 Jennifer Cao
Jennifer Cao Lyndah Chow
Lyndah Chow Steven Dow
Steven Dow- 1Flint Animal Cancer Center, College of Veterinary Medicine and Biomedical Sciences, Colorado State University, Fort Collins, CO, United States
- 2Department of Microbiology, Immunology, and Pathology, College of Veterinary Medicine and Biomedical Sciences, Colorado State University, Fort Collins, CO, United States
- 3Department of Clinical Sciences, College of Veterinary Medicine and Biomedical Sciences, Colorado State University, Fort Collins, CO, United States
Cancer progression and metastasis due to tumor immune evasion and drug resistance is strongly associated with immune suppressive cellular responses, particularly in the case of metastatic tumors. The myeloid cell component plays a key role within the tumor microenvironment (TME) and disrupts both adaptive and innate immune cell responses leading to loss of tumor control. Therefore, strategies to eliminate or modulate the myeloid cell compartment of the TME are increasingly attractive to non-specifically increase anti-tumoral immunity and enhance existing immunotherapies. This review covers current strategies targeting myeloid suppressor cells in the TME to enhance anti-tumoral immunity, including strategies that target chemokine receptors to deplete selected immune suppressive myeloid cells and relieve the inhibition imposed on the effector arms of adaptive immunity. Remodeling the TME can in turn improve the activity of other immunotherapies such as checkpoint blockade and adoptive T cell therapies in immunologically “cold” tumors. When possible, in this review, we have provided evidence and outcomes from recent or current clinical trials evaluating the effectiveness of the specific strategies used to target myeloid cells in the TME. The review seeks to provide a broad overview of how myeloid cell targeting can become a key foundational approach to an overall strategy for improving tumor responses to immunotherapy.
1 Introduction
Local immune suppression and dysregulation are common features of cancer and are closely associated with tumor metastasis and resistance to therapy. The interaction between cancer and the host immune system is a key factor in determining tumor control or progression (1–3). Tumor infiltrating leukocytes, particularly monocytes, myeloid derived suppressor cells (MDSCs) and neutrophils create a tumor microenvironment (TME) that is inhospitable to effector cells such as CD4 and CD8 T cells and NK cells (4–8). Myeloid lineage cells such as dendritic cells (DCs), tumor associated macrophages (TAMs) and MDSCs can serve a dichotomous role within the TME, though in general they are largely immune suppressive (9–12). These myeloid cells can promote tumor growth by exerting immune suppressive pressure, including secreted cytokines and growth factors promoting angiogenesis, direct cellular signaling or recruitment of Tregs and other immune suppressive cells such as TAMs, MDSCs, tumor associated neutrophils (TANs) and DCs (13). Myeloid cells in the TME can also assume a tumoricidal phenotype, as is the case with activated M1 macrophages producing free radicals and cytokines that stimulate the activation of effector T cells (14), or antigen presenting DCs that promote the expansion and activation of effector CD4 and CD8 T cells.
The net outcome of the dynamic interplay in the TME is determined in part by secreted factors and cell signaling from tumor and stromal cells and by the resident immune cells within the TME, which perpetuate either a suppressive or stimulatory immune landscape (1, 4, 10, 12). Targeting of myeloid immune suppressor cells to reduce or eliminate their immune suppressive impacts on adaptive immunity can turn the tide between cancer and the host’s immunity, thereby increasing tumor control and improving the efficacy of other treatments. In this review we summarize past and current strategies including relevant clinical trials that target myeloid cells in the TME as cancer immunotherapy strategies. Although this is not intended to be a fully comprehensive review of all strategies and trials, the goal is to emphasize that the myeloid cell component of the TME presents many opportunities for development of new immune based therapeutics.
2 Immune suppressive myeloid cells, origins, and key functions
2.1 Origin and differentiation of immune suppressor cells
Immune suppressor cells in the tumor microenvironment can be characterized by their cell type of origin. Myeloid derived suppressor cells (MDSC) are comprised of both neutrophil derived MDSC (PMN-MDSC) and monocyte derived MDSC (M-MDSC) with potent immune suppressive activity (15). Tumor associated macrophages (TAM) are derived from inflammatory monocytes recruited from the bloodstream in response to chemokines produced by tumor cells and the tumor stroma, including also myeloid cells themselves, and can be clearly distinguished phenotypically and functionally from MDSC (16) (17). The distinction between tumor associated neutrophils (TAN) and PMN-MDSC is somewhat more complicated, in that they share many phenotypic characteristics (18). Tolerogenic DCs are dendritic cells exposed to polarizing cytokines and surface molecules secreted by tumor cells and stromal cells within the tumor microenvironment (9) This population of immune suppressive DC suppress effector T cell responses, thereby contributing to an overall immune suppressed and hostile environment for infiltrating T cells.
The immune suppressive TAMs, DCs, TANs and MDSCs are recruited to the tumor by a variety of cellular and soluble factors within the tumor milieu, where they suppress effector functions of T cells and NK cells (Figure 1). The various mechanisms employed by immune suppressive myeloid cells become potential targets for new immunotherapies designed to reprogram the TME. Among the mechanisms employed by TAMs and MDSCs to suppress effector T cells and NK cells include upregulated expression of immune suppressive checkpoint molecules such as PD-L1 (19, 20), VISTA (21, 22), and B7-H3 ) (21, 23–25). Other mechanisms include secretion of immune suppressive cytokines such as IL10 and TGFβ, and VEGF (6, 26).
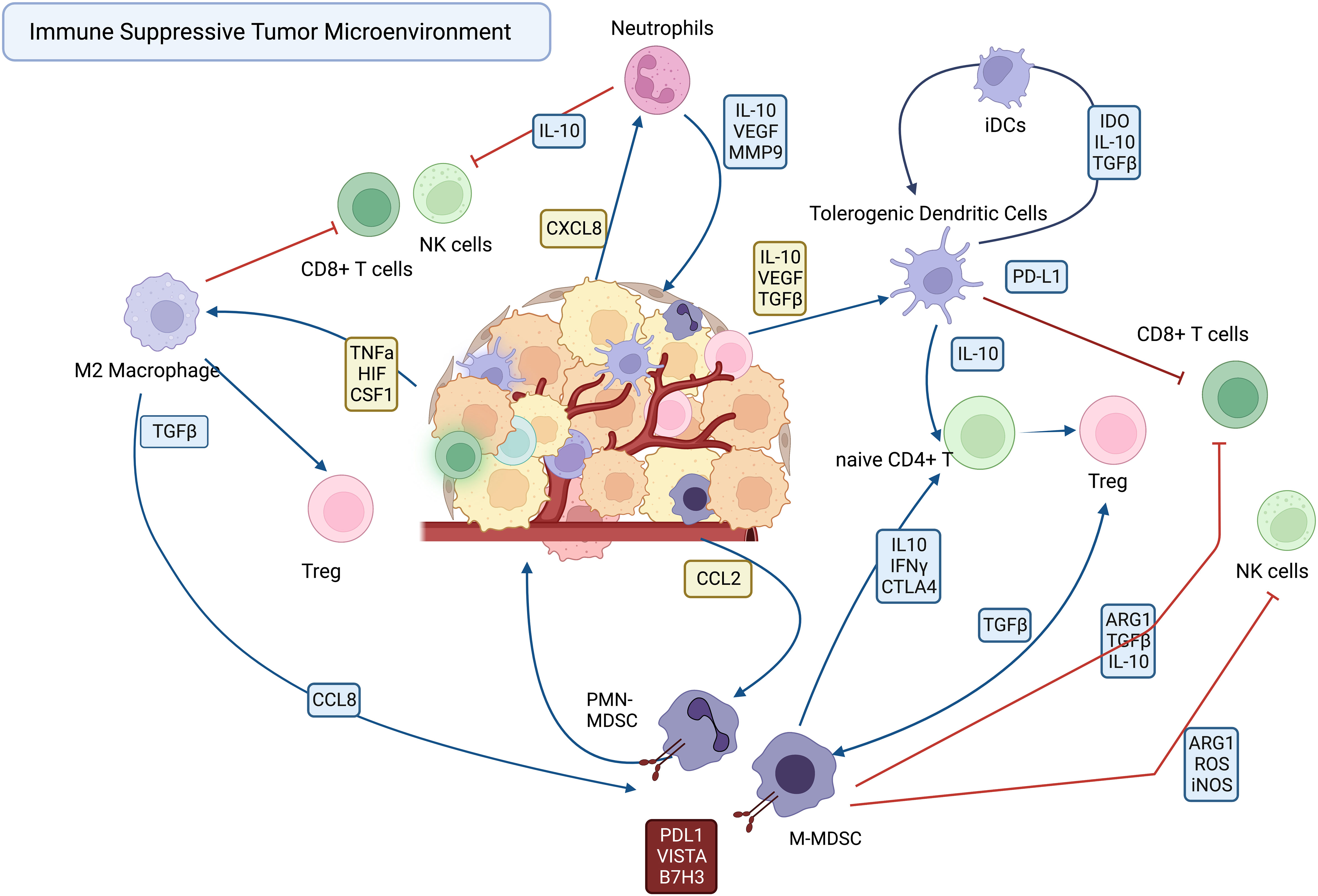
Figure 1 Cellular interactions in the immune suppressive tumor microenvironment.
2.2 Function of TAMs in the tumor microenvironment
Immunologically “cold” tumors evade immune surveillance through a variety of mechanisms. Down regulation of tumor associated antigens (TAA) (27, 28), maintaining inflammation leading to immune exhaustion (6, 26) and increasing angiogenesis to tumor sites (29, 30) are all methods used by tumors to persist and metastasize while evading detection by the immune system.
There is strong clinical evidence linking TAMs to cancer immune suppression. For example, the density of TAMs infiltrating tumors is strongly correlated with poor overall survival in many breast, ovarian, bladder, gastric, thyroid and colorectal cancers (31). TAMs have a relatively short half-life and are therefore must be replaced continuously by inflammatory monocytes recruited from the bloodstream primarily in response to the chemokine CCL2, produced by tumor cells, tumor fibroblasts, and by myeloid cells (32). This dependence of TAMs on continuous monocyte replacement opens a window of opportunity for therapeutic intervention and depletion of TAMS.
Once within the tumor, the differentiation of monocytes to TAMs is guided by either pro-inflammatory or anti-inflammatory factors produced within the TME. The overwhelming majority of TAMs in most tumors exist in a state that most closely resembles that of what has been defined experimentally as M2 polarization, which results in a macrophage that is generally immune suppressive and tumor growth and metastasis promoting (3). The M2 polarization state of TAMS is driven by a a diverse array of cytokines (eg, IL-10, TGF-b), chemokines (CXCL4, CCL5), growth factors (VEGF, M-CSF) and by local tumor hypoxia (33). Tumor cells can also directly contribute metabolically to M2 polarization by secretion of lactic acid and hypoxia-inducible factor (HIF1α) (34). Tumor cells also co-opt TAM signaling to promote tumor growth locally, and to become more invasive for generating metastases. For example, tumor secretion of TNF-α induces the chemokines CCL2 and CCL8 by TAMs which recruits additional CCR2+ monocytes to the TME (35). In another example, CCL8 produced by TAMs also upregulate tumor cell secretion of colony stimulating factor 1 (CSF-1) which is crucial to macrophage and DC survival and differentiation through signaling via CSF-1R (36, 37). One of the most important consequences of the accumulation of TAMs is the impact on T cell effector functions. For example, TGF-β signaling drives CD4 T cell differentiation towards immune suppressive Th2 and Treg phenotypes (38). TGF-β signaling also suppresses the effector functions of CD8 T cells and NK cells and decreases migration of DCs into the tumor tissues (9). Within the TME, T cells responding to TAM secreted factors exhibit upregulated expression of immune suppressive immune checkpoint molecules such as PD-1, CTLA-4, Lag3, and TIM3 (39). The expression of the ligand for PD-1 (PDL-1) is often higher on TAMs than it is on tumor cells, and PDL-1 signaling to TAM directly can reduce their ability to phagocytose tumor cells (40).
Metabolically, TAMs can reprogram the TME by producing enzymes that directly alter T cell signaling, or deplete necessary amino acids needed for T cell survival and proliferation. For example, TAM production of arginase 1 (Arg-1) leads to the depletion of L-arginine in turn leading to dysfunction of tumor infiltrating lymphocytes by TCR ζ chain downregulation (1, 12, 41). In another example, TAM (and tumor cells) can overproduce the enzyme indoleamine dehydrogenase (IDO), which depletes the TME of tryptophan, a necessary amino acid for T cell survival (42).
2.3 Function of immune suppressive DCs in the tumor microenvironment
Like TAMs, DCs in the TME exist primarily in an immune suppressive state and by inactivating effector T cells can promote more rapid tumor growth and metastasis (9). Immature DCs that reside in the TME recognize tumor cells and the products of tumor cell necrosis through damage-associated molecular patterns (DAMPs) which induce DC phagocytosis and processing of tumor antigens. This process matures DCs to serve their primary role as antigen presenting cells, and stimulates migration to lymph nodes, and ultimately leading to activation or inactivation of both CD4 and CD8 effector T cells (43). Classically differentiated DCs secrete proinflammatory cytokines such as IL-12 to activate IFNγ producing T cells and NK cells, which drives differentiation of Th1 T cells and activated CD8+ cytotoxic T cells (44). However, in the TME most DCs exist in an immature state and become toleragenic DCs (tDCs) following sustained exposure to cytokines such as VEGF, IL10 and TGFβ (9, 43). tDCs induce T cell anergy through checkpoint molecule signaling, including signaling via CD28 and PDL-1 to their cognate receptors CTLA-4 and PD-1 on T cells (45). tDCs also promote the generation of regulatory T cells (Tregs) from naïve CD4+ T cells by secretion of IL10 and TGF-β (43).
2.4 MDSCs in the tumor microenvironment
MDSCs are derived from immature monocytes and neutrophils, mobilized from the bone marrow in response to cytokines associated with chronic inflammation, including IL-3, GM-CSF, and G-CSF (46). The two primary populations of MDSC are defined as monocytic MDSC (M-MDSC) and neutrophilic MDSC (PMN-MDSC), which have both distinct and overlapping molecular and functional characteristics. For example, they exhibit distinct gene expression profiles, and unique immunologic functions such as production of arginase (PMN-MDSC) or reactive nitrogen or oxygen intermediates (M-MDSC) (47). Following their mobilization from the bone marrow and entry into the bloodstream, MDSC are recruited into tumor tissues in part by following chemokine gradients such as CCL2 and CXCL8, and other cytokines secreted by tumor cells and immune cells within the TME (48). Once in the TME, MDSC can be induced to undergo further differentiation to become more immune suppressive, by factors such as TGF-b produced by Tregs. MDSCs can also accumulate in secondary lymphoid tissues including the spleen and lymph nodes where they contribute to systemic immune suppression and further promote tumor progression (38, 49).
The expansion and differentiation of Tregs within the TME, is promoted in part by MDSC expression of tumor derived peptides on MHCI and MHCII molecules (50). As another mechanism of MDSC polarization, histamine released by mast cells binds to histamine receptor 1 on MDSCs inducing secretion of Arg-1 and nitric oxide synthase (iNOS) which inhibits T cell proliferation (51). Recent studies have correlated the abundance of MDSCs with poor prognosis and poor response to immune checking inhibitor (ICI) therapy in patients with various cancer types including breast, colorectal, lung and prostate cancers (52–54).
M-MDSC were originally defined in tumor-bearing mice as immature bone marrow derived cells that suppressed multiple T cell functions (55). The population of M-MDSC overall is considered to be more immune suppressive than PMN-MDSCs despite making up only 10-20% of the total MDSC population (56). Mechanisms of M-MDSC-mediated immune suppression include production of suppressive cytokines IL10 and TGF-β (4); They also promote T cell apoptosis by TCR-ζ chain downregulation through secretion of iNOS, arginase, and and reactive oxygen species (ROS) in mouse models. Secretion of iNOS also inhibits NK cells, thereby reducing antibody-dependent cell-mediated cytotoxicity (ADCC) (57). In a clinical setting, patients with non-small cell lung carcinoma (NSCLC), circulating tyrosine kinase receptor TIE2hi expressing M-MDSCs were found to suppress antigen-specificT cell responses and their presence was linked to poor patient outcomes (58). In contrast, patients with NSCLC treated with anti-PD-1 checkpoint blockade that had lower frequencies of both M-MDSCs and PMN-MDSCs had longer overall progression free survival (59).
PMN-MDSCs comprise the majority of MDSC populations (60). These PMN-MDSC are metabolically distinctive from mature neutrophils and promote early tumor dissemination and establishment of the pre-metastatic niche in the lungs and other sites (61). They also migrate more effectively and exert significantly greater immune suppressive activity compared to normal neutrophils. Mechanisms by which PMN-MDSCs inhibit T cell function include reactive nitrogen intermediates in mice and dogs, and ROS in humans (60, 62). Patients with primary and metastatic lung cancers exhibited high numbers of tumor infiltrating PMN-MDSCs, which was associated with suppressed NK cell activation and cytolytic activity, thought to be mediated by both cell-cell contact with PMN-MDSC and production of soluble factors within the TME (54).
2.5 Immune modulative TANs in the tumor microenvironment
Immune suppressive TANs and PMN-MDSCs are recruited to the TME primarily by the chemokine CXCL8, which signals via the chemokine receptors CXCL1 and CXCL2 (63). Given their common origins in the bone marrow and their shared need for growth factors and cytokines such as G-CSF, IL-6, and IL-17, it is somewhat difficult to definitely distinguish TANs from PMN-MDSCs (47, 63). Within tumor tissues, TANs are classified as either N1 or N2 populations, analogous to M1 and M2 macrophages (64). Populations of N1 TANs exhibit antitumor activity, whereas N2 TANs inhibit T cell proliferation and promote tumor growth (65). TGF-β secreted by tumor cells is one mechanism that polarizes TANs to the N2 phenotype (64, 66, 67). Within the TME, N2 TANs promote angiogenesis and play a role in establishing the pre-metastatic niche through secretion of VEGF and by expression of metalloproteinase-9 (MMP-9) which decreases the bioavailability of anti-angiogenic molecules (68, 69). High circulating numbers of N2 TANs have been associated with increased tumor metastatic progression, and genetic instability in tumors including melanoma and bronchioloalveolar carcinoma (70–72). Depletion of N2 TANs in animal models leads to increased numbers of effector CD8 T cells (73)’ and promotes their infiltration into the tumor (63). Secretion of TGF-β and IL-10 by N2 TANs stimulates MDSC expansion, further augmenting the immune suppressive nature of the TME. Importantly, the mechanisms by which N2 TANs suppress tumor immunity may in many cases be distinct and different in mice versus humans (74).
In addition to suppressing T cells, both PMN-MDSC and N2 TANs produce neutrophil extracellular traps (NETs). These complex webs, comprised of extruded DNA molecules studded with chromatin and other nuclear proteins, can promote tumor metastasis by trapping migrating tumor cells within tumor blood vessels, and then facilitating the survival of these early metastatic tumor cells (75, 76). The NETS secreted by TANs and PMN-MDSC also interrupt the cytotoxic activities of CD8+ T cells and NK cells (77).
3 Therapeutic targeting of immune suppressive macrophages and MDSC
3.1 Direct depletion of myeloid cells (TAM, MDSC) in the TME
Depletion of immune suppressive myeloid cell populations within the TME is one method to overcome the immune suppressive pressure they exert, particularly given that these cells, especially TAMs, can be quite numerous in the TME, in some cases comprising over 50% of the entire tumor cell population (78). Below we provide examples of the multiple strategies designed to deplete TME populations of immune suppressive TAMs.
3.1.1 Colony-stimulating factor 1 receptor blockade
One approach that has been extensively investigated is TAM depletion via blocking signaling by the essential macrophage growth factor receptor colony-stimulating factor 1 receptor (CSF-1R). CSF-1R is expressed by TAMs and binds to the primary ligands CSF-1 and IL-34 (79). CSF-1R signaling is crucial to macrophage differentiation and survival (79, 80). The density of CSF-1R+ TAMs in tumors correlates with poor outcomes in many tumor types, including colon adenocarcinoma, pancreatic cancer, classical Hodgkin lymphoma, leiomyosarcoma, hepatocellular carcinoma and breast cancer (81–86). CSF-1R is also expressed by other immune cells within the TME such as DC, MDSCs and neutrophils, and blocking CSF-1R signaling may therefore also deplete these cells in addition to TAMs (87). Disruption of CSF-1R signaling has been achieved by use of small molecules and monoclonal antibodies (87, 88). Multiple clinical trials are ongoing to evaluate the effects of CSF-1/CSF-1R blockade on TAM populations and tumor control in many both solid tumors and hematologic cancers (Table 1). To date clinical trials for CSF-1/CSF-1R blockade have been completed in non-pancreatic neuroendocrine tumors (89), Hodgkin lymphoma (90), ovarian cancer (NCT03166891, NCT03901118), non-Hodgkin lymphoma (NCT03974243) and hepatocellular carcinoma (NCT03245190). The accumulated results from these trials indicates safety and tolerability of the CSF-1R inhibitors, but limited efficacy, suggesting either refined dosage or timing of CSF-1/CSF-1R blockade, or the need to employ with other combination therapies (91).
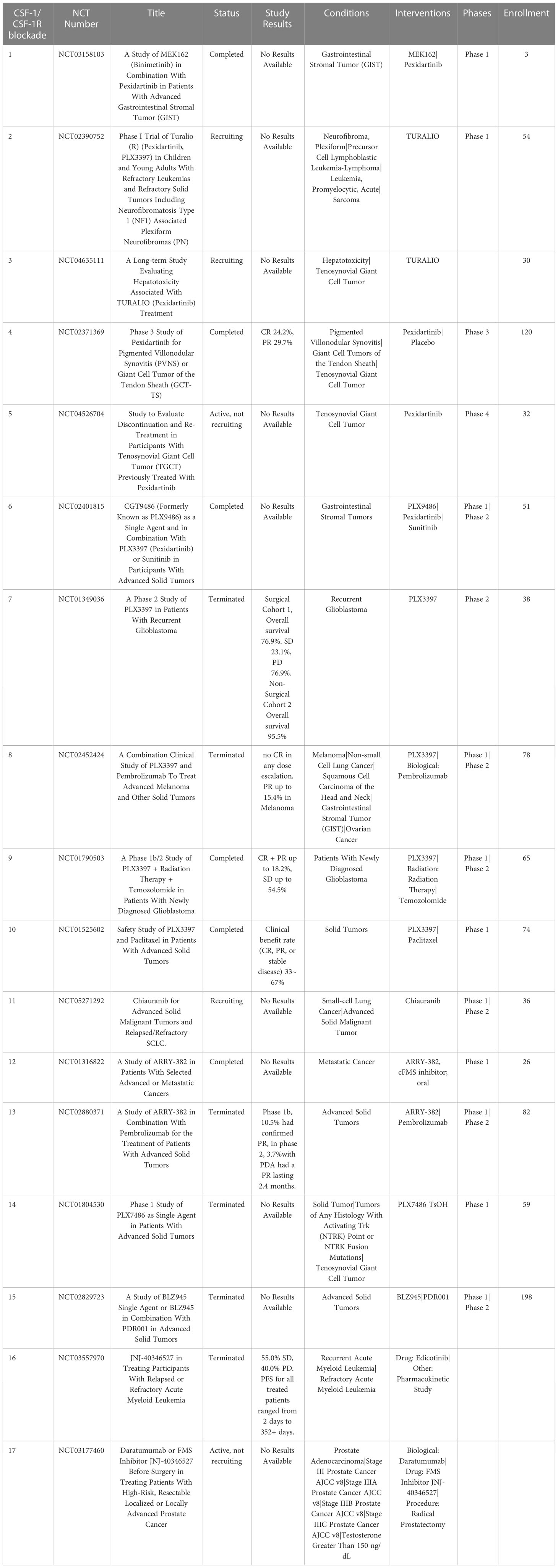
Table 1 US clinical trials in cancer using Colony-stimulating factor 1 receptor (CSF-1R) blockade as intervention.
3.1.1.1 Pexidartinib (PLX3397, TURALIO)
The small molecule drug PLX3397 targets CSF1R signaling and reprograms intra-tumoral immune suppressive myeloid cells (92), and has been shown to convert immune suppressive M-DSCs to a more proinflammatory tumoricidal phenotype (93, 94). PLX3397was approved by the FDA in 2019 for use in the treatment of diffuse type tenosynovial giant cell tumors (dt-TGCT), a rare and often unresectable non-life-threatening cancer of the tendon sheath that is driven by CSF-1 expressing TAMs (95). CSF-1 activation in dt-TGCT leads to recruitment of CSF-1R+ macrophages which make up a large bulk of the tumor mass (96). This specific tumor type is well-suited for targeting by CSF-1/CSF-1R pathway blockade; and treatment with anti-CSF-1R antibodies has shown significant reduction of CSF-1R+ TAMs within tumor tissues (97). In a phase III double blind clinical trial, 14.8% of patients with unresectable dt-TGCT treated with PLX3397 had a complete response (CR) and 24.6% had a partial response (PR) per RECIST criteria compared to zero response in the placebo control group (98).
Current clinical trials are investigating the effectiveness of PLX3397 in multiple cancer types including melanoma, prostate cancer, recurrent glioblastoma multiforme (GBM) and hematological malignancies (99–101). Preclinical use of orally administered PLX3397 for the treatment of recurrent GBM in phase II trials did not show statistically significant improvement in progression free survival of patients compared to historical controls, and there were no partial or complete responses observed in their 38-patient cohort (92, 102). In a phase II trial with 20 patients with relapsed Hodgkin lymphoma, single agent PLX3397 treatment showed an objective overall response rate (ORR) of 5% (103). Thus, the value of CSF-1R inhibition alone for treatment of tumors such as GBM may be limited.
Trials investigating the use of PLX3397 in combination with other agents are ongoing breast cancer (NCT01042379) and unresectable sarcomas and malignant peripheral nerve sheath tumors (NCT02584647). A Phase II trial in patients with advanced melanoma and other solid tumors in combination with PD-1 blocking antibody pembrolizumab (NCT02452424) was terminated early due to insufficient evidence of clinical efficacy (101).
3.1.1.2 Chiauranib (CS2164)
The small molecule drug chiauranib is a CSF-1R inhibitor that also selectively inhibits kinases related to angiogenesis, including VEGF, PDGFR, and c-kit (104). Chiauranib binds to the ATP site in VEGFR2 and inhibits kinase activity, as well as reducing phosphorylation of ERK1/2, thus decreasing expression of genes related to tumor angiogenesis. Chiauranib has shown efficacy in preclinical mouse models of hepatocellular carcinoma, colorectal cancer, and non-Hodgkin lymphoma (NHL) (105–107). Initial dose escalation trials demonstrated that 67% of patients achieved stable disease, with acceptable safety and tolerability (104). Current clinical trials are ongoing, with one phase II trial reported currently in the US (NCT05271292), evaluating chiauranib as a single agent to treat advanced solid malignant tumors.
3.1.1.3 Additional small molecule inhibitors of CSF-1R
Other small molecule CSF-1R inhibitors include ARRY-382, PLX7486, BLZ945 and JNJ-40346527 (edicotinib), and all are currently being evaluated in clinical trials for treatment of Hodgkin lymphoma (cHL) (87). A phase I study with ARRY382 for treatment of advanced solid tumors showed 15% stable disease with no objective responses observed out of 26 patients when administered in combination withpembrolizumab (NCT02880371). Phase I and II clinical studies of the drug JNJ-40346527 in patients with refractory Hodgkin lymphoma found that 11 of 20 patients (55.0%) had stable disease (SD) with progression free survival (PFS) times for all treated patients ranging from 2 days to 352 days (90).
3.1.1.4 Monoclonal antibodies targeting CSF-1 or CSF-R1
Monoclonal antibodies targeting CSF-1R in clinical development include emactuzumab, AMG820, IMC-CS4, cabiralizumab, MCS110 (lacnotuzumab) and PD036324 (Table 2). MCS110 and PD036324 target the CSF-1 (ligand) as opposed to the CSF-R1 receptor (87). Phase Ia/Ib trials with emactuzumab as either a single agent or in combination with paclitaxel in patients with metastatic solid tumors including mesothelioma, soft tissue sarcoma, ovarian, breast, pancreatic, endometrial cancer and dt-TGCT have been conducted. Study outcomes in the monotherapy group did not reveal any patients with objective tumor responses, with 13% of patients exhibiting SD. When administered in combination with paclitaxel, 7% of patients had PR with 43% showing SD (108). This study also demonstrated a significant reduction in the numbers of CSF-1R+ TAMs in both monotherapy and combination groups (101). The first human trial of AMG820 showed increased serum CSF-1 concentrations and decreased numbers of macrophages (109). Patients with relapsed or refractory advanced solid tumors treated with AMG820 experienced a 32% SD rate, while one patient with NSCLC experienced a PR. All the agents in trials targeting CSF-R1 have generally been well-tolerated to date, suggesting that sustained CSF-1R blockade treatment for weeks to months is safe. However, to date none of the CSF-1 or CSF-1R targeted agents has demonstrated significant antitumor activity clinically (110).
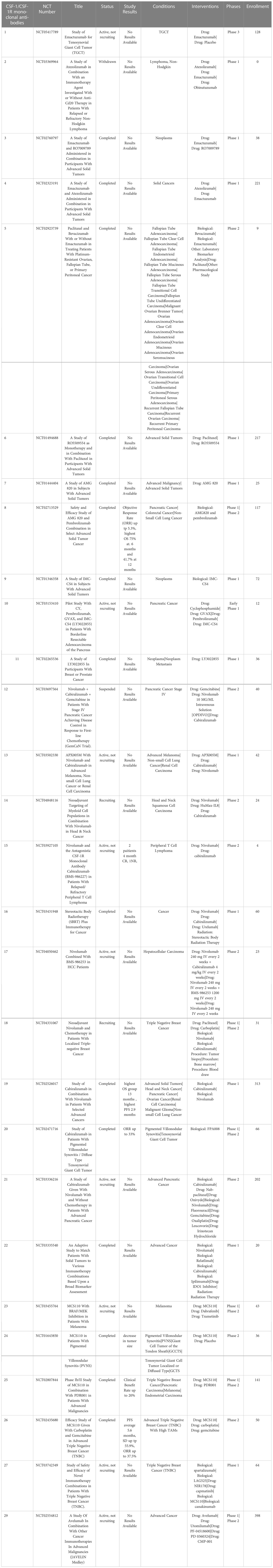
Table 2 US clinical trials in cancer using Monoclonal antibodies targeting CSF-1 or CDF-1R.
3.1.2 Trabectedin as myeloid cell depleting chemotherapy
Trabectedin an alkaloid drug that binds a minor groove of DNA and blocks the cell cycle and DNA repair pathways (111). It has been shown to selectively reduce TAMs in tumors without affecting the infiltration of T cells (112). Treatment with trabectedin also inhibits local differentiation of monocytes into TAMs (113). Use of trabectedin in multiple preclinical animal tumor models demonstrated depletion of TAMs and reduction of tumor growth, suppression of angiogenesis, and reduced concentrations of IL6, CCL2 and CXCL8 (114). Current phase II clinical trials of trabectedin are ongoing for treatment of soft tissue sarcoma, bone tumors and small round-cell sarcomas, administered in combination with low-dose radiation therapy (NCT05131386). Trabectedin is FDA approved for treatment of unresectable or metastatic liposarcoma and leiomyosarcoma (115) (Table 3).
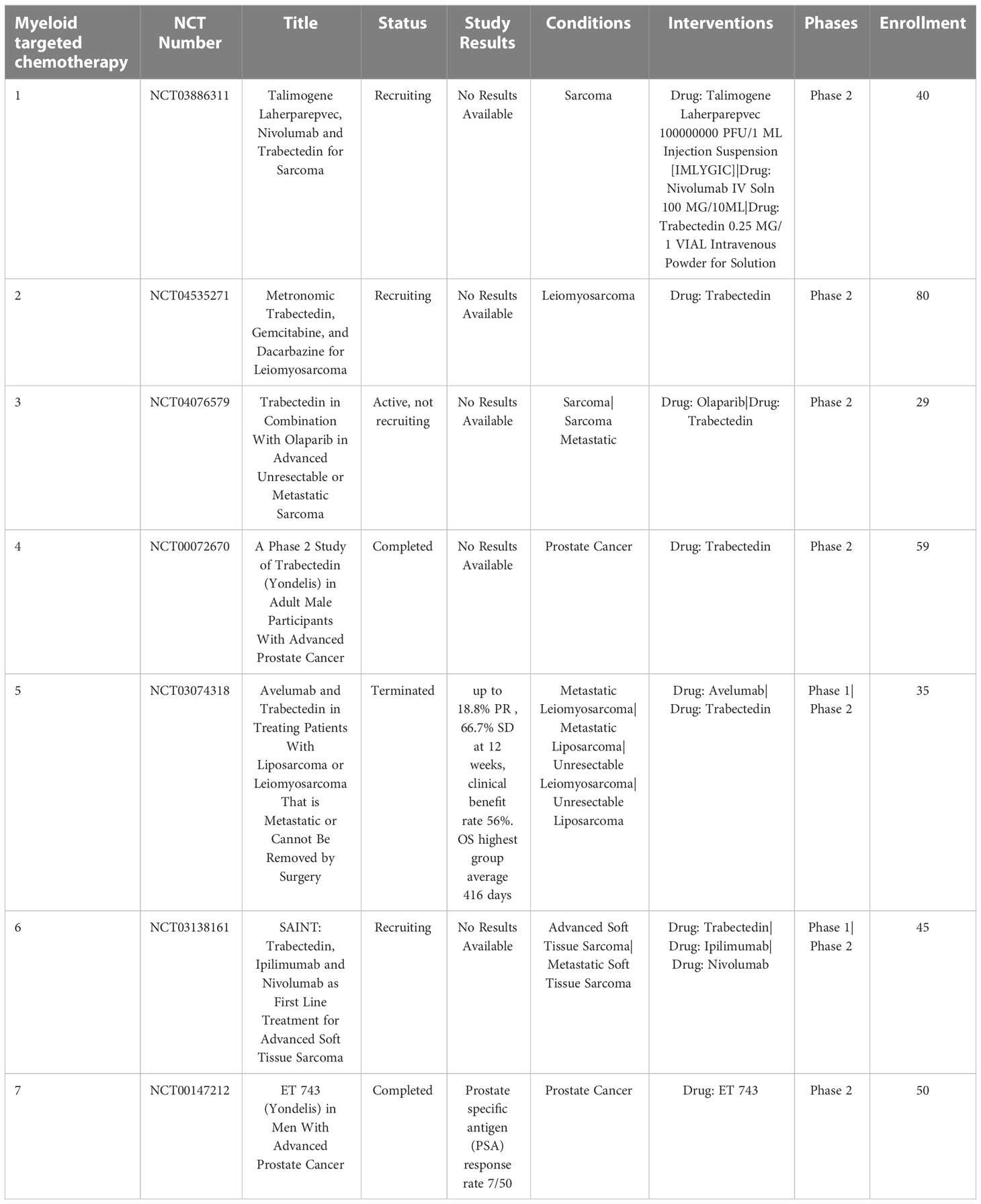
Table 3 US clinical trials using Trabectedin as myeloid cell depleting chemotherapy for cancer.
3.2 Chemokine receptor antagonists for monocyte and neutrophil migration inhibition
Chemokine receptor antagonists can reduce the infiltration of monocytes and MDSCs into the TME. The chemokine CCL2 binds to the receptor CCR2 expressed on inflammatory monocytes (116), which signals to circulating monocytes to promote extravasation from the vasculature and into inflamed tissues (32). Many tumors secrete large amounts of CCL2, thereby recruiting circulating inflammatory monocytes into tumor tissues where they then differentiate into M2 TAMs (32, 117, 118). CCL2 may also play a minor role in PMN-MDSC recruitment, though the primary chemokine driving TAN recruitment is CXCL8 (119). There have been numerous preclinical studies in rodent models assessing inhibitors of the CCL2-CCR2 axis using either small molecule CCL2 inhibitors or monoclonal antibodies, and most have demonstrated inhibition of tumor growth and/or decreased metastatic burden (120). In these models, CCL2-CCR2 signaling blockade has been shown to suppress tumor growth through multiple pathways including depletion of TAMs and M-MDSC and increasing infiltrating T cells (32, 118, 120).
3.2.1 CCR2 targeted antibodies
Carlumab (CNTO888) is a CCL2 neutralizing antibody that has been evaluated in multiple cancer models as either a single agent immunotherapy or in combination with chemotherapy (121). Pre-clinical mouse models evaluating carlumab have demonstrated increased IFNγ production by NK cells and antitumoral CD8+ T cells when combined with anticancer vaccines (122). Carlumab has demonstrated positive clinical responses when used in combination with chemotherapeutic drug docetaxel (123); phase II trials have been completed but Carlumab has since been discontinued (NCT00992186).
MLN1202 (plozalizumab) is a CCR2 blocking monoclonal antibody currently undergoing phase II clinical trials for treatment of metastatic bone cancer (NCT01015560). Results so far show that MLN1202 is relatively well tolerated with only 7.14% of patients experiencing severe adverse events (SAE). A phase I trial of MLN1202 in combination with nivolumab was terminated early due to serious adverse events (NCT02723006), which may suggest limited potential for MLN1202 as single or combined immunotherapy agent (124, 125) (Table 4).
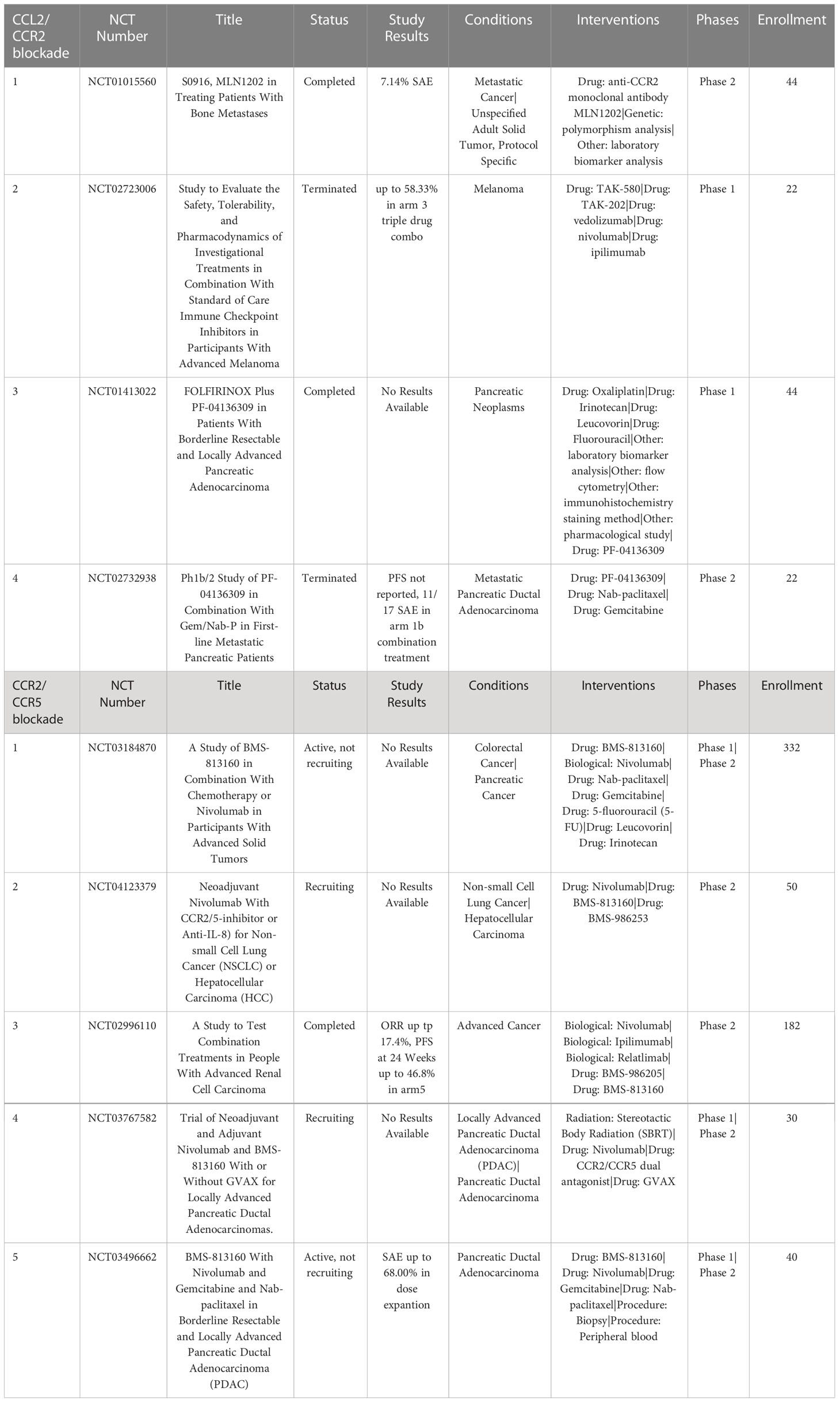
Table 4 US clinical trials in cancer targeting CCR2 or CCR5 axis.
3.2.2 CCL2 inhibitors
Bindarit is a small molecule drug that inhibits the synthesis of CCL2 and has been shown to induce tumor regression in preclinical studies by inhibiting TAM and MDSC infiltration of the TME in breast cancer, prostate cancer, and osteosarcoma animal models (126–129). A second CCL2 inhibitor mNOX-36 has been shown in a rat model of GBM to significantly inhibit tumor growth (130). The safety of mNOX-36 is currently being evaluated in Phase I trials (Table 4).
3.2.3 CCR2 inhibitors
RS 504393 is a small molecule CCR2 antagonist that has shown activity in blocking M-MDSCs and TAM recruitment into tumors following gemcitabine treatment in a mouse model of bladder cancer (131). Another CCR2 inhibitor (BMS CCR2 22), is a high affinity CCR2 antagonist that decreases TAM density as demonstrated in mouse metastatic hepatic cancer models. When combined with FOLFOX (folinic acid, fluorouracil oxaliplatin) chemotherapy regimine, administration of BMS CCR2 22 significantly increased efficacy and improved overall survival in mice with colon adenocarcinomas (117, 118). A third CCR2 antagonist, 747 is a natural product derived from the tree Abies georgei (132). The drug 747 is considered a selective CCR2 antagonist and has been shown to inhibit TAM recruitment and increase density of CD8+ tumor infiltrating lymphocytes as well as increase inflammatory cytokines such as IFN-γ in rodent mode. Treatment with 747 also increased tumor apoptosis when combined with sorafenib, a tyrosine kinase inhibitor, thereby potentiating antitumor activity by depleting TAMs (133).
A fourth selective CCR2 inhibitor (PF-04136309) has demonstrated antitumor activity in an orthotopic mouse model of pancreatic cancer (134). Phase Ib clinical trials in patients with pancreatic cancer evaluated treatment with PF-04136309 in combination with the chemotherapy regimen FOLFIRINOX (folinic acid, fluorouracil, irinotecan hydrochloride, and oxaliplatin) and demonstrated a 49% response rate, compare to no responding patients treated with FOLFIRINOX alone. In addition, administration of PF-04136309 in combination with FOLFIRINOX significantly decreased the numbers of CCR2+ monocytes in bone marrow samples, compared to FOLFIRINOX alone treated patients (135). A phase I study in patients with metastatic pancreatic cancer found that PF-04136309 given in combination with chemotherapy gemcitabine and nab-paclitaxel significantly decreased CD14+CCR2+ monocytes in circulation. However, the high incidence of pulmonary toxicity in patients treated with PF-04136309 led to a discontinuation of further clinical evaluation (136).
3.2.4 Dual CCR2/CCR5 inhibitor for myeloid cell targeting
BMS-813160 is a dual CCR2/CCR5 inhibitor which has been investigated in phase I and phase II trials as combination therapy (137). In ongoing phase II clinical trials for treatment of NSCLC and hepatocellular cancer, BMS-813160 is being administered in combination with nivolumab and the anti-CXCL8 drug BMS-986253 (NCT04123379) (138). BMS-813260 is also being investigated in phase II trials for pancreatic ductal carcinoma and colorectal cancer, administered in combination with either nivolumab or chemotherapy (139) (Table 4).
3.2.5 Repurposed angiotensin receptor antagonists for CCR2 inhibition
Losartan, a type 1 angiotensin II receptor (AT1R) blocker (ARB), has been found to exert off-target activity as a potent, non-competitive CCR2 antagonist (140). In a mouse syngeneic breast cancer model, losartan suppressed lung metastatic tumor burden significantly (141). In this model, the reduced metastatic burden was associated with a significant decrease in CD11b+/Ly6C+ monocytes recruited to the lungs (140). In studies in a dog model of metastatic osteosarcoma, the combination high dose losartan (10mg/kg PO BID) with the non-selective tyrosine kinase inhibitor toceranib demonstrated a response rate (PR) of 25% and clinical benefit rate of 50% (142). A similar phase I clinical trial is underway for pediatric osteosarcoma using the combination of losartan with the non-selective tyrosine kinase inhibitor sunitinib (NCT03900793). There are also multiple other clinical trials currently evaluating losartan in combination with radiation therapy and chemotherapy or immunotherapy. A phase II clinical trial of losartan in combination with nivolumab is currently underway in patients with localized pancreatic cancer (NCT03563248). In addition, losartan is being evaluated in combination with radiation therapy and chemotherapy in pancreatic cancer (NCT03563248, NCT04106856). A recent study also indicates that losartan treatment can reduce cerebral edema following immunotherapy in a rodent GBM model (143) (Table 5).
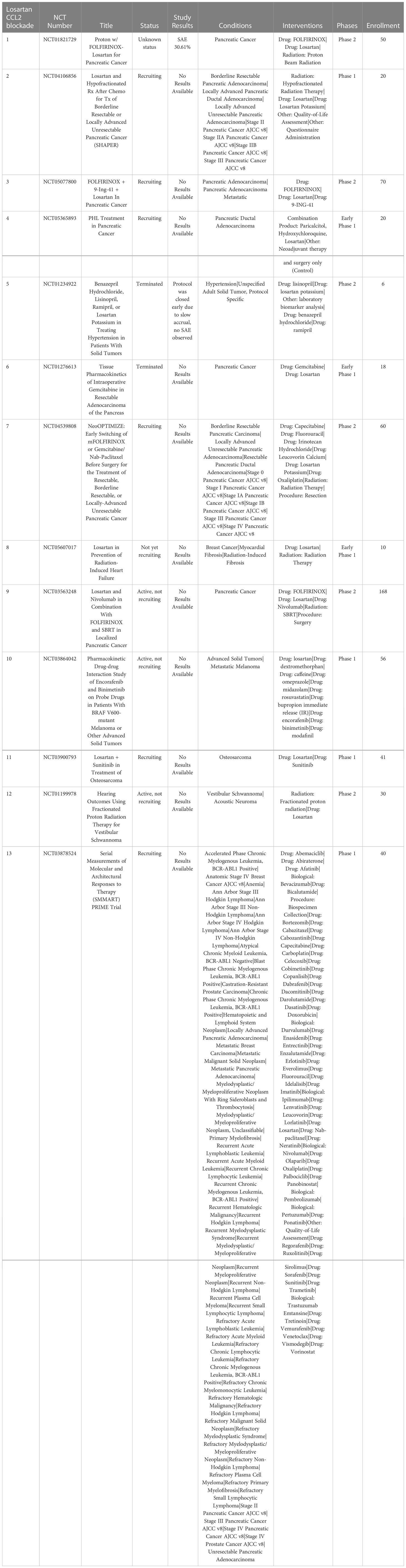
Table 5 US clinical trials using Repurposed angiotensin receptor antagonists for CCR2 inhibition in cancer.
3.2.6 CXCR1 blockade to deplete PMN-MDSC and TANs
The chemokine CXCL8, which signals via CXCR1 and CXCR2, triggers the recruitment of PMN-MDSC and TANs into the TME (144). The receptors CXCR1 and CXCR2 are primarily expressed on neutrophils (145). CXCR1 is very selective for CXCL8, whereas CXCR2 also binds other chemokines. Signaling by CXCR1 and CXCR2 are major mechanisms for recruiting neutrophils and PMN-MDSC into the TME which then differentiate into TANs or PMN-MDSCs (146). High expression of CXCL8 by tumors has been correlated with poor prognosis in many tumor types (147). Thus, CXCR1 and CXCR2 antagonists have been evaluated as strategies to deplete the TME of immune suppressive N2 TANs and PMN-MDSC (48, 148) (Table 6).
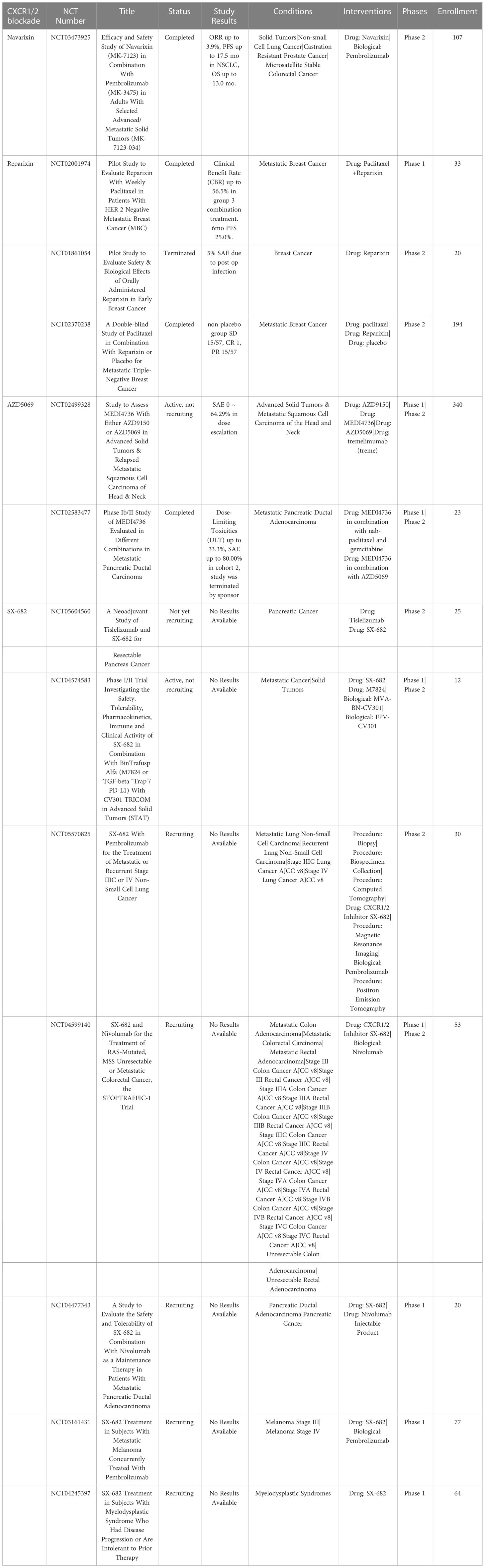
Table 6 US clinical trials using CXCR1 antagonists for cancer treatment.
3.2.6.1 CXCR1 antagonist navarixin
The selective CXCR1 antagonist navarixin was originally developed for treatment of chronic obstructive pulmonary disease (COPD), asthma and psoriasis (149). A current phase II clinical trial of navarixin in combination with pembrolizumab is underway in patients with either PD-1 positive refractory NSCLC, castration resistant prostate cancer, or microsatellite stable colorectal cancer (NCT03473925) (150).
3.2.6.2 CXCR1 antagonist reparixin
Reparixin is a small molecule dual antagonist of both CXCR1 and CXCR2 (151, 152). Reparixin was originally evaluated as a drug to prevent graft rejection for pancreatic islet cells (153). In vitro studies with reparixin in thyroid cancer found that it also exhibits direct anti-tumor activity (154). In a phase I clinical trial in patients with HER-2 negative metastatic breast cancer, reparixin was well tolerated in combination with paclitaxel chemotherapy (155). However, phase II double blinded clinical trials in triple negative breast cancer patients demonstrated no improvement reparixin in combination with paclitaxel exhibited no additional clinical benefit compared to treatment with paclitaxel alone (NCT02370238) (156).
3.2.6.3 CXCR1/2 antagonist ladarixin
Ladarixin, like reparixin is a dual CXCR11/2 antagonist (157). Preclinical evaluation of ladarixin demonstrated significant activity in a mouse model of pancreatic ductal adenocarcinomaimproved activity compared to either agent alone (148, 158). In an animal model of uveal melanoma administration of ladarixin repolarized TAMs to a M1 phenotype and inhibited tumor cell migration (157). Ladarixin has been used in clinical trials for diabetes, however clinical trials for cancers have not been reported.
3.2.6.4 CXCR2 antagonist AZD5069
AZD5069 is a highly selective small molecule antagonist of CXCR2 receptors that has been shown to inhibit neutrophil migration in patients with COPD (159). It is currently in clinical trials to deplete TANs in the TME in patients with metastatic pancreatic ductal adenocarcinoma and in relapsed metastatic squamous cell carcinoma of the head and neck in combination with ICI (160, 161). In addition, AZD5069 is being evaluated in combination with the androgen receptor antagonist enzalutamide in phase I/II trials in patients with metastatic castration resistant prostate cancer (mCRPC) in the UK (NCT03177187). The combination treatment was well tolerated with no dose limiting toxicities observed. The study observed that 2 out of 15 patients experienced a PR and 10 of 15 patients experienced SD, with responses lasting 2-16 months. Another trial demonstrated that AZD5069 has antitumor activity and depleted TAN density in patients with mCRPC (162).
3.2.6.5 Dual CXCR1/2 antagonist SX-682
SX-682 is another dual CXCR1/2 antagonist, which in rodent models of head and neck cancer have demonstrated suppression of PMN-MDSC accumulation and enhanced tumor infiltration with adoptively transferred NK cells (163, 164). SX-682 is currently being tested in phase I clinical trials in combination with ICI for metastatic melanoma (NCT03161431), and in phase II trials for pancreatic cancer, lung cancer, colon and rectal adenocarcinoma (NCT05604560, NCT05570825, NCT04599140).
3.3 CXCR4 blockade to inhibit tumor angiogenesis and metastases
Signaling by the chemokine receptor CXCR4 after binding the chemokine CXCL12 (SCF-1) triggers increased tumor proliferation, survival, and chemotaxis (165). Notably, CXCR4 is overexpressed by many different types of cancers, where it plays a role in tumor metastasis, and also a critical role in mobilizing and recruiting MDSC from bone marrow. Blockade of the CXCR4 signaling is hypothesized to not only decrease tumor angiogenesis but also decrease the number of cancer stem cells and increase mobilization and recruitment of effector T cells into the TME (166) (Table 7).
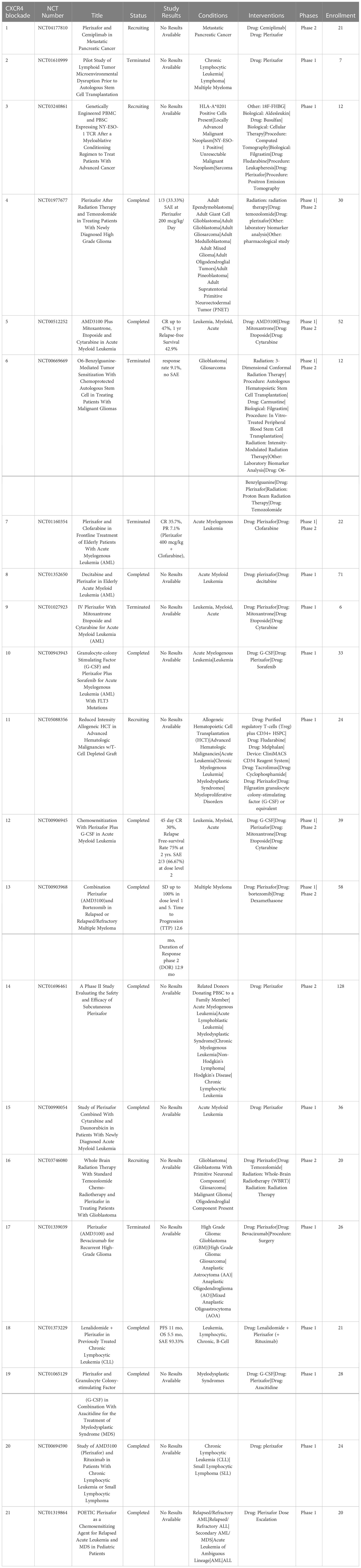
Table 7 US clinical trials using CXCR4 targeting drugs for cancer treatment.
3.3.1 AMD3100
AMD3100 (plerixafor) is currently the only FDA approved CXCR4 inhibitor. This drug was initially developed for treatment and prevention of HIV, but has now also been approved for treatment of non-Hodgkin lymphoma (NHL) and multiple myeloma (MM) (167, 168). Use of AMD3100 in combination with the anti-VEGFR2 antibody ramucirumab in a mouse model of colorectal cancer significantly reduced recruitment of immune suppressive monocytes, as the study demonstrated that depletion of immune suppressive Ly6Clow monocytes by CXCR4 blockade was associated with enhanced treatment efficacy of ramucirumab (169, 170). Administration of AMD3100 was also associated with increased CD8+ T cell infiltration and synergistic activity when combined with ICI (171) Use of AMD3100 in NHL and MM suppressed tumor growth and metastasis and was associated with converting Tregs to a Th1 phenotype and enhancing CD8+ T cell infiltration (172).
Another mechanism of AMD3100 antitumor activity was to block CXCR4+ tumor cells from interacting with CXCL12 produced by cancer associated fibroblasts (173). Use of AMD3100 in combination with ICI in patients with microsatellite unstable pancreatic or colorectal cancer demonstrated enhanced B cell and T cell antitumor responses (174). Clinical trials evaluating AMD3100 include phase II trials for metastatic pancreatic cancer, phase I and II trials for glioma, and phase I and II trials for hematopoietic malignancies. The proposed mechanism targeted in these trials is to sensitize the TME to chemotherapy by blocking the CXCR4 and CXCR/2 axes (175). Other applications of AMD3100 is as a hematopoietic stem cell (HSC) mobilizing agent (typically in combination with G-CSF) for hematopoietic stem cell transplantation (176).
3.3.2 BPRCX807
BPRCX807 is a selective CXCR4 antagonist that has shown activity in mouse models of hepatocellular carcinoma (177). In these models BPRCX807 prevented tumor cell migration and limited the development of metastases. Another activity of BPRCX807 is to reprogram immune suppressive TAMs to a more an immunostimulatory M1 phenotype, while at the same time promoting CD8+ T cell infiltration into tumors (177). Early mouse studies provide support for further investigation of CXCR4 blockade as a combination agent along with ICI (172).
4 Metabolic reprogramming to target myeloid suppressor cells
4.1 IDO inhibitors
The enzyme indoleamine 2,3-dioxygenase 1 (IDO1) converts the essential amino acid tryptophan (Trp) to kynurenine (Kyn), thereby leading to an overall depletion of this critical amino acid within the TME and tumor draining lymph nodes (178). Overexpression of IDO1 is considered an important driver of tumor associated immune suppression and a key to establishing immune tolerance of cancer antigens (179, 180). High intratumoral IDO1 expression is correlated with poor prognosis in melanoma, ovarian cancer, colorectal cancer, and lung cancers (181, 182). In ovarian cancer, high IDO1 expression also correlates with increased drug resistance (183). A high ratio of tryptophan to kynurenine in blood is also associated with a poorer prognosis in some cancers (184–187).
High levels of IDO1 expression by cancer cells can also drive MDSC expansion (188); Moreover, MDSCs also overexpress IDO1, triggering a positive feedback loop that reinforces and sustains the immune suppressive TME (189). Local depletion of tryptophan by IDO leads to cell cycle arrest and apoptosis of effector T cells in tumor tissues (190). In addition, IDO1 positive MDSCs also contribute to T cell exhaustion through IL-6 secretion. The local buildup of kynurenine concentrations within the TME also triggers deleterious alterations in the metabolic properties of tumor infiltrating T cells and converts CD4 effector cells to Tregs (190–193). Evaluation of IDO inhibitors in preclinical models demonstrated a reduction of IDO1+ MDSCs within the TME and measurable reduction of Kyn concentrations (178). Taken together, these properties make IDO1 a promising target for reversing immune suppression through metabolic reprogramming of the TME.
4.1.1 Epacadostat as an IDO synthesis inhibitor
Van den Eynde et al. summarized the many clinical trials evaluating epacadostat up to 2020 and in their paper discussed why the outcomes of these trials have been largely negative. The majority of these trials have evaluated epacadostat in combination with checkpoint blockade (CTLA4, PD-L1 or PD-1) and have to date failed to demonstrate any meaningful clinical benefit. It was concluded therefore that epacadostat did not improve ICI, as confirmed in at least 12 clinical trials (194, 195). Due to these poor results, remaining clinical trials with epacadostat have been withdrawn, downsized or suspended.
4.1.2 Navoximod (GDC-0919)
Navoximod has been evaluated clinically as a monotherapy or in combination with atezolizumab (NCT02048709, NCT02471846, NCT05469490, and these trials demonstrated that the navoximod was well tolerated and decreased plasma Kyn concentrations in a dose dependent manner (181). However, there was no clear tumor response benefit in the navoximod combination therapy arm when compared to treatment with atezolizumab alone (196).
4.2 Repurposed beta blockers as MDSC depleting agents
In addition to stimulation of cortisol release, chronic stress from inflammation in cancer is associated with prolonged activation of the sympathetic nervous system (53). Chronic adrenergic activation and release of catecholamines, primarily norepinephrine (Nor), has been associated with MDSC mobilization from the bone marrow and acquisition of greater immune suppressive properties, leading to both systemic and local immune suppression (197, 198). A consequence of increased Nor concentrations is higher concentrations of both MDSCs and TAMs in tumor tissues. For example, activation of β2-adrenergic receptor (β-AR) signaling was shown to upregulate STAT3 and NFk-b signaling pathways which drive development of immune suppressive MDSC and TAMs (199). Activation of β-AR signaling has been shown to polarize macrophages to an immunosuppressive M2 phenotype in a rodent breast cancer model (200, 201). Adrenergic signaling in tumor cells themselves can also be triggered by tumor hypoxia (202).
4.2.1 Propranolol as non-selective β-blocker
Use of non-selective beta blockers such as propranolol have been investigated for their ability to reprogram immune suppressive cells within the TME (200, 203). Blocking β-AR signaling by MDSCs with propranolol can prevent their mobilization from the bone marrow (204). In addition, propranolol treatment reprograms MDSCs to a less immune suppressive state by blocking STAT3 signaling (205). This effect has been demonstrated in rodent models, where treatment with propranolol reduces MDSC mobilization and accumulation within the TME, accompanied by inhibition of tumor growth and metastasis (206). In rodent models, treatment with propranolol blocked the accumulation of M2 macrophages in metastatic breast cancer and inhibited metastases (53). In a spontaneous melanoma mouse model, propranolol treatment significantly reduced intratumoral accumulation of neutrophils, immune suppressive inflammatory (CD11c-Ly6ChiLy6G-) macrophages and DCs in both the primary tumor and metastatic lesions (207). Multiple rodent studies and recent clinical trials in dogs and human patients have demonstrated the ability of propranolol 3008 treatment to improve responses to radiation therapy for glioma, 3009 breast cancer, and pancreatic cancer (200, 208) (Table 8).
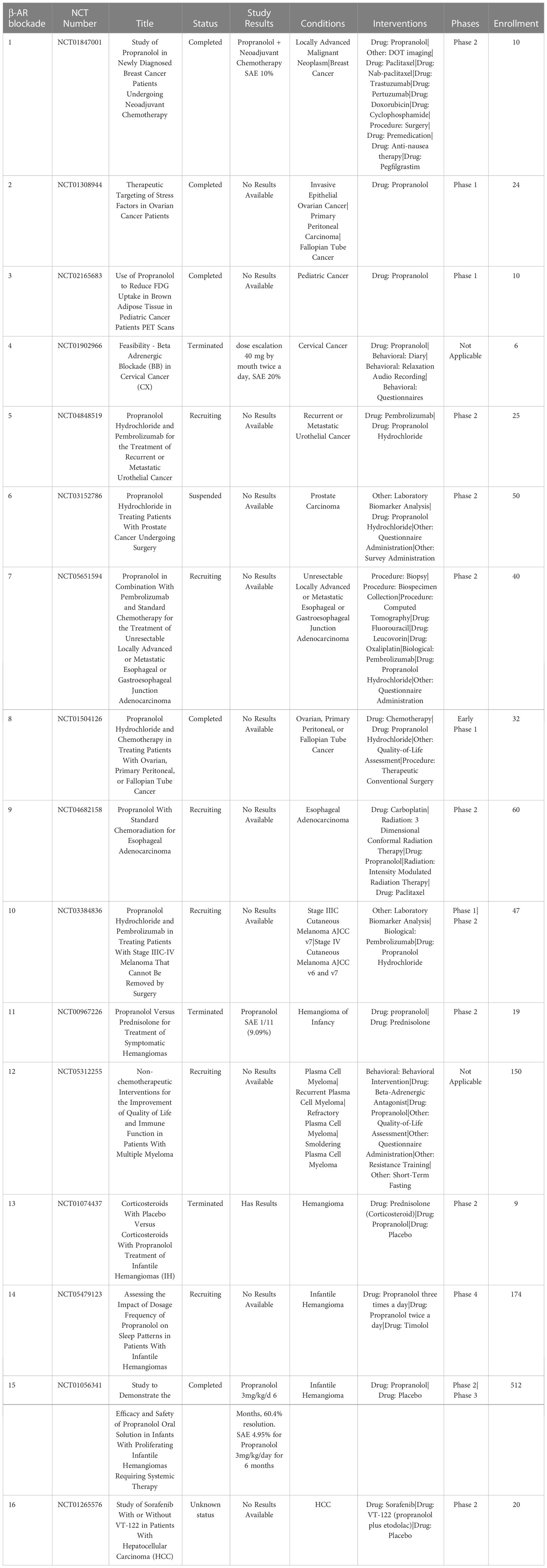
Table 8 US clinical trials using propranolol as cancer treatment.
In a phase II trial in patients with metastatic breast cancer it was found that in tumor tissues from propranolol treated patients there was upregulated expression of genes associated with classical dendritic cells and an increase in M1 macrophage polarization, along with an increase in CD69+ activated TAMs (209). Phase I trials of propranolol with pembrolizumab in patients with metastatic and locally advanced melanoma showed encouraging responses and the combination therapy to be well tolerated (210). In the USA there are currently 17 trials investigating propranolol in breast cancer, cervical cancer, prostate, esophageal, infantile hemangioma and hepatocellular carcinoma. Further studies are warranted to elucidate the clinical benefit of propranolol as a repurposed immunotherapy for TME reprogramming (Table 8).
4.3 Tyrosine kinase inhibitors
Tyrosine kinase inhibitors (TKIs), especially early generation non-specific TKIs such as sunitinib, have been shown to alter the immune suppressive TME, in part by reprogramming TAMs from M2 to M1 phenotypes, by reducing total TAM infiltrates and by blocking the accumulation of MDSCs and TANs (211–213). Tyrosine kinase receptors are extremely diverse family of receptors and there are >40 FDA approved TKI drugs. These TKIs are categorized according to the main receptor targeting sites which include, anaplastic lymphoma kinase (ALK), epidermal growth factor receptor (EGFR), FMS-like tyrosine kinase 3 (FLT3), vascular endothelial growth factor (VEGFR), and tropomyosin receptor kinase (TRK) (214). The positive clinical benefits observed when TKIs are combined with ICI indicate that the TKI impact on the TME is substantial and complementary to ICI therapy. The list of multi-target TKIs is quite extensive, therefore few are selected here for discussion to illustrate their potential as immunotherapy drugs.
4.3.1 VEGFR targeted TKIs
4.3.1.1 Sunitinib (SU011248, Sutent)
Sunitinib is a small molecule inhibitor that targets multiple kinases, with inhibitory effects against signaling by VEGFR, PDGFR and c-kit (215). It is an FDA approved agent for treatment of renal cell carcinoma and gastrointestinal stromal tumors (216). To date there are currently 270 US trials of sunitinib to treat, alone or in combination, many different cancers, including breast, hepatic, lung, and renal cancers. Early generation, multi-function TKIs such as sunitinib have been shown to exert impressive immune modulatory effects (217, 218). For example, sunitinib has been shown to deplete both MDSC and Tregs, in part by inhibiting STAT3 signaling (219); and in clinical trials positive responses to treatment with sunitinib has been associated with Treg depletion (220, 221).
4.3.1.2 Sorafenib (Nexavar, BAY 43-9006)
Sorafenib is another small molecule multi-kinase inhibitor, which in hepatocellular carcinoma has shown clinical benefit and antitumoral activity that is associated with immune remodeling of the TME (222). For example, treatment with sorafenib has been reported to selectively decrease Tregs numbers without impacting effector T cell numbers (223). Sorafenib has been shown to regulate the differentiation DCs in the TME (224) and to repolarize M2 TAMs to an M1 phenotype through inhibition of miR-101 expression and reduction of TGF-β secretion. Sorafenib has also been reported to induce secretion of pro-inflammatory cytokines such as IL-12 by TAMs (225, 226) and to decrease expression of PD-L1 on MDCS and plasmacytoid DCs (227, 228). There are 430 clinical trials registered in the US using sorafenib in cancer patients ranging from phase I to phase IV clinical trials, with many focused on renal cell carcinoma.
4.3.1.3 Lenvatinib (E7080, Lenvima)
Lenvatinib is another multitarget TKI that has shown in phase III trials clinical benefit as reflected by significantly increased overall survival times in patients with hepatocellular carcinoma (227). There are currently 128 registered lenvatinib clinical trials in the USA, with multiple phase I through phase III trials for treatment of thyroid cancer, renal cell carcinoma, hepatocellular carcinoma and melanoma. The antitumor activity of lenvatinib is also heavily linked to its anti-angiogenic properties (229). In addition, Lenvatinib has been shown to reduce TAMs and increase IFNγ secreting CD8 effector T cells in tumor tissues in a mouse model of colon carcinoma (230).
4.3.2 EGFR targeted TKI
EGFR targeted TKIs disrupt the immune suppressive TME by several mechanism including blocking cancer cell migration and nutrient delivery through targeting of endothelial cells and suppressing pericyte coverage (231). Highly proliferative cancer stem cells also express EGFR and can be inhibited by EGFR targeted TKIs (232). For example, EGFR-mutated NSCLC is known to be especially sensitive to treatment with EGFR TKIs (233); and these TKIS are therefore often a first line treatment for this cancer (233, 234). Many EGFR TKI drugs have been developed, and first-generation drugs such as gefitinib, erlotinib, and afatinib are approved for the treatment of EGFR mutated NSCLC. Currently third generation EGFR TKI drugs are being investigated as monotherapy and in combination with chemotherapy (213). There are currently around 8 FDA approved EGFR targeted TKI (235) with over 1200 total clinical trials in the US ranging from phase I to phase IV.
5 Future opportunities for myeloid cells as targets in cancer immunotherapy
Many different strategies targeting immune suppressor cells within the TME to reverse or ameliorate immune suppression have been evaluated. To date, the most successful strategies have been those targeting MDSCs, including the use of multi-function TKIs and repurposed beta blockers. For reprogramming TAMs, the most studied targets to date have been CSF-R1 inhibitors, either as biologics or targeted agents, though clinical responses to date have not been impressive (201, 236, 237). Other strategies have been even less successful, including the use of arginase and IDO inhibitors to reprogram metabolic pathways used by TAMs and tumor cells (194, 238). In the future, the most successful rational strategies will likely employ drugs and biologics targeting multiple different complementary pathways of tumor immune evasion, to block non-redundant mechanisms and pathways. Such combination strategies may also include creative uses of radiation therapy to enhance tumor immunogenicity, while MDSC or inflammatory monocyte targeted drugs can be used to relieve radiation induced inflammatory responses. Other gains will undoubtedly be realized when newer drugs and biologics with greater activity or more specific targeting of myeloid cell pathways enter the clinic. Thus, it is likely that we will see greater use of myeloid targeted agents as part of a more comprehensive strategy and platform for cancer immunotherapy (235).
Author contributions
JC, LC, SD collection and assembly of data, conception design and manuscript writing and revision. JC and LC are equal contribution first authors. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Charles Shipley Family Foundation, The V Foundation grant DCORC2021-003 awarded to JC and SD and NIH 1U01CA224182-01 awarded to SD.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Labani-Motlagh A, Ashja-Mahdavi M, Loskog A. The tumor microenvironment: A milieu hindering and obstructing antitumor immune responses. Front Immunol (2020), 940. doi: 10.3389/fimmu.2020.00940
2. Talaat IM, Kim B. A brief glimpse of a tangled web in a small world: Tumor microenvironment. Front Med (2022) 9:1002715. doi: 10.3389/fmed.2022.1002715
3. Barnestein R, Galland L, Kalfeist L, Ghiringhelli F, Ladoire S, Limagne E. Immunosuppressive tumor microenvironment modulation by chemotherapies and targeted therapies to enhance immunotherapy effectiveness. Oncoimmunology (2022) 11:2120676. doi: 10.1080/2162402x.2022.2120676
4. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12:253–68. doi: 10.1038/nri3175
5. Pramanik A, Bhattacharyya S. Myeloid derived suppressor cells and innate immune system interaction in tumor microenvironment. Life Sci (2022) 305:120755. doi: 10.1016/j.lfs.2022.120755
6. Jiang Y, Li Y, Zhu B. T-Cell exhaustion in the tumor microenvironment. Cell Death Dis (2015) 6:e1792. doi: 10.1038/cddis.2015.162
7. Lin Y, Xu J, Lan H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J Hematol Oncol (2019), 76. doi: 10.1186/s13045-019-0760-3
8. Shaul ME, Fridlender ZG. Tumour-associated neutrophils in patients with cancer. Nat Rev Clin Oncol (2019) 16:601–20. doi: 10.1038/s41571-019-0222-4
9. Shurin GV, Ma Y, Shurin MR. Immunosuppressive mechanisms of regulatory dendritic cells in cancer. Cancer Microenviron (2013) 6:159–67. doi: 10.1007/s12307-013-0133-3
10. Wei SY. Yin-yang regulating effects of cancer-associated genes, proteins, and cells: An ancient Chinese concept in vogue in modern cancer research. Biosci Trends (2017) 11:612–8. doi: 10.5582/bst.2017.01259
11. Clappaert EJ, Murgaski A, Van Damme H, Kiss M, Laoui D. Diamonds in the rough: Harnessing tumor-associated myeloid cells for cancer therapy. Front Immunol (2018) 9:2250. doi: 10.3389/fimmu.2018.02250
12. Cendrowicz E, Sas Z, Bremer E, Rygiel TP. The role of macrophages in cancer development and therapy. Cancers (Basel) (2021), 1946. doi: 10.3390/cancers13081946
13. Ostrand-Rosenberg S. Immune surveillance: A balance between protumor and antitumor immunity. Curr Opin Genet Dev (2008) 18:11–8. doi: 10.1016/j.gde.2007.12.007
14. Aminin D, Wang Y-M. Macrophages as a “weapon” in anticancer cellular immunotherapy. Kaohsiung J Med Sci (2021) 37:749–58. doi: 10.1002/kjm2.12405
15. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol (2021) 21:485–98. doi: 10.1038/s41577-020-00490-y
16. Davidov V, Jensen G, Mai S, Chen S-H, Pan P-Y. Analyzing one cell at a TIME: Analysis of myeloid cell contributions in the tumor immune microenvironment. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.01842
17. Movahedi K, Laoui D, Gysemans C, Baeten M, Stangé G, Van den Bossche J, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res (2010) 70:5728–39. doi: 10.1158/0008-5472.CAN-09-4672
18. Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun (2016) 7:12150. doi: 10.1038/ncomms12150
19. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A (2002) 99:12293–7. doi: 10.1073/pnas.192461099
20. Shi L, Chen S, Yang L, Li Y. The role of PD-1 and PD-L1 in T-cell immune suppression in patients with hematological malignancies. J Hematol Oncol (2013) 6:74. doi: 10.1186/1756-8722-6-74
21. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse ig superfamily ligand that negatively regulates T cell responses. J Exp Med (2011) 208:577–92. doi: 10.1084/jem.20100619
22. ElTanbouly MA, Croteau W, Noelle RJ, Lines JL. VISTA: a novel immunotherapy target for normalizing innate and adaptive immunity. Semin Immunol (2019) 42:101308. doi: 10.1016/j.smim.2019.101308
23. Miyamoto T, Murakami R, Hamanishi J, Tanigaki K, Hosoe Y, Mise N, et al. B7-H3 suppresses antitumor immunity via the CCL2-CCR2-M2 macrophage axis and contributes to ovarian cancer progression. Cancer Immunol Res (2022) 10:56–69. doi: 10.1158/2326-6066.Cir-21-0407
24. Sun Y, Wang Y, Zhao J, Gu M, Giscombe R, Lefvert AK, et al. B7-H3 and B7-H4 expression in non-small-cell lung cancer. Lung Cancer (2006) 53:143–51. doi: 10.1016/j.lungcan.2006.05.012
25. Ni L, Dong C. New checkpoints in cancer immunotherapy. Immunol Rev (2017) 276:52–65. doi: 10.1111/imr.12524
26. Zhang Z, Liu S, Zhang B, Qiao L, Zhang Y, Zhang Y. T Cell dysfunction and exhaustion in cancer. Front Cell Dev Biol (2020) 8:17. doi: 10.3389/fcell.2020.00017
27. Hicklin DJ, Marincola FM, Ferrone S. HLA class I antigen downregulation in human cancers: T-cell immunotherapy revives an old story. Mol Med Today (1999) 5:178–86. doi: 10.1016/s1357-4310(99)01451-3
28. Campoli M, Chang CC, Ferrone S. HLA class I antigen loss, tumor immune escape and immune selection. Vaccine (2002) 20 Suppl 4:A40–5. doi: 10.1016/s0264-410x(02)00386-9
29. Pluda JM. Tumor-associated angiogenesis: Mechanisms, clinical implications, and therapeutic strategies. Semin Oncol (1997) 24:203–18.
30. Veikkola T, Alitalo K. VEGFs, receptors and angiogenesis. Semin Cancer Biol (1999) 9:211–20. doi: 10.1006/scbi.1998.0091
31. Zhang Q-W, Liu L, Gong C-Y, Shi H-S, Zeng Y-H, Wang X-Z, et al. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PloS One (2012) 7:e50946. doi: 10.1371/journal.pone.0050946
32. Jin JK, Lin JT, Xu AK, Lou JA, Qian C, Li XM, et al. CCL2: An important mediator between tumor cells and host cells in tumor microenvironment. Front Oncol (2021) 11:722916. doi: 10.3389/fonc.2021.722916
33. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol (2002) 23:549–55. doi: 10.1016/S1471-4906(02)02302-5
34. Keeley T, Costanzo-Garvey DL, Cook LM. Unmasking the many faces of tumor-associated neutrophils and macrophages: Considerations for targeting innate immune cells in cancer. Trends Cancer (2019) 5:789–98. doi: 10.1016/j.trecan.2019.10.013
35. Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell (2019) 35:588–602.e10. doi: 10.1016/j.ccell.2019.02.009
36. Green CE, Liu T, Montel V, Hsiao G, Lester RD, Subramaniam S, et al. Chemoattractant signaling between tumor cells and macrophages regulates cancer cell migration, metastasis and neovascularization. PloS One (2009) 4:e6713. doi: 10.1371/journal.pone.0006713
37. Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer (2004) 4:71–8. doi: 10.1038/nrc1256
38. Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell (2008) 13:23–35. doi: 10.1016/j.ccr.2007.12.004
39. De Sanctis F, Adamo A, Cane S, Ugel S. Targeting tumour-reprogrammed myeloid cells: the new battleground in cancer immunotherapy. Semin Immunopathology (2022) 26:1–24. doi: 10.1007/s00281-022-00965-1
40. Hartley GP, Chow L, Ammons DT, Wheat WH, Dow SW. Programmed cell death ligand 1 (PD-L1) signaling regulates macrophage proliferation and activation. Cancer Immunol Res (2018) 6:1260–73. doi: 10.1158/2326-6066.CIR-17-0537
41. Rodriguez PC, Zea AH, DeSalvo J, Culotta KS, Zabaleta J, Quiceno DG, et al. L-arginine consumption by macrophages modulates the expression of CD3ζ chain in T lymphocytes. J Immunol (2003) 171:1232–9. doi: 10.4049/jimmunol.171.3.1232
42. Hasan MN, Capuk OA-O, Patel SA-O, Sun D. The role of metabolic plasticity of tumor-associated macrophages in shaping the tumor microenvironment immunity. Cancers (Basel) (2022) 14(14):3331. doi: 10.3390/cancers14143331
43. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol (2020) 20:7–24. doi: 10.1038/s41577-019-0210-z
44. Bottcher JP, Reis e Sousa C. The role of type 1 conventional dendritic cells in cancer immunity. Trends Cancer (2018) 4:784–92. doi: 10.1016/j.trecan.2018.09.001
45. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: A moving target in immunotherapy. Blood (2018) 131:58–67. doi: 10.1182/blood-2017-06-741033
46. Wu YZ, Yi M, Niu MK, Mei Q, Wu KM. Myeloid-derived suppressor cells: An emerging target for anticancer immunotherapy. Mol Cancer (2022) 21. doi: 10.1186/s12943-022-01657-y
47. Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, et al. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol (2010) 22:238–44. doi: 10.1016/j.coi.2010.01.021
48. Han ZJ, Li YB, Yang LX, Cheng HJ, Liu X, Chen H. Roles of the CXCL8-CXCR1/2 axis in the tumor microenvironment and immunotherapy. Molecules (2021) 27:137. doi: 10.3390/molecules27010137
49. Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol (2008) 181:5791–802. doi: 10.4049/jimmunol.181.8.5791
50. Centuori SM, Trad M, LaCasse CJ, Alizadeh D, Larmonier CB, Hanke NT, et al. Myeloid-derived suppressor cells from tumor-bearing mice impair TGF-β-induced differentiation of CD4+ CD25+ FoxP3+ tregs from CD4+ CD25– FoxP3– T cells. J leukocyte Biol (2012) 92:987–97. doi: 10.1189/jlb.0911465
51. Martin RK, Saleem SJ, Folgosa L, Zellner HB, Damle SR, Nguyen GKT, et al. Mast cell histamine promotes the immunoregulatory activity of myeloid-derived suppressor cells. J Leukocyte Biol (2014) 96:151–9. doi: 10.1189/jlb.5A1213-644R
52. Sieminska I, Baran J. Myeloid-derived suppressor cells as key players and promising therapy targets in prostate cancer. Front Oncol (2022) 12:862416. doi: 10.3389/fonc.2022.862416
53. An JL, Feng LF, Ren JL, Li YF, Li GR, Liu C, et al. Chronic stress promotes breast carcinoma metastasis by accumulating myeloid-derived suppressor cells through activating beta-adrenergic signaling. Oncoimmunology (2021) 10:2004659. doi: 10.1080/2162402x.2021.2004659
54. Yang Z, Guo J, Weng L, Tang W, Jin S, Ma W. Myeloid-derived suppressor cells-new and exciting players in lung cancer. J Hematol Oncol (2020) 13:10. doi: 10.1186/s13045-020-0843-1
55. Bennett JA, Rao VS, Mitchell MS. Systemic bacillus calmette-guerin (BCG) activates natural suppressor cells. Proc Natl Acad Sci U S A (1978) 75:5142–4. doi: 10.1073/pnas.75.10.5142
56. Condamine T, Mastio J, Gabrilovich DI. Transcriptional regulation of myeloid-derived suppressor cells. J Leukoc Biol (2015) 98:913–22. doi: 10.1189/jlb.4RI0515-204R
57. Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer (2019) 120:16–25. doi: 10.1038/s41416-018-0333-1
58. Lauret Marie Joseph E, Laheurte C, Jary M, Boullerot L, Asgarov K, Gravelin E, et al. Immunoregulation and clinical implications of ANGPT2/TIE2(+) m-MDSC signature in non-small cell lung cancer. Cancer Immunol Res (2020) 8:268–79. doi: 10.1158/2326-6066.CIR-19-0326
59. Koh J, Kim Y, Lee KY, Hur JY, Kim MS, Kim B, et al. MDSC subtypes and CD39 expression on CD8(+) T cells predict the efficacy of anti-PD-1 immunotherapy in patients with advanced NSCLC. Eur J Immunol (2020) 50:1810–9. doi: 10.1002/eji.202048534
60. Tumino N, Besi F, Martini S, Di Pace AL, Munari E, Quatrini L, et al. Polymorphonuclear myeloid-derived suppressor cells are abundant in peripheral blood of cancer patients and suppress natural killer cell anti-tumor activity. Front Immunol (2022) 12:803014. doi: 10.3389/fimmu.2021.803014
61. Patel S, Fu S, Mastio J, Dominguez GA, Purohit A, Kossenkov A, et al. Unique pattern of neutrophil migration and function during tumor progression. Nat Immunol (2018) 19:1236–47. doi: 10.1038/s41590-018-0229-5
62. Youn JI, Collazo M, Shalova IN, Biswas SK, Gabrilovich DI. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biol (2012) 91:167–81. doi: 10.1189/jlb.0311177
63. Fridlender ZG, Albelda SM. Tumor-associated neutrophils: Friend or foe? Carcinogenesis (2012) 33:949–55. doi: 10.1093/carcin/bgs123
64. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
65. Masucci MT, Minopoli M, Carriero MV. Tumor associated neutrophils. their role in tumorigenesis, metastasis, prognosis and therapy. Front Oncol (2019) 9. doi: 10.3389/fonc.2019.01146
66. Kargl J, Zhu XD, Zhang HJ, Yang GHY, Friesen TJ, Shipley M, et al. Neutrophil content predicts lymphocyte depletion and anti-PD1 treatment failure in NSCLC. JCI Insight (2019) 4:e130850. doi: 10.1172/jci.insight.130850
67. Emmons TR, Giridharan T, Singel KL, Khan ANH, Ricciuti J, Howard K, et al. Mechanisms driving neutrophil-induced T-cell immunoparalysis in ovarian cancer. Cancer Immunol Res (2021) 9:790–810. doi: 10.1158/2326-6066.CIR-20-0922
68. Piccard H, Muschel RJ, Opdenakker G. On the dual roles and polarized phenotypes of neutrophils in tumor development and progression. Crit Rev Oncol Hematol (2012) 82:296–309. doi: 10.1016/j.critrevonc.2011.06.004
69. Deryugina EI, Zajac E, Juncker-Jensen A, Kupriyanova TA, Welter L, Quigley JP. Tissue-infiltrating neutrophils constitute the major in vivo source of angiogenesis-inducing MMP-9 in the tumor microenvironment. Neoplasia (2014) 16:771–88. doi: 10.1016/j.neo.2014.08.013
70. Schaider H, Oka M, Bogenrieder T, Nesbit M, Satyamoorthy K, Berking C, et al. Differential response of primary and metastatic melanomas to neutrophils attracted by IL-8. Int J Cancer (2003) 103:335–43. doi: 10.1002/ijc.10775
71. Haqqani AS, Sandhu JK, Birnboim HC. Expression of interleukin-8 promotes neutrophil infiltration and genetic instability in mutatect tumors. Neoplasia (2000) 2:561–8. doi: 10.1038/sj.neo.7900110
72. Bellocq A, Antoine M, Flahault A, Philippe C, Crestani B, Bernaudin JF, et al. Neutrophil alveolitis in bronchioloalveolar carcinoma: Induction by tumor-derived interleukin-8 and relation to clinical outcome. Am J Pathol (1998) 152:83–92.
73. Li P, Lu M, Shi J, Hua L, Gong Z, Li Q, et al. Dual roles of neutrophils in metastatic colonization are governed by the host NK cell status. Nat Commun (2020) 11:4387. doi: 10.1038/s41467-020-18125-0
74. Eruslanov EB, Singhal S, Albelda SM. Mouse versus human neutrophils in cancer: A major knowledge gap. Trends Cancer (2017) 3:149–60. doi: 10.1016/j.trecan.2016.12.006
75. Teijeira A, Garasa S, Migueliz I, Cirella A, Melero I. Cxcr1 and Cxcr2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. J Immunother Cancer (2020) 8:A453–A. doi: 10.1136/jitc-2020-SITC2020.0755
76. Teijeira A, Garasa S, Ochoa MC, Villalba M, Olivera I, Cirella A, et al. IL8, neutrophils, and NETs in a collusion against cancer immunity and immunotherapy. Clin Cancer Res (2021) 27:2383–93. doi: 10.1158/1078-0432.Ccr-20-1319
77. Zhang Y, Wang CX, Yu MX, Zhao XY, Du JW, Li YY, et al. Neutrophil extracellular traps induced by activated platelets contribute to procoagulant activity in patients with colorectal cancer. Thromb Res (2019) 180:87–97. doi: 10.1016/j.thromres.2019.06.005
78. Pathria P, Louis TL, Varner JA. Targeting tumor-associated macrophages in cancer. Trends Immunol (2019) 40:310–27. doi: 10.1016/j.it.2019.02.003
79. Chitu V, Stanley ER. Colony-stimulating factor-1 in immunity and inflammation. Curr Opin Immunol (2006) 18:39–48. doi: 10.1016/j.coi.2005.11.006
80. Stanley ER, Chitu V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harbor Perspect Biol (2014) 6:a021857. doi: 10.1101/cshperspect.a021857
81. Wang X, Zhang J, Hu B, Qian F. High expression of CSF-1R predicts poor prognosis and CSF-1R(high) tumor-associated macrophages inhibit anti-tumor immunity in colon adenocarcinoma. Front Oncol (2022) 12:850767. doi: 10.3389/fonc.2022.850767
82. Candido JB, Morton JP, Bailey P, Campbell AD, Karim SA, Jamieson T, et al. CSF1R(+) macrophages sustain pancreatic tumor growth through T cell suppression and maintenance of key gene programs that define the squamous subtype. Cell Rep (2018) 23:1448–60. doi: 10.1016/j.celrep.2018.03.131
83. Koh YW, Park C, Yoon DH, Suh C, Huh J. CSF-1R expression in tumor-associated macrophages is associated with worse prognosis in classical Hodgkin lymphoma. Am J Clin Pathol (2014) 141:573–83. doi: 10.1309/AJCPR92TDDFARISU
84. Espinosa I, Beck AH, Lee CH, Zhu S, Montgomery KD, Marinelli RJ, et al. Coordinate expression of colony-stimulating factor-1 and colony-stimulating factor-1-related proteins is associated with poor prognosis in gynecological and nongynecological leiomyosarcoma. Am J Pathol (2009) 174:2347–56. doi: 10.2353/ajpath.2009.081037
85. Jia JB, Wang WQ, Sun HC, Zhu XD, Liu L, Zhuang PY, et al. High expression of macrophage colony-stimulating factor-1 receptor in peritumoral liver tissue is associated with poor outcome in hepatocellular carcinoma after curative resection. Oncologist (2010) 15:732–43. doi: 10.1634/theoncologist.2009-0170
86. Lin EY, Pollard JW. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res (2007) 67:5064–6. doi: 10.1158/0008-5472.CAN-07-0912
87. Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Ruttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer (2017) 5:53. doi: 10.1186/s40425-017-0257-y
88. Denny WA, Flanagan JU. Small-molecule CSF1R kinase inhibitors; Review of patents 2015-present. Expert Opin Ther Pat (2021) 31:107–17. doi: 10.1080/13543776.2021.1839414
89. Lu X, Yan S, Koral KA, Chen Z. Surufatinib for the treatment of advanced extrapancreatic neuroendocrine tumors. Expert Rev Anticancer Ther (2021) 21:917–26. doi: 10.1080/14737140.2021.1944110
90. von Tresckow B, Morschhauser F, Ribrag V, Topp MS, Chien C, Seetharam S, et al. An open-label, multicenter, phase I/II study of JNJ-40346527, a CSF-1R inhibitor, in patients with relapsed or refractory Hodgkin lymphoma. Clin Cancer Res (2015) 21:1843–50. doi: 10.1158/1078-0432.CCR-14-1845
91. Holmgaard RB, Brachfeld A, Gasmi B, Jones DR, Mattar M, Doman T, et al. Timing of CSF-1/CSF-1R signaling blockade is critical to improving responses to CTLA-4 based immunotherapy. Oncoimmunology (2016) 5:e1151595. doi: 10.1080/2162402X.2016.1151595
92. Yan D, Kowal J, Akkari L, Schuhmacher AJ, Huse JT, West BL, et al. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene (2017) 36:6049–58. doi: 10.1038/onc.2017.261
93. Erkes DA, Rosenbaum SR, Field CO, Chervoneva I, Villanueva J, Aplin AE. PLX3397 inhibits the accumulation of intra-tumoral macrophages and improves bromodomain and extra-terminal inhibitor efficacy in melanoma. Pigment Cell Melanoma Res (2020) 33:372–7. doi: 10.1111/pcmr.12845
94. Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, et al. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res (2014) 74:153–61. doi: 10.1158/0008-5472.CAN-13-1816
95. Benner B, Good L, Quiroga D, Schultz TE, Kassem M, Carson WE, et al. Pexidartinib, a novel small molecule CSF-1R inhibitor in use for tenosynovial giant cell tumor: A systematic review of pre-clinical and clinical development. Drug Des Devel Ther (2020) 14:1693–704. doi: 10.2147/DDDT.S253232
96. Somerhausen N, van den Rijn M. Tenosynovial giant cell tumour, diffuse type. WHO classification tumours soft Tissue Bone (2013) 4:102–3.
97. Cassier PA, Italiano A, Gomez-Roca CA, Le Tourneau C, Toulmonde M, Cannarile MA, et al. CSF1R inhibition with emactuzumab in locally advanced diffuse-type tenosynovial giant cell tumours of the soft tissue: A dose-escalation and dose-expansion phase 1 study. Lancet Oncol (2015) 16:949–56. doi: 10.1016/S1470-2045(15)00132-1
98. Tap WD, Gelderblom H, Palmerini E, Desai J, Bauer S, Blay JY, et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): A randomised phase 3 trial. Lancet (2019) 394(10197):478–87. doi: 10.1016/S0140-6736(19)30764-0
99. Rao R, Han R, Ogurek S, Xue CB, Wu LM, Zhang LG, et al. Glioblastoma genetic drivers dictate the function of tumor-associated macrophages/microglia and responses to CSF1R inhibition. Neuro-Oncology (2022) 24:584–97. doi: 10.1093/neuonc/noab228
100. Wesolowski R, Sharma N, Reebel L, Rodal MB, Peck A, West BL, et al. Phase ib study of the combination of pexidartinib (PLX3397), a CSF-1R inhibitor, and paclitaxel in patients with advanced solid tumors. Ther Adv Med Oncol (2019) 11:1758835919854238. doi: 10.1177/1758835919854238
101. Wainberg ZA, Eisenberg PD, Sachdev JC, Weise AM, Kaufman DR, Hutchinson M, et al. Phase 1/2a study of double immune suppression blockade by combining a CSF1R inhibitor (pexidartinib/PLX3397) with an anti PD-1 antibody (pembrolizumab) to treat advanced melanoma and other solid tumors. J Clin Oncol (2016) 34:TPS465–TPS. doi: 10.1200/jco.2016.34.4_suppl.tps465
102. Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An ivy foundation early phase clinical trials consortium phase II study. Neuro Oncol (2016) 18:557–64. doi: 10.1093/neuonc/nov245
103. Moskowitz CH, Younes A, de Vos S, Bociek RG, Gordon LI, Witzig TE, et al. CSF1R inhibition by PLX3397 in patients with relapsed or refractory Hodgkin lymphoma: results from a phase 2 single agent clinical trial. Blood (2012) 120:1638. doi: 10.1182/blood.V120.21.1638.1638
104. Sun Y, Yang L, Hao X, Liu Y, Zhang J, Ning Z, et al. Phase I dose-escalation study of chiauranib, a novel angiogenic, mitotic, and chronic inflammation inhibitor, in patients with advanced solid tumors. J Hematol Oncol (2019) 12:9. doi: 10.1186/s13045-018-0695-0
105. Zhou Y, Fu C, Kong Y, Pan D, Wang Y, Huang S, et al. Antitumor and immunomodulatory effects of a novel multitarget inhibitor, CS2164, in mouse hepatocellular carcinoma models. Anticancer Drugs (2019) 30:909–16. doi: 10.1097/cad.0000000000000791
106. Yin H, Xie J, Jiang P, Jiang X, Duan D, Qi J, et al. Chiauranib selectively inhibits colorectal cancer with KRAS wild-type by modulation of ROS through activating the p53 signaling pathway. Am J Cancer Res (2020) 10:3666–85.
107. Deng M, Zhao H, Chen Q, Zhao J, Shi Y, Yu L, et al. CS2164 suppresses acute myeloid leukemia cell growth via inhibiting VEGFR2 signaling in preclinical models. Eur J Pharmacol (2019) 853:193–200. doi: 10.1016/j.ejphar.2019.03.041
108. Gomez-Roca CA, Italiano A, Le Tourneau C, Cassier PA, Toulmonde M, D'Angelo SP, et al. Phase I study of emactuzumab single agent or in combination with paclitaxel in patients with advanced/metastatic solid tumors reveals depletion of immunosuppressive M2-like macrophages. Ann Oncol (2019) 30:1381–92. doi: 10.1093/annonc/mdz163
109. Papadopoulos KP, Gluck L, Martin LP, Olszanski AJ, Tolcher AW, Ngarmchamnanrith G, et al. First-in-Human study of AMG 820, a monoclonal anti-Colony-Stimulating factor 1 receptor antibody, in patients with advanced solid tumors. Clin Cancer Res (2017) 23:5703–10. doi: 10.1158/1078-0432.CCR-16-3261
110. Lin CC. Clinical development of colony-stimulating factor 1 receptor (CSF1R) inhibitors. J Immunother Precis Oncol (2021) 4:105–14. doi: 10.36401/JIPO-20-32
111. Grosso F, Jones RL, Demetri GD, Judson IR, Blay J-Y, Le Cesne A, et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: A retrospective study. Lancet Oncol (2007) 8:595–602. doi: 10.1016/S1470-2045(07)70175-4
112. Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell (2013) 23:249–62. doi: 10.1016/j.ccr.2013.01.008
113. Allavena P, Belgiovine C, Digifico E, Frapolli R, D’Incalci M. Effects of the anti-tumor agents trabectedin and lurbinectedin on immune cells of the tumor microenvironment. Front Oncol (2022) 12:851790. doi: 10.3389/fonc.2022.851790
114. Allavena P, Signorelli M, Chieppa M, Erba E, Bianchi G, Marchesi F, et al. Anti-inflammatory properties of the novel antitumor agent yondelis (trabectedin): inhibition of macrophage differentiation and cytokine production. Cancer Res (2005) 65:2964–71. doi: 10.1158/0008-5472.CAN-04-4037
115. Barone A, Chi DC, Theoret MR, Chen H, He K, Kufrin D, et al. FDA Approval summary: Trabectedin for unresectable or metastatic liposarcoma or leiomyosarcoma following an anthracycline-containing regimen. Clin Cancer Res (2017) 23:7448–53. doi: 10.1158/1078-0432.CCR-17-0898
116. O’Connor T, Heikenwalder M. CCL2 in the tumor microenvironment. Tumor Microenvironment: Role Chemokines - Pt B (2021) 1302:1–14. doi: 10.1007/978-3-030-62658-7_1
117. Grossman JG, Nywening TM, Belt BA, Panni RZ, Krasnick BA, DeNardo DG, et al. Recruitment of CCR2(+) tumor associated macrophage to sites of liver metastasis confers a poor prognosis in human colorectal cancer. Oncoimmunology (2018) 7:e1470729. doi: 10.1080/2162402X.2018.1470729
118. Xu M, Wang Y, Xia R, Wei Y, Wei X. Role of the CCL2-CCR2 signalling axis in cancer: Mechanisms and therapeutic targeting. In: Cell proliferation. John Wiley and Sons Inc (2021), e13115.
119. Oo MW, Kawai H, Takabatake K, Tomida S, Eguchi T, Ono K, et al. Resident stroma-secreted chemokine CCL2 governs myeloid-derived suppressor cells in the tumor microenvironment. JCI Insight (2022) 7:e148960. doi: 10.1172/jci.insight.148960
120. Qin R, Ren WH, Ya GQ, Wang B, He J, Ren SX, et al. Role of chemokines in the crosstalk between tumor and tumor-associated macrophages. Clin Exp Med (2022) 10:590941. doi: 10.1007/s10238-022-00888-z
121. Loberg RD, Ying C, Craig M, Day LL, Sargent E, Neeley C, et al. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression In vivo. Cancer Res (2007) 67:9417–24. doi: 10.1158/0008-5472.Can-07-1286
122. Zhu X, Fujita M, Snyder LA, Okada H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. J Neurooncol (2011) 104:83–92. doi: 10.1007/s11060-010-0473-5
123. Pienta KJ, Machiels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest New Drugs (2013) 31:760–8. doi: 10.1007/s10637-012-9869-8
124. Vela M, Aris M, Llorente M, Garcia-Sanz JA, Kremer L. Chemokine receptor-specific antibodies in cancer immunotherapy: achievements and challenges. Front Immunol (2015) 6:12. doi: 10.3389/fimmu.2015.00012
125. Yumimoto K, Sugiyama S, Mimori K, Nakayama KI. Potentials of c-c motif chemokine 2-C-C chemokine receptor type 2 blockers including propagermanium as anticancer agents. Cancer Sci (2019) 110:2090–9. doi: 10.1111/cas.14075
126. Mora E, Guglielmotti A, Biondi G, Sassone-Corsi P. Bindarit: an anti-inflammatory small molecule that modulates the NFkappaB pathway. Cell Cycle (2012) 11:159–69. doi: 10.4161/cc.11.1.18559
127. Steiner JL, Davis JM, McClellan JL, Guglielmotti A, Murphy EA. Effects of the MCP-1 synthesis inhibitor bindarit on tumorigenesis and inflammatory markers in the C3(1)/SV40Tag mouse model of breast cancer. Cytokine (2014) 66:60–8. doi: 10.1016/j.cyto.2013.12.011
128. Zollo M, Di Dato V, Spano D, Martino D, Liguori L, Marino N, et al. Targeting monocyte chemotactic protein-1 synthesis with bindarit induces tumor regression in prostate and breast cancer animal models. Clin Exp metastasis (2012) 29:585–601. doi: 10.1007/s10585-012-9473-5
129. Chen P, Zhou J, Li J, Zhang Q, Zuo Q. TIPE1 suppresses osteosarcoma tumor growth by regulating macrophage infiltration. Clin Trans Oncol (2019) 21:334–41. doi: 10.1007/s12094-018-1927-z
130. Cho HR, Kumari N, Thi Vu H, Kim H, Park C-K, Choi SH. Increased antiangiogenic effect by blocking CCL2-dependent macrophages in a rodent glioblastoma model: Correlation study with dynamic susceptibility contrast perfusion MRI. Sci Rep-Uk (2019) 9:1–12. doi: 10.1038/s41598-019-47438-4
131. Mu X-Y, Wang R-J, Yao Z-X, Zheng Z, Jiang J-T, Tan M-Y, et al. RS 504393 inhibits m-MDSCs recruiting in immune microenvironment of bladder cancer after gemcitabine treatment. Mol Immunol (2019) 109:140–8. doi: 10.1016/j.molimm.2019.02.014
132. Wu X, Singh R, Hsu DK, Zhou Y, Yu S, Han D, et al. A small molecule CCR2 antagonist depletes tumor macrophages and synergizes with anti–PD-1 in a murine model of cutaneous T-cell lymphoma (CTCL). J Invest Dermatol (2020) 140:1390–400.e4. doi: 10.1016/j.jid.2019.11.018
133. Yao W, Ba Q, Li X, Li H, Zhang S, Yuan Y, et al. A natural CCR2 antagonist relieves tumor-associated macrophage-mediated immunosuppression to produce a therapeutic effect for liver cancer. EBioMedicine (2017) 22:58–67. doi: 10.1016/j.ebiom.2017.07.014
134. Farina S, Yang H, Tu GH, Gamelin EC, Lin JC, Wang C, et al. Abstract LB-194: Targeting tumor associated myeloid cells with CCR2 inhibitor PF-04136309 enhances gemcitabine/paclitaxel and doxorubicin anti-tumor activity. Cancer Res (2017) 77:LB–194-LB-. doi: 10.1158/1538-7445.AM2017-LB-194
135. Wang-Gillam A, Nywening TM, Sanford DE, Lockhart AC, Suresh R, Tan BR, et al. Phase IB study of FOLFIRINOX plus PF-04136309 in patients with borderline resectable and locally advanced pancreatic adenocarcinoma (PC). Am Soc Clin Oncol (2015) 338. doi: 10.1200/jco.2015.33.3_suppl.338
136. Noel M, O’Reilly EM, Wolpin BM, Ryan DP, Bullock AJ, Britten CD, et al. Phase 1b study of a small molecule antagonist of human chemokine (CC motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Investigational New Drugs (2020) 38:800–11. doi: 10.1007/s10637-019-00830-3
137. Cherney RJ, Anjanappa P, Selvakumar K, Batt DG, Brown GD, Rose AV, et al. BMS-813160: A potent CCR2 and CCR5 dual antagonist selected as a clinical candidate. ACS Medicinal Chem Letters (2021) 12:1753–8. doi: 10.1021/acsmedchemlett.1c00373
138. Venturini N, Marron T, Casanova-Acebes M, Mandeli J, Doroshow D, Lucas N, et al. 629 neoadjuvant nivolumab combined with CCR2/5 inhibitor or anti-IL-8 antibody in non-small cell lung cancer and hepatocellular carcinoma. BMJ Specialist Journals (2022).
139. Le D, Gutierrez ME, Saleh M, Chen E, Mallick AB, Pishvaian MJ, et al. Abstract CT124: A phase Ib/II study of BMS-813160, a CC chemokine receptor (CCR) 2/5 dual antagonist, in combination with chemotherapy or nivolumab in patients (pts) with advanced pancreatic or colorectal cancer. Cancer Res (2018) 78:CT124–CT. doi: 10.1158/1538-7445.AM2018-CT124
140. Regan DP, Coy JW, Chahal KK, Chow L, Kurihara JN, Guth AM, et al. The angiotensin receptor blocker losartan suppresses growth of pulmonary metastases via AT1R-independent inhibition of CCR2 signaling and monocyte recruitment. J Immunol (2019) 202:3087–102. doi: 10.4049/jimmunol.1800619
141. Coulson R, Liew SH, Connelly AA, Haines L, Palmer E, Kurihara JN, et al. The angiotensin receptor blocker, losartan, inhibits mammary tumor development and progression to invasive carcinoma. Oncotarget (2017) 8:18640–56. doi: 10.18632/oncotarget.15553
142. Regan DP, Chow L, Das S, Haines L, Palmer E, Kurihara JN, et al. Losartan blocks osteosarcoma-elicited monocyte recruitment, and combined with the kinase inhibitor toceranib, exerts significant clinical benefit in canine metastatic osteosarcoma. Clin Cancer Res (2022) 28:662–76. doi: 10.1158/1078-0432.Ccr-21-2105
143. Datta M, Chatterjee S, Perez EM, Gritsch S, Roberge S, Duquette M, et al. Losartan controls immune checkpoint blocker-induced edema and improves survival in glioblastoma mouse models. Proc Natl Acad Sci (2023) 120:e2219199120. doi: 10.1073/pnas.2219199120
144. Ha H, Debnath B, Neamati N. Role of the CXCL8-CXCR1/2 axis in cancer and inflammatory diseases. Theranostics (2017) 7:1543–88. doi: 10.7150/thno.15625
145. SenGupta S, Hein LE, Parent CA. The recruitment of neutrophils to the tumor microenvironment is regulated by multiple mediators. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.734188
146. Yuen KC, Liu LF, Gupta V, Madireddi S, Keerthivasan S, Li CF, et al. High systemic and tumor-associated IL-8 correlates with reduced clinical benefit of PD-L1 blockade. Nat Med (2020) 26:693–+. doi: 10.1038/s41591-020-0860-1
147. Fousek K, Horn LA, Palena C. Interleukin-8: A chemokine at the intersection of cancer plasticity, angiogenesis, and immune suppression. Pharmacol Therapeut (2021) 219. doi: 10.1016/j.pharmthera.2020.107692
148. Che JX, Song R, Chen BH, Dong XW. Targeting CXCR1/2: The medicinal potential as cancer immunotherapy agents, antagonists research highlights and challenges ahead. Eur J Med Chem (2020) 185:111853. doi: 10.1016/j.ejmech.2019.111853
149. Porter DW, Bradley M, Brown Z, Canova R, Charlton S, Cox B, et al. The discovery of potent, orally bioavailable pyrazolo and triazolopyrimidine CXCR2 receptor antagonists. Bioorganic Medicinal Chem Letters (2014) 24:72–6. doi: 10.1016/j.bmcl.2013.11.074
150. Ronchetti L, Boubaker NS, Barba M, Vici P, Gurtner A, Piaggio G. Neutrophil extracellular traps in cancer: Not only catching microbes. J Exp Clin Cancer Res (2021) 40:1–9. doi: 10.1186/s13046-021-02036-z
151. Bizzarri C, Pagliei S, Brandolini L, Mascagni P, Caselli G, Transidico P, et al. Selective inhibition of interleukin-8-induced neutrophil chemotaxis by ketoprofen isomers. Biochem Pharmacol (2001) 61:1429–37. doi: 10.1016/S0006-2952(01)00610-4
152. Allegretti M, Bertini R, Cesta MC, Bizzarri C, Di Bitondo R, Di Cioccio V, et al. 2-arylpropionic CXC chemokine receptor 1 (CXCR1) ligands as novel noncompetitive CXCL8 inhibitors. J medicinal Chem (2005) 48:4312–31. doi: 10.1021/jm049082i
153. Citro A, Cantarelli E, Maffi P, Nano R, Melzi R, Mercalli A, et al. CXCR1/2 inhibition enhances pancreatic islet survival after transplantation. J Clin Invest (2012) 122:3647–51. doi: 10.1172/JCI63089
154. Liotti F, De Pizzol M, Allegretti M, Prevete N, Melillo RM. Multiple anti-tumor effects of reparixin on thyroid cancer. Oncotarget (2017) 8:35946. doi: 10.18632/oncotarget.16412
155. Schott AF, Goldstein LJ, Cristofanilli M, Ruffini PA, McCanna S, Reuben JM, et al. Phase ib pilot study to evaluate reparixin in combination with weekly paclitaxel in patients with HER-2–negative metastatic breast CancerReparixin and weekly paclitaxel in metastatic breast cancer. Clin Cancer Res (2017) 23:5358–65. doi: 10.1158/1078-0432.CCR-16-2748
156. Goldstein LJ, Mansutti M, Levy C, Chang JC, Henry S, Fernandez-Perez I, et al. A randomized, placebo-controlled phase 2 study of paclitaxel in combination with reparixin compared to paclitaxel alone as front-line therapy for metastatic triple-negative breast cancer (fRida). Breast Cancer Res Treat (2021) 190:265–75. doi: 10.1007/s10549-021-06367-5
157. Kemp DM, Pidich A, Larijani M, Jonas R, Lash E, Sato T, et al. Ladarixin, a dual CXCR1/2 inhibitor, attenuates experimental melanomas harboring different molecular defects by affecting malignant cells and tumor microenvironment. Oncotarget (2017) 8:14428–42. doi: 10.18632/oncotarget.14803
158. Piro G, Carbone C, Agostini A, Esposito A, De Pizzol M, Novelli R, et al. CXCR1/2 dual-inhibitor ladarixin reduces tumour burden and promotes immunotherapy response in pancreatic cancer. Br J Cancer (2022) 128:331–41. doi: 10.1038/s41416-022-02028-6
159. Nicholls DJ, Wiley K, Dainty I, MacIntosh F, Phillips C, Gaw A, et al. Pharmacological characterization of AZD5069, a slowly reversible CXC chemokine receptor 2 antagonist. J Pharmacol Exp Ther (2015) 353:340–50. doi: 10.1124/jpet.114.221358
160. Steele CW, Karim SA, Leach JD, Bailey P, Upstill-Goddard R, Rishi L, et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell (2016) 29:832–45. doi: 10.1016/j.ccell.2016.04.014
161. Anderson EM, Thomassian S, Gong J, Hendifar A, Osipov A. Advances in pancreatic ductal adenocarcinoma treatment. Cancers (2021) 13:5510. doi: 10.3390/cancers13215510
162. Guo C, Sharp A, Vogl U, Colombo I, Stathis A, Jain S, et al. 454O a phase (Ph) I/II trial of the CXCR2 antagonist AZD5069 in combination with enzalutamide (ENZA) in patients (pts) with metastatic castration resistant prostate cancer (mCRPC). Ann Oncol (2022) 33:S745. doi: 10.1016/j.annonc.2022.07.583
163. Greene S, Robbins Y, Mydlarz WK, Huynh AP, Schmitt NC, Friedman J, et al. Inhibition of MDSC trafficking with SX-682, a CXCR1/2 inhibitor, enhances NK-cell immunotherapy in head and neck cancer ModelsMyeloid cell inhibition enhances NK cellular immunotherapy. Clin Cancer Res (2020) 26:1420–31. doi: 10.1158/1078-0432.CCR-19-2625
164. Sun L, Clavijo PE, Robbins Y, Patel P, Friedman J, Greene S, et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight (2019) 4:e126853. doi: 10.1172/jci.insight.126853
165. Chatterjee S, Behnam Azad B, Nimmagadda S. The intricate role of CXCR4 in cancer. Adv Cancer Res (2014) 124:31–82. doi: 10.1016/b978-0-12-411638-2.00002-1
166. Luker GD, Yang J, Richmond A, Scala S, Festuccia C, Schottelius M, et al. At The bench: Pre-clinical evidence for multiple functions of CXCR4 in cancer. J leukocyte Biol (2021) 109:969–89. doi: 10.1002/JLB.2BT1018-715RR
167. Micallef IN, Stiff PJ, Nademanee AP, Maziarz RT, Horwitz ME, Stadtmauer EA, et al. Plerixafor plus granulocyte colony-stimulating factor for patients with non-Hodgkin lymphoma and multiple myeloma: Long-term follow-up report. Biol Blood Marrow Transplant (2018) 24:1187–95. doi: 10.1016/j.bbmt.2018.01.039
168. Uy GL, Rettig MP, Motabi IH, McFarland K, Trinkaus KM, Hladnik LM, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood (2012) 119:3917–24. doi: 10.1182/blood-2011-10-383406
169. Jung K, Heishi T, Incio J, Huang Y, Beech EY, Pinter M, et al. Targeting CXCR4-dependent immunosuppressive Ly6Clow monocytes improves antiangiogenic therapy in colorectal cancer. Proc Natl Acad Sci (2017) 114:10455–60. doi: 10.1073/pnas.1710754114
170. Dmello RS, To SQ, Chand AL. Therapeutic targeting of the tumour microenvironment in metastatic colorectal cancer. Int J Mol Sci (2021) 22:2067. doi: 10.3390/ijms22042067
171. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A (2013) 110:20212–7. doi: 10.1073/pnas.1320318110
172. Bule P, Aguiar SI, Aires-Da-Silva F, Dias JNR. Chemokine-directed tumor microenvironment modulation in cancer immunotherapy. Int J Mol Sci (2021) 22:9804. doi: 10.3390/ijms22189804
173. Shen B, Zheng M-Q, Lu J-W, Jiang Q, Wang T-H, Huang X-E. CXCL12-CXCR4 promotes proliferation and invasion of pancreatic cancer cells. Asian Pacific J Cancer Prev (2013) 14:5403–8. doi: 10.7314/APJCP.2013.14.9.5403
174. Fearon DT, Janowitz T. AMD3100/Plerixafor overcomes immune inhibition by the CXCL12–KRT19 coating on pancreatic and colorectal cancer cells. Brit J Cancer (2021) 125:149–51. doi: 10.1038/s41416-021-01315-y
175. Biasci D, Smoragiewicz M, Connell CM, Wang Z, Gao Y, Thaventhiran JE, et al. CXCR4 inhibition in human pancreatic and colorectal cancers induces an integrated immune response. Proc Natl Acad Sci (2020) 117:28960–70. doi: 10.1073/pnas.2013644117
176. Liu T, Li X, You S, Bhuyan SS, Dong L. Effectiveness of AMD3100 in treatment of leukemia and solid tumors: From original discovery to use in current clinical practice. Exp Hematol Oncol (2015) 5:19. doi: 10.1186/s40164-016-0050-5
177. Song J-S, Chang C-C, Wu C-H, Dinh TK, Jan J-J, Huang K-W, et al. A highly selective and potent CXCR4 antagonist for hepatocellular carcinoma treatment. Proc Natl Acad Sci (2021) 118:e2015433118. doi: 10.1073/pnas.2015433118
178. Le Naour J, Galluzzi L, Zitvogel L, Kroemer G, Vacchelli E. Trial watch: IDO inhibitors in cancer therapy. OncoImmunology (2020), 1777625. doi: 10.1080/2162402X.2020.1777625
179. Liu M, Wang X, Wang L, Ma X, Gong Z, Zhang S, et al. Targeting the IDO1 pathway in cancer: From bench to bedside. J Hematol Oncol (2018) 11:1–12. doi: 10.1186/s13045-018-0644-y
180. Li F, Zhang R, Li S, Liu J. IDO1: An important immunotherapy target in cancer treatment. Int immunopharmacology (2017) 47:70–7. doi: 10.1016/j.intimp.2017.03.024
181. Jung KH, LoRusso P, Burris H, Gordon M, Bang YJ, Hellmann MD, et al. Phase I study of the indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor navoximod (GDC-0919) administered with PD-L1 inhibitor (atezolizumab) in advanced solid tumors. Clin Cancer Res (2019) 25:3220–8. doi: 10.1158/1078-0432.CCR-18-2740
182. Nandre R, Verma V, Gaur P, Patil V, Yang XD, Ramlaoui Z, et al. IDO vaccine ablates immune-suppressive myeloid populations and enhances antitumor effects independent of tumor cell IDO status. Cancer Immunol Res (2022) 10:571–80. doi: 10.1158/2326-6066.Cir-21-0457
183. Niu N, Shen W, Zhong Y, Bast RC Jr., Jazaeri A, Sood AK, et al. Expression of B7–H4 and IDO1 is associated with drug resistance and poor prognosis in high-grade serous ovarian carcinomas. Hum pathology (2021) 113:20–7. doi: 10.1016/j.humpath.2021.04.003
184. Suzuki Y, Suda T, Furuhashi K, Suzuki M, Fujie M, Hahimoto D, et al. Increased serum kynurenine/tryptophan ratio correlates with disease progression in lung cancer. Lung cancer (2010) 67:361–5. doi: 10.1016/j.lungcan.2009.05.001
185. Lee SH, Mahendran R, Tham SM, Thamboo TP, Chionh BJ, Lim YX, et al. Tryptophan–kynurenine ratio as a biomarker of bladder cancer. BJU Int (2021) 127:445–53. doi: 10.1111/bju.15205
186. Mandarano M, Orecchini E, Bellezza G, Vannucci J, Ludovini V, Baglivo S, et al. Kynurenine/tryptophan ratio as a potential blood-based biomarker in non-small cell lung cancer. Int J Mol Sci (2021) 22:4403. doi: 10.3390/ijms22094403
187. de Jong RA, Nijman HW, Boezen HM, Volmer M, Klaske A, Krijnen J, et al. Serum tryptophan and kynurenine concentrations as parameters for indoleamine 2, 3-dioxygenase activity in patients with endometrial, ovarian, and vulvar cancer. Int J Gynecologic Cancer (2011) 21:1320–7. doi: 10.1097/IGC.0b013e31822017fb
188. Schafer CC, Wang Y, Hough KP, Sawant A, Grant SC, Thannickal VJ, et al. Indoleamine 2,3-dioxygenase regulates anti-tumor immunity in lung cancer by metabolic reprogramming of immune cells in the tumor microenvironment. Oncotarget (2016) 7:75407–24. doi: 10.18632/oncotarget.12249
189. Li F, Zhao Y, Wei L, Li S, Liu J. Tumor-infiltrating treg, MDSC, and IDO expression associated with outcomes of neoadjuvant chemotherapy of breast cancer. Cancer Biol Ther (2018) 19:695–705. doi: 10.1080/15384047.2018.1450116
190. Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, et al. Tumor-expressed IDO recruits and activates MDSCs in a treg-dependent manner. Cell Rep (2015) 13:412–24. doi: 10.1016/j.celrep.2015.08.077
191. Xu K, Fu Y, Gao H, Bai M, Liu H, Duan Y. L-tryptophan activates the aryl hydrocarbon receptor and induces cell cycle arrest in porcine trophectoderm cells. Theriogenology (2021) 171:137–46. doi: 10.1016/j.theriogenology.2021.05.012
192. Yoshida J, Ishibashi T, Nishio M. G1 cell cycle arrest by amlodipine, a dihydropyridine Ca2+ channel blocker, in human epidermoid carcinoma A431 cells. Biochem Pharmacol (2007) 73:943–53. doi: 10.1016/j.bcp.2006.12.011
193. Labadie BW, Bao R, Luke JJ. Reimagining IDO pathway inhibition in cancer immunotherapy via downstream focus on the tryptophan–Kynurenine–Aryl hydrocarbon AxisTrp–Kyn–AhR immunotherapy. Clin Cancer Res (2019) 25:1462–71. doi: 10.1158/1078-0432.CCR-18-2882
194. Van den Eynde BJ, van Baren N, Baurain J-F. Is there a clinical future for IDO1 inhibitors after the failure of epacadostat in melanoma? Annu Rev Cancer Biol (2020) 4:241–56. doi: 10.1146/annurev-cancerbio-030419-033635
195. Komiya T, Huang CH. Updates in the clinical development of epacadostat and other indoleamine 2, 3-dioxygenase 1 inhibitors (IDO1) for human cancers. Front Oncol (2018) 8:423. doi: 10.3389/fonc.2018.00423
196. Jung KH, LoRusso P, Burris H, Gordon M, Bang Y-J, Hellmann MD, et al. Phase I study of the indoleamine 2, 3-dioxygenase 1 (IDO1) inhibitor navoximod (GDC-0919) administered with PD-L1 inhibitor (Atezolizumab) in advanced solid TumorsNavoximod and atezolizumab in advanced solid tumors. Clin Cancer Res (2019) 25:3220–8. doi: 10.1158/1078-0432.CCR-18-2740
197. Sloan EK, Priceman SJ, Cox BF, Yu S, Pimentel MA, Tangkanangnukul V, et al The sympathetic nervous system induces a metastatic switch in primary breast cancer. doi: 10.1158/0008-5472.CAN-10-0522
198. Qiao G, Chen M, Mohammadpour H, MacDonald CR, Bucsek MJ, Hylander BL, et al. Chronic adrenergic stress contributes to metabolic dysfunction and an exhausted phenotype in t cells in the tumor microenvironment. Cancer Immunol Res (2021) 9:651–64. doi: 10.1158/2326-6066.CIR-20-0445
199. Braadland PR, Ramberg H, Grytli HH, Taskén KA. β-adrenergic receptor signaling in prostate cancer. Front Oncol (2015) 4:375. doi: 10.3389/fonc.2014.00375
200. Borgatti A, Dickerson EB, Lawrence J. Emerging therapeutic approaches for canine sarcomas: Pushing the boundaries beyond the conventional. Veterinary Comp Oncol (2020) 18:9–24. doi: 10.1111/vco.12554
201. Fjaestad KY, Romer AMA, Goitea V, Johansen AZ, Thorseth ML, Carretta M, et al. Blockade of beta-adrenergic receptors reduces cancer growth and enhances the response to anti-CTLA4 therapy by modulating the tumor microenvironment. Oncogene (2022) 41:1364–75. doi: 10.1038/s41388-021-02170-0
202. Barathova M, Grossmannova K, Belvoncikova P, Kubasova V, Simko V, Skubla R, et al. Impairment of hypoxia-induced CA IX by beta-blocker propranolol–impact on progression and metastatic potential of colorectal cancer cells. Int J Mol Sci (2020) 21:8760. doi: 10.3390/ijms21228760
203. Jin M-Z, Jin W-L. The updated landscape of tumor microenvironment and drug repurposing. Signal transduction targeted Ther (2020) 5:1–16. doi: 10.1038/s41392-020-00280-x
204. Cao M, Huang W, Chen Y, Li G, Liu N, Wu Y, et al. Chronic restraint stress promotes the mobilization and recruitment of myeloid-derived suppressor cells through β-adrenergic-activated CXCL5-CXCR2-Erk signaling cascades. Int J Cancer (2021) 149:460–72. doi: 10.1002/ijc.33552
205. Iñigo-Marco I, Alonso MM. Destress and do not suppress: targeting adrenergic signaling in tumor immunosuppression. J Clin Invest (2019) 129:5086–8. doi: 10.1172/JCI133115
206. MacDonald C, Ministero S, Pandey M, Robinson D, Forti Hong E, Hylander B, et al. Comparing thermal stress reduction strategies that influence MDSC accumulation in tumor bearing mice. Cell Immunol (2021) 361:104285. doi: 10.1016/j.cellimm.2021.104285
207. Jean Wrobel L, Bod L, Lengagne R, Kato M, Prevost-Blondel A, Le Gal FA. Propranolol induces a favourable shift of anti-tumor immunity in a murine spontaneous model of melanoma. Oncotarget (2016) 7:77825–37. doi: 10.18632/oncotarget.12833
208. Ammons DT, Guth A, Rozental AJ, Kurihara J, Marolf AJ, Chow L, et al. Reprogramming the Canine Glioma Microenvironment with Tumor Vaccination plus Oral Losartan and Propranolol Induces Objective Responses. Cancer Treat. Commun (2022) 2:1657–67. doi: 10.1158/2767-9764.CRC-22-0388
209. Hiller JG, Cole SW, Crone EM, Byrne DJ, Shackleford DM, Pang JMB, et al. Preoperative β-blockade with propranolol reduces biomarkers of metastasis in breast cancer: A phase II randomized trial. Clin Cancer Res (2020) 26:1803–11. doi: 10.1158/1078-0432.CCR-19-2641
210. Gandhi S, Pandey MR, Attwood K, Ji W, Witkiewicz AK, Knudsen ES, et al. Phase I clinical trial of combination propranolol and pembrolizumab in locally advanced and metastatic melanoma: Safety, tolerability, and preliminary evidence of antitumor activity. Clin Cancer Res (2021) 27:87–95. doi: 10.1158/1078-0432.CCR-20-2381
211. Phan TT, Ho TT, Nguyen HT, Nguyen HT, Tran TB, Nguyen ST. The prognostic impact of neutrophil to lymphocyte ratio in advanced non-small cell lung cancer patients treated with EGFR TKI. Int J Gen Med (2018) 11:423–30. doi: 10.2147/IJGM.S174605
212. Yun NK, Rouhani SJ, Bestvina CM, Ritz EM, Gilmore BA, Tarhoni I, et al. Neutrophil-to-Lymphocyte ratio is a predictive biomarker in patients with epidermal growth factor receptor (EGFR) mutated advanced non-small cell lung cancer (NSCLC) treated with tyrosine kinase inhibitor (TKI) therapy. Cancers (2021) 13:1426. doi: 10.3390/cancers13061426
213. Tan C-S, Kumarakulasinghe NB, Huang Y-Q, Ang YLE, Choo JR-E, Goh B-C, et al. Third generation EGFR TKIs: Current data and future directions. Mol Cancer (2018) 17:1–14. doi: 10.1186/s12943-018-0778-0
214. Huang L, Jiang S, Shi Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J Hematol Oncol (2020) 13:143. doi: 10.1186/s13045-020-00977-0
215. Mena AC, Pulido EG, Guillén-Ponce C. Understanding the molecular-based mechanism of action of the tyrosine kinase inhibitor: Sunitinib. Anticancer Drugs (2010) 21 Suppl 1:S3–11. doi: 10.1097/01.cad.0000361534.44052.c5
216. Abdel-Aziz AK, Abdel-Naim AB, Shouman S, Minucci S, Elgendy M. From resistance to sensitivity: Insights and implications of biphasic modulation of autophagy by sunitinib. Front Pharmacol (2017) 8. doi: 10.3389/fphar.2017.00718
217. Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther (2005) 315:971–9. doi: 10.1124/jpet.105.084145
218. London CA. Tyrosine kinase inhibitors in veterinary medicine. Topics companion Anim Med (2009) 24:106–12. doi: 10.1053/j.tcam.2009.02.002
219. Hao Z, Sadek I. Sunitinib: The antiangiogenic effects and beyond. OncoTargets Ther (2016) 9:5495. doi: 10.2147/OTT.S112242
220. Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res (2009) 15:2148–57. doi: 10.1158/1078-0432.CCR-08-1332
221. London C, Regan D, Chow L, Weishaar K, Gardner H, Thamm D, et al. 840 triple-drug oral immunotherapy targeting myeloid cells for treatment of metastatic osteosarcoma evaluated in spontaneous canine model. J ImmunoTherapy Cancer (2022) 10:A876–A. doi: 10.1136/jitc-2022-SITC2022.0840
222. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc J-F, et al. Sorafenib in advanced hepatocellular carcinoma. New Engl J Med (2008) 359:378–90. doi: 10.1056/NEJMoa0708857
223. Cabrera R, Ararat M, Xu Y, Brusko T, Wasserfall C, Atkinson MA, et al. Immune modulation of effector CD4+ and regulatory T cell function by sorafenib in patients with hepatocellular carcinoma. Cancer Immunol Immunother (2013) 62:737–46. doi: 10.1007/s00262-012-1380-8
224. Chuang H-Y, Chang Y-F, Liu R-S, Hwang J-J. Serial low doses of sorafenib enhance therapeutic efficacy of adoptive T cell therapy in a murine model by improving tumor microenvironment. PloS One (2014) 9:e109992. doi: 10.1371/journal.pone.0109992
225. Keating GM. Sorafenib: A review in hepatocellular carcinoma. Targeted Oncol (2017) 12:243–53. doi: 10.1007/s11523-017-0484-7
226. Bruix J, Raoul J-L, Sherman M, Mazzaferro V, Bolondi L, Craxi A, et al. Efficacy and safety of sorafenib in patients with advanced hepatocellular carcinoma: subanalyses of a phase III trial. J hepatology (2012) 57:821–9. doi: 10.1016/j.jhep.2012.06.014
227. Kudo M, Finn RS, Qin S, Han K-H, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet (2018) 391:1163–73. doi: 10.1016/S0140-6736(18)30207-1
228. Hatanaka T, Naganuma A, Kakizaki S. Lenvatinib for hepatocellular carcinoma: A literature review. Pharmaceuticals (2021) 14:36. doi: 10.3390/ph14010036
229. Capozzi M, De Divitiis C, Ottaiano A, von Arx C, Scala S, Tatangelo F, et al. Lenvatinib, a molecule with versatile application: From preclinical evidence to future development in anti-cancer treatment. Cancer Manag Res (2019) 11:3847–60. doi: 10.2147/cmar.S188316
230. Kato Y, Tabata K, Kimura T, Yachie-Kinoshita A, Ozawa Y, Yamada K, et al. Lenvatinib plus anti-PD-1 antibody combination treatment activates CD8+ T cells through reduction of tumor-associated macrophage and activation of the interferon pathway. PloS One (2019) 14:e0212513. doi: 10.1371/journal.pone.0212513
231. Tan H-Y, Wang N, Lam W, Guo W, Feng Y, Cheng Y-C. Targeting tumour microenvironment by tyrosine kinase inhibitor. Mol Cancer (2018) 17:43. doi: 10.1186/s12943-018-0800-6
232. Xu Y, Afify SM, Du J, Liu B, Hassan G, Wang Q, et al. The efficacy of PI3Kγ and EGFR inhibitors on the suppression of the characteristics of cancer stem cells. Sci Rep (2022) 12:347. doi: 10.1038/s41598-021-04265-w
233. Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. New Engl J Med (2020) 382:41–50. doi: 10.1056/NEJMoa1913662
234. Nan X, Xie C, Yu X, Liu J. EGFR TKI as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer. Oncotarget (2017) 8:75712–26. doi: 10.18632/oncotarget.20095
235. Pottier C, Fresnais M, Gilon M, Jérusalem G, Longuespée R, Sounni NE. Tyrosine kinase inhibitors in cancer: Breakthrough and challenges of targeted therapy. Cancers (Basel) (2020) 12:731. doi: 10.3390/cancers12030731
236. Law AM, Valdes-Mora F, Gallego-Ortega D. Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells (2020) 9:561. doi: 10.3390/cells9030561
237. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell (2014) 25:846–59. doi: 10.1016/j.ccr.2014.05.016
Keywords: cancer, immune suppresion, tumor associate macrophages (TAM), myeloid derived suppressor cells (MDSC), tumor associated neutrophils (TAN), cancer immune therapy, tumor microenvironment, dendritc cells
Citation: Cao J, Chow L and Dow S (2023) Strategies to overcome myeloid cell induced immune suppression in the tumor microenvironment. Front. Oncol. 13:1116016. doi: 10.3389/fonc.2023.1116016
Received: 05 December 2022; Accepted: 17 March 2023;
Published: 11 April 2023.
Edited by:
Satoshi Watanabe, Niigata University, JapanReviewed by:
Abhilasha Purohit, National Institutes of Health (NIH), United StatesBennett David Elzey, Purdue University, United States
Copyright © 2023 Cao, Chow and Dow. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven Dow, sdow@colostate.edu
†These authors have contributed equally to this work and share first authorship