 Blaž Škrlj
Blaž Škrlj Enja Kokalj
Enja Kokalj Nada Lavrač
Nada Lavrač- 1Jožef Stefan International Postgraduate School, Ljubljana, Slovenia
- 2Jožef Stefan Institute, Ljubljana, Slovenia
- 3University of Nova Gorica, Vipava, Slovenia
PubMed is the largest resource of curated biomedical knowledge to date, entailing more than 25 million documents. Large quantities of novel literature prevent a single expert from keeping track of all potentially relevant papers, resulting in knowledge gaps. In this article, we present CHEMMESHNET, a newly developed PubMed-based network comprising more than 10,000,000 associations, constructed from expert-curated MeSH annotations of chemicals based on all currently available PubMed articles. By learning latent representations of concepts in the obtained network, we demonstrate in a proof of concept study that purely literature-based representations are sufficient for the reconstruction of a large part of the currently known network of physical, empirically determined protein–protein interactions. We demonstrate that simple linear embeddings of node pairs, when coupled with a neural network–based classifier, reliably reconstruct the existing collection of empirically confirmed protein–protein interactions. Furthermore, we demonstrate how pairs of learned representations can be used to prioritize potentially interesting novel interactions based on the common chemical context. Highly ranked interactions are qualitatively inspected in terms of potential complex formation at the structural level and represent potentially interesting new knowledge. We demonstrate that two protein–protein interactions, prioritized by structure-based approaches, also emerge as probable with regard to the trained machine-learning model.
1 Introduction
To this date, textual data remain one of the most widely accessible sources of information. Contemporary databases of, for example, biomedical knowledge, such as the PubMed database (Web, 2012), can consist of tens of millions of annotated scientific documents, offering, for example, detailed insights into various aspects of disease development, and the potential links between the diseases (Hristovski et al., 2005; Venkatasubbaiah et al., 2020). Albeit most of such knowledge can be accessed, it is not necessarily directly useful to a researcher, given that manual inspection of thousands of articles is not the most optimal way of uncovering, for example, how two fields of molecular biology interlink or what are the potentially interesting novel biomarkers related to a given pair of domains.
To remedy this shortcoming, the field of literature-based discovery (LBD) emerged (Swanson, 1990; Smalheiser and Swanson, 1994), exploring how contemporary computational methods can be exploited for faster and more efficient generation of potentially interesting associations, bridging different, albeit well-established concepts. Most recent LBD approaches benefit from word embeddings (Mikolov et al., 2013). A study by Tshitoyan et al. (2019) showed that latent knowledge regarding future discoveries is to a large extent embedded in past publications by retrieving information from the scientific literature with the usage of word2vec embeddings (Mikolov et al., 2013). The recent approach by Lavrač et al. (2020) explored how word embeddings (Joulin et al., 2016) can be used for identification of novel bridging terms in the field of plant biology. A similar approach was also explored in the context of COVID-19–related biomarker discovery (Martinc et al., 2020). An approach by Crichton et al. (2020) proposed graph-based neural network methods to perform open and closed LBD and demonstrated improved performance on existing tasks.
The purpose of this work is multifold, and its contributions can be summarized as follows.
(1) We propose CHEMMESHNET, a network of paper annotations (MeSH terms of chemicals) constructed from more than 30 million PubMed documents.
(2) The annotations, present in CHEMMESHNET, were embedded in a low-dimensional vector space via network node representation learning, which enables their direct use in downstream tasks such as discovery of novel associations.
(3) We demonstrate that protein representations, obtained exclusively based on document annotations, can be used to reliably reconstruct a large part of the currently known human proteome (Oughtred et al., 2020).
(4) The quantitative reconstruction results indicate that representations, based on the singular value decomposition (SVD) of a normalized graph Laplacian matrix can already offer sufficient expressive power, while scaling seamlessly to millions of links on an off-the-shelf hardware.
(5) The obtained protein representations are finally used to prioritize the space of potentially interesting novel protein–protein interactions. The top-ranked interactions are analyzed qualitatively at the level of protein structure.
The rest of this work is structured as follows. In Section 2, we present a brief outline of the related work, followed by the presentation of the proposed methodology in Section 3. The evaluation is presented in Section 4. Results are presented in Sections 5 and 6. Followed by the discussion in Section 7 and conclusions in Section 8.
2 Related Work
This section discusses the relevant related work, spanning the fields of literature-based discovery (LBD) and network representation learning. It also presents the PubMed database of biomedical articles as it is the key resource for LBD considered in this work.
The field of literature-based discovery (LBD) was conceptualized in the 1990s, when Swanson (1990) and Smalheiser and Swanson (1994) developed early LBD approaches (e.g., the so-called ABC model) to detect interesting bridging terms (b-terms), aimed at uncovering new cross-domain relations among previously unrelated concepts in separate domain corpora of interest, surveyed also by Bruza and Weeber (2008). Initial LBD works explored how lexical statistics can offer novel insights (Lindsay and Gordon, 1999). LBD has led to the discovery of potential treatments in several domains, including multiple sclerosis (Kostoff et al., 2008) and has been applied successfully in drug development and repurposing (Deftereos et al., 2011). The recent surveys offer extensive overviews of the promising approaches of LBD and their implications (Smalheiser, 2012, 2017; Sebastian et al., 2017; Thilakaratne et al., 2019). In terms of evaluation of LBD systems, Yetisgen-Yildiz and Pratt (2008) offer a comprehensive overview of the existing evaluation strategies, emphasizing that rigorous inspection of the discovered knowledge is a critical component of every LBD system.
The proposed CHEMMESHNET-based discovery focuses exclusively on biomedical knowledge discovery from the MeSH (Medical Subject Heading) terms network. Similarly, the work of Kastrin et al. (2016) was one of the first to explore how networks of co-occurring MeSH terms can be used for novel discovery. Their work served as the basis for the idea proposed in this article, where the MeSH term networks are analyzed via a node embedding–based methodology. Other promising approaches were developed for better understanding of cancer development (Pyysalo et al., 2019) using a tool LION LBD that enables researchers to navigate published information and supports hypothesis generation and testing.
A part of the proposed methodology relates to network representation learning, revolving around the notion of node embedding. In the recent years, instead of designing algorithms for direct link prediction (Kastrin et al., 2016) and similar tasks, development of a methodology which first projects individual nodes into a latent space (embedding) where one can directly measure similarity and, for example, predict links, has been actively explored. Methods such as DeepWalk (Perozzi et al., 2014), node2vec (Grover and Leskovec, 2016), and similar ones explore how random walk-based sampling schemes can offer compressed node representations. The node embedding methods are commonly black-box, that is, real-valued latent representations without any interpretability, many times obtained efficiently via closed-form expressions. Apart from node classification and link prediction, some other uses of node embeddings include, for example, community detection (Škrlj et al., 2020b). For a more detailed overview, the interested reader can refer to the work of Zhang et al. (2018).
Finally, we discuss the main source of knowledge used in this work—the PubMed database of biomedical articles1 (Sayers et al., 2020). The database receives more than 3 billion search queries each year and represents the central body of knowledge related to the biomedical domain. The current version used in this work comprises more than 30 million scientific publications, all annotated with MeSH terms. This part of annotation is of key focus to the proposed CHEMMESHNET, as it offers direct insight into into, for example, key compounds relevant for a given article, which we posit is a rich resource of human-annotated information that can be further exploited for literature-based discovery, albeit at the MeSH term graph level.
3 CHEMMESHNET: Construction of Concept Networks From PubMed
We begin the description of the proposed methodology by first discussing the construction process of the network based on paper annotations, followed by the description of network construction and filtering. A schematic overview of the proposed approach is shown in Figure 1 and small subnetwork is shown in Figure 2.
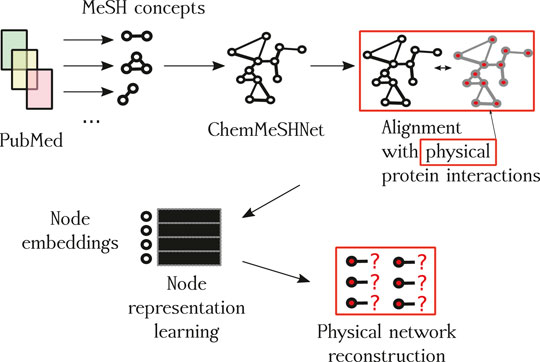
FIGURE 1. Overview of the proposed approach. The first contribution is the CHEMMESHNET, a network of term co-occurrences. Once constructed, the network is aligned with the space of empirically validated protein interactions (red squares), where the subset of proteins present in the annotations (nodes) of CHEMMESHNET are embedded into a low-dimensional space (node embeddings), and used to learn which links are actually present and which are not. Once trained, the best-performing classifier predicted scores for potential new interactions, which we also discuss as a part of qualitative evaluation.
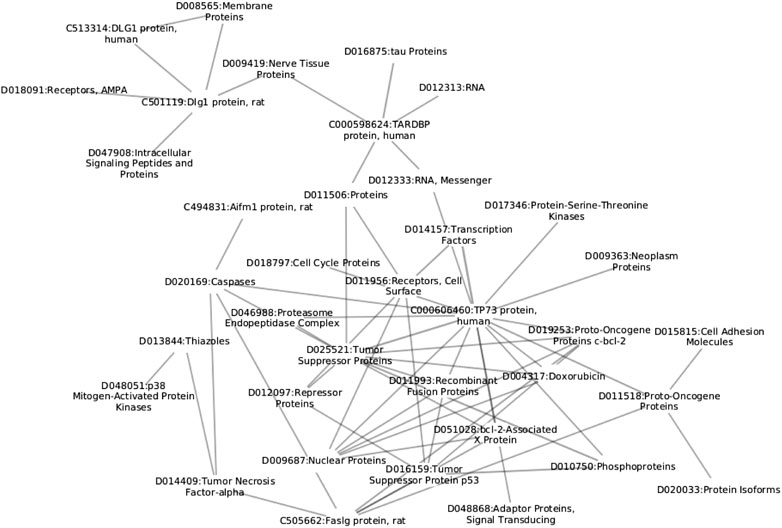
FIGURE 2. Example of MeSH chemical term subnetwork. The reader can observe different levels of information all interlinked within the same structure; from the cellular level (D018797), to protein level (C000598624).
3.1 Extraction of Annotations and Network Construction
In the first part of the proposed approach, we download XML representations for all available PubMed articles2. The XML files include abstracts and the annotations, which are the key focus of the remainder of this work. Each PubMed article has metadata, containing information on, for example, publisher, date, authors, but also the expert-curated space of chemicals. According to the annotators, these well-defined annotations offer insight into the key concepts related to the given article. All annotations of this type are also known as the MeSH tags. However, note that the considered set of MeSH tags does not entail all possible MeSH tags—they entail only the ones describing “chemicals”, such as proteins, compounds, and some processes. For each PubMed article (currently, there are more than 30 million articles available), we extracted these annotations and constructed a network. For example, an article with three annotations results in a triangle graph, where each of the terms is associated with all others. Intuitively, this step entails the common context. Such cliques are obtained for each PubMed article and joined into a single network by linking the common nodes. Further, each edge is weighted based on cumulative co-occurrence across all the documents. Schematic overview of this step is shown in Figure 1. The number of documents that have the sufficient mentioned annotation tags was 14,671,298. Note that albeit constructed from cliques, the obtained network is modular. The claim is inspected via the analysis of degree distribution in the following sections.
3.2 Embedding the Space of Concepts
The obtained network could be used for direct mining of the associations; however, such endeavor could be computationally expensive and prohibitive to multiple downstream tasks of interest. Hence, the nodes of the obtained network, that is, the article annotations, are then embedded into a low-dimensional vector space in which their semantic relations are preserved and can be directly computed.
The field of node embedding has grown in the last years; however, development of methods that scale to tens of millions of links on an off-the-shelf workstation remains an interesting research endeavor. The embedding approach employed in this work was largely inspired by the branch of methods that revolve around spectral graph decomposition, that is, the analysis of the meaning and potential implications of, for example, the largest eigenvalues of graph Laplacians and their corresponding eigenvectors (see, e.g., the work of Zhang et al. (2018)). The considered node embedding method is in one of the simplest (linear) algorithms for obtaining the representations. To embed all annotations, we implemented the node embedding as the following two-step procedure.
1. Normalized graph Laplacian. In the first step, we compute the normalized graph Laplacian. Let
2. Sparse Singular Value Decomposition. In the second step, the L is decomposed into three matrices:
The final part of this step includes re-multiplication of the first d diagonal entries of
Let
3.3 Formulating the Reconstruction Problem
The key task addressed in this work is network reconstruction. We formulate the task as follows. Let
To our knowledge, such endeavor is novel and was not tested before at such a scale. The main implication of being able to exploit purely literature-based representations of physical entities, such as proteins, is to learn potentially relevant associations between them potentially interesting discovery opportunities. We next discuss the evaluation of the proposed method that was implemented in order to be able to prioritize potential interactions between existing protein pairs from the largest currently available network of physical protein interactions.
4 Reconstruction Evaluation
The following section focuses on the evaluation of the proposed approach. The two main types of evaluation considered are described as follows.
The first type of evaluation focuses on the exploration of how well the network of empirically proven protein–protein interactions can be reconstructed based solely on the node (protein) representations learned from the constructed CHEMMESHNET. To quantify to what extent the relations between the proteins are learnable, we consider the task as link prediction and evaluate it as such. Here, we first generate a data set that captures the existing links as well as false ones, that is, pairs of protein representations that are not known to form complexes (do not interact). We obtain such node pairs by considering nodes linked with a shortest path of length, at least two. This constraint assumes that nodes that are relatively distant from one another are not expected to interact in this setting. We believe that the path-based negative sampling remains a better option to randomly selecting node pairs in terms of the amount of sampled false negatives. The constructed data set for each valid interaction considers three randomly sampled interactions at a given length that were not among the ground truth ones, that is, the negative samples.
The second type of evaluation concerns representation evaluation, which includes performance measurement of various machine-learning algorithms for the task of link prediction. The learners used in this work are the extreme gradient boosting machines (XGB) (Chen and Guestrin, 2016), random forests (RF) (Breiman, 2001), the recently introduced self-attention networks (SAN) (Škrlj et al., 2020a), Skope Rules3, a majority (Dummy) classifier, and a logistic regression classifier (LR). Data splitting was conducted via the scikit-learn library (Pedregosa et al., 2011) (Dummy and LR classifiers were implemented via this library as well). The SciPy library (Virtanen et al., 2020) was used to implement the embedding steps.
5 Quantitative Results Evaluation
In this section, we present the main results of this work. We begin by describing the constructed network of annotations, followed by the quantitative reconstruction experiments. In Figure 3, we show the node degree distribution of the obtained network with annotated node names.
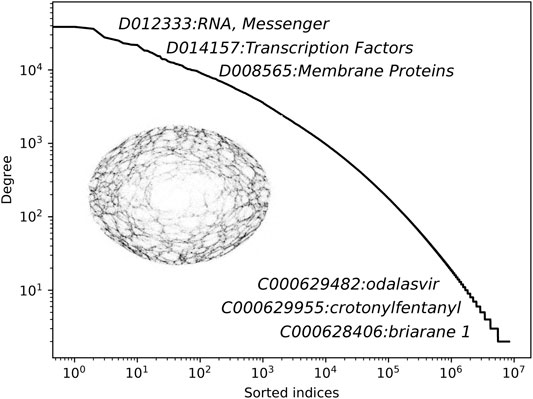
FIGURE 3. The degree distribution of the constructed CHEMMESHNET with some of the representative annotations. Note that for the purpose of this study, the constructed CHEMMESHNET consists only of MeSH terms, annotated as chemicals.
It can be observed that the distribution follows a linear trend in the log space, indicating that only a handful of nodes are very well connected (hubs), whereas the remaining ones are not. The distribution demonstrates that albeit clique-based construction was considered, the resulting network is far from being a regular graph. Additional statistics of the constructed network are shown in Table 1. The clustering coefficient measures how nodes in a graph tend to cluster together and is computed as the ratio between the number of closed triplets and the number of all triplets. The network density is computed as the number of actual connections, divided by all possible connections. The mean degree corresponds to the average number of connections of a node.

TABLE 1. Statistical overview of the constructed MeSH network of chemicals. This network is the prunned version of the one constructed directly from the MeSH annotations, as MeSH terms that are too rare: frequency
Next we present the results of the reconstruction experiment in Figure 4.
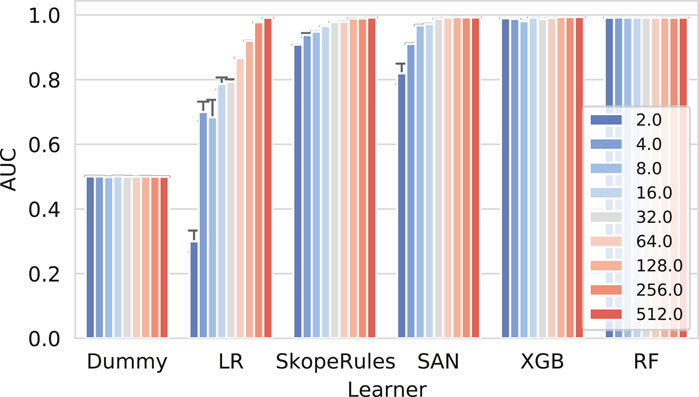
FIGURE 4. Network reconstruction benchmark results for individual learners. The more complex classifiers such as the self-attention networks (SAN), extreme gradient boosting (XGB), and Random Forests perform well when considering different amounts of training data (10, 50, and 90%). On the contrary, the logistic regression classifier (LR) performs adequately (AUC65) only if enough data were used. The impact of the embedding dimensionality can also be observed. Only high-enough dimensions result in good predictive performance, consistently for all models.
Here, a clear separation between the more complex models (neural networks and tree ensembles) and simpler ones (LR) can be observed. These results indicate that simple linear combinations of embedding dimensions are not sufficient to learn the difference between the true and false edges; however, even LR in some cases performs with AUC score
6 Qualitative Results Evaluation
We conducted the qualitative evaluation as follows. For 1 million interactions that were not used for training and evaluation of the method, we selected the top 10,000. From these, we considered only the ones that have predicted structural interfaces, indicating a potentially interesting physical interaction underpinning the ML-based prediction. For each of the interactions we obtained a score that was the SAN’s prediction, that is, the probability of the interaction. Hence, for this step, SAN was trained on the whole positive–negative interaction sample data set and used to predict probabilities of interactions for node pairs that were not considered during training.
6.1 Analysis of the Predicted Interactions
The task of link prediction resulted in an extensive list of ranked interactions, which was compared to the data in two major protein–protein interaction databases that collect the information from various reliable, curated sources and are also complemented with computational predictions. The considered databases are the Interactome INSIDER and STRINGdb, from which we obtained all the interactions pertaining to human proteins. They include 112,956 and 11,759,454 annotated protein–protein interactions, respectively. After individually matching them to our ranked list, we obtained intersections of sizes 98 and 1,692, respectively. The two databases (Interactome INSIDER and STRINGdb) represent two different aspects of protein interactions. The STRINGdb, being notably larger, includes binary interactions, that is, interactions where only the information about whether an interaction occurs or not is an interaction known. However, such interactions are not necessarily as informative as the ones on the structural level. The Interactome INSIDER offers direct exploration of such interactions at the single atom resolution, potentially offering more detailed information regarding actual protein–protein binding sites. It is noteworthy that the average probability of these interactions in both cases is higher than 99%. The list of the top 55 manually curated interactions and their probabilities of the intersection between our ranked list and the Interactome INSIDER database is shown in the Appendix in Table A1. In the following subsections, we evaluate in detail two high-probability protein–protein interactions proposed by our classifier (highlighted rows in Table 3).
6.2 Interaction Between PTPN1 and CAPN1
The first example considers Calpain-1 catalytic subunit (UniProt: P07384) and tyrosine–protein phosphatase non-receptor type 1 (UniProt: P18031). We identified a predicted structural interface between the two proteins via the Interactome INSIDER tool (Meyer et al., 2018), which offers insight into the ECLAIR-based structural predictions4.
The interaction interface is dark-colored in subfigures showing the structures (leftmost part of Figure 5). The first protein PTPN1—tyrosine–protein phosphatase—acts as a regulator of endoplasmic reticulum unfolded protein response. The second protein CAPN1 is a calcium-regulated non-lysosomal thiol-protease, which catalyzes limited proteolysis of substrates involved in cytoskeletal remodeling and signal transduction. The interface between the two proteins spans 468 amino acids, where numerous amino acids are predicted with very high confidence. As PTPN1 governs a signaling pathway, which modulates cell reorganization and cell–cell repulsion, the predicted interaction could offer novel insights into some of the key mechanisms of cell-to-cell signaling.
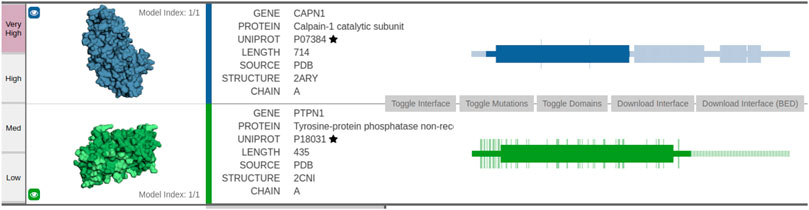
FIGURE 5. Visualization of the predicted interaction interface as obtained by the Interactome INSIDER tool. The sequential view of the amino acid sequences with annotated domain information (rectangles) is shown in the rightmost part of the figure. Note the overlap between the first domain of PTPN1 and the first domain of CAPN1—this domain overlap represents a potentially interesting protein interaction interface as predicted via ECLAIR.
6.3 Interaction Between EP300 and HIF1A
The second interaction we discuss in more detail is between histone acetyltransferase p300 (UniProt: Q09472) and hypoxia-inducible factor 1-alpha (UniProt: Q16665). This association was highlighted in the task of link prediction described in Section 4. We evaluated it by retrieving the relevant information from the Interactome INSIDER tool and the STRING database of protein functional interactions (Szklarczyk et al., 2020).
Protein EP300 functions as histone acetyltransferase and regulates transcription via chromatin remodeling, whereas protein HIF1A functions as a master transcriptional regulator of the adaptive response to hypoxia. We first analyzed the interaction using the Interactome INSIDER tool (Figure 6)5. The interface between the two proteins consists of 75 amino acid residues. The association of the two domains was obtained via co-crystallization experiments (leftmost part of Figure 6).
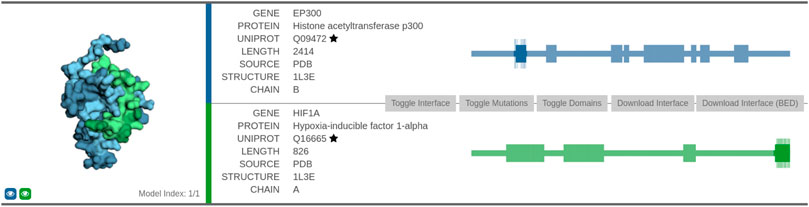
FIGURE 6. Visualization of the predicted interaction interface as obtained by the Interactome INSIDER tool. The sequential view of the amino acid sequences with the annotated domain information (rectangles) is shown in the rightmost part of the figure. The segments that form the interaction surface are shown in dark color, and the relevant amino acid residues are represented as vertical lines along the highlighted segment.
Furthermore, we also explored the confidence of the association provided by the STRING database. The STRING interaction scores represent an approximate confidence, given all the available evidence, their range is between 0 and 1. In the case of EP300 and HIF1A, the overall confidence is very high (0.998), and it is based on the following factors: the score that the interaction was experimentally determined (0.974) was obtained from an annotated database (0.900), was obtained via text-mining (0.614), and that their genes are co-expressed (0.055). The STRINGdb combines scores from separate interaction source channels by adding the probabilities together while simultaneously accounting for false discovery rate separately for each channel. Note that the interaction was not present among the ones from the BioGrid at the time of writing, indicating the proposed method’s capability to find well-represented interactions.
6.4 Analysis of False Positives
In the following table we present 25 selected false-positive results (prediction probability
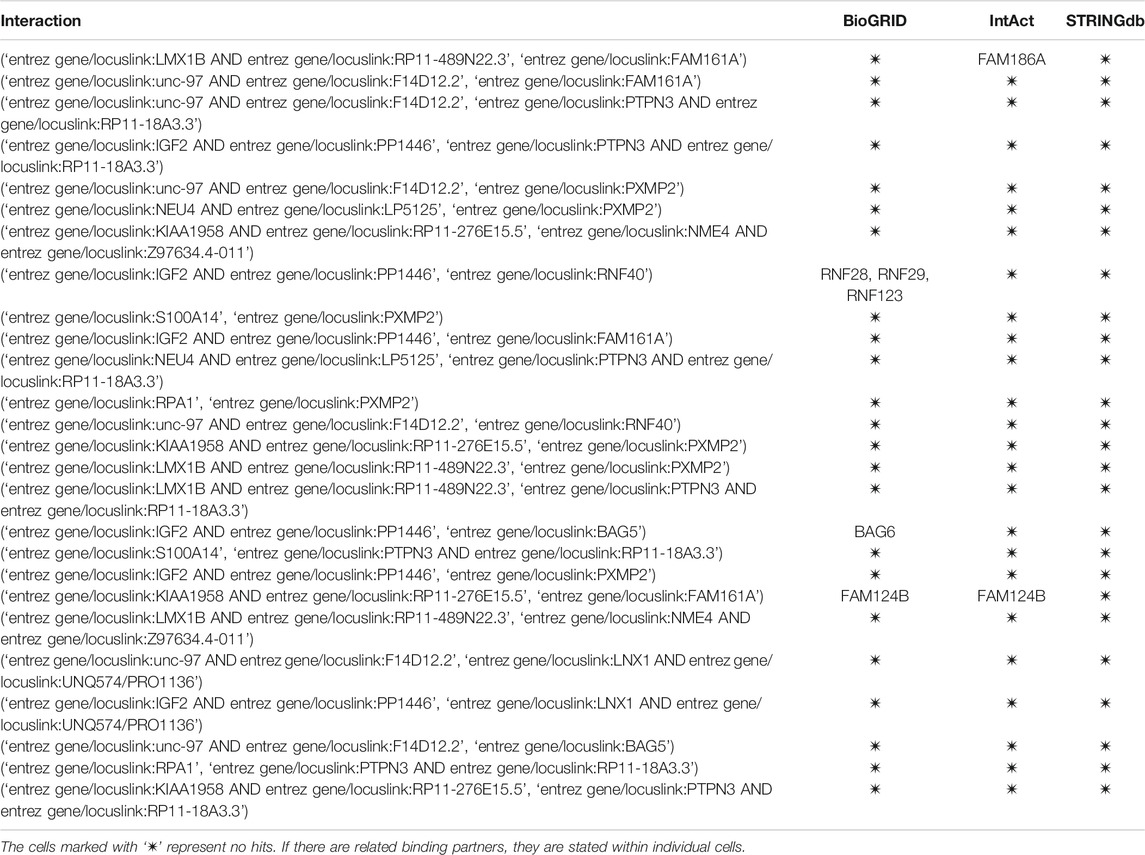
TABLE 2. Assessment of false-positive interactions. Each of the 25 interactions was manually assessed in the three stated databases for possible presence.
The first observation when additionally assessing the false-positive interaction is that none of the predicted interactions could be found in any of the databases via manual curation. The reasons for this result are the following. First, the high confidence criteria in STRINGdb are too stringent to unveil novel, potentially unproven interactions. Second, the manual curation could be extended to direct exploration of novel protein interaction articles; however, such manual curation was beyond the scope of this study. In terms of the observed similar interaction partners, we noticed the following: the FAM family of proteins (FAM124B) was identified as structurally similar to the query protein. The alignment of the two proteins is shown in Figure 7. The alignment was obtained with the Clustal Omega aligner Madeira et al. (2019).

FIGURE 7. Alignment of FAM124A (present) and FAM124B identified via manual curation.
It can be observed that parts of the two sequences overlap (approximately the residues 300–400). The overlap could indicate a similar binding site, showing a potential interaction of also FAM124B with the same protein as FAM124A. Overall, we observed that false-positive results offer an additional gateway to obtaining potentially interesting candidate interactions (apart from sampling out-of-training-distribution interactions).
7 Discussion
In the following section we discuss the main findings, ranging from the implications of the proposed method’s capability to reconstruct the physical networks to the discovery of novel interactions based on contextual representations of PubMed (chemicals) annotations.
The first part of this article focuses on the notion of network reconstruction. Here, we demonstrate that the existing BioGrid network of physical protein–protein interactions (Oughtred et al., 2020) can be adequately reconstructed by using more involved machine-learning models, such as tree ensembles or a deep neural network adapted for propositional data (SAN). We also demonstrated that simpler linear models do not perform well in this setting. The capability to reconstruct a physical network based solely on literature-based representations is a novel idea and was then extended as follows. We conducted an additional experiment, where the SAN model (one of the best-performing ones) was used to learn to estimate a probability of a given prediction. The model was trained on the collection of training interactions from the first experiment and used to predict probabilities for interactions, previously unseen by the model.
We qualitatively discuss such interactions, as they potentially represent novel knowledge. We demonstrated that one of the interactions estimated to occur (by SAN) with a high probability was in fact prioritized also by structure-only tools such as the Interactome INSIDER (Meyer et al., 2018), giving additional confidence to this interaction. We finally discuss the biological context of the interaction alongside possible implications of being able to rank millions of potentially interesting interactions. For the top prioritized interaction, we have shown that it could play a role in the cell regulation via the endoplasmic reticulum. Further, the computational predictions at the structural level obtained via Interactome INSIDER agree/confirm the predicted interaction, demonstrating the complementarity between the proposed machine learning–based and more conventional structure-based methodologies.
The focus of this work was on the relation between the literature-based MeSH tags and the existing empirical protein interaction networks. We believe that the use of biomedical named entity recognition methods, such as for example (Settles, 2005) could be additionally used for construction of knowledge graphs, extending the currently explored solution based on chemical annotations.
The considered work operates in the space of MeSH tags. There are multiple reasons for this design choice. First, memory-wise, dynamic construction of MeSH pairs is not as expensive as storing texts (possible multiple copies) during embedding construction. Second, MeSH terms are based on the whole article’s content, and not only the abstract. On the contrary, for the considered quantity of articles, the only sensible source of text are the abstracts (only a small percentage of the article is freely accessible). Thus, creation of embeddings based solely on abstracts could be problematic. We believe, however, that the two methodologies are compatible; both embedding-based and MeSH-based representations could be jointly used to learn potentially contextual representations which still maintain the information based on expensive manual curation.
This study focused on the chemicals-only part of the MeSH space. We believe that a natural extension of this work could include all MeSH tags, potentially offering richer context and applicability beyond protein/chemical networks. The current implementation of CHEMMESHNET offers direct construction of that network too; however, subsequent machine-learning experiments were beyond our computing capabilities to finish in reasonable time. The interested reader can find the instructions on how to create the extended MeSH network on the repository.
8 Conclusion
In this work, we presented CHEMMESHNET, a network derived from PubMed, comprising chemical-related MeSH tags. The network was hypothesized to be expressive enough to reconstruct existing physical protein–protein interaction relations, which we demonstrate quantitatively via link prediction. Further, we show how a machine-learning model, trained to recognize interactions, can be used to prioritize previously unseen interactions. We show for a pair of highly ranked interactions their overlap with the existing structure-based predictions, showcasing the added value of the proposed approach. Further, we performed extensive error analysis (manual inspection) of predicted interactions, demonstrating that this type of analysis is a potential source of novel interactions. We finally discussed the results in the context of biomedical knowledge discovery.
The proposed CHEMMESHNET serves as a freely available, mining-ready resource and is the key contribution of this article. Albeit being very expensive, we believe that it could be further extended to the space of all MeSH tags (not just chemicals). Such extensions are however potentially spatially more expensive and are therefore left for further work.
Data Availability Statement
The data sets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material. The code and the constructed MeSH networks are available at: https://gitlab.com/skblaz/chemmeshnet.
Author Contributions
BŠ and EK implemented the approach and wrote the core idea of the paper. NL supervised and guided the research, as well as wrote the paper.
Funding
The first two authors are funded via the junior research grants from the Slovenian Research Agency (ARRS). The work of the last author is funded by the Slovenian Research Agency (ARRS) core research program P2-0103 and research grant N2-0078 (financed under the ERC Complementary Scheme).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
APPENDIX
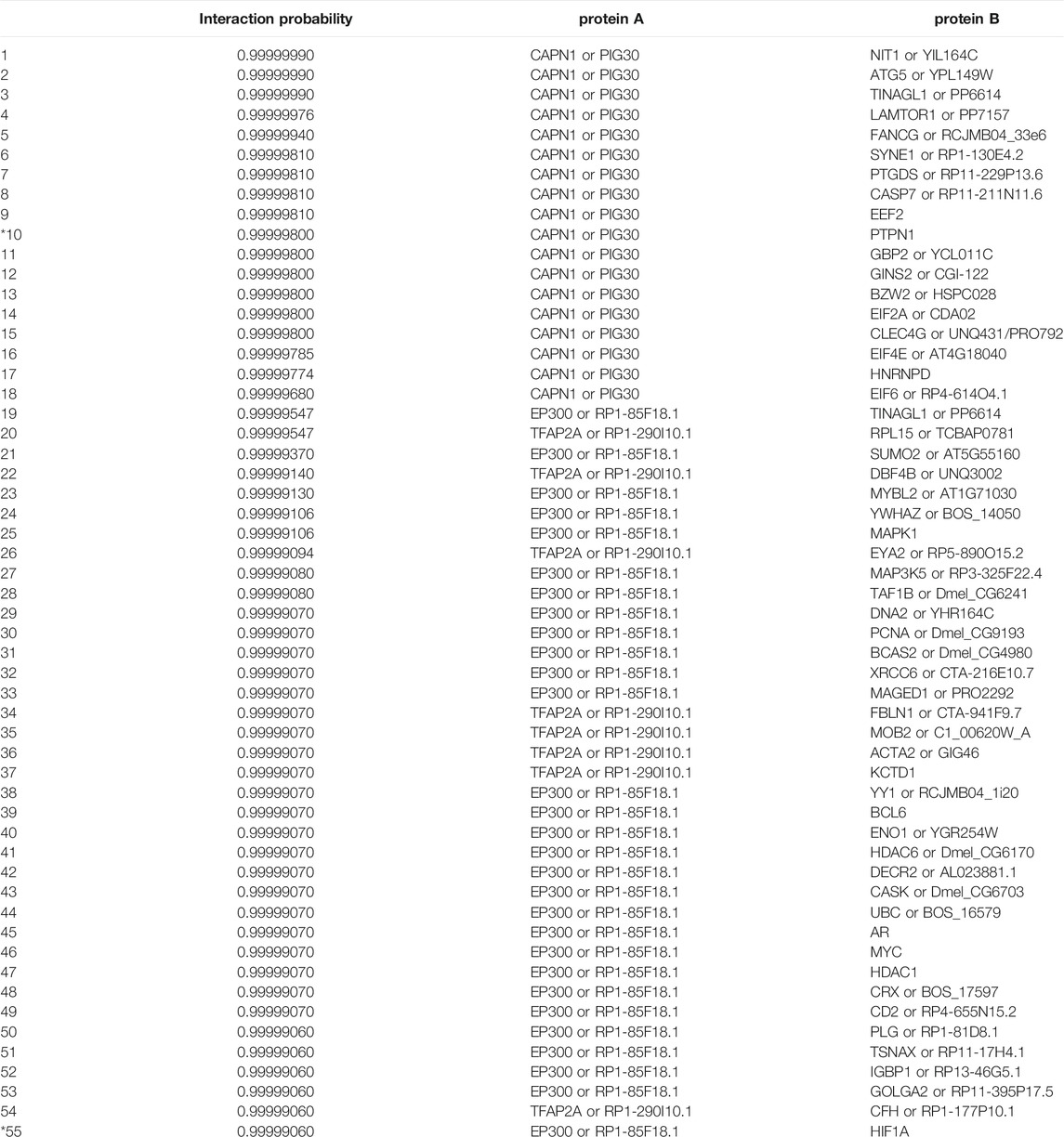
TABLE A1. The top 55 protein–protein interactions and their probabilities from the intersection between our list of ranked interactions and the Interactome INSIDER database. The two marked interactions (10th and 55th) are discussed in detail in Sections 6.2 and 6.3. The ‘or’ statements in the protein columns separate different names (synonyms) for the same protein.
Footnotes
1https://pubmed.ncbi.nlm.nih.gov/
2https://pubmed.ncbi.nlm.nih.gov/
3https://github.com/scikit-learn-contrib/skope-rules
4http://interactomeinsider.yulab.org/PPI_pair.html?interaction=P07384_P18031&mutcode
5http://interactomeinsider.yulab.org/PPI_pair.html?interaction=Q09472_Q16665&mutcode
References
Bruza, P., and Weeber, M. (2008). Literature-based discovery. Berlin Heidelberg: Springer Science & Business Media.
Chen, T., and Guestrin, C. (2016). “XGBoost: a scalable tree boosting system,” in Proceedings of the 22nd ACM SIGKDD international conference on knowledge discovery and data mining. Editors B. Krishnapuram, and M. Shah. (New York, USA: ACM, KDD), Vol. 16, 785–794.
Crichton, G., Baker, S., Guo, Y., and Korhonen, A. (2020). Neural networks for open and closed literature-based discovery. PLoS One 15, 1–16. doi:10.1371/journal.pone.0232891
Deftereos, S. N., Andronis, C., Friedla, E. J., Persidis, A., and Persidis, A. (2011). Drug repurposing and adverse event prediction using high-throughput literature analysis. Wiley Interdiscip Rev Syst Biol Med. 3, 323–334. doi:10.1002/wsbm.147
Grover, A., and Leskovec, J. (2016). “node2vec: scalable feature learning for networks,” in Proceedings of the 22nd ACM SIGKDD international conference on Knowledge discovery and data mining, San Francisco California USA, August, 2016, 855–864.
Hristovski, D., Peterlin, B., Mitchell, J. A., and Humphrey, S. M. (2005). Using literature-based discovery to identify disease candidate genes. Int. J. Med. Inform. 74, 289–298. doi:10.1016/j.ijmedinf.2004.04.024
Joulin, A., Grave, E., Bojanowski, P., and Mikolov, T. (2016). Bag of tricks for efficient text classification
Kastrin, A., Rindflesch, T. C., and Hristovski, D. (2016). Link prediction on a network of co-occurring mesh terms: towards literature-based discovery. Methods Inf. Med. 55, 340–346. doi:10.3414/ME15-01-0108
Kostoff, R. N., Briggs, M. B., and Lyons, T. J. (2008). Literature-related discovery (LRD): potential treatments for multiple sclerosis. Technol. Forecast. Soc. Change 75, 239–255. doi:10.1016/j.techfore.2007.11.002
Lavrač, N., Martinc, M., Pollak, S., Novak, M. P., and Cestnik, B. (2020). Bisociative literature-based discovery: Lessons learned and new word embedding approach. New Generation Comput. 38(4), 1–28. doi:10.1007/s00354-020-00108-w
Lindsay, R. K., and Gordon, M. D. (1999). Literature-based discovery by lexical statistics. J. Am. Soc. Inf. Sci. 50, 574–587. doi:10.1002/(sici)1097-4571(1999)50:7<574::aid-asi3>3.0.co;2-q
Madeira, F., Park, Y. M., Lee, J., Buso, N., Gur, T., Madhusoodanan, N., et al. (2019). The embl-ebi search and sequence analysis tools apis in 2019. Nucleic Acids Res. 47, W636–W641. doi:10.1093/nar/gkz268
Martinc, M., Škrlj, B., Pirkmajer, S., Lavrač, N., Cestnik, B., Marzidovšek, M., et al. (2020). “Covid-19 therapy target discovery with context-aware literature mining,” in International conference on discovery science. Editors A. Appice, G. Tsoumakas, Y. Manolopoulos, and S. MatwinThessaloniki, Greece, October 19–21, 2020. (Springer), 109–123.
Meyer, M. J., Beltrán, J. F., Liang, S., Fragoza, R., Rumack, A., Liang, J., et al. (2018). Interactome insider: a structural interactome browser for genomic studies. Nat. Methods 15, 107–114. doi:10.1038/nmeth.4540
Mikolov, T., Chen, K., Corrado, G., and Dean, J. (2013). Efficient estimation of word representations in vector space. arXiv preprint arXiv:1301.3781.
Oughtred, R., Rust, J., Chang, C., Breitkreutz, B.-J., Stark, C., Willems, A., et al. (2020). The bioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 30 (1), 187–200. doi:10.1002/pro.3978
Pedregosa, F., Varoquaux, G., Gramfort, A., Michel, V., Thirion, B., Grisel, O., et al. (2011). Scikit-learn: machine learning in Python. J. Machine Learn. Res. 12, 2825–2830. doi:10.5555/1953048.2078195
Perozzi, B., Al-Rfou, R., and Skiena, S. (2014). “Proceedings of the 20th ACM SIGKDD international conference on Knowledge discovery and data mining,” in Deepwalk: online learning of social representations. . Editors S. Macskassy, and C. Perlich. (New York, NY, United States: Association for Computing Machinery), 701–710.
Pyysalo, S., Baker, S., Ali, I., Haselwimmer, S., Shah, T., Young, A., et al. (2019). Lion lbd: a literature-based discovery system for cancer biology. Bioinformatics 35, 1553–1561. doi:10.1093/bioinformatics/bty845
Sayers, E. W., Beck, J., Brister, J. R., Bolton, E. E., Canese, K., Comeau, D. C., et al. (2020). Database resources of the national center for biotechnology information. Nucleic Acids Res. 48, D9–D16. doi:10.1093/nar/gkz899
Sebastian, Y., Siew, E.-G., and Orimaye, S. O. (2017). Emerging approaches in literature-based discovery: techniques and performance review. Knowledge Eng. Rev. 32, e12. doi:10.1017/s0269888917000042
Settles, B. (2005). ABNER: an open source tool for automatically tagging genes, proteins, and other entity names in text. Bioinformatics 21, 3191–3192. doi:10.1093/bioinformatics/bti475
Škrlj, B., Dzeroski, S., Lavrac, N., and Petkovic, M. (2020a). “Feature importance estimation with self-attention networks,” in Ecai 2020–24th European conference on artificial intelligence, 29 august-8 september 2020, santiago de Compostela, Spain, august 29 - september 8, 2020 - including 10th conference on prestigious applications of artificial intelligence (PAIS 2020). Editors G. D. Giacomo, A. Catalá, B. Dilkina, M. Milano, S. Barro, A. Bugarínet al. (IOS Press), Vol. 325, 1491–1498. doi:10.3233/FAIA200256
Škrlj, B., Kralj, J., and Lavrač, N. (2020b). Embedding-based silhouette community detection. Machine Learn. 109, 2161–2193.
Smalheiser, N. R. (2012). Literature-based discovery: beyond the ABCs. J. Am. Soc. Inf. Sci. 63, 218–224. doi:10.1002/asi.21599
Smalheiser, N. R. (2017). Rediscovering Don Swanson: the past, present and future of literature-based discovery. J. Data Inf. Sci. 2, 43–64. doi:10.1515/jdis-2017-0019
Smalheiser, N., and Swanson, D. (1994). Assessing a gap in the biomedical literature: Magnesium deficiency and neurologic disease. Neurosci. Res. Commun. 15, 1–9.
Swanson, D. R. (1990). Medical literature as a potential source of new knowledge. Bull. Med. Libr. Assoc. 78, 29.
Szklarczyk, D., Gable, A. L., Nastou, K. C., Lyon, D., Kirsch, R., Pyysalo, S., et al. (2020). The string database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49, D605–D612. doi:10.1093/nar/gkaa1074
Thilakaratne, M., Falkner, K., and Atapattu, T. (2019). A systematic review on literature-based discovery: General overview, methodology, and statistical analysis. ACM Comput. Surv. (Csur) 52, 1–34. doi:10.1145/3365756
Tshitoyan, V., Dagdelen, J., Weston, L., Dunn, A., Rong, Z., Kononova, O., et al. (2019). Unsupervised word embeddings capture latent knowledge from materials science literature. Nature 571, 95–98. doi:10.1038/s41586-019-1335-8
Venkatasubbaiah, M., Dwarakanadha Reddy, P., and Satyanarayana, S. V. (2020). Literature-based review of the drugs used for the treatment of covid-19. Curr. Med. Res. Pract. 10, 100–109. doi:10.1016/j.cmrp.2020.05.013
Virtanen, P., Gommers, R., Oliphant, T. E., Haberland, M., Reddy, T., Cournapeau, D., et al. (2020). Scipy 1.0: fundamental algorithms for scientific computing in python. Nat. Methods 17, 261–272. doi:10.1038/s41592-019-0686-2
Yetisgen-Yildiz, M., and Pratt, W. (2008). Evaluation of literature-based discovery systems. Berlin, Heidelberg: Springer Berlin Heidelberg, 101–113.
Keywords: literature-based discovery, knowledge graphs, PubMed, data-mining, machine-learning, representation learning
Citation: Škrlj B, Kokalj E and Lavrač N (2021) PubMed-Scale Chemical Concept Embeddings Reconstruct Physical Protein Interaction Networks. Front. Res. Metr. Anal. 6:644614. doi: 10.3389/frma.2021.644614
Received: 21 December 2020; Accepted: 08 February 2021;
Published: 13 April 2021.
Edited by:
Neil R. Smalheiser, University of Illinois at Chicago, United StatesReviewed by:
Gus Hahn-Powell, University of Arizona, United StatesAhmed Abdeen Hamed, Norwich University, United States
Copyright © 2021 Škrlj, Kokalj and Lavrač. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Blaž Škrlj, Ymxhei5za3JsakBpanMuc2k=