 Ethan J. Kilmister
Ethan J. Kilmister Lauren Hansen
Lauren Hansen Paul F. Davis
Paul F. Davis Sean R. R. Hall
Sean R. R. Hall Swee T. Tan
Swee T. Tan- 1Gillies McIndoe Research Institute, Wellington, New Zealand
- 2Wellington Regional Plastic, Maxillofacial and Burns Unit, Hutt Hospital, Wellington, New Zealand
- 3Department of Surgery, The Royal Melbourne Hospital, The University of Melbourne, Melbourne, VIC, Australia
Treatment of vascular anomalies (VAs) is mostly empirical and, in many instances unsatisfactory, as the pathogeneses of these heterogeneous conditions remain largely unknown. There is emerging evidence of the presence of cell populations expressing stemness-associated markers within many types of vascular tumors and vascular malformations. The presence of these populations in VAs is supported, in part, by the observed clinical effect of the mTOR inhibitor, sirolimus, that regulates differentiation of embryonic stem cells (ESCs). The discovery of the central role of the renin-angiotensin system (RAS) in regulating stem cells in infantile hemangioma (IH) provides a plausible explanation for its spontaneous and accelerated involution induced by β-blockers and ACE inhibitors. Recent work on targeting IH stem cells by inhibiting the transcription factor SOX18 using the stereoisomer R(+) propranolol, independent of β-adrenergic blockade, opens up exciting opportunities for novel treatment of IH without the β-adrenergic blockade-related side effects. Gene mutations have been identified in several VAs, involving mainly the PI3K/AKT/mTOR and/or the Ras/RAF/MEK/ERK pathways. Existing cancer therapies that target these pathways engenders the exciting possibility of repurposing these agents for challenging VAs, with early results demonstrating clinical efficacy. However, there are several shortcomings with this approach, including the treatment cost, side effects, emergence of treatment resistance and unknown long-term effects in young patients. The presence of populations expressing stemness-associated markers, including transcription factors involved in the generation of induced pluripotent stem cells (iPSCs), in different types of VAs, suggests the possible role of stem cell pathways in their pathogenesis. Components of the RAS are expressed by cell populations expressing stemness-associated markers in different types of VAs. The gene mutations affecting the PI3K/AKT/mTOR and/or the Ras/RAF/MEK/ERK pathways interact with different components of the RAS, which may influence cell populations expressing stemness-associated markers within VAs. The potential of targeting these populations by manipulating the RAS using repurposed, low-cost and commonly available oral medications, warrants further investigation. This review presents the accumulating evidence demonstrating the presence of stemness-associated markers in VAs, their expression of the RAS, and their interaction with gene mutations affecting the PI3K/AKT/mTOR and/or the Ras/RAF/MEK/ERK pathways, in the pathogenesis of VAs.
Introduction
Vascular anomalies (VAs) consist of a heterogenous group of disorders with infantile hemangioma (IH) being the most common, affecting 4–10% of infants (1). VAs may cause disfigurement and/or functional problems (2).
There has been a recent paradigm shift in the treatment of IH using β-blockers (3, 4) and angiotensin-converting enzyme (ACE) inhibitors (5, 6). One proposed mechanism of these agents is by targeting stem cells by modulating the renin-angiotensin system (RAS) (1, 7). For most other VAs, treatment remains empirical and unsatisfactory, although sirolimus (rapamycin), an mTOR inhibitor, is increasingly used for complex vascular anomalies (8).
Nomenclature applied to VAs has confused clinicians and patients, leading to incorrect diagnosis, improper treatment and misdirected research efforts (9). In 1982, Mulliken and Glowacki (10) first proposed a biologic classification for VAs that differentiates (infantile) hemangioma from vascular malformations. IH is characterized by an initial postnatal rapid proliferation followed by a slow involution, in which the cellular elements are gradually replaced by fibrofatty tissues (10). Vascular malformations are present at birth, grow commensurately throughout childhood and do not regress (10).
A biologic classification of VAs was created by the International Society for the Study of Vascular Anomalies in 1997, categorizing VAs into vascular tumors and vascular malformations (11). Increasing knowledge of VAs led to a revision of the classification in 2014 which further subcategorizes vascular tumors into benign, locally aggressive or borderline, and malignant tumors. Vascular malformations are categorized as simple malformations which may be high-flow or low-flow (2), combined, of major named vessels, or those associated with other anomalies (9) (Figure 1). However, the term hemangioma continues to be erroneously applied to different types of VAs, despite them being distinct biological entities (12).
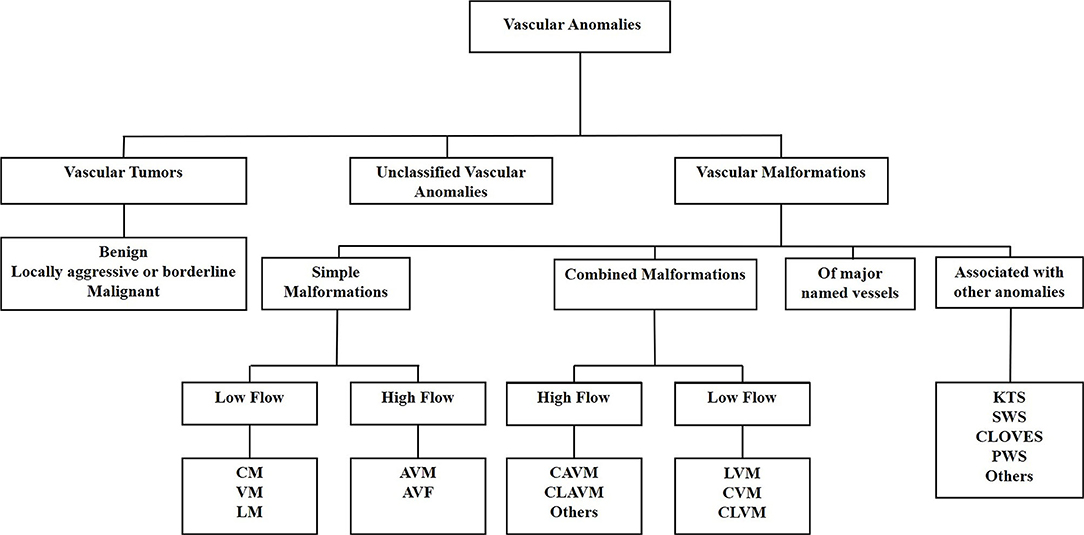
Figure 1. The International Society for the Study of Vascular Anomalies classification of vascular anomalies. Vascular anomalies (VAs) are categorized as vascular tumors, vascular malformations and unclassified VAs. Vascular tumors are categorized as being benign, locally aggressive or borderline, or malignant. Vascular malformations are categorized as simple malformations, combined malformations, of major named vessels, or are associated with other anomalies. Simple and combined malformations can be either high- or low-flow. AVF, arterio-venous fistula; AVM, arterio-venous malformation; CAVM, capillary-arteriovenous malformation; CLAVM, capillary-lymphatic-arteriovenous malformation; CLOVES, congenital lipomatous overgrowth-vascular malformation—epidermal nevi-spinal anomaly syndrome; CLVM, capillary-lymphatic-venous malformation; CM, capillary malformation; CVM, capillary-venous malformation; KTS, Klippel-Trénaunay syndrome; LM, lymphatic malformation; LVM, lymphatic venous malformation; PWS, port-wine stain; SWS, Sturge-Weber syndrome; VM, venous malformation.
This review discusses accumulating evidence implicating a role for populations of cells that express stemness-associated markers, the RAS, and the influence of gene mutations on the pathobiology of VAs, underscoring commonalities between VAs. This includes a potential stem cell origin, genetic mutations, treatment options such as sirolimus, and newer targeted therapies in development and their potential shortcomings.
Gene Mutations in Vascular Anomalies and Overgrowth Syndromes
Gene mutations have been identified in some VAs, although not in IH despite intensive research (9, 13). A missense mutation in TIE2 (Figure 2) that causes an arginine-to-tryptophan substitution at position 849 on chromosome 9, was discovered in familial venous malformation (VM) in 1996 (14). TIE2 (TEK or HYK), an endothelial-specific receptor in the tyrosine kinase sub-family (15), is critical for the regulation of vasculogenesis, vascular remodeling, endothelial cell integrity and survival, upon binding angiopoietin 1 (Ang-1) and angiopoietin 2 (Ang-2) (16). Additional germline and somatic mutations have since been identified in both familial and sporadic VM (15, 17–19).
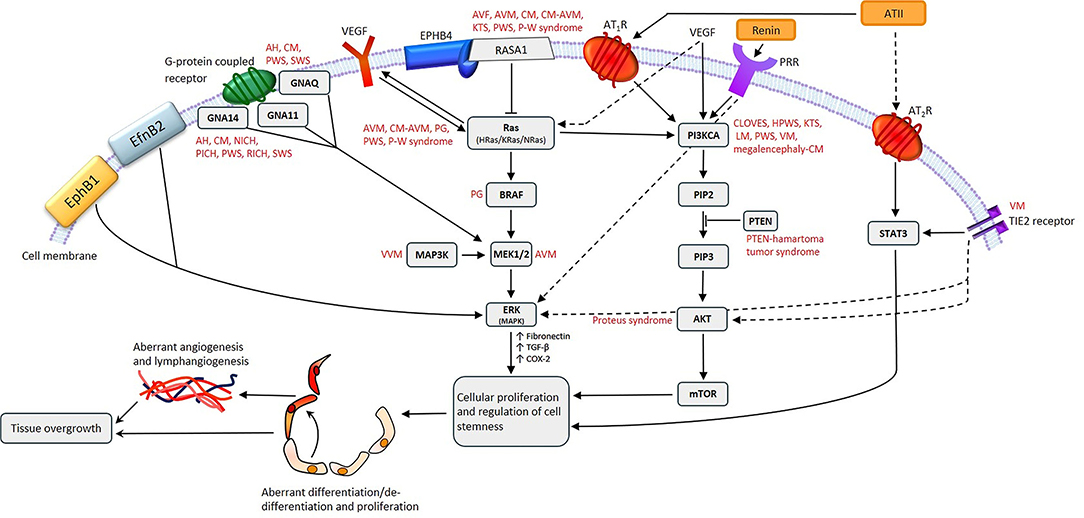
Figure 2. A proposed model for the role of gene mutations involving the Ras/BRAF/MEK/ERK and the PI3KCA/AKT/mTOR pathways by their interaction with different components of the renin-angiotensin system, leading to the induction and/or maintenance of cells that express stemness-associated markers in vascular anomalies. AH, anastomosing hemangioma; ATII, angiotensin II; AT1R, angiotensin II receptor 1; AT2R, angiotensin II receptor 2; PRR, pro-renin receptor; AVF, arteriovenous fistula; AVM, arterio-venous malformation; CM, capillary malformation; CM-AVM, capillary malformation—arterio-venous malformation; CM-AVM2, capillary malformation—arterio-venous malformation 2; CLOVES, congenital lipomatous overgrowth with vascular—epidermal and skeletal anomalies; HPWS, hypertrophic port-wine stain; P-W syndrome, Parkes-Weber syndrome; PWS, port-wine stain; KTS, Klippel-Trénaunay syndrome; LM, lymphatic malformation; NICH, non-involuting congenital hemangioma; PG, pyogenic granuloma; PICH, partially involuting congenital hemangioma; RICH, rapidly involuting congenital hemangioma; SWS, Sturge-Weber syndrome; VEGF, vascular endothelial growth factor; VM, venous malformation; VVM, verrucous venous malformation.
Glomuvenous malformation, characterized by the presence of glomus cells that represent immature vascular smooth muscle cells (vSMCs) in the walls of affected venous channels, is associated with mutations in the glomulin gene on chromosome 1p21-22, with 157delAAGAA being present in 48.8% of 23 affected families (20).
Fifty-four percent of VMs without detectable TIE2 mutations are caused by somatic activating mutations in the PIK3CA gene (Figure 2), which encodes a catalytic subunit of the phosphatidylinositol 3-kinase (PI3K) enzyme p110α (21). Somatic mutations in PIK3CA are also associated with lymphatic malformation (LM) (22) (Figure 2). Disorders which feature LM, including overgrowth syndromes such as congenital lipomatous overgrowth with vascular, epidermal and skeletal anomalies (CLOVES) and Klippel-Trénaunay syndrome (KTS), are also frequently associated with somatic mutations in the PIK3CA gene (22) (Figure 2). Mutations affecting PIK3CA also cause a megalencephaly-capillary malformation (23) (Figure 2).
Somatic mutations in the MAP2K1 gene, which encodes MEK1, are associated with arterio-venous malformation (AVM) (24) (Figure 2), and constitutively increased MEK1 activity is observed in various cancer types (25, 26).
Capillary malformation-arteriovenous malformation (CM-AVM) is an autosomal dominant vascular malformation, associated with mutations in the RASA1 gene (Figure 2) which have also been identified in capillary malformation (CM), AVM, arterio-venous fistula, and Parkes-Weber syndrome (27) (Figure 2). Germline loss-of-function mutations in EPHB4 are present in CM-AVM2, the second type of CM-AVM characterized by intra- and extra-cranial AVMs, multifocal CMs and telangiectasias (28).
Congenital hemangiomas are rare vascular tumors which are fully developed in utero. They are subcategorized into rapidly involuting congenital hemangioma (RICH), non-involuting congenital hemangioma (NICH) and partially involuting congenital hemangioma (PICH) (29, 30). RICH, NICH and PICH are associated with somatic activating mutations in GNAQ and GNA11 (31) (Figure 2). GNAQ, GNA11, and GNA14 mutations are present in anastomosing hemangioma (32) (Figure 2). GNAQ mutations have also been identified in Sturge-Weber syndrome (SWS) and CM (33, 34) (Figure 2). The Gln209 missense mutation in GNAQ and GNA11 has also been demonstrated in uveal melanoma, in which it constitutively activates MAPK signaling (35).
Somatic activating mutations in GNA14, HRas, KRas, NRas, and GNA11 are associated with certain vascular tumors (36) (Figure 2), and somatic mutations in MAPK3 are associated with verrucous venous malformation (VVM) (37) (Figure 2). Furthermore, mutations in both BRAF and Ras genes have been demonstrated in sporadic and secondary pyogenic granuloma (PG) (38) (Figure 2), and mutations in PTEN are associated with PTEN-hamartoma tumor syndrome (39) (Figure 2). A somatic activating mutation in AKT has been associated with Proteus syndrome (40) (Figure 2).
The identification of a range of somatic and some germline mutations affecting different genes in different types of VAs and overgrowth syndromes have improved our understanding of these challenging conditions. The occurrence of identical mutations in multiple VAs suggests it is not the type of mutation that dictates which type of VA develops, but rather what endothelial cell lineage or stem cell type the genetic alteration occurs in during embryogenesis (41).
This review analyzes mutations in each type of VA, how they may directly influence embryonic stem cell (ESC)-like cells, and the possible interactions between these mutations and the RAS in regulating the stemness of the primitive populations within VAs (Figure 2).
Induced Pluripotent Stem Cells
Introduction of transcription factors OCT4, SOX2, c-MYC, and KLF4 into adult mouse (42) and human (43) fibroblasts induces pluripotent stem cell (iPSC) formation. This can also be achieved with NANOG and LIN28 in place of c-MYC and KLF4 (44). The master regulators of pluripotency—OCT4, SOX2, c-MYC, and KLF4—are known as the “Yamanaka factors.” The expression of stemness-associated markers by various types of VAs (45–49) warrants further investigation including functional studies to confirm if they possess ESC properties.
Populations of Cells Expressing Stemness-Associated Markers in Vascular Anomalies
Populations of cells expressing stemness-associated markers have been identified in different types of vascular tumors including IH (50) and PG (51), and vascular malformations including VM (46), LM (45), VVM (47), AVM (48), and port-wine stain (PWS) (49)—the most common form of CM. The observation of the proliferative nature of vascular malformations challenges the prevailing notion that they consist of quiescent non-proliferating endothelium. It is interesting to speculate that gene mutations (Figure 2) may play a key role in the induction and/or maintenance of the primitive population by influencing aberrant proliferation and differentiation, or de-differentiation of mature cells by upregulating the Yamanaka factors (43).
The Renin-Angiotensin System and Its Bypass Loops
Populations of cells expressing stemness-associated markers within several types of VAs express components of the RAS (7, 52, 53). The classical RAS (Figure 3) is an endocrine cascade that is crucial for blood volume and blood pressure homeostasis (54). It is also a critical regulator of hematopoietic and mesenchymal stem cells (55). Renin, an aspartyl protease converted from its precursor pro-renin, cleaves angiotensinogen (AGT) to form angiotensin I (ATI) (54, 56). Angiotensin-converting enzyme (ACE) converts ATI to angiotensin II (ATII) (54, 56, 57), the physiologically active component of the cascade that exerts its physiologic effects by binding to ATII receptor 1 (AT1R) and ATII receptor 2 (AT2R) (54). There are also bypass loops of the RAS comprising enzymes such as cathepsins B, D and G and chymase (57, 58). Cathepsin B belongs to a family of cysteine proteases that converts pro-renin to renin (57). Cathepsin D, an aspartyl protease and a renin analog, cleaves AGT to form ATI (57). Cathepsin G, a serine protease, produces ATII directly from AGT or from ATI while chymase, a mast cell protease, converts ATI to ATII (57) (Figure 3). Cathepsins B, D, and G have been demonstrated in IH (59) and LM (60).
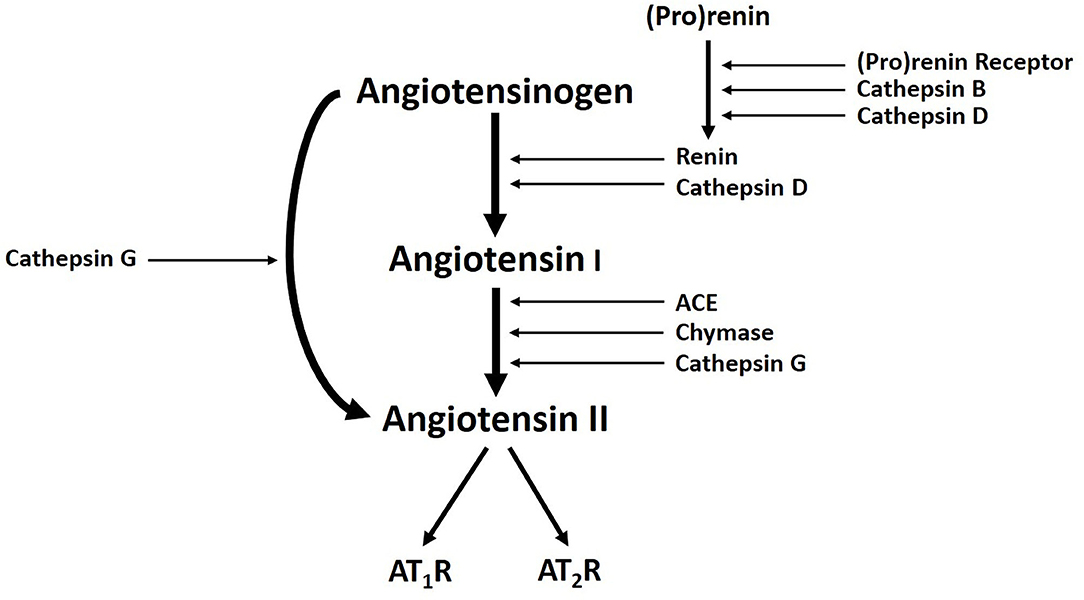
Figure 3. A Schema demonstrating the classical renin-angiotensin system, with cathepsins B, D, and G, acting as bypass loops. Activation of pro-renin occurs upon binding with pro-renin receptor. Renin then converts angiotensinogen into angiotensin I (ATI), which is cleaved by angiotensin-converting enzyme (ACE) to produce the active peptide angiotensin II (ATII). The actions of ATII are mediated through interactions with ATII receptor 1 (AT1R) and ATII receptor 2 (AT2R). Cathepsin B and cathepsin D contribute to renin activation. Cathepsin D and chymase mediates conversion of angiotensinogen into ATI. Cathepsin G promotes generation of ATII from ATI or directly from angiotensinogen. Reproduced with permission from Expert Rev Clin Pharmacol (6).
Vascular Tumors
Infantile Hemangioma
IH is characterized by rapid proliferation during infancy followed by slow involution over the subsequent 5–10 years (1). The identification of endothelial progenitor cells (EPCs) that co-express CD133 and CD34 in proliferating IH tissues (61) and the peripheral circulation (62) of IH patients, led to the proposal of EPCs as the origin for IH (1). However, the expression of markers associated with primitive hematopoietic cells and the transcription factors brachyury and GATA-2 by the endothelium of proliferating IH indicates an up-stream primitive mesodermal origin (63). This suggests the immature capillaries within proliferating IH are derived from a hemogenic endothelium, downstream of hemangioblasts (64), capable of undergoing mesenchymal (65, 66), neuronal (67), endothelial (67), and hematopoietic (68) differentiation. The expression of the stemness-associated markers OCT4, SSEA-4 and pSTAT3 on the endothelium of proliferating IH, implies the presence of an ESC-like population (50). STAT3 signaling plays an important role in the maintenance and stemness of cancer stem cells (69), and its activation maintains ESCs in an undifferentiated state (70). The ESC markers NANOG, SALL4 and CD133 have also been demonstrated on cells within the interstitium of proliferating IH (50).
The distinctive expression of glucose transporter-1 (GLUT-1) in IH (71) and syncytiotrophoblast membranes of the human placenta (72) and the unique co-expression of placental antigens FCγRIII, Lewis Y antigen and merosin, and type 3-iodothyronine deiodinase on IH, led to the speculation of a placental embolic origin of IH (1, 73–75). The presence of human chorionic gonadotrophin, human placental lactogen, but not human leucocyte antigen-G or cytokeratin 7, on proliferating endothelium of IH, suggests a placental chorionic villous mesenchymal core origin of IH (76).
The endothelium of the microvessels within proliferating IH expresses embryonic hemoglobin ζ (HBZ) and erythropoietin receptor (EPOR) (68). Erythropoietin is a crucial growth factor for the proliferation, differentiation and survival of erythroid progenitor cells (77). EPOR is found with HBZ in extra-embryonic yolk sac blood islands (77) – the location of primitive hematopoiesis during the first trimester of pregnancy (78, 79). Cell culture of IH explants in vitro form enucleated erythrocytes which express the erythrocyte-specific marker glycophorin A (68). These findings suggest that the endothelium of IH is a functional hemogenic endothelium which may be a site of primitive extra-medullary erythropoiesis (68).
The endothelium of proliferating IH also expresses the neural crest markers p75 (a neurotrophin receptor), SOX9 and SOX10 (63), NG2 and nestin (80), suggesting a primitive mesoderm with a neural crest phenotype (63). The segmental distribution of the IH seen in PHACES syndrome (63, 81), is reminiscent of the migratory paths of neural crest cells during embryogenesis (82).
Accelerated involution of proliferating IH induced by propranolol (83), acebutolol (84), other β-blockers (85, 86) and captopril (5), suggests a role for the RAS in the pathogenesis for IH (1, 6). The CD34+ hemogenic endothelium of proliferating IH expresses components of the RAS: ACE and AT2R, but not AT1R (7). Pro-renin receptor (PRR), another component of the RAS, is expressed on both endothelial and non-endothelial cell populations of IH. Renin has been demonstrated to promote proliferation of the endothelial cell population that may be blocked via inhibition of the canonical Wnt signaling pathway, using the Wnt receptor blocker dickkopf-1 (87). These findings suggest there are interactions between renin, PRR and Wnt signaling (88). This is unsurprising given PRR is a part of the Wnt/frizzled receptor complex (89).
Explants of proliferating IH form blast-like structures in media upon administration of ATII in a dose-dependent manner (90), which could explain the accelerated involution of proliferating IH induced by propranolol (7). This is supported by the observation that serum renin levels are five times higher in the first 3 months of life and remain three times higher at 3–12 months old, when compared to adults. Serum renin levels then taper to normal adult levels from 8 years of age (91). This gradual decline in serum renin levels reflects the programmed biologic behavior of IH (1, 92). Patients with proliferating IH treated with surgery or propranolol have significantly decreased plasma renin levels (82), whereas surgery and captopril administration significantly reduce the mean levels of ATII. However, none of these interventions significantly affect the mean serum levels of ACE (93). The significant changes in renin and mean levels of ATII following these interventions further evidences the crucial role of the RAS.
The observation that the angiotensin peptides significantly influence cellular proliferation of IH underscores a central role of the RAS in the pathobiology of IH. While explants of IH treated with an ACE or AT2R antagonist demonstrate a significant reduction in the nuclear expression of Ki67, a marker of cellular proliferation, the addition of an AT2R agonist increases the number of Ki67+ cells (90). ACE is expressed on the endothelium of IH (7). The ACE inhibitor ramipril blocked AT1-induced proliferation on IH explant in vitro (90). This shows that ATI and ATII promote cellular proliferation in proliferating IH through AT2R signaling (90). This suggests that conversion of ATI to the vasoactive peptide ATII occurs in a paracrine fashion within IH, i.e., the presence of a local (paracrine) RAS (7).
Phosphorylated forms of STAT1, STAT3, and STAT5 have been demonstrated in proliferating IH (94). This is significant as the STAT family consists of intracellular signaling molecules with multiple influences on stem cell populations, particularly hematopoietic stem cells (95). They translocate to the nucleus to influence gene expression upon latent STATs undergoing Janus-kinase (JAK)-mediated phosphorylation and subsequent dimerization (96). STAT activation decreases markedly and is not expressed by some cells in involuted IH. This decrease may reflect depleting stem cell numbers present within involuting IH as it transitions to a fibro-fatty residuum, and suggests that reduced STAT expression decreases stem cell maintenance (94). Binding of ATII to AT2R increases activation of STAT3 (94). This may explain the widespread activation of STAT3 in proliferating IH, underscoring the role of the RAS in spontaneous involution and accelerated involution of IH induced by RAS modulators (94). Functional work is needed to determine if accelerated involution induced by RAS-modulating therapies is due to decreased STAT3 signaling which has been thought to cause the loss of stem cell maintenance (94).
Propranolol is administered as a 1:1 racemic mixture of both R(+) and S(–) stereoisomers (97). Recent evidence demonstrates that the R stereoisomer of propranolol which is inactive against the β-adrenergic receptor, inhibits the growth of bEnd.3 hemangioma cells in vivo (98). R-propranolol is a small molecule inhibitor of the transcription factor SOX18 (97). Mutations in SOX18 cause hypotrichosis-lymphedema-telangiectasia and renal syndrome (HLTRS), which feature vascular and lymphatic defects (99). Long-term use of propranolol is effective for treating HLTRS (100), providing further evidence propranolol does not only act via β-adrenergic pathways (97). Induced differentiation of hemangioma-derived stem cells (HemSC) into hemangioma endothelial cells with VEGF-B, increases SOX18 expression. Treatment of HemSC with propranolol significantly reduces VEGF-B-induced upregulation of all endothelial markers investigated to a magnitude similar to the SOX18 inhibitor Sm4 (97). Moreover, when repeated with purified R(+) and S(−) enantiomers, only R-propranolol reproduces the endothelial cell marker inhibition seen with racemic propranolol. This indicates there may be a SOX18-dependent pathway, independent of β-adrenergic blockade, influencing HemSC differentiation (97). This suggests the possibility of treating IH independent of β-adrenergic blockade, thereby avoiding side effects of β-blocker therapy.
These findings have led to the proposal that IH originates from aberrant proliferation and differentiation of primitive mesoderm-derived hemogenic endothelium (63) with a neural crest phenotype (101) and a placental chorionic villous mesenchymal core origin (76), regulated by the RAS (3). Further functional studies are needed to fully determine the role of the RAS in the programmed biologic behavior of IH, and its accelerated involution induced by β-blockers and ACE inhibitors (3, 6, 102).
Pyogenic Granuloma
Populations of cells that express stemness-associated markers have been identified in the endothelium of the microvessels and the interstitium of PG, another common type of benign vascular tumor (51). The primitive subpopulation on the endothelium expresses the ESC markers OCT4, SOX2, NANOG, and pSTAT3, whereas a subpopulation within the interstitium expresses SOX2, NANOG, and pSTAT3 (51). These subpopulations in PG also express components of the RAS (52). The expression of ACE on the endothelium of the microvessels suggests paracrine conversion of ATI to ATII, which may impact cellular proliferation and angiogenesis within PG (54, 103). It has been proposed that the presence of AT1R on the microvessels within PG may contribute to immature microvessel formation by influencing the ESC-like population to differentiate down an endothelial phenotype (52, 104). If cells expressing stemness-associated markers preferentially differentiate toward an endothelial phenotype under the influence of the RAS, this could be targeted by RAS modulators (52).
Somatic gain of function mutations of HRas (Figure 2) have been identified in PG (105) with Ras signaling being associated with microvessel proliferation and angiogenesis (105). Further, KRas activation increases VEGF synthesis (106) (Figure 2), which at increased levels stimulates mobilization of hematopoietic stem cells and hematopoietic progenitor cells, and a cell population capable of rapid endothelial colony formation. This suggests an immature endothelial cell phenotype within PG (107). These mobilized hematopoietic stem cells and hematopoietic progenitor cells are comparable to the KDR+/CD34+/AC133+ cells seen in G-CSF mobilized blood, bone marrow and umbilical cord (108). It is interesting to speculate whether elevated VEGF expression caused by Ras mutations leads to increased levels of circulating precursor cells, and whether these are a source of the previously identified primitive cells within PG.
Vascular Malformations
Venous Malformation
Venous malformation (VM) represents the most common vascular malformation (109), whereby affected veins consist of a thin endothelial cell lining surrounded by sparsely and erratically distributed vSMCs which are sometimes absent (21). This, and the disordered extracellular matrix deposition, lead to dysfunctional basement membrane development and irregular endothelial cell monolayer arrangement, forming irregularly shaped ectatic vessels prone to thrombosis (21, 110). There are four subtypes of VM: sporadic VM, cutaneo-mucosal VM (CMVM), multifocal VM, and blue rubber bleb nevus syndrome (BRBN) (9).
Populations of cells that express the stemness-associated markers OCT4, NANOG, SOX2, SALL4, pSTAT3, and CD44 have been demonstrated within the endothelium of the lesional vessels and the interstitium in both subcutaneous VM (SCVM) and intramuscular VM (IMVM) (46). The endothelial sub-population which expresses OCT4, sitting atop the stem cell hierarchy, may differentiate and give rise to the interstitial subpopulation (46). c-kit, a stem cell growth factor receptor, has also been identified in the smaller lesional vessels in BRBN (111). This suggests that the growth of VM is associated with the c-kit signaling axis (111).
ACE and PRR have been demonstrated on the endothelium of the lesional vessels in VM (112). PRR binds pro-renin which facilitates increased local conversion of AGT to ATI (112) (Figure 3). PRR also functions in downstream signaling through the MAP kinases ERK1 and ERK2 to upregulate TGF-β1, PAI1, collagens, fibronectin (87, 113, 114) and cyclooxygenase-2 (115) (Figure 2). Some of these downstream effectors, particularly TGF-β1 and fibronectin, play a role in cellular growth, proliferation and differentiation (116) (Figure 2). These functions highlight the potential role of PRR in VM pathobiology (112).
The proangiogenic effects of ATII occur through its interactions with AT1R (117) (Figure 2). The endothelium of SCVM and IMVM expresses AT1R, and the endothelium of smaller vessels stains more strongly than that of dilated vessels by immunohistochemical staining (112). ATII has proangiogenic effects, when acting through AT1R, providing a possible explanation for the increased density of abnormal venous channels in VM (112). Further, AT2R is expressed on both endothelial and non-endothelial cells (112) and signaling through this receptor stimulates blast cell proliferation and their differentiation into either endothelial or hematopoietic progenitor cells (104).
Germline or sporadic gain of function mutations in TIE2 have been identified in ~60% of VM cases (18, 118) (Figure 2). PIK3CA mutations have also been demonstrated in about 25% of VM cases (119) (Figure 2). Although co-occurring mutations in TIE2 and PIK3CA are rare, they both result in PIK3 signaling and subsequent AKT activation (41) (Figure 2). TIE2, through its interaction with its ligand Ang-1, is involved in the recruitment of both pericytes and vSMCs (120). Ang-1, a glycoprotein, is a member of the angiopoietin family of growth factors and promotes vascular quiescence and structural integrity (121). Given the functional role of Ang-1 in vascular quiescence, we speculate that TIE2 dysregulation plays a role in the loss of quiescence, hence challenging the notion that VM is not a proliferative lesion. Dysfunction in TIE2 precipitates activation of STATs (120) and activation of the AKT phosphorylation pathway (122) (Figure 2). As a result, the affected vessels lack vSMCs, resulting in ‘blow-out' of the affected channels in VM (123). STAT1 is involved in inhibition of endothelial cell growth, while STAT3 is involved in angiogenesis and endothelial activation (124). Both are induced by the TIE2 mutant variant R849W; however, STAT3 is implicated in the pathogenesis of many cancer types (124). STATs have also been identified in proliferating IH and their role in stem cell maintenance has been suggested (94). Given subpopulations of cells that express stemness-associated markers have been identified in VM, it would be interesting to investigate whether the stem cell maintenance function of STATs also play a role in the pathogenesis of VM. Additionally, components of the RAS have been implicated in STAT expression, as the binding of ATII to AT2R activates STAT3 (94) (Figure 2). This provides some evidence for the interaction between genetic mutations, including those of TIE2, and the RAS, suggesting they act synergistically on STAT pathways to influence stemness. R849W, the most common germline mutation affecting TIE2, causes an arginine-to-tryptophan substitution at position 849 (19). It causes partial hyper-phosphorylation of TIE2 and requires a second somatic mutation for phenotypic penetration (19). TIE2 mutations have been implicated in ~60% of sporadic VM cases (125). Sixty percent of these have been attributed to L914F, a somatic mutation that causes a phenylalanine-to-leucine substitution at position 914 resulting in dysfunctional vascular development and cell migration (126). Approximately half of the remaining 40% of sporadic VM cases are attributed to mutations in PIK3CA (Figure 2), and the resultant PI3K signaling dysfunction (21), leading to upregulation of the protein kinase AKT (21). Somatic mutations in this gene have been identified in overgrowth syndromes including VM, and many cancer types (21). Increased AKT signaling has been suggested to promote endothelial cell survival and proliferation in vSMC-deficient malformed veins typically found in VM (119, 126–128).
There is increasing evidence demonstrating interactions between gene mutations and the RAS in VM (129, 130). Cheng et al. (131) demonstrate the role of renin in activating the PI3K signaling pathway (Figure 2). ATII, acting downstream of renin also activates the PI3K pathway by binding to AT1R, as well as stimulating G protein subunit q (131) (Figure 2). Further research is needed to determine whether the same interaction occurs between RAS and the PI3K signaling pathway in VM.
The expression of components of the RAS by ESC-like subpopulations within VM may underscore the observed effect of propranolol, a RAS inhibitor (132), and celecoxib, a COX-2 inhibitor which causes upregulation of PRR (133), on a patient with troublesome VM (134). The clinical response to β-blockers has been hypothesized to be caused by a reduction in plasma renin levels (135), while COX-2 inhibitors potentially act to prevent ATII-induced expression of PRR (133). Additionally, pharmaceuticals targeting the PI3K pathway, such as sirolimus (an mTOR inhibitor), have proved effective in refractive cases of VM (21). Lesion growth and vascular volume of VM have been shown to decrease in mice following sirolimus treatment, whilst radiological lesion size, bleeding, pain and esthetic impairment are reduced following administration of sirolimus in a small number of patients (136, 137). Sirolimus binds to and interferes with the intracellular receptor on mTOR, FKBP12 (138). Given mTOR's role in cellular growth and proliferation, the response of VM to sirolimus challenges the notion that vascular malformations contain a quiescent endothelium (10).
Verrucous Venous Malformation
VVM, formerly known as verrucous hemangioma (9), is a subtype of VM that presents clinically as a vascular malformation but with histopathological characteristics of a vascular tumor (139). A significant proportion of these non-hereditary, congenital lesions contain a somatic missense mutation in MAP3K (37). These lesions are present at birth and grow commensurately throughout infancy (140). They eventually form friable, dark black-blue, hyperkeratotic plaques which bleed easily with minor trauma (140). The endothelium of VVM expresses GLUT-1, the transcription factor OCT4 and brachyury—a marker of primitive mesoderm, and also ACE—the central component of the RAS (9).
Lymphatic Malformation
LM affects 1:5,000 births (141) and may be identified prenatally, at birth, or during childhood (142). LMs fall under three of the four groups of vascular malformations: simple malformations, combined malformations, and malformations of major named vessels (9). LMs are further categorized into microcystic or macrocystic, or mixed subtypes (9). LM commonly affects the cervicofacial and axillary regions (9). LMs consist of thin-walled dilated channels or cysts with a lymphatic morphology lined by flat endothelial cells and few pericytes (9) surrounded by stroma (141). Somatic activating mutations in PIK3CA have been identified in both isolated LMs and those associated with several overgrowth syndromes (22) (Figure 2).
A cell population that expresses the stemness-associated markers OCT4, NANOG, SOX2, KLF4, and c-MYC on the D2-40+ endothelium of the lesion vessels, and c-MYC and SOX2 within the fibrous stroma have been demonstrated in microcystic LM (45). Further, endothelial cells lining aberrant lymphatic vessels within LM and in surrounding stroma express CD133 (143). Isolated CD133+ cells express NANOG and OCT4, and markers found on circulating endothelial precursor cells such as CD90, CD146, c-kit and VEGFR2 (143). These CD133+ cells also express the lymphatic endothelial markers D2-40 and VEGFR3 (45, 143). CD133 is expressed by hematopoietic stem cells and EPCs, but not ESCs (45, 144). These CD133+ progenitor cells are multipotent, and are able to differentiate into bone, smooth muscle, fat, and lymphatic endothelial cells in vitro (143). In vivo, CD133+ LM cells differentiate into lymphatic endothelial cells giving rise to dilated lymphatic channels characteristic of human LMs (143). Further, LM-derived cells demonstrate increased proliferation, migration, and resistance to apoptosis, in vitro (145). The presence of progenitor cells capable of multipotent differentiation supports a potential role for aberrant ESC-like cells in the pathogenesis of LM. We speculate that these aberrant progenitor cells are downstream to a more primitive ESC-like population, which may include a cell population that expresses stemness-associated markers in microcystic LM (45).
Arterio-Venous Malformation
AVM consists of a tangle of arteries and veins providing direct connections between high-flow and low-flow vessels (146) forming the central nidus, bypassing the normal capillary network (146).
Although the pathogenesis of AVM remains unclear, there is evidence showing that AVM may arise from errors in angiogenesis during 4th−6th week of gestation that leads to aberrant vessel remodeling (147–149). It has been proposed that a gene mutation causes aberrant signaling in pathways, such as the PI3K/AKT and mTOR pathways, which are associated with angiogenesis and vascular development (24) (Figure 2). Ras (not to be confused with the RAS), a series of GTPases and a variety of tyrosine kinase receptors, modulate the PI3K/AKT pathway to enable generation of lipid products (24, 150, 151). These lipid products contribute to normal vascular development and angiogenesis, and play a role in cellular survival, proliferation, and cytoskeletal re-organization (24, 150, 151). The mTOR complex lies downstream of PI3K signaling, and collectively the PI3K/AKT/mTOR pathway is a major regulator of cell survival (150–152) (Figure 2). Ras proteins also play a role in the activation of the MAPK cascade (24). The role of MAPK is extensive in regulating gene expression and cell proliferation, differentiation and survival (24, 153). Several studies have revealed pathogenic variants in the Ras/MAPK pathways in patients with AVM (24, 154, 155) (Figure 2). Further, somatic mutations have been identified in MAP2K1, which encodes for MEK1, in AVM (24) (Figure 2). MEK inhibitors are in clinical use for the treatment of several cancer types (156, 157), and result in clinical improvement in patients with central conducting lymphatic anomalies with recurrent ARAF mutations (158). These mutations in MAPK's negative regulatory domain (24) are similar to those seen in malignant melanoma (159) and lung cancer (160, 161). The potential use of this targeted therapy in AVM warrants investigation.
Populations of cells that express the stemness-associated markers OCT4, SOX2, KLF4, and c-MYC have been identified on the endothelium and media of the lesional vessels, as well as cells within the stroma of the nidus of AVM (48). Zheng et al. (162) propose that the PI3K/AKT pathway lies downstream of ATII receptors in ESCs, as ATII upregulates phospho-Akt, and show that ATII promotes differentiation of mouse ESCs to form SMCs through activation of ATR1 (162). We propose that an interaction may exist between gene mutations and the cells that express stemness-associated markers that may influence the pathophysiology of AVM (Figure 2), similar to VM. The precise relationship between the ESC-like population and the RAS warrants further investigation including the expression of components of RAS, particularly ATII, in AVM. Further, over-activation of the MAPK pathway affects SOX2 in pre-gestational models, increasing cellular proliferation and impairing terminal differentiation (163). Increased clonogenic capacity of SOX2 may be prolonged, in part, by the MAPK pathway (163) and errors causing dysregulation in this pathway, including the aforementioned pathogenic variants, contribute to the inexorable nature of AVM.
Port-Wine Stain
PWS, or nevus flammeus, is the most common type of CM (164). It affects about 26 million people globally (164, 165) and is characterized by an increased number of dilated venular-capillary vessels within the dermis (9). Fifteen to Twenty percent of PWS affecting the V1 dermatome of the trigeminal nerve are associated with SWS (165). PWS is also part of KTS and Parkes-Weber syndrome (166).
The standard treatment of PWS is pulsed dye laser requiring multiple treatment sessions (167), with persistence remaining a challenge (168). Surgical de-bulking is used for hypertrophic PWS (HPWS) with modest results (169).
There are two main hypotheses for the pathogenesis of PWS: denervation and gene mutations (165). Most facial PWS show trigeminal nerve dermatomal distribution, along V2 (32%), combined V1-V2 (41%), combined V2-V3 (5%), and involvement of V1-3 (10%) (170). Microvessels within the middle and deep dermis of PWS lack normal innervation (171, 172). Also, the nerve fibers in PWS do not respond to epinephrine administration in vitro, implying a defective sympathetic basal tone as a cause for the vessel dilation seen in PWS (165, 173). “Denervation” leads to decreased basal sympathetic tone, and the loss or decrease of neuronal factors has been postulated to cause PWS (165). Alternatively, it is interesting to speculate that aberrant neural differentiation from sympathetic neuronal progenitor cells may have caused deficient innervation and sympathetic tone.
There may be a link between the gene mutations and the ESC-like population in PWS (49). Mutations in GNAQ (R183Q) (33) and PI3K (G1049N) (165, 174) have been demonstrated in PWS, and aberrant MAPK and/or PI3K signaling during embryonic development may contribute to PWS formation and SWS (165) (Figure 2). Further, a somatic mutation in GNAQ activates ERK via MEK, which may contribute to PWS development (33). Like ERK, c-Jun N-terminal kinases (JNK) are activated in PWS, which is also associated with the development of PWS (175). Activation of ERK via Ras signaling induces phosphorylation of KLF4 (176) and c-MYC (177). Activation of JNK via the Rac pathway induces expression of Wnt and BMP-signaling, both of which are critical in the self-renewal capability of ESCs. Wnt signaling upregulates the transcription factors OCT4, SOX2, and NANOG, whilst BMP limits NANOG activity to cause differentiation of ESCs (178) (Figure 4). VEGF, which is overexpressed in PWS (179), significantly increases Ras signaling which activates ERK, thus increasing the expression of stem cell markers which may bolster and sustain the resident ESC-like population in PWS (49) (Figure 4).
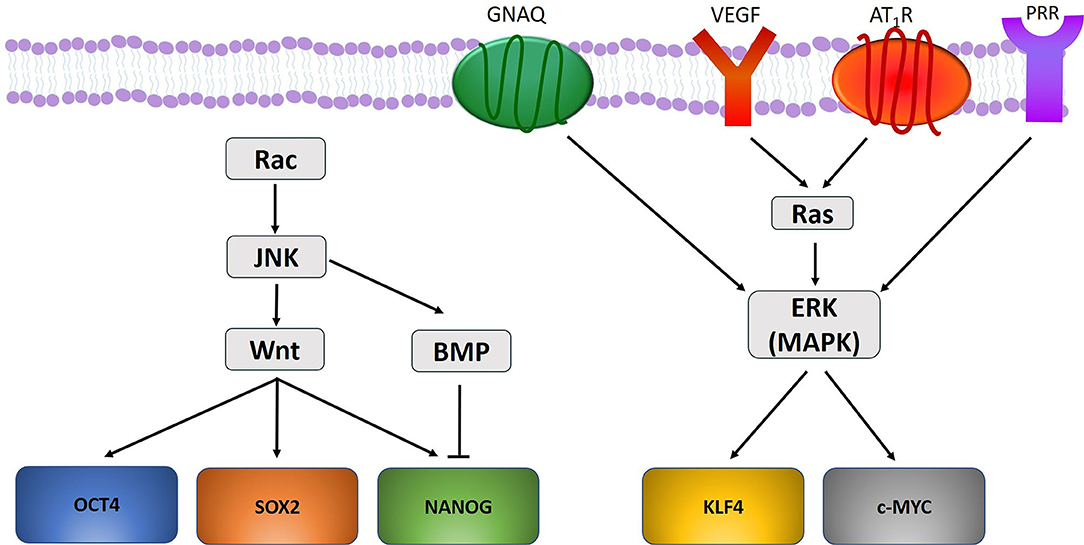
Figure 4. A proposed model portraying how gene mutations identified within vascular anomalies affect the expression of the stemness-associated markers OCT4, SOX2, NANOG, KLF4, and c-MYC. OCT4, SOX2, and NANOG are upregulated through Wnt signaling, and NANOG is down regulated through BMP-signaling. KLF4 and c-MYC are upregulated by ERK activation under the influence of GNAQ, VEGF, and RAS signaling via activation of angiotensin II receptor 1 (AT1R) and pro-renin receptor (PRR).
Mutations in EphB4, RASA1, TIE2, and co-expression of Eph receptor B1 (EphB1) and ephrin B2 (EfnB2) lead to MAPK activation (165) (Figure 2). EphB1 forward signaling and EfnB2 reverse signaling activate MAPK pathways (165). Nguyen et al. (165) propose that EphB1 and EfnB2 co-expression lead to MAPK activation. A mutation in PI3K causes activation of the AKT/mTOR pathway, as does the overexpression of VEGF-A and VEGFR2 (165). Defective signaling in the MAPK and PI3K pathways leads to abnormal cellular proliferation, survival, migration, cytoskeletal arrangement and vascular permeability (165). These changes ultimately lead to the development of PWS/SWS (165).
RASA1 mutations have been demonstrated in familial AVM, SWS, KTS and Parkes-Weber syndrome (166, 180, 181) (Figure 2), suggesting that germline mutations in RASA1 are associated with familial susceptibility to these VAs (165). A somatic mutation in PIKC3A (G1049N) has been demonstrated in HPWS (174) (Figure 2). PIKC3A (G1049N) mutation increases endothelial cell proliferation in vitro (182), which may occur in PWS (165, 182). Somatic mutations in SMARCA4, EPHA3, KRas, NRas, MAP2K1, and PDGFR-β have also been identified in PWS (165, 174) and they may act as “second hit” mutations for germline mutations such as RASA1, to create the PWS phenotype (165).
Populations of cells expressing the stemness-associated markers OCT4, SOX2, KLF4 and c-MYC but not NANOG, have been demonstrated in HPWS (49). It is proposed that the absence of NANOG in PWS supports the potential role of JNK in PWS, since JNK activates BMP, a NANOG suppressor (49). The presence of primitive populations within PWS is also supported by evidence demonstrating that endothelial cells within PWS co-express CD133 and CD166, markers shown to also be co-expressed by cells with EPC-like function (183). These cells displaying an EPC phenotype also express both the venous and arterial markers EphB1 and EfnB2, respectively. Co-expression of these markers on normal human dermal endothelial cells induces morphologic characteristics of PWS in vitro (183). It is thought that co-expression of EphB1 and EfnB2 prevents normal differentiation of arterioles and venules from the primitive capillary plexus, resulting in a pre-determined fate to form venule-like channels (183). PWS may result from these differentiation-impaired EPCs that co-express EphB1 and EfnB2, to develop into venule-like structures that progressively dilate (165). Further, cells from connective tissues, hair follicles and glands in PWS also have a GNAQ mutation, suggesting pluripotent cells may give rise to multiple lineages in PWS (184). The presence of GNAQ mutation in cells within different tissue types suggests these cells originate from an upstream ESC-like subpopulation recently identified (49).
mTOR Inhibition in Vascular Malformations
Sirolimus is increasingly used for complex vascular anomalies. mTOR is a conserved serine/threonine protein kinase that regulates cellular proliferation, cell growth, motility and survival, protein synthesis, and transcription (185, 186). It consists of two functionally distinct complexes—mTORC1 and mTORC2, each processing different substrates (187). mTOR coordinates normal cellular growth through regulation of ribosome creation and protein synthesis by integrating signals from the PI3K/AKT pathway. Increased activation of these pathways has been demonstrated in overgrowth syndromes which include VAs (150).
Sirolimus was first administered to an infant with kaposiform hemangioendothelioma (KHE) with Kasabach-Merritt phenomenon (KMP) refractory to other therapies (8), based on the high lymphatic component within this tumor, and that the PI3K/AKT/mTOR pathway is activated in angiogenesis and lymphangiogenesis. Sirolimus has also been used to treat lymphangioleiomyomatosis, tuberous sclerosis and neurofibromatosis (8). Subsequent discovery of somatic mutations in the PI3K/mTOR (PIK3CA) pathway (188), and both germline and somatic mutations in related pathways in VAs, provide further rationale for inhibiting mTOR in the treatment of isolated complex VAs not associated with overgrowth syndromes (126, 189, 190).
Since its initial use in KHE, several case reports, retrospective case series and clinical trials have shown positive results with sirolimus (137, 150, 191, 192). A recent systematic review (193) analyzing two randomized controlled trials, two non-randomized prospective trials, and 69 retrospective case reports and case series, involving a total of 373 patients, provides evidence of the efficacy of sirolimus in VAs. It shows oral sirolimus is highly effective in treating vascular tumors associated with KMP, with 95.5% of patients showing clinical improvement and 93% showing normalization of coagulopathy. Reduced size of VM is observed in 89.9% of patients, and clinical improvement of LM in 94.9% of patients, but improvements have not been observed in AVM (193).
Sirolimus is associated with potential side effects, which can be broad and multi-system, most commonly oral mucositis, dyslipidemia, leukopenia, gastrointestinal symptoms, paronychia and eczema (193). Serious infection and increased risk of lymphoproliferative disease have also been reported (193, 194).
mTOR pathways regulate somatic cell reprogramming, with short-course treatment using sirolimus showing enhanced somatic cell reprogramming, whereas longer-course treatment and mTOR-knockout decreases reprogramming (195). mTOR activation in human somatic cells with ectopic expression of OCT4, SOX2, KLF4, and c-MYC, significantly increases production of iPSCs (185). mTOR inhibition induces a paused pluripotent state in mouse blastocysts which have globally suppressed transcription, but maintain their gene expression signature and pluripotency. mTOR therefore regulates developmental timing at the peri-implantation stage (196). Based on the mechanism of action of sirolimus and its association with improved clinical outcomes in VAs, it is interesting to speculate whether mTOR also affects reprogramming and proliferation of the cells that express stemness-associated markers within VAs. This also raises the possibility that sirolimus may reduce or inhibit stemness conferred by the presence of these transcription factors in VAs in a manner dependent on the dose and duration of treatment.
Targeted Therapies for Vascular Anomalies
The discovery of germline and somatic mutations and associated variants in VAs and overgrowth syndromes (Table 1), underscores an exciting prospect of targeted therapy using new and existing agents to target these mutations and related pathways for these challenging conditions (197).
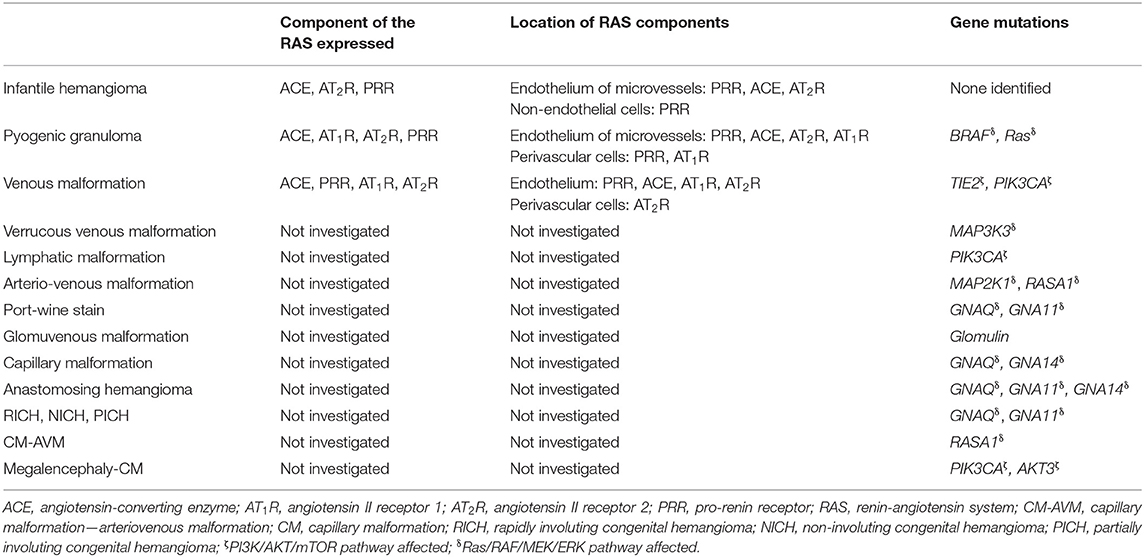
Table 1. Expression and location of components of the renin-angiotensin system, and gene mutations in vascular anomalies.
Gene mutations affecting the PI3K/AKT/mTOR and the Ras/RAF/MEK/ERK pathways are the two main pathways implicated in the pathogenesis of VAs. They control cellular growth, differentiation and transcription and they interact at multiple levels (154) (Figure 2).
A recent report on the treatment of patients with CLOVES (part of the PIK3CA-related overgrowth syndromes) using the PIK3CA inhibitor BYL719, shows promising results. This targeted therapy improved symptoms in all patients, with a decrease in the size of the intractable VAs, improvement of congestive heart failure and cardiac hemihypertrophy, and scoliosis (198). No significant side effects were reported (138, 193). Several other PI3K inhibitors are being clinically investigated for various cancer types (199). In a pre-clinical model of AVM using BRAF-mutated zebrafish, treatment with the orally active inhibitor of mutated BRAF, vemurafenib, used clinically to treat BRAF-mutated metastatic melanoma, restores normal blood flow (154).
Targeting mutations in MEK1/2 (MAP2K1) in VAs with MEK inhibitors has been proposed. MEK inhibitors are currently used to target the MAPK2K1/ERK pathway in multiple cancer types, including advanced soft tissue sarcoma and metastatic melanoma (200, 201). Blockade of AKT and PI3K signaling may inhibit metastatic gastric cancer (202). Repurposing cancer therapeutics that target mutations affecting components of these key pathways in VAs warrants further investigation.
Clinical trials are underway investigating miransertib (ARQ092), a pan-AKT inhibitor, in cancer (41). Low-dose miransertib reduces levels of phospho-AKT by about half in 83% of tissue samples from patients with Proteus syndrome (203) which is caused by a gain of function mutations in the AKT pathway (40). This inhibitor also demonstrates an anti-proliferative effect in fibroblasts isolated from PIK3CA-related overgrowth syndromes (204). Based on these findings, a phase I/II clinical trial is investigating the use of miransertib in PIK3CA-related overgrowth syndromes and other VA.
There may be pitfalls in targeting specific mutations within the PI3K/AKT/mTOR or RAF/MEK/ERK pathways, as they interact via Ras (205) (Figure 2). Kras mutations which are common in non-small cell lung cancer (NSCLC), cause activation of the RAF/MEK/ERK pathway (206). This has led to the development of small molecule inhibitors that target MEK (207). However, these therapies are ineffective in NSCLC due to subsequent activation of the PI3K/AKT/mTOR pathway (208). Further, activating mutations in PIK3CA increase resistance to MEK inhibitors, and a mutation in PTEN, an inhibitor of PIK3CA, causes complete resistance (209). Therefore, a cautionary approach in targeting specific mutations in these pathways should be taken, as resistance may develop because alternative pathways are activated. It is important that research efforts continue to look broadly for an effective, safer and affordable treatment for VAs.
Even if existing targeted cancer therapies can be effectively repurposed for the treatment of VAs including overgrowth syndromes, the cost of such agents may be prohibitive. For example, the cost for a full-course of trastuzumab is USD50,000 (210). Another consideration is the potential long-term side effects of potentially life-long treatment for young patients with congenital anomalies. This underscores the need to better understand the etiology of this group of challenging conditions. The identification of the expression of the RAS by primitive populations in many types of VAs (Table 1) opens up an exciting prospect of novel therapeutic targeting of these primitive populations by manipulation of the RAS using low-cost, off-patent and commonly available oral medications.
Conclusion
This review presents accumulating evidence demonstrating the presence of populations of cells that express stemness-associated markers in a growing number of vascular tumors and vascular malformations. As sirolimus targets cellular proliferation, survival and stemness via mTOR inhibition, its observed effect on some vascular tumors and vascular malformations, including overgrowth syndromes, may be attributed to its action on these primitive populations within these VAs. Gene mutations identified in VAs predominantly affect either the PI3K/AKT/mTOR or the Ras/RAF/MEK/ERK pathways. Existing cancer therapies that target these pathways open the possibility of repurposing these agents for the management of some of these challenging VAs. However, there are potential drawbacks, including the cost, potential long-term side effects in young patients, and the emergence of treatment resistance.
The discovery of cells that express stemness-associated markers in IH and the potential regulatory role of the RAS in its pathogenesis, underscore its spontaneous and accelerated involution induced by β-blockers and ACE inhibitors. Recent work demonstrating the effectiveness of targeting IH stem cells through inhibiting SOX18 using R-propranolol, may lead to more effective treatment of IH without the side effects of β-adrenergic blockade.
The observation of the expression of components of the RAS by populations of cells that also express stemness-associated markers in many types of VAs, opens up an exciting area of research. The stemness-associated markers expressed by cell populations and their cellular location within VAs are shown in Table 2. The expression of some or all transcription factors involved in generation of iPSCs by the cell populations in various types of VAs warrants further investigation including functional work to determine if they possess ESC properties. Studies are needed to determine whether mutations affecting the PI3K/AKT/mTOR or the Ras/RAF/MEK/ERK pathways, through their interaction with components of the RAS expressed on the primitive cells in VAs, could induce and/or maintain these cells in the primitive state. Further investigation into the precise interaction between the RAS and these pathways affected by the aforementioned mutations, and its effect on the stem cells may lead to improved understanding of the pathogenesis of these hitherto enigmatic conditions. This may provide immense opportunities for repurposing existing low-cost commonly available oral medications for the treatment of these challenging conditions.
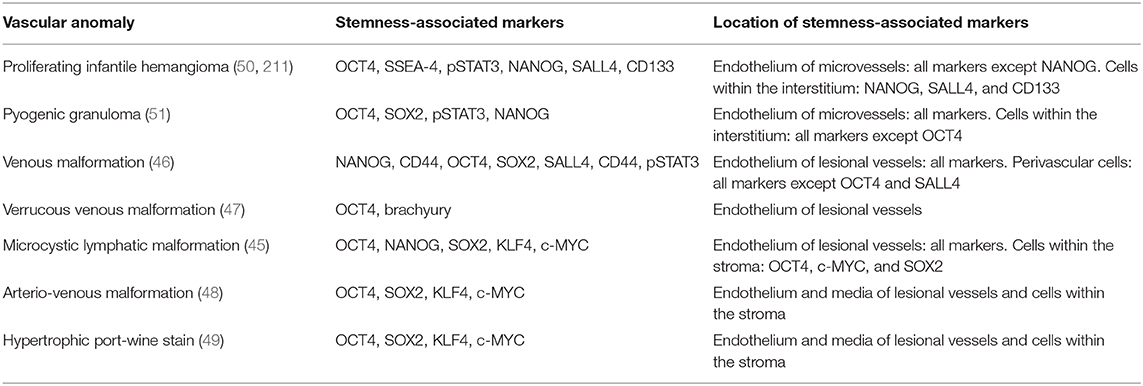
Table 2. Expression and location of stemness-associated markers in vascular anomalies.
Author Contributions
EK and LH drafted the manuscript. ST, PD, and SH critically revised the manuscript. All authors approved the manuscript.
Funding
EK and LH are supported by the Kristen Deane Scholarship and the Deane Endowment Trust Scholarship, respectively.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Itinteang T, Withers AH, Davis PF, Tan ST. Biology of infantile hemangioma. Front Surg. (2014) 1:38. doi: 10.3389/fsurg.2014.00038
2. Muller-Wille R, Wildgruber M, Sadick M, Wohlgemuth WA. Vascular anomalies (Part II): interventional therapy of peripheral vascular malformations. Rofo. (2018) 190:927–37. doi: 10.1055/s-0044-101266
3. Tan CE, Itinteang T, Leadbitter P, Marsh R, Tan ST. Low-dose propranolol regimen for infantile haemangioma. J Paediatr Child Health. (2015) 51:419–24. doi: 10.1111/jpc.12720
4. Itinteang T, Withers AHJ, Leadbitter P, Day DJ, Tan ST. Reply: pharmacologic therapies for Infantile hemangioma is there a rational basis? Plast Reconstr Surg. (2012) 129:725e−7e. doi: 10.1097/PRS.0b013e318245eb11
5. Tan ST, Itinteang T, Day DJ, O'Donnell C, Mathy JA, Leadbitter P. Treatment of infantile haemangioma with captopril. Br J Dermatol. (2012) 167:619–24. doi: 10.1111/j.1365-2133.2012.11016.x
6. Koh SP, Leadbitter P, Smithers F, Tan ST. β-blocker therapy for infantile haemangioma. Expert Rev Clin Pharmacol. (2020) 13:899–915. doi: 10.1080/17512433.2020.1788938
7. Itinteang T, Brasch HD, Tan ST, Day DJ. Expression of components of the renin–angiotensin system in proliferating infantile haemangioma may account for the propranolol-induced accelerated involution. J Plast Reconstr Aesthetic Surg. (2011) 64:759–65. doi: 10.1016/j.bjps.2010.08.039
8. Hammill AM, Wentzel M, Gupta A, Nelson S, Lucky A, Elluru R, et al. Sirolimus for the treatment of complicated vascular anomalies in children. Pediatr Blood Cancer. (2011) 57:1018–24. doi: 10.1002/pbc.23124
9. Wassef M, Blei F, Adams D, Alomari A, Baselga E, Berenstein A, et al. Vascular anomalies classification: recommendations from the international society for the study of vascular anomalies. Pediatrics. (2015) 136:e203–14. doi: 10.1542/peds.2014-3673
10. Mulliken JB, Glowacki J. Hemangiomas and vascular malformations in infants and children: a classification based on endothelial characteristics. Plast Reconstr Surg. (1982) 69:412–22. doi: 10.1097/00006534-198203000-00002
11. Enjolras O. Classification and management of the various superficial vascular anomalies: hemangiomas and vascular malformations. J Dermatol. (1997) 24:701–10. doi: 10.1111/j.1346-8138.1997.tb02522.x
12. Steele L, Zbeidy S, Thomson J, Flohr C. How is the term haemangioma used in the literature? An evaluation against the revised ISSVA classification. Pediatr Dermatol. (2019) 36:628–33. doi: 10.1111/pde.13885
13. Nguyen H-L, Boon L, Vikkula M. The genetic basis of vascular anomalies. In: Trenor C, III, Adams D, editors. Vascular Anomalies. Cham: Springer (2020). p. 17–29.
14. Shu W, Lin Y, Hua R, Luo Y, He N, Fang L, et al. Cutaneomucosal venous malformations are linked to the TIE2 mutation in a large chinese family. Exp Dermatol. (2012) 21:456–7. doi: 10.1111/j.1600-0625.2012.01492.x
15. Du Z, Zheng J, Zhang Z, Wang Y. Review of the endothelial pathogenic mechanism of TIE2-related venous malformation. J Vasc Surg Venous Lymphat Disord. (2017) 5:740–8. doi: 10.1016/j.jvsv.2019.11.001
16. Khan AA, Sandhya VK, Singh P, Parthasarathy D, Kumar A, Advani J, et al. Signaling network map of endothelial TEK tyrosine kinase. J Signal Transduct. (2014) 2014:173026. doi: 10.1155/2014/173026
17. Wang H, Zhang Y, Toratani S, Okamoto T. Transformation of vascular endothelial cells by a point mutation in the Tie2 gene from human intramuscular haemangioma. Oncogene. (2004) 23:8700–4. doi: 10.1038/sj.onc.1208006
18. Limaye N, Wouters V, Uebelhoer M, Tuominen M, Wirkkala R, Mulliken JB, et al. Somatic mutations in angiopoietin receptor gene TEK cause solitary and multiple sporadic venous malformations. Nat Genet. (2009) 41:118–24. doi: 10.1038/ng.272
19. Wouters V, Limaye N, Uebelhoer M, Irrthum A, Boon LM, Mulliken JB, et al. Hereditary cutaneomucosal venous malformations are caused by TIE2 mutations with widely variable hyper-phosphorylating effects. Eur J Hum Genet. (2010) 18:414–20. doi: 10.1038/ejhg.2009.193
20. Brouillard P, Ghassibé M, Penington A, Boon LM, Dompmartin A, Temple IK, et al. Four common glomulin mutations cause two thirds of glomuvenous malformations (“familial glomangiomas”): evidence for a founder effect. J Med Genet. (2005) 42:e13. doi: 10.1136/jmg.2004.024174
21. Limaye N, Kangas J, Mendola A, Godfraind C, Schlogel MJ, Helaers R, et al. Somatic activating PIK3CA mutations cause venous malformation. Am J Hum Genet. (2015) 97:914–21. doi: 10.1016/j.ajhg.2015.11.011
22. Luks VL, Kamitaki N, Vivero MP, Uller W, Rab R, Bovee JV, et al. Lymphatic and other vascular malformative/overgrowth disorders are caused by somatic mutations in PIK3CA. J Pediatr. (2015) 166:1048–54.e1-5. doi: 10.1016/j.jpeds.2014.12.069
23. Riviere JB, Mirzaa GM, O'Roak BJ, Beddaoui M, Alcantara D, Conway RL, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. (2012) 44:934–40. doi: 10.1038/ng.2331
24. Couto JA, Huang AY, Konczyk DJ, Goss JA, Fishman SJ, Mulliken JB, et al. Somatic MAP2K1 mutations are associated with extracranial arteriovenous malformation. Am J Hum Genet. (2017) 100:546–54. doi: 10.1016/j.ajhg.2017.01.018
25. Marks JL, Gong Y, Chitale D, Golas B, McLellan MD, Kasai Y, et al. Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma. Cancer Res. (2008) 68:5524–8. doi: 10.1158/0008-5472.CAN-08-0099
26. Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, et al. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. (1994) 265:966–70. doi: 10.1126/science.8052857
27. Revencu N, Boon LM, Mendola A, Cordisco MR, Dubois J, Clapuyt P, et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mutat. (2013) 34:1632–41. doi: 10.1002/humu.2243128
28. Amyere M, Revencu N, Helaers R, Pairet E, Baselga E, Cordisco M, et al. Germline loss-of-function mutations in EPHB4 cause a second form of capillary malformation-arteriovenous malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation. (2017) 136:1037–4829. doi: 10.1161/CIRCULATIONAHA.116.026886
29. Berenguer B, Mulliken JB, Enjolras O, Boon LM, Wassef M, Josset P, et al. Rapidly involuting congenital hemangioma: clinical and histopathologic features. Pediatr Dev Pathol. (2003) 6:495–510. doi: 10.1007/s10024-003-2134-6
30. Enjolras O, Mulliken JB, Boon LM, Wassef M, Kozakewich HP, Burrows PE. Noninvoluting congenital hemangioma: a rare cutaneous vascular anomaly. Plast Reconstr Surg. (2001) 107:1647–54. doi: 10.1097/00006534-200106000-00002
31. Ayturk Ugur M, Couto Javier A, Hann S, Mulliken John B, Williams KL, Huang August Y, et al. Somatic activating mutations in GNAQ and GNA11 are associated with congenital hemangioma. Am J Hum Genet. (2016) 98:789–95. doi: 10.1016/j.ajhg.2016.03.009
32. Liau JY, Tsai JH, Lan J, Chen CC, Wang YH, Lee JC, et al. GNA11 joins GNAQ and GNA14 as a recurrently mutated gene in anastomosing hemangioma. Virchows Arch. (2019) 476:475–81. doi: 10.1007/s00428-019-02673-y
33. Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. (2013) 368:1971–9. doi: 10.1056/NEJMoa1213507
34. Couto JA, Huang L, Vivero MP, Kamitaki N, Maclellan RA, Mulliken JB, et al. Endothelial cells from capillary malformations are enriched for somatic GNAQ Mutations. Plast Reconstr Surg. (2016) 137:77e−82e. doi: 10.1097/PRS.0000000000001868
35. Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O'Brien JM, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. (2009) 457:599–602. doi: 10.1038/nature07586
36. Lim YH, Bacchiocchi A, Qiu J, Straub R, Bruckner A, Bercovitch L, et al. GNA14 Somatic mutation causes congenital and sporadic vascular tumors by MAPK activation. Am J Hum Genet. (2016) 99:443–50. doi: 10.1016/j.ajhg.2016.06.010
37. Couto JA, Vivero MP, Kozakewich HPW, Taghinia AH, Mulliken JB, Warman ML, et al. A somatic MAP3K3 mutation is associated with verrucous venous malformation. Am J Hum Genet. (2015) 96:480–6. doi: 10.1016/j.ajhg.2015.01.007
38. Groesser L, Peterhof E, Evert M, Landthaler M, Berneburg M, Hafner C. BRAF and RAS mutations in sporadic and secondary pyogenic granuloma. J Invest Dermatol. (2016) 136:481–6. doi: 10.1038/JID.2015.376
39. Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. (1997) 16:64–7. doi: 10.1038/ng0597-64
40. Lindhurst MJ, Sapp JC, Teer JK, Johnston JJ, Finn EM, Peters K, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. (2011) 365:611–9. doi: 10.1056/NEJMoa1104017
41. Le Cras T, Boscolo E. Cellular and molecular mechanisms of PIK3CA-related vascular anomalies. Vasc Biol. (2019) 1:H33–40. doi: 10.1530/VB-19-0016
42. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. (2006) 126:663–76. doi: 10.1016/j.cell.2006.07.024
43. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. (2007) 131:861–72. doi: 10.1016/j.cell.2007.11.019
44. Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. (2007) 318:1917–20. doi: 10.1126/science.1151526
45. Eady EK, Brasch HD, de Jongh J, Marsh RW, Tan ST, Itinteang T. Expression of embryonic stem cell markers in microcystic lymphatic malformation. Lymphat Res Biol. (2019) 17:496–503. doi: 10.1089/lrb.2018.0046
46. Tan E, Siljee SD, Brasch HD, Enriquez S, Tan ST, Itinteang T. Embryonic stem cell-like subpopulations in venous malformation. Front Med. (2017) 4:162. doi: 10.3389/fmed.2017.00162
47. Laing EL, Brasch HD, Steel R, Jia J, Itinteang T, Tan ST, et al. Verrucous hemangioma expresses primitive markers. J Cutan Pathol. (2013) 40:391–6. doi: 10.1111/cup.12078
48. Luke Krishnan CS, Brasch HD, Patel J, Bockett N, Paterson E, Davis PF, et al. Expression of induced pluripotent stem cell markers in extracranial arterio venous malformation. Front Surg. (2021) Under Revision.
49. William J, Brasch HD, Bockett N, Patel J, Paterson E, Davis PF, et al. Embryonic stem cell-like population in hypertrophic port wine stain. J Vasc Anom. (2021). doi: 10.1097/JOVA.0000000000000006
50. Itinteang T, Tan ST, Brasch HD, Steel R, Best HA, Vishvanath A, et al. Infantile haemangioma expresses embryonic stem cell markers. J Clin Pathol. (2012) 65:394–8. doi: 10.1136/jclinpath-2011-200462
51. Blackwell MG, Itinteang T, Chibnall AM, Davis PF, Tan ST. Expression of embryonic stem cell markers in pyogenic granuloma. J Cutan Pathol. (2016) 43:1096–101. doi: 10.1111/cup.12786
52. Papali'i-Curtin JC, Brasch HD, van Schaijik B, de Jongh J, Marsh RW, Tan ST, et al. Expression of components of the renin-angiotensin system in pyogenic granuloma. Front Surg. (2019) 6:13. doi: 10.3389/fsurg.2019.00013
53. Tan EMS, Brasch HD, Davis PF, Itinteang T, Tan ST. Embryonic stem cell-like population within venous malformation expresses the renin-angiotensin system. Plast Reconstr Surg Glob Open. (2019) 7:e2170. doi: 10.1097/GOX.0000000000002170
54. Nguyen Dinh Cat A, Touyz RM. A new look at the renin–angiotensin system—Focusing on the vascular system. Peptides. (2011) 32:2141–50. doi: 10.1016/j.peptides.2011.09.010
55. Durik M, Seva Pessoa B, Roks AJ. The renin-angiotensin system, bone marrow and progenitor cells. Clin Sci (Lond). (2012) 123:205–23. doi: 10.1042/CS20110660
56. Nehme A, Zouein FA, Zayeri ZD, Zibara K. An update on the tissue renin angiotensin system and its role in physiology and pathology. J Cardiovasc Dev Dis. (2019) 6:14. doi: 10.3390/jcdd6020014
57. Munro MJ, Wickremesekera AC, Davis PF, Marsh R, Tan ST, Itinteang T. Renin-angiotensin system and cancer: a review. Integr Cancer Sci Ther. (2017) 4:1–6. doi: 10.15761/ICST.1000231
58. Roth I, Wickremesekera AC, Wickremesekera SK, Davis PF, Tan ST. Therapeutic targeting of cancer stem cells via modulation of the renin-angiotensin system. Front Oncol. (2019) 9:745. doi: 10.3389/fonc.2019.00745
59. Itinteang T, Chudakova DA, Dunne JC, Davis PF, Tan ST. Expression of cathepsins B, D, and G in infantile hemangioma. Front Surg. (2015) 2:26. doi: 10.3389/fsurg.2015.00026
60. Mehrotra S, van Schaijik B, Boyes K, Bockett N, Brasch HD, Itinteang T, Tan ST. Expression of cathepsins B, D and G in microcystic lymphatic malformation. Lymph Biol Res. (2020) doi: 10.1089/lrb.2020.0047. [Epub ahead of print].
61. Yu Y, Flint AF, Mulliken JB, Wu JK, Bischoff J. Endothelial progenitor cells in infantile hemangioma. Blood. (2004) 103:1373–5. doi: 10.1182/blood-2003-08-2859
62. Kleinman ME, Tepper OM, Capla JM, Bhatt KA, Ceradini DJ, Galiano RD, et al. Increased circulating AC133+ CD34+ endothelial progenitor cells in children with hemangioma. Lymphat Res Biol. (2003) 1:301–7. doi: 10.1089/153968503322758102
63. Itinteang T, Tan ST, Brasch H, Day DJ. Primitive mesodermal cells with a neural crest stem cell phenotype predominate proliferating infantile haemangioma. J Clin Pathol. (2010) 63:771–6. doi: 10.1136/jcp.2010.079368
64. Itinteang T, Tan ST, Brasch H, Day DJ. Haemogenic endothelium in infantile haemangioma. J Clin Pathol. (2010) 63:982–6. doi: 10.1136/jcp.2010.081257
65. Yu Y, Fuhr J, Boye E, Gyorffy S, Soker S, Atala A, et al. Mesenchymal stem cells and adipogenesis in hemangioma involution. Stem Cells. (2006) 24:1605–12. doi: 10.1634/stemcells.2005-0298
66. Itinteang T, Vishvanath A, Day DJ, Tan ST. Mesenchymal stem cells in infantile haemangioma. J Clin Pathol. (2011) 64:232–6. doi: 10.1136/jcp.2010.085209
67. Khan ZA, Boscolo E, Picard A, Psutka S, Melero-Martin JM, Bartch TC, et al. Multipotential stem cells recapitulate human infantile hemangioma in immunodeficient mice. J Clin Invest. (2008) 118:2592–9. doi: 10.1172/JCI33493
68. Itinteang T, Tan ST, Brasch HD, Vishvanath A, Day DJ. Primitive erythropoiesis in infantile haemangioma. Br J Dermatol. (2011) 164:1097–100. doi: 10.1111/j.1365-2133.2010.10187.x
69. Tyagi N, Marimuthu S, Bhardwaj A, Deshmukh SK, Srivastava SK, Singh AP, et al. p-21 activated kinase 4 (PAK4) maintains stem cell-like phenotypes in pancreatic cancer cells through activation of STAT3 signaling. Cancer Lett. (2016) 370:260–7. doi: 10.1016/j.canlet.2015.10.028
70. Matsuda T, Nakamura T, Nakao K, Arai T, Katsuki M, Heike T, et al. STAT3 activation is sufficient to maintain an undifferentiated state of mouse embryonic stem cells. EMBO J. (1999) 18:4261–9. doi: 10.1093/emboj/18.15.4261
71. North PE, Waner M, Mizeracki A, Mihm MC Jr. GLUT1: a newly discovered immunohistochemical marker for juvenile hemangiomas. Hum Pathol. (2000) 31:11–22. doi: 10.1016/s0046-8177(00)80192-6
72. Jansson T, Wennergren M, Illsley NP. Glucose transporter protein expression in human placenta throughout gestation and in intrauterine growth retardation. J Clin Endocrinol Metab. (1993) 77:1554–62. doi: 10.1210/jcem.77.6.8263141
73. North PE, Waner M, Mizeracki A, Mrak RE, Nicholas R, Kincannon J, et al. A unique microvascular phenotype shared by juvenile hemangiomas and human placenta. Arch Dermatol. (2001) 137:559–70.
74. Huang SA, Tu HM, Harney JW, Venihaki M, Butte AJ, Kozakewich HP, et al. Severe hypothyroidism caused by type 3 iodothyronine deiodinase in infantile hemangiomas. N Engl J Med. (2000) 343:185–9. doi: 10.1056/NEJM200007203430305
75. Barnes CM, Huang S, Kaipainen A, Sanoudou D, Chen EJ, Eichler GS, et al. Evidence by molecular profiling for a placental origin of infantile hemangioma. Proc Natl Acad Sci USA. (2005) 102:19097–102. doi: 10.1073/pnas.0509579102
76. Itinteang T, Tan ST, Guthrie S, Tan CE, McIntyre BC, Brasch HD, et al. A placental chorionic villous mesenchymal core cellular origin for infantile haemangioma. J Clin Pathol. (2011) 64:870–4. doi: 10.1136/jclinpath-2011-200191
77. Kertesz N, Wu J, Chen TH, Sucov HM, Wu H. The role of erythropoietin in regulating angiogenesis. Dev Biol. (2004) 276:101–10. doi: 10.1016/j.ydbio.2004.08.025
78. Qiu C, Olivier EN, Velho M, Bouhassira EE. Globin switches in yolk sac-like primitive and fetal-like definitive red blood cells produced from human embryonic stem cells. Blood. (2008) 111:2400–8. doi: 10.1182/blood-2007-07-102087
79. Schechter AN. Hemoglobin research and the origins of molecular medicine. Blood. (2008) 112:3927–38. doi: 10.1182/blood-2008-04-078188
80. Spock CL, Tom LK, Canadas K, Sue GR, Sawh-Martinez R, Maier CL, et al. Infantile hemangiomas exhibit neural crest and pericyte markers. Ann Plast Surg. (2015) 74:230–6. doi: 10.1097/SAP.0000000000000080
81. Lee G, Kim H, Elkabetz Y, Al Shamy G, Panagiotakos G, Barberi T, et al. Isolation and directed differentiation of neural crest stem cells derived from human embryonic stem cells. Nat Biotechnol. (2007) 25:1468–75. doi: 10.1038/nbt1365
82. Metry DW, Hawrot A, Altman C, Frieden IJ. Association of solitary, segmental hemangiomas of the skin with visceral hemangiomatosis. Arch Dermatol. (2004) 140:591–6. doi: 10.1001/archderm.140.5.591
83. Leaute-Labreze C, Dumas de la Roque E, Hubiche T, Boralevi F, Thambo JB, Taieb A. Propranolol for severe hemangiomas of infancy. N Engl J Med. (2008) 358:2649–51. doi: 10.1056/NEJMc0708819
84. Bigorre M, Van Kien AK, Valette H. Beta-blocking agent for treatment of infantile hemangioma. Plast Reconstr Surg. (2009) 123:195e−6e. doi: 10.1055/s-0034-1376259
85. de Graaf M, Raphael MF, Breugem CC, Knol MJ, Bruijnzeel-Koomen CAFM, Kon M, et al. Treatment of infantile haemangiomas with atenolol: Comparison with a historical propranolol group. J Plast Reconstr Aesthet Surg. (2013) 66:1732–40. doi: 10.1016/j.bjps.2013.07.035
86. Ni N, Langer P, Wagner R, Guo S. Topical timolol for periocular hemangioma: Report of further study. Arch Ophthalmol. (2011) 129:373–9. doi: 10.1001/archophthalmol.2011.24
87. Sakoda M, Ichihara A, Kaneshiro Y, Takemitsu T, Nakazato Y, Nabi AN, et al. (Pro) Renin receptor–mediated activation of mitogen-activated protein kinases in human vascular smooth muscle cells. Hypertens Res. (2007) 30:1139. doi: 10.1291/hypres.30.1139
88. van Schaijik B, Tan ST, Marsh RW, Itinteang T. Expression of (pro)renin receptor and its effect on endothelial cell proliferation in infantile hemangioma. Pediatr Res. (2019) 86:202–7. doi: 10.1038/s41390-019-0430-8
89. Balakumar P, Jagadeesh G. Potential cross-talk between (pro)renin receptors and Wnt/frizzled receptors in cardiovascular and renal disorders. Hypertens Res. (2011) 34:1161–70. doi: 10.1038/hr.2011.113
90. Itinteang T, Marsh R, Davis PF, Tan ST. Angiotensin II causes cellular proliferation in infantile haemangioma via angiotensin II receptor 2 activation. J Clin Pathol. (2015) 68:346. doi: 10.1136/jclinpath-2014-202794
91. Fiselier TJ, Lijnen P, Monnens L, van Munster P, Jansen M, Peer P. Levels of renin, angiotensin I and II, angiotensin-converting enzyme and aldosterone in infancy and childhood. Eur J Pediatr. (1983) 141:3–7. doi: 10.1007/BF00445660
92. Chang LC, Haggstrom AN, Drolet BA, Baselga E, Chamlin SL, Garzon MC, et al. Growth characteristics of infantile hemangiomas: implications for management. Pediatrics. (2008) 122:360–7. doi: 10.1542/peds.2007-2767
93. Sulzberger L, Baillie R, Itinteang T, de Jong S, Marsh R, Leadbitter P, et al. Serum levels of renin, angiotensin-converting enzyme and angiotensin II in patients treated by surgical excision, propranolol and captopril for problematic proliferating infantile haemangioma. J Plast Reconstr Aesthet Surg. (2016) 69:381–6. doi: 10.1016/j.bjps.2015.10.020
94. Sulzberger L, Tan EMS, Davis PF, Brasch HD, Tan ST, Itinteang T. Phosphorylated forms of STAT1, STAT3, and STAT5 are expressed in proliferating but not involuted infantile hemangioma. Front Surg. (2018) 5:31. doi: 10.3389/fsurg.2018.00031
95. Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. (2009) 458:904–8. doi: 10.1038/nature07815
96. Levy DE, Darnell JE Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. (2002) 3:651–62. doi: 10.1038/nrm909
97. Overman J, Fontaine F, Wylie-Sears J, Moustaqil M, Huang L, Meurer M, et al. R-propranolol is a small molecule inhibitor of the SOX18 transcription factor in a rare vascular syndrome and hemangioma. eLife. (2019) 8:e43026. doi: 10.7554/eLife.43026
98. Sasaki M, North PE, Elsey J, Bubley J, Rao S, Jung Y, et al. Propranolol exhibits activity against hemangiomas independent of beta blockade. NPJ Precis Oncol. (2019) 3:27. doi: 10.1038/s41698-019-0099-9
99. Moalem S, Brouillard P, Kuypers D, Legius E, Harvey E, Taylor G, et al. Hypotrichosis-lymphedema-telangiectasia-renal defect associated with a truncating mutation in the SOX18 gene. Clin Genet. (2015) 87:378–82. doi: 10.1111/cge.12388
100. Wunnemann F, Kokta V, Leclerc S, Thibeault M, McCuaig C, Hatami A, et al. Aortic dilatation associated with a de novo mutation in the SOX18 gene: expanding the clinical spectrum of hypotrichosis-lymphedema-telangiectasia syndrome. Can J Cardiol. (2016) 32:135 e1–7. doi: 10.1016/j.cjca.2015.04.004
101. Itinteang T, Davis PF, Tan ST. Infantile hemangiomas exhibit neural crest and pericyte markers. Ann Plast Surg. (2015) 74:383.
102. Ardekani GS, Ebrahimi S, Amini M, Aslani FS, Handjani F, Omrani GR, et al. Topical captopril as a novel agent against hypertrophic scar formation in New Zealand white rabbit skin. Wounds. (2008) 20:101–6.
103. Fyhrquist F, Saijonmaa O. Renin-angiotensin system revisited. J Intern Med. (2008) 264:224–36. doi: 10.1111/j.1365-2796.2008.01981.x
104. Zambidis ET, Soon Park T, Yu W, Tam A, Levine M, Yuan X, et al. Expression of angiotensin-converting enzyme (CD143) identifies and regulates primitive hemangioblasts derived from human pluripotent stem cells. Blood. (2008) 112:3601–14. doi: 10.1182/blood-2008-03-144766
105. Lim YH, Douglas SR, Ko CJ, Antaya RJ, McNiff JM, Zhou J, et al. Somatic activating RAS mutations cause vascular tumors including pyogenic granuloma. J Invest Dermatol. (2015) 135:1698–700. doi: 10.1038/jid.2015.55
106. Kranenburg O, Gebbink MFBG, Voest EE. Stimulation of angiogenesis by Ras proteins. Biochimica et biophysica acta. (2004) 1654:23–37. doi: 10.1016/j.bbcan.2003.09.004
107. Moore MA, Hattori K, Heissig B, Shieh JH, Dias S, Crystal RG, et al. Mobilization of endothelial and hematopoietic stem and progenitor cells by adenovector-mediated elevation of serum levels of SDF-1, VEGF, and angiopoietin-1. Ann NY Acad Sci. (2001) 938:36–45. doi: 10.1111/j.1749-6632.2001.tb03572.x
108. Peichev M, Naiyer AJ, Pereira D, Zhu Z, Lane WJ, Williams M, et al. Expression of VEGFR-2 and AC133 by circulating human CD34(+) cells identifies a population of functional endothelial precursors. Blood. (2000) 95:952–8. doi: 10.1182/blood.V95.3.952.003k27_952_958
109. Vikkula M, Boon LM, Mulliken JB. Molecular genetics of vascular malformations. Matrix Biol. (2001) 20:327–35. doi: 10.1016/s0945-053x(01)00150-0
110. Kim Y-W, Lee B-B. Epidemiologic aspect of congenital vascular malformation. In: Kim Y-W, Lee B-B, Yakes WF, Do Y-S, editors. Congenital Vascular Malformations: A Comprehensive Review of Current Management. Berlin, Heidelberg: Springer (2017). p. 31–4.
111. Mogler C, Beck C, Kulozik A, Penzel R, Schirmacher P, Breuhahn K. Elevated expression of c-kit in small venous malformations of blue rubber bleb nevus syndrome. Rare Tumors. (2010) 2:e36. doi: 10.4081/rt.2010.e36
112. Siljee S, Keane E, Marsh R, Brasch HD, Tan ST, Itinteang T. Expression of the components of the renin-angiotensin system in venous malformation. Front Surg. (2016) 3:24. doi: 10.3389/fsurg.2016.00024
113. Huang Y, Wongamorntham S, Kasting J, McQuillan D, Owens RT, Yu L, et al. Renin increases mesangial cell transforming growth factor-β1 and matrix proteins through receptor-mediated, angiotensin II-independent mechanisms. Kidney Int. (2006) 69:105–13. doi: 10.1038/sj.ki.5000011
114. Huang Y, Noble N, Zhang J, Xu C, Border W. Renin-stimulated TGF-β1 expression is regulated by a mitogen-activated protein kinase in mesangial cells. Kidney Int. (2007) 72:45–52. doi: 10.1038/sj.ki.5002243
115. Kaneshiro Y, Ichihara A, Takemitsu T, Sakoda M, Suzuki F, Nakagawa T, et al. Increased expression of cyclooxygenase-2 in the renal cortex of human prorenin receptor gene-transgenic rats. Kidney Int. (2006) 70:641–6. doi: 10.1038/sj.ki.5001627
116. Poniatowski ŁA, Wojdasiewicz P, Gasik R, Szukiewicz D. Transforming growth factor beta family: insight into the role of growth factors in regulation of fracture healing biology and potential clinical applications. Mediators Inflamm. (2015) 2015:17. doi: 10.1155/2015/137823
117. Deshayes F, Nahmias C. Angiotensin receptors: a new role in cancer? Trends Endocrinol Metab. (2005) 16:293–9. doi: 10.1016/j.tem.2005.07.009
118. Soblet J, Limaye N, Uebelhoer M, Boon LM, Vikkula M. Variable somatic TIE2 mutations in half of sporadic venous malformations. Mol Syndromol. (2013) 4:179–83. doi: 10.1159/000348327
119. Castel P, Carmona FJ, Grego-Bessa J, Berger MF, Viale A, Anderson KV, et al. Somatic PIK3CA mutations as a driver of sporadic venous malformations. Sci Transl Med. (2016) 8:332ra42. doi: 10.1126/scitranslmed.aaf1164
120. Korpelainen EI, Karkkainen M, Gunji Y, Vikkula M, Alitalo K. Endothelial receptor tyrosine kinases activate the STAT signaling pathway: mutant Tie-2 causing venous malformations signals a distinct STAT activation response. Oncogene. (1999) 18:1–8. doi: 10.1038/sj.onc.1202288
121. Brindle NPJ, Saharinen P, Alitalo K. Signaling and functions of angiopoietin-1 in vascular protection. Circ Res. (2006) 98:1014–23. doi: 10.1161/01.RES.0000218275.54089.12
122. Goines J, Li X, Cai Y, Mobberley-Schuman P, Metcalf M, Fishman SJ, et al. A xenograft model for venous malformation. Angiogenesis. (2018) 21:725–35. doi: 10.1007/s10456-018-9624-7
123. Steiner F, FitzJohn T, Tan ST. Surgical treatment for venous malformation. J Plast Reconstr Aesthet Surg. (2013) 66:1741–9. doi: 10.1016/j.bjps.2013.07.033
124. Huang Y-H, Wu M-P, Pan S-C, Su W-C, Chen Y-W, Wu L-W. STAT1 activation by venous malformations mutant Tie2-R849W antagonizes VEGF-A-mediated angiogenic response partly via reduced bFGF production. Angiogenesis. (2013) 16:207–22. doi: 10.1007/s10456-012-9313-x
125. Greene AK, Goss JA. Vascular anomalies: from a clinicohistologic to a genetic framework. Plast Reconstr Surg. (2018) 141:709e−17e. doi: 10.1097/PRS.0000000000004294
126. Uebelhoer M, Natynki M, Kangas J, Mendola A, Nguyen HL, Soblet J, et al. Venous malformation-causative TIE2 mutations mediate an AKT-dependent decrease in PDGFB. Hum Mol Genet. (2013) 22:3438–48. doi: 10.1093/hmg/ddt198
127. Castillo SD, Tzouanacou E, Zaw-Thin M, Berenjeno IM, Parker VE, Chivite I, et al. Somatic activating mutations in Pik3ca cause sporadic venous malformations in mice and humans. Sci Transl Med. (2016) 8:332ra43. doi: 10.1126/scitranslmed.aad9982
128. Morris PN, Dunmore BJ, Tadros A, Marchuk DA, Darland DC, D'Amore PA, et al. Functional analysis of a mutant form of the receptor tyrosine kinase Tie2 causing venous malformations. J Mol Med (Berl). (2005) 83:58–63. doi: 10.1007/s00109-004-0601-9
129. Eklund L, Olsen BR. Tie receptors and their angiopoietin ligands are context-dependent regulators of vascular remodeling. Exp Cell Res. (2006) 312:630–41. doi: 10.1016/j.yexcr.2005.09.002
130. Yang H, Raizada MK. Role of phosphatidylinositol 3-kinase in angiotensin II regulation of norepinephrine neuromodulation in brain neurons of the spontaneously hypertensive rat. J Neurosci. (1999) 19:2413–23. doi: 10.1523/JNEUROSCI.19-07-02413.1999
131. Cheng WH, Lu PJ, Hsiao M, Hsiao CH, Ho WY, Cheng PW, et al. Renin activates PI3K-Akt-eNOS signalling through the angiotensin AT(1) and Mas receptors to modulate central blood pressure control in the nucleus tractus solitarii. Br J Pharmacol. (2012) 166:2024–35. doi: 10.1111/j.1476-5381.2012.01832.x
132. Bühler FR, Laragh JH, Baer L, Vaughan ED, Brunner HR. Propranolol inhibition of renin secretion. N Engl J Med. (1972) 287:1209–14. doi: 10.1056/NEJM197212142872401
133. Wang F, Lu X, Peng K, Zhou L, Li C, Wang W, et al. COX-2 mediates angiotensin II-induced (pro)renin receptor expression in the rat renal medulla. Am J Physiol Renal Physiol. (2014) 307:F25–32. doi: 10.1152/ajprenal.00548.2013
134. Abematsu T, Okamoto Y, Nakagawa S, Kurauchi K, Kodama Y, Nishikawa T, et al. Rectosigmoid colon venous malformation successfully treated with propranolol and celecoxib. J Pediat Surg. (2015) 3:331–3. doi: 10.1016/j.epsc.2015.06.009
135. Holmer SR, Hengstenberg C, Mayer B, Engel S, Löwel H, Riegger G, et al. Marked suppression of renin levels by β-receptor blocker in patients treated with standard heart failure therapy: a potential mechanism of benefit from β-blockade. J Intern Med. (2001) 249:167–72. doi: 10.1046/j.1365-2796.2001.00786.x
136. Boscolo E, Limaye N, Huang L, Kang KT, Soblet J, Uebelhoer M, et al. Rapamycin improves TIE2-mutated venous malformation in murine model and human subjects. J Clin Invest. (2015) 125:3491–504. doi: 10.1172/JCI76004
137. Hammer J, Seront E, Duez S, Dupont S, Van Damme A, Schmitz S, et al. Sirolimus is efficacious in treatment for extensive and/or complex slow-flow vascular malformations: a monocentric prospective phase II study. Orphanet J Rare Dis. (2018) 13:191. doi: 10.1186/s13023-018-0934-z
138. Ballou LM, Lin RZ. Rapamycin and mTOR kinase inhibitors. J Chem Biol. (2008) 1:27–36. doi: 10.1007/s12154-008-0003-5
139. Tennant LB, Mulliken JB, Perez-Atayde AR, Kozakewich HP. Verrucous hemangioma revisited. Pediatr Dermatol. (2006) 23:208–15. doi: 10.1111/j.1525-1470.2006.00219.x
140. Imperial R, Helwig EB. Verrucous hemangioma: a clinicopathologic study of 21 cases. Arch Dermatol. (1967) 96:247–53. doi: 10.1001/archderm.96.3.247
141. Perkins JA, Manning SC, Tempero RM, Cunningham MJ, Edmonds JL Jr, Hoffer FA, et al. Lymphatic malformations: current cellular and clinical investigations. Otolaryngol Head Neck Surg. (2010) 142:789–94. doi: 10.1016/j.otohns.2010.02.025
142. Perkins JA, Manning SC, Tempero RM, Cunningham MJ, Edmonds JL Jr, Hoffer FA, et al. Lymphatic malformations: review of current treatment. Otolaryngol Head Neck Surg. (2010) 142:795–803. doi: 10.1016/j.otohns.2010.02.026
143. Wu JK, Kitajewski C, Reiley M, Keung CH, Monteagudo J, Andrews JP, et al. Aberrant lymphatic endothelial progenitors in lymphatic malformation development. PLoS ONE. (2015) 10:e0117352. doi: 10.1371/journal.pone.0117352
144. Zhao W, Ji X, Zhang F, Li L, Ma L. Embryonic stem cell markers. Molecules. (2012) 17:6196–236. doi: 10.3390/molecules17066196
145. Lokmic Z, Mitchell GM, Koh Wee Chong N, Bastiaanse J, Gerrand Y-W, Zeng Y, et al. Isolation of human lymphatic malformation endothelial cells, their in vitro characterization and in vivo survival in a mouse xenograft model. Angiogenesis. (2014) 17:1–15. doi: 10.1007/s10456-013-9371-8
146. Arteriovenous Malformation (AVM) of the Brain. (2019). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK430685/ (accessed December 15, 2019).
147. Kohout MP, Hansen M, Pribaz JJ, Mulliken JB. Arteriovenous malformations of the head and neck: natural history and management. Plast Reconstr Surg. (1998) 102:643–54. doi: 10.1097/00006534-199809030-00006
148. Fowell C, Jones R, Nishikawa H, Monaghan A. Arteriovenous malformations of the head and neck: current concepts in management. Br J Oral Maxillofac Surg. (2016) 54:482–7. doi: 10.1016/j.bjoms.2016.01.034
149. Suami H, Lee B-B. Embryological background of congenital vascular malformations. In: Kim Y-W, Lee B-B, Yakes WF, Do Y-S, editors. Congenital Vascular Malformations: A Comprehensive Review of Current Management. Berlin, Heidelberg: Springer (2017) p. 7–14.
150. Adams DM, Trenor CC III, Hammill AM, Vinks AA, Patel MN, Chaudry G, et al. Efficacy and safety of sirolimus in the treatment of complicated vascular anomalies. Pediatrics. (2016) 137:e20153257. doi: 10.1542/peds.2015-3257
151. Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. (2011) 36:320–8. doi: 10.1016/j.tibs.2011.03.006
152. Vignot S, Faivre S, Aguirre D, Raymond E. mTOR-targeted therapy of cancer with rapamycin derivatives. Ann Oncol. (2005) 16:525–37. doi: 10.1093/annonc/mdi113
153. Dard L, Bellance N, Lacombe D, Rossignol R. RAS signalling in energy metabolism and rare human diseases. Biochim Biophys Acta Bioenerg. (2018) 1859:845–67. doi: 10.1016/j.bbabio.2018.05.003
154. Al-Olabi L, Polubothu S, Dowsett K, Andrews KA, Stadnik P, Joseph AP, et al. Mosaic RAS/MAPK variants cause sporadic vascular malformations which respond to targeted therapy. J Clin Invest. (2018) 128:1496–508. doi: 10.1172/JCI98589
155. Lekwuttikarn R, Lim YH, Admani S, Choate KA, Teng JMC. Genotype-Guided medical treatment of an arteriovenous malformation in a child. JAMA Dermatol. (2019) 155:256–7. doi: 10.1001/jamadermatol.2018.4653
156. Falchook GS, Lewis KD, Infante JR, Gordon MS, Vogelzang NJ, DeMarini DJ, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. (2012) 13:782–9. doi: 10.1016/S1470-2045(12)70269-3
157. Britten CD. PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer Chemother Pharmacol. (2013) 71:1395–409. doi: 10.1007/s00280-013-2121-1
158. Li D, March ME, Gutierrez-Uzquiza A, Kao C, Seiler C, Pinto E, et al. ARAF recurrent mutation causes central conducting lymphatic anomaly treatable with a MEK inhibitor. Nat Med. (2019) 25:1116–22. doi: 10.1038/s41591-019-0479-2
159. Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet. (2012) 44:133. doi: 10.1038/ng.1026
160. Arcila ME, Drilon A, Sylvester BE, Lovly CM, Borsu L, Reva B, et al. MAP2K1 (MEK1) mutations define a distinct subset of lung adenocarcinoma associated with smoking. Clin Cancer Res. (2015) 21:1935–43. doi: 10.1158/1078-0432.CCR-14-2124
161. Bansal A, Ramirez RD, Minna JD. Mutation analysis of the coding sequences of MEK-1 and MEK-2 genes in human lung cancer cell lines. Oncogene. (1997) 14:1231. doi: 10.1038/sj.onc.1200947
162. Zheng X, Wu Y, Zhu L, Chen Q, Zhou Y, Yan H, et al. Angiotensin II promotes differentiation of mouse embryonic stem cells to smooth muscle cells through PI3-kinase signaling pathway and NF-κB. Differentiation. (2013) 85:41–54. doi: 10.3390/ijms20082026
163. Haston S, Pozzi S, Carreno G, Manshaei S, Panousopoulos L, Gonzalez-Meljem JM, et al. MAPK pathway control of stem cell proliferation and differentiation in the embryonic pituitary provides insights into the pathogenesis of papillary craniopharyngioma. Development. (2017) 144:2141–52. doi: 10.1242/dev.150490
164. Jacobs AH, Walton RG. The incidence of birthmarks in the neonate. Pediatrics. (1976) 58:218–22.
165. Nguyen V, Hochman M, Mihm MC Jr, Nelson JS, Tan W. The pathogenesis of port wine stain and sturge weber syndrome: complex interactions between genetic alterations and aberrant MAPK and PI3K activation. Int J Mol Sci. (2019) 20:2243. doi: 10.3390/ijms20092243
166. Eerola I, Boon LM, Mulliken JB, Burrows PE, Dompmartin A, Watanabe S, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. (2003) 73:1240–9. doi: 10.1086/379793
167. Jeon H, Bernstein LJ, Belkin DA, Ghalili S, Geronemus RG. Pulsed dye laser treatment of port-wine stains in infancy without the need for general anesthesia. JAMA Dermatol. (2019) 155:435–41. doi: 10.1001/jamadermatol.2018.5249
168. Gao L, Phan S, Nadora DM, Chernova M, Sun V, Preciado SM, et al. Topical rapamycin systematically suppresses the early stages of pulsed dye laser-induced angiogenesis pathways. Lasers Surg Med. (2014) 46:679–88. doi: 10.1002/lsm.22296
169. Minkis K, Geronemus RG, Hale EK. Port wine stain progression: a potential consequence of delayed and inadequate treatment? Lasers Surg Med. (2009) 41:423–6. doi: 10.1002/lsm.20788
170. Tallman B, Tan OT, Trainor S, Morelli JG, Weston WL, Piepenbrink J, et al. Location of port-wine stains and the likelihood of ophthalmic and/or central nervous system complications. Pediatrics. (1991) 87:323.
171. Rydh M, Malm M, Jernbeck J, Dalsgaard CJ. Ectatic blood vessels in port-wine stains lack innervation: possible role in pathogenesis. Plast Reconstr Surg. (1991) 87:419–22. doi: 10.1097/00006534-199103000-00003
172. Smoller BR, Rosen S. Port-wine stains. A disease of altered neural modulation of blood vessels? Arch Dermatol. (1986) 122:177–9. doi: 10.1001/archderm.1986.01660140067019
173. Rosen S, Smoller B. Pathogenesis of port wine stains. A new hypothesis. Med Hypotheses. (1987) 22:365–8. doi: 10.1016/0306-9877(87)90031-4
174. Lian CG, Sholl LM, Zakka LR, O TM, Liu C, Xu S, et al. Novel genetic mutations in a sporadic port-wine stain. JAMA Dermatol. (2014) 150:1336–40. doi: 10.1001/jamadermatol.2014.1244
175. Frigerio A, Wright K, Wooderchak-Donahue W, Tan OT, Margraf R, Stevenson DA, et al. Genetic variants associated with port-wine stains. PLoS ONE. (2015) 10:e0133158. doi: 10.1371/journal.pone.0133158
176. Riverso M, Montagnani V, Stecca B. KLF4 is regulated by RAS/RAF/MEK/ERK signaling through E2F1 and promotes melanoma cell growth. Oncogene. (2017) 36:3322–33. doi: 10.1038/onc.2016.481
177. Tsai WB, Aiba I, Long Y, Lin HK, Feun L, Savaraj N, et al. Activation of Ras/PI3K/ERK pathway induces c-Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deiminase resistance in melanoma cells. Cancer Res. (2012) 72:2622–33. doi: 10.1158/0008-5472.CAN-11-3605
178. Yi F, Pereira L, Hoffman JA, Shy BR, Yuen CM, Liu DR, et al. Opposing effects of Tcf3 and Tcf1 control Wnt stimulation of embryonic stem cell self-renewal. Nat Cell Biol. (2011) 13:762–70. doi: 10.1038/ncb2283
179. Vural E, Ramakrishnan J, Cetin N, Buckmiller L, Suen JY, Fan CY. The expression of vascular endothelial growth factor and its receptors in port-wine stains. Otolaryngol Head Neck Surg. (2008) 139:560–4. doi: 10.1016/j.otohns.2008.07.015
180. Hershkovitz D, Bercovich D, Sprecher E, Lapidot M. RASA1 mutations may cause hereditary capillary malformations without arteriovenous malformations. Br J Dermatol. (2008) 158:1035–40. doi: 10.1111/j.1365-2133.2008.08493.x
181. Cai R, Liu F, Hua C, Yu Z, Ramien M, Malic C, et al. A novel RASA1 mutation causing capillary malformation-arteriovenous malformation (CM-AVM): the first genetic clinical report in East Asia. Hereditas. (2018) 155:24. doi: 10.1186/s41065-018-0062-8
182. di Blasio L, Puliafito A, Gagliardi PA, Comunanza V, Somale D, Chiaverina G, et al. PI3K/mTOR inhibition promotes the regression of experimental vascular malformations driven by PIK3CA-activating mutations. Cell Death Dis. (2018) 9:45. doi: 10.1038/s41419-017-0064-x
183. Tan W, Wang J, Zhou F, Gao L, Yin R, Liu H, et al. Coexistence of Eph receptor B1 and ephrin B2 in port-wine stain endothelial progenitor cells contributes to clinicopathological vasculature dilatation. Br J Dermatol. (2017) 177:1601–11. doi: 10.1111/bjd.15716
184. Tan W, Nadora DM, Gao L, Wang G, Mihm MC Jr, Nelson JS. The somatic GNAQ mutation (R183Q) is primarily located within the blood vessels of port wine stains. J Am Acad Dermatol. (2016) 74:380–3. doi: 10.1016/j.jaad.2015.09.063
185. Lee S, Kang KW, Kim J, Lee B, Jung JH, Park Y, et al. CXCR2 ligands and mTOR activation enhance reprogramming of human somatic cells to pluripotent stem cells. Stem Cells Dev. (2019) 29:119–32. doi: 10.1089/scd.2019.0188
186. Lipton JO, Sahin M. The neurology of mTOR. Neuron. (2014) 84:275–91. doi: 10.1016/j.neuron.2014.09.034
187. Jewell JL, Guan K-L. Nutrient signaling to mTOR and cell growth. Trends Biochem Sci. (2013) 38:233–42. doi: 10.1016/j.tibs.2013.01.004
188. Kurek KC, Luks VL, Ayturk UM, Alomari AI, Fishman SJ, Spencer SA, et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet. (2012) 90:1108–15. doi: 10.1016/j.ajhg.2012.05.006
189. Revencu N, Boon LM, Mulliken JB, Enjolras O, Cordisco MR, Burrows PE, et al. Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Human Mutat. (2008) 29:959–65. doi: 10.1002/humu.20746
190. Osborn AJ, Dickie P, Neilson DE, Glaser K, Lynch KA, Gupta A, et al. Activating PIK3CA alleles and lymphangiogenic phenotype of lymphatic endothelial cells isolated from lymphatic malformations. Hum Mol Genet. (2015) 24:926–38. doi: 10.1093/hmg/ddu505
191. Vlahovic AM, Vlahovic NS, Haxhija EQ. Sirolimus for the treatment of a massive capillary-lymphatico-venous malformation: a case report. Pediatrics. (2015) 136:e513–6. doi: 10.1542/peds.2014-3469
192. Schroeder U, Lauten M, Stichtenoth G, Gebhard MP, Buchholz M, Kaiser MM. Laryngomalacia and complicated, life-threatening mTOR-positive Kaposiform hemangioendothelioma cured by Supraglottoplasty and sirolimus. Klin Padiatr. (2014) 226:362–8. doi: 10.1055/s-0034-1372587
193. Freixo C, Ferreira V, Martins J, Almeida R, Caldeira D, Rosa M, et al. Efficacy and safety of sirolimus in the treatment of vascular anomalies: a systematic review. J Vasc Surg. (2020) 71:318–27. doi: 10.1016/j.jvs.2019.06.217
194. Nee R, Hurst FP, Dharnidharka VR, Jindal RM, Agodoa LY, Abbott KC. Racial variation in the development of posttransplant lymphoproliferative disorders after renal transplantation. Transplantation. (2011) 92:190–5. doi: 10.1097/TP.0b013e3182200e8a
195. Aarts M, Georgilis A, Beniazza M, Beolchi P, Banito A, Carroll T, et al. Coupling shRNA screens with single-cell RNA-seq identifies a dual role for mTOR in reprogramming-induced senescence. Genes Dev. (2017) 31:2085–98. doi: 10.1101/gad.297796.117
196. Bulut-Karslioglu A, Biechele S, Jin H, Macrae TA, Hejna M, Gertsenstein M, et al. Inhibition of mTOR induces a paused pluripotent state. Nature. (2016) 540:119–23. doi: 10.1038/nature20578
197. Damme A, Seront E, Dekeuleneer V, Boon L, Vikkula M. New and emerging targeted therapies for vascular malformations. Am J Clin Dermatol. (2020) 21:657–68. doi: 10.1007/s40257-020-00528-w
198. Venot Q, Blanc T, Rabia SH, Berteloot L, Ladraa S, Duong JP, et al. Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature. (2018) 558:540–6. doi: 10.1038/s41586-018-0217-9
199. Keppler-Noreuil KM, Parker VE, Darling TN, Martinez-Agosto JA. Somatic overgrowth disorders of the PI3K/AKT/mTOR pathway & therapeutic strategies. Am J Med Genet C Semin Med Genet. (2016) 172:402–21. doi: 10.1002/ajmg.c.31531
200. Atkinson V, Sandhu S, Hospers G, Long GV, Aglietta M, Ferrucci PF, et al. Dabrafenib plus trametinib is effective in the treatment of BRAF V600-mutated metastatic melanoma patients: analysis of patients from the dabrafenib plus trametinib Named Patient Program (DESCRIBE II). Melanoma Res. (2019) 30:261–7 doi: 10.1097/CMR.0000000000000654
201. Subbiah V, Meyer C, Zinner R, Meric-Bernstam F, Zahurak ML, O'Connor A, et al. Phase Ib/II study of the safety and efficacy of combination therapy with multikinase VEGF inhibitor pazopanib and MEK inhibitor trametinib in advanced soft tissue sarcoma. Clin Cancer Res. (2017) 23:4027–34. doi: 10.1158/1078-0432.CCR-17-0272
202. Peng X, Zhou J, Li B, Zhang T, Zuo Y, Gu X. Notch1 and PI3K/Akt signaling blockers DAPT and LY294002 coordinately inhibit metastasis of gastric cancer through mutual enhancement. Cancer Chemother Pharmacol. (2019) 85:309–20. doi: 10.1007/s00280-019-03990-4
203. Keppler-Noreuil KM, Sapp JC, Lindhurst MJ, Darling TN, Burton-Akright J, Bagheri M, et al. Pharmacodynamic study of miransertib in individuals with proteus syndrome. Am J Hum Genet. (2019) 104:484–91. doi: 10.1016/j.ajhg.2019.01.015
204. Lindhurst MJ, Yourick MR, Yu Y, Savage RE, Ferrari D, Biesecker LG. Repression of AKT signaling by ARQ 092 in cells and tissues from patients with Proteus syndrome. Sci Rep. (2015) 5:17162. doi: 10.1038/srep17162
205. Sakata K, Kato S, Fox JC, Shigemori M, Morimatsu M. Autocrine signaling through Ras regulates cell survival activity in human glioma cells: potential cross-talk between Ras and the phosphatidylinositol 3-kinase-Akt pathway. J Neuropathol Exp Neurol. (2002) 61:975–83. doi: 10.1093/jnen/61.11.975
206. Yoon YK, Kim HP, Han SW, Oh DY, Im SA, Bang YJ, et al. KRAS mutant lung cancer cells are differentially responsive to MEK inhibitor due to AKT or STAT3 activation: implication for combinatorial approach. Mol Carcinog. (2010) 49:353–62. doi: 10.1002/mc.20607
207. Singer G, Oldt R III, Cohen Y, Wang BG, Sidransky D, Kurman RJ, et al. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J Natl Cancer Inst. (2003) 95:484–6. doi: 10.1093/jnci/95.6.484
208. Meng J, Peng H, Dai B, Guo W, Wang L, Ji L, et al. High level of AKT activity is associated with resistance to MEK inhibitor AZD6244 (ARRY-142886). Cancer Biol Ther. (2009) 8:2073–80. doi: 10.4161/cbt.8.21.9844
209. Wee S, Jagani Z, Xiang KX, Loo A, Dorsch M, Yao YM, et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. (2009) 69:4286–93. doi: 10.1158/0008-5472.CAN-08-4765
210. Nelson KM, Gallagher PC. Biosimilars lining up to compete with Herceptin–opportunity knocks. Expert Opin Ther Pat. (2014) 24:1149–53. doi: 10.1517/13543776.2014.964683
Keywords: vascular anomalies, vascular tumor, vascular malformation, embryonic stem cells, induced pluripotent stem cells, renin-angiotensin system, gene mutations, stemness-associated markers
Citation: Kilmister EJ, Hansen L, Davis PF, Hall SRR and Tan ST (2021) Cell Populations Expressing Stemness-Associated Markers in Vascular Anomalies. Front. Surg. 7:610758. doi: 10.3389/fsurg.2020.610758
Received: 27 September 2020; Accepted: 31 December 2020;
Published: 09 February 2021.
Edited by:
Ramin Shayan, O'Brien Institute, AustraliaReviewed by:
Fatih Zor, Wake Forest School of Medicine, United StatesJune Wu, Columbia University Irving Medical Center, United States
Copyright © 2021 Kilmister, Hansen, Davis, Hall and Tan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Swee T. Tan, c3dlZS50YW5AZ21yaS5vcmcubno=