 Lenka Sedlarikova1†
Lenka Sedlarikova1† Eva Kriegova
Eva Kriegova- 1Department of Immunology, Faculty of Medicine and Dentistry, Palacky University and University Hospital, Olomouc, Czechia
- 2Department of Hemato-Oncology, Faculty of Medicine and Dentistry, Palacky University and University Hospital, Olomouc, Czechia
Inhibitors of B-cell receptor signaling, ibrutinib and idelalisib, and BCL-2 antagonist, venetoclax, have become the mainstay of treatment for chronic lymphocytic leukemia (CLL). Despite significant efficacy in most CLL patients, some patients develop resistance to these agents and progress on these drugs. We provide a state-of-the-art overview of the acquired resistance to novel agents. In 80% of patients with ibrutinib failure, acquired mutations in BTK and PLCG2 genes were detected. No distinct unifying resistance-associated mutations or deregulated signaling pathways have been reported in idelalisib failure. Acquired mutations in the BCL2 gene were detected in patients who had failed on venetoclax. In most cases, patients who have progressed on ibrutinib and venetoclax experience resistance-associated mutations, often present at low allelic frequencies. Resistance-associated mutations tend to occur between the second and fourth years of treatment and may already be detected several months before clinical relapse. We also discuss the development of next-generation agents for CLL patients who have acquired resistant mutations to current inhibitors.
Introduction
In the last decade, the great shift in the therapeutic management of chronic lymphocytic leukemia (CLL) is attributed to the approval of two B-cell receptor (BCR) signaling pathway inhibitors (ibrutinib and idelalisib) and the BCL-2 antagonist venetoclax. These novel agents have shown significant clinical efficacy in high-risk patients with relapsed/refractory (R/R) disease with 17p deletion and/or TP53 mutation and complex karyotype as well as in previously untreated patients with/without poor-risk features (1–4). Despite the induction of long-term remission in most CLL patients, in some patients, the treatment fails. The number of patients who progress or develop clinical resistance is expected to increase with the growing number of patients indicated for this treatment and due to the long-term administration of these agents. Therefore, understanding the mechanisms driving resistance and identification of involved driver mutations and signaling pathways is a current need. These findings will help to design new targeted therapies to overcome resistance and to find drug combinations that will prevent the development of resistance or will help to avoid relapse. This manuscript provides an up-to-date overview of the acquired resistance-associated mutations in CLL patients treated with ibrutinib, idelalisib, and venetoclax. These mutations emerge during the treatment course and predispose to the loss of function of the drug and disease progression. Moreover, we summarize recent findings in ways on how to manage the patients who have acquired resistant mutations to current inhibitors.
Resistance-Associated Mutations in Ibrutinib Treatment
So far, most information about resistance-associated mutations has been described for patients treated with ibrutinib, an oral agent inactivating Bruton's tyrosine kinase (BTK). This antiproliferative and proapoptotic agent, acting via formation of an irreversible covalent bond at the C481 position of BTK, inhibits both B-cell receptor (BCR) and NF-κB pathways [Figure 1; (5, 6)]. Ibrutinib has been shown to be highly effective not only in R/R CLL patients with del(17p) and/or TP53 mutation or complex karyotype (1, 2) but also in previously untreated patients with/without poor-risk features (3, 4). Primary resistance with no initial response to ibrutinib has been observed rarely and its mechanism is not yet understood (7–9). Acquired secondary resistance to ibrutinib occurs in 8–13% of CLL cases who responded well to the treatment initiation (7, 8) and are most commonly caused by the occurrence of resistant-associated mutations, as reported below (Figure 2).
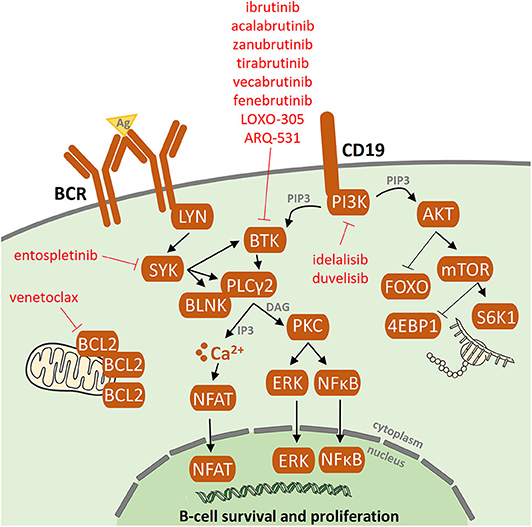
Figure 1. B-cell receptor (BCR) signaling pathway and BCR inhibitors. Following antigen binding to the B-cell receptors, BCR signaling is chronically activated in CLL cells. As a consequence, the activated BCR pathway results in the production of second messengers and the activation of NF-kB and subsequent pro-proliferation and antiapoptotic pathways. Targets of the BCR inhibitors: ibrutinib, idelalisib, acalabrutinib, zanubrutinib, tirabrutinib, fenebrutinib, vecabrutinib, LOXO-305, ARQ-531, and entospletinib are depicted.
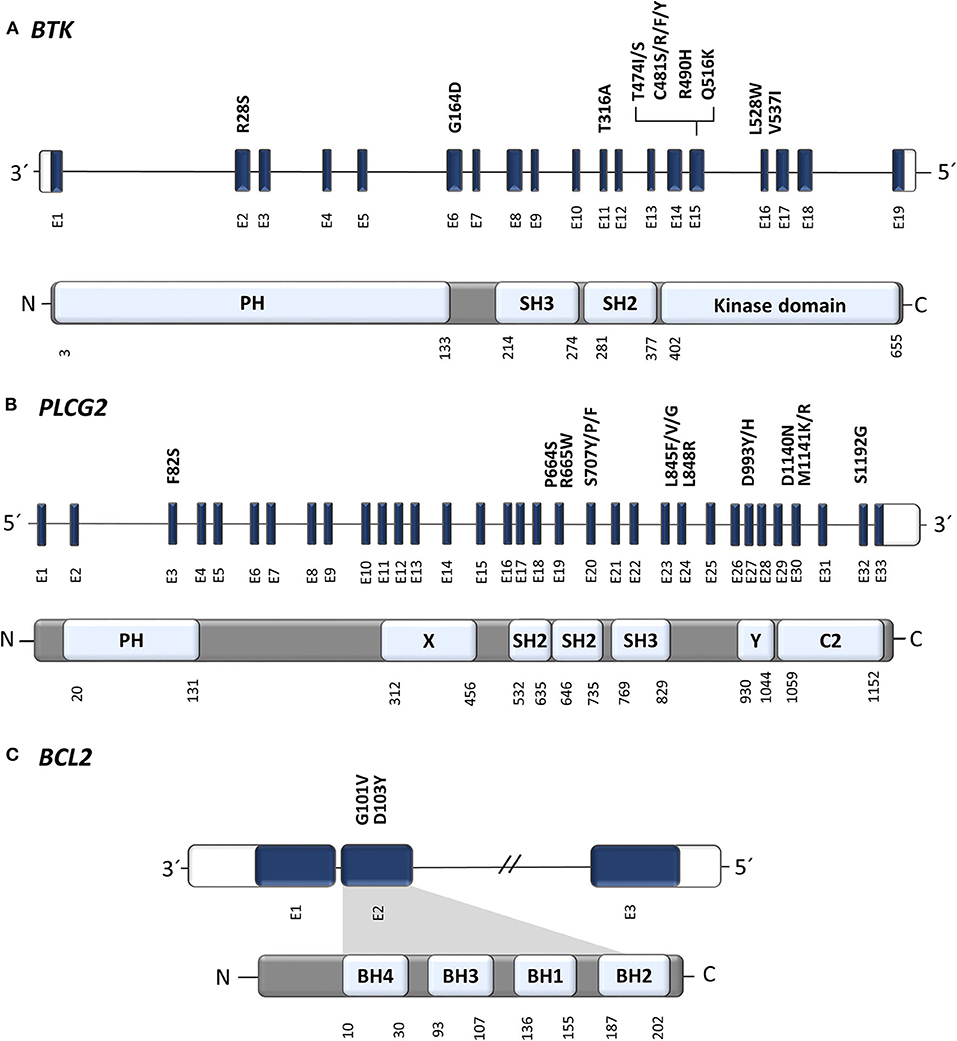
Figure 2. Position of known resistance-associated mutations in (A) BTK, (B) PLCG2, and (C) BCL2 genes. E, exon; PH, Pleckstrin homology domain; SH2/3, Src homology domain 2/3 (autoinhibitory domain); C2, Ca binding motive; X, catalytic domain; Y, catalytic domain; BH1-4, BCL-2 homology domain 1-4. Gene structure has been visualized according to a genome browser Ensembl (Ensembl release 99—January 2020, EMBL-EBI); protein domains and amino acid positions according UniProt Knowledgebase (2020_01 release—February 2020, UniProt Consortium).
In 2014, a study using whole-exome sequencing discovered acquired mutations within the BTK gene in 5/6 high-risk CLL patients relapsing on ibrutinib (10). A recent study on 30 CLL patients with residual lymphocytosis treated with ibrutinib for 3 years confirmed the presence of BTK mutations in 57% of CLL patients, and the presence of BTK mutations was associated with subsequent relapse (11). A number of studies have confirmed the presence of this mutation in CLL patients relapsing on ibrutinib and have shown that those mutations were not present prior to drug administration (12–14). The most common mutation (C481S) was found at the position of the binding site for ibrutinib thus reducing ibrutinib affinity for BTK (15, 16). More variants in the BTK gene, such as C481R, C481F, and C481Y, as well as less frequent variants at other gene positions (R28S, G164D, T316A, T474I/S, R490H, Q516K, L528W, and V537I), were revealed in later studies [Figure 2, Table 1; (12, 17–21)]. However, mutations outside of the kinase domain are rare (19). Functional characterization of these mutations has shown that the increase in ibrutinib dose is not sufficient to overcome the effect of the C481S/R/F/Y mutations (16). The BTK mutations usually develop between the second and fourth year of ibrutinib treatment (median 34.3 months, range 14–76.8 months) [Figure 3; (17)].
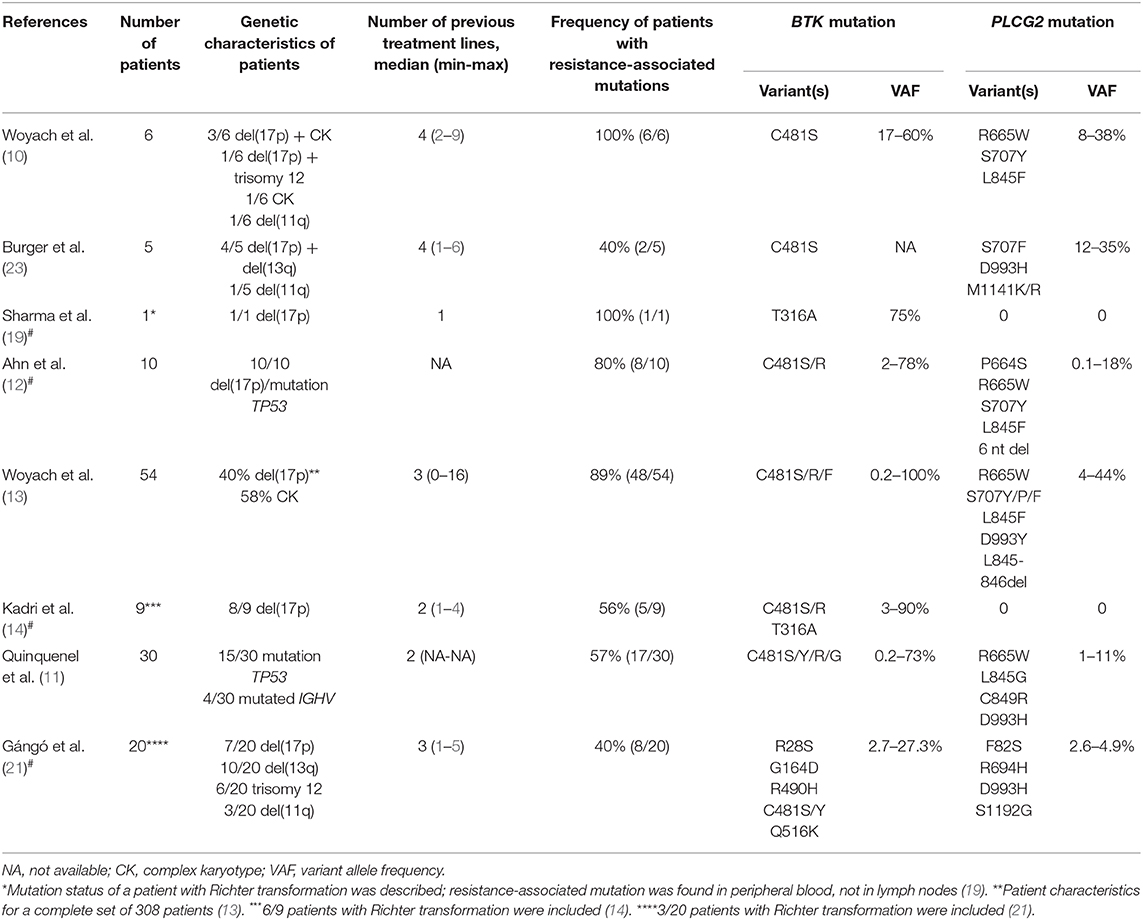
Table 1. Overview of selected studies describing the incidence of resistance-associated mutations in CLL patients treated with ibrutinib including selected studies describing resistance-associated mutations in patients with Richter transformation#.
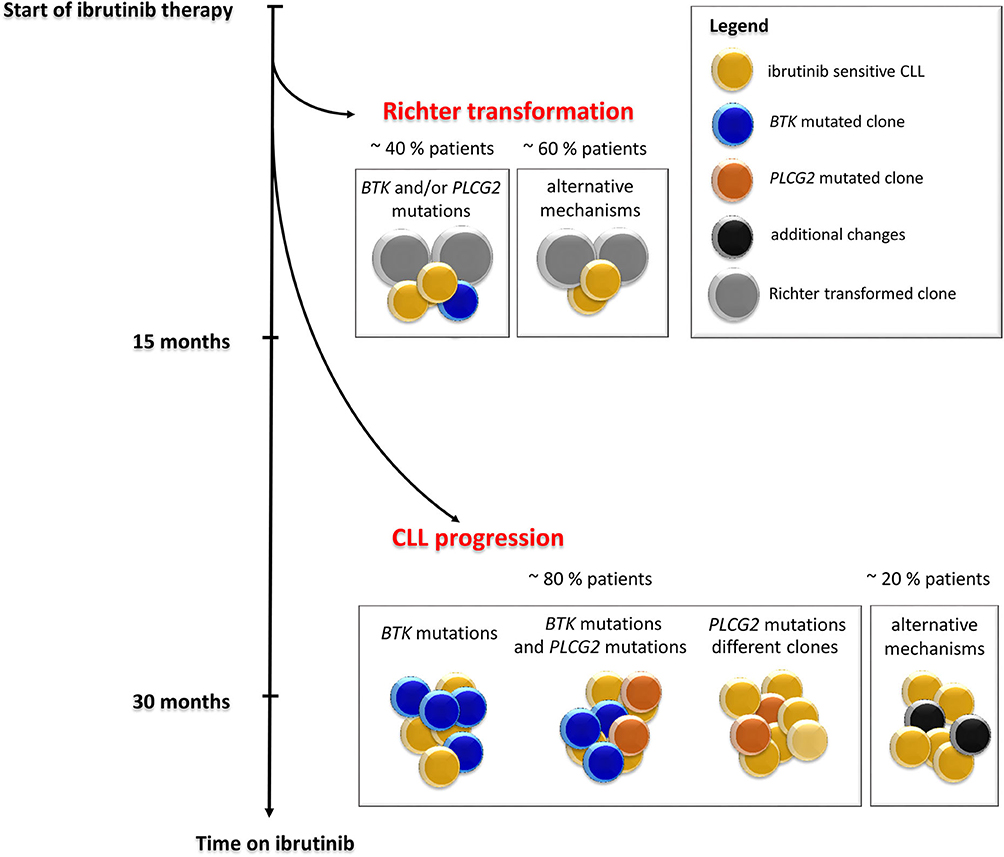
Figure 3. Timeline and frequency of occurrence of resistance-associated mutations emergence in CLL patients who progressed on ibrutinib treatment. Resistance-associated mutations in BTK and PLCG2 genes usually develop between the second and fourth year of ibrutinib treatment. Mutated clones may occur at low allelic frequencies and may be detected several months before clinical relapse. Richter transformation is an early event that usually develops during the first 15 months of treatment with ibrutinib. Adapted from Ahn et al. (12).
PLCG2 gain-of-function mutations are the second most frequent mutations found in CLL patients who failed on the ibrutinib treatment (Figure 2). PLCG2 is the gene encoding phospholipase Cγ2, the protein immediately downstream of BTK. These mutations mostly have an activating effect resulting in continuous BCR signaling independently on BTK activation (10, 22). PLCG2 mutation hotspots are located in several domains of the gene and often co-occur with hotspot BTK mutations (11–13, 21, 23). Moreover, different PLCG2 mutations are usually found in multiple subclones with low allelic burden (12, 13, 17). A recent study confirmed PLCG2 mutations in 13% of ibrutinib treated patients with residual lymphocytosis (11). However, the exact contribution of the PLCG2 to the clinical resistance of CLL patients remains not fully understood (22). Similarly, as BTK mutations, also PLCG2 mutations usually occur between the second and fourth year on ibrutinib (median 35.1 months, range 17.4–64.6 months) [Figure 3; (17)].
Although mutations in BTK and PLCG2 genes are detected in ~80% of CLL patients who failed on ibrutinib (10, 12, 13), for 20% of patients, ibrutinib resistance-associated mutations remain unknown (13, 14, 23). These data further support the presence of alternative mechanisms of drug resistance other than BTK/PLCG2 mutations in a subset of patients who are still under investigation. To date, several candidate loci/mutations that may contribute to resistance have been described. These include del(8p) and SF3B1, PCLO, EP300, MLL2, and EIF2A mutations (10, 14, 23). More candidate genetic factors associated with resistance for ibrutinib-treated patients involve BCL6 rearrangements, MYC gene abnormalities, del(17p), del(18p), 2p gain, XPO1 overexpression, complex karyotype, epigenetic changes, and changes in the cell microenvironment (1, 14, 18, 24). However, it remains unclear whether these aberrations contribute causally to clinical resistance.
BTK and/or PLCG2 Mutations Clonality
Studies of follow-up samples revealed that mutations within the BTK and PLCG2 genes are often present several months before clinical relapse is observed [Figure 3; (11–13)]. Those resistance-associated mutations were detected as early as 9.3 months prior to clinical progression (13). Moreover, Ahn et al. revealed that the resistance-associated mutations may be detected using highly sensitive approaches 15 months prior to disease progression (12).
The same BTK mutations have also been reported for CLL patients who experienced Richter transformation on ibrutinib treatment. In this group of patients, the resistance-associated mutations appeared in a smaller portion of patients (~40%) earlier, mostly within 15 months after ibrutinib treatment initiation [Figure 3; (13, 14)].
Nearly half of the patients who progress on ibrutinib carry BTK mutations in minor subclones with low variant allele frequencies (VAF) below 10% (12, 13). Nevertheless, cases with variants >80% VAF were also no exception (25). Clonal development and subclonal heterogeneity of resistance-associated mutations, i.e., the detection of multiple independent subclones carrying different variants of resistance-associated mutations with distinct growth rates, are also frequently observed in ibrutinib-treated patients (12, 23). In particular, it applies to PLCG2 gene variants with lower VAF. However, the precise mechanism by which BTK and/or PLCG2 mutations drive clinical resistance when present at such low allelic frequencies has not been elucidated yet.
In the recent study of Gángó et al., an association between reduction or even elimination of TP53 mutated clones and the presence of BTK mutations has been reported (21). Vice versa, in patients with persisting TP53 mutated subclones, no BTK mutations were detected in this study. The authors speculate that a longer duration of ibrutinib treatment could create conditions for the survival of subclones with BTK mutations by eliminating subclones with TP53 mutations or that the elimination of TP53 subclones enables the expansion of subclones harboring BTK mutations (21). The loss of preexisting mutations in TP53 and BIRC3 genes in patients who gained BTK mutations was also observed in another recent study (20). The relationship between TP53 and BTK mutations should be confirmed on a larger patient cohort.
Another study showed that resistant mutated subclones and disease progression may occur only in specific compartments (13, 14). For some patients, Woyach et al. reported the presence of resistance-associated mutations only in lymph nodes without a presence in a corresponding peripheral blood sample (13). For these patients, the disease progression was also observed only in the lymph nodes. Therefore, it is necessary to choose the right collection material for the analysis of resistance-associated mutations, and even a negative result from peripheral blood may not mean that the patient does not have a subclone with a resistance-associated mutation in another location (25).
Resistance-Associated Mutations in Idelalisib Treatment
Idelalisib is a phosphatidylinositol 3-kinase δ isoform (PI3Kδ) inhibitor, a member of the BCR inhibitor family, which not only inhibits PI3K signaling but also selectively induces apoptosis in CLL cells [Figure 1; (26)]. Idelalisib was approved for the treatment of patients with R/R CLL and high-risk patients with TP53 disruption (27, 28). Regardless of its clinical efficacy, disease progression during idelalisib treatment is observed in some CLL patients (29–31).
Despite intensive research, the biological mechanism of the disease progression in idelalisib-treated patients remains unknown and no resistance-associated mutations in specific gene(s) or signaling pathway alterations have been found so far (29–31). A whole-exome sequencing study in a small cohort of 13 CLL patients who progressed on idelalisib treatment revealed that no mutations occurred in the PI3K signaling pathway or in any related signaling pathway (31).
These results indicate that there is probably no single pathway or specific mutation associated with idelalisib resistance thus deserving future investigations. A recent study investigating in vivo mouse models of resistance to PI3K inhibitors identified upregulation of genes from the integrin receptor complex as a possible mechanism of resistance (30). This suggests that resistance to idelalisib may be more likely mediated by dysregulation of survival signaling rather than by recurrent mutations. Nevertheless, it remains to be determined to what extent this mechanism of resistance plays a role in human CLL.
Resistance-Associated Mutations in Venetoclax Treatment
Venetoclax is an oral BH3 mimetic and highly selective inhibitor of the BCL-2 antiapoptotic protein, capable of restoring apoptosis tumor cells with high overall response rates as well as in heavily pretreated, high-risk CLL patients. Venetoclax is primarily available for CLL patients with TP53 disruption, for patients who failed on ibrutinib or were not suitable for the treatment with BCR signaling inhibitors, as well as for patients refractory to chemoimmunotherapy (32–34).
There is already growing evidence about the role of acquired mutations leading to the progression and failure of venetoclax. A recent study reported G101V mutation in the BCL2 gene in 7 of 15 (47%) CLL patients progressing on venetoclax (35). Functional analysis demonstrated that G101V mutation disrupts the bond of venetoclax to BCL-2, therefore preventing the drug from competing with proapoptotic molecules to bind with BCL-2. The G101V mutation was absent at baseline, first detected 19–42 months after the initiation of venetoclax treatment and 25 months prior to clinical relapse, and persisted in five of seven patients for more than 6 months after the discontinuation of venetoclax therapy (35). The G101V mutation showed a high variability in VAF ranging from 1.4 to 70% (35).
Another recent study confirmed G101V in three of four CLL patients treated with venetoclax and found a second BCL2 variant, D103Y, also within the BH3-binding site (36). The D103Y was acquired 39 months after the initiation of venetoclax therapy, at first with a low VAF of 7%, increasing to 18% over the next 5 months of venetoclax treatment in the reported patient. At a later date, this patient became positive also for G101V mutation with 14% VAF. NGS analysis showed that these two mutations exist as independent subclones with different growth dynamics, and they were not detected in the control group of 546 CLL patients not treated with venetoclax (36).
A whole-exome sequencing study in a small cohort of eight patients with del(17p) progressing on venetoclax identified a number of candidate resistance-associated aberrations, such as homozygous deletions of CDKN2A/B resulting in the loss of cell cycle control in three patients and mutations in the antiproliferative BTG1 gene in two patients (37). Analysis of pretreatment samples revealed that these aberrations developed after the treatment initiation. In this study, the spectrum of other mutations in CLL-associated genes has been identified, such as BRAF, NOTCH1, RB1, SF3B1, and TP53 mutations. Nevertheless, their causal relationship to resistance has not yet been established.
Current Developments to Circumvent Failure on Novel Agents
Current studies have provided evidence that the early and sensitive detection of BTK, PLCG2, and BCL2 mutations may be predictive of an impending relapse, as they occur several months before clinical relapse (35, 38). Therefore, the emergence of the resistance-associated mutations in patients receiving long-term treatment with BCR and BCL-2 inhibitors should be tested at regular intervals using highly sensitive ultradeep NGS approaches. As the resistance-associated mutations occur often at low VAF, it is necessary to standardize sequencing parameters in order to minimize the probability of false-positive and false-negative results (39–42).
In the case of a positive result for BTK, PLCG2, and BCL2 mutations, the treatment should not be discontinued but rather different appropriate inhibitors or alternative combination therapies should be considered (43, 44). Therefore, understanding the nature of disease progression and emergence of clinical resistance has important implications, especially in the development of new treatment strategies that would help overcome resistance before it develops or as it emerges (43).
There are currently several options, clinically proven as well as in experimental settings, how to circumvent failure on ibrutinib and other novel agents (Figure 4). The most promising is to target alternative molecules or signaling pathways involved in the survival of malignant cells (Figure 5). Another therapeutic intervention could be the use of different combination therapies to reverse relapse (43). Very promising is the combination of ibrutinib with anti-CD20 antibodies (28). In addition, the combination of ibrutinib with venetoclax represents a great potential in the treatment of CLL, as there is increasing evidence that CLL cells previously treated with BCR inhibitors show an increased dependence on BCL-2 expression (45, 46). This is further supported by the observation of the changes in BCL-2 family protein levels in CLL cells when treated with ibrutinib (45) and acalabrutinib (47). Additionally, venetoclax complements ibrutinib and acalabrutinib–mediated apoptosis in CLL cells (45, 47). The synergic effect of both drugs acting by different mechanisms has led to a deep therapeutic effect in CLL (45, 46, 48); however, the exact mechanism of their interaction in CLL should be further elucidated.
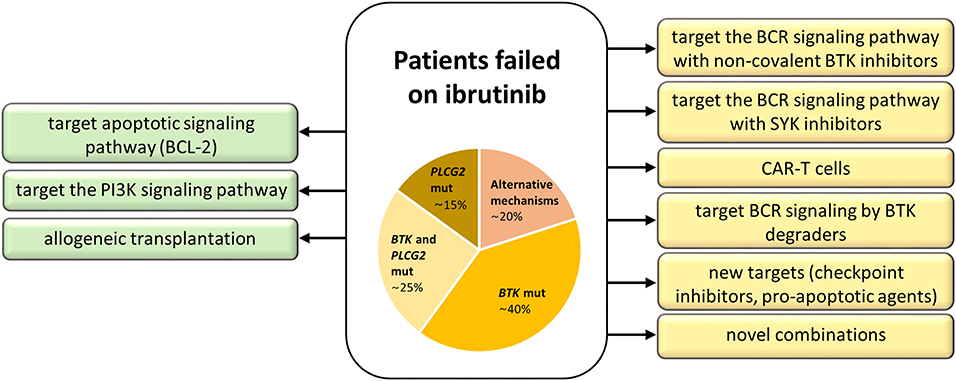
Figure 4. Treatment strategies for patients with acquired resistance to ibrutinib. Approved treatment options are on the left (green), experimental ones on the right (yellow).
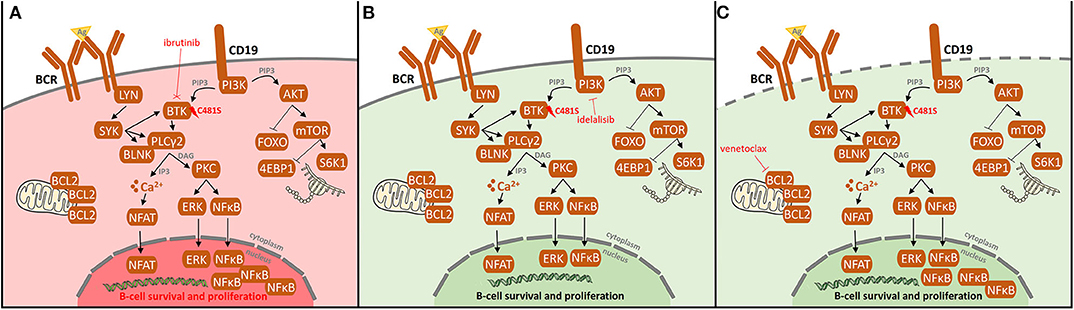
Figure 5. Resistance-associated BTK (C481) mutation and possibilities of its overcoming in ibrutinib-treated CLL patients. (A) After the occurrence of BTK (C481) mutation, ibrutinib does not bind to BTK causing the loss of its therapeutic effect leading to BCR and NF-kB pathway activation, both signaling pathways pivotal for maintenance and proliferation of CLL cells. Additionally, activating mutations in PLCG2 gene may result in continuous BCR signaling independently on BTK activation. To overcome the resistance by mutations in BTK/PLCG2 genes, other pathways may be targeted. (B) Idelalisib targets phosphatidylinositol 3-kinase (PI3K), resulting in the downregulation of PI3K/mTOR signaling pathway, irrespectively on the BTK mutation status. (C) Venetoclax targets BCL-2, thus leading to the apoptosis of CLL cells. As the previous treatment with BCR inhibitors results in an increased dependence on BCL-2 expression together with the fact that venetoclax complements ibrutinib-mediated apoptosis, a deep therapeutic effect on venetoclax is achieved.
Next-Generation Inhibitors and Experimental Treatment Options
New covalent BTK inhibitors with high selectivity to BTK, such as acalabrutinib (ACP-196), zanubrutinib (BGB-3111), and tirabrutinib (ONO/GS-4059), have high therapeutic potential [Figure 1; (49–51)]. However, like ibrutinib, they covalently bind to BTK and are, therefore, not suitable for the treatment of patients with resistance-associated BTK hotspot mutation (C481S/R/F/Y). Compared to ibrutinib, the recently approved acalabrutinib (52) shows a more acceptable safety profile and modulation of BCL-2 family proteins contributing to cell death induced by venetoclax after acalabrutinib treatment in CLL (47). Similarly to ibrutinib, acquired resistance to acalabrutinib was mainly associated with BTK mutations (53). Zanubrutinib is the next potent and highly selective inhibitor of BTK, currently approved for mantle cell lymphoma treatment and tested in clinical trials for CLL (52). In four CLL patients progressing on zanubrutinib treatment, Handunnetti et al. identified BTK C481 and L528W mutations, both of them absent prior to zanubrutinib treatment (54).
Importantly, a new generation of non-covalent BTK inhibitors is currently being tested such as fenebrutinib (GDC-0853), ARQ-531, LOXO-305, and vecabrutinib (SNS-062) (51, 52, 55). The first clinical studies with fenebrutinib have shown that non-covalent selective inhibition of BTK may be effective in CLL patients with acquired resistance to ibrutinib therapy (56). But in vitro mutagenesis of the BTK gene has shown that mutations in the kinase domain (L512M, E513G, F517L, and L547P) reduce the effect of these new non-covalent BTK inhibitors (57). Also, ARQ-531, an ATP-competitive non-covalent reversible inhibitor, is able to overcome BTK (C481S) and PLCG2 (R665W, S707P, S707F, R742P, and L845fs) mutations as shown in animal models (58). Similarly, the reversible BTK inhibitor LOXO-305 shows great potential in overcoming acquired resistance to irreversible BTK inhibitors in preclinical CLL models; phase 1 clinical trial of LOXO-305 is currently ongoing (59).
Among experimental treatment options in patients who failed on ibrutinib belong SYK (Spleen tyrosine kinase) inhibitors, chimeric antigen receptor T (CAR-T) cell therapy, anti-PD-1 treatment, and BTK degraders (Figure 4). SYK is a kinase upstream of both BTK and PI3Kδ in the BCR signaling pathway. The SYK inhibitor entospletinib shows a clinical activity for R/R CLL patients who have relapsed on BTK or PI3Kδ inhibitors, even in the presence of BTK and PLCG2 mutations (44). CAR-T cell therapy (60) represents another treatment opportunity for CLL patients failing BCR or BCL-2 inhibitor therapies, currently available only in clinical trials (52). Further, response to PD-1 blockade with pembrolizumab has been observed in patients with Richter transformation who had progression after prior therapy with ibrutinib, but not in R/R CLL patients (61). Another emerging approach evidenced to have an effect on both wild-type and mutated BTK (C481S) in preclinical studies is the novel agent BTK degrader, MT-802, causing ubiquitination of BTK, and subsequent degradation through proteasome (62). A recent study on animal models has shown the possibility to overcome resistance to ibrutinib by preventing FOXO3a nuclear export and PI3K/AKT activation (63). These promising data and the development of new approaches to overcome acquired resistance to current BCR and BCL-2 inhibitors show great hope not only for CLL patients.
Conclusion
With increasing experience in the treatment with novel agents, it has become extremely important to (i) detect genetic aberrations associated with resistance and progression and (ii) understand the mechanisms of resistance development and disease progression in patients treated with these agents. Resistance-associated mutations in BTK, PLCG2, and BCL2 have the potential to be used as a biomarker for future relapse or disease progression and hence their detection could facilitate early therapeutic intervention therapy to prevent the relapse. In addition, understanding the mechanism of resistance may also help find a way on how to prevent resistance before it develops or to overcome as it emerges.
In the near future, we can expect an increase in the number of patients indicated for inhibitor therapy due to the excellent performance of these agents, not only in CLL. Since patients are treated for longer with these drugs, it will certainly be reflected in the increased incidence of disease progression and failure on those agents; hence, the challenge of managing resistance and identifying which patients are at risk for relapse is of the highest importance. There is a long way to learn on how to manage a resistant disease that we will encounter more and more often in the era of treatment with novel targeted agents.
Data Availability Statement
All datasets generated for this study are included in the article.
Author Contributions
LS and EK wrote the manuscript. LS and AP prepared the figures and revisions. TP, PT, and AP revised the manuscript critically. EK contributed to conceptualization, study design, writing, and editing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Ministry of Health of the Czech Republic (MH CZ VES16-32339A), in part by FNOl 00098892, and the Internal Grant Agency of Palacky University (IGA UP_2020_016).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. (2013) 369:32–42. doi: 10.1056/NEJMoa1215637
2. Byrd JC, Hillmen P, O'Brien S, Barrientos JC, Reddy NM, Coutre S, et al. Long-term follow-up of the RESONATE phase 3 trial of ibrutinib vs ofatumumab. Blood. (2019) 133:2031–42. doi: 10.1182/blood-2018-08-870238
3. Burger JA, Tedeschi A, Barr PM, Robak T, Owen C, Ghia P, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. (2015) 373:2425–37. doi: 10.1056/NEJMoa1509388
4. Barr PM, Robak T, Owen C, Tedeschi A, Bairey O, Bartlett NL, et al. Sustained efficacy and detailed clinical follow-up of first-line ibrutinib treatment in older patients with chronic lymphocytic leukemia: extended phase 3 results from RESONATE-2. Haematologica. (2018) 103:1502–10. doi: 10.3324/haematol.2018.192328
5. Herman SE, Mustafa RZ, Gyamfi JA, Pittaluga S, Chang S, Chang B, et al. Ibrutinib inhibits BCR and NF-κB signaling and reduces tumor proliferation in tissue-resident cells of patients with CLL. Blood. (2014) 123:3286–95. doi: 10.1182/blood-2014-02-548610
6. Yosifov DY, Wolf C, Stilgenbauer S, Mertens D. From biology to therapy: the CLL success story. Hemasphere. (2019) 3:e175. doi: 10.1097/HS9.0000000000000175
7. Kaur V, Swami A. Ibrutinib in CLL: a focus on adverse events, resistance, and novel approaches beyond ibrutinib. Ann Hematol. (2017) 96:1175–84. doi: 10.1007/s00277-017-2973-2
8. Puła B, Gołos A, Górniak P, Jamroziak K. Overcoming ibrutinib resistance in chronic lymphocytic leukemia. Cancers. (2019) 11:E1834. doi: 10.3390/cancers11121834
9. Woyach JA, Johnson AJ. Targeted therapies in CLL: mechanisms of resistance and strategies for management. Blood. (2015) 126:471–7. doi: 10.1182/blood-2015-03-585075
10. Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib. N Engl J Med. (2014) 370:2286–94. doi: 10.1056/NEJMoa1400029
11. Quinquenel A, Fornecker LM, Letestu R, Ysebaert L, Fleury C, Lazarian G, et al. Prevalence of BTK and PLCG2 mutations in a real-life CLL cohort still on ibrutinib after 3 years: a FILO group study. Blood. (2019) 134:641–4. doi: 10.1182/blood.2019000854
12. Ahn IE, Underbayev C, Albitar A, Herman SE, Tian X, Maric I, et al. Clonal evolution leading to ibrutinib resistance in chronic lymphocytic leukemia. Blood. (2017) 129:1469–79. doi: 10.1182/blood-2016-06-719294
13. Woyach JA, Ruppert AS, Guinn D, Lehman A, Blachly JS, Lozanski A, et al. BTKC481S-mediated resistance to ibrutinib in chronic lymphocytic leukemia. J Clin Oncol. (2017) 35:1437–43. doi: 10.1200/JCO.2016.70.2282
14. Kadri S, Lee J, Fitzpatrick C, Galanina N, Sukhanova M, Venkataraman G, et al. Clonal evolution underlying leukemia progression and richter transformation in patients with ibrutinib-relapsed CLL. Blood Adv. (2017) 1:715–27. doi: 10.1182/bloodadvances.2016003632
15. Cheng S, Guo A, Lu P, Ma J, Coleman M, Wang YL. Functional characterization of BTK(C481S) mutation that confers ibrutinib resistance: exploration of alternative kinase inhibitors. Leukemia. (2015) 29:895–900. doi: 10.1038/leu.2014.263
16. Hamasy A, Wang Q, Blomberg KE, Mohammad DK, Yu L, Vihinen M, et al. Substitution scanning identifies a novel, catalytically active ibrutinib-resistant BTK cysteine 481 to threonine (C481T) variant. Leukemia. (2017) 31:177–85. doi: 10.1038/leu.2016.153
17. Jones D, Woyach JA, Zhao W, Caruthers S, Tu H, Coleman J, et al. PLCG2 C2 domain mutations co-occur with BTK and PLCG2 resistance mutations in chronic lymphocytic leukemia undergoing ibrutinib treatment. Leukemia. (2017) 31:1645–7. doi: 10.1038/leu.2017.110
18. Maddocks KJ, Ruppert AS, Lozanski G, Heerema NA, Zhao W, Abruzzo L, et al. Etiology of ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA Oncol. (2015) 1:80–7. doi: 10.1001/jamaoncol.2014.218
19. Sharma S, Galanina N, Guo A, Lee J, Kadri S, Van Slambrouck C, et al. Identification of a structurally novel BTK mutation that drives ibrutinib resistance in CLL. Oncotarget. (2016) 7:68833–41. doi: 10.18632/oncotarget.11932
20. Kanagal-Shamanna R, Jain P, Patel KP, Routbort M, Bueso-Ramos C, Alhalouli T, et al. Targeted multigene deep sequencing of Bruton tyrosine kinase inhibitor-resistant chronic lymphocytic leukemia with disease progression and Richter transformation. Cancer. (2019) 125:559–74. doi: 10.1002/cncr.31831
21. Gángó A, Alpár D, Galik B, Marosvári D, Kiss R, Fésüs V, et al. Dissection of subclonal evolution by temporal mutation profiling in chronic lymphocytic leukemia patients treated with ibrutinib. Int J Cancer. (2020) 146:85–93. doi: 10.1002/ijc.32502
22. Liu TM, Woyach JA, Zhong Y, Lozanski A, Lozanski G, Dong S, et al. Hypermorphic mutation of phospholipase C, γ2 acquired in ibrutinib-resistant CLL confers BTK independency upon B-cell receptor activation. Blood. (2015) 126:61–8. doi: 10.1182/blood-2015-02-626846
23. Burger JA, Landau DA, Taylor-Weiner A, Bozic I, Zhang H, Sarosiek K, et al. Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat Commun. (2016) 7:11589. doi: 10.1038/ncomms11589
24. Maffei R, Fiorcari S, Martinelli S, Potenza L, Luppi M, Marasca R. Targeting neoplastic B cells and harnessing microenvironment: the “double face” of ibrutinib and idelalisib. J Hematol Oncol. (2015) 8:60. doi: 10.1186/s13045-015-0157-x
25. Lampson BL, Brown JR. Are BTK and PLCG2 mutations necessary and sufficient for ibrutinib resistance in chronic lymphocytic leukemia? Expert Rev Hematol. (2018) 11:185–94. doi: 10.1080/17474086.2018.1435268
26. Hoellenriegel J, Meadows SA, Sivina M, Wierda WG, Kantarjian H, Keating MJ, et al. The phosphoinositide 3'-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. (2011) 118:3603–12. doi: 10.1182/blood-2011-05-352492
27. Brown JR, Byrd JC, Coutre SE, Benson DM, Flinn IW, Wagner-Johnston ND, et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110δ, for relapsed/refractory chronic lymphocytic leukemia. Blood. (2014) 123:3390–7. doi: 10.1182/blood-2013-11-535047
28. Burger JA, Wiestner A. Targeting B cell receptor signalling in cancer: preclinical and clinical advances. Nat Rev Cancer. (2018) 18:148–67. doi: 10.1038/nrc.2017.121
29. Arnason JE, Brown JR. Targeting B cell signaling in chronic lymphocytic leukemia. Curr Oncol Rep. (2017) 19:61. doi: 10.1007/s11912-017-0620-7
30. Scheffold A, Jebaraj BMC, Tausch E, Bloehdorn J, Ghia P, Yahiaoui A, et al. IGF1R as druggable target mediating PI3K-δ inhibitor resistance in a murine model of chronic lymphocytic leukemia. Blood. (2019) 134:534–47. doi: 10.1182/blood.2018881029
31. Ghia P, Ljungström V, Tausch E, Agathangelidis A, Scheffold A, Scarfo L, et al. Whole-exome sequencing revealed no recurrent mutations within the PI3K pathway in relapsed chronic lymphocytic leukemia patients progressing under idelalisib treatment. Blood. (2016) 128:2770. doi: 10.1182/blood.V128.22.2770.2770
32. Stilgenbauer S, Eichhorst B, Schetelig J, Coutre S, Seymour JF, Munir T, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. (2016) 17:768–78. doi: 10.1016/S1470-2045(16)30019-5
33. Jones JA, Mato AR, Wierda WG, Davids MS, Choi M, Cheso, et al. Venetoclax for chronic lymphocytic leukaemia progressing after ibrutinib: an interim analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol. (2018) 19:65–75. doi: 10.1016/S1470-2045(17)30909-9
34. Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. (2016) 374:311–22. doi: 10.1056/NEJMoa1513257
35. Blombery P, Anderson MA, Gong JN, Thijssen R, Birkinshaw RW, Thompson ER, et al. Acquisition of the recurrent Gly101Val mutation in BCL2 confers resistance to venetoclax in patients with progressive chronic lymphocytic leukemia. Cancer Discov. (2019) 9:342–53. doi: 10.1182/blood-2018-120761
36. Tausch E, Close W, Dolnik A, Bloehdorn J, Chyla B, Bullinger L, et al. Venetoclax resistance and acquired BCL2 mutations in chronic lymphocytic leukemia. Haematologica. (2019) 104:e434–7. doi: 10.3324/haematol.2019.222588
37. Herling CD, Abedpour N, Weiss J, Schmitt A, Jachimowicz RD, Merkel O, et al. Clonal dynamics towards the development of venetoclax resistance in chronic lymphocytic leukemia. Nat Commun. (2018) 9:727. doi: 10.1038/s41467-018-03170-7
38. Sutton LA. Mechanisms of resistance to targeted therapies in chronic lymphocytic leukemia. HemaSphere. (2019) 3:40–3. doi: 10.1097/HS9.0000000000000240
39. Jennings LJ, Arcila ME, Corless C, Kamel-Reid S, Lubin IM, Pfeifer J, et al. Guidelines for validation of next-generation sequencing-based oncology panels: a joint consensus recommendation of the association for molecular pathology and College of American Pathologists. J Mol Diagn. (2017) 19:341–65. doi: 10.1016/j.jmoldx.2017.01.011
40. Bacher U, Shumilov E, Flach J, Porret N, Joncourt R, Wiedemann G, et al. Challenges in the introduction of next-generation sequencing (NGS) for diagnostics of myeloid malignancies into clinical routine use. Blood Cancer J. (2018) 8:113. doi: 10.1038/s41408-018-0148-6
41. Merker JD, Devereaux K, Iafrate AJ, Kamel-Reid S, Kim AS, Moncur JT, et al. Proficiency testing of standardized samples shows very high interlaboratory agreement for clinical next-generation sequencing-based oncology assays. Arch Pathol Lab Med. (2019) 143:463–71. doi: 10.5858/arpa.2018-0336-CP
42. Petrackova A, Vasinek M, Sedlarikova L, Dyskova T, Schneiderova P, Novosad T, et al. Standardization of sequencing coverage depth in NGS: recommendation for detection of clonal and subclonal mutations in cancer diagnostics. Front Oncol. (2019) 9:851. doi: 10.3389/fonc.2019.00851
43. Fürstenau M, Hallek M, Eichhorst B. Sequential and combination treatments with novel agents in chronic lymphocytic leukemia. Haematologica. (2019) 104:2144–54. doi: 10.3324/haematol.2018.208603
44. Awan FT, Thirman MJ, Patel-Donnelly D, Assouline S, Rao AV, Ye W, et al. Entospletinib monotherapy in patients with relapsed or refractory chronic lymphocytic leukemia previously treated with B-cell receptor inhibitors: results of a phase 2 study. Leuk Lymphoma. (2019) 60:1972–7. doi: 10.1080/10428194.2018.1562180
45. Deng J, Isik E, Fernandes SM, Brown JR, Letai A, Davids MS. Bruton's tyrosine kinase inhibition increases BCL-2 dependence and enhances sensitivity to venetoclax in chronic lymphocytic leukemia. Leukemia. (2017) 31:2075–84. doi: 10.1038/leu.2017.32
46. Bose P, Gandhi V. Recent therapeutic advances in chronic lymphocytic leukemia. F1000Res. (2017) 6:1924. doi: 10.12688/f1000research.11618.1
47. Patel VK, Lamothe B, Ayres ML, Gay J, Cheung JP, Balakrishnan K, et al. Pharmacodynamics and proteomic analysis of acalabrutinib therapy: similarity of on-target effects to ibrutinib and rationale for combination therapy. Leukemia. (2018) 32:920–30. doi: 10.1038/leu.2017.321
48. Tam CS, Siddiqi T, Allan JN, Kipps TJ, Flinn IW, Kuss BJ, et al. Ibrutinib (Ibr) plus Venetoclax (Ven) for first-line treatment of Chronic Lymphocytic Leukemia (CLL)/Small Lymphocytic Lymphoma (SLL): results from the MRD Cohort of the Phase 2 CAPTIVATE Study. Blood. (2019) 134:35. doi: 10.1182/blood-2019-121424
49. Byrd JC, Harrington B, O'Brien S, Jones JA, Schuh A, Devereux S, et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med. (2016) 374:323–32. doi: 10.1056/NEJMoa1509981
50. Walter HS, Rule SA, Dyer MJ, Karlin L, Jones C, Cazin B, et al. A phase 1 clinical trial of the selective BTK inhibitor ONO/GS-4059 in relapsed and refractory mature B-cell malignancies. Blood. (2016) 127:411–9. doi: 10.1182/blood-2015-08-664086
51. Thompson PA, Burger JA. Bruton's tyrosine kinase inhibitors: first and second generation agents for patients with Chronic Lymphocytic Leukemia (CLL). Expert Opin Investig Drugs. (2018) 27:31–42. doi: 10.1080/13543784.2018.1404027
52. Iovino L, Shadman M. Novel therapies in chronic lymphocytic leukemia: a rapidly changing landscape. Curr Treat Options Oncol. (2020) 21:24. doi: 10.1007/s11864-020-0715-5
53. Woyach J, Huang Y, Rogers K, Bhat SA, Grever MR, Lozanski A, et al. Resistance to acalabrutinib in CLL Is mediated primarily by BTK mutations. Blood. (2019) 134 (Suppl. 1):504. doi: 10.1182/blood-2019-127674
54. Handunnetti SM, Pek Sang Tang C, Nguyen T, Zhou X, Thompson E, Sun H, et al. BTK Leu528Trp - a potential secondary resistance mechanism specific for patients with chronic lymphocytic leukemia treated with the next generation BTK inhibitor. Blood. (2019) 134 (Suppl. 1):170. doi: 10.1182/blood-2019-125488
55. Crawford JJ, Johnson AR, Misner DL, Belmont LD, Castanedo G, Choy R, et al. Discovery of GDC-0853: a potent, selective, and noncovalent Bruton's tyrosine kinase inhibitor in early clinical development. J Med Chem. (2018) 61:2227–45. doi: 10.1021/acs.jmedchem.7b01712
56. Reiff SD, Muhowski EM, Guinn D, Lehman A, Fabian CA, Cheney C, et al. Noncovalent inhibition of C481S Bruton tyrosine kinase by GDC-0853: a new treatment strategy for ibrutinib-resistant CLL. Blood. (2018) 132:1039–49. doi: 10.1182/blood-2017-10-809020
57. Wang S, Mondal S, Zhao C, Berishaj M, Ghanakota P, Batlevi CL, et al. Noncovalent inhibitors reveal BTK gatekeeper and auto-inhibitory residues that control its transforming activity. JCI Insight. (2019) 4:e127566. doi: 10.1172/jci.insight.127566
58. Reiff SD, Mantel R, Smith LL, Greene JT, Muhowski EM, Fabian CA, et al. The BTK inhibitor ARQ 531 targets ibrutinib-resistant CLL and Richter transformation. Cancer Discov. (2018) 8:1300–15. doi: 10.1158/2159-8290.CD-17-1409
59. Gomez EB, Lippincott I, Rosendahal MS, Rothenberg SM, Andrews SW, Brandhuber BJ. Loxo-305, a highly selective and non-covalent next generation BTK inhibitor, inhibits diverse BTK C481 substitution mutations. Blood. (2019) 134(Suppl. 1):4644. doi: 10.1182/blood-2019-126114
60. Lemal R, Tournilhac O. State-of-the-art for CAR T-cell therapy for chronic lymphocytic leukemia in 2019. J Immunother Cancer. (2019) 7:202. doi: 10.1186/s40425-019-0686-x
61. Ding W, LaPlant BR, Call TG, Parikh SA, Leis JF, He R, et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood. (2017) 129:3419–27. doi: 10.1182/blood-2017-02-765685
62. Buhimschi AD, Armstrong HA, Toure M, Jaime-Figueroa S, Chen TL, Lehman A, et al. Targeting the C481S ibrutinib-resistance mutation in Bruton's tyrosine kinase using PROTAC-mediated degradation. Biochemistry. (2018) 57:3564–75. doi: 10.1021/acs.biochem.8b00391
Keywords: CLL, targeted therapy, resistance-associated mutations, treatment resistance, genetic aberrations
Citation: Sedlarikova L, Petrackova A, Papajik T, Turcsanyi P and Kriegova E (2020) Resistance-Associated Mutations in Chronic Lymphocytic Leukemia Patients Treated With Novel Agents. Front. Oncol. 10:894. doi: 10.3389/fonc.2020.00894
Received: 07 March 2020; Accepted: 06 May 2020;
Published: 25 June 2020.
Edited by:
Dimitar G. Efremov, International Centre for Genetic Engineering and Biotechnology, ItalyReviewed by:
Lydia Scarfò, San Raffaele Hospital, ItalyBartosz Michał Puła, Institute of Hematology and Transfusiology, Poland
Copyright © 2020 Sedlarikova, Petrackova, Papajik, Turcsanyi and Kriegova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eva Kriegova, eva.kriegova@email.cz
†These authors have contributed equally to this work