 Magda Zanelli1*
Magda Zanelli1* Alessandra Bisagni1
Alessandra Bisagni1 Francesca Sanguedolce2Giuseppe Broggi3
Francesca Sanguedolce2Giuseppe Broggi3 Valentina Fragliasso4Maurizio Zizzo5Andrea Palicelli1Giovanni Martino6,7
Valentina Fragliasso4Maurizio Zizzo5Andrea Palicelli1Giovanni Martino6,7 Camilla Cresta6Cecilia Caprera6Matteo Corsi6Pietro Gentile8
Camilla Cresta6Cecilia Caprera6Matteo Corsi6Pietro Gentile8 Fabrizio Gozzi8
Fabrizio Gozzi8 Domenico Trombetta9
Domenico Trombetta9 Paola Parente9
Paola Parente9 Rosario Caltabiano3
Rosario Caltabiano3 Nektarios Koufopoulos10
Nektarios Koufopoulos10 Luca Cimino8,11
Luca Cimino8,11 Alberto Cavazza1Giulio Fraternali Orcioni12Stefano Ascani6
Alberto Cavazza1Giulio Fraternali Orcioni12Stefano Ascani6- 1Pathology Unit, Azienda USL-IRCCS di Reggio Emilia, Reggio Emilia, Italy
- 2Pathology Unit, Policlinico Riuniti, University of Foggia, Foggia, Italy
- 3Department of Medical and Surgical Sciences and Advanced Technologies “G.F. Ingrassia” Anatomic Pathology, University of Catania, Catania, Italy
- 4Laboratory of Translational Research, Azienda USL-IRCCS di Reggio Emilia, Reggio Emila, Italy
- 5Surgical Oncology Unit, Azienda USL-IRCCS di Reggio Emilia, Reggio Emilia, Italy
- 6Pathology Unit, Azienda Ospedaliera Santa Maria di Terni, University of Perugia, Terni, Italy
- 7Hematology, Centro di Ricerca Emato-Oncologica (C.R.E.O.), University of Perugia, Perugia, Italy
- 8Ocular Immunology Unit, Azienda USL-IRCCS di Reggio Emilia, Reggio Emilia, Italy
- 9Laboratory Oncology, Fondazione IRCCS Casa Sollievo della Sofferenza San Giovanni Rotondo, San Giovanni Rotondo, Italy
- 10Second Department of Pathology, Medical School, National and Kapodistrian University of Athens, Attikon University Hospital, Athens, Greece
- 11Department of Surgery, Medicine, Dentistry and Morphological Sciences, University of Modena and Reggio Emilia, Modena, Italy
- 12Pathology Unit, Azienda Ospedaliera di Cuneo, Cuneo, Italy
Myeloproliferative neoplasms (MPNs) are classified into Philadelphia (Ph) chromosome–positive chronic myeloid leukemia (CML) and Ph-negative MPNs. BCR::ABL1 translocation is the key genetic event of CML, whereas JAK2/MPL/CALR mutations are molecular aberrations of Ph-negative MPNs. Despite initially considered mutually exclusive genetic aberrations, the co-occurrence of BCR::ABL1 and JAK2 has been reported in a limited number of cases. The two genetic alterations may be identified either at the same time or JAK2 aberration may be detected in patients with a previous CML treated with tyrosine kinase inhibitors or, finally, BCR::ABL1 translocation occurs in patients with a history of JAK2-positive MPN. This combination of genomic alterations is potentially confounding with clinical manifestations often misinterpreted either as disease progression or drug resistance, therefore leading to inappropriate patient’s treatment. Our systematic review aims to improve hematologist and pathologist knowledge on this rare subset of patients. Starting from the presentation of two additional cases from our routine daily practice, we focus mainly on clinical, laboratory, and bone marrow histological findings, which may represent useful clues of BCR::ABL1 and JAK2 co-occurrence. The interaction between JAK2 and BCR::ABL1 clones during the disease course as well as therapy and outcome are presented.
1 Introduction
Myeloproliferative neoplasms (MPNs) are clonal disorders of hematopoietic stem cells with proliferation of one or more of the hematopoietic lineages, classified into distinct categories depending on different features, including the underlying genetic abnormalities (1–3). MPNs are classified into Philadelphia (Ph)–positive t(9;22) (q34.1;q11.2) chronic myeloid leukemia (CML) and Ph-negative MPNs. BCR::ABL1 gene translocation is the key molecular event defining CML. The reciprocal rearrangement and fusion of the BCR gene on chromosome 22 and ABL gene on chromosome 9 leads to the production of an oncoprotein that can be p190, p210, or p230 depending on the breakpoint of BCR::ABL. The presence of BCR::ABL1 translocation is strictly associated with CML, being an essential requisite for CML development and diagnosis. On the other hand, mutations in the Janus kinase 2 (JAK2) gene on exon 14, mostly at codon 617 (JAK2 V617F) or other activating JAK2 mutations in exon 12 or, more rarely, mutations in the myeloproliferative leukemia (MPL) gene or in exon 9 of Calreticulin (CALR) gene are observed in Ph-negative MPNs, which may have different clinical and morphological features presenting as either polycythemia vera (PV), primary myelofibrosis (PMF), or essential thrombocythemia (ET). The gain-of-function JAK2 mutation leads to the activation of the JAK/STAT signaling pathway, resulting in cellular proliferation and resistance to apoptosis. The JAK2 V617F mutation is detected in PV (95% of cases), PMF (60%), and ET (50%) (1–3). Although BCR::ABL1 translocation and JAK2 mutation were initially considered to be mutually exclusive, more recently the simultaneous presence of these genetic alterations has been reported (4–53). The co-occurrence of JAK2 V617F mutation and BCR::ABL1 translocation in the same patient is a rare event with a frequency ranging from 0.2% to 2.5% according to different studies (21, 38, 54). The two genetic alterations may be identified either simultaneously or JAK2 mutation may be found in the setting of a previously diagnosed CML treated with a tyrosine kinase inhibitor (TKI) or finally BCR::ABL1 translocation may develop in patients with a long history of JAK2-positive MPN. Current scientific literature reports 85 cases of coexistence of BCR::ABL1 and JAK2 (4–53). Most reports are isolated cases or small case series and, given the paucity of data, clinical and pathological characteristics and prognosis of patients harboring concurrent BCR::ABL1 and JAK2 abnormalities have not been systematically evaluated. In the present report, starting from the presentation of two cases with a long history of JAK2-positive PMF subsequently developing CML with BCR::ABL1 translocation acquisition, we performed a systematic review with the purpose to improve our understanding of this particular setting of MPNs with co-occurring BCR::ABL1 translocation and JAK2 mutation, focusing on clinical and laboratory signs and on bone marrow (BM) morphological features, which may represent clues of coexistence of Ph-negative MPNs and CML. Interaction between JAK2 and BCR::ABL1 clones during the disease course is also discussed as well as treatment and outcome.
2 Case descriptions
2.1 Case 1
A 56-year-old woman presented to our center because of increasing white blood cell count (WBC 48.1 × 109/L). The patient had a 13-year history of a Ph-negative, JAK2-positive MPN consistent with pre-fibrotic PMF. At the time of PMF diagnosis the patient had splenomegaly (spleen: 14 cm in diameter), with platelet (PTL) count up to 800 × 109/L, whereas WBC and hemoglobin (Hb) level were within normal limits. Peripheral blood (PB) smear showed dacryocytes, poikilocytes, erythroblasts, and neutrophils with hyper-segmented nuclei. BM aspirate revealed giant megakaryocytes (MKs) and small MKs with hyperchromatic nuclei. BM biopsy showed an hypercellular marrow (80% cellularity) with reduced erythropoiesis and hyperplastic, normal-maturing granulopoiesis; variably sized MKs, including large elements with bulbous nuclei admixed with small-sized forms with hyperchromatic nuclei, were increased and showed loose and dense clusters (Figures 1, 2). Reticulin fibrosis grade 0 was observed. Chromosomal analysis demonstrated a normal female karyotype (46XX) and molecular testing revealed V617F mutation in JAK2 gene (allelic burden: 83.98%), whereas CALR and MPL mutations were absent as well as BCR::ABL1 translocation. Due to repeated cerebral thrombo-embolic episodes, the patient received antiaggregant therapy with acetylsalicylic acid (ASA) and hydroxyurea (HU), followed 8 years later by interferon (IFN) alpha with good control of the disease.
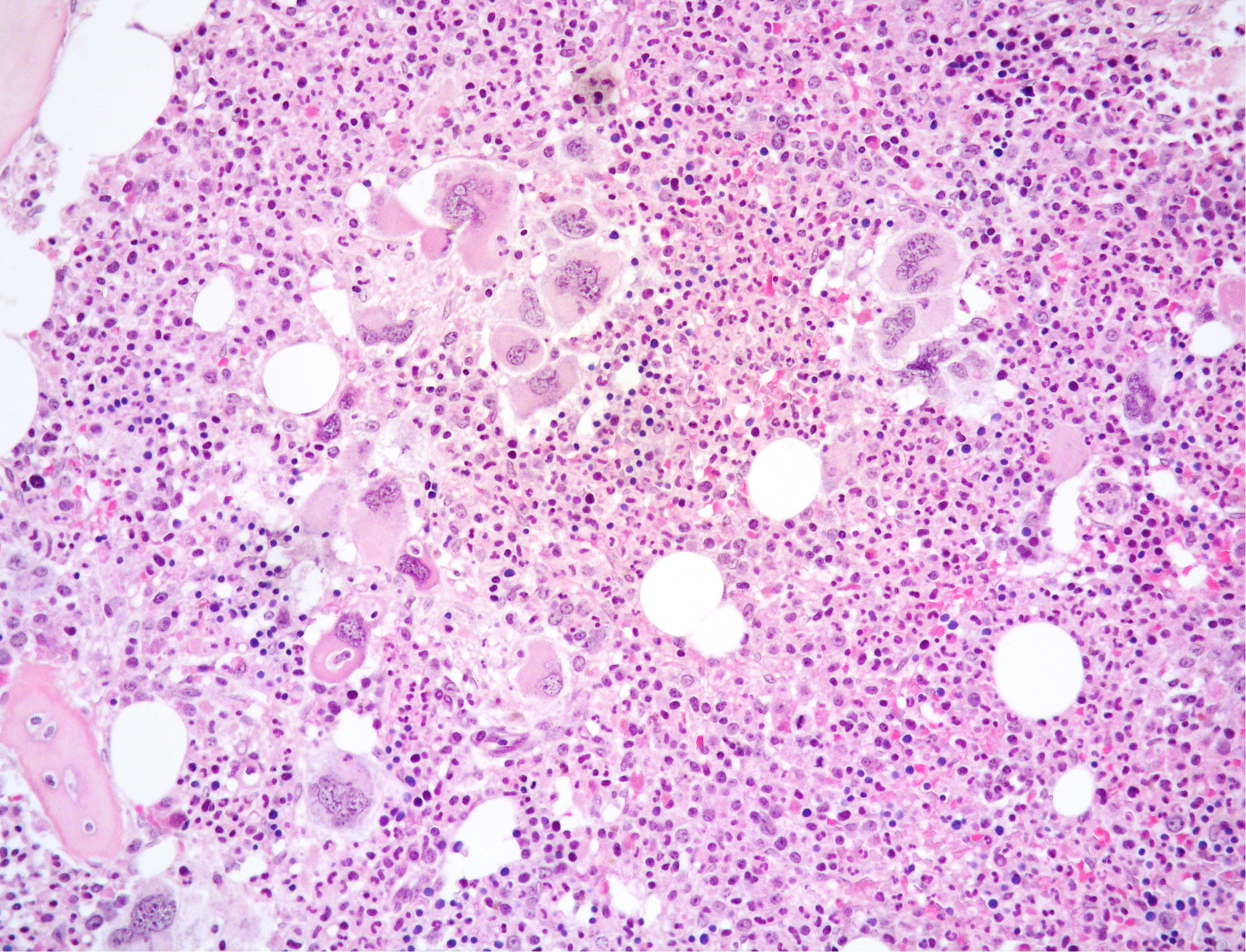
Figure 1 Medium power view of BM biopsy showing hypercellular marrow with normal-maturing prevalent granulopoiesis and clusters of variably sized MKs including large forms with bulbous nuclei (Hematoxylin and eosin, 200x magnification).
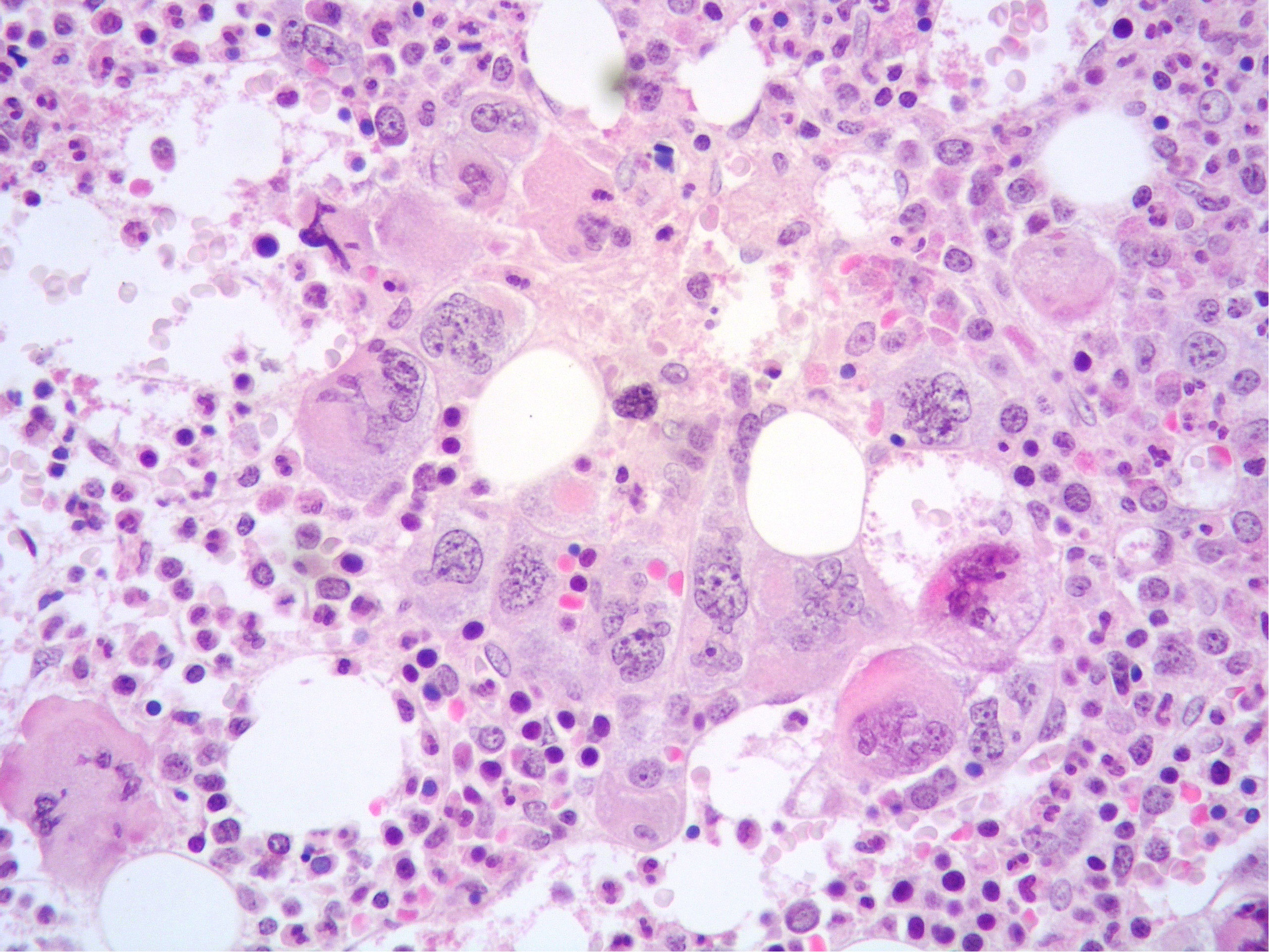
Figure 2 High power view of BM highlighting a dense cluster of variably-sized MKs (Hematoxylin and eosin, 400× magnification).
At the time of referral to our center, blood tests showed progressively increasing leukocytosis up to 48 × 109/L, with normal Hb level (13.3 g/dL) and normal PTL count (416 × 109/L). Chromosomal analysis demonstrated Ph chromosome; fluorescent in-situ hybridization (FISH) analysis detected a BCR::ABL1 fusion in 99% of interphase nuclei indicative of t(9:22)(q34;q11) translocation; quantitative reverse transcriptase–polymerase chain reaction (RT-PCR) for BCR::ABL1 fusion transcripts demonstrated 177% BCR::ABL1 fusion transcript of p210 variant. At this time, JAK2 V617F remained positive with an allelic burden of 21.48%; MPL and CALR remained negative. BM biopsy revealed hypercellularity with high myeloid: erythroid ratio and increased forms of intermediate maturation (metamyelocytes and band neutrophils); MKs were increased in number and predominantly small sized with scarce tendency to form loose clusters (Figure 3); no increase in CD34-positive hematopoietic precursors was noted. Reticulin fibrosis grade 0 was present. BM histopathology combined with clinical data and molecular results were suggestive of CML chronic phase (CP) with concomitant JAK2 mutation and BCR::ABL1 rearrangement. The patient was started on TKI Imatinib at 400 mg/die with a complete cytogenetic response (CCyR) and BCR::ABL1 transcript lower than 10%, 3 months later. Despite a good response of the Ph-positive MPN, JAK2 V617 allelic burden increased up to 89.83%. The marrow was hypercellular with granulocytic predominance and an increase in variably sized MKs with clustering. Grade 0 fibrosis was present. The pathological features were consistent with persistency of the JAK2-positive MPN. IFN-alpha (3 MU/every 10 days) was therefore re-introduced in association with Imatinib (400 mg/die) and ASA, obtaining a good control of the disease with deep molecular response (DMR) of CML, but JAK2 still positive at a constant allelic burden (20%) at 7 years from CML diagnosis.
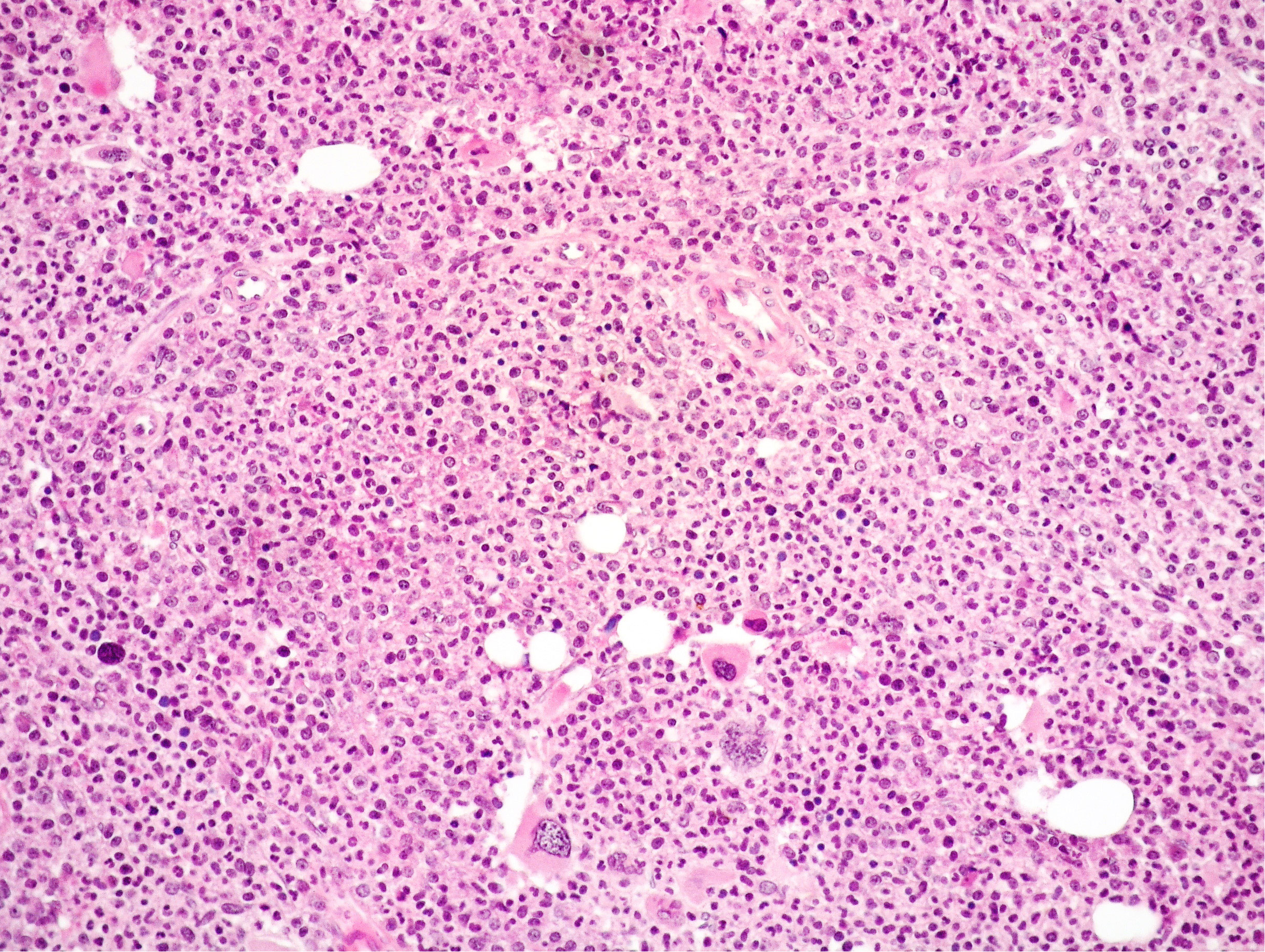
Figure 3 Medium power view of BM biopsy showing hypercellularity with high myeloid: erythroid ratio and mainly small MKs with scarce tendency to form clusters (Hematoxylin and eosin, 200× magnification).
2.2 Case 2
An 82-year-old woman was referred to our center because of increasing leukocytosis (WBC 53.78 × 109/L) with mild anemia (Hb 10.7 g/dL) and normal PTL count (399 × 109/L). The patient had been diagnosed JAK2 V617F–positive (allelic burden: 27%) PMF 7 year earlier at another institution and had been treated with HU for few weeks only, due to drug intolerance (cutaneous edema). The patient had voluntarily interrupted hematologic follow-up for approximately 6 years before presenting to our center. At the time of initial PMF diagnosis, BCR::ABL1 was negative. At the time the patient was referred to us, BM biopsy showed a markedly hypercellular marrow (95% cellularity) with significantly increased myeloid to erythroid ratio, normally maturing myeloid lineage and no increase in CD34-positive hematopoietic precursors. The erythroid lineage was markedly reduced as well as MKs, which were predominantly of small size. Grade 3 reticulin fibrosis was present. Quantitative RT-PCR identified BCR::ABL1 p210 fusion gene (95%). Karyotyping showed 46XX and cytogenetic analysis revealed t(9;22)(q34;q11). FISH analysis identified BCR::ABL1 fusion in 90% of interphase nuclei. JAK2 V617F mutation was confirmed (allelic burden: 30%). Splenomegaly (spleen: 14.5 cm in diameter) was identified. Therefore, the final diagnosis was CML in CP, arising in the background of a JAK2-positive MPN with grade 3 reticulin fibrosis consistent with fibrotic PMF. The patient was started on Imatinib at 100 mg/die, subsequently increased to 200 mg/die. Four months later, BCR::ABL1 fusion gene declined to 18%. Despite a discrete CML control, due to increasing thrombocytosis (PTL count > 1.000 × 109/L), a new BM biopsy was performed 5 months later. The marrow was hypercellular (80% cellularity) with a significant prevalence of the myeloid lineage and an increase in variably sized MKs even with hyperchromatic nuclei and a moderate tendency of dense clustering. Grade 3 reticulin fibrosis was present. JAK2 V617F mutation was confirmed with 15% allelic burden. The pathological features were in keeping with persistency of the JAK2-positive MPN with fibrosis. Molecular analysis showed persistence of BCR::ABL1 fusion gene (15%). In addition to Imatinib (200 mg/die), the patient was put on recombinant IFN alpha-2b (750.000 U/die). Twelve months later, PTL count decreased to 500 × 109/L with 5% JAK2 allelic burden and a major molecular response (MR3) was achieved (BCR-ABL1 fusion gene: 0.07%). The patient is still on treatment with both drugs with a good control of the disease at 24 months from CML diagnosis.
3 Methods of literature review
Our systematic literature review was carried out adhering to the Preferred Reported Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines. The search was conducted using PubMed/MEDLINE, EMBASE, Scopus, Cochrane Library (Cochrane Database of Systematic Reviews, Cochrane Central Register of Controlled Trials (CENTRAL), and Web of Science (Science and Social Science Citation Index) databases, with the following non-MeSH/MeSH terms “JAK2” AND BCR::ABL1 coexistent” [Mesh]. The search was performed from 2005 when JAK2 gene was first discovered to August 2023. The criteria for inclusion were as follows: (1) MPNs harboring concurrent JAK2 mutation and BCR::ABL1 translocation; (2) clinically relevant dual driver mutations representing coexistent Ph-positive CML and Ph-negative JAK2-positive MPN; (3) retrospective, observational case-control studies, case reports and/or series, literature review. The exclusion criteria were as follows: (1) studies not published in English; (2) lack of concurrent JAK2 mutation and BCR::ABL1 translocation (3) clinically non-relevant, incidentally detected BCR-ABL1 translocation, in the context of a Ph-negative JAK2-positive MPN; (4) clinically non-relevant, incidentally detected JAK2 mutation, in the context of a Ph-positive MPN. Two independent reviewers (M. Zanelli, AB) identified papers on the basis of title, abstract, and key words; then, they evaluated whether the selected papers met the inclusion criteria by reading the article full texts. Moreover, reference lists from each retrieved article were checked to find additional reports. From selected papers, the following information was collected: author’s surname, year of publication, patient’s age and sex, disease course, clinical and laboratory findings suggestive of a second coexistent MPN, BM histological features suggestive of a second coexistent MPN, interaction between JAK2 and BCR::ABL1 clones, treatment, and outcome. A third independent reviewer (AS) revised all collected results and solved discrepancies.
4 Results of literature data including our cases
4.1 Study selection and characteristics
The final literature search, performed in August 2023, identified 678 potential items of interest (Figure 4). After removing irrelevant publications (615), 65 records were further analyzed. Fifteen of these were excluded according to exclusion criteria, while 50 full-text articles were assessed for eligibility and included into qualitative synthesis. The included articles were case reports/case series (4–53).
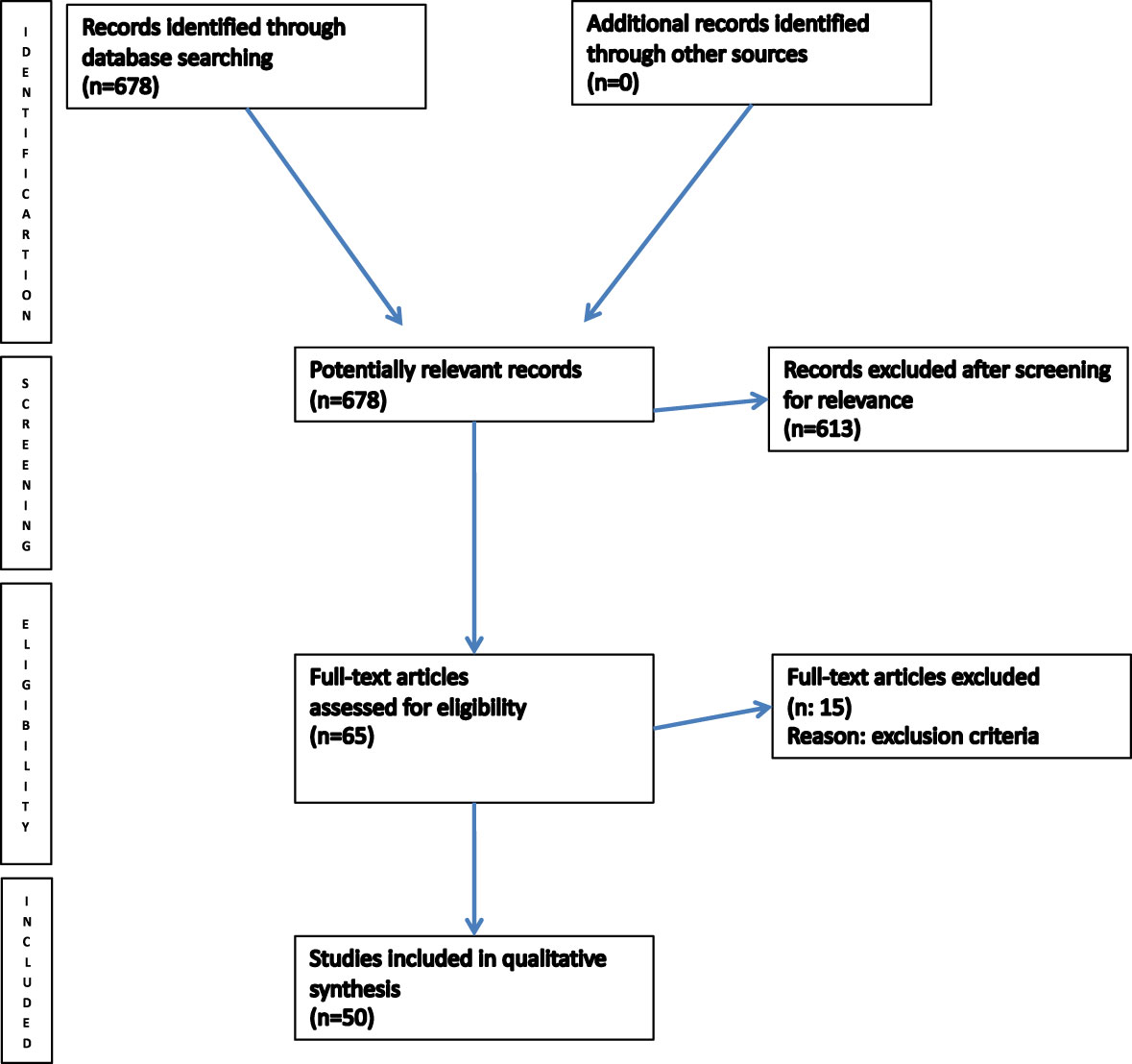
Figure 4 PRISMA flow chart of literature search.
4.2 Epidemiology
To date, 50 studies have analyzed MPNs with concomitant JAK2 and BCR::ABL1, with a total of 85 cases (4–53). Including our two cases, a total of 87 cases have been described so far. Patient characteristics, course of the disease, clinical and laboratory data, as well as BM histological features suggestive of concomitant JAK2 and BCR::ABL1, interaction between JAK2 and BCR::ABL1 clones, treatment and outcome, are summarized in Supplementary Table S1.
Patients with coexistent JAK2 and BCR::ABL1 were aged between 24 and 87 years with an average of 60.1 years. Males were slightly more frequently affected than females (46/87; 52.87%). Including our cases, 28 patients had PV, 24 PMF, 20 ET, 10 MPNs not otherwise specified (MPN, NOS), three post-polycythemia vera myelofibrosis (PPV-MF), one post-essential thrombocythemia myelofibrosis (PET-MF) and in one case the initial diagnosis was unavailable.
Three different settings of patients were observed: Group 1: Ph-negative MPN preceded CML in 43 cases (43/87; 49.42%) (6, 8, 13, 15–17, 21–23, 27, 28, 31, 32, 35, 37–39, 41–43, 45–51), Group 2: CML preceded Ph-negative MPN in 20 cases (20/87; 22.98%) (4, 5, 9, 10, 15, 18, 19, 22, 24–26, 38, 39, 46, 48) and Group 3: CML and Ph-negative MPN were diagnosed concomitantly in 24 cases (24/87; 27.58%) (7, 10–12, 14, 15, 20, 22, 23, 29, 30, 33, 34, 36, 37, 39, 40, 44). Data on the distribution of Ph-negative MPNs in the three groups of patients are summarized in Table 1. Data on different types of TKIs used in this setting of patients and data on the limited number of cases receiving allogenic stem-cell transplantation (allo-SCT) are summarized in Tables 2 and 3, respectively. Data on the frequency of cases showing an opposite growth between BCR::ABL1 and JAK2 clones under TKI treatment in the three groups are showed in Figure 5.
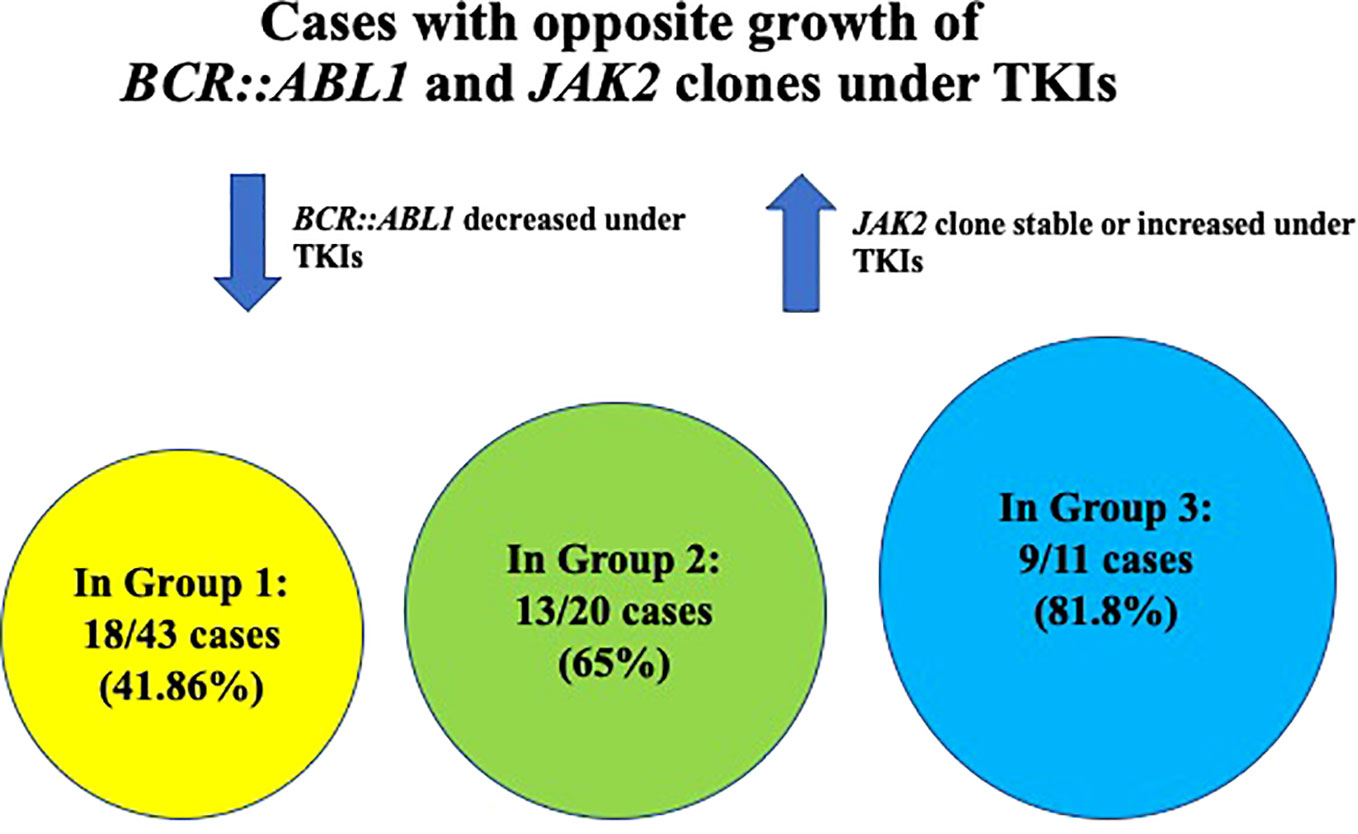
Figure 5 Graphic showing the frequency of cases with opposite growth of BCR::ABL1 and JAK2 clones under TKI treatment in the three different groups of patients.
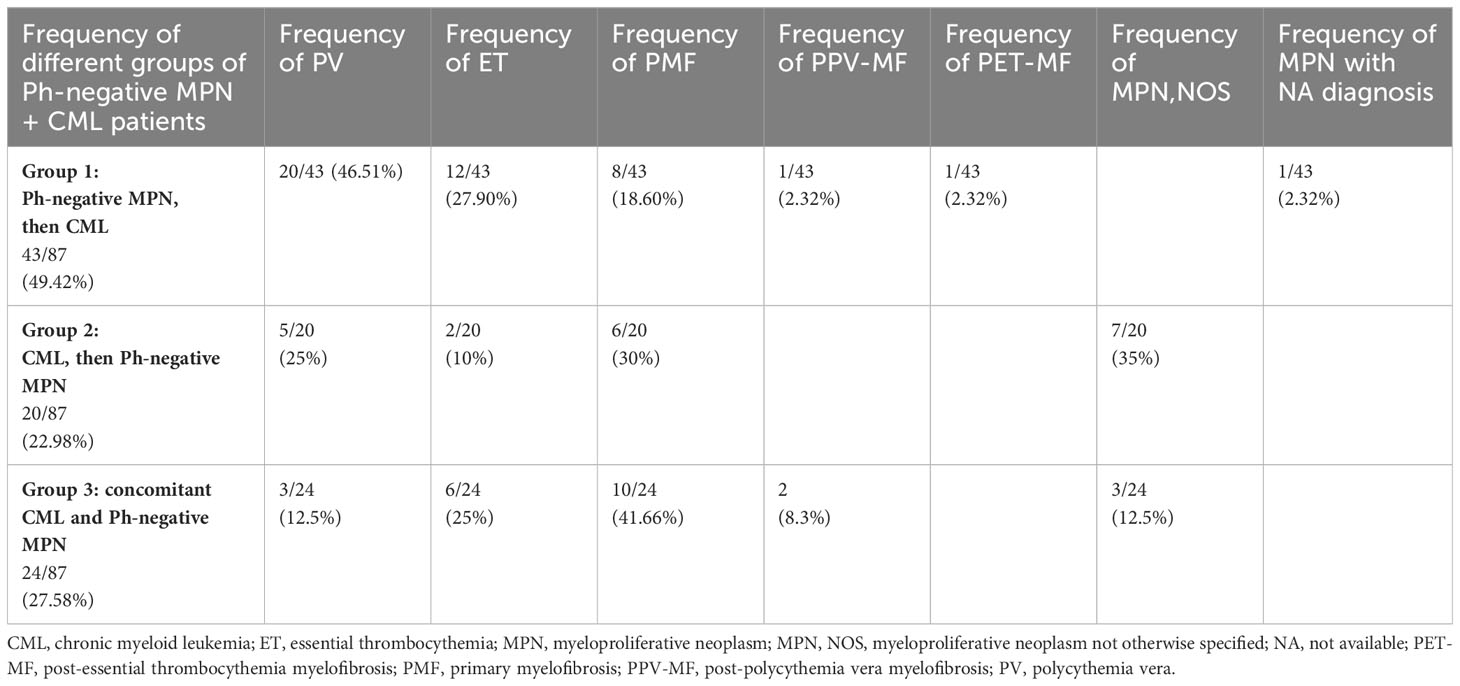
Table 1 Distribution of Ph-negative MPNs in the three groups of patients with concomitant or sequential JAK2-positive MPN and CML (see Supplementary Material).
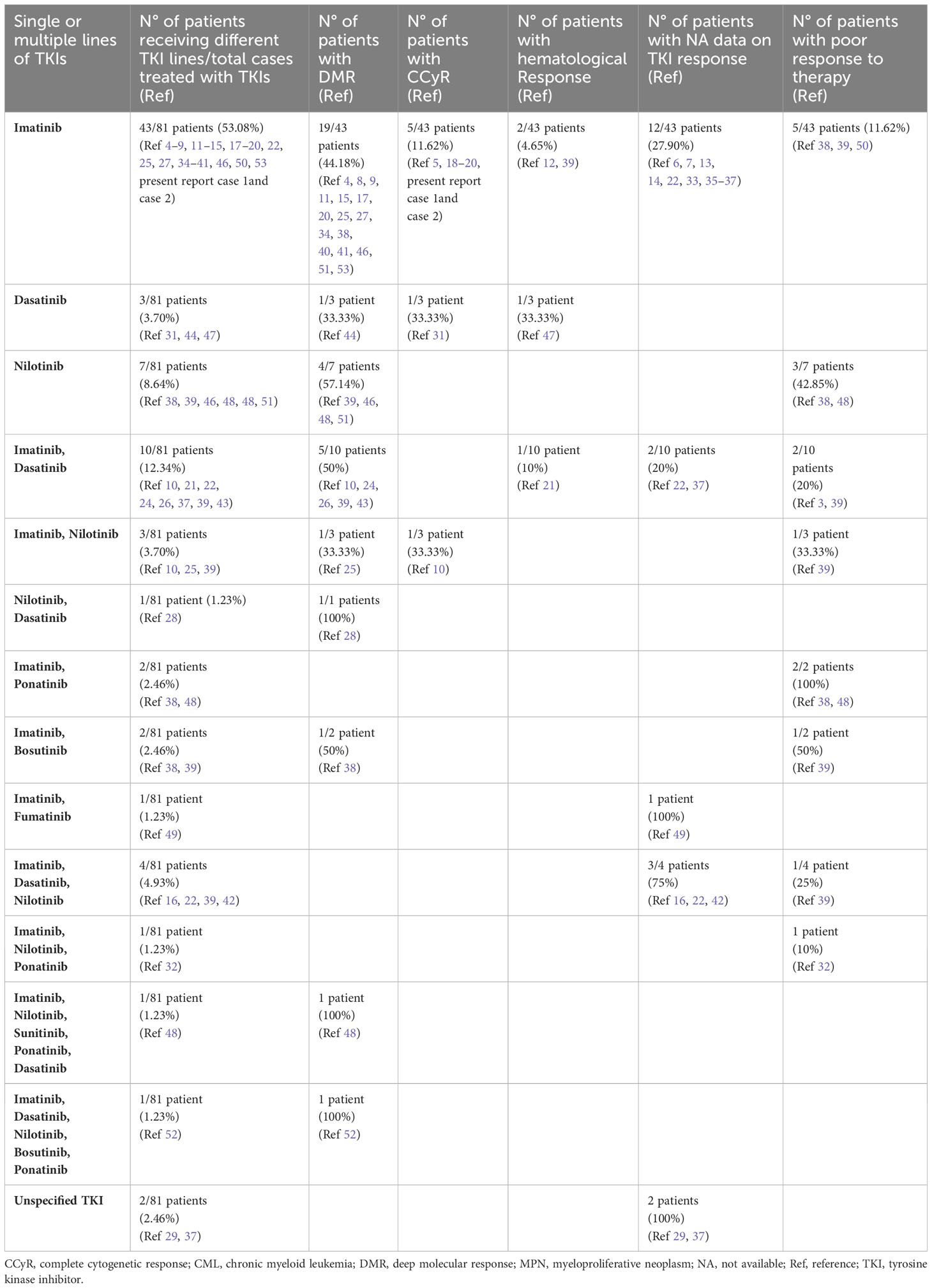
Table 2 Data on different TKIs used in concomitant or sequential JAK2-positive MPNs and CML (see Supplementary Material).
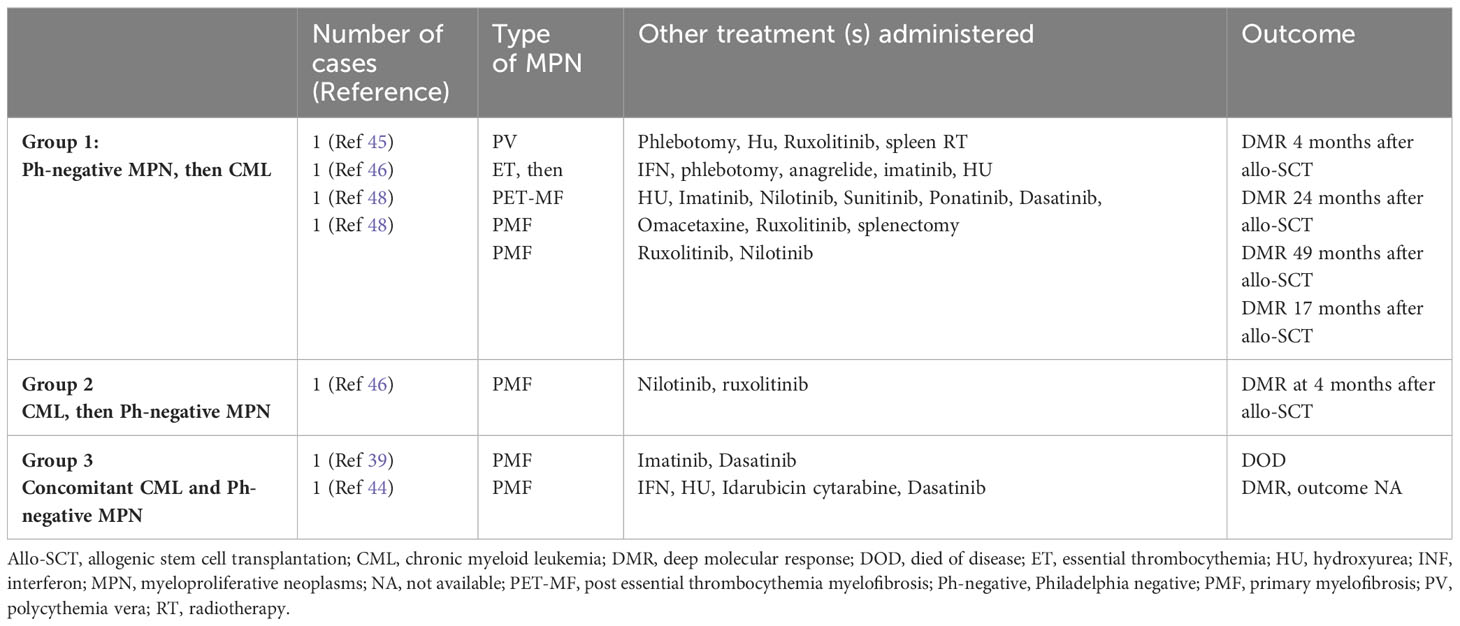
Table 3 Allo-SCT in concomitant or sequential JAK2-positive MPNs and CML (see Supplementary Material).
4.3 Group 1 (Ph-negative MPN preceding CML): clinical symptoms and laboratory signs at second disease (CML) occurrence
In Group 1 (43 cases of Ph-negative MPN preceding CML), the Ph-negative MPNs were distributed as follows: 20 PV, 12 ET, 8 PMF, 1 PPV-MF, 1 PET-MF; one case with unavailable MPN diagnosis.
At CML occurrence, WBC increase was the most common laboratory sign, observed in 38 of 42 cases with available data (4–43, 45–51), followed by anemia in 17/42 (6, 16, 17, 23, 27, 31, 37–39, 45, 50); basophilia in 17–42 (6, 13, 17, 23, 27, 35, 38, 41, 45, 47–50); PTL decrease in 9/42 (13, 27, 37–39, 45); leukoerythroblastosis in 7/42 (17, 31, 43, 45, 48); PTL increase in 6/42 (6, 22, 23, 37, 39, 48); eosinophilia in 6/42 (13, 41, 45, 47, 48), and LDH increase in 2/42 (27, 48). In this group, splenomegaly was detected in just under half of cases (20/42) (6, 8, 13, 16, 17, 23, 27, 28, 31, 32, 43, 45, 48–51) and hepatomegaly in 4/42 (6, 27, 49); systemic symptoms were observed only in a minority of patients as follows: fatigue/feeling unwell in 7/42 (6, 16, 31, 48, 51); weight loss (31, 48) and shortness of breath in 2/42 (6); dyspnea on exertion in 1/42 (27) and decreased appetite in 1/42 (48).
4.4 Group 1: histological features of BM biopsy performed at second disease (CML) occurrence
In Group 1, BM histology performed at the time of CML occurrence in 35/43 cases showed the following features: CML, CP in the majority of cases (13/35) (15–17, 21, 27, 32, 38, 39, 42, 43, 51); CML accelerated phase (AP) in 3/35 (6, 39, 50); CML+fibrosis in 7/35 (31, 41, 47–49), hence a total of 23/35 cases showed histological features reminiscent of CML; PET-MF histology in 3/35 (22, 23, 46); PPV-MF histology in 2/35 (48, 51); PMF histology in 1/35 (28); whereas hybrid features were observed in the remaining cases as follows: PPV-MF+CML in 3/35 (39, 45) with hybrid MKs both hypo-lobate and large hyper-lobate detected in 1/3 (45); PET-MF+CML in 2/35 (38, 39) with hybrid MKs in 1/2 (38); CML with hybrid MKs in 1/35 cases (48).
Of note, our case n°1 with CML (CP) histology was re-biopsied after having achieved a CCyR of CML with TKI; at this time, JAK2 allele burden increased and histology switched to a PMF phenotype. Our case n°2 was re-biopsied at the time of CML partial MR and the histology switched from CML+fibrosis to a phenotype suggestive of JAK2+MPN with fibrosis, in particular the MKs from small-sized, non-clustering forms changed to variably sized, clustering MKs.
4.5 Group 1: BCR::ABL1 and JAK2 clone interaction
In Group 1, the following interaction between BCR::ABL1 and JAK2 clones were observed at the time of CML occurrence and during the course of the disease: the majority of cases (18/43; 41.86%) showed an opposite growth of the two genomic alterations with BCR::ABL1 decrease under TKI and JAK2 increase. In particular, 13/18 cases were positive for both BCR::ABL1 and JAK2 at CML occurrence and, under TKI, BCR::ABL1 decreased (resulting often in CMR of CML) and JAK2 increased (8, 13, 15, 21, 28, 31, 38, 39, 41, 49, 51); additionally, in 2/13 cases, JAK2 increase was clinically associated with concomitant hematocrit (HCT) increase (8) or with concomitant PTL and WBC increase (31); in 4/18 cases, BCR::ABL1 was positive and JAK2 negative at CML occurrence and, under TKI, an opposite growth of the two genomic alteration was seen, with BCR::ABL1 decrease and JAK2 increase (16, 17, 27, 43); in all these four cases, JAK2 increase was associated with clinical PV recurrence; in 1/18, at CML occurrence BCR::ABL1 was positive and JAK2 not evaluated and under TKI, an opposite growth of BCR::ABL1 and JAK2 was observed similarly to the above mentioned cases (46). In 3/43 cases, BCR::ABL1 and JAK2 were both positive at CML occurrence and presented a similar growth either remaining both positive and at the same allele burden before and after TKI (1/3 cases) (39), or both increasing during the disease course, without TKI (1/3 cases) (45) or both decreasing under TKI (1/3 cases). Finally, in 21/43 cases the interaction between BCR::ABL1 and JAK2 during the course of the disease was unavailable (6, 22, 23, 32, 35, 37–39, 42, 48, 50, 51).
4.6 Group 1: treatment and outcome
In this paragraph, as well as in the following paragraphs on treatment for Groups 2 and 3, the type of Ph-negative MPN preceding CML is indicated in brackets for each single case.
TKI therapy was administered in 41/43 cases; of 2/43 not receiving TKI, ASA, HU, and thalidomide were administered in 1/2 (ET) with unavailable outcome (23), whereas phlebotomy, HU, ruxolitinib, spleen radiotherapy, and allo-SCT in 1/2 (PV), with undetectable genomic markers at 4 months after SCT (45). TKI alone was administered in 1/41 cases (PMF) with DMR of CML (28). In the remaining cases, TKI was combined with other treatments as follows: TKI and phlebotomy in 3/41 cases with CCyR of CML in 1/3 (PV) and DMR of CML in 1/3 (PV) with no further data on outcome (8, 51), whereas in 1/3 cases (PV), outcome was unavailable (16); TKI and HU in 3/41 cases, with death of disease (DOD) in 1/3 (PV) (39) and unavailable outcome in 2/3 cases (1 PMF and 1 ET) (13, 37); TKI and IFN in 1/41 cases (PV) with death for AML 34 months after CML (15); TKI, HU and IFN in 3/41 cases with DMR of CML and good control of disease at 24 months from CML in 1/3 cases (PMF, present report case 2), good control of blood parameters in 1/3 (ET), which is under evaluation for SCT (22) and outcome unavailable in 1/3 (PET-MF) (37); TKI, HU and phlebotomy in 2/41 with DMR of CML and good control of PV in both cases (17, 27); TKI, HU and ASA in 1/41 (ET) with complete hematologic remission (47); TKI, HU, ASA, and phlebotomy in 2/41 (PV) with DMR of CML (41, 43); TKI, HU, and pipobroman in 1/41 cases (PV) with only hematologic response (21); TKI, HU, IFN, and ASA in 1/41 (PMF) with good control of diseases at 7 years from CML (PMF, present report case 1); TKI and ruxolitinib in 2/41, with DMR of CML in 1/2 (ET and PET-MF) and unavailable outcome in 1/2 (PPV-MF) (37, 38); TKI, ruxolitinib and phlebotomy in 1/41 (PV), with unavailable outcome (49); TKI, IFN and ruxolitinib in 1/41 (PV and PPV-MF) with suboptimal response and unavailable outcome (38); TKI, HU and ruxolitinib in 3/41 with DMR of CML in 1/3, but patient alive with disease (PMF and PET-MF) in 2/3 cases (39) and scarce response to therapy in 1/3 (PMF) (50); TKI, HU, ruxolitinib and phlebotomy in 1/41 (PV), with death for sepsis (48); TKI, HU, IFN, ruxolitinib in 1/41 (ET) with patient under evaluation for radiotherapy and SCT (49); TKI, HU, IFN, ruxolitinib, and phlebotomy in 1/41 (PV) with patient alive with disease (39); TKI and anagrelide in 1/41 (initial diagnosis unavailable) with remission (39); TKI, HU, and anagrelide in 1/41 (ET) with no response to therapy (48); TKI, phlebotomy and anagrelide in 1/41 (PV) with unavailable outcome (6); TKI, HU, anagrelide, IFN and ASA in 1/41 (ET) with unavailable outcome (42); TKI, HU, anagrelide, ruxolitinib, and ASA in 1/41 (PV) with CCyR of CML (31); TKI, HU, anagrelide, radioactive phosphorus in 1/41 (PV), with death for cerebral event 4 months after CML diagnosis (6); TKI, anagrelide, ruxolitinib and cytarabine in 1/41 (ET) with evolution to CML (BP) and death (38); TKI, HU, anagrelide, cytarabine and ruxolitinib in 1/41 (ET) with evolution to CML (BP) and poor response to therapy (32); TKI, HU, thalidomide and ruxolitinib in 1/41 (PV) with death for disease (39); TKI, phlebotomy, pipobroman, HU, spleen radiotherapy, IFN and ruxolitinib in 1/41 (PV) with death for gastric cancer (35); TKI, ruxolitinib and allo-SCT in 1/41 (PMF) with no detectable genomic markers 17 months after CML (48); TKI, omacetaxine, HU, ruxolitinib, splenectomy and allo-SCT in 1/41 (PMF) with no detectable genomic markers 49 months after SCT (48); TKI, IFN, anagrelide, HU, phlebotomy and allo-SCT in 1/41 (ET) with complete remission of both diseases at 24 months after allo-SCT (46).
4.7 Group 2 (CML preceding Ph-negative MPN): clinical symptoms and laboratory signs at second disease (Ph-negative MPN) occurrence
In Group 2 (20 cases), the Ph-negative MPNs occurring as second disease after CML, were distributed as follows: seven cases were JAK2+MPN, NOS; six cases PMF; five cases PV and two cases ET.
Laboratory signs detected in six cases of JAK2+MPN, NOS with available data occurring after CML were as follows: PTL increase in 4/6 cases (26, 38, 39); WBC increase in 3/6 (19, 22) and LDH increase in 1/6 (26). Of note in in 2/4 cases with PTL increase and in 1/3 cases with WBC increase, these laboratory signs were present despite good MR of CML (22, 26, 38).
Laboratory signs observed in five cases of PMF with available data occurring after CML were as follows: WBC increase in 3/5 (4, 15); PTL increase in 2/5 (10, 46); PTL decrease in 2/5 (4, 48); LDH increase in 2/5 (4, 15); anemia and immature myeloid cells in PB in 1/5 (48). In this group, splenomegaly was detected in 3/5 cases (4, 15, 48). Of note, in 1/2 cases with PTL increase, this laboratory sign was present despite good MR of CML (46).
Laboratory signs detected in the 5 cases of PV occurring after CML were as follows: red blood cell (RBC) increase and HCT increase in 2/5 cases (5, 24); WBC, Hb level and PTL increase in 1/5 cases (18); HCT and Hb increase in 1/5 (53), whereas HCT and Hb increase were associated with low erythropoietin (EPO) level in 1/5 (52).
Laboratory signs detected in the two cases of ET occurring after CML were as follows: PTL increase in 2/2 cases despite CCyR of CML (25); WBC increase in 1/2 despite CCyR of CML (25).
4.8 Group 2: histological features of BM biopsy performed at second disease (Ph-negative MPN) occurrence
BM biopsy performed at the time of Ph-negative MPN occurrence showed different features according to the type of Ph-negative MPN.
BM biopsy performed in six cases of PMF occurring after CML, showed the typical PMF histology (4, 9, 10, 15, 46, 48). Of note, in 2/6 cases, PMF histological features became clearly evident after CMR of CML with TKI; at the time PMF histology was detected, BCR::ABL1 was negative and JAK2 positive, at a constant allele burden in one case (4) and at an increased allele burden in the other case (9). Moreover, in 1/2 cases with PMF histology obvious after TKI, the presence of fibrosis at initial CML diagnosis could have been a possible clue for PMF, although fibrosis alone is not sufficient for PMF diagnosis as fibrosis may be found even in CML; in this case, BCR::ABL1 and JAK2 were both present from initial CML diagnosis, however PMF features became obvious after CMR of CML (4).
BM biopsy in five cases of JAK2-positive MPN, NOS occurring after CML with available histology, showed histological features consistent with Ph-negative MPN in all cases, in particular increase of large, clustering MKs was detected (15, 22, 38, 39). Of note, in 2/5 cases Ph-negative phenotype became evident after TKI, with BCR::ABL1 decrease and JAK2 increase (15, 22); in addition, in one of these two cases, BM performed at the time of initial CML diagnosis retrospectively revised showed not only MKs with hypo-lobate nuclei, typical of CML, but even large and occasionally clustering MKs with bulbous nuclei (22).
BM biopsy, available in 1/5 cases of PV occurring after CML, showed typical PV histological features (18). BM biopsy, available 1/2 cases of ET occurring after CML, showed MK increase (25).
4.9 Group 2: BCR::ABL1 and JAK2 clone interaction
In Group 2, the majority of cases (13/20; 65%) showed an opposite growth of BCR::ABL1 and JAK2 as follows: in 4/20 cases, BCR::ABL1 and JAK2 were coexistent at the time of CML and under TKI, BCR::ABL1 decreased whether JAK2 increased (4, 5, 9, 26); in 11/20 cases, only BCR::ABL1 was present at CML diagnosis and JAK2 was either negative (6/11 cases) (18, 25, 48, 52, 53) or not evaluated (5/11 cases) (19, 22, 38, 46); under TKI, BCR::ABL1 decreased with CMR and JAK2 progressively increased. In 3/20, both BCR::ABL1 and JAK2 decreased under TKI showing a similar growth (10, 15, 24) and in 2/20 cases clone interaction was unavailable (15, 39).
4.10 Group 2: treatment and outcome
TKI treatment was administered in 19/20 cases and of these cases, TKI alone was administered in 2/19 cases (PMF), both of which achieved DMR of CML (9, 10). TKI and HU were administered in 3/19 cases, of which, 1/3 (MPN, NOS) achieved CCyR of CML (26), 1/3 (MPN, NOS) achieved CMR of CML and good control of PTL count (38) and 1/3 (MPN, NOS) died of disease (CML-BP), one year after diagnosis (38). TKI and IFN were administered in 2/19 cases, of which one (MPN, NOS) achieved CCyR of CML (19) and one (MPN, NOS) was alive with disease (39); TKI and hydroxycarbamide in 1/19 (PMF) with DMR of CML (4); TKI, IFN, HU and phlebotomy in 2/19, of which one (PV) achieved CCyR of CML and one (PV) achieved negativity of both genomic markers (5, 24); TKI, IFN, HU, phlebotomy and ASA in 2/19 (PV) with DMR of CML and good control of disease (52, 53); TKI, anagrelide and HU in 1/19 (MPN, NOS) with DMR of CML (15); TKI, IFN, HU and cytosine arabinoside in 1/19 (PV) with DMR of CML (18); TKI, anagrelide, HU and IFN in 1/19 (ET) with CCyR of CML, good hematological parameters, but JAK2 constantly positive (25); TKI, HU and ruxolitinib in 1/19 (PMF) with scarce response to therapy (48); TKI and anagrelide in 1/19 (ET) with CCyR of CML and good clinical conditions, but JAK2 constantly positive at 3 years from CML diagnosis (25); TKI, ruxolitinib and allo-SCT in 1/19 (PMF), with complete remission at 4 months after allo-SCT (46); TKI, HU and spleen radiotherapy in 1/19 (MPN, NOS), with death for pneumonia (22). In 1/20 cases (PMF), HU and IFN were administered without TKI and outcome is unavailable (15).
4.11 Group 3 (coexistent CML and Ph-negative MPN): clinical and laboratory signs
Of Group 3 (24 cases of concomitant CML and Ph-negative MPN), 10 cases were coexistent PMF+CML; six cases coexistent ET+CML; three cases coexistent MPN, NOS+CML, three cases coexistent PV+CML, and two cases coexistent PPV-MF+CML.
Clinical symptoms and laboratory signs observed in eight cases of coexistent PMF+CML with available data were as follows: WBC increase in 6/8 (12, 33, 39, 40, 44); anemia in 5/8 (7, 14, 39, 44); PTL increase in 5/8 (12, 14, 20, 40, 44); splenomegaly in 5/8 (7, 14, 20, 40, 44); basophilia in 2/8 (12, 33), high LDH in 2/8 (33 ,44), weight loss 1/8 (40), night sweats in 1/8 (40), pruritus in 1/8 (40), hepatomegaly in 1/8 (40) and leukoerythroblastosis in 1/8 (33).
Laboratory signs observed in six cases of coexistent ET+CML were as follows: WBC increase in 4/6 cases (23, 30, 36, 39); PTL increase in 5/6 (20, 23, 30, 34); basophilia in 2/6 (23, 36) and anemia in 1/6 (23).
Laboratory signs detected in three cases of coexistent MPN, NOS+CML were as follows: WBC increase in 1/3 cases (15); anemia in 1/3 (29), and, finally, high PTL despite TKI in 1/3 cases (36).
Laboratory signs and clinical symptoms observed in three cases of coexistent PV+CML were as follows: high WBC in 3/3 cases (11, 22, 39); high HB level in 1/3 (11), high HCT in 1/3 (11), EPO level below normal level in 1/3 (11), basophilia and eosinophilia in 1/3 cases (22) as well as splenomegaly (22). Of note, after TKI, despite WBC decrease, Hb level and HCT were found to be increased in 1/3 cases (22).
Laboratory signs detected in two cases of coexistent PPV-MF+CML were as follows: splenomegaly in 2/2 cases (37); anemia in 1/2 (37) and WBC increase in 1/2 (37).
4.12 Group 3: histological features of BM biopsy
In Group 3, BM histology showed the following features subdivided according to the type of Ph-negative MPN associated with CML.
BM biopsy of the 10 cases of coexisting CML+PMF showed the following features: a mixed histology CML+PMF in 4/10 cases (33, 39, 40); BM fibrosis in 2/10 cases (7, 10); BM fibrosis+MK hyperplasia in 2/10 (12, 20); PMF histology in 1/10 (44); CML + fibrosis in 1/10 (14). Of note, 1/3 cases with mixed histology CML+PMF and the case with CML+fibrosis were re-biopsied after TKI and showed a switch to PMF histology (14).
BM biopsy was available in 5/6 cases with coexistent CML+ET. A mixed histology of CML+ET was present in 2/5 cases (36, 39), CML (CP) in 1/5 (20) and MK hyperplasia in 1/5 (23), whereas in 1/5, a CML histology was observed pre-TKI therapy, with a shift to ET-histology post-TKI therapy (34).
BM biopsy was available in 2/3 cases with coexistent CML+PV; a mixed histology of CML+PV was present in 1/2 (39) and MF histology in 1/2 (22).
BM biopsy of the three cases of coexistent CML+JAK2-positive MPN, NOS showed a mixed histology combining CML features+ clustering large MKs in 2/3 (15, 29) and panmyelosis with MK clustering in 1/3 (36).
BM biopsy data were incomplete in two cases of coexistent CML+PPV-MF and only grades 1 and 2 fibrosis was reported (37).
4.13 Group 3: BCR::ABL1 and JAK2 clone interaction
In group 3, BCR::ABL1 and JAK2 were detected at diagnosis, but clone interaction during the course of the disease was unavailable in 13/24 cases (14, 29, 30, 34, 36, 37, 39, 44). In 9/11 cases (81.8%) with available data on clone interaction, BCR::ABL1 and JAK2 showed an opposite growth as, under TKI, BCR::ABL1 decreased and JAK2 either remained constant or increased (7, 10, 11, 15, 20, 22, 33, 40); in 1/22 cases, BCR::ABL1 and JAK2 showed an opposite growth, but in this case, under HU, JAK2 decreased and BCR::ABL1 remained positive (23). Finally, in 1/24 cases, JAK2 and BCR::ABL1 showed a similar growth as both decreased; however JAK2 decrease was achieved with a combined TKI plus HU treatment (12).
4.14 Group 3: treatment and outcome
TKI treatment was administered in 21/23 cases with available therapy data; among these cases, TKI was the only therapy in 3/21 cases (1 PMF, 2 MPN, NOS) (10, 15, 29) and in 1/3 bortezomib and radiotherapy were also administered due to concomitant multiple myeloma (29); TKI and phlebotomy in 2/21 (PV) (11, 39); TKI and HU were administered in 5/21 cases (4 PMF, 1 ET) (7, 12, 14, 36, 40) with the adjunct of ASA in 1/5 (ET) (36); TKI, IFN and phlebotomy in 1/21 (PV) (22); TKI and IFN in 1/21 (PPV-MF) (37); TKI and lenalidomide in 1/21 (33); TKI, hydroxycarbamide and ASA in 1/21 (PMF) (20); TKI and ASA in 2/21 (ET) (20, 36); TKI, HU and ruxolitinib in 1/21 (PMF) (39); TKI, IFN and HU in 1/21 (ET) (34); TKI, anagrelide and HU in 1/21 (ET) (39); TKI and allo-HSCT in 1/21 (PMF) (39) and TKI, IFN, HU and allo-SCT in 1/21 (PMF) (44). In 1/23 cases (ET), HU and ASA without TKI were administered (23) and 1/23 (PPV-MF), HU and ruxolitinib without TKI (37). DMR of CML was reported in seven cases (7, 11, 15, 20, 34, 40, 44) and CCyR of CML in two cases (10, 20), but no further data on the outcome of these cases were reported. Data on outcome were available in 5/24 cases as follows: 2/5 cases were alive in hematological remission and three cases DOD. Of the two cases alive in hematological remission, one case (PV) received TKI and phlebotomy (39) and one (PMF) TKI and HU (12). Of the DOD cases, 1/3 (PMF) received TKI and allo-SCT (39), 1/3 (ET) received TKI, HU and anagrelide (39) and 1/3 (PMF) received TKI, ruxolitinib and HU (39).
5 Discussion
JAK2 and BCR::ABL1 are driver genomic alterations leading to different types of MPNs. Despite historically considered mutually exclusive driver mutant genes, the coexistence of JAK2 V617 (or rarely JAK2 exon 12) and BCR::ABL1 has been reported mainly as isolated case descriptions or small case series (4–53). In 2016, Martin-Cabrera et al. reported the incidence of 0.2% of BCR::ABL1/JAK2 V617F double-positive cases in a large cohort of 10.875 MPN cases (54). In 2018, Soderquist et al. estimated a frequency of 0.4% among 1570 MPNs tested for both genomic alterations (39). The previously reported higher frequency of 2.5% by Pieri et al. might be overestimated and likely related to the different cohorts selected in these studies as Pieri et al. evaluated only BCR::ABL1-positive cases, whereas Martin-Cabrera et al. and Soderquist et al. selected all MPN cases (21, 39, 54). Rare cases with coexistence of CALR and BCR::ABL1 have been reported, but their discussion goes beyond the scope of the present paper (55, 56).
In our systematic review, we selected cases with clinically relevant dual driver mutations representing co-existent Ph-positive CML and Ph-negative MPN with JAK2 mutation (4–53). It is well-known that very low level of BCR::ABL1 transcripts may be found in some healthy individuals without clinical features of CML (57). Some Ph-negative MPNs, particularly ET, have been detected to have very low level of BCR::ABL1 transcripts of no clinical and pathologic significance (38) and likewise, the presence of JAK2 mutation, usually at low allelic burden, in the context of a CML may not change the clinical features of CML (58, 59). These situations likely represent examples of clonal hematopoiesis of indeterminate potential (CHIP) (38, 59).
In our systematic review, the majority of BCR::ABL1/JAK2 V617F double-positive cases fell into the group of Ph-negative MPN preceding CML (49.42%), followed by the group of CML and Ph-negative MPN occurring simultaneously (27.58%) and by the group of CML preceding Ph-negative MPN (22.98%). When Ph-negative MPN preceded CML, PV was the most commonly involved disease (20/43; 46.51%), followed by ET (12/43; 27.90%) and PMF (8/43; 18.60%). On the other hand, when CML preceded Ph-negative MPN, the most frequently detected was MPN, NOS (7/20; 35%) followed by PMF (6/20; 30%), PV (5/20; 25%) and ET (2/20; 10%). When Ph-negative MPN and CML occurred simultaneously, PMF was the most frequent disease (10/24; 41.66%) followed by ET (6/24; 25%) and by MPN, NOS (3/24; 12.5%) and PV (3/24; 12.5%) (see Table 1). Of note, MPNs preceding CML are more often non-fibrotic forms (PV, ET), whereas when CML is diagnosed before MPN, fibrosis is observed in most MPN cases at the time of diagnosis. It might be speculated that the onset of a MPN in the context of a CML under TKIs may be masked by the myelosuppressive effect of TKIs and, hence, only the fibrotic evolution gives sufficient force for the clinical and histological emergence of the MPN clone. On the other hand, the different distribution of MPNs may represent a real biological difference. More studies on large number of cases are needed for the understanding of this issue.
Hematologists and pathologists may face this confounding combination of genomic alterations, which can lead to clinical manifestations easily misinterpreted either as disease progression or resistance to treatment, hence, causing an inadequate management of the patient.
A modification in symptoms or laboratory data, during the course of Ph-negative MPNs, is commonly suspected to represent a sign of disease progression as Ph-negative MPNs may undergo myelofibrotic progression or transformation to acute leukemia or, more rarely, develop unusual types of progression with the occurrence of either monocytosis with the acquisition of (myelodysplastic/myeloproliferative) MDS/MPN-like features or leukocytosis with the acquisition of chronic neutrophilic leukemia–like features (1–3, 60). Likewise, a change in clinical manifestations in patients affected by CML is often interpreted as drug-resistance or a possible evolution to acute leukemia (1–3). However, when facing a change in the clinical course of either CML or Ph-negative MPN, hematologists and pathologists need to exclude even the possibility of a combination of BCR::ABL1 translocation and JAK2 mutation.
Clues to the likelihood of the coexistence of CML and Ph-negative MPN may be obtained from laboratory exams, modification in clinical symptoms as well from BM morphology. The appearance of constitutional symptoms, splenomegaly, PTL increase, RBC increase or cytopenia in patients with CML in MR should lead clinicians to rule out a coexisting Ph-negative MPN and mutations of JAK2, MPL, CALR should be evaluated. Likewise, in patients affected by a known Ph-negative MPN, an unexpected WBC or PTL increase or a change in symptoms should alert the clinician to exclude the emergence of CML. BM histology may reveal a shift of phenotype from CML to Ph-negative MPN or vice versa during treatment. It is well known that MK size and clustering may allow the distinction between the different types of MPNs with small-sized, hypo-lobate, non-clustering MKs more commonly found in CML and large, hyper-lobate clustering forms more typical of Ph-negative MPNs. The appearance of large hyper-lobate and clustering MKs with or without fibrosis as well as the identification of MKs with hybrid features (both small-sized, hypo-lobate and large-sized hyper-lobate forms) in the marrow of patients with a known CML should prompt testing for mutated JAK2/CALR/MPL. On the other hand, patients with a known Ph-negative MPN showing BM histological features reminiscent of CML should undergo cytogenetic and molecular testing to identify Ph chromosome and BCR::ABL1 transcripts. Routinely performed BM biopsies during the course of Ph-negative and Ph-positive MPNs are essential to identify a possible shift of phenotype. The role of well-trained hematopathologists in the evaluation of BM biopsies is essential for the correct diagnosis; moreover, a strict integration between pathological features and molecular data is critical in particular in this setting of patients.
The clonal composition of MPNs harboring both BRC::ABL1 and JAK2 V617F has been discussed in various reports, with some studies favoring the presence of two independent clones (8) and others supporting the hypothesis that the two genetic events occur in the same clone (15–17). Despite being rather evident that each of the two theories may be possible, the presence of two independent clones seems to represent the more common event. Interestingly, we observed an opposite growth of BCR::ABL1 and JAK2 under TKI treatment, with BCR::ABL1 decrease and JAK2 increase in the majority of patients of our systematic review (81.8% in concomitant CML and Ph-negative MPN; 65% in CML preceding Ph-negative MPN; 41.86% in MPN preceding CML). The increase in JAK2 mutation burden during Ph-positive clone suppression by TKI treatment favors the hypothesis of two independent clones. Unfortunately, data on outcome in this subset of patients carrying both BCR::ABL1 and JAK2 are often incomplete or unavailable; therefore, it is rather difficult to draw conclusions on the optimal treatment. Whether a TKI associated with JAK2-inhibitor represents an optimal therapeutic strategy requires further evaluation. In those patients with dual anomalies, CML seems to be rather easy to manage, with often a good response of BCR::ABL1 burden to different types of TKIs. The JAK2-positive MPNs have been treated with various therapies including HU, IFN, anagrelide and JAK2-inhibitors such as ruxolitinib, but the paucity of follow-up data does not allow to understand which is the optimal management strategy. Promising results with allo-SCT have been recently reported (see Table 3, Supplementary Materials), although data on a larger number of cases are needed (46).
Author contributions
MZa: Conceptualization, Methodology, Writing – original draft, Writing – review & editing. AB: Data curation, Methodology, Writing – original draft. FS: Data curation, Methodology, Writing – original draft. GB: Data curation, Methodology, Writing – original draft. VF: Data curation, Methodology, Writing – original draft. MZi: Data curation, Writing – original draft. AP: Data curation, Writing – original draft. GM: Data curation, Writing – original draft. CaC: Data curation, Writing – original draft. CeC: Data curation, Writing – original draft. MC: Data curation, Writing – original draft. PG: Data curation, Writing – original draft. FG: Data curation, Writing – original draft. DT: Data curation, Writing – original draft. PP: Data curation, Writing – original draft. RC: Data curation, Writing – original draft. NK: Data curation, Writing – original draft. LC: Data curation, Writing – original draft. AC: Data curation, Writing – original draft. GF: Formal analysis, Investigation, Writing – original draft. SA: Conceptualization, Investigation, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The study was partially supported by the Italian Ministry of Health- Ricerca Corrente Annual Program 2024. We are grateful to Dr Francesca Sabrina Vinci, Dr Giovanni Mattia and Dr Virginia Dolcini of Grant Office & Research Administration, Azienda USL-IRCCS Reggio Emilia for their support. MZ is grateful to her husband Luigi Pizzuti for the informatic support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1329298/full#supplementary-material
References
1. WHO Classification of Tumours Editorial Board. WHO Classification of Tumours Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: IARC (2017).
2. Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: myeloid and histiocytic/ dendritic neoplasms. Leukemia (2022) 36:1703–19. doi: 10.1038/s41375-022-01613-1
3. Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood (2022) 140:1200–28. doi: 10.1182/blood.2022015850
4. Kramer A, Reiter A, Kruth J, Erben P, Hochhaus A, Muller M, et al. JAK2-V617F mutation in a patient with Philadelphia-chromosome-positive chronic myeloid leukemia. Lancet Oncol (2007) 8:658–60. doi: 10.1016/S1470-2045(07)70206-1
5. Inami M, Inokuchi K, Okabe M, Kosaka F, Mitamura Y, Yamaguchi H. Polycythemia associate with the JAK2V617F mutation emerged during treatment of chronic myelogenous leukemia. Leukemia (2007) 21:1103–4. doi: 10.1038/sj.leu.2404591
6. Mirza I, Frantz C, Clarke G, Voth AJ, Turner R. Transformation of polycythemia vera to chronic myelogenous leukemia. Arch Pathol Lab Med (2007) 131:1719–24. doi: 10.5858/2007-131-1719-TOPVTC
7. Bornhauser M, Mohr B, Oelschlaegel U, Bornhauser P, Jacki S, Ehninger G, et al. Concurrent JAK2 (V617F) mutation and BCR-ABL translocation within committed myeloid progenitors in myelofibrosis. Leukemia (2007) 21:1824–6. doi: 10.1038/sj.leu.2404730
8. Bocchia M, Vannucchi AM, Gozzetti A, Guglielmelli P, Poli G, Crupi R, et al. Insights into JAK2-V617F mutation in CML. Lancet Oncol (2007) 8(10):864–6. doi: 10.1016/S1470-2045(07)70295-4
9. Hussein K, Bock O, Seegers A, Flasshove M, Henneke F, Buesche G, et al. Myelofibrosis evolving during imatinib treatment of a chronic myeloproliferative disease with coexisting BCR-ABL translocation and JAK2V617 mutation. Blood (2007) 109(9):4106–7. doi: 10.1182/blood-2006-12-061135
10. Kim YK, Shin MG, Kim HR, Yang DH, Cho SH, Lee JJ, et al. Simultaneous occurrence of the JAK2V617F mutation and BCR-ABL gene rearrangement in patients with chronic myeloproliferative disorders. Leukemia Res (2008) 32:993–1003. doi: 10.1016/j.leukres.2007.10.018
11. Cambier N, Renneville A, Cazaentre T, Soenen V, Cossement C, Giraudier S, et al. JAK2V617F-positive polycythemia vera and Philadelphia chromosome-positive chronic myeloid leukemia: one patient with two distinct myeloproliferative disorders. Leukemia (2008) 22(7):1454–5. doi: 10.1038/sj.leu.2405088
12. De Mello Chonchon MR, Lima Costa J, Yoshinaga Novaes MM, Dorlhiac-Llacer PE, De Alencar Fischer Chamone D, Bendit I. Simultaneous detection of JAK2V617F mutation and BCR-ABL translocation in a patient with chronic myelogenous leukemia. Int J Hematol (2008) 88(2):243–5. doi: 10.1007/s1285-008-0131-2
13. Jallades L, Hayette S, Tigaud I, Johnston A, Coffier B, Magaud JP, et al. Emergence of therapy-unrelated CML on a background of BCR-ABL-negative JAK2V617F-positive chronic idiopathic myelofibrosis. Leuk Res (2008) 32:1608–10. doi: 10.1016/j.leukres.2008.03.004
14. Pardini S, Fozza C, Contini S, Rimini E, Ottaviani E, Amabile M, et al. A cases of coexistence between JAK2V617F and BCR/ABL. Eur J Haematol (2008) 1(1):75–6. doi: 10.1111/j.1600-0609.2008.01063x
15. Hussein K, Bock O, Theophile K, Seegers A, Arps H, Basten O, et al. Chronic myeloproliferative diseases with concurrent BCR-ABL junction and JAK2V617F mutation. Leukemia (2008) 22:1059–62. doi: 10.1038/sj.leu.2404993
16. Pingali SRK, Mathiason MA, Lovrich SD, Go RS. Emergence of chronic myelogenous leukemia from a background of myeloproliferative disorder: JAK2V617F as a potential risk factor for BCR-ABL translocation. Clin Lymphoma Myeloma (2009) 9(5):E25–9. doi: 10.3816/CLM.2009.n.080
17. Bee PC, Gan GG, Nadarajan VS, Latiff NA, Menaka N. A man with concomitant polycythaemia vera and chronic myeloid leukemia: the dynamics of the two disorders. Int J Hematol (2010) 91:16–139. doi: 10.1007/s12185-009-0471-6
18. Tefferi A, Levitt R, Lasho TL, Knudson RA, Ketterling RP. Postimatinib therapy emergence of a new JAK2V617F clone and subsequent development of overt polycythemia vera in a patient with chronic myelogenous leukaemia. Eur J Haematol (2010) 85(1):86–7. doi: 10.1111/j.1600-0609.2010.01458.x
19. Caocci G, Atzeni S, Orrù N, Littera R, Culurgioni F, Marongiu F, et al. Response to imatinib in a patient with chronic myloid leukemia simultaneously expressing p190 BCR-ABL oncoprotein and JAK2V617F. Leuk Res (2010) 34:e27–9. doi: 10.1016/j.leukres.2009.08.009
20. Veronese L, Tchirkov A, Richard-Pebrel C, Ledoux-Pion A, Fleury J, Cheleteix C, et al. A thrombocytosis occurring in Philadelphia positive CML in molecular response to imatinib can reaveal an underlying JAK2V617 myeloproliferative neoplasm. Leuk Res (2010) 34:e94–6. doi: 10.1016/j.leukres.2009.09.25
21. Pieri L, Spolverini A, Scappini B, Occhini U, Birtolo S, Bosi A, et al. Concomitant occurrence of BCR-ABL and JAK2V617F mutation. Blood (2011) 118(12):3445–6. doi: 10.1182/blood-2011-07-365007
22. Hummel JM, Kletecka MCF, Sanks JK, Chiselite MD, Roulston D, Smith LB. Concomitant BCR-ABL1 translocation and JAK2V61F mutation in three patients with myeloproliferative neoplasms. Diagn Mol Pathol (2012) 21(3):176–83. doi: 10.1097/PDM.0b013e318246975e
23. Xiao X, Zhang Y, Zhang GS, Zheng WL, Xiao L, Liu SF. Coexistence of JAK2V617F mutation and BCR-ABL1 transcript in two Chinese patients with chronic myelogenous leukemia. Acta Haematol (2012) 127:47–9. doi: 10.1159/000331564
24. Inokuchi K, Yamaguchi H, Tamai H, Dan K. Disappearance of both the BCR-ABL1 fusion gene and the JAK2V617F mutation with Dasatinib therapy in a patient with Imatinib-resistant chronic myelogenous leukemia. J Clin Exp Hematopathol (2012) 52(2):145–7. doi: 10.3960/jslrt.52.145
25. Lee YJ, Moon JH, Shin HC, Seo JW, Han SA, Seo SK, et al. Two CML patients who subsequently developed features of essential thrombocythemia with JAK2V617F mutation while in complete cytogenetic remission after treatment with imatinib mesylate. Int J Hematol (2013) 97:804–7. doi: 10.1007/s12185-013-1326-8
26. Pastore F, Schneider S, Christ O, Hiddemann W, Spiekermann K. Impressive thrombocytosis evolving in a patient with a BCR-ABL positive CML in major molecular response during dasatinib treatment unmasks an additional JAK2V617F. Exp Hematol Oncol (2013) 2(1):24. doi: 10.1186/2162-3619-2-24
27. Ursuleac I, Colita AC, Adam T, Jardan C, Coriu D. The concomitant occurrence of JAK2V617F mutation and BCR/ABL transcript with phenotypic expression-an overlapping myeloproliferative disorder or two distinct diseases?-case report. J Med Life (2013) 15(6):34–7.
28. Yamada O, Mahfoudhi E, Plo I, Ozaki K, Nakatake M, Akiyama M, et al. Emergence of a BCR-ABL translocation in a patient with the JAK2 V617F mutation: evidence for secondary a cquisition of BCR-ABL in the JAK2 V617F clone. JCO (2014) 32(21):e76–9. doi: 10.1200/JCO.2012.47.8669
29. Maerki J, Katava G, Siegel D, Silberberg J, Bhattacharyya PK. Unusual case of simultaneous presentation of plasma cell myeloma, chronic mylogenous leukemia and a JAK2 positive myeloproliferative disorder. Case Rep Hematol (2014) 2014:738428. doi: 10.1155/2014/738428
30. Quin YW, Yang Y, Li S, Wang C. Coexistence of JAK2V617F mutation and BCR-ABL translocation in a pregnant woman with essential thrombocythemia. Indian J Hematol Blood Transfus (2014) 30(1):S331–4. doi: 10.1007/s12288-014-0385-1
31. Zhou A, Knoche EM, Engle EK, Fisher DAC, Oh ST. Concomitant JAK2V617F-positive polycythemia vera and BCR-ABL-positive chronic myelogenous leukemia treated with ruxolitinib and dasatinib. Blood Cancer J (2015) 5:e351. doi: 10.1038/bcj.2015.77
32. Chen Z, Wang W, Verstovsek S, Cortes JE, Medeiros LJ, Hu S. Chronic myelogenous leukemia in patients with MPL or JAK2 mutation-positive myeloproliferative neoplasm. Int J Lab Hem (2015) 37:e150–2. doi: 10.1111/ijlh.12398
33. Wang Y, Xia J, Wu X, Hu Y. A patient with BCR-ABL and JAK2 V617F double-positive myeloproliferative neoplasm with overlapping clinical phenotypes. Am J Med Sci (2015) 350(1):68–9. doi: 10.1097/MAJ.0000000000000448
34. Pagnano KB, Delamain MT, Magnus MM, Vassallo J, DE Souza CA, DE Almeida D, et al. Concomitant essential thrombocythemia with JAK2 V617F mutation in a patient with chronic myeloid leukemia with major molecular response with imatinib and long-term follow-up. Oncol Lett (2016) 12(1):485–7. doi: 10.3892/ol.2016.4631
35. Paz DL, Ianotto JC, Chauveau A, Guibourg B, Lelucq L, Lippert E, et al. Expansion of a BCR-ABL clone in a JAK2 V617F myeloproliferative neoplasm treated by ruxolitinib. Ann Hematol (2016) 95:349–50. doi: 10.1007/s00277-015-2518-5
36. Darling HS, Kumar R, Kapoor R, Singh J, Verma T. BCR-ABL and JAK2V617F mutation co-existence, rare or just unexplored. Indian J Hematol Blood Transfus (2017) 33(4):633–5. doi: 10.1007/s12288-0781-4
37. Kandarpa M, Wu YM, Robinson D, Burke PW, Chinnaiyan AM, Talpaz M. Clinical characteristics and whole exome/transcriptome sequencing of coexisting chronic myeloid leukemia and myelofibrosis. AJH (2017) 92:555–61. doi: 10.1002/ajh.24728
38. Boddu P, Chihara D, Masarova L, Pemmaraju N, Patel KP, Verstovsek S. The co-occurrence of driver mutations in chronic myeloproliferative neoplasms. Ann Hematol (2018) 97(11):2071–80. doi: 10.1007/s00277-018-3402x
39. Soderquist CR, Ewalt MD, Czuchlewski DR, Geyer JT, Rogers HJ, His ED. Myeloproliferative neoplasms with concurrent BCR-ABL1 translocation and JAK2V617F mutation: a multi-institutional study from the bone marrow pathology group. Mod Path (2018) 31(5):690–704. doi: 10.1038/modpathol.2017.182
40. Bader G, Dreiling B. Concurrent JAK2-positive myeloproliferative disorder and chronic myelogenous leukemia: a novel entity? A case report with review of the literature. J Invest Med High (2019) 7:2324709619832322. doi: 10.1177/2324709619832322
41. Swaminathan M, Patel KP, Huynh-Lu J, Tang G, Zuo Z, Miranda R, et al. Unique case of myeloproliferative neoplasm with two rare clonal abnormalities: rare JAK2 Exon12 mutation and rare e14a3 (b3a3) BCR-ABL fusion transcript. Acta Haematol (2019) 141:23–7. doi: 10.1159/000494427
42. Tirrò E, Stella S, Massimino M, Zammit V, Pennisi MS, Vitale SR, et al. Colony-forming cell assay detecting the co-expression of JAK2V617F and BCR-ABL1 in the same clone: a case report. Acta Haematol (2019) 141:261–7. doi: 10.1159/000496821
43. Lorenzo M, Grille S, Stevenazzi M. Emergence of BCR-ABL1 chronic myeloid leukemia in a JAK2-V617F polycythemia vera. J Hematol (2020) 9(1-2):23–9. doi: 10.14740/jh591
44. Yue Y, Wei W, Guo Y, Wang F, Dong W, Liu Y, et al. Successful treatment of a patient with chronic myelogenous leukemia with concurrent Janus Kinase 2 (JAK2) R79rS mutation and breakpoint cluster region-ABL1 (BCR-ABL1) fusion: a case report and literature review. Am J Case Rep (2020) 21:e925151. doi: 10.12659/AJCR.925151
45. Zhao Y, McCracken J, Wang E. Sequential development of JAK2V617F and BCR-ABL1: a “double-hit” myeloproliferative neoplasm with morphologic transition. J Hematopath (2021) 14:251–3. doi: 10.1007/s12308-021-00458-4
46. Sorà F, Chiusolo P, Autore F, Giammarco S, Laurenti L, Innocenti I, et al. Is allogenic transplantation an option in patients affected by concurrent myelofibrosis and chronic myeloid leukemia (CML)? Mediterr J Hematol Infect Dis (2021) 13(1):e2021062. doi: 10.4084/MJHID.2021.062
47. De Bruyne S, Steel E, Petrick M, Denys B, Vandepoele K, Van Roy N, et al. Development of chronic myeloid leukemia in a patient previously diagnosed with a JAK2-positive myeloproliferative neoplasm. Clin Chem Lab Med (2021) 59(10):e392–4.
48. Zhao Y, Reddi D, McCracken J, Iranzad N, Rehder C, Neff J, et al. Sequential development of JAK2V617F mutation and BCR-ABL1 fusion in individual patients with myeloproliferative neoplasms. A linear clonal evolution or parallel clonal competition? Arch Pathol Lab Med (2022) 146:710–7. doi: 10.5858/arpa.2021-0096-OA
49. Zhang Y, Bi H, Wang Y, Chen L, Pan J, Xu P, et al. BCR-ABL1 is a secondary event after JAK2V617F in a patient with essential thrombocythemia who develop chronic myeloid leukemia. Blood Sci (2022) 4:199–204. doi: 10.1097/BS90000000000000129
50. Ryu J, Chu D, Park B, Kim M, Cho YU, Hwang SH, et al. Rare case of accelerated-phase chronic myeloid leukemia diagnosed during treatment for JAK2 V617F-positive primary myelofibrosis. Lab Med (2022) 53:e140–4. doi: 10.1093/labmed/Imac011
51. Hochman MJ, Smith BD, Karantanos T, Branstein EM, Gojo I, Jain T, et al. Chronic myeloid leukemia (CML) evolves from Philadelphia chromosome-negative myeloproliferative neoplasms (MPNs) with unexpected frequency. Int J Hematol (2023) 117:456–62. doi: 10.1007/s12185-022-03463-0
52. Tosoni L, Pizzano U, Mullai R, Morelli G, Franzoni A, Damante G, et al. JAK2 V617F-mutated polycythemia vera developing in a patient with a 20-year-long chronic myeloid leukemia at the time of first molecular response. Ann Hematol (2023) 102:1279–80. doi: 10.1007/s00277-023-05187-5
53. Lapietra G, Limongi MZ, Buffolino S, Nanni M, Ballarò D, Martelli M, et al. Late appearance of JAK2 mutated polycythemia vera in a patient with typical chronic myeloid leukemia on imatinib: speculations about role of therapeutic pressure and of secondary genetic events. Leuk Res (2023) 26:107035. doi: 10.1016/j.leukres.2023.107035
54. Martin-Cabrera P, Haferlach C, Kern W, Schnitter S, Haferlach T. BCR-ABL1-positive and JAK2 V617F-positive clone in 23 patients with both aberrations reveal biologic and clinical importance. BJH (2017) 176:131–43. doi: 10.1111/bjh.13932
55. Yoon SY, Jeong SY, Kim C, Lee MY, Kim J, Kim KH, et al. Philadelphia+ chronic myeloid leukemia with CALR mutation: a case report and literature review. Cancer Res Treat (2020) 52(3):987–91. doi: 10.4143/crt.2019.703
56. Guidotti F, Gardellini A, Feltri M, Zancanella M, Saccà V, Alberio F, et al. Concurrent chronic myeloid leukemia and CALR-mutated chronic myeloproliferative neoplasm. Blood Cells molecules Dis (2020) 81:102395. doi: 10.1016/j.bemd.2019.102395
57. Biernaux C, Loos M, Sels A, Huez G, Stryckman P. Detection of major bcr-abl gene expression at a very low level in blood cells of some healthy individuals. Blood (1995) 86(8):3118–22.
58. Campiotti L, Appio L, Solbiati F, Ageno W, Venco A. JAK2-V617F mutation and Philadelphia positive chronic myeloid leukemia. Leuk Res (2009) 33:e212–3. doi: 10.1016/j.leukres.2009.06.011
59. Tarantini F, Cumbo C, Zagaria A, Parciante E, Anelli L, Coccaro N, et al. JAK2 V617F in chronic myeloid leukemia: driving force or passive bystander? Hematology (2022) 27(1):842–6. doi: 10.1080/16078454.2022.2108902
Keywords: BCR::ABL1, JAK2, chronic myeloid leukemia, myeloproliferative neoplasm, polycythemia vera, primary myelofibrosis, essential thrombocythemia
Citation: Zanelli M, Bisagni A, Sanguedolce F, Broggi G, Fragliasso V, Zizzo M, Palicelli A, Martino G, Cresta C, Caprera C, Corsi M, Gentile P, Gozzi F, Trombetta D, Parente P, Caltabiano R, Koufopoulos N, Cimino L, Cavazza A, Fraternali Orcioni G and Ascani S (2024) Co-occurrence of JAK2-V617 F mutation and BCR::ABL1 translocation in chronic myeloproliferative neoplasms: a potentially confounding genetic combination. Front. Oncol. 13:1329298. doi: 10.3389/fonc.2023.1329298
Received: 28 October 2023; Accepted: 22 December 2023;
Published: 12 January 2024.
Edited by:
Giuseppe Gaetano Loscocco, University of Florence, ItalyReviewed by:
Massimiliano Bonifacio, University of Verona, ItalySusanne Saussele, University of Heidelberg, Germany
Copyright © 2024 Zanelli, Bisagni, Sanguedolce, Broggi, Fragliasso, Zizzo, Palicelli, Martino, Cresta, Caprera, Corsi, Gentile, Gozzi, Trombetta, Parente, Caltabiano, Koufopoulos, Cimino, Cavazza, Fraternali Orcioni and Ascani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Magda Zanelli, magda.zanelli@ausl.re.it