
- 1 Laboratory of Molecular Neuropathology, Centre for Integrative Biology, University of Trento, Trento, Italy
- 2 Neuroscience Institute, National Research Council, Pisa, Italy
- 3 Laboratory of Developmental Neurobiology, Centre for Integrative Biology, University of Trento, Trento, Italy
Epilepsy is characterized by spontaneous recurrent seizures and comprises a diverse group of syndromes with different etiologies. Epileptogenesis refers to the process whereby the brain becomes epileptic and can be related to several factors, such as acquired structural brain lesions, inborn brain malformations, alterations in neuronal signaling, and defects in maturation and plasticity of neuronal networks. In this review, we will focus on alterations of brain development that lead to an hyperexcitability phenotype in adulthood, providing examples from both animal and human studies. Malformations of cortical development (including focal cortical dysplasia, lissencephaly, heterotopia, and polymicrogyria) are frequently epileptogenic and result from defects in cell proliferation in the germinal zone and/or impaired neuronal migration and differentiation. Delayed or reduced arrival of inhibitory interneurons into the cortical plate is another possible cause of epileptogenesis. GABAergic neurons are generated during early development in the ganglionic eminences, and failure to pursue migration toward the cortex alters the excitatory/inhibitory balance resulting in aberrant network hyperexcitability. More subtle defects in the developmental assembly of excitatory and inhibitory synapses are also involved in epilepsy. For example, mutations in the presynaptic proteins synapsins and SNAP-25 cause derangements of synaptic transmission and plasticity which underlie appearance of an epileptic phenotype. Finally, there is evidence that defects in synapse elimination and remodeling during early “critical periods” can trigger hyperexcitability later in life. Further clarification of the developmental pathways to epilepsy has important implications for disease prevention and therapy.
Introduction
Epilepsy is one of the most common neurological disorders, characterized by the repeated occurrence of spontaneous bursts of neuronal overactivity, known as seizures. Seizures typically arise in restricted regions of the brain and may remain confined to these areas or spread to the whole cerebral hemispheres. The hippocampal formation and cerebral cortex are considered the most epileptogenic regions of the brain (Pitkanen and Sutula, 2002; Avanzini and Franceschetti, 2003). The behavioral manifestations of seizures, as well as the severity of the epileptic condition, strictly relate to the brain regions that are affected by overactivity. Epilepsy comprises a large group of syndromes with different etiologies. A large series of recent studies demonstrated that several developmental factors (including congenital brain malformations, altered neuronal signaling during embryonic life, and defects in postnatal maturation of neuronal networks) contribute to epileptogenesis, leading to the concept of epilepsy as a neurodevelopmental disorder.
In the first part of the review, we will focus on those alterations in embryonic development of the cerebral cortex that lead to an hyperexcitability phenotype in postnatal life. Taking examples from both animal and human studies, we will describe the role of a number of key developmental genes controlling the migration of projection (excitatory) and local circuit (inhibitory) neurons, and describe how their altered function may result in epileptogenesis. In the second part of the review, we will describe the defects in the developmental assembly of excitatory and inhibitory synapses that are also involved in epileptogenesis. Specifically, we will focus on defects in synapse elimination and remodeling during early “critical periods” that may trigger hyperexcitability later in life. The deep understanding of the complex developmental processes involved in epileptogenesis may have important implications for disease prevention and therapy.
Developmental Defects of the Cerebral Cortex Lead to Epilepsy
The development of the mammalian cerebral cortex can be subdivided into three partially overlapping phases (Rubenstein, 2000, 2011; Lui et al., 2011). During the first phase, stem cells located in the ventricular and subventricular zones of the telencephalon proliferate and differentiate into neuronal precursors or glial cells. During the second phase, neurons migrate from their place of origin and reach their final destination in the cerebral cortex (Marin and Rubenstein, 2003; Kriegstein et al., 2006; Fishell, 2007; Fishell and Hanashima, 2008). The mature cerebral cortex is organized in six layers (Molyneaux et al., 2007) and contains two major types of projection neurons. The vast majority (∼80%) are glutamatergic (excitatory) neurons extending their long axon into the ipsilateral or contralateral cortex (cortico-cortical neurons, located in layers 2/3) or toward subcortical regions (cortico-fugal neurons, located in layers 5/6). The remainder (∼20%) are GABAergic local circuit neurons (inhibitory interneurons) that establish synaptic contacts with excitatory neurons located in their proximity.
Migration of glutamatergic and GABAergic cortical neurons occurs in two different ways. Glutamatergic projection neurons are generated from neuronal precursors located in the neocortical neuroepithelium via asymmetric cell divisions of cortical primary progenitors (radial glia) located in the ventricular and subventricular zones (Malatesta et al., 2003; Kriegstein et al., 2006; Hansen et al., 2010). Asymmetric divisions generate immature projection neurons, that migrate toward the cortical plate along radial glial processes (Rakic, 2007) and hence reach their final destination in specific cortical layers through the interaction with local molecular cues, such as Reelin (Frotscher, 2010). Cortical neurons are generated in an inside-out pattern, layer 6 neurons being the first to be born. The majority of GABAergic interneurons are instead generated outside of the cerebral cortex, in the ganglionic eminences of the basal forebrain (Wonders and Anderson, 2006). The immature interneurons initially migrate tangentially along the subventricular zone of the basal forebrain, and then change direction by following a radial or an oblique path to enter the cortical plate, from where they reach their final destination into the layers of the cerebral cortex (Kriegstein and Noctor, 2004).
After completion of neuronal migration, the third, last phase of cortical development involves a complex series of apoptotic and synaptogenic events aimed at finely regulating the number of mature neurons and their connections, ultimately leading to the shaping of cortical circuits.
The embryonic development of the cerebral cortex is a complex process, tightly controlled by a series of gene expression cascades (Guillemot et al., 2006). Alterations of these gene regulatory pathways during development may lead to cortical malformations, resulting in malfunction during postnatal life. Accordingly, several cortical abnormalities have been identified that are caused by mutations in key genes involved in the different phases of cortical development. Cortical malformations may result from abnormal neuronal proliferation, migration defects of both excitatory and inhibitory neurons, or altered synaptogenesis/circuit formation, and are usually epileptogenic (Barkovich et al., 2005; Guerrini and Parrini, 2010; Manzini and Walsh, 2011).
Defects of neuronal and glial proliferation during embryonic development result in epileptogenic cortical lesions. Tuberous sclerosis complex (TSC) is a genetic disorder characterized by the widespread development of benign tumors (hamartomas) in multiple organ systems including the brain. Cortical tubers, subependymal nodules, and subependymal giant cell astrocytomas represent the typical lesions observed in TSC. These malformations result in early-onset seizures, that are often accompanied by intellectual disability and autism. TSC results from mutations of TSC1 (hamartin) or TSC2 (tuberin) genes that lead to hamartomatous growths of neuronal and glial cells (Holmes and Stafstrom, 2007).
Neuronal migration disorders are a heterogeneous group of neurological conditions characterized by abnormal neuronal positioning in the cerebral cortex. Smooth cortex/lissencephaly (absent or reduced convolutions resulting in cortical thickening and smooth cerebral surface) and heterotopias (typically, ectopic nodules of gray matter located in a periventricular or subcortical position) have been associated to mutations in a number of genes regulating cortical neuron migration and are characterized by severe neurological impairment and epilepsy (Guerrini and Parrini, 2010). Migration defects of both excitatory and inhibitory neurons contribute to these conditions and lead to an altered excitation/inhibition balance and aberrant network hyperexcitability.
Following the initial generation and migration of excitatory and inhibitory neurons, immature neural networks are transformed into organized circuits through a process of refinement that is largely controlled by electrical activity (Katz and Shatz, 1996). During sensitive phases of the early postnatal life, called “critical periods,” initially exuberant connections are eliminated and the remaining synapses undergo a functional maturation (Katz and Shatz, 1996; Berardi et al., 2000). Perturbations in this developmental refinement of neuronal circuitry during critical periods may trigger hyperexcitability and epilepsy later in life. In the following sections, we will detail some of the most significant molecular mechanisms involved in neuronal proliferation, neuronal migration, and synaptic refinement (schematically illustrated in Figure 1 and summarized in Table 1), whose perturbation during embryonic or postnatal development results in epileptogenesis.
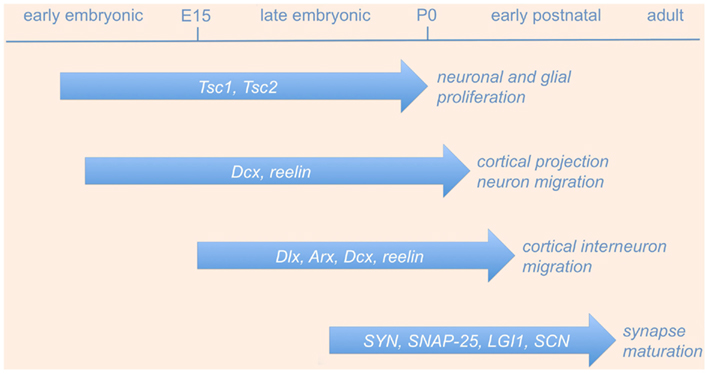
Figure 1. Genes involved in neurodevelopmental causes of epilepsy. The picture shows a schematic time-line of the most significant gene functions involved in neuronal proliferation, neuronal migration, and synaptic refinement, whose perturbation during embryonic or postnatal development results in epileptogenesis (see text and Table 1 for details). Abbreviations are as in the text.
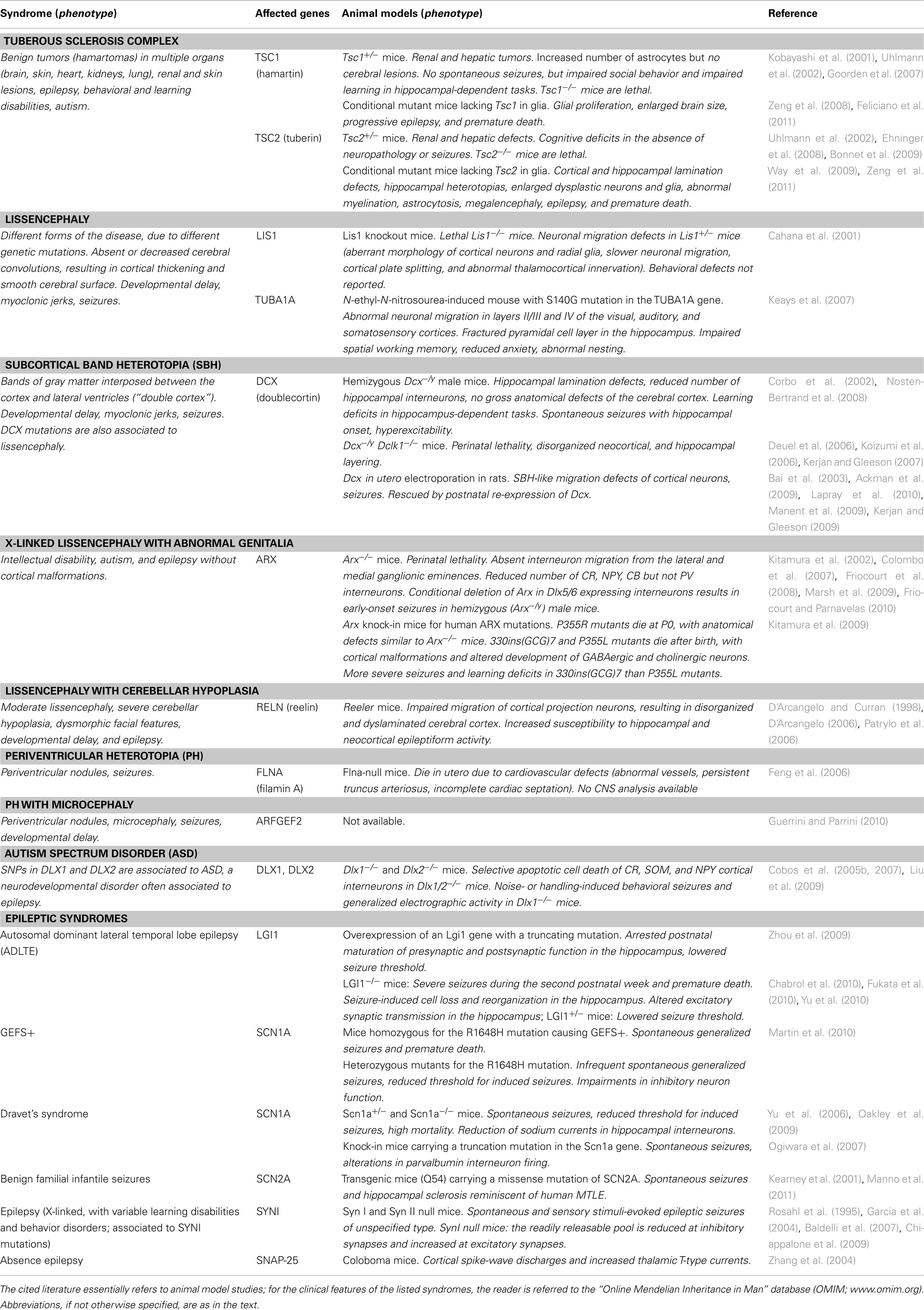
Table 1. Genes involved in neurodevelopmental epileptic syndromes and their corresponding mouse models.
Neuronal Proliferation Defects
Tuberous sclerosis complex is a neurocutaneous syndrome characterized by benign tumors, early-onset epilepsy, intellectual disability, and autism (Holmes and Stafstrom, 2007). TSC results from loss-of-function mutations of TSC1 or TSC2 genes, which are crucially involved in the control of neuronal and glial cell proliferation during embryonic development. TSC1 encodes a protein (hamartin) containing two coiled-coil domains, while TSC2 encodes a GTPase activating protein (tuberin) that inhibits small G-proteins belonging to the Ras-related super-family. Hamartin and tuberin are both expressed in neurons and astrocytes of specific central nervous system (CNS) regions such as forebrain, cerebellum, and brainstem, where they form a protein–protein complex that constitutively inhibits mammalian target of rapamycin (mTOR), a serine–threonine kinase positively regulating protein synthesis, cell proliferation, and survival. The function of the TSC1/2 complex is controlled by multiple intracellular signaling pathways converging on AKT, a pro-survival and pro-oncogenic kinase that directly phosphorylates TSC2 inhibiting its function. Loss of TSC1/2 function leads to activation of the mTOR cascade and results in increased cell proliferation; conversely, inhibition of mTOR function (e.g., by rapamycin) results in growth suppression and reduced cell size (Jozwiak, 2006; Holmes and Stafstrom, 2007). The occurrence of tubers in animal models of TSC has been a point of controversy in the field. In contrast with the main feature of the human disease, Tsc1+/− or Tsc2+/− mice do not develop tubers (Kobayashi et al., 2001; Uhlmann et al., 2002; Goorden et al., 2007; Ehninger et al., 2008; Bonnet et al., 2009). However, conditional inactivation of Tsc1 or Tsc2 in astrocytes leads to tuber-like lesions and severe seizures in mice (Zeng et al., 2008, 2011; Way et al., 2009). More recently, in utero electroporation was used to induce homozygous (Tsc1−/−) cell clones on a TSC1 heterozygous (Tsc1+/−) background. This strategy resulted in cortical tuber-like lesions and a lower seizure threshold, suggesting that cells inside the tubers might have an additional somatic mutation that might contribute to the pathological phenotype (Feliciano et al., 2011). These studies clearly indicate that altering TSC1/2 signaling in specific CNS cell types is at the origin of TSC, but also point out the difficulty of modeling TSC in mice.
The histopathological features of cortical tubers in TSC reflect the antiproliferative role normally exerted by the TSC1/2 signaling complex. Cortical tubers in TSC patients are characterized by giant, dysplastic, and heterotopic neurons with aberrant dendrites and axons, as well as by proliferating astrocytes (Holmes and Stafstrom, 2007). Cortical hyperexcitability arises in the proximity of tubers, but its causes remain largely unknown (Major et al., 2009). Several hypotheses have been proposed to explain epileptogenesis in TSC. Increased expression of NMDA glutamate receptors and decreased expression of the GABA synthetic enzyme glutamic acid decarboxylase (GAD65), GABA vesicular transporter (vGAT), and GABA receptor subunits have been described in cortical tubers from human TSC patients (White et al., 2001), suggesting that excitation/inhibition imbalance in cortical circuits may contribute to epileptogenesis. Another intriguing hypothesis is that cortical tubers may alter thalamocortical connectivity during early brain development, thus resulting in hyperexcitable cortical circuits (Holmes and Stafstrom, 2007).
Seizure suppression in TSC remains a difficult task to be achieved. Infantile spasms in TSC often respond to vigabatrin (a GABA-transaminase inhibitor), but not to other antiepileptic drugs (AEDs), and the surgical removal of tubers remains in many cases the only therapeutic option (Holmes and Stafstrom, 2007). For these reasons, much emphasis has been put to the potential beneficial effects of the mTOR inhibitor rapamycin as a novel anticonvulsant and antiepileptogenic drug. Indeed, rapamycin is able to reduce seizures and prevent epileptogenesis in various animal models. For example, rapamycin treatment blocked epilepsy progression in conditional mutant mice lacking TSC1 or the TSC-positive regulator PTEN (Zeng et al., 2008; Sunnen et al., 2011), and reversed learning deficits in a Tsc2+/− mice (Ehninger et al., 2008). Importantly, rapamycin administered to chronically epileptic rats following kainic acid (KA; Zeng et al., 2009) or pilocarpine (Huang et al., 2010) treatment has been shown to suppress acquired epilepsy, even though these results have not been replicated in mice (Buckmaster and Lew, 2011). However, some authors pointed out that rapamycin treatment in animal models is still far to be optimal, since seizures may reappear after treatment cessation, and continuous rapamycin exposure might severely affect animal growth and health (Sunnen et al., 2011). In this respect, valid alternative strategies might be represented by a high-dose pulse treatment (Raffo et al., 2011) or even prenatal exposure (to be applied in cases of familial TSC predisposition; Anderl et al., 2011), that have been successfully tested in rodents. According to these results obtained in animal models, preliminary findings in human patients are encouraging: rapamycin has been shown to induce the regression of astrocytomas in a small group (n = 5) of TSC cases (Franz et al., 2006), and to reduce seizure frequency in a single young TSC patient (Muncy et al., 2009). However, it is important to point out that while the effects of rapamycin on tumor growth in TSC patients are now well documented and reproduced in many cases, its efficacy on seizure control and other neurological deficits needs to be further investigated.
Migration Defects of Cortical Projection Neurons
The classification and neuropathological features of genetic neuronal migration disorders have been described in other reviews (Guerrini and Marini, 2006; Guerrini and Parrini, 2010). Different forms of lissencephaly (“smooth brain”) and heterotopias have been associated to mutations in genes involved in cortical neuron migration (LIS1, DCX, ARX, TUBA1A, RELN, FLNA, and ARFGEF2). Here we will focus on two of these genes, doublecortin (DCX) and reelin (RELN), whose function in neuronal migration and epilepsy has been investigated in more detail.
Doublecortin
Dominant, X-linked DCX mutations cause lissencephaly or subcortical band heterotopia (SBH; “double cortex”), syndromes characterized by mental retardation and epilepsy in humans. Specifically, DCX mutations cause lissencephaly in hemizygous males, whereas heterozygous females show a mosaic phenotype with a normal cortex and a second band of heterotopic neurons beneath the cortex (“double cortex”; des Portes et al., 1998; Matsumoto et al., 2001). DCX encodes a 40-kDa microtubule-associated protein (doublecortin) expressed in migrating neuroblasts and containing two conserved tandem repeats required for tubulin binding and microtubule stabilization (des Portes et al., 1998; Gleeson et al., 1998; Gleeson and Walsh, 2000). How doublecortin influences microtubule dynamics during brain embryonic development remains largely unclear.
Mice lacking Dcx (hemizygous Dcx−/Y male and heterozygous Dcx+/− female mice) have been generated in the attempt to reproduce the human phenotype of DCX mutations (Corbo et al., 2002). Dcx−/Y male mice present hippocampal lamination defects and a reduced number of hippocampal interneurons, but have no gross anatomical defects at the level of the cerebral cortex (Corbo et al., 2002; Nosten-Bertrand et al., 2008). Similar defects are observed also in Dcx+/− female mice. Importantly, Dcx−/Y male mice have hippocampal-dependent learning deficits (Corbo et al., 2002), are hyperexcitable and show spontaneous seizures with hippocampal onset (Nosten-Bertrand et al., 2008).
Severe migration defects of cortical projection neurons and cortical malformation have been instead detected in mice lacking both doublecortin and doublecortin-like kinase 1 (Dclk1; Deuel et al., 2006; Koizumi et al., 2006; Kerjan and Gleeson, 2007). More importantly, Dcx RNA interference (RNAi) performed by in utero electroporation results in migration defects of pyramidal cortical neurons and spontaneous seizures in adult rats (Bai et al., 2003; Ackman et al., 2009; Lapray et al., 2010), similar to those observed in human SBH. Manent et al. (2009) tested whether the postnatal re-expression of Dcx could rescue these anatomical and neurological deficits. To this aim, the Dcx knockdown RNAi vector was in utero electroporated into cortical neurons together with a tamoxifen-inducible Dcx expression vector system. In this way, it was possible to knockdown Dcx in cortical neurons at embryonic day (E)14 and then re-activate it between postnatal day (P)0 and P10 by treating newborn pups with tamoxifen. Postnatal re-expression of Dcx induced in this way was able to reduce neuronal migration defects and seizures in this model of SBH (Kerjan and Gleeson, 2009; Manent et al., 2009).
Dcx and doublecortin-like kinase have also been implicated in the migration of cortical interneurons (Friocourt et al., 2007; see section below). Taken together, these data indicate that doublecortin is required for hippocampal lamination as well as migration of neocortical pyramidal neurons and interneurons; as a consequence of this important role, an altered function of doublecortin during embryonic forebrain development results in epilepsy in the postnatal life.
Reelin
Mutation of the RELN gene, coding for the extracellular glycoprotein Reelin, causes a neuronal migration disorder called lissencephaly with cerebellar hypoplasia (Zaki et al., 2007; Guerrini and Parrini, 2010). Reelin binds several receptors, including a complex composed by the apolipoprotein E receptor 2 (ApoER2) and the very low-density lipoprotein receptor (VLDLR) (Honda et al., 2011). Following receptor binding, Reelin activates the intracellular protein Disabled homolog 1 (Dab1), whose phosphorylation enhances signal transduction pathways leading to axonal growth, neuronal migration, and synaptic plasticity (Frotscher, 2010; Honda et al., 2011). Reelin-activated pathways include the PI3K/Akt/Gsk-3β and Notch cascades (Honda et al., 2011). During embryonic development, Reelin is mainly expressed by Cajal–Retzius cells in the marginal zone of the neocortex and outer molecular layer of the hippocampus, whereas in the adult brain it is expressed in GABAergic interneurons (Alcantara et al., 1998). Mice lacking Reelin (reeler mice) show a dramatic impairment of migration of cortical projection neurons, that results in a highly disorganized and dyslaminated cerebral cortex (D’Arcangelo and Curran, 1998; D’Arcangelo, 2006). The final outcome is an increased susceptibility to hippocampal and neocortical epileptiform activity (Patrylo et al., 2006). During embryogenesis, the firstly generated cortical neurons migrate from the germinal zone and form the preplate. At the arrival of subsequently generated neurons, the preplate splits into two layers: the Cajal–Retzius layer (expressing Reelin) and the subplate. Successive waves of neurons migrate past the subplate, but not past the Cajal–Retzius layer, that will form the most superficial layer of the mature cerebral cortex. In the absence of Reelin, neuronal migration does not occur properly and neocortical lamination is perturbed. The preplate does not split, and newly generated neurons pile up behind the previously generated ones, resulting in an inverted layering of the neocortex. Several hypotheses have been proposed to explain the role of Reelin in neuronal cortical migration: (i) Reelin might function as a chemo-attractant for migrating neurons; (ii) it might control the preplate splitting that initiates the inside-out lamination; (iii) it might determine the detachment of neurons from radial glial processes and their subsequent differentiation (D’Arcangelo and Curran, 1998; Cooper, 2008; Gaiano, 2008). More recent studies suggest that Reelin and Notch signaling cooperate to regulate neuronal migration in the neocortex (Hashimoto-Torii et al., 2008).
Reelin is also implicated in the migration of excitatory neurons of the hippocampus; a marked dispersion of projection neurons is observed in the stratum pyramidale and dentate granule cell layer in reeler mice (D’Arcangelo and Curran, 1998). Granule cell dispersion (GCD) is often observed in human mesial temporal lobe epilepsy (MTLE), and recent studies investigated the role of Reelin in this form of epileptic condition. In the adult hippocampus, Reelin-positive interneurons (Cajal–Retzius cells) are normally found in the CA1 and CA3 regions, in the hilus, and along the hippocampal fissure, both in mouse (Heinrich et al., 2006) and human samples (Haas et al., 2002). Indeed, GCD correlates with hippocampal Reelin loss in MTLE patients, as shown by studies performed on hippocampal tissues resected from adult subjects who underwent temporal lobectomy for intractable MTLE (Haas et al., 2002). GCD and loss of Reelin was also observed in rodent models of MTLE (Heinrich et al., 2006; Gong et al., 2007; Antonucci et al., 2008, 2009; Duveau et al., 2010). The causative role of Reelin deficiency in GCD is confirmed by the observation that GCD is induced by the infusion of Reeling-blocking antibody, and prevented by the administration of exogenous Reelin (Haas and Frotscher, 2010). It remains still to be determined how Reelin loss and subsequent GCD contribute to epileptogenesis, and whether they are a cause or an effect of seizures (Haas and Frotscher, 2010).
Migration Defects of Cortical Interneurons
Cortical GABAergic interneurons are generated in ganglionic eminences of the basal forebrain, from where they migrate to reach their final location (Wonders and Anderson, 2006). Several genes have been implicated in interneuron differentiation and migration; the role of specific interneuron types in epilepsy is a developing field, which may help us to better understand the complex neurodevelopmental processes underlying seizure onset and control. Here we will focus on Dlx, Arx, Dcx, and Reelin genes, whose role in interneuron migration and possible implications in epileptogenesis has been extensively studied in mouse models.
Dlx
Migration and differentiation of cortical GABAergic interneurons has been shown to depend on the action of Dlx homeobox transcription factors, namely Dlx1/2 and Dlx5/6. Dlx genes are expressed in GABAergic neuron progenitors of the developing forebrain and in subsets of mature GABAergic interneurons of the adult cerebral cortex (Cobos et al., 2005b, 2007; Wang et al., 2010). Loss-of-function mutations of pairs of mouse Dlx genes (Dlx1/2 or Dlx 5/6) severely affect the migration and differentiation of distinct subpopulations of cortical interneurons. Specifically, Dlx1/2 have a crucial role in the migration and survival of calretinin (CR), somatostatin (SOM), and neuropeptide Y (NPY) positive interneurons (Anderson et al., 1997; Cobos et al., 2005b, 2007) whereas Dlx5/6 preferentially control parvalbumin-positive interneurons (Wang et al., 2010).
Migration defects and subsequent reduced number of cortical interneurons result in epilepsy in Dlx mutant mice. Mice lacking Dlx1 (Dlx1−/− mice) show selective apoptotic cell death of CR, SOM, and NPY cortical interneurons, resulting in decreased synaptic inhibition at 1 month of age. Accordingly, adult Dlx1−/− mice show noise- or handling-induced behavioral seizures and generalized electrographic activity (Cobos et al., 2005b). Mice homozygous for both Dlx5 and Dlx6 (Dlx5/6−/− mice) show a normal layering of the cerebral cortex and a normal positioning of MGE-derived cortical interneurons, but die perinatally. A reduced number of PV cortical interneurons is observed in these mutants, and the surviving PV interneurons show an increased dendritic branching (Wang et al., 2010). Conversely, heterozygous Dlx5/6+/− mice do not present interneuron loss nor other gross anatomical abnormalities, but develop epilepsy; this has been ascribed to functional deficit and aberrant connectivity of PV neurons (Wang et al., 2010).
Taken together, these studies clearly demonstrate that Dlx genes regulate the development and function of forebrain interneurons, which are crucial to maintain the inhibitory tone of the cerebral cortex and other brain structures. The reduced function of Dlx transcription factors (or of their transcriptional targets, such as GAD and vGAT; Stuhmer et al., 2002; Rubenstein, 2011) might increase the excitation/inhibition ratio thus predisposing the cerebral cortex to hyperexcitability and epilepsy.
Arx
The aristaless-related homeobox gene (Arx) belongs to the family of paired-class homeobox transcription factors, and its expression is controlled by the Dlx genes in cells derived from basal ganglia progenitor domains (Cobos et al., 2005a). Arx plays a pivotal role in the development of the CNS. ARX large deletions, protein truncating mutations and missense mutations in the homeobox region lead to X-linked lissencephaly with abnormal genitalia, while other ARX mutations cause intellectual disability, autism, and epilepsy without cortical malformations (Kitamura et al., 2002). Arx expression has been detected in the developing lateral and medial ganglionic eminence and later in cortical progenitors and migrating interneurons (Colombo et al., 2004; Friocourt et al., 2006). Studies in animal models showed that Arx is critical for radial migration of cortical progenitors and is crucially involved in the development and migration of GABAergic interneurons (Colombo et al., 2007; Friocourt et al., 2008; Marsh et al., 2009). In Arx-null embryos, interneuron migration from the lateral and medial ganglionic eminences is nearly absent, whereas migration through the cortical layers is only partially impaired. As a consequence, CR, NPY, and calbindin (CB) interneurons are severely reduced (Kitamura et al., 2002; Colombo et al., 2007). No loss of PV interneurons was instead observed in hemizygous Arx−/− mice suggesting the Arx loss results in specific interneuron subtype deficits (Friocourt and Parnavelas, 2010). These studies also revealed that abnormal expression of transcription factors potentially important for neuronal differentiation and migration occurs in Arx−/− mice.
Dcx
Cortical interneuron migration is also controlled by both Dcx and Dclk. Dcx knockdown by RNAi slows the migration of interneurons from the ganglionic eminence to the cerebral cortex (Friocourt et al., 2007), and cortices of Dcx, Dclk, and Dcx/Dclk mutant mice contain less CB-positive interneurons as compared to control mice (Friocourt et al., 2007; Nosten-Bertrand et al., 2008). An increased number of CR-positive interneurons is detected in the dentate gyrus of Dcx mutant mice (Nosten-Bertrand et al., 2008), whereas no defects of PV interneurons are detected in Dcx, Dclk, and Dcx/Dclk mutant mice (Kerjan and Gleeson, 2009).
Reelin
A large subset of cortical and hippocampal GABAergic interneurons express Reelin. These interneurons derive from the caudal ganglionic eminence (CGE), and occupy superficial layer of the cerebral cortex (Miyoshi et al., 2010) and strata lacunosum moleculare and deep radiatum of the mature hippocampus (Tricoire et al., 2011). The role of Reelin in interneuron migration and positioning in forebrain areas has been debated. Some authors showed that in reeler mice, GABAergic interneurons migrate normally into the developing forebrain, but fail to acquire proper layer position in the mature cerebral cortex (Hammond et al., 2006; Yabut et al., 2007). Using transplants to analyze the mechanisms underlying the positioning of cortical interneurons, other authors instead found that layer acquisition by these cells does not directly depend on Reelin signaling (Pla et al., 2006). Interestingly, a recent study showed that neuronal activity is essential for the migration and morphology of CGE-derived Reelin- and CR-positive interneurons (De Marco García et al., 2011). To suppress excitability in CGE-derived interneurons, the authors electroporated in utero the inward rectifying potassium channel Kir2.1 under the control of a Dlx5/6 enhancer element at E15.5, and then analyzed the positioning and morphology of Reelin and CR interneurons. Results showed that neuronal activity is required before P3 for the correct migration of CGE-derived interneurons, and that after P3, glutamate-mediated activity controls the development of their connections (De Marco García et al., 2011). These findings indicate that the genetic program underlying the development of Reelin-expressing interneurons is strongly modulated by activity, and may have implications for the role of Reelin in neurodevelopmental epileptic syndromes.
Gaba and Glutamate Control Neuronal Migration in the Forebrain
Studies performed on corticohippocampal organotypic co-cultures from EGFP-expressing mouse embryos demonstrated that GABA and glutamate may modulate neuronal migration in the developing forebrain. Specifically, GABA and glutamate modulate the migration of hippocampal pyramidal neurons respectively acting on GABAA and NMDA receptors (Manent et al., 2005), while glutamate controls the migration of GABAergic interneurons via AMPA receptor activation (Manent et al., 2006). These results led the authors to postulate that the migrations of glutamatergic and GABAergic interneurons are inter-dependent: glutamate released from pioneer glutamatergic neurons controls the migration of GABAergic interneurons, which in turn would facilitate glutamate neuron migration via GABA release (Manent and Represa, 2007). In keeping with these observation, the same authors showed that prenatal exposure to some AEDs acting on GABA signaling (such as vigabatrin, valproate, and lamotrigine) results in hippocampal and cortical dysplasias in the developing embryos (Manent et al., 2007, 2008), thus raising serious concerns about the possible consequences of AEDs use during pregnancy.
Developmental Remodeling of Neuronal Circuits
In the previous sections we have considered how defects in the early stages of brain development (i.e., neuronal proliferation and migration) can induce an epileptic phenotype. Following the initial assembly of excitatory and inhibitory neurons, immature neural networks are transformed into organized circuits that subserve adult brain function (Katz and Shatz, 1996). This process of network refinement is largely controlled by electrical activity. In particular, work in the sensory cortices has clearly established the existence of so-called “critical periods,” during which patterns of activity generated by sensory experience play a critical role for maturation of cortical function (Berardi et al., 2000). During these sensitive phases of development, initially exuberant connections are eliminated and neuronal dendrites are correspondingly pruned and narrowed. The remaining synapses functionally mature (Katz and Shatz, 1996). In particular, in the visual system, synapse elimination and remodeling depends upon the amount and patterning of neural activity within the visual pathway (Fagiolini et al., 1994; Caleo et al., 2007).
Perturbations in this developmental refinement of neuronal circuitry during critical periods may trigger hyperexcitability and epilepsy later in life. One clear example comes from a study on blockade of hippocampal activity during early development (Galvan et al., 2000). In the rodent hippocampus, CA3 pyramidal neurons display an exuberant growth of axon collaterals during postnatal weeks 2 and 3; this developmental time period corresponds to a critical phase, when CA3 networks have a marked propensity to generate epileptic seizures (Swann and Brady, 1984). Exuberant CA3 axons are then remodeled with maturation, so that about half of the branches are eliminated (Gomez-Di Cesare et al., 1997). To prevent this axonal remodeling, Swann and colleagues infused tetrodotoxin (a blocker of voltage-gated sodium channels) into the rat hippocampus for about 10 days starting from P12 (Galvan et al., 2000). This transient blockade of neuronal activity resulted in the establishment of a chronic epileptic focus in adulthood, with prolonged electrographic seizures originating from the infused hippocampus (Galvan et al., 2000). These results are consistent with the idea that blockade of neuronal activity during early critical phases can enhance seizure susceptibility later in life by preventing developmental remodeling of neural circuits. Indeed, the immature brain is more prone to seizures than the adult brain (Ben-Ari and Holmes, 2006).
Recently, a persistent immaturity of glutamatergic circuitries in the hippocampus was found to underlie seizure susceptibility in autosomal dominant lateral temporal lobe epilepsy (ADLTE; Zhou et al., 2009). This study investigated the function of LGI1 (leucine-rich, glioma-inactivated 1), a gene that is implicated in about half the cases of ADLTE (Kalachikov et al., 2002; Striano et al., 2011). LGI1 encodes a protein that localizes to glutamatergic synapses, binds its receptors ADAM22 and ADAM23 (disintegrin and metalloproteinase domain 22 and 23), and copurifies with presynaptic and postsynaptic regulatory molecules (Fukata et al., 2006; Schulte et al., 2006). LGI1 expression increases in the rodent hippocampus exactly during the third postnatal week, when glutamatergic synapses are pruned and mature (Fukata et al., 2006; Zhou et al., 2009). In particular, excitatory neurons downregulate their presynaptic vesicular release probability and reduce their postsynaptic NMDA-receptor subunit NR2B; during this same period, dendritic arbors and spines are pruned and remodeled. Anderson and colleagues generated mice expressing a truncated form of the LGI1 gene, found in ADLTE patients, and showed that hippocampal glutamatergic synapses did not properly mature in these mice (Zhou et al., 2009). Specifically, (i) the normal, age-dependent decrease of probability of glutamate release was blocked in mutant mice; (ii) developmental changes in glutamate receptor composition were impaired by mutated LGI1; and (iii) dendrites of adult mutant mice remained immature, with a high density of branches and spine protrusions (Zhou et al., 2009). Importantly, mutant LGI1 mice displayed a lower threshold for the development of pharmacologically induced seizures. There was no significant effect of mutated LGI1 on GABAergic neurotransmission (Zhou et al., 2009; Yu et al., 2010). All together, these data suggest that mutated LGI1 blocks the normal functional maturation of both presynaptic and postsynaptic compartments and halts structural pruning, thus maintaining a high density of excitatory synaptic inputs converging onto hippocampal neurons and leading to a hyperexcitability phenotype (Caleo, 2009; Zhou et al., 2009). Other reports are consistent with the idea that LGI1 regulates the development of glutamatergic synapses (Fukata et al., 2010; Yu et al., 2010). In keeping with this findings, recent data show that heterozygous mice lacking LGI1 (LGI1+/− mice) display a lower threshold to auditory stimuli induced seizures, and homozygous LGI1−/− mice develop spontaneous recurrent seizures followed hippocampal neuronal loss, mossy fiber sprouting, astrocyte reactivity, and GCD (Chabrol et al., 2010).
Epileptogenic Ion Channel Mutations: Focus on SCN2A
In the last years, a number of epileptogenic mutations have been identified in different genes (Noebels, 2003). The majority of these genes code for voltage- or ligand-gated ion channels. Mutations in different subunits of sodium, potassium, and calcium channels underlie different forms of genetic epilepsies (“channelopathies”; Kullmann, 2010; Mantegazza et al., 2010). Mutations of the nicotinic acetylcholine receptor subunits are associated with autosomal dominant nocturnal frontal lobe epilepsy (De Fusco et al., 2000). Mutations in the voltage-gated potassium channel subunit genes KCNQ2 and KCNQ3 are associated with benign familial neonatal seizures (Leppert and Singh, 1999). Mutations in the GABAA receptor, which is the primary mediator of synaptic inhibition, have also been found to contribute to several idiopathic epilepsies and febrile seizures (Galanopoulou, 2010). The functional consequences of these epileptogenic mutations have been amply studied, and the data have provided significant knowledge on the pathogenic mechanisms that lead to epilepsy. For more details on these findings, the reader is referred to a series of excellent reviews (Noebels, 2003; Kullmann, 2010; Mantegazza et al., 2010).
Mutations in voltage-gated sodium channel genes (Nav) are the most common genetic cause of familial epilepsy. Specifically, mutations in the Nav1.1 alpha subunit gene (SCN1A) are responsible for “generalized epilepsy with febrile seizures plus” (GEFS+; Scheffer and Berkovic, 1997) and Dravet’s syndrome (Mantegazza et al., 2010; Meisler et al., 2010). Missense mutations in Nav1.2 alpha subunit (SCN2A) are found in patients with benign familial neonatal-infantile convulsions (Avanzini et al., 2007). Functional studies demonstrate that Nav1.2 mutations result in modifications of the gating properties of the channel, with a net amplification of the sodium current and resulting greater depolarization, consistent with the hyperexcitability phenotype (Scalmani et al., 2006).
It is important to point out that it is often difficult to predict the epileptic phenotype based solely from the change in the behavior of the mutated channel. First, it is crucial to determine cell-specific patterns of expression of the mutated subunit, as expression in excitatory vs. inhibitory neurons can lead to completely opposite effects on excitability status of the neuronal network (Yu et al., 2006). Second, the cellular background (i.e., the modifier effect of other channels expressed in the same neuron) has to be considered (Glasscock et al., 2007). “Humanized” mouse models (in which human mutated channel sequences are inserted in genetically engineered mice) offer an unique opportunity to study the modulatory role of the genetic background and of the interactions between different genes. One example is represented by transgenic mice (Q54) carrying a human, epileptogenic missense mutation of the sodium channel SCN2A (Nav1.2; Kearney et al., 2001). Mice carrying the Q54 transgene on a SJL strain background exhibit severe spontaneous seizures originating in the hippocampus with onset at about 4 weeks, and progressive hippocampal sclerosis with extensive cell loss and gliosis in areas CA1, CA3, and hilus (Kearney et al., 2001; Manno et al., 2011). However, on a pure C57BL/6J background, onset of seizures is delayed and the epileptic phenotype is mild. Two modifier loci responsible for the difference in severity between strains C57BL/6J and SJL have been mapped, and the evidence points to the voltage-gated potassium channel gene Kcnv2 as one modifier (Bergren et al., 2005, 2009). These data indicate that severity of the epileptic condition may be significantly impacted by gene interactions.
Finally, in the study of channelopathies it is important to underlie the possible effects of the complex interaction between mutated genes and the environment. As we have described in the previous section, early environmental conditions shape neuronal connectivity, and the resulting changes in network organization can potently affect clinical expression of a mutated channel. In keeping with this notion, Q54 transgenic mice (carrying an epileptogenic mutated SCN2A; see above) exposed to environmental enrichment from birth show a dramatic reduction of spontaneous seizures and hippocampal cell loss (Manno et al., 2011). The data indicate that an enriched housing from birth may have profound antiepileptic and neuroprotective effects. Environmental enrichment may exert these actions by up-regulation of neurotrophic factors, plastic rearrangements in excitatory/inhibitory circuits, and stimulation of neurogenesis (Dhanushkodi and Shetty, 2008; Sale et al., 2009). Thus, changes in network organization due to environmental influences can halt epileptogenic changes and dampen hyperexcitability of neural networks.
Epileptogenic Alterations of Presynaptic Protein Function
In addition to channelopathies, defects in the control of neurotransmitter release account for a wide variety of epileptic syndromes. Presynaptic proteins that have been found to be involved in epilepsy include the synaptic vesicle (SV) protein SV2A (Crowder et al., 1999; Janz et al., 1999), the SV-anchoring phosphoproteins synapsins (Rosahl et al., 1995; Baldelli et al., 2007) and the plasma membrane fusion protein SNAP-25 (Zhang et al., 2004). Not all disruptions of the neurotransmitter machinery are equally epileptogenic, as shown by the lack of seizures in mice lacking other presynaptic proteins, such as synaptotagmin (Fernandez-Chacon et al., 2001) or synaptobrevin/VAMP (Schoch et al., 2001). Data obtained in mice mutant for synapsins and SNAP-25 are particularly interesting, since they suggest that hyperexcitability might result from developmental alterations causing an unbalance in the activity of excitatory and inhibitory neurons.
Synapsins
Synapsins (Syns) comprise a family of neuron-specific proteins encoded by three distinct genes (SYNI, SYNII, and SYNIII) located in chromosome X, 3, and 22, respectively. The Syns are phosphoproteins implicated in the regulation of neurotransmitter release and synapse formation. In particular, Syns are responsible for the formation and maintenance of SV pools within the nerve terminal (Benfenati et al., 1992) and their expression has been causally related to the formation and maturation of synaptic connectivity during neural development and synaptogenesis (Valtorta et al., 1995; Bonanomi et al., 2005). Despite the absence of gross defects in brain morphology, Syn I and Syn II (but not Syn III) knockout (KO) mice exhibit early-onset spontaneous and sensory stimuli-evoked epileptic seizures (Rosahl et al., 1995). Syn I mutations can also contribute to human epilepsy (Garcia et al., 2004). In Syn KO mice, spontaneous recurrent seizures develop after 2 months of age and the animals become more susceptible with age (Rosahl et al., 1995). This latent period of “epileptogenesis” suggests that alterations in the developmental assembly of neural networks may underlie hyperexcitability in these mice.
Several theories have been put forward to explain the nexus between the molecular function of Syns at the neuronal level and the onset of the epileptic phenotype. Since Syns are involved in crucial steps of presynaptic physiology and play a role in the formation of the synaptic connectivity during development, it is likely that mutations affecting their expression and/or phosphorylation-dependent functions will result in significant changes in synaptic transmission, plasticity and development which could be potentially related to the appearance of an epileptic phenotype. Thus, the epileptic phenotype of Syn KO animals might be due to a defect in the overall brain connectivity (Chin et al., 1995), or to an impairment in synaptogenesis within confined neuronal populations (Gitler et al., 2004; Bonanomi et al., 2005). There is also support for the idea that lack of Syns produces an imbalance in the activity of excitatory and inhibitory neurons. Indeed, Syn I KO reduces the readily releasable pool at inhibitory synapses, and increases it at excitatory synapses (Baldelli et al., 2007; Chiappalone et al., 2009).
SNAP-25
A further example of presynaptic protein involved in epilepsy is SNAP-25. SNAP-25, together with syntaxin and synaptobrevin/VAMP, is a member of the SNARE (soluble NSF attachment protein receptor) complex which contributes to the formation of the fusion complex required for SV exocytosis. Beside participating in SV fusion, SNAP-25 has been proposed to regulate exocytosis at additional steps. Through its interaction with other proteins, such as synaptotagmin or complexins (Jahn et al., 2003) or through putative calcium binding sites (Sorensen et al., 2002), SNAP-25 appears to participate in the calcium-dependent regulation of exocytosis. SNAP-25 has also been shown to interact with and modulate the activity of various voltage-activated calcium channels (Atlas, 2001; Pozzi et al., 2008; Condliffe et al., 2010). The possible involvement of SNAP-25 in neuronal hyperexcitability and epilepsy comes from the neurological mouse mutant Coloboma (Cm/+), bearing an autosomal dominant deletion on chromosome 2, including the SNAP-25 gene (Hess et al., 1992, 1994). Cm/+ mice show 50% reduction in SNAP-25 mRNA and protein, and impaired evoked neurotransmitter release (Raber et al., 1997; Wilson, 2000). Interestingly, Cm/+ mice show robust cortical cortical spike-wave discharges and increased thalamic T-type currents (Zhang et al., 2004), two typical features of “absence” epilepsy (Tsakiridou et al., 1995; Coenen and Van Luijtelaar, 2003).
Similarly to what already described for Syns, the role of SNAP-25 in the etiopathology of epilepsy may involve alterations of synaptic formation/refinement during development. The epileptic phenotype of SNAP-25 mutant mice may be produced by a defect in the overall brain connectivity consequent to the reduced expression of the protein, which is known to play a crucial role in neurite extension (Osen-Sand et al., 1996). Alternatively, it may be the consequence of unbalances in neurotransmitter release due to deficits in the SNARE complex. Interestingly, SNAP-25 is expressed at much higher levels at excitatory vs. inhibitory synapses (Verderio et al., 2004; Bragina et al., 2007). Thus, hyperexcitability could result from perturbations of the processes that balance the developmental assembly of inhibitory and excitatory circuits. Furthermore, recent studies have shown a role of SNAP-25 in the regulation of calcium homeostasis in neurons. In particular, it has been shown that phosphorylated SNAP-25 negatively modulates calcium dynamics by inhibiting voltage-gated calcium channels (Verderio et al., 2004; Pozzi et al., 2008). Thus, reduced levels of SNAP-25 may lead to increased calcium currents (Condliffe et al., 2010) and consequently to the onset of epileptic discharges.
Conclusions and Future Directions
In this review, we have highlighted some of the neurodevelopmental pathways that lead to a chronic epileptic condition in adulthood. Abnormal development of the cerebral cortex (due to perturbations of neuronal proliferation and/or migration) is a frequent cause of epilepsy. In addition, more subtle alterations in the assembly and fine-tuning of neuronal networks may also lead to an hyperexcitability phenotype later in life. A better understanding of the biology of these epileptogenic mechanisms has important implications for the development of novel therapeutic approaches.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by the Italian Ministry of University and Research (PRIN 2008 grant # 200894SYW2_002 to Yuri Bozzi), Italian Ministry of Health (grant RF-TAA-2008-1141282 to Yuri Bozzi and Young Investigator Grant to Matteo Caleo), Tuscany Region (Health Program 2009 to Matteo Caleo), and Compagnia di San Paolo (Matteo Caleo).
References
Ackman, J. B., Aniksztejn, L., Crepel, V., Becq, H., Pellegrino, C., Cardoso, C., Ben-Ari, Y., and Represa, A. (2009). Abnormal network activity in a targeted genetic model of human double cortex. J. Neurosci. 29, 313–327.
Alcantara, S., Ruiz, M., D’Arcangelo, G., Ezan, F., De Lecea, L., Curran, T., Sotelo, C., and Soriano, E. (1998). Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. J. Neurosci. 18, 7779–7799.
Anderl, S., Freeland, M., Kwiatkowski, D. J., and Goto, J. (2011). Therapeutic value of prenatal rapamycin treatment in a mouse brain model of tuberous sclerosis complex. Hum. Mol. Genet. 20, 4597–4604.
Anderson, S. A., Eisenstat, D. D., Shi, L., and Rubenstein, J. L. (1997). Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science 278, 474–476.
Antonucci, F., Bozzi, Y., and Caleo, M. (2009). Intrahippocampal infusion of botulinum neurotoxin E (BoNT/E) reduces spontaneous recurrent seizures in a mouse model of mesial temporal lobe epilepsy. Epilepsia 50, 963–966.
Antonucci, F., Di Garbo, A., Novelli, E., Manno, I., Sartucci, F., Bozzi, Y., and Caleo, M. (2008). Botulinum neurotoxin E (BoNT/E) reduces CA1 neuron loss and granule cell dispersion, with no effects on chronic seizures, in a mouse model of temporal lobe epilepsy. Exp. Neurol. 210, 388–401.
Atlas, D. (2001). Functional and physical coupling of voltage-sensitive calcium channels with exocytotic proteins: ramifications for the secretion mechanism. J. Neurochem. 77, 972–985.
Avanzini, G., and Franceschetti, S. (2003). Cellular biology of epileptogenesis. Lancet Neurol. 2, 33–42.
Avanzini, G., Franceschetti, S., and Mantegazza, M. (2007). Epileptogenic channelopathies: experimental models of human pathologies. Epilepsia 48(Suppl. 2), 51–64.
Bai, J., Ramos, R. L., Ackman, J. B., Thomas, A. M., Lee, R. V., and Loturco, J. J. (2003). RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat. Neurosci. 6, 1277–1283.
Baldelli, P., Fassio, A., Valtorta, F., and Benfenati, F. (2007). Lack of synapsin I reduces the readily releasable pool of synaptic vesicles at central inhibitory synapses. J. Neurosci. 27, 13520–13531.
Barkovich, A. J., Kuzniecky, R. I., Jackson, G. D., Guerrini, R., and Dobyns, W. B. (2005). A developmental and genetic classification for malformations of cortical development. Neurology 65, 1873–1887.
Ben-Ari, Y., and Holmes, G. L. (2006). Effects of seizures on developmental processes in the immature brain. Lancet Neurol. 5, 1055–1063.
Benfenati, F., Valtorta, F., Rubenstein, J. L., Gorelick, F. S., Greengard, P., and Czernik, A. J. (1992). Synaptic vesicle-associated Ca2+/calmodulin-dependent protein kinase II is a binding protein for synapsin I. Nature 359, 417–420.
Berardi, N., Pizzorusso, T., and Maffei, L. (2000). Critical periods during sensory development. Curr. Opin. Neurobiol. 10, 138–145.
Bergren, S. K., Chen, S., Galecki, A., and Kearney, J. A. (2005). Genetic modifiers affecting severity of epilepsy caused by mutation of sodium channel Scn2a. Mamm. Genome 16, 683–690.
Bergren, S. K., Rutter, E. D., and Kearney, J. A. (2009). Fine mapping of an epilepsy modifier gene on mouse chromosome 19. Mamm. Genome 20, 359–366.
Bonanomi, D., Menegon, A., Miccio, A., Ferrari, G., Corradi, A., Kao, H. T., Benfenati, F., and Valtorta, F. (2005). Phosphorylation of synapsin I by cAMP-dependent protein kinase controls synaptic vesicle dynamics in developing neurons. J. Neurosci. 25, 7299–7308.
Bonnet, C. S., Aldred, M., von Ruhland, C., Harris, R., Sandford, R., and Cheadle, J. P. (2009). Defects in cell polarity underlie TSC and ADPKD-associated cystogenesis. Hum. Mol. Genet. 18, 2166–2176.
Bragina, L., Candiracci, C., Barbaresi, P., Giovedi, S., Benfenati, F., and Conti, F. (2007). Heterogeneity of glutamatergic and GABAergic release machinery in cerebral cortex. Neuroscience 146, 1829–1840.
Buckmaster, P. S., and Lew, F. H. (2011). Rapamycin suppresses mossy fiber sprouting but not seizure frequency in a mouse model of temporal lobe epilepsy. J. Neurosci. 31, 2337–2347.
Cahana, A., Escamez, T., Nowakowski, R. S., Hayes, N. L., Giacobini, M., von Holst, A., Shmueli, O., Sapir, T., McConnell, S. K., Wurst, W., Martinez, S., and Reiner, O. (2001). Targeted mutagenesis of Lis1 disrupts cortical development and LIS1 homodimerization. Proc. Natl. Acad. Sci. U.S.A. 98, 6429–6434.
Caleo, M., Restani, L., Gianfranceschi, L., Costantin, L., Rossi, C., Rossetto, O., Montecucco, C., and Maffei, L. (2007). Transient synaptic silencing of developing striate cortex has persistent effects on visual function and plasticity. J. Neurosci. 27, 4530–4540.
Chabrol, E., Navarro, V., Provenzano, G., Cohen, I., Dinocourt, C., Rivaud-Péchoux, S., Fricker, D., Baulac, M., Miles, R., Leguern, E., and Baulac, S. (2010). Electroclinical characterization of epileptic seizures in leucine-rich, glioma-inactivated 1-deficient mice. Brain 133, 2749–2762.
Chiappalone, M., Casagrande, S., Tedesco, M., Valtorta, F., Baldelli, P., Martinoia, S., and Benfenati, F. (2009). Opposite changes in glutamatergic and GABAergic transmission underlie the diffuse hyperexcitability of synapsin I-deficient cortical networks. Cereb. Cortex 19, 1422–1439.
Chin, L. S., Li, L., Ferreira, A., Kosik, K. S., and Greengard, P. (1995). Impairment of axonal development and of synaptogenesis in hippocampal neurons of synapsin I-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 92, 9230–9234.
Cobos, I., Borello, U., and Rubenstein, J. L. (2007). Dlx transcription factors promote migration through repression of axon and dendrite growth. Neuron 54, 873–888.
Cobos, I., Broccoli, V., and Rubenstein, J. L. (2005a). The vertebrate ortholog of Aristaless is regulated by Dlx genes in the developing forebrain. J. Comp. Neurol. 483, 292–303.
Cobos, I., Calcagnotto, M. E., Vilaythong, A. J., Thwin, M. T., Noebels, J. L., Baraban, S. C., and Rubenstein, J. L. (2005b). Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat. Neurosci. 8, 1059–1068.
Coenen, A. M., and Van Luijtelaar, E. L. (2003). Genetic animal models for absence epilepsy: a review of the WAG/Rij strain of rats. Behav. Genet. 33, 635–655.
Colombo, E., Collombat, P., Colasante, G., Bianchi, M., Long, J., Mansouri, A., Rubenstein, J. L., and Broccoli, V. (2007). Inactivation of Arx, the murine ortholog of the X-linked lissencephaly with ambiguous genitalia gene, leads to severe disorganization of the ventral telencephalon with impaired neuronal migration and differentiation. J. Neurosci. 27, 4786–4798.
Colombo, E., Galli, R., Cossu, G., Gecz, J., and Broccoli, V. (2004). Mouse orthologue of ARX, a gene mutated in several X-linked forms of mental retardation and epilepsy, is a marker of adult neural stem cells and forebrain GABAergic neurons. Dev. Dyn. 231, 631–639.
Condliffe, S. B., Corradini, I., Pozzi, D., Verderio, C., and Matteoli, M. (2010). Endogenous SNAP-25 regulates native voltage-gated calcium channels in glutamatergic neurons. J. Biol. Chem. 285, 24968–24976.
Cooper, J. A. (2008). A mechanism for inside-out lamination in the neocortex. Trends Neurosci. 31, 113–119.
Corbo, J. C., Deuel, T. A., Long, J. M., Laporte, P., Tsai, E., Wynshaw-Boris, A., and Walsh, C. A. (2002). Doublecortin is required in mice for lamination of the hippocampus but not the neocortex. J. Neurosci. 22, 7548–7557.
Crowder, K. M., Gunther, J. M., Jones, T. A., Hale, B. D., Zhang, H. Z., Peterson, M. R., Scheller, R. H., Chavkin, C., and Bajjalieh, S. M. (1999). Abnormal neurotransmission in mice lacking synaptic vesicle protein 2A (SV2A). Proc. Natl. Acad. Sci. U.S.A. 96, 15268–15273.
D’Arcangelo, G. (2006). Reelin mouse mutants as models of cortical development disorders. Epilepsy Behav. 8, 81–90.
D’Arcangelo, G., and Curran, T. (1998). Reeler: new tales on an old mutant mouse. Bioessays 20, 235–244.
De Fusco, M., Becchetti, A., Patrignani, A., Annesi, G., Gambardella, A., Quattrone, A., Ballabio, A., Wanke, E., and Casari, G. (2000). The nicotinic receptor beta 2 subunit is mutant in nocturnal frontal lobe epilepsy. Nat. Genet. 26, 275–276.
De Marco García, N. V., Karayannis, T., and Fishell, G. (2011). Neuronal activity is required for the development of specific cortical interneuron subtypes. Nature 472, 351–355.
des Portes, V., Pinard, J. M., Billuart, P., Vinet, M. C., Koulakoff, A., Carrie, A., Gelot, A., Dupuis, E., Motte, J., Berwald-Netter, Y., Catala, M., Kahn, A., Beldjord, C., and Chelly, J. (1998). A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell 92, 51–61.
Deuel, T. A., Liu, J. S., Corbo, J. C., Yoo, S. Y., Rorke-Adams, L. B., and Walsh, C. A. (2006). Genetic interactions between doublecortin and doublecortin-like kinase in neuronal migration and axon outgrowth. Neuron 49, 41–53.
Dhanushkodi, A., and Shetty, A. K. (2008). Is exposure to enriched environment beneficial for functional post-lesional recovery in temporal lobe epilepsy? Neurosci. Biobehav. Rev. 32, 657–674.
Duveau, V., Madhusudan, A., Caleo, M., Knuesel, I., and Fritschy, J. M. (2010). Impaired reelin processing and secretion by Cajal–Retzius cells contributes to granule cell dispersion in a mouse model of temporal lobe epilepsy. Hippocampus 21, 935–944.
Ehninger, D., Han, S., Shilyansky, C., Zhou, Y., Li, W., Kwiatkowski, D. J., Ramesh, V., and Silva, A. J. (2008). Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat. Med. 14, 843–848.
Fagiolini, M., Pizzorusso, T., Berardi, N., Domenici, L., and Maffei, L. (1994). Functional postnatal development of the rat primary visual cortex and the role of visual experience: dark rearing and monocular deprivation. Vision Res. 34, 709–720.
Feliciano, D. M., Su, T., Lopez, J., Platel, J. C., and Bordey, A. (2011). Single-cell Tsc1 knockout during corticogenesis generates tuber-like lesions and reduces seizure threshold in mice. J. Clin. Invest. 121, 1596–1607.
Feng, Y., Chen, M. H., Moskowitz, I. P., Mendonza, A. M., Vidali, L., Nakamura, F., Kwiatkowski, D. J., and Walsh, C. A. (2006). Filamin A (FLNA) is required for cell–cell contact in vascular development and cardiac morphogenesis. Proc. Natl. Acad. Sci. U.S.A. 103, 19836–19841.
Fernandez-Chacon, R., Konigstorfer, A., Gerber, S. H., Garcia, J., Matos, M. F., Stevens, C. F., Brose, N., Rizo, J., Rosenmund, C., and Sudhof, T. C. (2001). Synaptotagmin I functions as a calcium regulator of release probability. Nature 410, 41–49.
Fishell, G. (2007). Perspectives on the developmental origins of cortical interneuron diversity. Novartis Found. Symp. 288, 21–35; discussion 35–44, 96–28.
Fishell, G., and Hanashima, C. (2008). Pyramidal neurons grow up and change their mind. Neuron 57, 333–338.
Franz, D. N., Leonard, J., Tudor, C., Chuck, G., Care, M., Sethuraman, G., Dinopoulos, A., Thomas, G., and Crone, K. R. (2006). Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann. Neurol. 59, 490–498.
Friocourt, G., Kanatani, S., Tabata, H., Yozu, M., Takahashi, T., Antypa, M., Raguenes, O., Chelly, J., Ferec, C., Nakajima, K., and Parnavelas, J. G. (2008). Cell-autonomous roles of ARX in cell proliferation and neuronal migration during corticogenesis. J. Neurosci. 28, 5794–5805.
Friocourt, G., Liu, J. S., Antypa, M., Rakic, S., Walsh, C. A., and Parnavelas, J. G. (2007). Both doublecortin and doublecortin-like kinase play a role in cortical interneuron migration. J. Neurosci. 27, 3875–3883.
Friocourt, G., and Parnavelas, J. G. (2010). Mutations in ARX result in several defects involving GABAergic neurons. Front. Cell. Neurosci. 4:4. doi:10.3389/fncel.2010.00004
Friocourt, G., Poirier, K., Rakic, S., Parnavelas, J. G., and Chelly, J. (2006). The role of ARX in cortical development. Eur. J. Neurosci. 23, 869–876.
Frotscher, M. (2010). Role for Reelin in stabilizing cortical architecture. Trends Neurosci. 33, 407–414.
Fukata, Y., Adesnik, H., Iwanaga, T., Bredt, D. S., Nicoll, R. A., and Fukata, M. (2006). Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science 313, 1792–1795.
Fukata, Y., Lovero, K. L., Iwanaga, T., Watanabe, A., Yokoi, N., Tabuchi, K., Shigemoto, R., Nicoll, R. A., and Fukata, M. (2010). Disruption of LGI1-linked synaptic complex causes abnormal synaptic transmission and epilepsy. Proc. Natl. Acad. Sci. U.S.A. 107, 3799–3804.
Gaiano, N. (2008). Strange bedfellows: Reelin and Notch signaling interact to regulate cell migration in the developing neocortex. Neuron 60, 189–191.
Galanopoulou, A. S. (2010). Mutations affecting GABAergic signaling in seizures and epilepsy. Pflugers Arch. 460, 505–523.
Galvan, C. D., Hrachovy, R. A., Smith, K. L., and Swann, J. W. (2000). Blockade of neuronal activity during hippocampal development produces a chronic focal epilepsy in the rat. J. Neurosci. 20, 2904–2916.
Garcia, C. C., Blair, H. J., Seager, M., Coulthard, A., Tennant, S., Buddles, M., Curtis, A., and Goodship, J. A. (2004). Identification of a mutation in synapsin I, a synaptic vesicle protein, in a family with epilepsy. J. Med. Genet. 41, 183–186.
Gitler, D., Takagishi, Y., Feng, J., Ren, Y., Rodriguiz, R. M., Wetsel, W. C., Greengard, P., and Augustine, G. J. (2004). Different presynaptic roles of synapsins at excitatory and inhibitory synapses. J. Neurosci. 24, 11368–11380.
Glasscock, E., Qian, J., Yoo, J. W., and Noebels, J. L. (2007). Masking epilepsy by combining two epilepsy genes. Nat. Neurosci. 10, 1554–1558.
Gleeson, J. G., Allen, K. M., Fox, J. W., Lamperti, E. D., Berkovic, S., Scheffer, I., Cooper, E. C., Dobyns, W. B., Minnerath, S. R., Ross, M. E., and Walsh, C. A. (1998). Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 92, 63–72.
Gleeson, J. G., and Walsh, C. A. (2000). Neuronal migration disorders: from genetic diseases to developmental mechanisms. Trends Neurosci. 23, 352–359.
Gomez-Di Cesare, C. M., Smith, K. L., Rice, F. L., and Swann, J. W. (1997). Axonal remodeling during postnatal maturation of CA3 hippocampal pyramidal neurons. J. Comp. Neurol. 384, 165–180.
Gong, C., Wang, T. W., Huang, H. S., and Parent, J. M. (2007). Reelin regulates neuronal progenitor migration in intact and epileptic hippocampus. J. Neurosci. 27, 1803–1811.
Goorden, S. M. I., van Woerden, G. M., van der Weerd, L., Cheadle, J. P., and Elgersma, Y. (2007). Cognitive deficits in Tsc1((/() mice in the absence of cerebral lesions and seizures. Ann. Neurol. 62, 648–655.
Guerrini, R., and Marini, C. (2006). Genetic malformations of cortical development. Exp. Brain Res. 173, 322–333.
Guillemot, F., Molnar, Z., Tarabykin, V., and Stoykova, A. (2006). Molecular mechanisms of cortical differentiation. Eur. J. Neurosci. 23, 857–868.
Haas, C. A., Dudeck, O., Kirsch, M., Huszka, C., Kann, G., Pollak, S., Zentner, J., and Frotscher, M. (2002). Role for reelin in the development of granule cell dispersion in temporal lobe epilepsy. J. Neurosci. 22, 5797–5802.
Haas, C. A., and Frotscher, M. (2010). Reelin deficiency causes granule cell dispersion in epilepsy. Exp. Brain Res. 200, 141–149.
Hammond, V., So, E., Gunnersen, J., Valcanis, H., Kalloniatis, M., and Tan, S. S. (2006). Layer positioning of late-born cortical interneurons is dependent on Reelin but not p35 signaling. J. Neurosci. 26, 1646–1655.
Hansen, D. V., Lui, J. H., Parker, P. R., and Kriegstein, A. R. (2010). Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 464, 554–561.
Hashimoto-Torii, K., Torii, M., Sarkisian, M. R., Bartley, C. M., Shen, J., Radtke, F., Gridley, T., Sestan, N., and Rakic, P. (2008). Interaction between Reelin and Notch signaling regulates neuronal migration in the cerebral cortex. Neuron 60, 273–284.
Heinrich, C., Nitta, N., Flubacher, A., Muller, M., Fahrner, A., Kirsch, M., Freiman, T., Suzuki, F., Depaulis, A., Frotscher, M., and Haas, C. A. (2006). Reelin deficiency and displacement of mature neurons, but not neurogenesis, underlie the formation of granule cell dispersion in the epileptic hippocampus. J. Neurosci. 26, 4701–4713.
Hess, E. J., Collins, K. A., Copeland, N. G., Jenkins, N. A., and Wilson, M. C. (1994). Deletion map of the coloboma (Cm) locus on mouse chromosome 2. Genomics 21, 257–261.
Hess, E. J., Jinnah, H. A., Kozak, C. A., and Wilson, M. C. (1992). Spontaneous locomotor hyperactivity in a mouse mutant with a deletion including the Snap gene on chromosome 2. J. Neurosci. 12, 2865–2874.
Holmes, G. L., and Stafstrom, C. E. (2007). Tuberous sclerosis complex and epilepsy: recent developments and future challenges. Epilepsia 48, 617–630.
Honda, T., Kobayashi, K., Mikoshiba, K., and Nakajima, K. (2011). Regulation of cortical neuron migration by the Reelin signaling pathway. Neurochem. Res. 36, 1270–1279.
Huang, X., Zhang, H., Yang, J., Wu, J., Mcmahon, J., Lin, Y., Cao, Z., Gruenthal, M., and Huang, Y. (2010). Pharmacological inhibition of the mammalian target of rapamycin pathway suppresses acquired epilepsy. Neurobiol. Dis. 40, 193–199.
Janz, R., Goda, Y., Geppert, M., Missler, M., and Sudhof, T. C. (1999). SV2A and SV2B function as redundant Ca2+ regulators in neurotransmitter release. Neuron 24, 1003–1016.
Jozwiak, J. (2006). Hamartin and tuberin: working together for tumour suppression. Int. J. Cancer 118, 1–5.
Kalachikov, S., Evgrafov, O., Ross, B., Winawer, M., Barker-Cummings, C., Martinelli Boneschi, F., Choi, C., Morozov, P., Das, K., Teplitskaya, E., Yu, A., Cayanis, E., Penchaszadeh, G., Kottmann, A. H., Pedley, T. A., Hauser, W. A., Ottman, R., and Gilliam, T. C. (2002). Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat. Genet. 30, 335–341.
Katz, L. C., and Shatz, C. J. (1996). Synaptic activity and the construction of cortical circuits. Science 274, 1133–1138.
Kearney, J. A., Plummer, N. W., Smith, M. R., Kapur, J., Cummins, T. R., Waxman, S. G., Goldin, A. L., and Meisler, M. H. (2001). A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience 102, 307–317.
Keays, D. A., Tian, G., Poirier, K., Huang, G.-J., Siebold, C., Cleak, J., Oliver, P. L., Fray, M., Harvey, R. J., Molnar, Z., Pinon, M. C., Dear, N., Valdar, W., Brown, S. D. M., Davies, K. E., Rawlins, J. N.P., Cowan, N. J., Nolan, P., Chelly, J., and Flint, J. (2007). Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell 128, 45–57.
Kerjan, G., and Gleeson, J. G. (2007). Genetic mechanisms underlying abnormal neuronal migration in classical lissencephaly. Trends Genet. 23, 623–630.
Kitamura, K., Itou, Y., Yanazawa, M., Ohsawa, M., Suzuki-Migishima, R., Umeki, Y., Hohjoh, H., Yanagawa, Y., Shinba, T., Itoh, M., Nakamura, K., and Goto, Y. (2009). Three human ARX mutations cause the lissencephaly-like and mental retardation with epilepsy-like pleiotropic phenotypes in mice. Hum. Mol. Genet. 18, 3708–3724.
Kitamura, K., Yanazawa, M., Sugiyama, N., Miura, H., Iizuka-Kogo, A., Kusaka, M., Omichi, K., Suzuki, R., Kato-Fukui, Y., Kamiirisa, K., Matsuo, M., Kamijo, S., Kasahara, M., Yoshioka, H., Ogata, T., Fukuda, T., Kondo, I., Kato, M., Dobyns, W. B., Yokoyama, M., and Morohashi, K. (2002). Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat. Genet. 32, 359–369.
Kobayashi, T., Minowa, O., Sugitani, Y., Takai, S., Mitani, H., Kobayashi, E., Noda, T., and Hino, O. (2001). A germ-line Tsc1 mutation causes tumor development and embryonic lethality that are similar, but not identical to, those caused by Tsc2 mutation in mice. Proc. Natl. Acad. Sci. U.S.A. 98, 8762–8767.
Koizumi, H., Tanaka, T., and Gleeson, J. G. (2006). Doublecortin-like kinase functions with doublecortin to mediate fiber tract decussation and neuronal migration. Neuron 49, 55–66.
Kriegstein, A., Noctor, S., and Martinez-Cerdeno, V. (2006). Patterns of neural stem and progenitor cell division may underlie evolutionary cortical expansion. Nat. Rev. Neurosci. 7, 883–890.
Kriegstein, A. R., and Noctor, S. C. (2004). Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 27, 392–399.
Lapray, D., Popova, I. Y., Kindler, J., Jorquera, I., Becq, H., Manent, J. B., Luhmann, H. J., and Represa, A. (2010). Spontaneous epileptic manifestations in a DCX knockdown model of human double cortex. Cereb. Cortex 20, 2694–2701.
Leppert, M., and Singh, N. (1999). Benign familial neonatal epilepsy with mutations in two potassium channel genes. Curr. Opin. Neurol. 12, 143–147.
Liu, X., Novosedlik, N., Wang, A., Hudson, M. L., Cohen, I. L., Chudley, A. E., Forster-Gibson, C. J., Lewis, S. M. E., and Holden, J. J. A. (2009). The DLX1 and DLX2 genes and susceptibility to autism spectrum disorders. Eur. J. Hum. Genet. 17, 228–235.
Lui, J. H., Hansen, D. V., and Kriegstein, A. R. (2011). Development and evolution of the human neocortex. Cell 146, 18–36.
Major, P., Rakowski, S., Simon, M. V., Cheng, M. L., Eskandar, E., Baron, J., Leeman, B. A., Frosch, M. P., and Thiele, E. A. (2009). Are cortical tubers epileptogenic? Evidence from electrocorticography. Epilepsia 50, 147–154.
Malatesta, P., Hack, M. A., Hartfuss, E., Kettenmann, H., Klinkert, W., Kirchhoff, F., and Gotz, M. (2003). Neuronal or glial progeny: regional differences in radial glia fate. Neuron 37, 751–764.
Manent, J. B., Demarque, M., Jorquera, I., Pellegrino, C., Ben-Ari, Y., Aniksztejn, L., and Represa, A. (2005). A noncanonical release of GABA and glutamate modulates neuronal migration. J. Neurosci. 25, 4755–4765.
Manent, J. B., Jorquera, I., Ben-Ari, Y., Aniksztejn, L., and Represa, A. (2006). Glutamate acting on AMPA but not NMDA receptors modulates the migration of hippocampal interneurons. J. Neurosci. 26, 5901–5909.
Manent, J. B., Jorquera, I., Franco, V., Ben-Ari, Y., Perucca, E., and Represa, A. (2008). Antiepileptic drugs and brain maturation: fetal exposure to lamotrigine generates cortical malformations in rats. Epilepsy Res. 78, 131–139.
Manent, J. B., Jorquera, I., Mazzucchelli, I., Depaulis, A., Perucca, E., Ben-Ari, Y., and Represa, A. (2007). Fetal exposure to GABA-acting antiepileptic drugs generates hippocampal and cortical dysplasias. Epilepsia 48, 684–693.
Manent, J. B., and Represa, A. (2007). Neurotransmitters and brain maturation: early paracrine actions of GABA and glutamate modulate neuronal migration. Neuroscientist 13, 268–279.
Manent, J. B., Wang, Y., Chang, Y., Paramasivam, M., and Loturco, J. J. (2009). Dcx reexpression reduces subcortical band heterotopia and seizure threshold in an animal model of neuronal migration disorder. Nat. Med. 15, 84–90.
Manno, I., Macchi, F., Caleo, M., and Bozzi, Y. (2011). Environmental enrichment reduces spontaneous seizures in the Q54 transgenic mouse model of temporal lobe epilepsy. Epilepsia 52, e113–e117.
Mantegazza, M., Curia, G., Biagini, G., Ragsdale, D. S., and Avoli, M. (2010). Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. 9, 413–424.
Manzini, M. C., and Walsh, C. A. (2011). What disorders of cortical development tell us about the cortex: one plus one does not always make two. Curr. Opin. Genet. Dev. 21, 333–339.
Marin, O., and Rubenstein, J. L. (2003). Cell migration in the forebrain. Annu. Rev. Neurosci. 26, 441–483.
Marsh, E., Fulp, C., Gomez, E., Nasrallah, I., Minarcik, J., Sudi, J., Christian, S. L., Mancini, G., Labosky, P., Dobyns, W., Brooks-Kayal, A., and Golden, J. A. (2009). Targeted loss of Arx results in a developmental epilepsy mouse model and recapitulates the human phenotype in heterozygous females. Brain 132, 1563–1576.
Martin, M. S., Dutt, K., Papale, L. A., Dubé, C. M., Dutton, S. B., de Haan, G., Shankar, A., Tufik, S., Meisler, M. H., Baram, T. Z., Goldin, A. L., and Escayg, A. (2010). Altered function of the SCN1A voltage-gated sodium channel leads to gamma-aminobutyric acid-ergic (GABAergic) interneuron abnormalities. J. Biol. Chem. 285, 9823–9834.
Matsumoto, N., Leventer, R. J., Kuc, J. A., Mewborn, S. K., Dudlicek, L. L., Ramocki, M. B., Pilz, D. T., Mills, P. L., Das, S., Ross, M. E., Ledbetter, D. H., and Dobyns, W. B. (2001). Mutation analysis of the DCX gene and genotype/phenotype correlation in subcortical band heterotopia. Eur. J. Hum. Genet. 9, 5–12.
Meisler, M. H., O’Brien, J. E., and Sharkey, L. M. (2010). Sodium channel gene family: epilepsy mutations, gene interactions and modifier effects. J. Physiol. 588, 1841–1848.
Miyoshi, G., Hjerling-Leffler, J., Karayannis, T., Sousa, V. H., Butt, S. J., Battiste, J., Johnson, J. E., Machold, R. P., and Fishell, G. (2010). Genetic fate mapping reveals that the caudal ganglionic eminence produces a large and diverse population of superficial cortical interneurons. J. Neurosci. 30, 1582–1594.
Molyneaux, B. J., Arlotta, P., Menezes, J. R., and Macklis, J. D. (2007). Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 8, 427–437.
Muncy, J., Butler, I. J., and Koenig, M. K. (2009). Rapamycin reduces seizure frequency in tuberous sclerosis complex. J. Child Neurol. 24, 477.
Nosten-Bertrand, M., Kappeler, C., Dinocourt, C., Denis, C., Germain, J., Phan Dinh Tuy, F., Verstraeten, S., Alvarez, C., Metin, C., Chelly, J., Giros, B., Miles, R., Depaulis, A., and Francis, F. (2008). Epilepsy in Dcx knockout mice associated with discrete lamination defects and enhanced excitability in the hippocampus. PLoS ONE 3, e2473.
Oakley, J. C., Kalume, F., Yu, F. H., Scheuer, T., and Catterall, W. A. (2009). Temperature- and age-dependent seizures in a mouse model of severe myoclonic epilepsy in infancy. Proc. Natl. Acad. Sci. U.S.A. 106, 3994–3999.
Ogiwara, I., Miyamoto, H., Morita, N., Atapour, N., Mazaki, E., Inoue, I., Takeuchi, T., Itohara, S., Yanagawa, Y., Obata, K., Furuichi, T., Hensch, T. K., and Yamakawa K. (2007). Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J. Neurosci. 27, 5903–5914.
Osen-Sand, A., Staple, J. K., Naldi, E., Schiavo, G., Rossetto, O., Petitpierre, S., Malgaroli, A., Montecucco, C., and Catsicas, S. (1996). Common and distinct fusion proteins in axonal growth and transmitter release. J. Comp. Neurol. 367, 222–234.
Patrylo, P. R., Browning, R. A., and Cranick, S. (2006). Reeler homozygous mice exhibit enhanced susceptibility to epileptiform activity. Epilepsia 47, 257–266.
Pitkanen, A., and Sutula, T. P. (2002). Is epilepsy a progressive disorder? Prospects for new therapeutic approaches in temporal-lobe epilepsy. Lancet Neurol. 1, 173–181.
Pla, R., Borrell, V., Flames, N., and Marín, O. (2006). Layer acquisition by cortical GABAergic interneurons is independent of Reelin signaling. J. Neurosci. 26, 6924–6934.
Pozzi, D., Condliffe, S., Bozzi, Y., Chikhladze, M., Grumelli, C., Proux-Gillardeaux, V., Takahashi, M., Franceschetti, S., Verderio, C., and Matteoli, M. (2008). Activity-dependent phosphorylation of Ser187 is required for SNAP-25-negative modulation of neuronal voltage-gated calcium channels. Proc. Natl. Acad. Sci. U.S.A. 105, 323–328.
Raber, J., Mehta, P. P., Kreifeldt, M., Parsons, L. H., Weiss, F., Bloom, F. E., and Wilson, M. C. (1997). Coloboma hyperactive mutant mice exhibit regional and transmitter-specific deficits in neurotransmission. J. Neurochem. 68, 176–186.
Raffo, E., Coppola, A., Ono, T., Briggs, S. W., and Galanopoulou, A. S. (2011). A pulse rapamycin therapy for infantile spasms and associated cognitive decline. Neurobiol. Dis. 43, 322–329.
Rakic, P. (2007). The radial edifice of cortical architecture: from neuronal silhouettes to genetic engineering. Brain Res. Rev. 55, 204–219.
Rosahl, T. W., Spillane, D., Missler, M., Herz, J., Selig, D. K., Wolff, J. R., Hammer, R. E., Malenka, R. C., and Sudhof, T. C. (1995). Essential functions of synapsins I and II in synaptic vesicle regulation. Nature 375, 488–493.
Rubenstein, J. L. (2000). Intrinsic and extrinsic control of cortical development. Novartis Found. Symp. 228, 67–75; discussion 75–82, 109–113.
Rubenstein, J. L. (2011). Annual research review: development of the cerebral cortex: implications for neurodevelopmental disorders. J. Child Psychol. Psychiatry 52, 339–355.
Sale, A., Berardi, N., and Maffei, L. (2009). Enrich the environment to empower the brain. Trends Neurosci. 32, 233–239.
Scalmani, P., Rusconi, R., Armatura, E., Zara, F., Avanzini, G., Franceschetti, S., and Mantegazza, M. (2006). Effects in neocortical neurons of mutations of the Na(v)1.2 Na+ channel causing benign familial neonatal-infantile seizures. J Neurosci 26, 10100–10109.
Scheffer, I. E., and Berkovic, S. F. (1997). Generalized epilepsy with febrile seizures plus: a genetic disorder with heterogeneous clinical phenotypes. Brain 120, 479–490.
Schoch, S., Deak, F., Konigstorfer, A., Mozhayeva, M., Sara, Y., Sudhof, T. C., and Kavalali, E. T. (2001). SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 294, 1117–1122.
Schulte, U., Thumfart, J. O., Klocker, N., Sailer, C. A., Bildl, W., Biniossek, M., Dehn, D., Deller, T., Eble, S., Abbass, K., Wangler, T., Knaus, H. G., and Fakler, B. (2006). The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvbeta1. Neuron 49, 697–706.
Sorensen, J. B., Matti, U., Wei, S. H., Nehring, R. B., Voets, T., Ashery, U., Binz, T., Neher, E., and Rettig, J. (2002). The SNARE protein SNAP-25 is linked to fast calcium triggering of exocytosis. Proc. Natl. Acad. Sci. U.S.A. 99, 1627–1632.
Striano, P., Busolin, G., Santulli, L., Leonardi, E., Coppola, A., Vitiello, L., Rigon, L., Michelucci, R., Tosatto, S. C., Striano, S., and Nobile, C. (2011). Familial temporal lobe epilepsy with psychic auras associated with a novel LGI1 mutation. Neurology 76, 1173–1176.
Stuhmer, T., Anderson, S. A., Ekker, M., and Rubenstein, J. L. (2002). Ectopic expression of the Dlx genes induces glutamic acid decarboxylase and Dlx expression. Development 129, 245–252.
Sunnen, C. N., Brewster, A. L., Lugo, J. N., Vanegas, F., Turcios, E., Mukhi, S., Parghi, D., D’Arcangelo, G., and Anderson, A. E. (2011). Inhibition of the mammalian target of rapamycin blocks epilepsy progression in NS-Pten conditional knockout mice. Epilepsia 52, 2065–2075.
Swann, J. W., and Brady, R. J. (1984). Penicillin-induced epileptogenesis in immature rat CA3 hippocampal pyramidal cells. Brain Res. 314, 243–254.
Tricoire, L., Pelkey, K. A., Erkkila, B. E., Jeffries, B. W., Yuan, X., and McBain, C. J. (2011). A blueprint for the spatiotemporal origins of mouse hippocampal interneuron diversity. J. Neurosci. 31, 10948–10970.
Tsakiridou, E., Bertollini, L., De Curtis, M., Avanzini, G., and Pape, H. C. (1995). Selective increase in T-type calcium conductance of reticular thalamic neurons in a rat model of absence epilepsy. J. Neurosci. 15, 3110–3117.
Uhlmann, E. J., Apicelli, A. J., Baldwin, R. L., Burke, S. P., Bajenaru, M. L., Onda, H., Kwiatkowski, D., and Gutmann, D. H. (2002). Heterozygosity for the tuberous sclerosis complex (TSC) gene products results in increased astrocyte numbers and decreased p27-Kip1 expression in TSC2+/− cells. Oncogene 21, 4050–4059.
Valtorta, F., Iezzi, N., Benfenati, F., Lu, B., Poo, M. M., and Greengard, P. (1995). Accelerated structural maturation induced by synapsin I at developing neuromuscular synapses of Xenopus laevis. Eur. J. Neurosci. 7, 261–270.
Verderio, C., Pozzi, D., Pravettoni, E., Inverardi, F., Schenk, U., Coco, S., Proux-Gillardeaux, V., Galli, T., Rossetto, O., Frassoni, C., and Matteoli, M. (2004). SNAP-25 modulation of calcium dynamics underlies differences in GABAergic and glutamatergic responsiveness to depolarization. Neuron 41, 599–610.
Wang, Y., Dye, C. A., Sohal, V., Long, J. E., Estrada, R. C., Roztocil, T., Lufkin, T., Deisseroth, K., Baraban, S. C., and Rubenstein, J. L. (2010). Dlx5 and Dlx6 regulate the development of parvalbumin-expressing cortical interneurons. J. Neurosci. 30, 5334–5345.
Way, S. W., McKenna, J. III, Mietzsch, U., Reith, R. M., Wu, H. C., and Gambello, M. J. (2009). Loss of Tsc2 in radial glia models the brain pathology of tuberous sclerosis complex in the mouse. Hum. Mol. Genet. 18, 1252–1265.
White, R., Hua, Y., Scheithauer, B., Lynch, D. R., Henske, E. P., and Crino, P. B. (2001). Selective alterations in glutamate and GABA receptor subunit mRNA expression in dysplastic neurons and giant cells of cortical tubers. Ann. Neurol. 49, 67–78.
Wilson, M. C. (2000). Coloboma mouse mutant as an animal model of hyperkinesis and attention deficit hyperactivity disorder. Neurosci. Biobehav. Rev. 24, 51–57.
Wonders, C. P., and Anderson, S. A. (2006). The origin and specification of cortical interneurons. Nat. Rev. Neurosci. 7, 687–696.
Yabut, O., Renfro, A., Niu, S., Swann, J. W., Marín, O., and D’Arcangelo, G. (2007). Abnormal laminar position and dendrite development of interneurons in the reeler forebrain. Brain Res. 1140, 75–83.
Yu, F. H., Mantegazza, M., Westenbroek, R. E., Robbins, C. A., Kalume, F., Burton, K. A., Spain, W. J., Mcknight, G. S., Scheuer, T., and Catterall, W. A. (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9, 1142–1149.
Yu, Y. E., Wen, L., Silva, J., Li, Z., Head, K., Sossey-Alaoui, K., Pao, A., Mei, L., and Cowell, J. K. (2010). Lgi1 null mutant mice exhibit myoclonic seizures and CA1 neuronal hyperexcitability. Hum. Mol. Genet. 19, 1702–1711.
Zaki, M., Shehab, M., El-Aleem, A. A., Abdel-Salam, G., Koeller, H. B., Ilkin, Y., Ross, M. E., Dobyns, W. B., and Gleeson, J. G. (2007). Identification of a novel recessive RELN mutation using a homozygous balanced reciprocal translocation. Am. J. Med. Genet. A 143A, 939–944.
Zeng, L. H., Rensing, N. R., and Wong, M. (2009). The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J. Neurosci. 29, 6964–6972.
Zeng, L. H., Rensing, N. R., Zhang, B., Gutmann, D. H., Gambello, M. J., and Wong, M. (2011). Tsc2 gene inactivation causes a more severe epilepsy phenotype than Tsc1 inactivation in a mouse model of tuberous sclerosis complex. Hum. Mol. Genet. 20, 445–454.
Zeng, L. H., Xu, L., Gutmann, D. H., and Wong, M. (2008). Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann. Neurol. 63, 444–453.
Zhang, Y., Vilaythong, A. P., Yoshor, D., and Noebels, J. L. (2004). Elevated thalamic low-voltage-activated currents precede the onset of absence epilepsy in the SNAP25-deficient mouse mutant coloboma. J. Neurosci. 24, 5239–5248.
Keywords: cortex, hippocampus, GABA, glutamate, critical period, sodium channels
Citation: Bozzi Y, Casarosa S and Caleo M (2012) Epilepsy as a neurodevelopmental disorder. Front. Psychiatry 3:19. doi: 10.3389/fpsyt.2012.00019
Received: 15 November 2011; Accepted: 23 February 2012;
Published online: 19 March 2012.
Edited by:
Daniela Tropea, Trinity College Dublin, IrelandReviewed by:
Beatriz Cubelos, Centro de Biología Molecular Severo Ochoa, SpainGraziella Di Cristo, Centre Hospitalier Universitaire Sainte-Justine/University of Montreal, Canada
Copyright: © 2012 Bozzi, Casarosa and Caleo. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Matteo Caleo, Neuroscience Institute, National Research Council, Via G. Moruzzi 1, 56123 Pisa, Italy. e-mail: caleo@in.cnr.it