 Viraj Muthye1,2
Viraj Muthye1,2 James D. Wasmuth1,2*
James D. Wasmuth1,2*- 1Faculty of Veterinary Medicine, University of Calgary, Calgary, AB, Canada
- 2Host-Parasite Interactions Research Training Network, University of Calgary, Calgary, AB, Canada
Introduction: Molecular mimicry is a strategy used by parasites to evade the host’s immune system and facilitate transmission to a new host. To date, high-throughput examples of molecular mimicry have been limited to comparing protein sequences. However, recent advances in the prediction of tertiary structural models, led by Deepmind’s AlphaFold, enable the comparison of thousands of proteins from parasites and their hosts at the structural level, allowing for the identification of more mimics. Here, we present the first proteome-level search for tertiary structure similarity between proteins from Plasmodium falciparum, a malaria-causing parasite, and humans.
Methods: We assembled a database of experimentally-characterized protein tertiary structures (from the Protein Data Bank) and AlphaFold-generated protein tertiary structures from P. falciparum, human, and 15 negative control species, i.e., species not infected by P. falciparum. We aligned human and control structures to the parasite structures using Foldseek.
Results: We identified molecular mimicry in three proteins that have been previously proposed as mediators of Plasmodium-human interactions. By extending this approach to all P. falciparum proteins, we identified an additional 41 potential mimics that are supported by additional experimental data.
Discussion: Our findings demonstrate a valuable application of AlphaFold-derived tertiary structural models, and we discuss key considerations for its effective use in other host-parasite systems.
1 Introduction
Parasites encounter host defenses at various points in their life cycle and employ a wide range of strategies for evading their host’s immune response and successfully transmitting to a new host (Chulanetra and Chaicumpa, 2021). These host-parasite interactions may be mediated by parasite-derived molecules—including proteins, lipids, sugars—that unexpectedly resemble host-derived molecules. This is termed ‘molecular mimicry’, which was originally defined as the sharing of antigens between parasite and host (Damian, 1964). One of the earliest reports of molecular mimicry came from the parasitic nematode Ascaris lumbricoides, which possesses A- and B-like blood group antigens in its polysaccharides (Oliver-González, 1944). The definition of molecular mimicry has adapted to keep up with molecular and genomic technologies and is now widely considered to similarity between proteins at the level of primary structure (amino acid sequence) and tertiary structure [summarized in (Tayal et al., 2022)]. An assumption is that molecular mimicry confers a fitness benefit to the pathogen. However, the term molecular mimicry is also used to explain the cross-reactivity between exogenous and self-peptides and is the theoretical framework for understanding autoimmunity (Getts et al., 2013). Related to both these definitions, molecular mimicry might also result in heterologous immunity, in which the infection from one parasite protects against infection by other parasites with similar antigenic molecules (Balbin et al., 2023).
Here, our focus is on molecular mimicry that confers a fitness advantage to the parasite, by either co-opting or disrupting the function of the mimicked host protein. Examples of molecular mimicry come from most branches of life. For instance, pathogenic bacterium Escherichia coli injects the TccP protein into host cells, which targets the polymerization of host actin. TccP contains multiple repeated motifs that mimic an internal regulatory element present in host N-WASP (neural Wiskott–Aldrich syndrome protein), which results in the activation of N-WASP (Sallee et al., 2008). This promotion of actin polymerization results in the creation of structures on epithelial cells that promote pathogen survival in the intestine. In another example, the myxoma virus decreases the number of activated macrophages by expressing its M128L protein on the host cell surface (Cameron et al., 2005). M128L shares significant sequence similarity with host CD47 and competes with it to bind with its receptor SIRPα. Within eukaryotic pathogens, the apicomplexan Babesia microti expresses the BmP53 protein which contains a domain that resembles thrombospondin (TSP1), a component of platelet cells (Mousa et al., 2017). The BmP53 TSP-1 is immunologically cross-reactive with human, and it is proposed that BmP3 helps cloak the extra-cellular stages from the immune system.
To the best of our knowledge, the first study to identify host-parasite molecular mimicry at a genome-scale across multiple species was by Ludin and colleagues (Ludin et al., 2011). They considered the protein sequences from eight species of eukaryotic parasites, the host (human), and seven non-pathogenic, eukaryotic, negative control species. Their approach identified multiple potential instances of mimicry in these parasites. For example, they detected a 14-amino acid motif in multiple PfEMP1 proteins in Plasmodium falciparum that was identical to the heparin-binding domain in human vitronectin, a protein with multiple roles in human including cell-adhesion. The approach was repeated to find ninety-four potential mimicry proteins in a tapeworm-fish system (Hebert et al., 2015). It was also adapted and expanded for use with 62 pathogenic bacteria and identified approximately 100 potential mimics (Doxey and McConkey, 2013). These approaches rely on two proteins sharing enough sequence similarity to be detected by the sequence alignment software, e.g., BLAST (Altschul et al., 1990). However, these approaches depend on two proteins sharing a reasonably high level of local sequence similarity. For instance, several viruses express proteins with tertiary structure similarity, but undetectable sequence similarity, to human Bcl-2, and interfere with regulation of apoptosis (Kvansakul et al., 2007; Westphal et al., 2007). Similarly, in Plasmodium falciparum, a search of parasite proteins targeted to host extracellular vesicles revealed that at least eight shared unexpected and significant tertiary structure similarity with host proteins (Armijos-Jaramillo et al., 2021).
The opportunity to detect host-parasite mimicry at the level of tertiary structure has been limited by the number of available tertiary protein structures. Even for a parasite as important as P. falciparum, the protein databank (PDB) contains structures from less than 4% of the protein-coding genes in its genome (Table 1). We expect that most, if not all, other bacterial and eukaryotic pathogen species will have worse coverage. Proteome-wide searches for host-parasite molecular mimicry at the level of tertiary structure depended on in silico predictions that were of inconsistent quality (Armijos-Jaramillo et al., 2021). The prediction of tertiary protein structures from amino acid sequences has seen a much-publicized boon, in no small part to the development of AlphaFold (Ronneberger et al., 2021). In an early large-scale application, the AlphaFold Protein Structure Database (AFdb) provided tertiary structure predictions for 16 model organisms and 32 pathogen species of global health concern (https://alphafold.com). Complementing the release of AlphaFold was Foldseek, a novel approach to aligning tertiary protein structures (van Kempen et al., 2023). Comparisons showed that Foldseek was nearly 20,000 times faster than existing protein structure aligners while maintaining accuracy [but see (Holm, 2022)]. These two major advances in structural bioinformatics—AlphaFold and Foldseek—have empowered us to investigate the usefulness of using tertiary protein structures for identifying instances of host-parasite molecular mimicry.
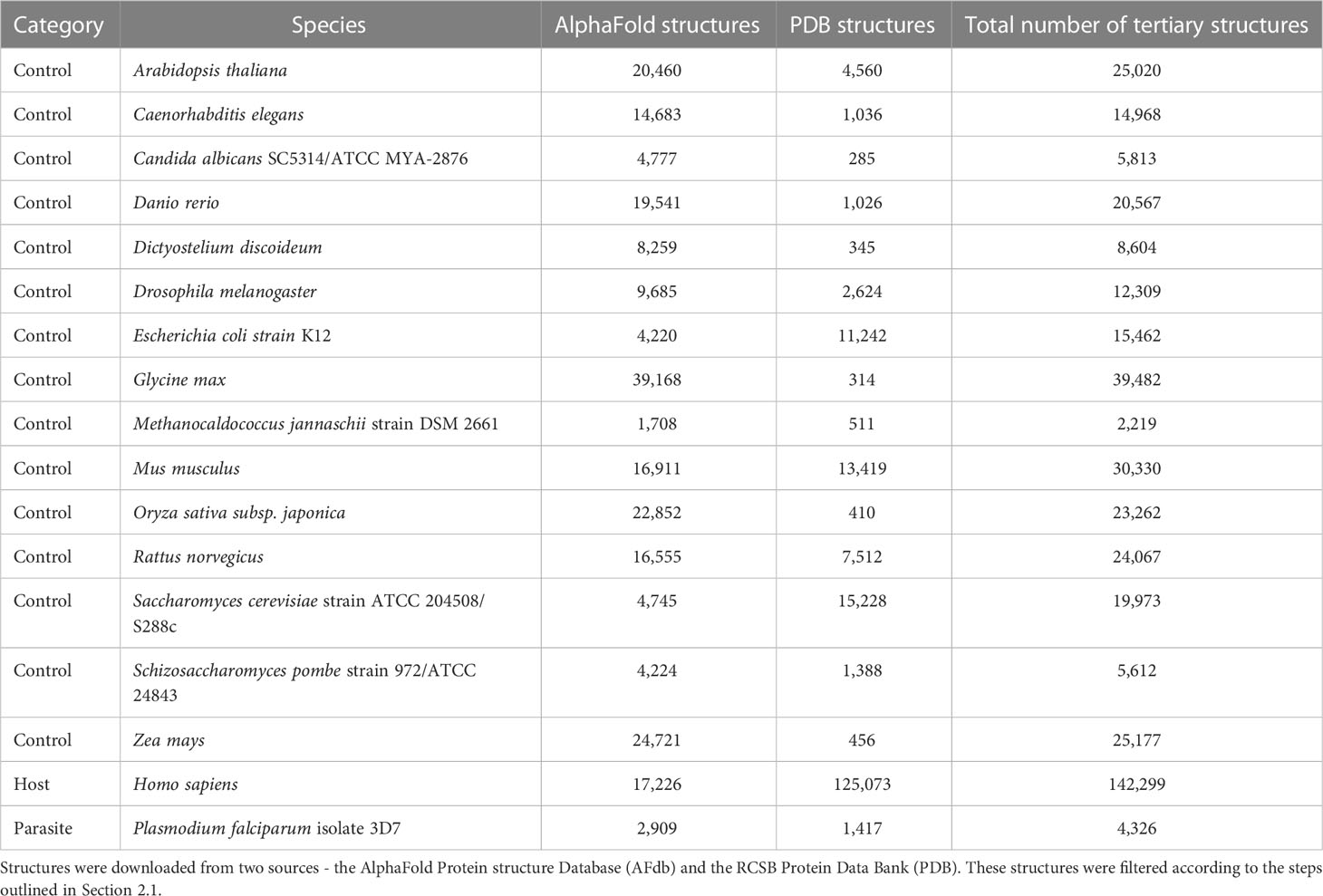
Table 1 The number of tertiary protein structures for each species used in this study.
Our parasitic species of study is Plasmodium falciparum. While our understanding of the mediators of host-parasite interactions is limited, as the leading cause of severe malaria in humans, P. falciparum might represent the current pinnacle of our knowledge. Furthermore, the protein tertiary structures are complemented with a broad range of curated -omics datasets available on PlasmoDB, which can help with candidate prioritization (Aurrecoechea et al., 2008; Amos et al., 2022). Proteins expressed by P. falciparum mediate interactions with its human host at multiple stages in its life cycle (Acharya et al., 2017). Molecular mimicry plays a role at both the liver and blood stages for immune evasion and cytoadherence. For instance, RIFIN, a prominent erythrocyte surface protein expressed by P. falciparum, binds to human LILRB1 which inhibits stimulation of the immune response. RIFIN does this by mimicking MHC Class I, the activating ligand of LILRBA (Harrison et al., 2020). Meanwhile, the circumsporozoite protein (CSP), which promotes invasion of human liver cells, has an 18 amino acid region that is similar to a cytoadhesive region in mammalian thrombospondin (Robson et al., 1988; Cerami et al., 1992).
In this study, our goal was to identify P. falciparum proteins which share tertiary structure similarity with human proteins but not detectable sequence similarity. First, we examined P. falciparum proteins which are known or have been implicated to directly interact with human biomolecules. We found new potential instances of molecular mimicry. Second, we extended our approach to consider all P. falciparum proteins and leveraged experimental datasets to filter the candidate mimics. Overall, our study highlights the advantages of using tertiary protein structures for identifying instances of molecular mimicry.
2 Methods and materials
2.1 Compiling the datasets of tertiary protein structures
We downloaded tertiary protein structures for Plasmodium falciparum 3D7 (parasite), human (host), and 15 negative control species, i.e., species that are not infected by P. falciparum (Tables 1, 2; File S1). Protein structures for these species were downloaded from two sources: 1) the RCSB Protein Data Bank (PDB) (experimentally determined protein structures, last accessed 06/16/2022), and 2) the AlphaFold Protein Structure Database (AFdb) (computationally predicted protein structures, https://alphafold.ebi.ac.uk/). The structures downloaded from both sources were processed before analysis (explained below).
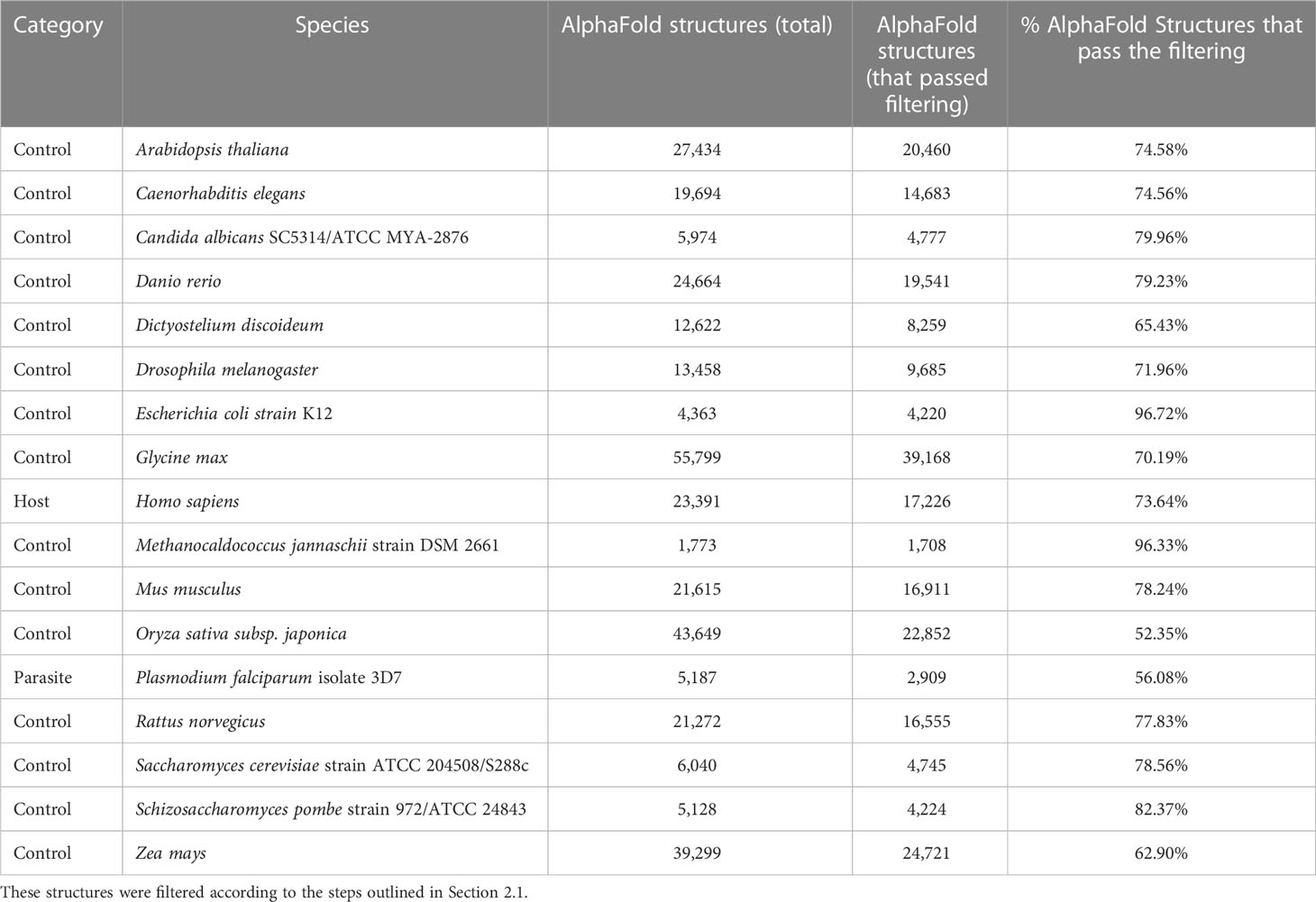
Table 2 The number of tertiary protein structures from the AlphaFold Protein structure Database (AFdb) for each species used in this study.
2.1.1 Processing the PDB structures
Several PDB structures were composed of chains from multiple source organisms. We separated such structures into individual chains and extracted the appropriate chains corresponding to each species. For instance, the PDB structure 7F9N is composed of four chains (A to D), of which two chains (A and B) are from P falciparum Rifin (RIF, PF3D7_1000500) and two chains (C and D) are from the human leukocyte-associated immunoglobulin-like receptor 1 protein (LAIR1, Q6GTX8) (Figure 1A). We included only chains A and B for P. falciparum. Additionally, multiple structure chains were chimeric or ambiguous, i.e., mapping to multiple source organisms. For instance, the PDB structure 4O2X has two chains (A and B) which map to both P. falciparum and Escherichia coli strain K12. Such chains were discarded to not confound downstream analysis.
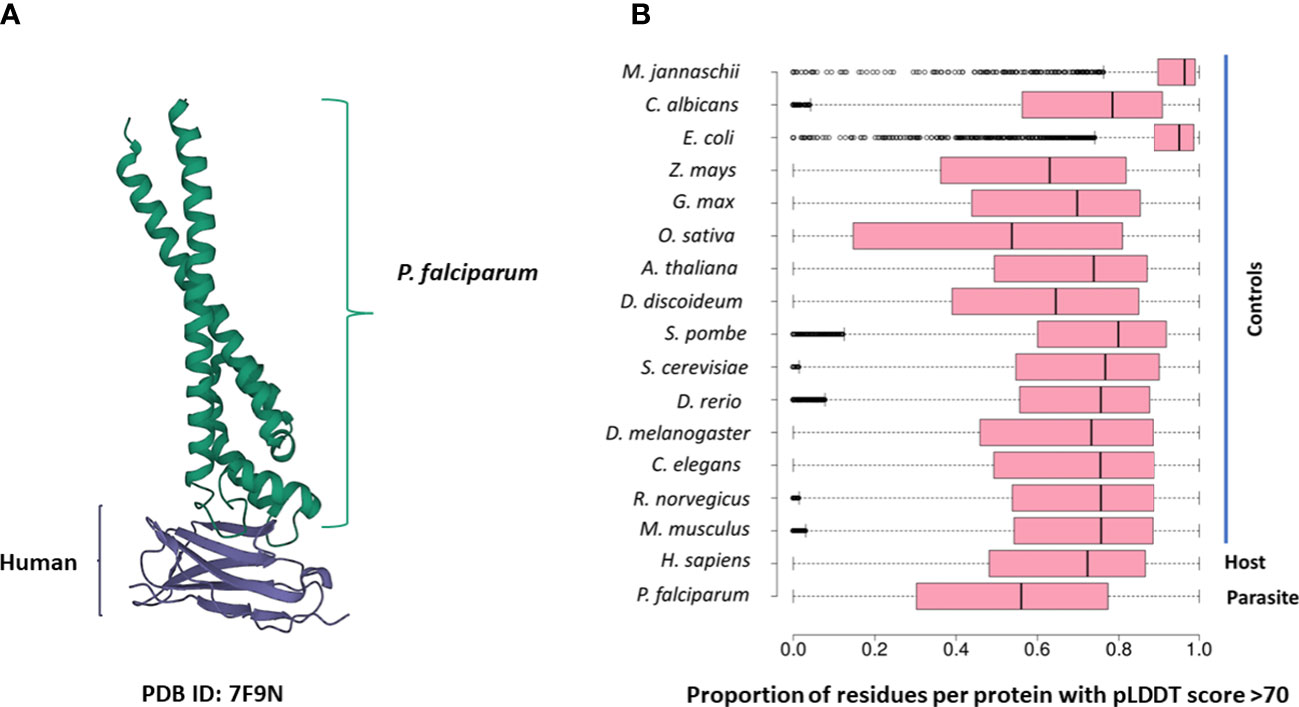
Figure 1 (A) The 7F9N protein tertiary structure from the RCSB Protein Data Bank. The chains from the P. falciparum RIFIN protein (RIF, PF3D7_1000500) are colored green and the chains from the human LAIR protein (LAIR1, Q6GTX8) are colored in purple. (B) Box-plot representing the overall prediction accuracy for each protein in the AFdb. Each point in this distribution represents the proportion of all residues with a pLDDT score above 70 for one AlphaFold structure. The median value is the parallel bar within the box. The limits of the box are the 25th and 75th percentiles, the whiskers extend 1.5 times the interquartile range, and the dots are the outliers.
2.1.2 Processing the AlphaFold structures
AlphaFold assigns a score to each residue in the predicted structure called the ‘pLDDT’ score. This score is a measure of prediction confidence for that residue. The pLDDT scores range from 0 (low confidence of prediction) to 100 (high confidence of prediction). Regions with a pLDDT score above 90 are modelled with high accuracy, between 70-90 are modelled well, and 50-70 are modelled with low confidence. The AlphaFold database suggests that regions with pLDDT scores less than 50 should not be interpreted as this low score could be indicative of intrinsic protein disorder. For each AlphaFold structure, we calculated the proportion of the total residues with a pLDDT score of more than 70. We retained predicted structures with at least half the residues of the structure modelled with a pLDDT score above 70 (Figure 1B; File S1).
2.2 Identification of Plasmodium falciparum proteins known to interact with human molecules
We performed a literature survey to identify P. falciparum proteins that are known to interact with human molecules. We started with a review of P. falciparum-human protein interactions (Acharya et al., 2017). Then, we identified all abstracts on PubMed using the query [‘plasmodium falciparum’ AND ‘interact*’ AND ‘protein*’ AND ‘human*’] from 2017 to 2022. This resulted in 648 abstracts (as of 8/8/2022 1:36 PM). We read all 648 abstracts to identify the P. falciparum proteins of interest. The PlasmoDB ID for each protein was mapped to Uniprot IDs using PlasmoDB (Release 52, 30 August 2022). Three large gene families (PfEMP1, RIFIN, and STEVOR) play an important role in host-parasite interactions and pathogenesis in P. falciparum. Proteins belonging to these three families were downloaded from PlasmoDB.
2.3 Identification of sequence and structure similarity between Plasmodium falciparum and human proteins
2.3.1 Analysis of protein sequence similarity
We analyzed sequence similarity between the proteins from P. falciparum, human, and 15 negative control species. We determined sequence similarity using three pairwise alignment search tools. SSEARCH36 implements the Smith-Waterman algorithm guaranteeing the optimal alignment. We used the following parameters for SSEARCH36 from Fasta36 ‘m 8 -s BL62 -f 12 -g 1’. BLASTP (Altschul et al., 1990) and DIAMOND (Buchfink et al., 2021) implement heuristic algorithms that are faster than SSEARCH36 but do not guarantee the optimal alignment. We used BLASTP with an e-value cut-off of 1e-3 and performed an ultra-sensitive DIAMOND BLASTP search with the same e-value cut-off. For both aligners, we searched using the BLOSUM45 and BLOSUM62 substitution matrices. All other parameters were left as default. Additionally, we used OrthoFinder version 2.5.4 to identify groups of orthologous proteins between all the 17 species used in this study, using DIAMOND as the aligner (Emms and Kelly, 2019). The results of this analysis were used to identify human proteins that had orthologs in only the other three vertebrates used in this study (mouse, rat, and zebrafish).
2.3.2 Analysis of protein structure similarity
We aligned all P. falciparum structures to a database consisting of human structures and control structures. The structural aligner used was Foldseek v4 (easy-search -s 9.5 –max-seqs 1000). We used an e-value cut-off of 0.01. Foldseek was also used to visualize structural alignments using the option ‘format-mode 3’. Individual structures were visualized using EzMol (Reynolds et al., 2018). Additionally, we used Foldseek to calculate the TM-score of the aligned regions (–format-output ‘query, target, alntmscore’). While a structure might have more than half its residues with a pLDDT score of at least 70, it is possible that a region of poorly modelled residues in such structures could be aligned to another structure. Thus, we calculated the average pLDDT score of the aligned residues as well as the proportion of the aligned residues with pLDDT of at least 70. This was done to ensure that the regions of the predicted structures in the alignments were modelled well.
2.3.3 Expression analysis
Expression analysis of P. falciparum proteins was carried out using the publicly available RNA-seq datasets available in PlasmoDB. We identified all P. falciparum proteins with expression in the 90th percentile in at least one of the stages in the intra-erythrocytic life cycle (young ring 8 hpi, late ring/early trophozoite 16 hpi, mid trophozoite 24 hpi, late trophozoite 32 hpi, early schizont 40 hpi, schizont 44 hpi, late schizont 48 hpi, and purified merozoites 0 hpi) using data from (Wichers et al., 2019). We also identified all P. falciparum proteins with expression in the 90th percentile in the ring and/or sporozoite stage using data from (Zanghì et al., 2018).
2.3.4 Analysis of protein subcellular localization
To identify P. falciparum proteins carried in extracellular vesicles (EV), we applied an approach used in (Armijos-Jaramillo et al., 2021). In this study, we define a parasite EV protein as one identified in at least one of the three studies in the abovementioned study (Mantel et al., 2013; Abdi et al., 2017; Sampaio et al., 2018). We extracted information on subcellular localization of P. falciparum and human proteins from Uniprot and PlasmoDB.
3 Results
3.1 Assembling and filtering the datasets of crystallized and computationally predicted tertiary protein structures
We compiled tertiary protein structures from Plasmodium falciparum 3D7 (parasite), human (host), and 15 negative control species, those not infected by the parasite (Tables 1, 2; File S1). These structures were downloaded from the RCSB Protein Data Bank (PDB) and the AlphaFold Protein Structure database (AFdb). All AlphaFold-generated structures were filtered using the pLDDT score, a per-residue metric of the confidence of prediction accuracy. Following the AlphaFold documentation, we considered structures to be high confidence if at least half their residues had a pLDDT score above 70. Through this filtering, we retained 56% of P. falciparum structures, 74% of human structures, and between 97% (E. coli) and 52% (Oryza sativa) for the control species (Tables 1, 2 and Figure 1B; File S1).
3.2 Investigating the effect of the source of tertiary structures on Foldseek alignments
We wanted to determine whether the source of the tertiary structure—crystallised (PDB) or computationally-predicted (AlphaFold)—affected the Foldseek search results. Following our filtering steps, 167 P. falciparum proteins were represented by structures from both PDB and AlphaFold and 159 aligned to at least one structure from the host and/or negative control species. For each of these 159 proteins, we compared the Foldseek results for their PDB and AlphaFold structures. For most of these proteins (107/159), the results for both PDB and AlphaFold queries agreed between 90 and 100%. For almost 10% of these proteins (14/159), the agreement between the results was less than 50% (File S2; Figure S1).
3.3 Structural analysis of parasite proteins experimentally known to interact with human proteins
We performed a literature review and identified 74 P. falciparum proteins that interact with human molecules at various stages in the parasite’s life cycle (File S3). We also included three large P. falciparum gene families which are thought to play a role in parasite virulence—PfEMP1 (61 proteins), RIFIN (158 proteins), and STEVOR (32 proteins) (File S3). Overall, 206 of these proteins were represented by at least one structure in our database of PDB and high-quality AlphaFold structures. To understand whether molecular mimicry plays a role in how these proteins interact with the host, we asked the question: do these 206 P. falciparum proteins share sequence and/or tertiary structural similarity with human proteins?
Of the 206 proteins, 171 did not have sequence or structure similarity to any human protein. We found that 31 of the remaining 35 P. falciparum proteins shared structural similarity to at least one human protein. Of these, three proteins aligned to human proteins which were restricted in vertebrates at the sequence-level (orthologs only in mouse, rat, and/or zebrafish). They were the parasite circumsporozoite protein (CSP, PF3D7_0304600) and two PfEMP1 proteins (PF3D7_0800100 and PF3D7_0617400). CSP was aligned to human thrombospondin (TSP1, P07996) (alignment TM-score greater than 0.6 for all alignments). Interestingly, as per previous sequence-based approaches, CSP mimics a cytoadhesive region in mammalian thrombospondin (Robson et al., 1988; Cerami et al., 1992).
From these 31 P. falciparum proteins which share structural similarity to at least one human protein, we removed all proteins which shared sequence similarity with human proteins – 18, 16, and 17 proteins that aligned to human proteins by BLASTP, DIAMOND, and SSEARCH36 respectively (Figure 2A). Interestingly, over one-third of these 31 P. falciparum proteins (11/31) did not have detectable sequence similarity to any human protein (Figure 2A). For these 11 proteins, we visually inspected their structural alignments with human proteins (File S4). The top human alignments for seven of these 11 proteins had a low TM-score (<0.2) and were not analysed further (File S4). Here, we present the biological relevance of their interactions for three of the remaining four proteins.
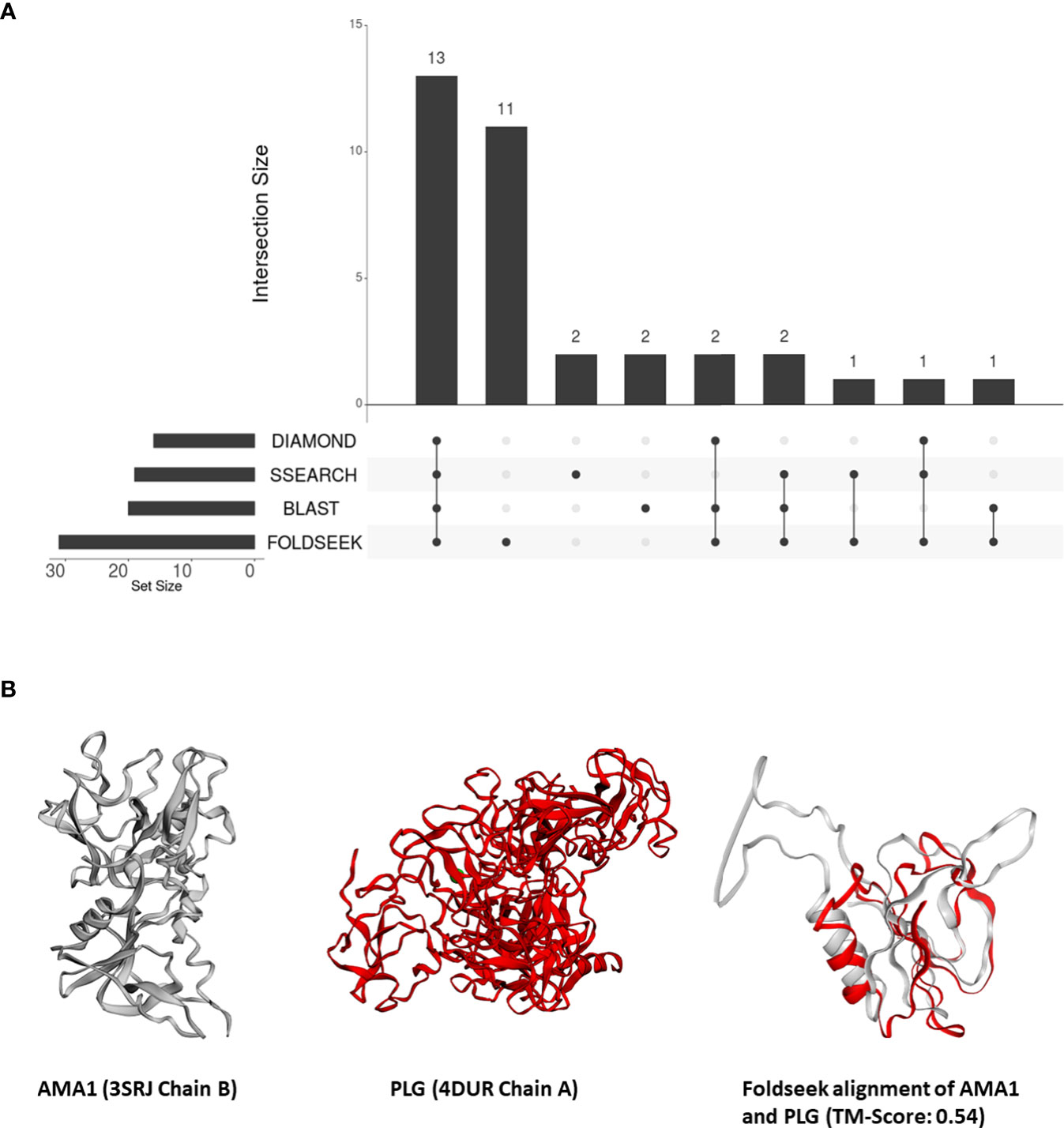
Figure 2 (A) An UpSet plot of the 206 parasite proteins that interact with human molecules and are represented by at least one structure in our database. The R package ‘UpSetR’ was used to generate the UpSet plot (Conway et al., 2017). This plot displays the number of these interacting parasite proteins aligned to human proteins by DIAMOND, BLAST, Foldseek, and/or SSEARCH36. (B) Tertiary structure alignments of the parasite AMA1 structure in grey (3SRJ chain B), the human PLG structure in red (4DUR chain A), and the region aligned by Foldseek.
3.3.1 P38
The 6-cysteine protein/merozoite surface protein P38 (P38, PF3D7_0508000) functions in red-blood cell invasion and binds to human glycophorin-A (GLPA, P02724) (Paul et al., 2017). P38 belongs to the 6-cysteine protein family and is expressed on the surface of the blood stages of the P. falciparum life cycle (Feller et al., 2013; Lyons et al., 2022). We found that P38 was structurally similar to a region containing an immunoglobulin domain in human titin (TTN, Q8WZ42). Specifically, the AlphaFold prediction for P. falciparum P38 was aligned to the human PDB structure 3LCY (‘Titin Ig tandem domains A164-A165’). The next match was the human receptor-type tyrosine-protein phosphatase F (PTPRF, P10586); P38 was structurally similar to a region containing two Ig-like domains. Ig-like domains are present in several adhesion proteins in human, where they play an important role in homophilic cell adhesion (Leshchyns’ka & Sytnyk, 2016). This suggests that P38, in addition to binding to GLPA, functions in adhesion owing to its similarity to the Ig-like domains.
3.3.2 AMA1
The apical membrane antigen (AMA1, PF3D7_1133400) is a potential vaccine target (Remarque et al., 2008). Previous studies have suggested that the interaction between AMA1 and the P. falciparum rhoptry neck protein (RON) is essential for merozoite invasion (Srinivasan et al., 2011). AMA1 also interacts with the human erythrocyte membrane transport protein Kx protein (Kato et al., 2005). However, a later study demonstrated that AMA1 is involved in the attachment of the merozoite to the host cell, but is dispensable for cell invasion (Bargieri et al., 2013).
In our analysis, AMA1 was aligned to the PAN protein domain in human plasminogen (PLG, P00747) (top human Foldseek alignment based on bit-score) (Figure 2B). While this similarity could not be detected by sequence-similarity searches, domains I and II of P. falciparum AMA1 adopt a PAN domain fold (Bai et al., 2005). These PAN modules are involved in diverse protein-protein and protein-carbohydrate interactions (Tordai et al., 1999). Human PLG, possesses a signal peptide and, as per Uniprot, is secreted and localized to the cell surface. AMA1, on the other hand, is surface-exposed in sporozoites (Swearingen et al., 2016), and is also predicted to possess a signal peptide. Thus, we posit that merozoite attachment to human cells is mediated by its structural similarity to human plasminogen.
3.3.3 CyRPA
The cysteine-rich protective antigen (CyRPA, PF3D7_0423800) interacts with two other parasite proteins (RH5 and Ripr) to form a complex on the surface of an invading merozoite (Ragotte et al., 2020). Understanding the similarity of CyRPA to human proteins is important because this complex is considered a potential vaccine target (Ragotte et al., 2020). CyRPA was represented by multiple structures from both the PDB and the AlphaFold Protein Structure Database and was similar to structures from several human proteins. The alignment with the best TM-score was to the region containing the EPTP protein domain (PF03736) in the human adhesion G-protein coupled receptor V1 protein (ADGRV1, Q8WXG9). This domain is likely a member of the 7-bladed beta-propeller fold (Scheel et al., 2002). The alignment with the second-best TM-score was to the human sialidase-1 protein. Both these results are interesting since CyRPA is known to form a 6-bladed beta-propeller fold that is similar to the sialidase fold, even though CyRPA has no sialidase activity (Favuzza et al., 2017).
In our earlier filtering step, we excluded AlphaFold structures if more than half of the predicted residues had low confidence (pLDDT<70). However, it is important to note that the retained structures may still contain regions with low confidence. The Foldseek algorithm considers the 3D neighbourhood of every residue, so we cannot mask out short regions of low confidence from the structural model directly. To address this, we checked the alignments of the four Plasmodium proteins described above and the aligned regions of three—AMA1, P38, and CyRPA—had a mean pLDDT greater than 70. Furthermore, the TM-scores for AMA1 and CyRPA was high (>0.5) and was above the noise cut-off for P38 (0.3) (File S4). Together this further supports the reliability of these alignments.
3.4 Large-scale analysis of the structural similarity between Plasmodium falciparum and human proteins
Our findings from the previous section demonstrated the effectiveness of our approach in identifying instances of unexpected structural similarities to human proteins that play a role in host-Plasmodium interactions. This motivated us to expand our approach and search all tertiary structures from P. falciparum against a database of 415,164 structures from human and 15 negative control species. Out of a total of 4,326 tertiary structures for Plasmodium, 3,649 (84%) aligned to a structure from either human or the negative control species. Out of the 3,350 (77%) could be aligned to a human structure, 59 structures aligned only to vertebrate proteins (human, mouse, rat, zebrafish) and 27 aligned to only mammalian proteins. Only eight structures aligned exclusively to a human structure. Nearly one-fifth of the P. falciparum proteins that shared structural similarity to human proteins (346 of 2,120) had no detectable sequence similarity to a human protein (Figure 3A; File S5). On average, 326 P. falciparum proteins had structural similarity but no sequence similarity to the control proteins.
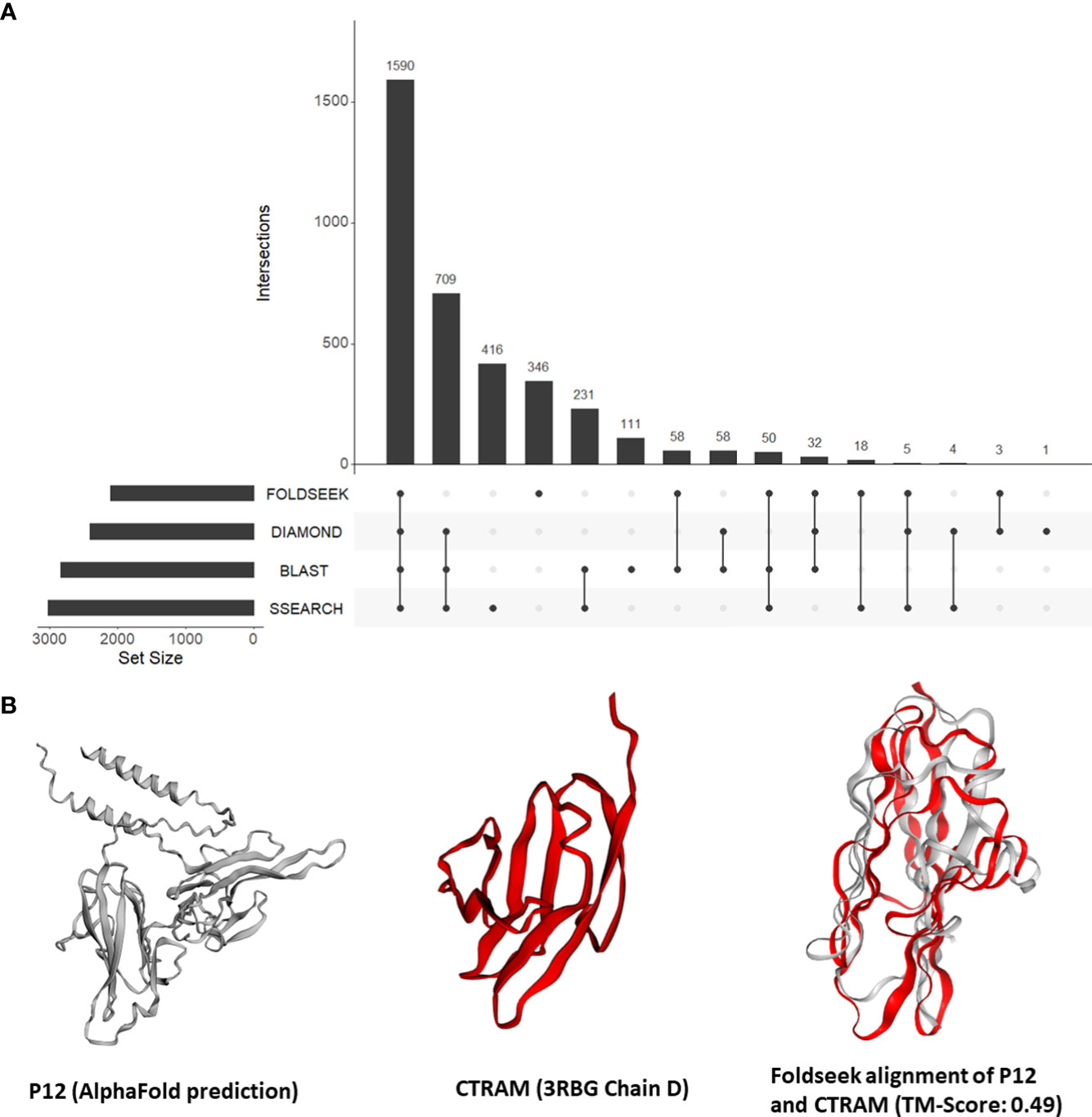
Figure 3 (A) An UpSet plot of all Plasmodium falciparum proteins which were aligned to human proteins by Foldseek, DIAMOND, BLASTP, and/or SSEARCH36. The R package ‘UpSetR’ was used to generate the UpSet plot (Conway et al., 2017). (B) Tertiary structure alignments of the parasite P12 structure in grey (AlphaFold prediction) and the human CTRAM structure in red (3RBG chain D), and the region aligned by Foldseek.
To prioritise the proteins, we categorized them with available annotations from PlasmoDB and Uniprot. In category 1, we analysed the predicted function of the aligned human protein, where the top scoring alignment was with a human protein with a GO term of interest: ‘immune system process’, ‘cell adhesion’, ‘cytoskeleton’, and/or ‘signalling’. In category 2, we examined the likely export of the P. falciparum protein from the cell, where the P. falciparum protein was predicted to contain a signal peptide. Finally, category 3 was gene expression, where we selected P. falciparum proteins whose genes were expressed in the 90th percentile in at least one of the human life cycle stages of the parasite—sporozoite and ring—or one of the intraerythrocytic stages (Zanghì et al., 2018; Wichers et al., 2019).
A total of 145 P. falciparum proteins could be placed in at least one of the categories, with 85 proteins in category 1, 41 in category 2, 76 in category 3, 31 in two categories, and 13 proteins in all three categories (Figure S2). The 44 P. falciparum proteins present in more than category represent instances of mimicry (File S4). Here, we present the biological relevance of a subset of these proteins.
First, we focused on the 13 P. falciparum proteins placed in all three categories (File S4). These 13 proteins include three known mediators of human-Plasmodium interactions mentioned in section 3.3 (CyRPA, P38, and AMA1). In addition, the P. falciparum 6-cysteine protein P12/merozoite surface protein P12 (P12, PF3D7_0612700) was structurally similar to the human cytotoxic and regulatory T-cell molecule (CRTAM, O95727) (alignment TM-score = 0.49) (Figure 3B). Interestingly, while the precise function of P12 is not known (Lyons et al., 2022), 6-cysteine proteins are one of the most abundant surface antigens in the P. falciparum have been attributed to virulence in related parasites (Wasmuth et al., 2012). P12 is localized on the surface of schizonts and merozoites. CRTAM is a host cell membrane protein and is involved in the development of cytotoxic CD4 and CD8 cells (Takeuchi et al., 2016). The expression of CRTAM increases during P. falciparum infection (Dooley et al., 2022). Furthermore, from StringDB, we found that eight of the 10 interacting partners of human CRTAM were mapped to ‘immune system process’ (GO:0002376) (Figure S3). Therefore, we posit that P12 mimics CRTAM to modulate the host immune system during infection.
We then expanded our focus to P. falciparum proteins found in two categories. The P. falciparum protein sporozoite surface protein 3 (SSP3, PF3D7_0812300) was aligned to human EGF-containing fibulin-like extracellular matrix protein 2 (EFEMP2, O95967) (alignment TM-score=0.58), with both proteins predicted to possess a signal peptide. EFEMP2 has been proposed to bind heparan sulfate (Djokic et al., 2013). This is interesting because multiple P. falciparum proteins, like the circumsporozoite protein (CS) (Frevert et al., 1993) and thrombospondin-related adhesive protein (TRAP) (Robson et al., 1995), are known to bind heparan sulfate. Our results suggest that SSP3 functions in targeting the sporozoites to the liver by mimicking the heparin-binding activity of EFEMP2.
Another P. falciparum protein was found to mimic a heparin-binding human protein. The uncharacterized P. falciparum protein (PF3D7_1118900), which possesses a signal peptide, was aligned to the region containing the MAM protein domain (PF00629) in human nephronectin (NPNT, Q6UXI9) (alignment TM-score=0.48). NPNT is a basement membrane protein which binds to heparin and basement membrane heparan sulfate proteoglycans through its MAM protein domain (Sato et al., 2013). Thus, our approach found a potential function for the uncharacterized P. falciparum protein. We propose that the uncharacterized P. falciparum protein mimics either the heparin-binding or the chondroitin sulfate-binding property of NPNT.
The P. falciparum glutaminyl-peptide cyclotransferase (PF3D7_1446900) was aligned to human autophagy-related protein 16-1 (ATG16L1, Q676U5) (alignment TM-score=0.47), a protein mapped to the GO term ‘positive regulation of autophagy’. The P. falciparum protein is possibly exposed on the surface of salivary gland sporozoites (Swearingen et al., 2016). ATG16L1 lacks a signal peptide and is mapped to the GO term ‘cytosol’ in Uniprot. From StringDB, we found that eight of 10 interacting proteins were associated with the Reactome Pathway ‘Macroautophagy’ (HSA-1632852) (Figure S4). Interestingly, while autophagy plays an important role in defence against intracellular parasites like P. falciparum, some parasites are known to manipulate this pathway for their own benefit; P. berghei growth is negatively affected when the human macroautophagy pathway is interrupted (Prado et al., 2015; Thieleke-Matos et al., 2016; Ghartey-Kwansah et al., 2020). Therefore, our results suggest that the P. falciparum protein mimics ATG16L1 to modulate the human macroautophagy pathway.
Additionally, four of these 145 P. falciparum proteins have been reported to be present in extracellular vesicles. Unlike the mimics identified in (Armijos-Jaramillo et al., 2021), our approach identified candidate mimics that can be identified using structure-similarity searches alone. These were the glideosome-associated connector (GAC, PF3D7_1361800), DNA/RNA-binding protein Alba 4 (ALBA4, PF3D7_1347500), high molecular weight rhoptry protein 3 (RhopH3, PF3D7_0905400), and pyridoxine biosynthesis protein PDX1 (PDX1, PF3D7_0621200). Interestingly, the parasite PDX1 protein was similar to the human copper homeostasis protein cutC homolog protein (CUTC, Q9NTM9) (alignment TM-score=0.69), which has copper binding activity (Li et al., 2010). While copper is an essential trace element for most organisms, excess copper is toxic for Plasmodium spp. (Rosenberg et al., 2006). This similarity to a copper binding protein suggests that PDX1 could function in copper sequestration in EVs.
4 Discussion
In this study, we present, to the best of our knowledge, the first genome-scale search of tertiary structural similarity between P. falciparum proteins and human proteins. We demonstrated the usefulness of our approach by showing that approximately 7% of the P. falciparum proteins had similarity to human proteins that could be detected only at the level of tertiary structure, and not primary sequence. We validated our approach on known mediators of host-parasite interactions and identified known similarities between CSP and thrombospondin-1 and AMA1 and the PAN protein domain in human plasminogen. Using available molecular and -omics datasets from PlasmoDB, we shortlisted a set of 44 instances of mimicry that are candidate mediators of host-parasite interactions, which improves our understanding of existing P. falciparum-human interactions.
We consider there to be six points to for the interpretation of the results of this analysis. First, the source of the structural model (PDB or AlphaFold) made a considerable difference (<50% agreement) for 10% of proteins. We emphasize that this is for AlphaFold structures that were filtered for high confidence; we anticipate that for lower confidence AlphaFold structures, an agreement with a PDB structure would be worse. Improvement in the AlphaFold software and its evolutionary models will lead to better structural models, but users must remain vigilant of accuracy metrics when using this promising resource. If a particular gene of interest has a protein structural model available in the PDB, we recommend incorporating it in the analysis. A recent notable study attempted to overcome this challenge by improving AlphaFold predictions for two parasites, Trypanosoma cruzi and Leishmania infantum (Wheeler, 2021). They attributed the poor accuracy of tertiary structure prediction for several parasite proteins to the low number of representative parasite sequences used to predict them and demonstrated that they could improve the AlphaFold predictions by increasing the number of parasite sequences used to model these structures.
Second, we noticed that the number of parasite proteins with structural similarity, but not sequence similarity, to human and negative control proteins was similar for most species studied. Some of these would be distant homologs which are not detected by sequence-based tools employed in this study. Indeed, the failure of homology detection has been shown to be one of the important reasons for the presence of lineage-specific genes (Weisman et al., 2020). In fact, multiple studies have demonstrated the effectiveness of using structure data to identify distant homologs that are missed by sequence-based approaches (Andorf et al., 2022; Monzon et al., 2022). Other proteins, however, would represent false positives of our approach based on our definition of ‘molecular mimics’, in which we define that a mimic confers benefit to the parasite. To this end, we used experimental data on P. falciparum from PDB to shortlist 44 proteins in P. falciparum that are structurally similar to human proteins and have a high probability of benefiting the parasite via its mimicry of a human protein. It is important to note that most parasites lack similar resources, making such an analysis challenging. This highlights the need to develop multi-omics resources, like PlasmoDB, for other parasites of global health concern.
Third, we discarded AlphaFold predictions with low prediction accuracy, so remove structures with a high level of intrinsic protein disorder. This is unfortunate, as protein-protein interactions are often mediated by disordered regions. For example, it has been proposed that viruses modulate host cellular processes by mimicking regions in disordered regions (Xue et al., 2014; Dolan et al., 2015; Garg et al., 2022). To potentially identify mimicry in disordered regions, it may be beneficial to consider a sequence k-mer based approach.
Fourth, the choice of structure aligner can impact the results. Foldseek, while being significantly faster than other available aligners, may trade off sensitivity (Holm, 2022; van Kempen et al., 2023). DALI, on the other hand, is arguably the most accurate but may not be suitable for large-scale structural similarity searches due to its longer run time.
Fifth, it is important to inspect the version of each protein sequence in the Uniprot database and its corresponding structure in the PDB and AlphaFold.The Uniprot database is updated routinely and identifiers assigned to a protein may change, the corresponding protein in the PDB or AlphaFold would probably not be updated as often as Uniprot.
And sixth, we acknowledge the need for validation. In this study, we identified Plasmodium proteins which structural resemble non-homologous human proteins. We use additional data to highlight those proteins which may be interacting with human proteins to the benefit of Plasmodium. The next step is validation, ideally through a biochemical assay, e.g., Yeast Two Hybrid or a mass spectrometry approach [reviewed in (Nicod et al., 2017)]. Computational approaches can be used to dock two proteins and predict their binding affinities. However, the accurate calculation of binding energies between two proteins is a laborious and error-prone process, lacking consistency between methods. For most of the predictions presented here, we have used AlphaFold models; a small change in side-chain orientation may be tolerated in structural alignments but can lead to a dramatic change in binding energies. We note that algorithms which refine structural models and use them for a range of purposes are being released at a frenetic rate. However, we remain cautious at introducing further technical uncertainty; a recent study showed that only 51% of PPIs could be corrected predicted from AlphaFold models (Bryant et al., 2022).
In conclusion, we present, to the best of our knowledge, the first genome-level search of tertiary structure similarity between P. falciparum and human proteins, leading to the identification of instances of molecular mimicry. Leveraging the extensive experimental data available for P. falciparum on PlasmoDB, we sought to provide biological relevance of the identified similarities. The catalogued list of 44 candidate mimics serves as candidates for experimental validation. It is worth noting that this list is a subset of the total identifiable mimics; containing those which could not be identified by commonly used sequence similarity approaches. Our findings contribute to a deeper understanding of molecular mechanisms underlying Plasmodium-human interactions and may have help guide vaccine and drug development against malaria.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
Conceived and designed the analyses: VM and JW. Performed the analyses: VM. Wrote the manuscript: VM and JW. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant (#04589-2020) to JW and an Eyes High Postdoctoral recruitment scholarship to VM. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
We thank Kaylee Rich, Constance Finney, and Justin MacCallum for their discussions on various aspects of the manuscript. We acknowledge the high-performance computing resources made available by the Faculty of Veterinary Medicine and Research Computing at the University of Calgary.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpara.2023.1162697/full#supplementary-material
References
Abdi A., Yu L., Goulding D., Rono M. K., Bejon P., Choudhary J., et al. (2017). Proteomic analysis of extracellular vesicles from a plasmodium falciparum Kenyan clinical isolate defines a core parasite secretome. Wellcome Open Res. 2, 50. doi: 10.12688/wellcomeopenres.11910.2
Acharya P., Garg M., Kumar P., Munjal A., Raja K. D. (2017). Host–parasite interactions in human malaria: clinical implications of basic research. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.00889
Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215 (3), 403–410. doi: 10.1016/S0022-2836(05)80360-2
Amos B., Aurrecoechea C., Barba M., Barreto A., Basenko E. Y., Bażant W., et al. (2022). VEuPathDB: the eukaryotic pathogen, vector and host bioinformatics resource center. Nucleic Acids Res. 50 (D1), D898–D911. doi: 10.1093/nar/gkab929
Andorf C. M., Sen S., Hayford R. K., Portwood J. L., Cannon E. K., Harper L. C., et al. (2022). FASSO: an AlphaFold based method to assign functional annotations by combining sequence and structure orthology. BioRxiv, 2022–11 doi: 10.1101/2022.11.10.516002
Armijos-Jaramillo V., Mosquera A., Rojas B., Tejera E. (2021). The search for molecular mimicry in proteins carried by extracellular vesicles secreted by cells infected with plasmodium falciparum. Communicat. Integr. Biol. 14 (1), 212–220. doi: 10.1080/19420889.2021.1972523
Aurrecoechea C., Brestelli J., Brunk B. P., Dommer J., Fischer S., Gajria B., et al. (2008). PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res. 37 (suppl_1), D539–D543. doi: 10.1093/nar/gkn814
Bai T., Becker M., Gupta A., Strike P., Murphy V. J., Anders R. F., et al. (2005). Structure of AMA1 from plasmodium falciparum reveals a clustering of polymorphisms that surround a conserved hydrophobic pocket. Proc. Natl. Acad. Sci. 102 (36), 12736–12741. doi: 10.1073/pnas.0501808102
Balbin C. A., Nunez-Castilla J., Stebliankin V., Baral P., Sobhan M., Cickovski T., et al. (2023). Epitopedia: identifying molecular mimicry between pathogens and known immune epitopes. ImmunoInformatics 9, 100023. doi: 10.1016/j.immuno.2023.100023
Bargieri D. Y., Andenmatten N., Lagal V., Thiberge S., Whitelaw J. A., Tardieux I., et al. (2013). Apical membrane antigen 1 mediates apicomplexan parasite attachment but is dispensable for host cell invasion. Nat. Commun. 4 (1), 2552. doi: 10.1038/ncomms3552
Bryant P., Pozzati G., Elofsson A. (2022). Improved prediction of protein-protein interactions using AlphaFold2. Nat. Commun. 13 (1), 1265. doi: 10.1038/s41467-022-28865-w
Buchfink B., Reuter K., Drost H.-G. (2021). Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18 (4), 366–368. doi: 10.1038/s41592-021-01101-x
Cameron C. M., Barrett J. W., Mann M., Lucas A., McFadden G. (2005). Myxoma virus M128L is expressed as a cell surface CD47-like virulence factor that contributes to the downregulation of macrophage activation in vivo. Virology 337 (1), 55–67. doi: 10.1016/j.virol.2005.03.037
Cerami C., Frevert U., Sinnis P., Takacs B., Clavijo P., Santos M. J., et al. (1992). The basolateral domain of the hepatocyte plasma membrane bears receptors for the circumsporozoite protein of plasmodium falciparum sporozoites. Cell 70 (6), 1021–1033. doi: 10.1016/0092-8674(92)90251-7
Chulanetra M., Chaicumpa W. (2021). Revisiting the mechanisms of immune evasion employed by human parasites. Front. Cell. Infect. Microbiol. 11. doi: 10.3389/fcimb.2021.702125
Conway J. R., Lex A., Gehlenborg N. (2017). UpSetR: an r package for the visualization of intersecting sets and their properties. Bioinf. (Oxford England) 33 (18), 2938–2940. doi: 10.1093/bioinformatics/btx364
Damian R. T. (1964). Molecular mimicry: antigen sharing by parasite and host and its consequences. Am. Nat. 98 (900), 129–149. doi: 10.1086/282313
Djokic J., Fagotto-Kaufmann C., Bartels R., Nelea V., Reinhardt D. P. (2013). Fibulin-3, -4, and -5 are highly susceptible to proteolysis, interact with cells and heparin, and form multimers. J. Biol. Chem. 288 (31), 22821–22835. doi: 10.1074/jbc.M112.439158
Dolan P. T., Roth A. P., Xue B., Sun R., Dunker A. K., Uversky V. N., et al. (2015). Intrinsic disorder mediates hepatitis c virus core-host cell protein interactions. Protein Science : A Publ. Protein Soc. 24 (2), 221–235. doi: 10.1002/pro.2608
Dooley N. L., Chabikwa T. G., Pava Z., Loughland J. R., Hamelink J., Berry K., et al. (2022). Malaria drives unique regulatory responses across multiple immune cell subsets. BioRxiv 11, 16. doi: 10.1101/2022.11.16.516822
Doxey A. C., McConkey B. J. (2013). Prediction of molecular mimicry candidates in human pathogenic bacteria. Virulence 4 (6), 453–466. doi: 10.4161/viru.25180
Emms D. M., Kelly S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20 (1), 238. doi: 10.1186/s13059-019-1832-y
Favuzza P., Guffart E., Tamborrini M., Scherer B., Dreyer A. M., Rufer A. C., et al. (2017). Structure of the malaria vaccine candidate antigen CyRPA and its complex with a parasite invasion inhibitory antibody. ELife 6, e20383. doi: 10.7554/eLife.20383
Feller T., Thom P., Koch N., Spiegel H., Addai-Mensah O., Fischer R., et al. (2013). Plant-based production of recombinant plasmodium surface protein pf38 and evaluation of its potential as a vaccine candidate. PloS One 8 (11), e79920. doi: 10.1371/journal.pone.0079920
Frevert U., Sinnis P., Cerami C., Shreffler W., Takacs B., Nussenzweig V. (1993). Malaria circumsporozoite protein binds to heparan sulfate proteoglycans associated with the surface membrane of hepatocytes. J. Exp. Med. 177 (5), 1287–1298. doi: 10.1084/jem.177.5.1287
Garg A., Dabburu G. R., Singhal N., Kumar M. (2022). Investigating the disordered regions (MoRFs, SLiMs and LCRs) and functions of mimicry proteins/peptides in silico. PloS One 17 (4), 1–11. doi: 10.1371/journal.pone.0265657
Getts D. R., Chastain E. M. L., Terry R. L., Miller S. D. (2013). Virus infection, antiviral immunity, and autoimmunity. Immunol. Rev. 255 (1), 197–209. doi: 10.1111/imr.12091
Ghartey-Kwansah G., Adu-Nti F., Aboagye B., Ankobil A., Essuman E. E., Opoku Y. K., et al. (2020). Autophagy in the control and pathogenesis of parasitic infections. Cell Bioscience 10 (1), 101. doi: 10.1186/s13578-020-00464-6
Harrison T. E., Mørch A. M., Felce J. H., Sakoguchi A., Reid A. J., Arase H., et al. (2020). Structural basis for RIFIN-mediated activation of LILRB1 in malaria. Nature 587 (7833), 309–312. doi: 10.1038/s41586-020-2530-3
Hebert F. O., Phelps L., Samonte I., Panchal M., Grambauer S., Barber I., et al. (2015). Identification of candidate mimicry proteins involved in parasite-driven phenotypic changes. Parasites Vectors 8, 225. doi: 10.1186/s13071-015-0834-1
Holm L. (2022). Dali server: structural unification of protein families. Nucleic Acids Res.50 (W1), W210–W215. doi: 10.1093/nar/gkac387
Kato K., Mayer D. C. G., Singh S., Reid M., Miller L. H. (2005). Domain III of plasmodium falciparum apical membrane antigen 1 binds to the erythrocyte membrane protein kx. Proc. Natl. Acad. Sci. U. S. A. 102 (15), 5552–5557. doi: 10.1073/pnas.0501594102
Kvansakul M., van Delft M. F., Lee E. F., Gulbis J. M., Fairlie W. D., Huang D. C. S., et al. (2007). A structural viral mimic of prosurvival bcl-2: a pivotal role for sequestering proapoptotic bax and bak. Mol. Cell 25 (6), 933–942. doi: 10.1016/j.molcel.2007.02.004
Leshchyns’ka I., Sytnyk V. (2016). Reciprocal interactions between cell adhesion molecules of the immunoglobulin superfamily and the cytoskeleton in neurons. Front. Cell Dev. Biol. 4. doi: 10.3389/fcell.2016.00009
Li Y., Du J., Zhang P., Ding J. (2010). Crystal structure of human copper homeostasis protein CutC reveals a potential copper-binding site. J. Struct. Biol. 169 (3), 399–405. doi: 10.1016/j.jsb.2009.10.012
Ludin P., Nilsson D., Mäser P. (2011). Genome-wide identification of molecular mimicry candidates in parasites. PloS One 6 (3), e17546. doi: 10.1371/journal.pone.0017546
Lyons F. M. T., Gabriela M., Tham W., Dietrich M. H., Craig A. (2022). Plasmodium 6-cysteine proteins: functional diversity, transmission- blocking antibodies and structural scaffolds. Front. Cell. Infect. Microbiol. 12, 1–20. doi: 10.3389/fcimb.2022.945924
Mantel P.-Y., Hoang A. N., Goldowitz I., Potashnikova D., Hamza B., Vorobjev I., et al. (2013). Malaria-infected erythrocyte-derived microvesicles mediate cellular communication within the parasite population and with the host immune system. Cell Host Microbe 13 (5), 521–534. doi: 10.1016/j.chom.2013.04.009
Monzon V., Paysan-Lafosse T., Wood V., Bateman A. (2022). Reciprocal best structure hits: using AlphaFold models to discover distant homologues. Bioinf. Adv. 2 (1), vbac072. doi: 10.1093/bioadv/vbac072
Mousa A. A., Roche D. B., Terkawi M. A., Kameyama K., Kamyingkird K., Vudriko P., et al. (2017). Human babesiosis: indication of a molecular mimicry between thrombospondin domains from a novel babesia microti BmP53 protein and host platelets molecules. PloS One 12 (10), e0185372. doi: 10.1371/journal.pone.0185372
Nicod C., Banaei-Esfahani A., Collins B. C. (2017). Elucidation of host-pathogen protein-protein interactions to uncover mechanisms of host cell rewiring. Curr. Opin. Microbiol. 39, 7–15. doi: 10.1016/j.mib.2017.07.005
Oliver-González J. (1944). The inhibition of human isoagglutinins by a polysaccharide from ascaris suum. J. Infect. Dis. 74 (2), 81–84. doi: 10.1093/infdis/74.2.81
Paul G., Deshmukh A., Kaur I., Rathore S., Dabral S., Panda A., et al. (2017). A novel Pfs38 protein complex on the surface of plasmodium falciparum blood − stage merozoites. Malaria J. 16, 1–15. doi: 10.1186/s12936-017-1716-0
Prado M., Eickel N., De Niz M., Heitmann A., Agop-Nersesian C., Wacker R., et al. (2015). Long-term live imaging reveals cytosolic immune responses of host hepatocytes against plasmodium infection and parasite escape mechanisms. Autophagy 11 (9), 1561–1579. doi: 10.1080/15548627.2015.1067361
Ragotte R. J., Higgins M. K., Draper S. J. (2020). The RH5-CyRPA-Ripr complex as a malaria vaccine target. Trends Parasitol. 36 (6), 545–559. doi: 10.1016/j.pt.2020.04.003
Remarque E. J., Faber B. W., Kocken C. H. M., Thomas A. W. (2008). Apical membrane antigen 1: a malaria vaccine candidate in review. Trends Parasitol. 24 (2), 74–84. doi: 10.1016/j.pt.2007.12.002
Reynolds C. R., Islam S. A., Sternberg M. J. E. (2018). EzMol: a web server wizard for the rapid visualization and image production of protein and nucleic acid structures. J. Mol. Biol. 430 (15), 2244–2248. doi: 10.1016/j.jmb.2018.01.013
Robson K. J., Frevert U., Reckmann I., Cowan G., Beier J., Scragg I. G., et al. (1995). Thrombospondin-related adhesive protein (TRAP) of plasmodium falciparum: expression during sporozoite ontogeny and binding to human hepatocytes. EMBO J. 14 (16), 3883–3894. doi: 10.1002/j.1460-2075.1995.tb00060.x
Robson K. J. H., Hall J. R. S., Jennings M. W., Harris T. J. R., Marsh K., Newbold C. I., et al. (1988). A highly conserved amino-acid sequence in thrombospondin, properdin and in proteins from sporozoites and blood stages of a human malaria parasite. Nature 335 (6185), 79–82. doi: 10.1038/335079a0
Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Ballard A. J., Cowie A., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi: 10.1038/s41586-021-03819-2
Rosenberg E., Litus I., Schwarzfuchs N., Sinay R., Schlesinger P., Golenser J., et al. (2006). Pfmdr2 confers heavy metal resistance to plasmodium falciparum*. J. Biol. Chem. 281 (37), 27039–27045. doi: 10.1074/jbc.M601686200
Sallee N. A., Rivera G. M., Dueber J. E., Vasilescu D., Mullins R. D., Mayer B. J., et al. (2008). The pathogen protein EspFU hijacks actin polymerization using mimicry and multivalency. Nature 454 (7207), 1005–1008. doi: 10.1038/nature07170
Sampaio N. G., Emery S. J., Garnham A. L., Tan Q. Y., Sisquella X., Pimentel M. A., et al. (2018). Extracellular vesicles from early stage plasmodium falciparum-infected red blood cells contain PfEMP1 and induce transcriptional changes in human monocytes. Cell. Microbiol. 20 (5), e12822. doi: 10.1111/cmi.12822
Sato Y., Shimono C., Li S., Nakano I., Norioka N., Sugiura N., et al. (2013). Nephronectin binds to heparan sulfate proteoglycans via its MAM domain. Matrix Biology : J. Int. Soc. Matrix Biol. 32 (3–4), 188–195. doi: 10.1016/j.matbio.2013.01.005
Scheel H., Tomiuk S., Hofmann K. (2002). A common protein interaction domain links two recently identified epilepsy genes. Hum. Mol. Genet. 11 (15), 1757–1762. doi: 10.1093/hmg/11.15.1757
Srinivasan P., Beatty W. L., Diouf A., Herrera R., Ambroggio X., Moch J. K., et al. (2011). Binding of plasmodium merozoite proteins RON2 and AMA1 triggers commitment to invasion. Proc. Natl. Acad. Sci. 108 (32), 13275–13280. doi: 10.1073/pnas.1110303108
Swearingen K. E., Lindner S. E., Shi L., Shears M. J., Harupa A., Hopp C. S., et al. (2016). Interrogating the plasmodium sporozoite surface: identification of surface-exposed proteins and demonstration of glycosylation on CSP and TRAP by mass spectrometry-based proteomics. PloS Pathog. 12 (4), e1005606. doi: 10.1371/journal.ppat.1005606
Takeuchi A., Badr M. E. S. G., Miyauchi K., Ishihara C., Onishi R., Guo Z., et al. (2016). CRTAM determines the CD4+ cytotoxic T lymphocyte lineage. J. Exp. Med. 213 (1), 123–138. doi: 10.1084/jem.20150519
Tayal S., Bhatia V., Mehrotra T., Bhatnagar S. (2022). ImitateDB: a database for domain and motif mimicry incorporating host and pathogen protein interactions. Amino Acids 54 (6), 923–934. doi: 10.1007/s00726-022-03163-3
Thieleke-Matos C., Lopes da Silva M., Cabrita-Santos L., Portal M. D., Rodrigues I. P., Zuzarte-Luis V., et al. (2016). Host cell autophagy contributes to plasmodium liver development. Cell. Microbiol. 18 (3), 437–450. doi: 10.1111/cmi.12524
Tordai H., Bányai L., Patthy L. (1999). The PAN module: the n-terminal domains of plasminogen and hepatocyte growth factor are homologous with the apple domains of the prekallikrein family and with a novel domain found in numerous nematode proteins. FEBS Lett. 461 (1–2), 63–67. doi: 10.1016/S0014-5793(99)01416-7
van Kempen M., Kim S. S., Tumescheit C., Mirdita M., Lee J., Gilchrist C. L. M., et al. (2023). Fast and accurate protein structure search with foldseek. Nat. Biotechnol, 1–4 doi: 10.1038/s41587-023-01773-0
Wasmuth J. D., Pszenny V., Haile S., Jansen E. M., Gast A. T., Sher A., et al. (2012). Integrated bioinformatic and targeted deletion analyses of the SRS gene superfamily identify SRS29C as a negative regulator of toxoplasma virulence. MBio 3 (6), e00321–e00312. doi: 10.1128/mBio.00321-12
Weisman C. M., Murray A. W., Eddy S. R. (2020). Many, but not all, lineage-specific genes can be explained by homology detection failure. PloS Biol. 18 (11), e3000862. doi: 10.1371/journal.pbio.3000862
Westphal D., Ledgerwood E. C., Hibma M. H., Fleming S. B., Whelan E. M., Mercer A. A. (2007). A novel bcl-2-like inhibitor of apoptosis is encoded by the parapoxvirus ORF virus. J. Virol. 81 (13), 7178–7188. doi: 10.1128/JVI.00404-07
Wheeler R. J. (2021). A resource for improved predictions of trypanosoma and leishmania protein three-dimensional structure. PloS One 16 (11), 1–12. doi: 10.1371/journal.pone.0259871
Wichers J. S., Scholz J. A. M., Strauss J., Witt S., Lill A., Ehnold L.-I., et al. (2019). Dissecting the gene expression, localization, membrane topology, and function of the plasmodium falciparum STEVOR protein family. MBio 10 (4), e01500–19. doi: 10.1128/mBio.01500-19
Xue B., Blocquel D., Habchi J., Uversky A. V., Kurgan L., Uversky V. N., et al. (2014). Structural disorder in viral proteins. Chem. Rev. 114 (13), 6880–6911. doi: 10.1021/cr4005692
Keywords: molecular mimicry, malaria, plasmodium, AlphaFold, host-parasite interactions, structural comparisons
Citation: Muthye V and Wasmuth JD (2023) Proteome-wide comparison of tertiary protein structures reveals molecular mimicry in Plasmodium-human interactions. Front. Parasitol. 2:1162697. doi: 10.3389/fpara.2023.1162697
Received: 09 February 2023; Accepted: 05 June 2023;
Published: 15 June 2023.
Edited by:
Bruce A. Rosa, Washington University in St. Louis, United StatesReviewed by:
Hao-Ching Wang, Taipei Medical University, TaiwanVinicio Armijos, University of the Americas, Ecuador
Copyright © 2023 Muthye and Wasmuth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James D. Wasmuth, andhc211dGhAdWNhbGdhcnkuY2E=