Medicines Research Centre, GlaxoSmithKline S.p.A., Verona, Italy
The basolateral nucleus (BLA) of the amygdala contributes to the consolidation of memories for emotional or stressful events. The nucleus contains a high density of CRF1 receptors that are activated by corticotropin-releasing factor (CRF). Modulation of the excitability of neurons in the BLA by CRF may regulate the immediate response to stressful events and the formation of associated memories. In the present study, CRF was found to increase the amplitude of field potentials recorded in the BLA following excitatory afferent stimulation, in vitro. The increase was mediated by CRF1 receptors, since it could be blocked by the selective, non-peptide antagonists, NBI30775 and NBI35583, but not by the CRF2-selective antagonist, astressin 2B. Furthermore, the CRF2-selective agonist, urocortin II had no effect on field potential amplitude. The increase induced by CRF was long-lasting, could not be reversed by subsequent administration of NBI35583, and required the activation of protein kinase C. This effect of CRF in the BLA may be important for increasing the salience of aversive stimuli under stressful conditions, and for enhancing the consolidation of associated memories. The results provide further justification for studying the efficacy of selective antagonists of the CRF1 receptor to reduce memory formation linked to emotional or traumatic events, and suggest that these compounds might be useful as prophylactic treatments for stress-related illnesses such as post-traumatic stress disorder.
Corticotropin-releasing factor (CRF) is a 41-amino acid peptide released from neurons within the CNS that modulates neural activity through activation of CRF1 and CRF2 receptors present in discrete brain areas (Holmes et al., 2003
). The peptide is known to contribute to the behavioural and autonomic response to stress (Croiset et al., 2000
; Koob and Henrichs, 1999
; Liu et al., 2004
; Makino et al., 2002
; Rainnie et al., 2004
), in part through modulation of amygdala function. The amygdala plays a key role in the central processing of stressful or emotionally charged stimuli (Davis, 1992
; Phelps and LeDoux, 2005
), and in particular, the basolateral complex of the amygdala (BLA) has been shown to mediate emotional arousal (Roozendaal et al., 2002
) and the consolidation of emotional memory (McGaugh, 2004
; Paré, 2003
). The BLA receives excitatory input from diverse regions of the CNS, including the thalamus (Blair et al., 2003
; Muller et al., 1997
; Turner and Herkenham, 1991
), lateral entorhinal cortex (Mcdonald and Mascagni, 1997
), hippocampus, frontal cortex (Pitkanen et al., 1997
), occipital cortex (Mcdonald and Mascagni, 1996
), and temporal cortex (Mascagni et al., 1993
), so is well placed to integrate sensory input with information already in memory. Repeated activation of the BLA, or repetitive administration of exogenous CRF receptor agonists results in altered sensitivity of BLA neurons to further stimulation, and the development of a chronic anxiety-like state (Sajdyk and Gehlert, 2000
). Activation of CRF receptors in the BLA has been shown to increase the excitability of projection neurons through the inhibition of a calcium-dependent potassium channel (Rainnie et al., 1992
); an effect that was most likely mediated by CRF1 receptors given their high density on these neurons (Chalmers et al., 1995
; Chen et al., 2000
; Van Pett et al., 2000
). This effect of CRF on projection neurons in the BLA contrasted with the peptide’s action in the central amygdala, where it reduced the excitability of recorded cells. CRF has long been known to have excitatory effects on neurons in the CNS (Siggins et al., 1985
), with notable effect on the firing of dopaminergic neurons in the ventral tegmental area (Korotkova et al., 2006
). Of particular relevance to the current study, CRF has been shown to induce a long-lasting increase in the excitability of neurons in the dentate gyrus of the hippocampus, in vivo (Wang et al., 2000
), an effect that was dependent on protein synthesis and suggests that the peptide may have profound effects on long-term neural function and plasticity.
Given the recent availability of selective CRF1 receptor ligands, in particular the non-peptide CRF1 receptor antagonists, NBI35583 (Guo et al., 2005
) and NBI30775 (Chen and Grigoriadis, 2005
), we have now investigated the pharmacology of CRF modulation of neural activity in the BLA following afferent stimulation, in vitro. These studies may help to understand how selective CRF1 receptor antagonists might be of benefit in the treatment of stress-related disorders.
All work was carried out in compliance with the Italian national legislation (DL 116/92) and European Directive 86/609.
Subjects and Brain Slice Preparation
In vitro brain slices were obtained from male Sprague-Dawley rats (80–100 g; 3–4 weeks of age). Rats were anaesthetised with ether or isoflurane and decapitated. The brain was removed and cooled rapidly in a modified artificial cerebrospinal fluid (ACSF) solution (0–6°C) bubbled continuously with 95% O2 and 5% CO2 to maintain pH (7.35–7.45). Coronal slices (400 μm) were cut using a Vibroslice (Campden Instruments) and placed in oxygenated ACSF at 21°C. Following incubation for at least 1 h, a slice was transferred to the recording chamber, where it was fully submerged and continuously perfused with oxygenated ACSF maintained at 33 ± 1°C. ACSF was made up as follows (in mM): 118 NaCl, 3 KCl, 1 MgCl2, 1 NaH2PO4, 2.5 CaCl, 11 D-glucose, and 25 NaHCO3.
Electrophysiological Recording
The basolateral complex of the amygdala, composed of the lateral and basolateral nuclei was evident as the region defined on its lateral border by the external capsule and its medial border by the longitudinal association bundle. Extracellular field potentials were evoked by stimulation (0.05 Hz, 0.1 ms duration) delivered to the lateral nucleus with a bipolar electrode and recorded with a glass micropipette containing ACSF (resistance 3–8 MΩ). Unless otherwise stated, the stimulus amplitude was set to produce a response that was ∼60–80% of the maximum field potential amplitude at baseline. Field potentials were amplified (Axoprobe-1A, Axon Instruments), bandpass filtered between 3 Hz and 3 kHz and digitised using a National Instruments interface running custom Labview software. Data are expressed as mean ± SEM; n = number of slices (in some cases where multiple slices were derived from the same rat, the number of rats is also indicated in the figure legend). The effect of treatment on stimulus-response and paired-pulse results were analysed using a two-way ANOVA. All other data were analysed using paired or unpaired Student’s t-tests, or ANOVA followed by a post-hoc test, as indicated.
Drugs
Drugs were dissolved in ACSF and introduced to the recording chamber at a flow rate of 2 ml/min. Human/rat CRF; the AMPA glutamate receptor antagonist, 6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX), the protein kinase A (PKA) inhibitor, N-[2-(p-Bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide dihydrochloride (H-89); the protein kinase C (PKC) inhibitor, 3-(N-[Dimethylamino]propyl-3-indolyl)-4-(3-indolyl)maleimide (BIS-I) were all obtained from Sigma-Aldrich (St. Louis, MO, USA). The selective CRF1 receptor antagonists NB30775, NBI 35583; the selective CRF2 receptor agonist, urocortin II, and antagonist, astressin 2B, were gifts from Neurocrine Inc. and the Salk Institute (USA).
Evoked Field Responses in the Basolateral Amygdala
Stimulation of the lateral amygdala evoked a characteristic field potential in the BLA. The field potentials were composed of a short latency (1 ms) negative-going potential, which was unaffected by CNQX (Figure 1
A), but which was inhibited by TTX (100 nM, Figure 1
B). This was followed by a second, longer latency (2–3 ms) negative going potential that could be inhibited by application of the AMPA/kainate receptor antagonist, CNQX (10 μM, n = 6, Figure 1
A), as well as by TTX (Figure 1
B).
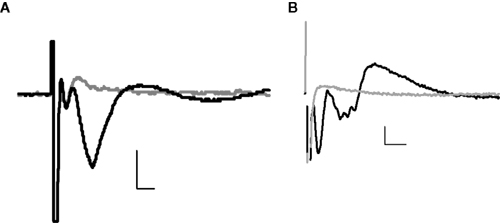
Figure 1. Example field potentials evoked in the BLA by stimulation of the lateral amygdala. (A) ±CNQX (10 μM); (B) ±TTX (100 nM). In each case, the black trace = baseline, grey trace = after drug; horizontal scale bars 2 ms, vertical 0.2 mV.
Effect of CRF
Application of CRF (0.1 μM) to the slice significantly enhanced the amplitude of the longer latency synaptic response (fPSP), but not the early response (Figure 2
).
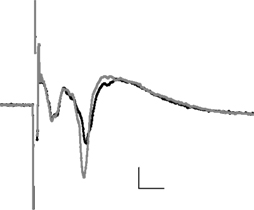
Figure 2. The averaged field potential (n = 20 responses) before and after application of CRF (0. 1 μM). The black trace = baseline, grey trace = after drug; horizontal scale bars 2 ms, vertical 0.2 mV.
Furthermore, the enhancement was sustained beyond the washout of the peptide (Figure 3
A). In order to define more clearly the component of the field response that was affected by CRF, a series of experiments was conducted in which TTX was applied at the end of the experiment and the residual non-biological component digitally subtracted from the responses (Figure 3
B). In this series, CRF (0.1 μM) again produced a significant increase in the amplitude of the fPSP component (44 ± 11%; p < 0.01, paired t-test, n = 7 slices, Figure 3
C).
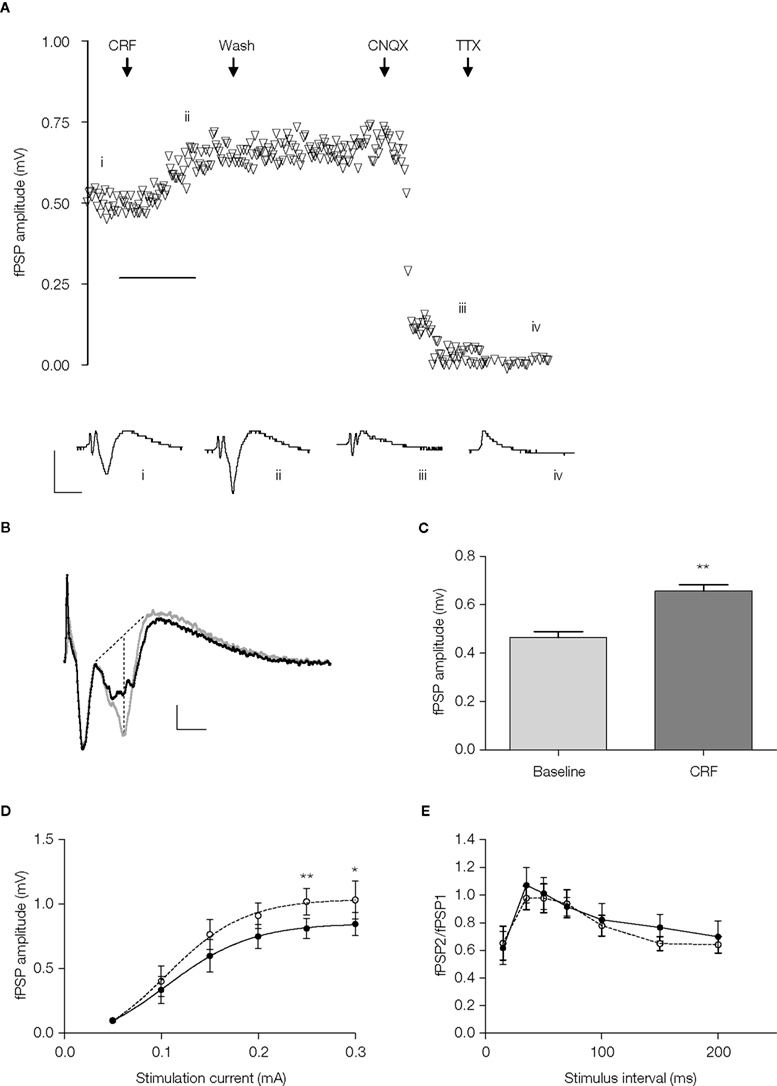
Figure 3. (A) Timecourse of fPSP amplitude and example traces (i) at baseline, (ii) following application of CRF (0.1 μM), (iii) following washout of the CRF and application of CNQX (10 μM), and (iv) following application of TTX (100 nM). Each symbol represents the fPSP amplitude for a single response. Horizontal scale bar 10 min. (B) Example of evoked field potentials before (black trace) and after (grey trace) the application of CRF (0.1 μM); the non-biological component that was not sensitive to TTX applied at the end of the experiment has been digitally removed. Dotted lines indicate the method for determining the fPSP amplitude. (C) The mean fPSP amplitude evoked by currents at 60–80% of maximum at baseline and after 15 min incubation with CRF (0.1 μM). Amplitude was determined from responses similar to that shown in (B). **p < 0.01, paired t-test, n = 7 slices, 5 rats. (D) Stimulus current-response relationship for evoked fPSPs in the absence (•) and presence (ο) of CRF (0.1 μM), statistical analysis was carried using a two-way repeated measures ANOVA (see text) followed by a post-hoc Bonferroni test (*p < 0.05, **p < 0.01, n = 7 slices, 5 rats). (E) Paired-pulse relationship for evoked fPSPs in the absence (•) and presence (ο) or CRF (0.1 μM), statistical analysis was carried out using a two-way repeated-measures ANOVA (see text; n = 8 slices, 5 rats).
The increase in fPSP produced by CRF was apparent over a range of stimulus intensities (repeated measures two-way ANOVA: significant effect of current, F(5,36) = 13, p < 0.0001; significant effect of CRF, F(1,36) = 29, p < 0.0001; no interaction between current and CRF, F(5,36) = 1.8, p = 0.14; n = 7 slices); although a post-hoc Bonferroni test showed that a significant increase occurred only at the highest stimulus intensities (Figure 3
D). In order to determine whether the effect of CRF was likely to be due to a pre- or post-synaptic action, responses to pairs of stimuli separated by a short interval were examined. CRF (0.1 μM) had no effect on the paired-pulse profile obtained with inter-pulse intervals of 20–200 ms (repeated measures two-way ANOVA: significant effect of interval, F(6,49) = 3.0, p < 0.05; no effect of CRF, F(1,49) = 1.96, p = 0.17, no interaction between interval and CRF, F(6,49) = 0.51, p = 0.79; n = 8 slices, Figure 3
E).
Determination of the Receptor Mediating the Effect of CRF
The CRF1 selective antagonist, NBI35583 significantly reduced the enhancement of fPSP amplitude induced by 0.1 μM CRF in a concentration-dependent manner, with a pIC50 of 6.7 ± 0.2 (data fitted to a sigmoidal curve of the form Y = min + {max − min [1 + 10^(LogIC50-X)]}; n = 6–12 slices per concentration; Figure 4
A). A second CRF1 selective antagonist, NBI30775 tested at a single concentration of 1 μM also significantly reduced the response to the peptide (CRF alone: 33 ± 6% increase, CRF + NBI 30775: 11 ± 4% increase, p < 0.05 unpaired t-test, n = 6–9 slices; Figure 4
B).
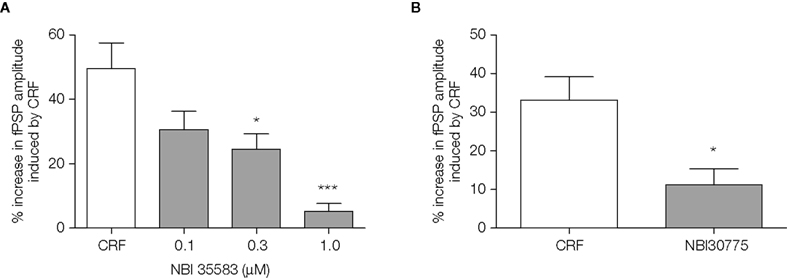
Figure 4. (A) The CRF1 receptor antagonists NBI 35583 (0.1–1.0 μM, 20 min before 0.1 μM CRF); data shown are the mean ± SEM from 12 control slices (11 rats) and 6–9 slices (3–6 rats) pretreated with antagonist at the concentrations indicated. *p < 0.05, ***p < 0.001, one-way ANOVA followed by Bonferroni t-test. (B) NBI 30775 (1 μM, 20 min before CRF) blocked the increase in fPSP amplitude induced by 0.1 μM CRF; data shown are the mean ± SEM from 9 control slices (7 rats) and 6 slices (6 rats) pretreated with the antagonist. *p < 0.05, unpaired t-test.
In further experiments, a selective CRF2 receptor agonist, urocortin II (0.1 μM; functional cAMP accumulation driven by mouse recombinant CRF2, EC50 = 0.14 nM, and by human recombinant CRF1, EC50 > 100 nM; Reyes et al., 2001) had no effect on fPSP amplitude alone (2 ± 4% increase, p > 0.05, paired t-test, n = 6 slices), while on the same slices, subsequent application of CRF (0.1 μM) enhanced fPSP amplitude (Figure 5
A). Finally, the CRF2 receptor-selective antagonist, astressin 2B (0.1 μM; binding affinity for human recombinant CRF2 receptors, IC50 = 1.3 nM, and hCRF1 IC50 > 500 nM; Rivier et al., 2002), applied 20 min before CRF, failed to block the effect of the latter on fPSP amplitude (CRF alone: 25 ± 4% increase, n = 3; CRF + astressin 2B: 29 ± 7% increase, n = 8 slices, p > 0.05 unpaired t-test; Figure 5
B).
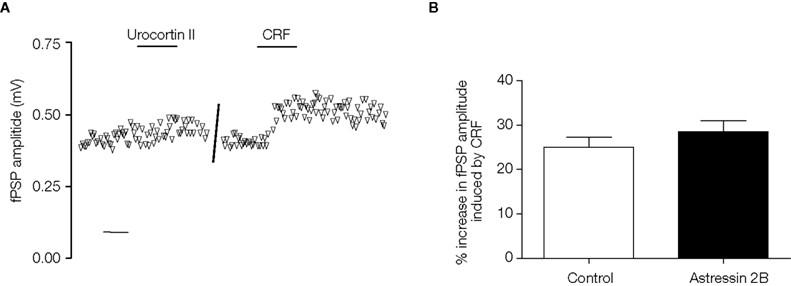
Figure 5. (A) Example of the timecourse of fPSP amplitude before and after application of the CRF2 receptor agonist, urocortin II (0.1 μM), and subsequently CRF (0.1 μM) on the same slice. Each symbol represents the fPSP amplitude for a single response. Scale bar 10 min. (B) The increase in fPSP amplitude induced by CRF (0.1 μM, n = 3 slices from 3 rats) is unaffected by the CRF2 receptor antagonist, astressin 2B (0.1 μM, applied 20 min before 0.1 μM CRF, n = 8 slices from 4 rats). Data shown are the mean ± SEM.
Determination of the Downstream Consequence of CRF Receptor Activation
The enhancement of evoked fPSPs by CRF (0.1 μM) above baseline remained significant 30 min after wash-out of the peptide (15 min treatment with CRF: 55 ± 12% increase over baseline, p < 0.001, n = 7 slices; 30 min after washout: 41 ± 11% increase over baseline, p < 0.01, n = 7 slices; one-way repeated measures ANOVA followed by Bonferroni test; Figure 6
). Furthermore, from this same analysis, the increase 30 min after washout was not significantly different to that at the end of the 15-min incubation with CRF. In a separate series of experiments, the antagonist, NBI35583 (1 μM) was added to the superfusion during the washout of CRF; however, again the fPSP remained significantly elevated after 30 min of washout (15 min treatment with CRF: 50 ± 12%, p < 0.01, n = 6 slices; 30 min of washout in the presence of 1 μM NBI 35583: 37 ± 7%, p < 0.05, n = 6 slices; one-way repeated measures ANOVA followed by Bonferroni test; Figure 6
). From this analysis, the increase 30 min after washout in the presence of NBI35583 was not significantly different to that at the end of the 15-min incubation with CRF.
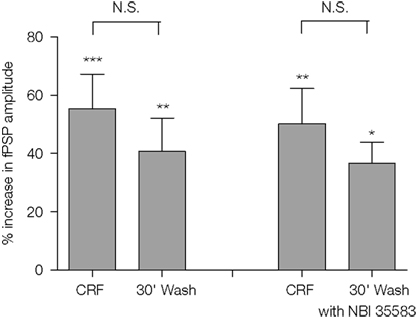
Figure 6. Comparison of the increase in fPSP amplitude following application of CRF (0.1 μM) and 30 min after wash-out of the peptide in the absence (n = 7 slices from 5 rats) or presence of NBI35583 (1 μM; n = 6 slices from 5 rats). Data shown are the mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001, N.S. not significant, one-way repeated measures ANOVA followed by Bonferroni multiple comparison.
In order to investigate the downstream intracellular pathways activated by CRF, the effects of the peptide were examined on slices pre-treated for 1 h with the selective PKA inhibitor H-89 or the selective PKC inhibitor BIS-I. As a control, CRF (0.1 μM) was applied to separate slices that had been obtained from the same rat, but which had not been exposed to the inhibitors. The enhancement of the fPSP by CRF in the presence of H-89 (10 μM) was similar to that in control slices (control: 33 ± 10% vs. H-89-treated: 28 ± 6%, n = 7 slices per group; Figure 6
); furthermore, H-89 did not affect the longevity of the increase 30 min after washout of CRF (control: 31 ± 11%; H-89: 29 ± 6%, n = 7 slices; Figure 7
). However, in the presence of BIS I (1.2 μM), the enhancement by CRF was significantly reduced both during application of the peptide (control: 35 ± 5%, BIS-I: 8 ± 3%, p < 0.05, one-way ANOVA followed by a Bonferroni test, n = 7 slices per group; Figure 6
) and 30 min after washout (control: 47 ± 7%; BIS-I: 9 ± 3%, p < 0.001, one-way ANOVA followed by a Bonferroni test, n = 7 slices; Figure 7
).
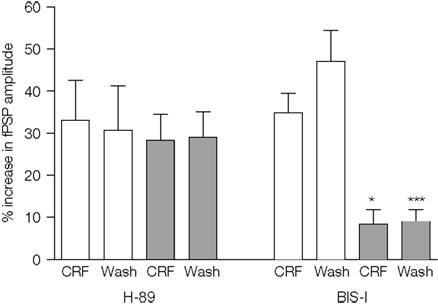
Figure 7. The effect of CRF (0.1 μM) on the amplitude of fPSPs evoked in control slices or slices that had been incubated with the protein kinase inhibitors, H-89 (10 μM; 7 slices from 5 rats per group) or BIS-I (1.2 μM; 7 slices from 4 rats per group). Control slices were obtained from the same rats as slices incubated with one or other of the inhibitors. Data shown are the mean ± SEM. *p < 0.05, ***p < 0.001, one-way ANOVA followed by Bonferroni comparison vs. the respective control.
This study examined the effects of rat/human CRF peptide on field potentials recorded in the BLA that were evoked by electrical stimulation within the lateral amygdala. The principal findings were: (1) CRF increased the amplitude of the presumed post-synaptic component of the field potential via activation of CRF1 receptors. (2) The increase due to CRF was maintained following washout of the peptide. (3) The increase was dependent on activation of PKC, but not PKA. These results suggest that activation of CRF1 receptors on neurons in the BLA leads to a long-lasting increase in their response to afferent excitatory transmission. These findings are discussed below.
Extracellular field potential recording was used to evaluate drug effects on the response of BLA neurons to afferent excitatory stimulation. The field potential responses observed in this study have been described by others (Aroniadou-Anderjaska et al., 2001
; Chapman and Bellavance, 1992
; Isoardi et al., 2004
) and are suggested to reflect two major physiological components. The first, a short latency negative-going potential, was unaffected by CNQX or CRF, but was abolished by TTX, consistent with the suggestion that it reflects the pre-synaptic fibre volley (Isoardi et al., 2004
). The second, longer latency, negative-going component was inhibited by the AMPA/kainate glutamate receptor antagonist, CNQX, and is presumed to reflect the excitatory post-synaptic response of neurons in the BLA (Isoardi et al., 2004
). This component was enhanced by CRF. Across several series of experiments described in the present study, the mean increase in the amplitude of the fPSP following application of CRF ranged from 25% (e.g. Figure 4
B, n = 8) to nearly 60% (e.g. Figure 5
, n = 7). This variation was also reflected within experiments such that the increase in fPSP amplitude due to CRF observed from individual BLA slices ranged from just 14–100%. In a subsequent study (manuscript in preparation), we found that the increase in fPSP following application of CRF was significantly reduced in brain slices obtained from rats previously exposed to behavioural stress, and furthermore was correlated with the trait anxiety level of the rat (determined from the animal’s behaviour on an elevated plus maze). Consequently, we suggest that different levels of stress in the groups of rats used in the present study and individual differences in trait anxiety might contribute to the range of increases observed with CRF in the present study. However, given the relatively small fPSP observed in the BLA in vitro, compared to more laminar structures such as the hippocampus, it is possible that methodological variations also contribute to the apparent variation in CRF effect across experiments.
The use of selective CRF1 or CRF2 receptor ligands confirmed that the enhancement of evoked fPSPs by CRF was mediated by CRF1, and not CRF2 receptors since the effects could be blocked by the non-peptide CRF1-selective antagonists, NBI30775 and NBI35583, but not by the selective CRF2 receptor peptide antagonist, astressin 2B. Furthermore, the peptide agonist, urocortin II had no effect on fPSP amplitude. The pIC50 determined for NBI35583 in the present study is similar to that determined from the inhibition of CRF-stimulated ACTH production by primary rat pituitary cells (pIC50 6.8 ± 0.3, n = 6; D. Grigoriadis, personal communication). These results are also consistent with the high density of CRF1 receptor mRNA and protein found in the BLA of the rat (Chalmers et al., 1995
; Van Pett et al., 2000
) and mouse (Chen et al., 2000
). The enhancement of fPSPs by CRF could arise through increased glutamate release or a change in the excitability of post-synaptic neurons. The absence of effect of CRF on the paired-pulse relationship is indicative of a post-synaptic effect, since enhanced glutamate release would be expected to affect the ratio of the amplitude of the second fPSP relative to the first in paired-pulse experiments (Sanchez-Prieto et al., 1996
). A drawback of field potential recording used in the present study is the complexity of the field response in the amygdala and the relatively small range of stimulation currents over which the response can be reliably measured. Consequently, we cannot rule out the possibility that the experiments were not sufficiently sensitive to detect modulation of the paired-pulse profile by CRF. It remains possible that CRF1 receptors are expressed on the terminals of glutamatergic afferents into the BLA, since CRF1 receptor mRNA is observed in the lateral amygdala and other structures afferent to the BLA, such as the hippocampus, entorhinal cortex, and thalamus (Van Pett et al., 2000
). Furthermore, CRF has been shown to enhance glutamate release in the rat locus coeruleus, in vivo (Singewald et al., 1996
). However, a study of the potentiating effect of CRF on afferent excitation in the lateral septum also showed that the peptide had no effect on paired-pulse facilitation, leading the authors to suggest a post-synaptic site of action (Liu et al., 2004
). This latter study investigated the effects of CRF on the response of individual neurons in the lateral septum, so is likely to have been more sensitive to effects on paired-pulse facilitation.
The enhancement of evoked fPSPs by CRF was maintained following washout of the peptide, and could not be reversed by application of the antagonist, NBI35583 during washout. A possible explanation for this observation might be slow kinetics of dissociation of the peptide from CRF receptors in the slice, such that application of the competitive antagonist was insufficient to displace CRF from the receptors during the 30-min washout. However, given the high concentration of NBI35583 used in these experiments, this explanation seems unlikely. Alternatively, CRF1 receptor activation may couple to a downstream effector that, once activated, is then independent of CRF receptor stimulation. A similarly prolonged effect of CRF in the BLA was reported by Rainnie et al. (1992)
, discussed further below. Whilst we have yet to identify the final effector mechanism responsible for the enhancement of fPSPs by CRF, it is likely to be downstream of PKC, since the effects of CRF could be prevented by prior application of the PKC inhibitor BIS-I, but not the PKA inhibitor H-89. Concentrations of the two inhibitors selective for PKC and PKA were chosen based on studies by Blank et al. (2003)
. CRF1 receptors have previously been reported to couple to both Gs and Gq G-proteins to activate PKA and PKC, respectively in the mouse hippocampus (Blank et al., 2003
; Eckart et al., 2002
). The modulation by CRF of serotonergic regulation of GABAergic transmission in prefrontal cortex has been shown to be mediated by CRF1 receptors coupled through PKC (Tan et al., 2004
). Furthermore, CRF-induced inhibition of adenylate cyclase is mediated by PKC in rat Leydig cells (Ulisse et al., 1990
).
Rainnie et al. (1992)
, using current clamp recording from neurons in the BLA, found that CRF (0.125 μM) reduced the slow after-hyperpolarisation (s-AHP) following action potentials evoked by current injection. The peptide also increased calcium spikes recorded in the presence of TTX and TEA, a voltage-gated potassium channel blocker. They did not determine whether these effects were mediated through activation of PKC, although they comment that a possible mechanism for the enhancement of calcium spikes by CRF might be the enhancement of calcium channel conductance, an effect that has been shown to be mediated by both PKC and PKA in mouse anterior pituitary tumour cells (Reisine and Guild, 1987
), and which was subsequently confirmed to occur also in the central nucleus of the amygdala (Yu and Shinnick-Gallagher, 1998
). The results of the study by Rainnie et al. (1992)
demonstrate a clear post-synaptic effect of CRF on neurons in the BLA at a concentration similar to that used in the present study; furthermore, the observed reduction in s-AHP and enhanced calcium spikes are consistent with the enhanced field potential response observed in the present study. Further studies combining intracellular recording from neurons in the BLA with afferent stimulation, similar to that conducted in the lateral septum by Liu et al. (2004)
, would be valuable to explore the mechanism underlying the enhancement of fPSPs in the present study.
The present results are reminiscent of the long-lasting increase in the excitability of neurons in the dentate gyrus of the hippocampus observed in vivo (Wang et al., 2000
). In that earlier study, the identity of the CRF receptor mediating the effects was not determined; however, the increase in evoked field responses was similar to that observed with electrically-induced long-term potentiation. The present results suggest that CRF-induced long-term plasticity may be a feature of several corticolimbic brain areas and thus may be an important regulator of learning and memory. Plasticity within the BLA is hypothesised to contribute to the development of anxiety-related disorders (Rainnie et al., 2004
; Shekhar et al., 2005
). Studies in vivo have shown that repeated stimulation of the BLA or local injection of a CRF1 agonist, urocortin, results in altered sensitivity of BLA neurons to further stimulation, the development of a chronic anxiety-like state (Sajdyk and Gehlert, 2000
), and the facilitation of inhibitory avoidance learning (Liang and Lee, 1988
). Antagonists of the CRF1 receptor have been shown to inhibit aversive memory consolidation (Roozendaal et al., 2002
), a process known to be dependent on BLA activity (McGaugh, 2004
; Paré, 2003
). For example, the CRF1 antagonist DMP696 has been shown to have no effect on the acquisition of contextual fear conditioning, butcould prevent the expression of conditioning (freezing) when the animals were tested 48 h later, suggesting reduced consolidation (Hubbard et al., 2007
). This has led to the hypothesis that the release of stress hormones, such as CRF, in the amygdala is a critical mechanism by which stressful or aversive memories are reinforced (Roozendaal et al., 2008
). The results of the present study provide further support for this hypothesis by providing evidence for a long-lasting enhancement of the responsivity of BLA neurons to afferent activation that is induced by CRF and mediated by CRF1 receptors. Thus, the acute release of CRF in the amygdala might contribute not only to increased anxiety in response to an aversive stimulus, but also a stronger consolidation of memory for that stimulus and its context. Confirmation in the present study that the effects of the CRF peptide in the BLA are mediated by CRF1 receptors contributes to the rationale to explore the therapeutic efficacy of CRF1 receptor antagonists in patients suffering from stress-related disorders. In particular, the results suggest that CRF1 receptor antagonists might be beneficial in the prophylactic treatment of illnesses such as post-traumatic stress disorder.
The authors are full time employees of Glaxosmithkline S.p.A.
We thank Dr Paul Chapman for practical advice regarding amygdala recordings and Dr Dimitri Grigoriadis at Neurocrine Biosciences Inc. for the provision of CRF1 receptor antagonists.
Blank, T., Nijholt, I., Grammatopoulus, D. K., Randeva, H. S., Hillhouse, E. W., and Spiess, J. (2003). Corticotropin-releasing factor receptors couple to multiple G-proteins to activate diverse intracellular signalling pathways in mouse hippocampus: role in neuronal excitability and associative learning. J. Neurosci. 23, 700–707.
Chen, Y., Brunson, K. L., Muller, M. B., Cariaga, W., and Baram, T. (2000). Immunocytochemical distribution of corticotropin-releasing hormone receptor type-1 (CRF1)-like immunoreactivity in the mouse brain: light microscopy analysis using an antibody directed against the C-terminus. J. Comp. Neurol. 420, 305–323.
Guo, Z., Tellew, J. E., Gross, R. S., Dyck, B., Grey, J., Haddach, M., Kiankarimi, M., Lanier, M., Li, B. F., Luo, Z., McCarthy, J. R., Moorjani, M., Saunders, J., Sullivan, R., Zhang, X., Zamani-Kord, S., Grigoriadis, D. E., Crowe, P. D., Chen, T., and Williams, J. P. (2005). Design and synthesis of tricyclic imidazo[4,5-b]pyridin-2-ones as corticotropin-releasing factor-1 antagonists. J. Med. Chem. 48, 5104–5107.
Reyes, T. M., Lewis, K., Perrin, M. H., Kunitake, K. S., Vaughan, J., Arias, C. A., Hogenesch, J. B., Gulyas, J., Rivier, J., Vale, W. W., and Sawchenko, P. E. (2001). Urocortin II: a member of the corticotropin-releasing factor (CRF) neuropeptide family that is selectively bound by type 2 CRF receptors. Proc. Natl. Acad. Sci. USA 98, 2843–2848.
Rivier, J., Gulyas, J., Kirby, D., Low, W., Perrin, M. H., Kunitake, K., DiGruccio, M., Vaughan, J., Reubi, J. C., Waser, B., Koerber, S. C., Martinez, V., Wang, L., Tache, Y., and Vale, W. (2002). Potent and long-acting corticotropin releasing factor (CRF) receptor 2 selective peptide competitive antagonists. J. Med. Chem. 45, 4737–4747.