 Maite Stratmann
Maite Stratmann Caterina Gagliardi
Caterina Gagliardi Melania Capasso
Melania Capasso- German Center for Neurodegenerative Diseases (DZNE), Venusberg Campus 1, Bonn, Germany
Proton channels are transmembrane proteins that enable selective proton (H+) transport. The voltage-gated proton channel Hv1 or HVCN1 is the only one found in mammalian cells, primarily in immune cells, where it facilitates rapid proton extrusion in response to membrane depolarization, mediating outward proton currents. Therefore, it is well equipped to support NADPH-oxidase function, facilitating the proton flux that maintains physiological pH and membrane potential for efficient reactive oxygen species (ROS) production. In the central nervous system (CNS), Hv1 is predominantly found in microglia. Its role in microglia homeostasis is yet to be elucidated; however, recent research has highlighted its involvement in neurological conditions, including demyelinating disease, spinal cord injury, stroke, and Parkinsonism. These studies have shown beneficial effects of Hv1 deletion, including improved neurological function, reduced microglial activation, enhanced myelination, and decreased neuroinflammation. This review explores the role of Hv1 in the CNS and its potential as a therapeutic target in neurodegenerative diseases.
Introduction
The voltage-gated proton channel Hv1 (HVCN1/VSOP) is a highly proton-selective transmembrane protein that plays a central role in regulating intracellular pH and membrane potential, particularly in immune responses (Decoursey et al., 2000; Kapus et al., 1993; He et al., 2021; Capasso et al., 2010; Wu et al., 2012; Wang et al., 2021; Ramsey et al., 2006; Sasaki et al., 2006; DeCoursey, 2010; Tomb et al., 2008; Tombola et al., 2010). For a review of the physiology of the channel please refer to (DeCoursey, 2010; DeCoursey, 2003).
In different immune cell types, Hv1 functions largely to support NADPH-oxidase complex activity (Figure 1). Upon activation, the complex transfers electrons across biological membranes to reduce molecular oxygen (O2) into superoxide (O2−) during the respiratory burst, contributing to cellular oxidative processes (Decoursey, 2010; Henderson et al., 1987; El Chemaly et al., 2010). The electron transfer depolarizes the membrane and lowers cytosolic pH, two phenomena that suppress NADPH-oxidase activity but open Hv1, enabling the extrusion of protons to restore membrane potential and pH and ensuring continued ROS generation (Ramsey et al., 2009; Morihata et al., 2000). In Hv1-deficient cells this feedback fails, leading to intracellular acidosis, impaired ROS production, and compromised immune responses (DeCoursey, 2003).
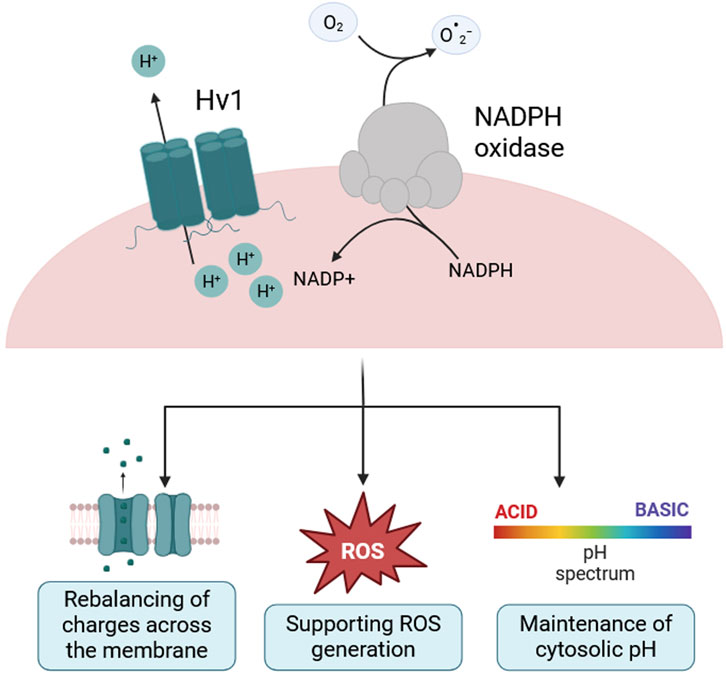
Figure 1. Hv1 supports NADPH oxidase activity, ROS generation and cytosolic pH homeostasis. Schematic of the voltage-gated proton channel Hv1 acting in concert with the NADPH oxidase complex. During NADPH oxidase activity, electrons are transferred from cytosolic NADPH to molecular oxygen to generate superoxide (
While extensively studied in peripheral immune cells (DeCoursey et al., 2000; Kapus et al., 1993; Capasso et al., 2010; Capasso, 2014), the role of Hv1 in microglia and more generally in the central nervous system (CNS) is still being elucidated, as previously reviewed by He et al. (2021). Microglia are the tissue-resident macrophages of the CNS and maintain its homeostasis by pruning synapses, clearing cellular debris, and responding to injury (Guzmán-Ruíz et al., 2024; Salter and Stevens, 2017). However, their dysregulation is implicated in a range of neurological diseases, largely through excessive ROS production and secretion of pro-inflammatory cytokines (Salter and Stevens, 2017). ROS generated by the NADPH oxidase play a dual role in microglial physiology. At low concentrations, they participate in signaling cascades regulating proliferation and stress responses and supporting microglia’s role during development. However, when produced in excess, ROS induce mitochondrial dysfunction and oxidative damage to lipids, proteins and nucleic acids that might lead to apoptosis (Tauffenberger and Magistretti, 2021). Thus, the fine-tuned regulation of ROS via Hv1 may be critical for maintaining a functional balance between physiological and pathological microglial activation (Haslund-Vinding et al., 2017).
Hv1 expression in the brain is both region- and age-dependent (Kawai et al., 2021). It is undetectable in neonates, detectable in adults, and significantly upregulated in aged brains, particularly in the striatum, where it correlates with markers of microglial activation CD11b, CX3CR1, CD68 (Kawai et al., 2021; Okochi et al., 2009). This age-related upregulation likely parallels the increased activation state of microglia observed during aging. Interestingly, in aged Hv1-deficient mice, increased cortical oxidative stress and altered microglial morphology have been reported, along with behavioral changes linked to anxiety and dysregulated GPCR signaling (Kawai et al., 2021). These findings suggest that Hv1 may influence both cellular and behavioral aspects of aging via region-specific effects on microglia.
Emerging findings now implicate Hv1 in a range of CNS pathologies characterized by oxidative stress and inflammation. In models of ischemic stroke (Wu et al., 2012; Yu et al., 2020; Yu et al., 2018; Li et al., 2019; Tian et al., 2016; Kawai et al., 2017; Kimura et al., 2025; Yang et al., 2025), traumatic and spinal cord injury (Liu et al., 2023; Ritzel et al., 2021; Li et al., 2020a; Li et al., 2020b; Li et al., 2023; Murugan et al., 2020), demyelination (Wang et al., 2021; Chen et al., 2020; Liu et al., 2015; Sun et al., 2024), pain (Peng et al., 2021; Zhang et al., 2022), and Parkinson’s disease (Neal et al., 2023), Hv1 deletion or pharmacological inhibition consistently reduced microglial activation, lowering ROS levels and improving neurological outcomes. These findings point to Hv1 as a potential therapeutic target for limiting microglia-mediated neuroinflammation and secondary neuronal damage, as we will illustrate in details in subsequent paragraphs.
Stroke and hypoperfusion
Stroke and cerebral hypoperfusion are major neurological insults involving restricted blood and oxygen supply to the brain, which trigger energy failure and excitotoxicity and are accompanied by activation of inflammatory processes. Central to this pathology is microglial overproduction of ROS, which drives neurotoxicity (Kimura et al., 2025; Zhu et al., 2022). Increasing evidence implicates Hv1 as a mediator of such damage: Wu and colleagues first demonstrated that Hv1 mediates NADPH-oxidase-dependent ROS generation in microglia and its deletion in a middle cerebral artery occlusion (MCOA) ischemic stroke model reduced infarct volume and neuronal death, improving neurological deficit scores 24 h post-injury (Wu et al., 2012). A follow-up study in a photothrombotic stroke mouse model confirmed these findings: Hv1 KO caused a shift towards an anti-inflammatory microglial phenotype, resulting in reduced brain injury and improved motor function (Tian et al., 2016). Interestingly, the role of Hv1 in stroke appeared age-dependent, as neuroprotection was evident in 6-month-old but not 9-week-old mice (Kawai et al., 2017), possibly due to aforementioned age-dependent changes in Hv1 expression.
Furthermore, in chronic cerebral hypoperfusion, using the bilateral carotid artery stenosis model, Hv1 KO mice displayed lower ROS levels, reduced cytokine release, and an anti-inflammatory microglial profile (lower CD68/CD16, higher CD206/Arg1). White matter integrity and working memory were preserved, likely due to improved oligodendrocyte precursor cell (OPC) proliferation and differentiation (Yu et al., 2020). Mechanistically, Hv1 KO suppressed PI3K/Akt activation in microglia, limiting pro-inflammatory polarization and enhancing OPC maturation (Yu et al., 2018). Indeed, in co-cultures, Hv1 KO microglia reduced OPC apoptosis and MAPK pathway activation under oxygen-glucose deprivation.
Pharmacological inhibition of Hv1 in a experimental stroke models also showed promise. Brain-targeting lipid nanoparticles carrying the Hv1 inhibitor YHV984 reduced infarct size when administered 1 h post-stroke, suppressing NLRP3 inflammasome activation and decreasing neuronal death (Yang et al., 2025). Behavioral outcomes and survival improved significantly; however, efficacy with delayed treatment remains unknown. Further insights into potential therapeutic strategies came from Li and colleagues, who suggested that Hv1 interacted functionally with the Na+/H+ exchanger NHE-1 in the context of stroke (Li et al., 2019). Their results indicated a potential synergistic effect of inhibiting both Hv1 and NHE, supporting co-targeting as a strategy for future investigation.
Collectively, these results converge on a shared mechanism through which Hv1 amplifies microglial-mediated damage in acute and chronic models of brain vascular pathology, which warrants further investigations.
Traumatic CNS injury
Traumatic CNS injury, including traumatic brain injury (TBI) and spinal cord injury (SCI), is trauma caused by external forces, which triggers acute and chronic secondary damage characterized by oxidative stress, acidosis, and neuroinflammation (Rubiano et al., 2015; Ryan et al., 2023; Venkatesh et al., 2019). Microglial Hv1 has emerged as a driver of these responses, promoting extracellular acidosis and ROS production. In a controlled cortical impact model of TBI, Hv1 was upregulated in the cortex and hippocampus within 24 h and remained elevated for several weeks (Ritzel et al., 2021). Hv1 KO mice exhibited attenuated ROS production, reduced expression of pro-inflammatory cytokines (TNF-α, IL-1β, IL-6), and improved neurological performance across multiple behavioral tests, including novel object recognition, Y-maze, and beam walk tests.
Mechanistic studies have implicated Hv1 in amplifying acidosis-driven damage via neuronal acid-sensing ion channels (Zeng et al., 2015). However, acidosis-induced cell swelling itself exacerbates Hv1 activity by stretching the membrane, thereby triggering the channel’s mechanosensitivity (Pathak et al., 2016). Additionally, Hv1 upregulation in microglia of the olfactory bulb after TBI led to disrupted network activity and synaptic loss, contributing to olfactory dysfunction (Liu et al., 2023). These deficits were rescued in Hv1 KO or NADPH-oxidase-deficient mice and ameliorated by pharmacological NADPH-oxidase inhibition.
Similar mechanisms were observed in SCI models. Following thoracic contusion, Hv1 KO mice showed improved white matter preservation, enhanced neuronal survival, and improved motor recovery over several weeks post-injury (Murugan et al., 2020). Reduced IL-1β expression, lower ROS, and diminished microglial activation were observed, along with decreased extracellular acidosis and leukocyte infiltration (Murugan et al., 2020; Li et al., 2021). Hv1 deletion also shifted microglia toward an anti-inflammatory phenotype, with reduced expression of the pro-inflammatory markers CD16/CD32 and elevated anti-inflammatory markers CD206 and Arg1 (Li et al., 2020b). This was accompanied by an increase in neuroprotective astrocytes, and their elevated expression of synaptogenic and neurotrophic factors (Li et al., 2020b; Li et al., 2023). These effects were linked to reduced astrocytic ROS and downstream STAT3 phosphorylation (Li et al., 2023), suggesting that Hv1 indirectly modulates astrocyte behavior through redox-sensitive signaling pathways.
Another mechanism linking Hv1 to secondary damage is inflammasome activation. After SCI, Hv1 KO mice show significantly lower levels of NLRP3 inflammasome components, which resulted in reduced neuronal pyroptosis in the lesion area (Li et al., 2020a). The same effect could be achieved with antioxidant treatment, indicating that Hv1-driven ROS is a key upstream trigger of inflammasome activation and cell death.
In vitro studies of neuron-microglia co-cultures treated with the Hv1 inhibitor 2CGI offer corroborating evidence of reduced TNF-α, ROS, and zinc-mediated neuronal damage and upregulation of neurotrophic factors (Hernandez-Espinosa et al., 2023). However, as 2CGI may also inhibit the NLRP3 inflammasome (Liu et al., 2025), more selective Hv1 inhibitors or genetic tools are needed to dissect Hv1-specific pathways.
Taken together, these studies establish Hv1 as a central regulator of microglia- and astrocyte-mediated secondary damage following CNS trauma, confirming it as a promising therapeutic target that should be investigated further, including with more specific pharmacological inhibitors.
Peripheral nerve injury and pain
Microglial activation and oxidative stress are central also to the pathogenesis of chronic pain and peripheral nerve injury. Recent research demonstrated that Hv1 is functionally expressed in spinal microglia and upregulated following spinal nerve transection (SNT), a model for peripheral nerve injury (Peng et al., 2021). Mice lacking Hv1 displayed reduced pain behaviors such as mechanical allodynia and thermal hyperalgesia compared to WT mice. Although microglial proliferation and p38 MAPK activation were intact in Hv1 KO mice, Hv1 deficiency led to lower ROS production, alongside lower astrocytic activation in the ipsilateral dorsal horn. Furthermore, IFN-γ levels in spinal astrocytes diminished in Hv1 KO mice; indeed, neutralizing IFN-γ in WT mice mimicked the protective phenotype of Hv1 deficiency, while exogenous IFN-γ reversed the Hv1 KO phenotype. These results suggest that Hv1 and IFN-γ in microglia-astrocyte interaction promote pain hypersensitivity.
Zhang and colleagues extended these findings by identifying Hv1 mRNA expression in dorsal root ganglion (DRG) neurons, challenging the previous notion of a microglia-specific expression in the CNS (Zhang et al., 2022). DRG Hv1 expression increased in response to several pain models, including complete Freud’s adjuvant (CFA)-induced inflammation, spared-nerve injury and formalin injection, as well as stimulation with TNF-α, IL-1β, and PMA. Activity- and injury-induced neuronal Hv1 expression promoted intracellular alkalization, ROS production, and pro-inflammatory cytokine release. Both genetic deletion and pharmacological inhibition of Hv1 with the selective inhibitor YHV984 reduced ROS levels, cytokine release, and pain-related behaviors. Mechanistically, YHV-984 normalized SHP-1/pAKT signaling in DRG neurons, preventing the CFA-induced decrease of SHP-1 expression and subsequent overactivation of the PI3K-pAKT pathway. Importantly, Hv1 inhibition also alleviated morphine-induced tolerance, a major obstacle in chronic pain treatment. Co-administration of YHV984 preserved morphine analgesic effects while preventing allodynia, drug tolerance, and DRG ROS production. These findings implicate Hv1 as a modulator of both injury- and opioid-related neuroimmune mechanisms.
Demyelinating diseases
Emerging evidence points to a role for Hv1 also in demyelinating diseases such as multiple sclerosis (MS), a chronic inflammatory disease of the CNS in which oxidative stress plays a central role. Post-mortem analyses of MS brains revealed oxidized lipids and DNA in active plaques, especially in oligodendrocytes and axonal spheroids. In a chronic experimental autoimmune encephalomyelitis (EAE) model, a distinct ROS-positive CD11b+ microglial subset was identified using single-cell RNA sequencing. Treating mice with acivicin, which enhances antioxidant capacity by inhibiting glutathione breakdown, reduced oxidative damage and neurodegeneration even 80 days post-disease onset, highlighting the therapeutic potential of targeting ROS production (Mendiola et al., 2020).
Liu and colleagues used a cuprizone-induced demyelination model in adult male mice to investigate Hv1 in demyelination (Liu et al., 2015). Hv1 KO mice exhibited reduced ROS levels, decreased microgliosis, increased oligodendrocyte progenitor proliferation, and enhanced oligodendrocyte maturation in the corpus callosum. Even 2 weeks after recovery, HV1 KO mice showed preserved myelin integrity and improved motor performance in the rotarod test. Similarly, in a lysophosphatidylcholine (LPC)-induced demyelination model of the corpus callosum, Hv1 KO decreased ROS production and lowered microglial activation, improving remyelination (Chen et al., 2020). Enhanced myelin repair was linked to increased oligodendrocyte maturation and decreased autophagy in microglia, alongside improved spatial memory in the Morris water maze. These findings underscore the role of Hv1 in microglial-mediated demyelination and support its therapeutic potential in promoting both protection from demyelination and regeneration.
Wang and colleagues further investigated Hv1 in microglial migration and myelin debris clearance (Wang et al., 2021). Genetic deletion or antibody-mediated neutralization of Hv1 enhanced microglial motility and debris clearance in vivo, however, the ability of the reported anti-Hv1 antibody to block proton channel activity warrants further investigation. The authors showed that Hv1 was not required for myelin phagocytosis in vitro but rather for proper lysosomal acidification in human kidney cell line 293T but not in bone marrow-derived macrophages, suggesting a cell-type-specific role. Moreover, systemic LPS exposure increased Hv1 expression in the corpus callosum, indicating that Hv1 reacts to inflammatory stimuli in a wider range of ways than only focal demyelination.
Following up on Hv1 involvement in neuroinflammation, Sun and colleagues tested the Hv1 inhibitor 2-GBI in an LPS-induced neuroinflammation model. Inhibition of Hv1 blocked HIF-1α-mediated aerobic glycolysis, resulting in reduced ROS production and suppression of pro-inflammatory signaling both in vitro and in vivo. Systemic 2-GBI treatment decreased hippocampal microglial activation and inflammation, which translated to improved cognition in the novel object recognition test and Y maze. Complementing these pharmacological findings, genetic approaches using Hv1 knockout, knockdown, and overexpression models demonstrated that Hv1 loss attenuates NF-κB–mediated cytokine production and PI3K/Akt/HIF-1α–dependent glycolytic reprogramming in microglia in a LPS-induced hippocampal neuroinflammation model (Sun et al., 2025). Hv1 deficiency disrupted the cellular redox state and altered metabolism, resulting in increased levels of glutamate and aspartate alongside decreased lactate and citrate, reflecting impaired glycolysis and TCA cycle flux. This metabolic reprogramming was accompanied by improved cognition in LPS-treated mice.
Collectively, these findings emphasize Hv1 involvement in demyelinating diseases and neuroinflammation. Genetic and pharmacological Hv1 inhibition reduced neuroinflammation and improved myelin clearance, protecting cognitive and motor function. However, it is important to consider the limitations of the cuprizone and LPC model, which mimics toxin-induced demyelination and lacks the autoimmune component of MS, an aspect in which Hv1 might also play a role. Further investigation using immune-driven models and human samples will be crucial to validate the role of Hv1 in disease progression and its therapeutic potential.
Other neurodegenerative diseases: Parkinsonism, Amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD) and Alzheimer’s disease (AD)
Recent evidence highlights the involvement of Hv1 in Parkinsonism, particularly in promoting microglial activation and dopaminergic neuron loss. Transcriptomic data from patient brains revealed its elevated expression, especially in men (Neal et al., 2023). In both acute 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and sub-chronic lipopolysaccharide (LPS)-induced Parkinson’s mouse models, Hv1 expression was upregulated in the striatum 2 days after treatment, and Hv1 deficiency conferred partial neuroprotection, with diminished loss of tyrosine hydroxylase-positive neurons (Neal et al., 2023). This was accompanied by decreased pro-inflammatory and oxidative markers (e.g., TNF-α, IL1b, IL6, Nos2, IFN-γ, gp91phox) and increased anti-inflammatory markers (e.g., Arg1, YM1). In vitro, Hv1 KO microglia failed to show typical LPS-induced inflammatory responses, and their conditioned media caused reduced dopaminergic neuron death. These findings suggest Hv1 promotes both basal and stimulus-induced pro-inflammatory activity and may contribute to dopaminergic neuron vulnerability. However, the MPTP model represents acute toxicity and may not reflect chronic PD pathophysiology. Investigating Hv1 in more relevant systems, such as iPSC-derived neuron-microglia co-cultures or overexpression of α-synuclein (Pinto-Costa et al., 2023), will be needed to clarify its role in chronic neurodegeneration.
Elevated Hv1 protein has been observed across multiple neurodegenerative contexts, including ALS and HD in mouse models, and, in the case of HD also in patient blood samples (Wang et al., 2021). Similarly transcriptomic analyses of human AD postmortem tissue show increased Hv1 expression, although functional studies in AD remain limited (Ou et al., 2021; Brooks and Mias, 2019; Seligmann et al., 2024). These converging findings indicate a broader upregulation of Hv1 across neurodegenerative disorders, consistent with its role in regulating microglial polarization and sustaining neuroinflammation, processes that are central to the pathophysiology of AD, ALS, and HD. Through supporting NADPH-oxidase-dependent ROS production, Hv1 may amplify oxidative stress, drive neuronal injury and death, and facilitate propagation of misfolded protein aggregates (Ma et al., 2017; Zhang et al., 2023). Given that chronic oxidative stress and persistent neuroinflammation are common hallmarks of these diseases (Ma et al., 2017; Zhang et al., 2023), Hv1 may act as a shared amplifier of pathogenic microglial responses and represent an important mechanistic link between microglial activation, ROS production, and neurodegeneration.
Hv1 has also been investigated in the context of aging and cognition (He et al., 2021), suggesting a potential contribution to age-related cognitive decline and neurodegenerative conditions such as dementia. Notably, Hv1 deficiency has been associated with improved cognitive performance in models of neuroinflammation (Sun et al., 2025), supporting a role for Hv1 in mediating inflammation-induced cognitive impairment. However, direct functional evidence in the context of aging and dementia remains limited.
Discussion, treatment perspectives, and future outlook
The voltage-gated proton channel Hv1 has emerged as a key regulator of oxidative stress in the CNS; its expression in microglia and its role in ROS production place it at the center of redox-sensitive inflammatory responses. In pathologies such as stroke, TBI, SCI, chronic pain, demyelinating diseases, and neurodegeneration, Hv1 is often upregulated in response to injury or disease-related stimuli.
Recent evidence challenges the traditional view that Hv1 uniformly promotes ROS generation. In primary microglia, Hv1 deletion paradoxically increased extracellular ROS production, likely via altered actin dynamics that enhance NADPH-oxidase assembly, independently of proton extrusion (Kawai et al., 2017). This implies that Hv1 may normally suppress excessive actin remodeling, indirectly limiting NADPH-oxidase complex assembly and ROS generation. Importantly, such mechanisms illustrate that Hv1 functions are not restricted to cytokine- or inflammation-driven pathways but also extend to cytoskeletal regulation and other non-inflammatory processes. Similarly, in vitro data from Hv1 KO cells showed an upregulation of genes linked to migration, such as ACTA2, SEMA6A, GPNMB, RAC2, LRP1 (Wang et al., 2021). These findings underscore the multifaceted role of Hv1 in microglia (Figure 2) and the need to investigate its role in relevant models and with genetic and pharmacological inhibition.
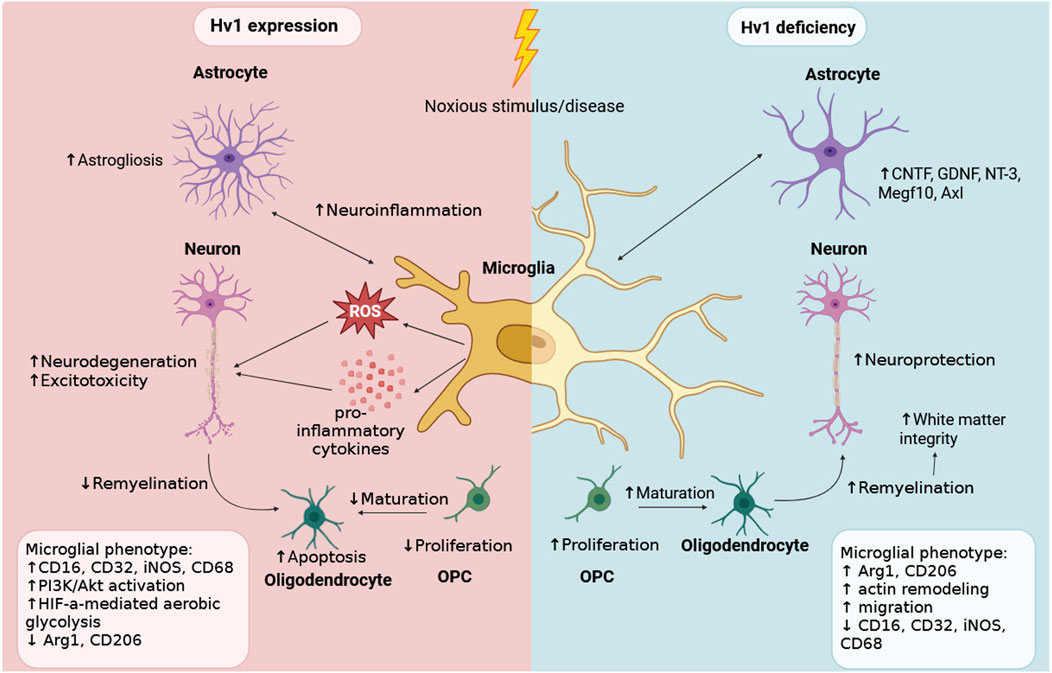
Figure 2. Hv1 expression in microglia modulates their inflammatory phenotype in neurological disorders. Schematic representation of the effects of Hv1 expression versus Hv1 deficiency in microglia upon noxious stimulation or disease conditions. In the presence of Hv1 (left), microglia release increased levels of ROS and pro-inflammatory cytokines, which promote astrogliosis, neuroinflammation, neurodegeneration, excitotoxicity, and impaired OPC maturation. As a result, oligodendrocyte apoptosis increases and remyelination decreases. Hv1-deficient microglia (right) produce less ROS and exhibit an anti-inflammatory phenotype, resulting in lower astrocyte activation, improved neuronal survival, higher OPC proliferation and maturation, increased white matter integrity as well as increased actin remodeling and migration. Created in BioRender. Gagliardi, C (2025) https://BioRender.com/vl1u9jq.
Hv1 presents a compelling therapeutic target due to its unique structure, restricted expression profile, and functional role in proton extrusion. Unlike the NADPH-oxidase complexes, which require assembly of multiple subunits, Hv1 is a simple voltage-gated homodimer, offering a structurally less complex and potentially more tractable drug target (Tomb et al., 2008). Hv1 inhibition can suppress excess ROS production without compromising basal homeostatic functions. Moreover, since Hv1 conducts significantly more protons per NADPH-oxidase electron transfer event (De and coursey, 2003), lower doses of Hv1 inhibitors are likely sufficient to reach therapeutic efficiency. Some Hv1 inhibitors, such as zinc or guanidine derivatives such as 2-guanidinobenzimidazole (2GBI), suffered from poor specificity, low potency, or limited bioavailability (He et al., 2021; Liu et al., 2025; Seredenina et al., 2015; Hong et al., 2014). Derivatives like CIGBI have modestly improved cell permeability and affinity but still face challenges related to specificity and therapeutic utility (Hong et al., 2014). Other inhibitors affect Hv1 indirectly by altering intracellular pH homeostasis, such as 4-aminopyridine, amiloride, and amantadine, or by modulating lipid rafts like epigallocatechin-3-gallate (Jin et al., 2013). More promising candidates have since emerged. YHV-984, a small molecule inhibitor, shows improved selectivity and efficacy in models of neuroinflammation and pain but has limited water solubility that limits its therapeutic potential (Zhang et al., 2022). To address this, lipid nanoparticles (LNPs) modified with the T7 peptide carrying a small molecule inhibitor (T7-LNP@YHV984) were developed, successfully delivering YHV-984 across the blood-brain barrier in an ischemic stroke model, where it accumulated in microglia, reduced neuroinflammation, and improved neuronal survival and behavioral outcomes.
Despite its promises, Hv1-targeted therapy faces several challenges. Hv1 function is context- and cell-dependent, and its inhibition may suppress protective microglial responses in early disease stages (Ekdahl et al., 2009). This highlights the need for precise control over timing, dosage, and cell-specific delivery. Current studies rely heavily on germline Hv1 knockout models (He et al., 2021), which may introduce developmental compensations that obscure the temporal or cell type-specific functions of Hv1 in the adult brain. Future work should prioritize conditional and inducible models to dissect the function of Hv1 in distinct cell populations and disease stages.
Moreover, the broader role of Hv1 in glial crosstalk, network-level signaling, and regional heterogeneity remains poorly understood. Its regulation in aging and chronic disease contexts also requires further investigation. There is increasing interest in whether Hv1 modulation might offer therapeutic benefit beyond acute CNS injuries, including in chronic neurodegenerative diseases or systemic conditions with neurological impact, such as diabetes or cancer.
In conclusion, Hv1 is emerging as a central modulator of microglial function, oxidative stress, and neuroinflammation. As new molecular and delivery platforms advance, Hv1 holds a significant potential as a therapeutic target across a wide range of acute and chronic neurological conditions.
Author contributions
MS: Conceptualization, Visualization, Writing – original draft. CG: Conceptualization, Visualization, Writing – review and editing. MC: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Brooks, L. R. K., and Mias, G. I. (2019). Data-driven analysis of age, sex, and tissue effects on gene expression variability in Alzheimer’s disease. Front. Neurosci. 13, 392. doi:10.3389/fnins.2019.00392
Capasso, M. (2014). Regulation of immune responses by proton channels. Immunology 143 (2), 131–137. doi:10.1111/imm.12326
Capasso, M., Bhamrah, M. K., Henley, T., Boyd, R. S., Langlais, C., Cain, K., et al. (2010). HVCN1 modulates BCR signal strength via regulation of BCR-dependent generation of reactive oxygen species. Nat. Immunol. 11 (3), 265–272. doi:10.1038/ni.1843
Chen, M., Yang, L. L., Hu, Z. W., Qin, C., Zhou, L. Q., Duan, Y. L., et al. (2020). Deficiency of microglial Hv1 channel is associated with activation of autophagic pathway and ROS production in LPC-induced demyelination mouse model. J. Neuroinflammation 17 (1), 333. doi:10.1186/s12974-020-02020-y
DeCoursey, T. E. (2003). Voltage-gated proton channels and other proton transfer pathways. Physiol. Rev. 83 (2), 475–579. doi:10.1152/physrev.00028.2002
DeCoursey, T. E. (2010). Voltage-gated proton channels find their dream job managing the respiratory burst in phagocytes. Physiol. (Bethesda) 25 (1), 27–40. doi:10.1152/physiol.00039.2009
DeCoursey, T. E., Cherny, V. V., Zhou, W., and Thomas, L. L. (2000). Simultaneous activation of NADPH oxidase-related proton and electron currents in human neutrophils. Proc. Natl. Acad. Sci. U. S. A. 97 (12), 6885–6889. doi:10.1073/pnas.100047297
Ekdahl, C. T., Kokaia, Z., and Lindvall, O. (2009). Brain inflammation and adult neurogenesis: the dual role of microglia. Neuroscience 158 (3), 1021–1029. doi:10.1016/j.neuroscience.2008.06.052
El Chemaly, A., Okochi, Y., Sasaki, M., Arnaudeau, S., Okamura, Y., and Demaurex, N. (2010). VSOP/Hv1 proton channels sustain calcium entry, neutrophil migration, and superoxide production by limiting cell depolarization and acidification. J. Exp. Med. 207 (1), 129–139. doi:10.1084/jem.20091837
Guzmán-Ruíz, M. A., Guerrero Vargas, N. N., Ramírez-Carreto, R. J., González-Orozco, J. C., Torres-Hernández, B. A., Valle-Rodríguez, M., et al. (2024). Microglia in physiological conditions and the importance of understanding their homeostatic functions in the arcuate nucleus. Front. Immunol. 15, 1392077. doi:10.3389/fimmu.2024.1392077
Haslund-Vinding, J., McBean, G., Jaquet, V., and Vilhardt, F. (2017). NADPH oxidases in oxidant production by microglia: activating receptors, pharmacology and association with disease. Br. J. Pharmacol. 174 (12), 1733–1749. doi:10.1111/bph.13425
He, J., Ritzel, R. M., and Wu, J. (2021). Functions and mechanisms of the voltage-gated Proton Channel Hv1 in brain and spinal cord injury. Front. Cell Neurosci. 15, 662971. doi:10.3389/fncel.2021.662971
Henderson, L. M., Chappell, J. B., and Jones, O. T. (1987). The superoxide-generating NADPH oxidase of human neutrophils is electrogenic and associated with an H+ channel. Biochem. J. 246 (2), 325–329. doi:10.1042/bj2460325
Hernandez-Espinosa, D. R., Gale, J. R., Scrabis, M. G., and Aizenman, E. (2023). Microglial reprogramming by Hv1 antagonism protects neurons from inflammatory and glutamate toxicity. J. Neurochem. 165 (1), 29–54. doi:10.1111/jnc.15760
Hong, L., Kim, I. H., and Tombola, F. (2014). Molecular determinants of Hv1 proton channel inhibition by guanidine derivatives. Proc. Natl. Acad. Sci. U. S. A. 111 (27), 9971–9976. doi:10.1073/pnas.1324012111
Jin, S., Park, M., and Song, J. H. (2013). (-)-Epigallocatechin-3-gallate inhibits voltage-gated proton currents in BV2 microglial cells. Eur. J. Pharmacol. 698 (1–3), 154–160. doi:10.1016/j.ejphar.2012.11.036
Kapus, A., Romanek, R., Qu, A. Y., Rotstein, O. D., and Grinstein, S. (1993). A pH-sensitive and voltage-dependent proton conductance in the plasma membrane of macrophages. J. general physiology 102 (4), 729–760. doi:10.1085/jgp.102.4.729
Kawai, T., Okochi, Y., Ozaki, T., Imura, Y., Koizumi, S., Yamazaki, M., et al. (2017). Unconventional role of voltage-gated proton channels (VSOP/Hv1) in regulation of microglial ROS production. J. Neurochem. 142 (5), 686–699. doi:10.1111/jnc.14106
Kawai, T., Takao, K., Akter, S., Abe, M., Sakimura, K., Miyakawa, T., et al. (2021). Heterogeneity of microglial proton channel in different brain regions and its relationship with aging. J. Neurochem. 157 (3), 624–641. doi:10.1111/jnc.15292
Kimura, S., Iwata, M., Takase, H., Lo, E. H., and Arai, K. (2025). Oxidative stress and chronic cerebral hypoperfusion: an overview from preclinical rodent models. J. Cereb. Blood Flow and Metabolism 45 (3), 381–395. doi:10.1177/0271678x241305899
Li, W., Ward, R., Dong, G., Ergul, A., and O'Connor, P. (2019). Neurovascular protection in voltage-gated proton channel Hv1 knock-out rats after ischemic stroke: interaction with Na(+)/H(+) exchanger-1 antagonism. Physiol. Rep. 7 (13), e14142. doi:10.14814/phy2.14142
Li, X., Yu, Z., Zong, W., Chen, P., Li, J., Wang, M., et al. (2020a). Deficiency of the microglial Hv1 proton channel attenuates neuronal pyroptosis and inhibits inflammatory reaction after spinal cord injury. J. Neuroinflammation 17 (1), 263. doi:10.1186/s12974-020-01942-x
Li, X., Liu, R., Yu, Z., He, D., Zong, W., Wang, M., et al. (2020b). Microglial Hv1 exacerbates secondary damage after spinal cord injury in mice. Biochem. Biophysical Res. Commun. 525, 208–215. doi:10.1016/j.bbrc.2020.02.012
Li, Y., Ritzel, R. M., He, J., Cao, T., Sabirzhanov, B., Li, H., et al. (2021). The voltage-gated proton channel Hv1 plays a detrimental role in contusion spinal cord injury via extracellular acidosis-mediated neuroinflammation. Brain, Behav. Immun. 91, 267–283. doi:10.1016/j.bbi.2020.10.005
Li, Y., Xie, Y., Liu, R., Wang, Z., Chen, P., Wang, M., et al. (2023). Knockout of microglial Hv1 proton channel reduces neurotoxic A1 astrocytes and neuronal damage via the ROS/STAT3 pathway after spinal cord injury. Glia 71 (10), 2418–2436. doi:10.1002/glia.24433
Liu, J., Tian, D., Murugan, M., Eyo, U. B., Dreyfus, C. F., Wang, W., et al. (2015). Microglial Hv1 proton channel promotes cuprizone-induced demyelination through oxidative damage. J. Neurochem. 135 (2), 347–356. doi:10.1111/jnc.13242
Liu, X., Lei, Z., Gilhooly, D., He, J., Li, Y., Ritzel, R. M., et al. (2023). Traumatic brain injury-induced inflammatory changes in the olfactory bulb disrupt neuronal networks leading to olfactory dysfunction. Brain, Behav. Immun. 114, 22–45. doi:10.1016/j.bbi.2023.08.004
Liu, X., He, H., Qi, M., Jiang, Z., Lin, B., Wang, X., et al. (2025). A small molecule directly targets NLRP3 to promote inflammasome activation and antitumor immunity. Cell Death Dis. 16 (1), 252. doi:10.1038/s41419-025-07578-0
Ma, M. W., Wang, J., Zhang, Q., Wang, R., Dhandapani, K. M., Vadlamudi, R. K., et al. (2017). NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 12 (1), 7. doi:10.1186/s13024-017-0150-7
Mendiola, A. S., Ryu, J. K., Bardehle, S., Meyer-Franke, A., Ang, K. K. H., Wilson, C., et al. (2020). Transcriptional profiling and therapeutic targeting of oxidative stress in neuroinflammation. Nat. Immunol. 21 (5), 513–524. doi:10.1038/s41590-020-0654-0
Morihata, H., Kawawaki, J., Sakai, H., Sawada, M., Tsutada, T., and Kuno, M. (2000). Temporal fluctuations of voltage-gated proton currents in rat spinal microglia via pH-dependent and -independent mechanisms. Neurosci. Res. 38 (3), 265–271. doi:10.1016/s0168-0102(00)00170-x
Murugan, M., Zheng, J., Wu, G., Mogilevsky, R., Zheng, X., Hu, P., et al. (2020). The voltage-gated proton channel Hv1 contributes to neuronal injury and motor deficits in a mouse model of spinal cord injury. Mol. Brain 13 (1), 143. doi:10.1186/s13041-020-00682-6
Neal, M. L., Beier, E. E., Hossain, M. M., Boyle, A., Zheng, J., Kim, C., et al. (2023). Voltage-gated proton channel Hv1 regulates neuroinflammation and dopaminergic neurodegeneration in Parkinson’s disease models. Antioxidants 12, 582. doi:10.3390/antiox12030582
Okochi, Y., Sasaki, M., Iwasaki, H., and Okamura, Y. (2009). Voltage-gated proton channel is expressed on phagosomes. Biochem. Biophysical Res. Commun. 382 (2), 274–279. doi:10.1016/j.bbrc.2009.03.036
Ou, G. Y., Lin, W. W., and Zhao, W. J. (2021). Construction of long noncoding RNA-associated ceRNA networks reveals potential biomarkers in alzheimer’s disease. J. Alzheimer's Dis. 82 (1), 169–183. doi:10.3233/jad-210068
Pathak, M. M., Tran, T., Hong, L., Joós, B., Morris, C. E., and Tombola, F. (2016). The Hv1 proton channel responds to mechanical stimuli. J. General Physiology 148 (5), 405–418. doi:10.1085/jgp.201611672
Peng, J., Yi, M. H., Jeong, H., McEwan, P. P., Zheng, J., Wu, G., et al. (2021). The voltage-gated proton channel Hv1 promotes microglia-astrocyte communication and neuropathic pain after peripheral nerve injury. Mol. Brain 14 (1), 99. doi:10.1186/s13041-021-00812-8
Pinto-Costa, R., Harbachova, E., La Vitola, P., and Di Monte, D. A. (2023). Overexpression-induced α-synuclein brain spreading. Neurotherapeutics 20 (1), 83–96. doi:10.1007/s13311-022-01332-6
Ramsey, I. S., Moran, M. M., Chong, J. A., and Clapham, D. E. (2006). A voltage-gated proton-selective channel lacking the pore domain. Nature 440 (7088), 1213–1216. doi:10.1038/nature04700
Ramsey, I. S., Ruchti, E., Kaczmarek, J. S., and Clapham, D. E. (2009). Hv1 proton channels are required for high-level NADPH oxidase-dependent superoxide production during the phagocyte respiratory burst. Proc. Natl. Acad. Sci. U. S. A. 106 (18), 7642–7647. doi:10.1073/pnas.0902761106
Ritzel, R. M., He, J., Li, Y., Cao, T., Khan, N., Shim, B., et al. (2021). Proton extrusion during oxidative burst in microglia exacerbates pathological acidosis following traumatic brain injury. Glia 69 (3), 746–764. doi:10.1002/glia.23926
Rubiano, A. M., Carney, N., Chesnut, R., and Puyana, J. C. (2015). Global neurotrauma research challenges and opportunities. Nature 527 (7578), S193–S197. doi:10.1038/nature16035
Ryan, A. K., Rich, W., and Reilly, M. A. (2023). Oxidative stress in the brain and retina after traumatic injury. Front. Neurosci. 17, 1021152. doi:10.3389/fnins.2023.1021152
Salter, M. W., and Stevens, B. (2017). Microglia emerge as central players in brain disease. Nat. Med. 23 (9), 1018–1027. doi:10.1038/nm.4397
Sasaki, M., Takagi, M., and Okamura, Y. (2006). A voltage sensor-domain protein is a voltage-gated proton channel. Science 312 (5773), 589–592. doi:10.1126/science.1122352
Seligmann, B., Camiolo, S., Hernandez, M., Yeakley, J. M., Sahagian, G., and McComb, J. (2024). Molecular gene expression testing to identify Alzheimer’s disease with high accuracy from fingerstick blood. J. Alzheimer’s Dis. 101 (3), 813–822. doi:10.3233/JAD-240174
Seredenina, T., Demaurex, N., and Krause, K. H. (2015). Voltage-gated proton channels as novel drug targets: from NADPH oxidase regulation to sperm biology. Antioxidants and Redox Signal. 23 (5), 490–513. doi:10.1089/ars.2013.5806
Sun, L., Wang, X., Guan, S., Chi, L., Liang, M., Lu, X., et al. (2024). Inhibition of voltage-gated Hv1 alleviates LPS-induced neuroinflammation via regulation of microglial metabolic reprogramming. Int. Immunopharmacol. 127, 111361. doi:10.1016/j.intimp.2023.111361
Sun, L., Wang, X., Guan, S., Zhang, P., Chen, D., and Luo, T. (2025). Deficiency of microglial Hv1 protects against lipopolysaccharide-induced neuroinflammation via the NF-κB signaling pathway and HIF1α-mediated metabolic reprogramming. FASEB J. 39 (17), e70894. doi:10.1096/fj.202402271rrr
Tauffenberger, A., and Magistretti, P. J. (2021). Reactive oxygen species: beyond their reactive behavior. Neurochem. Res. 46 (1), 77–87. doi:10.1007/s11064-020-03208-7
Tian, D. S., Li, C. Y., Qin, C., Murugan, M., Wu, L. J., and Liu, J. L. (2016). Deficiency in the voltage-gated proton channel Hv1 increases M2 polarization of microglia and attenuates brain damage from photothrombotic ischemic stroke. J. Neurochem. 139 (1), 96–105. doi:10.1111/jnc.13751
Tombola, F., Ulbrich, M. H., and Isacoff, E. Y. (2008). The voltage-gated proton channel Hv1 has two pores, each controlled by one voltage sensor. Neuron 58 (4), 546–556. doi:10.1016/j.neuron.2008.03.026
Tombola, F., Ulbrich, M. H., Kohout, S. C., and Isacoff, E. Y. (2010). The opening of the two pores of the Hv1 voltage-gated proton channel is tuned by cooperativity. Nat. Struct. Mol. Biol. 17 (1), 44–50. doi:10.1038/nsmb.1738
Venkatesh, K., Ghosh, S. K., Mullick, M., Manivasagam, G., and Sen, D. (2019). Spinal cord injury: pathophysiology, treatment strategies, associated challenges, and future implications. Cell Tissue Res. 377 (2), 125–151. doi:10.1007/s00441-019-03039-1
Wang, F., Ma, X. R., Wu, Y., Xu, Y. C., Gu, H. M., Wang, D. X., et al. (2021). Neutralization of Hv1/HVCN1 with antibody enhances microglia/macrophages myelin clearance by promoting their migration in the brain. Front. Cell Neurosci. 15, 768059. doi:10.3389/fncel.2021.768059
Wu, L. J., Wu, G., Sharif, M. R. A., Baker, A., Jia, Y., Fahey, F. H., et al. (2012). The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke. Nat. Neurosci. 15 (4), 565–573. doi:10.1038/nn.3059
Yang, Z., Jin, L., Li, L., Wu, Y., Liu, W., Feng, X., et al. (2025). Brain targeted lipid nanoparticles with Hv1 inhibitors alleviate neuroinflammation post-ischemic stroke. J. Nanobiotechnology 23 (1), 464. doi:10.1186/s12951-025-03540-6
Yu, Y., Yu, Z., Xie, M., Wang, W., and Luo, X. (2018). Hv1 proton channel facilitates production of ROS and pro-inflammatory cytokines in microglia and enhances oligodendrocyte progenitor cells damage from oxygen-glucose deprivation in vitro. Biochem. Biophysical Res. Commun. 498 (1), 1–8. doi:10.1016/j.bbrc.2017.06.197
Yu, Y., Luo, X., Li, C., Ding, F., Wang, M., Xie, M., et al. (2020). Microglial Hv1 proton channels promote white matter injuries after chronic hypoperfusion in mice. J. Neurochem. 152 (3), 350–367. doi:10.1111/jnc.14925
Zeng, W. Z., Liu, D. S., Liu, L., She, L., Wu, L. J., and Xu, T. L. (2015). Activation of acid-sensing ion channels by localized proton transient reveals their role in proton signaling. Sci. Rep. 5 (1), 14125. doi:10.1038/srep14125
Zhang, Q., Ren, Y., Mo, Y., Guo, P., Liao, P., Luo, Y., et al. (2022). Inhibiting Hv1 channel in peripheral sensory neurons attenuates chronic inflammatory pain and opioid side effects. Cell Res. 32 (5), 461–476. doi:10.1038/s41422-022-00616-y
Zhang, W., Xiao, D., Mao, Q., and Xia, H. (2023). Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target. Ther. 8 (1), 267. doi:10.1038/s41392-023-01486-5
Keywords: Hv1, microglia, ROS - reactive oxygen species, proton channel, neuroinflammation
Citation: Stratmann M, Gagliardi C and Capasso M (2025) Proton channel Hv1 modulates microglial responses to neurological disorders. Front. Biophys. 3:1681011. doi: 10.3389/frbis.2025.1681011
Received: 06 August 2025; Accepted: 26 September 2025;
Published: 08 October 2025.
Edited by:
Joao L. Carvalho-de-Souza, Midwestern University, United StatesReviewed by:
Naileth Gonzalez-Sanabria, The University of Chicago, United StatesTomoya Kubota, Osaka University, Japan
Garilyn Jentarra, Midwestern University, United States
Copyright © 2025 Stratmann, Gagliardi and Capasso. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Melania Capasso, bWVsYW5pYS5jYXBhc3NvQGR6bmUuZGU=