, Lee JH, Jahng JW, Park K. P2X7 receptor and NLRP3 inflammasome activation in head and neck cancer. Oncotarget. (2017) 8:48972–82. doi: 10.18632/oncotarget.16903
87. Weichand B, Popp R, Dziumbla S, Mora J, Strack E, Elwakeel E, et al. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1β. J Exp Med. (2017) 214:2695–713. doi: 10.1084/jem.20160392
88. Li L, Liu Y. Aging-related gene signature regulated by Nlrp3 predicts glioma progression. Am J Cancer Res. (2015) 5:442–9.
89. Feng X, Luo Q, Zhang H, Wang H, Chen W, Meng G, et al. The role of NLRP3 inflammasome in 5-fluorouracil resistance of oral squamous cell carcinoma. J Exp Clin Cancer Res. (2017) 36:81. doi: 10.1186/s13046-017-0553-x
90. Weng L, Mitoma H, Trichot C, Bao M, Liu Y, Zhang Z, et al. The E3 ubiquitin ligase tripartite motif 33 is essential for cytosolic RNA-induced NLRP3 inflammasome activation. J Immunol. (2014) 193:3676–82. doi: 10.4049/jimmunol.1401448
91. Daley D, Mani VR, Mohan N, Akkad N, Pandian G, Savadkar S, et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J Exp Med. (2017) 214:1711–24. doi: 10.1084/jem.20161707
92. Dupaul-Chicoine J, Arabzadeh A, Dagenais M, Douglas T, Champagne C, Morizot A, et al. The Nlrp3 inflammasome suppresses colorectal cancer metastatic growth in the liver by promoting natural killer cell tumoricidal activity. Immunity. (2015) 43:751–63. doi: 10.1016/j.immuni.2015.08.013
93. Aucagne R, Droin N, Paggetti J, Lagrange B, Largeot A, Hammann A, et al. Transcription intermediary factor 1γ is a tumor suppressor in mouse and human chronic myelomonocytic leukemia. J Clin Invest. (2011) 121:2361–70. doi: 10.1172/JCI45213
94. Herquel B, Ouararhni K, Khetchoumian K, Ignat M, Teletin M, Mark M, et al. Transcription cofactors TRIM24, TRIM28, and TRIM33 associate to form regulatory complexes that suppress murine hepatocellular carcinoma. Proc Natl Acad Sci USA. (2011) 108:8212–7. doi: 10.1073/pnas.1101544108
95. Herquel B, Ouararhni K, Martianov I, Le Gras S, Ye T, Keime C, et al. Trim24-repressed VL30 retrotransposons regulate gene expression by producing noncoding RNA. Nat Struct Mol Biol. (2013) 20:339–46. doi: 10.1038/nsmb.2496
96. Vincent DF, Gout J, Chuvin N, Arfi V, Pommier RM, Bertolino P, et al. Tif1γ suppresses murine pancreatic tumoral transformation by a Smad4-independent pathway. Am J Pathol. (2012) 180:2214–21. doi: 10.1016/j.ajpath.2012.02.006
97. Shi X, Mihaylova VT, Kuruvilla L, Chen F, Viviano S, Baldassarre M, et al. Loss of TRIM33 causes resistance to BET bromodomain inhibitors through MYC- and TGF-β-dependent mechanisms. Proc Natl Acad Sci USA. (2016) 113:E4558–66. doi: 10.1073/pnas.1608319113
98. Wang L, Lei Z, Liu X, Liu R, Zhang H. [Association of mutation and methylation in the promoter region of TIF1γ with non-small cell lung cancer]. Zhongguo Fei Ai Za Zhi. (2013) 16:227–32. doi: 10.3779/j.issn.1009-3419.2013.05.02
99. Aucagne R, Droin N, Solary E, Bastie JN, Delva L. [TIF1γ: a tumor suppressor gene in chronic myelomonocytic leukemia]. Med Sci. (2011) 27(8–9):696–8. doi: 10.1051/medsci/2011278006
100. Wang E, Kawaoka S, Roe JS, Shi J, Hohmann AF, Xu Y, et al. The transcriptional cofactor TRIM33 prevents apoptosis in B lymphoblastic leukemia by deactivating a single enhancer. ELife. (2015) 4:e06377. doi: 10.7554/eLife.06377
101. Kusy S, Gault N, Ferri F, Lewandowski D, Barroca V, Jaracz-Ros A, et al. Adult hematopoiesis is regulated by TIF1γ, a repressor of TAL1 and PU.1 transcriptional activity. Cell Stem Cell. (2011) 8:412–25. doi: 10.1016/j.stem.2011.02.005
102. Ransom DG, Bahary N, Niss K, Traver D, Burns C, Trede NS, et al. The zebrafish moonshine gene encodes transcriptional intermediary factor 1gamma, an essential regulator of hematopoiesis. PLoS Biol. (2004) 2:E237. doi: 10.1371/journal.pbio.0020237
103. Ohta M, Greenberger JS, Anklesaria P, Bassols A, Massague J. Two forms of transforming growth factor-beta distinguished by multipotential haematopoietic progenitor cells. Nature. (1987) 329:539–41. doi: 10.1038/329539a0
104. Kusy S, Romeo PH. [TIF1γ is a chief conductor of the hematopoietic system]. Med Sci. (2011) 27(8–9):698–700. doi: 10.1051/medsci/2011278007
105. Ferri F, Parcelier A, Petit V, Gallouet AS, Lewandowski D, Dalloz M, et al. TRIM33 switches off Ifnb1 gene transcription during the late phase of macrophage activation. Nat Commun. (2015) 6:8900. doi: 10.1038/ncomms9900
106. Gallouet AS, Ferri F, Petit V, Parcelier A, Lewandowski D, Gault N, et al. Macrophage production and activation are dependent on TRIM33. Oncotarget. (2017) 8:5111–22. doi: 10.18632/oncotarget.13872
107. Demy DL, Tauzin M, Lancino M, Le Cabec V, Redd M, Murayama E, et al. Trim33 is essential for macrophage and neutrophil mobilization to developmental or inflammatory cues. J Cell Sci. (2017) 130:2797–807. doi: 10.1242/jcs.203471
108. Chretien ML, Legouge C, Martin RZ, Hammann A, Trad M, Aucagne R, et al. Trim33/Tif1 γ is involved in late stages of granulomonopoiesis in mice. Exp Hematol. (2016) 44:727–39.e6. doi: 10.1016/j.exphem.2016.04.009
109. Doisne JM, Bartholin L, Yan KP, Garcia CN, Duarte N, Le Luduec JB, et al. iNKT cell development is orchestrated by different branches of TGF-beta signaling. J Exp Med. (2009) 206:1365–78. doi: 10.1084/jem.20090127
110. Tanaka S, Jiang Y, Martinez GJ, Tanaka K, Yan X, Kurosaki T, et al. Trim33 mediates the proinflammatory function of Th17 cells. J Exp Med. (2018) 215:1853–68. doi: 10.1084/jem.20170779
111. Duan MC, Zhong XN, Liu GN, Wei JR. The Treg/Th17 paradigm in lung cancer. J Immunol Res. (2014) 2014:730380. doi: 10.1155/2014/730380
112. Joerger M, Finn SP, Cuffe S, Byrne AT, Gray SG. The IL-17-Th1/Th17 pathway: an attractive target for lung cancer therapy? Exp Opin Ther Targets. (2016) 20:1339–56. doi: 10.1080/14728222.2016.1206891
113. Song Y, Yang JM. Role of interleukin (IL)-17 and T-helper (Th)17 cells in cancer. Biochem Biophys Res Commun. (2017) 493:1–8. doi: 10.1016/j.bbrc.2017.08.109
114. Guery L, Hugues S. Th17 cell plasticity and functions in cancer immunity. BioMed Res Int. (2015) 2015:314620. doi: 10.1155/2015/314620
115. Ostuni R, Kratochvill F, Murray PJ, Natoli G. Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol. (2015) 36:229–39. doi: 10.1016/j.it.2015.02.004
116. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. (2015) 27:462–72. doi: 10.1016/j.ccell.2015.02.015
Keywords: TIF1γ, cancer, TGFβ/Smad signaling, Wnt/β-catenin signaling, DHX33-NLRP3 signaling
Citation: Yu C, Ding Z, Liang H, Zhang B and Chen X (2019) The Roles of TIF1γ in Cancer. Front. Oncol. 9:979. doi: 10.3389/fonc.2019.00979
Received: 15 April 2019; Accepted: 13 September 2019;
Published: 02 October 2019.
Edited by:
Massimiliano Berretta, Centro di Riferimento Oncologico di Aviano (IRCCS), ItalyReviewed by:
Giuseppe Palma, National Cancer Institute G. Pascale Foundation (IRCCS), ItalyGermain Gillet, Université Claude Bernard Lyon 1, France
Copyright © 2019 Yu, Ding, Liang, Zhang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bixiang Zhang, Yml4aWFuZ3poYW5nQDE2My5jb20=; Xiaoping Chen, Y2hlbnhwY2hlbnhwQDE2My5jb20=
†These authors have contributed equally to this work