1
Department of Psychology, Yale University, New Haven, CT, USA
2
Department of Psychology, University of Canterbury, Christchurch, New Zealand
3
Interdepartmental Neuroscience Program, Yale University, New Haven, CT, USA
In both invertebrate and vertebrate models of synaptic plasticity, signaling via the putative ”retrograde messenger” nitric oxide (NO) has been hypothesized to serve as a critical link between functional and structural alterations at pre- and postsynaptic sites. However, while in vitro models of synaptic plasticity have consistently implicated NO signaling in linking postsynaptic induction mechanisms with accompanying presynaptic changes, a convincing role of such ”retrograde signaling” in mammalian memory formation has remained elusive. Using auditory Pavlovian fear conditioning, we show that synaptic plasticity and NO signaling in the lateral nucleus of the amygdala (LA) regulate the expression of the ERK-driven immediate early gene early growth response gene I (EGR-1) in regions of the auditory thalamus that are presynaptic to the LA. Further, antisense knockdown of EGR-1 in the auditory thalamus impairs both fear memory consolidation and the training-induced elevation of two presynaptically localized proteins in the LA. These findings indicate that synaptic plasticity and NO signaling in the LA during auditory fear conditioning promote alterations in ERK-driven gene expression in auditory thalamic neurons that are required for both fear memory consolidation as well as presynaptic correlates of fear memory formation in the LA, and provide general support for a role of NO as a ”retrograde signal” in mammalian memory formation.
In vertebrate models of synaptic plasticity, it is widely accepted that N-methyl-D-aspartate receptor (NMDAR)-driven recruitment of intracellular signaling pathways promotes long-term plastic change and memory through alterations of transcription and translation and accompanying morphological changes at both pre- and post-synaptic sites. Long-term potentiation (LTP) in area CA1 of the hippocampus, for example, which is known to be induced postsynaptically by NMDAR-mediated elevations in Ca2+ in the postsynaptic cell (Malenka and Nicoll, 1999
), has been shown to be accompanied not only by postsynaptic morphological alterations (Engert and Bonhoeffer, 1999
; Toni et al., 1999
), but also by corresponding presynaptic changes (Bonhoeffer et al., 1989
; Lisman and Harris, 1993
).
The fact that LTP is accompanied by presynaptic alterations while LTP induction is driven by NMDAR-mediated postsynaptic signaling has led to the long-held hypothesis that some signal must act in a retrograde manner to promote presynaptic aspects of plasticity (Bliss et al., 1986
). Of the various candidates for such a “retrograde messenger”, nitric oxide (NO) is perhaps the best characterized. A highly soluble gas generated in the postsynaptic cell by the Ca2+-regulated enzyme nitric oxide synthase (NOS), NO promotes the formation of cGMP and the activation of the cGMP-dependent protein kinase (PKG) (Bredt and Snyder, 1992
). In the postsynaptic cell, signaling via cGMP-PKG has been suggested to play a critical role in transcriptional regulation (Lu et al., 1999
). Presynaptically, activation of cGMP-PKG signaling has been shown to promote mobilization of synaptic vesicles in the presynaptic cell, leading to enhanced transmitter release (Ninan et al., 2006
), as well as structural changes in the presynaptic terminal (Antonova et al., 2001
; Wang et al., 2005
).
The role of the NO-cGMP-PKG signaling pathway has been extensively studied in hippocampal synaptic plasticity (Schuman and Madison, 1991
; Zhuo et al., 1994
; Arancio et al., 1996a
; Son et al., 1998
; Lu et al., 1999
; Monfort et al., 2002
) and memory formation (Chapman et al., 1992
; Bohme et al., 1993
; Holscher et al., 1996
; Zou et al., 1998
). However, while in vitro studies have convincingly supported a role for NO as a “retrograde messenger” in hippocampal synaptic plasticity (Arancio et al., 1996a
,b
), a corresponding role in hippocampal memory formation has remained elusive and controversial. This is likely due, in part, to the relative complexity of hippocampal-dependent learning tasks, which can make identification of the relevant synapses underlying acquisition of the task problematic. In the present study, we have used amygdala-dependent auditory Pavlovian fear conditioning to examine whether fear memory formation in the lateral nucleus of the amygdala (LA) is driven by NO-driven presynaptic alterations at LA synapses. In contrast to hippocampal-dependent learning tasks, auditory fear conditioning is a relatively simple associative learning task in which the relevant neural pathways and synapses have been well characterized (LeDoux, 2000
; Blair et al., 2001
; Maren, 2001
). Specifically, auditory fear conditioning has been shown to promote alterations in synaptic transmission between neurons originating in the auditory thalamus, including the medial division of the medial geniculate nucleus and posterior intralaminar nucleus (MGm/PIN), and the LA (Quirk et al., 1995
; Rogan et al., 1997
), and these alterations have been shown to be critical for fear memory formation (Schafe et al., 2005
).
We have recently shown that memory consolidation of auditory Pavlovian fear conditioning and associated synaptic plasticity at thalamic inputs to the LA requires NO signaling in the LA (Schafe et al., 2005
; Ota et al., 2008
). In other studies, we and others have shown that mRNA synthesis and signaling via the ERK/MAPK signaling cascade in MGm/PIN neurons are critical for fear memory formation (Apergis-Schoute et al., 2005
; Parsons et al., 2006
) and synaptic plasticity at thalamic inputs to the LA (Apergis-Schoute et al., 2005
). In the present study, we now show that NMDAR-driven synaptic plasticity and NO signaling in the LA regulate ERK-driven gene expression in MGm/PIN projection neurons that contributes to both fear memory consolidation and to alterations in presynaptically localized proteins at LA synapses. Our findings support the hypothesis that NMDAR-driven synaptic plasticity and NO signaling in the LA drive changes in gene expression in thalamic projection neurons that are critical for fear memory formation by promoting presynaptic aspects of plasticity at LA synapses, and support a more general role for NO-driven “retrograde signaling” in mammalian memory formation.
Subjects
Adult male Sprague Dawley rats (Harlan; N = 347) were housed individually in plastic cages and maintained on a 12-h light/dark cycle. Food and water were provided ad libitum throughout the experiment.
Surgical Procedures
Under a mixture of Ketamine (100 mg/kg) and Xylazine (6.0 mg/kg) anesthesia, rats were implanted bilaterally with 26-gauge stainless steel guide cannulas aimed at either the LA [Bregma −3.2 AP ± 5.0 ML, −8.0 DV] or the MGm/PIN [−5.5 AP, ± 2.8 ML, −6.6 DV]. Guide cannulas were fixed to screws in the skull using a mixture of acrylic and dental cement, and a 31-gauge dummy cannula was inserted into each guide cannula to prevent clogging. Rats were given Buprenex (0.2 mg/kg) as an analgesic and given at least 5 days to recover prior to experimental procedures. All procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Experimental Animals and were approved by the Yale University Institutional Animal Care and Use Committee.
Drugs and Intra-Cranial Infusions
The membrane-impermeable NO scavenger c-PTIO (Tocris) was dissolved in sterile saline at a concentration of 2 µg/µl. The MEK inhibitor U0126 (Promega) and the NOS inhibitor 7-Nitroindazole (7-Ni; EDM Chemicals) were dissolved in DMSO to produce a stock solution of 4 µg/µl and diluted 1:1 with ACSF prior to infusion into the brain. The NR2B-selective antagonist ifenprodil (Sigma) was dissolved at a concentration of 2 µg/µl in 2% HBC-saline solution. The PKG inhibitor Rp-8-Br-PET-cGMPS (Calbiochem) and the PKG activator 8-Br-cGMP (Calbiochem) were dissolved in distilled water at concentrations of either 2 µg/µL or 20 µg/µL, respectively. All drug or vehicle solutions were infused into either the LA or the MGm/PIN at a volume of 0.5 µl and a rate of 0.2 µl/min. Rats were habituated to dummy cannula removal and replacement on the day prior to each infusion. Following each infusion, the infusion cannulas were allowed to remain in the MGm/PIN or the LA for at least 1 min to allow for adequate diffusion.
Western Blotting
For Western blotting experiments involving non-cannulated rats (Figure 1
), rats were habituated to handling for 4 days before training. “Paired” rats received three conditioning trials consisting of a 20 s, 5 kHz, 75 dB tone that co-terminated with a 1 s, 0.5-mA footshock [ITI = 120 s]. “Tone Alone” control rats were placed in the conditioning chamber and exposed to three tones [20 s, 5 kHz, 75 dB] without receiving footshock. “Immediate Shock” control rats were placed in the conditioning chamber and immediately exposed to three 0.5 mA, 1-s footshocks [ISI = 500 ms]. This procedure allows the experimenter to control for shock-induced changes in gene expression in the absence of a context-shock association (Fanselow, 1980
). “Delayed Shock” controls were placed in the conditioning chamber and, following a 2.5-min delay, exposed to three 0.5 mA, 1-s footshocks [ITI = 120 s]. “Naïve” control rats were handled and sacrificed without exposure to the box. Two hours following training, rats were anesthetized with chloral hydrate (250 mg/kg) and decapitated. Brains were removed from the skull, frozen on dry ice, and stored at −80°C until processed.
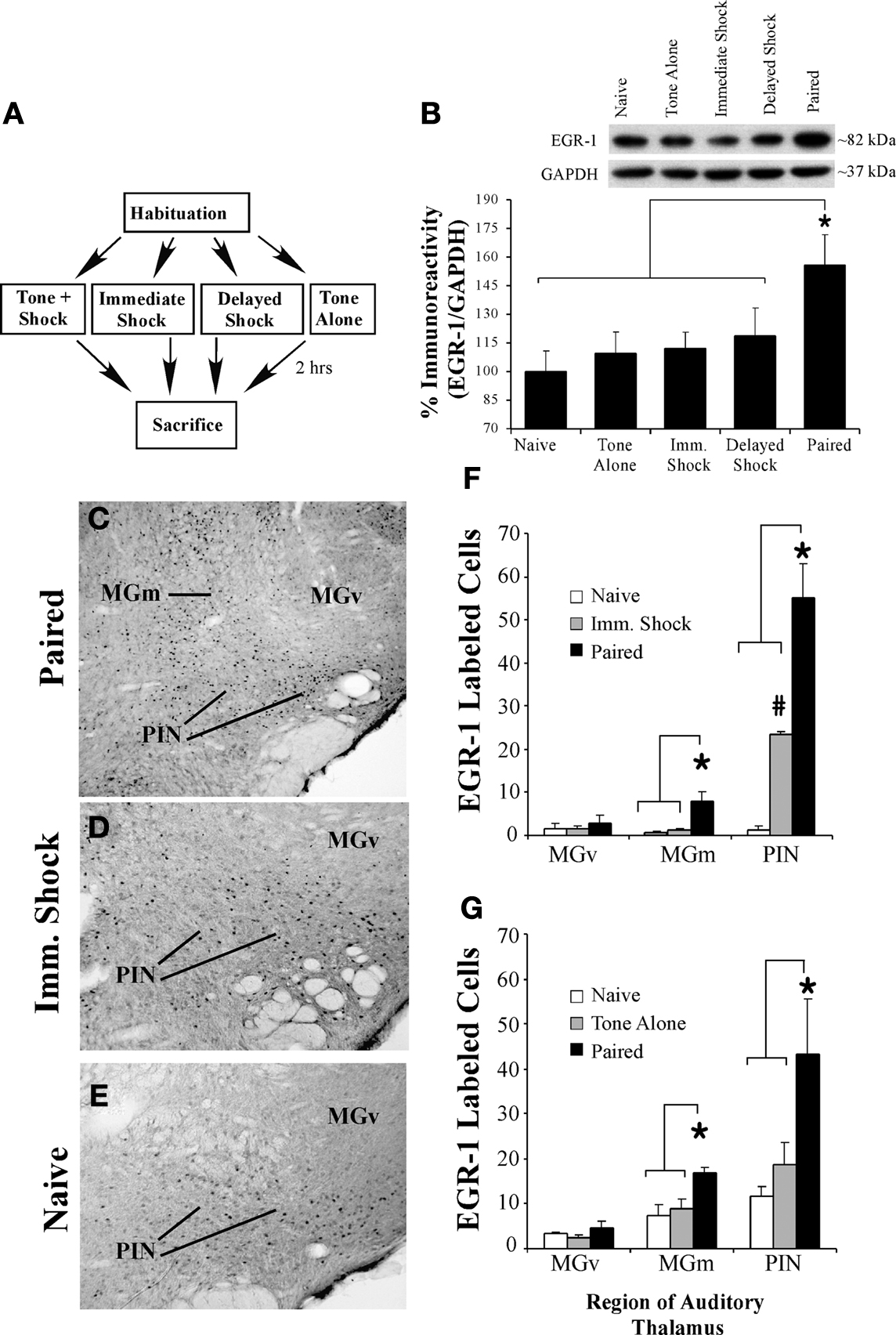
Figure 1. Fear conditioning regulates EGR-1 expression in the MGm/PIN. (A) Schematic of the behavioral protocol. (B) Western blot analysis of EGR-1 protein in the MGm/PIN of Paired (n = 8), Tone Alone (n = 8), Imm. Shock (n = 8), Delayed Shock (n = 8), and Naïve (n = 8) groups following fear conditioning. *p < 0.05 relative to Naïve, Tone Alone, Imm. Shock, and Delayed Shock groups. Representative blots can be seen in the inset. (C) Representative 10X photomicrograph of immunolabeled EGR-1 neurons in a Paired rat. (D–E) Representative 20X photomicrographs of immunolabeled EGR-1 neurons in an Imm. Shock and Naïve rat, respectively. (F) Quantification of EGR-1 labeled cells in the MGv, MGm, and the PIN of Paired (n = 3), Imm. Shock (n = 3), and Naïve (n = 3) groups. *p < 0.05 relative to Naive and Imm. Shock groups. #p < 0.05 relative to Naïve group. (G) Quantification of EGR-1 labeled cells in the MGv, MGm, and the PIN of Paired (n = 4), Tone Alone (n = 4), and Naïve (n = 4) groups. *p < 0.05 relative to Naive and Tone Alone groups.
For Western blotting experiments involving intra-MGm/PIN drug infusion (Figure 2
), rats with indwelling MGm/PIN cannulas received intra-MGm/PIN infusion of either vehicle (50% DMSO in ACSF) or U0126 (1 µg; 0.5 µl) followed 30 min later by fear conditioning consisting of three pairings of a 20 s, 5 kHz, 75 dB tone that co-terminated with a 1 s, 1.0 mA footshock [ITI = 120 s]. Two hours after training, rats were anesthetized with chloral hydrate (250 mg/kg; i.p.), decapitated, and brains were frozen at −80oC until processed. In other experiments (Figure 6
), rats received an intra-MGm/PIN infusion of either EGR-1 antisense ODN (250 pmol; 0.5 µl) or scrambled ODN (250 pmol; 0.5 µl). Animals were trained 90 min later with three pairings of a 20 s, 5 kHz, 75 dB tone that co-terminated with a 1 s, 1.0 mA footshock [ITI = 120 s] and anesthetized and decapitated 24 h later.
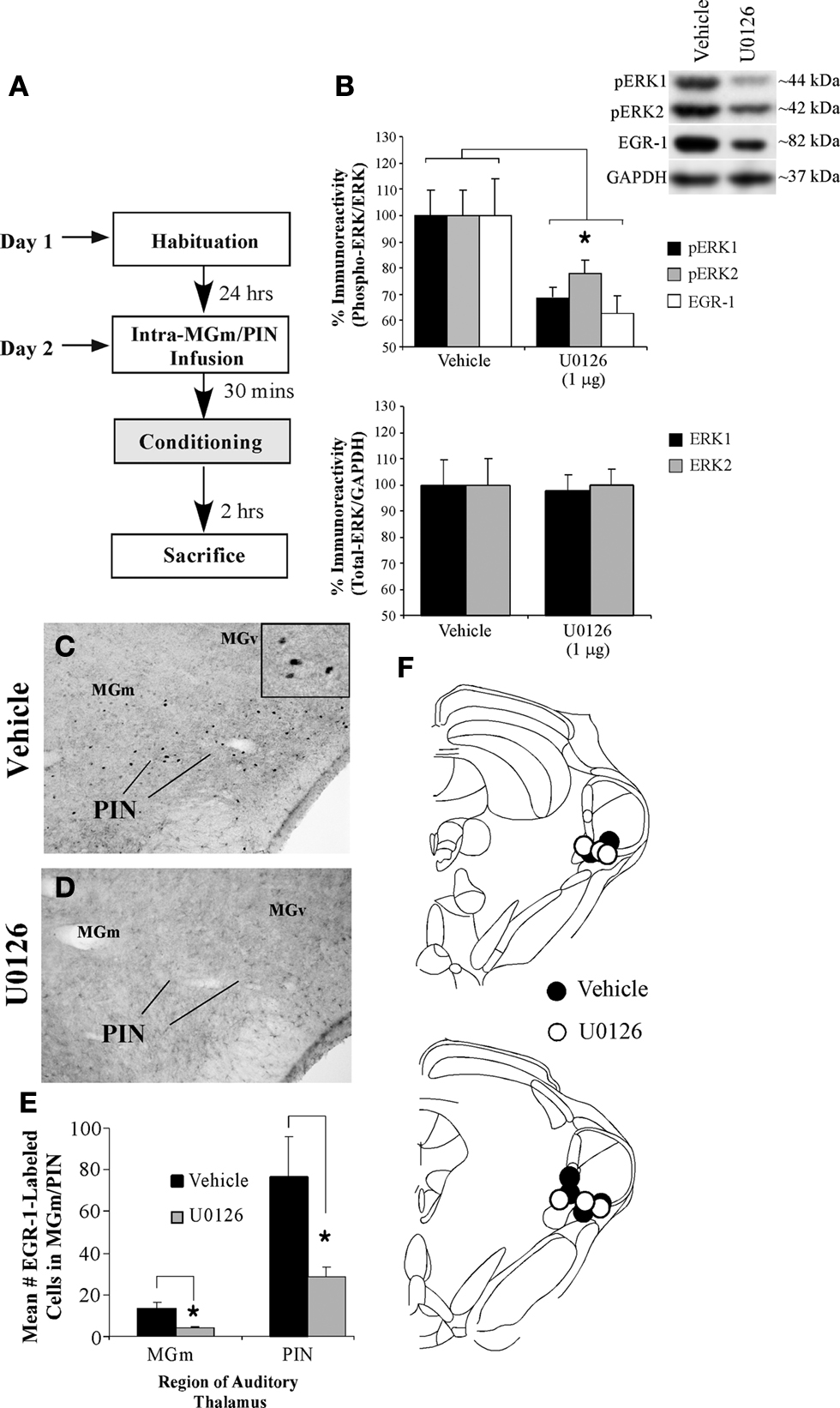
Figure 2. Training-induced expression of EGR-1 in the MGm/PIN is impaired by intra-MGm/PIN infusion of a MEK inhibitor. (A) Schematic of the behavioral protocol. (B) Western blot analysis of phospho-ERK1 (black bars), phospho-ERK2 (gray bars), and EGR-1 proteins (white bars) in the MGm/PIN of 50% DMSO vehicle (n = 5) and U0126-infused groups (n = 7) following fear conditioning. *p < 0.05 relative to vehicle groups. Representative blots can be seen in the inset. (C–D) Representative 20X photomicrographs of immunolabeled EGR-1 neurons in the MGm/PIN of rats receiving vehicle or U0126 infusion, respectively. Higher-level magnification of EGR-1 labeled neurons the vehicle-infused rat is shown in the inset of (C). (E) Quantification of EGR-1 labeled cells in the MGm/PIN of vehicle (n = 6) and U0126-treated rats (n = 6). *p < 0.05 relative to the vehicle group. (F) Cannula placements for rats given intra-MGm/PIN infusion of either vehicle (black circles) or U0126 (white circles). Panels adapted from Paxinos and Watson (1997)
.
For Western blotting experiments involving intra-LA drug infusion (Figure 3
), rats with indwelling LA cannulas received intra-LA infusion of either ifenprodil (1.0 µg; 0.5 µl), c-PTIO (1.0 µg; 0.5 µl), Rp-8-Br-PET-cGMPS (1.0 µg; 0.5 µl), 8-Br-cGMP (10 µg; 0.5 µl), or their respective vehicle solutions. Thirty minutes after drug infusions (or 1 h in the case of Rp-8-Br-PET-cGMPS and 8-Br-cGMP), rats were trained with three pairings of a 20 s, 5 kHz, 75 dB tone that co-terminated with a 1.0 s, 1.0-mA footshock. This slightly higher shock intensity (1.0 vs. 0.5 mA) was used for intra-LA infusion experiments, since previous work in our lab has suggested that LA-cannulated animals require higher shock intensities to acquire the same level of fear conditioning as non-cannulated rats. For experiments involving intra-LA infusion of 8-Br-cGMP, rats were trained with two tone-shock pairings with an 0.5-mA shock intensity in an effort to avoid ceiling effects that might obscure observation of training-induced elevations in EGR-1 protein above the level of vehicle controls (Ota et al., 2008
). Rats in all groups were anesthetized and sacrificed by decapitation 2 h following conditioning. Note that while we did not run separate groups of rats to verify memory impairment at 24 h, each of these compounds has been shown in previous work to impair either acquisition or consolidation of auditory fear conditioning (Schafe et al., 2000
, 2005
; Rodrigues et al., 2001
; Ota et al., 2008
).
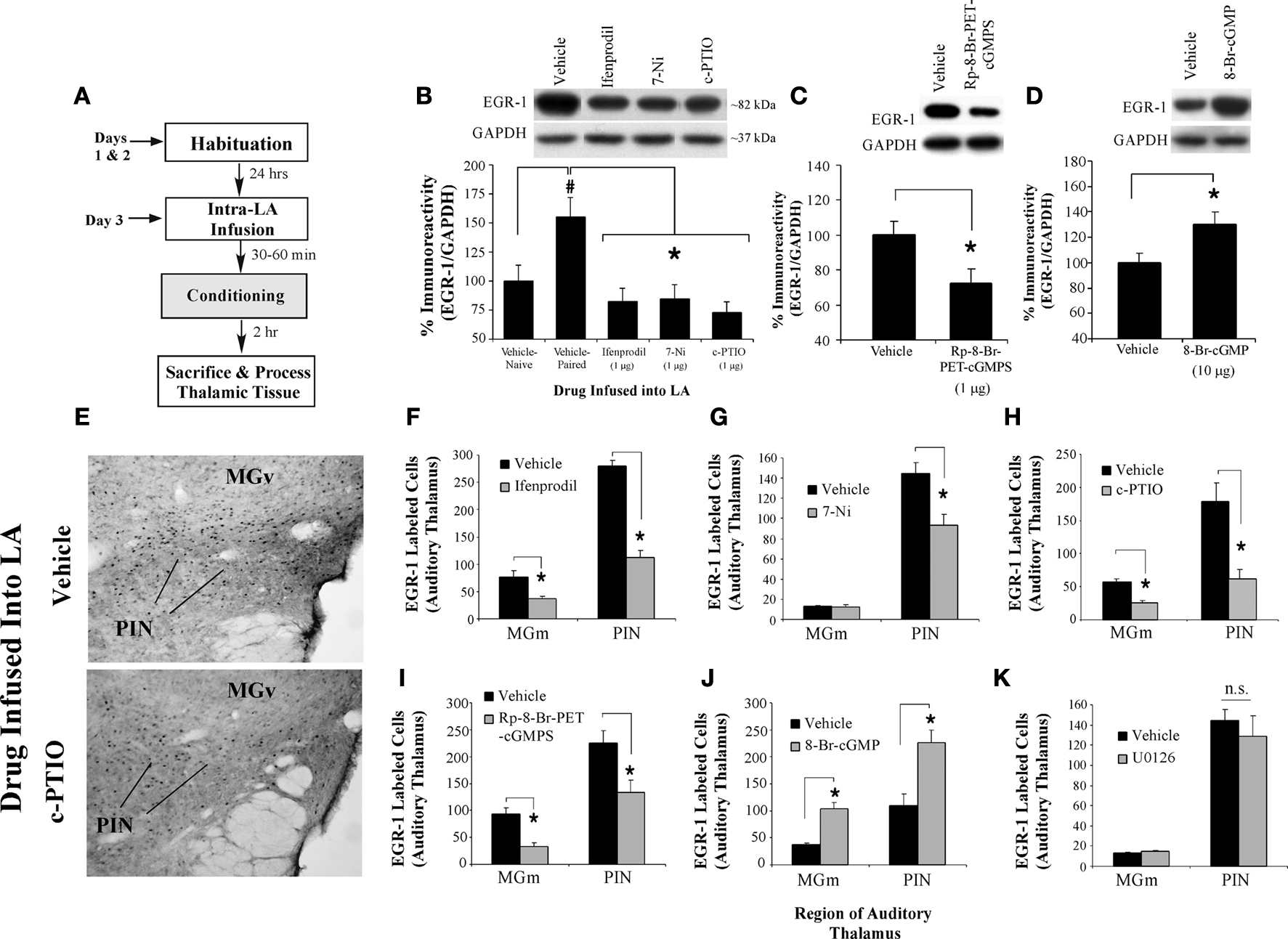
Figure 3. NMDAR-driven synaptic plasticity and NO signaling in the LA regulate training-induced EGR-1 expression in the MGm/PIN. (A) Schematic of the behavioral protocol. (B) Western blot analysis of EGR-1 protein in the MGm/PIN of rats given intra-LA infusion of vehicle (n = 8), ifenprodil (n = 8), 7-Ni (n = 7), or c-PTIO (n = 8). Representative blots can be seen in the inset. *p < 0.05 relative to the vehicle group. (C) Western blot analysis of EGR-1 protein in the MGm/PIN of rats given intra-LA infusion of vehicle (n = 9) or Rp-8-Br-PET-cGMPS (n = 13). Representative blots can be seen in the inset. *p < 0.05 relative to the vehicle group. (D) Western blot analysis of EGR-1 protein in the MGm/PIN of rats given intra-LA infusion of vehicle (n = 14) or 8-Br-cGMP (n = 14). Representative blots can be seen in the inset. *p < 0.05 relative to the vehicle group. (E) Representative 20X photomicrographs of EGR-1 immunolabled cells from the MGm/PIN in rats given intra-LA infusion of either vehicle (top) or c-PTIO (bottom). (F) Quantification of EGR-1 labeled cells in the MGm and PIN of rats given intra-LA infusion of vehicle (n = 4) or ifenprodil (n = 4). (G) Quantification of EGR-1 labeled cells in the MGm and PIN of rats given intra-LA infusion of vehicle (n = 5) or 7-Ni (n = 4). (H) Quantification of EGR-1 labeled cells in the MGm and PIN of rats given intra-LA infusion of vehicle (n = 3) or c-PTIO (n = 4). (I) Quantification of EGR-1 labeled cells in the MGm and PIN of rats given intra-LA infusion of vehicle (n = 3) or Rp-8-Br-PET-cGMPS (n = 5). (J) Quantification of EGR-1 labeled cells in the MGm and PIN of rats given intra-LA infusion of vehicle (n = 5) or 8-Br-cGMP (n = 5). (K) Quantification of EGR-1 labeled cells in the MGm and PIN of rats given intra-LA infusion of vehicle (n = 5) or U0126 (n = 4). *p < 0.05 relative to the vehicle group.
For Western blotting, punches containing the MGm/PIN or LA from the right side of the brain were obtained with a 1 mm punch tool from 400-µm-thick frozen sections taken on a sliding microtome. Punches were manually dounced in 100 µl of ice-cold hypotonic lysis buffer [10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 2.5 mM sodium pyrophosphate, 1 mM phenylmethylsulfonyl fluoride, 1 mM β-glycerophosphate, 1% Igepal CA-630, 1% protease inhibitor cocktail (Sigma) and 1 mM sodium orthovanadate]. Sample buffer was immediately added to the homogenates, and the samples were boiled for 4 min. Homogenates were electrophoresed on 10% Tris-HCl gels and blotted to Immobilon-P (Millipore, Bedford, MA, USA). For EGR-1 experiments, we loaded 20 µg of protein per lane. For synaptophysin and synapsin experiments, we loaded 5 µg per lane. Western blots were blocked in TTBS buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.05% Tween 20) with 5% dry milk and then incubated with either anti-phospho-ERK/MAPK antibody (1:1,000; Cell Signaling), total (unphosphorylated) ERK/MAPK antibody (1:1,000; Cell Signaling), anti-EGR-1 antibody (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-synapsin antibody (1:1,000; Cell Signaling), or anti-synaptophysin antibody (1:5,000; DakoCytomation). Blots were then incubated with the appropriate secondary antibody conjugated to horseradish peroxidase (Cell Signaling) and developed using West Dura chemiluminescent substrate (Pierce Laboratories, Rockford, IL, USA). Densitometry was conducted using Image J software. To control for inconsistencies in loading, optical densities were normalized to GAPDH protein (1:5,000; Abcam). Data were normalized to the average value of naïve, vehicle- or scrambled ODN-infused controls, and data were analyzed using Student’s t-tests or ANOVA.
Immunohistochemistry
Immunohistochemical experiments were carried out identically to their corresponding Western blotting experiments, with the exception that rats were sacrificed and perfused through the heart with ice-cold phosphate-buffered saline (PBS), followed by ice-cold 4% paraformaldehyde in 0.1 M phosphate buffer (PB). Brains were removed and post-fixed in 4% paraformaldehyde-PB for 12 h followed by cryoprotection in 20% glycerol-0.1 M PB for 48–72 h. Free-floating sections (40 µm) containing the MGm/PIN were cut using a sliding microtome. After blocking in PBS containing 2% bovine serum albumin (BSA; Sigma Fraction V)-0.1% Triton X-100, slices were incubated overnight at room temperature (RT) in anti-EGR-1 antibody (Santa Cruz Biotechnology, rabbit polyclonal; 1:2,000) in PBS-1% BSA-0.1% Triton X-100. After three washes in PBS, tissue sections were visualized using VectaStain ABC kit (Vector Laboratories, Burlingame, CA, USA) and developed using a DAB peroxidase substrate (Sigma, St. Louis, MO, USA). Sections were mounted on Fisherbrand electrostatic slides and coverslipped. Sections from comparable anterior-posterior levels were selected for scoring, approximately 5.8–6.0 mm posterior to Bregma. At this level, the MGm, MGv, and PIN nuclei are all well represented (see Figure 1
C). Cell counts were taken from the MGm, MGv, and PIN from at least three sections per rat from the right side of the brain and scored using Image J. Since every sixth section through the MGm/PIN was processed for immunohistochemistry, it was not necessary to correct for double-counting. Cell counts were analyzed using Student’s t-test or ANOVA.
Tract-Tracing and Double-Labeling Experiments
Rats received bilateral intra-LA guide cannula implants as described above. Following recovery, rats received a microinfusion (0.3 µl/side) of a 2% solution of fluorogold (FG; Fluorochrome, LLC) dissolved in physiological saline. Ten to fourteen days later, animals were randomly divided into three experimental groups. “Paired” rats received fear conditioning consisting of three pairings of a tone (5 kHz, 75 dB, 20 s) that co-terminated with a 1.0 mA, 1.0-s footshock [ITI = 120 s]. “Immediate shock” rats received three 1.0 mA, 1.0 s footshocks [ISI = 500 ms]. A third group of rats (“Naïve”) was given no training or shocks. Two hours after training, rats were anesthetized with chloral hydrate (250 mg/kg; i.p.) and perfused in the same manner as described above for immunohistochemistry.
Approximately seven random sections containing the MGm/PIN from the right side of the brain of each animal were assayed for the presence of FG. Sections were washed in PBS before being transferred to a blocking solution (1% albumin from bovine serum (BSA) and 0.1% Triton-X in PBS) for 1 h at RT. After blocking, sections were transferred to FG IgG solution (1:50,000; fluorogold-Rabbit, Fluorochrome LLC) and incubated overnight at RT. Sections were then washed in PBS and transferred to a secondary antibody solution (biotin-conjugated goat anti-rabbit IgG) and incubated at RT for 1 h. Sections were washed, transferred to an avidin-biotin complex (ABC) solution for an additional hour at RT, washed, and then developed using a Vector-NovaRED peroxidase substrate solution. After extensive washing, sections were then processed for EGR-1 immunolabeling. Methods for EGR-1 labeling were the same as described above, with the exception that the sections were developed in a Vector Blue alkaline phosphatase solution. Sections were then mounted on slides and coverslipped.
Thalamic sections between −5.8 and −6.3 relative to bregma were selected for scoring. We first counted the total number of FG labeled cells in the PIN, followed by total number of EGR-1 labeled cells. Finally, the total number of cells expressing both FG and EGR-1 label were counted, and this number was expressed as a percentage of the total number of FG cells in the sampled area. Results were then analyzed using ANOVA.
Oligodeoxynucleotide Design and Preparation
EGR-1 antisense and scrambled ODNs (Midland Certified Reagent Company, Midland, Texas) were designed under guidelines described in a previous study (Malkani et al., 2004
). The EGR-1 ODN encoded an antisense sequence for the EGR-1 mRNA sequence near the translation start site (GenBank accession number NM 012551, bases 348–365). The scrambled ODN served as a control and did not show significant homology to sequences in the GenBank database. Both ODNs contained phosphorothioate linkages on the bases of both the 5′ and 3′ ends and phosphodiester internal bonds. The following sequences were used (“∼” denotes a phosphorothioate linkage): 5′-G∼GTAGTTGTCCATGGTG∼G-3′ (antisense) and 5′-G∼TTGGAGTCGGTGGTTC∼A-3′ (scrambled).
Verification of ODN Diffusion and Knockdown
To verify diffusion of the EGR-1 oligonucleotide in the MGm/PIN, rats were first infused with a biotinylated EGR-1 antisense ODN (Midland). The biotin-EGR-1 antisense ODN was infused bilaterally at the dose and volume used in the behavioral experiments (250 pmol; 0.5 µl), and rats were sacrificed by perfusion 30 min later. Brains were sectioned at 40 µm and sections containing the MGm/PIN were processed using a standard ABC-DAB reaction to visualize the extent of the diffusion and cellular uptake of the ODN. To verify EGR-1 knockdown with the EGR-1 antisense ODN, rats received an intra-MGm/PIN infusion of either EGR-1 antisense ODN (250 pmol; 0.5 µl) or scrambled ODN (250 pmol; 0.5 µl) on opposite sides of the brain. Animals were trained 90 min later with a single pairing of a 30 s, 5 kHz, 75 dB tone that co-terminated with a 1.0 s, 1.5-mA footshock (as in the ODN behavioral experiments). Two hours later, animals were anesthetized and sacrificed by decapitation, and punches containing the MGm/PIN from scrambled and antisense ODN-infused sides of the brain were processed using Western blotting as described above.
Behavioral Procedures
Rats were habituated to the conditioning chamber and to dummy cannula removal for a minimum of 10–15 min. The following day, rats were given bilateral intra-MGm/PIN infusion of either EGR-1 antisense or scrambled ODN (250 pmol; 0.5 µl). The infusion cannulas were allowed to remain in the MGm/PIN for at least 2 min following the infusion to allow ODNs to diffuse throughout the MGm/PIN. Ninety min after ODN infusions, rats were trained with a single conditioning trial consisting of a 30 s, 5 kHz, 75 dB tone that co-terminated with a 1.0 s, 1.5-mA footshock. Testing for conditioned fear responses (freezing) was conducted at 3 h (short-term memory; STM) and 24 h (long-term memory; LTM) after conditioning. For each test, rats were placed in a distinctive environment consisting of a flat black plastic floor that had been washed with a distinctive peppermint soap and exposed to five conditioned stimulus (CS) tones (5 kHz, 75 dB, 30 s). Total seconds freezing during the CS presentations was scored for each rat, and this number was expressed as a percentage of the total CS presentation time. For analysis, freezing across each trial was averaged into a single score for each memory test. All data were analyzed with ANOVA and Duncan’s post hoc t-tests. Differences were considered significant if p < 0.05.
At the end of the behavioral experiment, rats were anesthetized with chloral hydrate (250 mg/kg) and perfused with 10% buffered formalin. Nissl staining and light microscopy were used to verify the location of the cannula tips within the MGm/PIN or the LA.
Auditory Fear Conditioning Regulates the Expression of the ERK-Driven Immediate Early gene EGR-1 in the MGm/PIN
Most recent studies that have sought to examine the cellular mechanisms by which fear memories are formed and stored have focused on postsynaptic events at LA synapses, including the recruitment of NMDAR-driven protein kinase signaling cascades that are thought to promote fear memory consolidation by engaging activators of transcription in the nucleus (Schafe and LeDoux, 2000
; Schafe et al., 2000
; Rodrigues et al., 2004
). We and others have recently reported, however, that intracellular signaling via the ERK/MAPK cascade and mRNA transcription in the MGm/PIN plays an equally critical role in memory consolidation of auditory fear conditioning (Apergis-Schoute et al., 2005
; Parsons et al., 2006
). Auditory fear conditioning has also been observed to regulate the activation of the transcription factor cAMP response element binding protein (CREB) in the MGm/PIN, and over-expression of CREB in the MGm/PIN enhances fear memory formation (Han et al., 2008
). Collectively, this pattern of findings suggests that ERK-driven transcriptional regulation in the MGm/PIN, in addition to that in the LA, is necessary for fear memory consolidation. Remarkably, however, signaling via ERK/MAPK in the MGm/PIN is also critical for synaptic plasticity in the LA, suggesting that the functional significance of ERK-driven transcriptional changes in the MGm/PIN may be to promote presynaptic aspects of plasticity at the level of the LA (Apergis-Schoute et al., 2005
). Together with our recent observations that NO signaling in the LA is required for fear memory consolidation (Schafe et al., 2005
; Ota et al., 2008
), we have hypothesized that synaptic plasticity and NO signaling in the LA drive changes in ERK-driven gene expression in MGm/PIN projection neurons that contribute to presynaptic aspects of plasticity in the LA and to fear memory formation.
As a first test of this hypothesis, we used a combination of Western blotting and immunohistochemistry to examine whether auditory fear conditioning regulates the expression of the ERK and CREB-driven IEG EGR-1 (zif-268, Krox-24) in the MGm/PIN (Impey et al., 2004
). In our Western blotting experiments (Figures 1
A,B), we observed a significant increase in EGR-1 protein expression in MGm/PIN homogenates from “Paired” rats relative to “Tone Alone”, “Immediate Shock”, “Delayed Shock”, and “Naïve” controls. The ANOVA revealed a significant effect for group [F(4,35) = 2.69, p < 0.05], with the Paired group being significantly different from each of the other groups (p < 0.05; Duncan’s test). There were no significant differences between the other groups (p > 0.05). The levels of the loading control GAPDH also failed to differ between groups, F(4,35) = 0.22, p > 0.05 (data not shown), suggesting that protein lysate loading was equivalent among the five groups.
The findings of our immunohistochemical experiments are depicted in Figures 1
C–G. Representative photomicrographs from “Paired”, “Immediate Shock”, and “Naïve” rats can be viewed in Figures 1
C–E, while cell counts are presented in Figures 1
F–G. We observed EGR-1 labeled cells throughout the MGm and PIN. Very few EGR-1 labeled cells were observed in the MGv (Figure 1
C). Cell counts from Naïve, Immediate Shock, and Paired rats are presented in Figure 1
F. Analysis of cell counts in the MGm revealed a significant effect, F(2,6) = 11.2, p < 0.01, with the Paired group being significantly different from Immediate Shock and Naïve controls (p < 0.05; Duncan’s test). No difference between Naïve and Immediate Shock groups was observed (p > 0.05). A similar increase was observed within the PIN, F(2,6) = 35.08, p < 0.01, with the Paired group being significantly different from Immediate Shock and Naïve controls (p < 0.05). Within the PIN, a significant difference between Naïve and Immediate Shock groups was also observed (p < 0.05). Within the MGv, no significant differences were observed, F(2,6) = 0.24, p > 0.05.
Cell counts from “Naïve”, “Tone Alone”, and “Paired” rats are presented in Figure 1
G. No significant differences were observed in the MGv, F(2,9) = 1.35, p > 0.05. In contrast, significant differences were observed within the MGm [F(2,9) = 5.71, p < 0.03] and PIN [F(2,9) = 4.65, p < 0.05]. Within each of these latter subnuclei, the Paired group was significantly different from both Tone Alone and Naïve controls (p < 0.05), while no significant differences between Naïve and Tone Alone groups were observed (p > 0.05).
Collectively the findings of our initial Western blotting and immunohistochemical experiments suggest that auditory Pavlovian fear conditioning regulates the expression of EGR-1 protein in the MGm/PIN in an associative manner.
Training-Induced Elevation in EGR-1 Expression in the MGm/PIN is Downastream of ERK/MAPK Activation
Previous studies have shown that inhibition of ERK/MAPK signaling or mRNA synthesis in the MGm/PIN impairs fear memory consolidation (Apergis-Schoute et al., 2005
; Parsons et al., 2006
). These findings suggest that ERK/MAPK signaling in MGm/PIN neurons drives changes in gene expression that are critical for fear memory formation. In this next experiment, we asked whether training-induced expression of EGR-1 in the MGm/PIN is downstream of activation of the ERK/MAPK signaling pathway.
In our experiments, rats were given bilateral intra-MGm/PIN infusion of the MEK inhibitor U0126 (1 µg/side), a dose that we have previously shown to be effective at impairing fear memory consolidation when infused into the MGm/PIN (Apergis-Schoute et al., 2005
). Thirty minutes later, rats were conditioned followed by sacrifice 2 h later by either decapitation or perfusion (Figure 2
A). In Western blotting experiments, EGR-1 protein expression in MGm/PIN tissue was examined in homogenates taken from around the cannula tips. To verify the effectiveness of U0126 in impairing ERK/MAPK activation in each sample, we also probed the blots with an anti-phospho-ERK antibody. In immunohistochemical experiments, brain slices containing the MGm/PIN were processed using an anti-EGR-1 antibody as above.
Relative to vehicle-infused controls, rats infused with U0126 exhibited significantly lower expression of both EGR-1 and activated ERK/MAPK in the MGm/PIN following fear learning (Figure 2
B, top). The ANOVA (drug group by protein with repeated measure for protein) revealed a significant effect for drug [F(1,10) = 27.74, p < 0.01]. The effect for protein [F(2,20) = 0.68, p > 0.05] and the drug by protein interaction [F(2,20) = 0.68, p > 0.05] were not significant. Further, both groups (vehicle, U0126) exhibited equivalent expression of total (unphosphorylated) ERK/MAPK (Figure 2
B, bottom), indicating that the reduction in EGR-1 and phospho-ERK proteins was not due to drug-induced alterations in total protein. The ANOVA (drug group by kinase with repeated measure for kinase) for total ERK revealed no significant effect of drug [F(1,10) = 0.07, p > 0.05], kinase [F(1,10) = 1.13, p > 0.05], or drug by kinase interaction [F(1,10) = 1.13, p > 0.05].
In our immunohistochemical experiments, rats given intra-MGm/PIN infusion of U0126 exhibited significantly lower expression of EGR-1 labeled cells in the MGm/PIN relative to vehicle-infused controls [Figures 2
C–E; MGm: t(10) = 2.46, p < 0.05; PIN: t(10) = 3.70, p < 0.02]. Cannula placements for animals infused with either vehicle or U0126 can be observed in Figure 2
F.
NMDAR-driven Synaptic Plasticity and no Signaling in the LA are Required for Training-induced Elevation in EGR-1 Expression in the MGm/PIN
Our initial findings showed that auditory fear conditioning regulates the expression of ERK-driven IEG EGR-1 in the MGm/PIN in an associative manner. In this next series of experiments, we used pharmacological methods combined with Western blotting and immunohistochemistry to ask whether synaptic plasticity in the LA is critical for promoting training-induced elevations in EGR-1 in the MGm/PIN. We further examined the possibility that NO signaling in the LA, which we have recently shown to be critical for fear memory consolidation (Schafe et al., 2005
; Ota et al., 2008
), is critical for linking synaptic plasticity in the LA to alterations in gene expression in the MGm/PIN.
In our Western blotting and immunohistochemistry experiments, rats received intra-LA infusion of either vehicle, the NR2B-selective antagonist ifenprodil (1 µg/side; 0.5 µl), the NOS inhibitor 7-Ni (1 µg/side; 0.5 µl), the membrane-impermeable NO scavenger c-PTIO (1 µg/side; 0.5 µl), the PKG inhibitor Rp-8-Br-PET-cGMPS (1 µg/side; 0.5 µl), or the PKG activator 8-Br-cGMP (10 µg/side; 0.5 µl) followed by fear conditioning and analysis of EGR-1 protein in the MGm/PIN (Figure 3
A). Using immunohistochemistry, we further examined the effect of intra-LA infusion of the MEK inhibitor U0126 (1 µg/side; 0.5 µl) on training-induced EGR-1 expression in the MGm/PIN. The dose of ifenprodil used in the present study has previously been shown to significantly impair fear memory acquisition when infused into the LA prior to fear conditioning (Rodrigues et al., 2001
). The doses of c-PTIO, 7-Ni, Rp-8-Br-PET-cGMPS, and U0126 have previously been shown to significantly impair fear memory consolidation when infused into the LA prior to fear conditioning; that is, fear acquisition and STM are intact, while LTM is impaired (Schafe et al., 2000
, 2005
; Ota et al., 2008
). The dose of 8-Br-cGMP, conversely, has been shown to significantly enhance fear memory consolidation when infused into the LA prior to fear conditioning; that is, acquisition and STM are similar to vehicle controls, while LTM is enhanced (Ota et al., 2008
).
The findings of our Western blotting experiments are depicted in Figures 3
B–D. Relative to vehicle controls, intra-LA infusion of either ifenprodil, 7-Ni, or c-PTIO significantly reduced training-induced expression of EGR-1 in the MGm/PIN (Figure 3
B). The ANOVA revealed a significant effect of group, F(3,27) = 11.85, p < 0.01, with all three drugs differing significantly from vehicle (p < 0.05). The levels of the loading control GAPDH, however, did not differ, F(3,27) = 0.789, p > 0.05 (data not shown). Training-induced expression of EGR-1 was similarly reduced by intra-LA infusion of the PKG inhibitor Rp-8-Br-PET-cGMPS, t(20) = 3.24, p < 0.01 (Figure 3
C), while intra-LA infusion of the PKG activator 8-Br-cGMP produced a significant increase in the training-induced expression of EGR-1 in the MGm/PIN, t(26) = 3.03, p < 0.01 (Figure 3
D). The levels of the loading control GAPDH did not differ in the MGm/PIN following intra-LA infusion of either Rp-8-Br-PET-cGMPS [t(20) = 0.003, p > 0.05] or 8-Br-cGMP [t(26) = 0.03, p > 0.05] (data not shown).
The findings of our immunohistochemistry experiments are depicted in Figures 3
E–K. Representative photomicrographs of EGR-1 immunolabel in the MGm/PIN from rats receiving intra-LA infusion of vehicle or c-PTIO can be viewed in Figure 3
E. As in our initial experiments, we observed EGR-1 labeled cells throughout the MGm and PIN, while very few EGR-1 labeled cells were observed in the MGv. Statistical analysis revealed that intra-LA infusion of ifenprodil significantly reduced training-induced expression of EGR-1 in the MGm [t(6) = 3.37, p < 0.05] and PIN [t(6) = 10.5, p < 0.01; Figure 3
F]. Intra-LA infusion of 7-Ni significantly reduced training-induced expression of EGR-1 in the PIN [t(7) = 3.19, p < 0.05], but not in the MGm [t(7) = 0.49, p > 0.05; Figure 3
G]. Intra-LA infusion of c-PTIO significantly impaired training-induced expression of EGR-1 in the MGm [t(5) = 5.67, p < 0.01] and PIN [t(5) = 3.79, p < 0.02; Figure 3
H]. Intra-LA infusion of Rp-8-Br-PET-cGMPS resulted in significant impairment of EGR-1 expression in the MGm [t(6) = 5.00, p < 0.01] and PIN [t(6) = 2.65, p < 0.05; Figure 3
I]. Conversely, intra-LA infusion of 8-Br-cGMP resulted in significant enhancement of EGR-1 expression in the MGm [t(8) = 5.81, p < 0.01] and PIN [t(8) = 3.68, p < 0.01; Figure 3
J].
In contrast to the other drugs, intra-LA infusion of U0126 appeared to have no effect on training-induced expression of EGR-1 in either the MGm [t(7) = 1.55, p > 0.05] or PIN [t(7) = 0.69, p > 0.05; Figure 3
K]. Thus, intra-LA infusion of drugs that affect either synaptic plasticity (ifenprodil) or NO signaling (7-Ni, c-PTIO, Rp-8-Br-PET-cGMPS, 8-Br-cGMP) significantly regulate training-induced expression of EGR-1 in the MGm/PIN, while intra-LA infusion of the MEK inhibitor U0126 does not.
Importantly, the reduction in EGR-1 expression in the MGm/PIN following intra-LA infusion of either ifenprodil, 7-Ni, or c-PTIO is not observed in naïve animals that do not receive fear conditioning (data not shown). In a separate experiment, rats given intra-LA infusion of vehicle (n = 7), ifenprodil (n = 7), 7-Ni (n = 7), or c-PTIO (n = 7) prior to sacrifice at the same time interval as the trained animals described above, exhibited no significant differences in levels of EGR-1 protein in the MGm/PIN [F(3,24) = 1.38, p > 0.05]. Further, relative to vehicle controls (n = 5), the reduction or enhancement in EGR-1 expression in the MGm/PIN following intra-LA infusion of Rp-8-Br-PET-cGMPS (n = 5) or 8-Br-cGMP (n = 6), respectively, was not observed in naïve animals that did not receive fear conditioning [F(2,13) = 0.31, p < 0.05]. In addition, levels of the loading control GAPDH did not differ between groups infused with vehicle vs. ifenprodil vs. 7-Ni vs. c-PTIO [F(3,24) = 1.08, p > 0.05] or between groups infused with vehicle vs. Rp-8-Br-PET-cGMPS vs. 8-Br-cGMP [F(2,13) = 2.00, p > 0.05]. Collectively, our findings suggest that the regulation in EGR-1 expression in the MGm/PIN following intra-LA infusion of ifenprodil, 7-Ni, c-PTIO, Rp-8-Br-PET-cGMPS, or 8-Br-cGMP cannot be attributed to infusion of these drugs alone, but rather to infusion in combination with fear conditioning.
MGm/PIN Neurons that Express EGR-1 after Fear Learning Project to the LA
Our initial experiments collectively showed that NMDAR-driven synaptic plasticity and NO signaling in the LA regulate ERK-driven elevations in EGR-1 in MGm/PIN neurons following auditory fear conditioning. Here, we used a combination of fluorogold (FG) tract-tracing and double-labeling immunohistochemical methods to ask whether cells that express EGR-1 in the MGm/PIN following fear learning project to the LA (Figure 4
A).
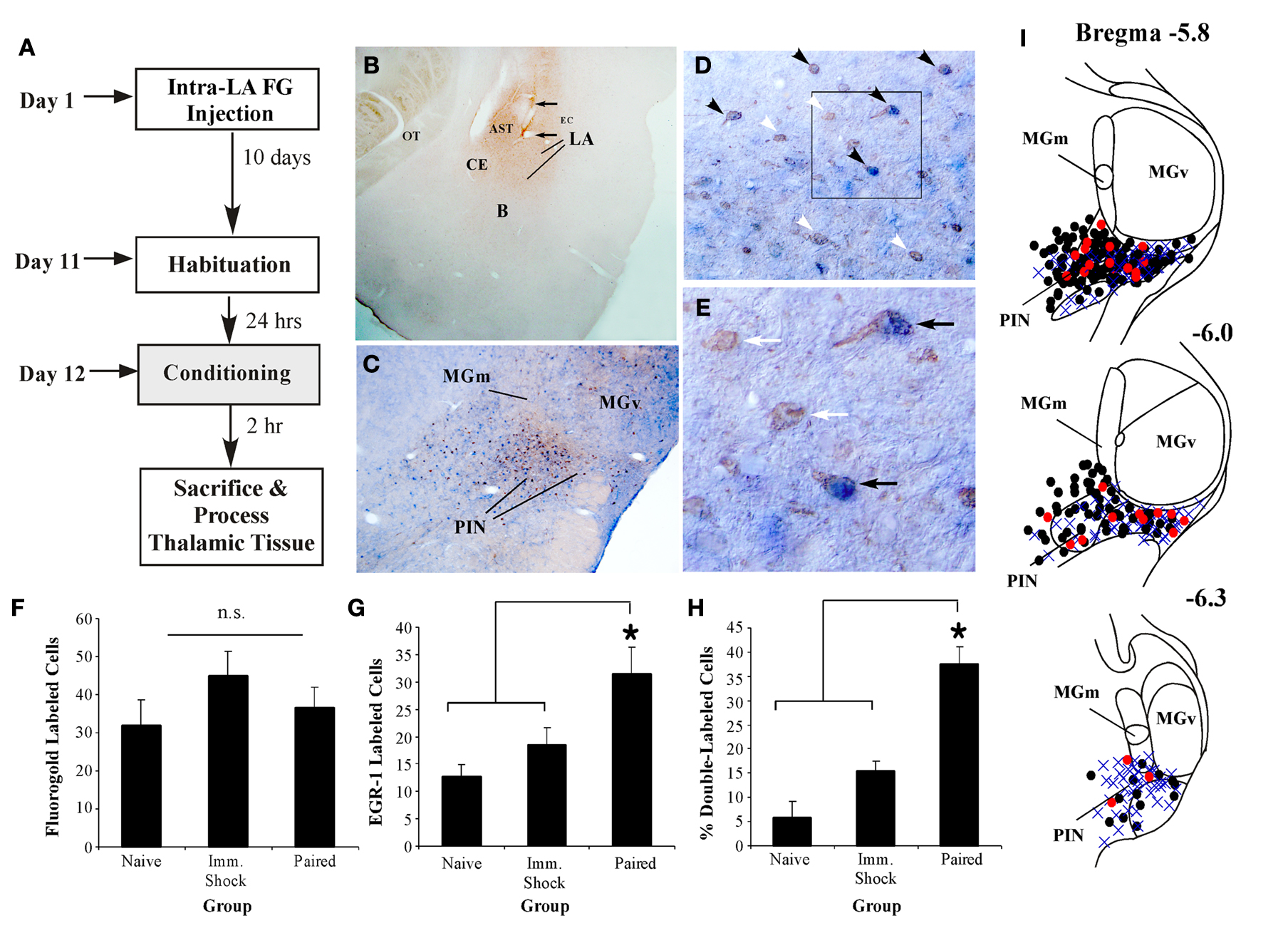
Figure 4. MGm/PIN neurons that express EGR-1 after fear learning project to the LA. (A) Schematic of the behavioral protocol. (B) Representative FG injection site in the LA. The arrows represent the cannula track. (C) Representative 20X image of cells in the MGm/PIN labeled for either FG (reddish-brown stain) or EGR-1 (blue stain). Here, FG is stained with a NovaRED peroxidase substrate (Vector), while EGR-1 is stained with Vector blue alkaline phosphatase substrate (see Materials and Methods). (D) Higher-magnification (20X) images of cells expressing FG, EGR-1, or both labels (black arrows). Single labeled cells are represented by white arrows. (E) Higher-magnification (40X) images of either single (white arrows) or double-labeled cells (black arrows) depicted in the box from (D). (F) Quantification of FG labeled cells in the PIN of Paired (n = 7), Imm. Shock (n = 6), and Naïve (n = 4) groups. n.s. = not significant. (G) Quantification of EGR-1 labeled cells in the PIN of Paired, Imm. Shock, and Naïve groups. *p < 0.05 relative to Naive and Imm. Shock groups. (H) Quantification of the percentage of double-labeled cells in the PIN of Paired, Imm. Shock, and Naïve groups. *p < 0.05 relative to Naive and Imm. Shock groups. (I) Dot diagrams representing the location of the total number FG-labeled cells (black circles), EGR-1-labeled cells (blue crosses), and double-labeled cells (red circles) within the PIN of Paired rats.
Figure 4
B shows a representative FG deposit in the LA, while representative sections from the MGm/PIN exhibiting FG and EGR-1 labeling in a paired rat are shown in Figures 4
C–E. Figure 4
C depicts the distribution of both labels in the MGm, PIN, and MGv. Consistent with previously documented projection patterns (LeDoux et al., 1990
), FG-labeled cells were observed throughout the MGm/PIN, with a particularly dense expression in the PIN region. In contrast, FG-labeled cells were sparse or notably absent in the MGv. Further, in agreement with our initial experiments, we observed EGR-1 labeled cells throughout the MGm and PIN. Very few EGR-1 labeled cells were observed in the MGv (Figure 4
C). Representative higher-magnification photomicrographs of single and double-labeled cells within the PIN are depicted in Figures 4
D–E. Double-labeled cells were clearly identifiable using the two labels. The reddish-brown FG label was largely restricted to the cytosol and proximal dendrites of MGm/PIN cells, while the blue EGR-1 label was restricted to the nucleus (Figure 4
E).
For this experiment, we restricted our analyses to counting cells in the PIN region, since this region exhibited the most robust FG staining. We found no significant differences in the total number of FG-labeled cells between naïve, immediate shock, and paired groups [Figure 4
F; F(2,14) = 1.01, p > 0.05]. In contrast, we observed significant elevations in EGR-1 labeling [Figure 4
G, F(2,14) = 5.04, p < 0.03], with the paired group being significantly different from immediate shock and naïve controls (p < 0.05). No statistically significant differences were observed between immediate shock and naïve groups (p > 0.05). The overall level of EGR-1 labeling in this experiment was substantially lower than that observed in our initial experiments (Figures 1 and 3
), likely due to the different sensitivities of the alkaline phosphatase and peroxidase detection systems used in these experiments. Despite this shift in sensitivity, however, we were able to reproduce the same pattern of results in EGR-1 labeling between naïve, immediate shock, and paired groups that we observed in our initial experiments (Figure 1
).
In our final analysis, we calculated the number of FG-labeled cells in the PIN that also expressed EGR-1 for each group, and expressed this value as a percentage of the total number of FG cells. The paired group exhibited a significantly higher percentage of double-labeled cells relative to immediate shock or naïve conditions [Figure 4
H, F(2,14) = 24.5, p < 0.01], with the paired group being significantly different from immediate shock and naïve controls (p < 0.05). No statistically significant differences were observed between immediate shock and naïve groups (p > 0.05). Interestingly, only about 40% of the LA-projecting cells in the PIN were observed to express EGR-1 after fear learning. This number is in agreement with previous anatomical reports that have indicated that only about 50% of LA projecting cells in MGm/PIN are glutamatergic (LeDoux and Farb, 1991
), suggesting that only a subset of MGm/PIN cells that project to the LA are likely contributing to excitatory synaptic transmission during auditory fear conditioning.
Figure 4
I depicts the locations of the total number FG-labeled cells (black circles), EGR-1 labeled cells (blue crosses), and double-labeled cells (red circles) at different anterior-posterior levels of the PIN in the paired group. Double-labeled cells were scattered throughout the PIN region and represented at each rostro-caudal level.
Training-induced Elevation in EGR-1 Expression in the MGm/PIN is Required for fear Memory Consolidation
Our initial series of experiments showed that auditory fear conditioning regulates the expression of EGR-1 in the MGm/PIN in an associative manner. We further showed that NMDAR-driven synaptic plasticity and NO signaling in the LA are critical for training-induced expression of EGR-1 in the MGm/PIN. In this series of experiments, we asked whether training-induced expression of EGR-1 in the MGm/PIN is obligatory for fear memory consolidation using localized antisense oligodeoxynucleotide (ODN) knockdown of EGR-1.
We first asked whether intra-MGm/PIN infusion of our EGR-1 antisense ODN does, in fact, produce a reliable knockdown of EGR-1 protein in the MGm/PIN (Figure 5
A). In these experiments, rats received intra-MGm/PIN infusion of either EGR-1 antisense or scrambled ODN (250 pmol; 0.5 µl) on opposite sides of the brain. Rats were then fear conditioned with a single tone-shock pairing (see Materials and Methods) and sacrificed 2 h later. Punches taken from around the cannula tips in the MGm/PIN were processed for EGR-1 protein using Western blotting. The analysis revealed that infusion of the EGR-1 antisense ODN produced a statistically significant (∼20%) knockdown of EGR-1 protein, t(9) = 2.26, p < 0.05, while the levels of the loading control GAPDH did not differ, t(9) = 0.83 (data not shown). While modest, this 20% knockdown of EGR-1 is comparable to what has been observed previously in the LA using this ODN (Malkani et al., 2004
). Further, it is important to note that our ODN-treated MGm/PIN punches contain both pre-existing EGR-1 as well as training-induced EGR-1, and it is only the latter pool that would be expected to be affected by infusion of the antisense ODN at this relatively short infusion to sacrifice interval.
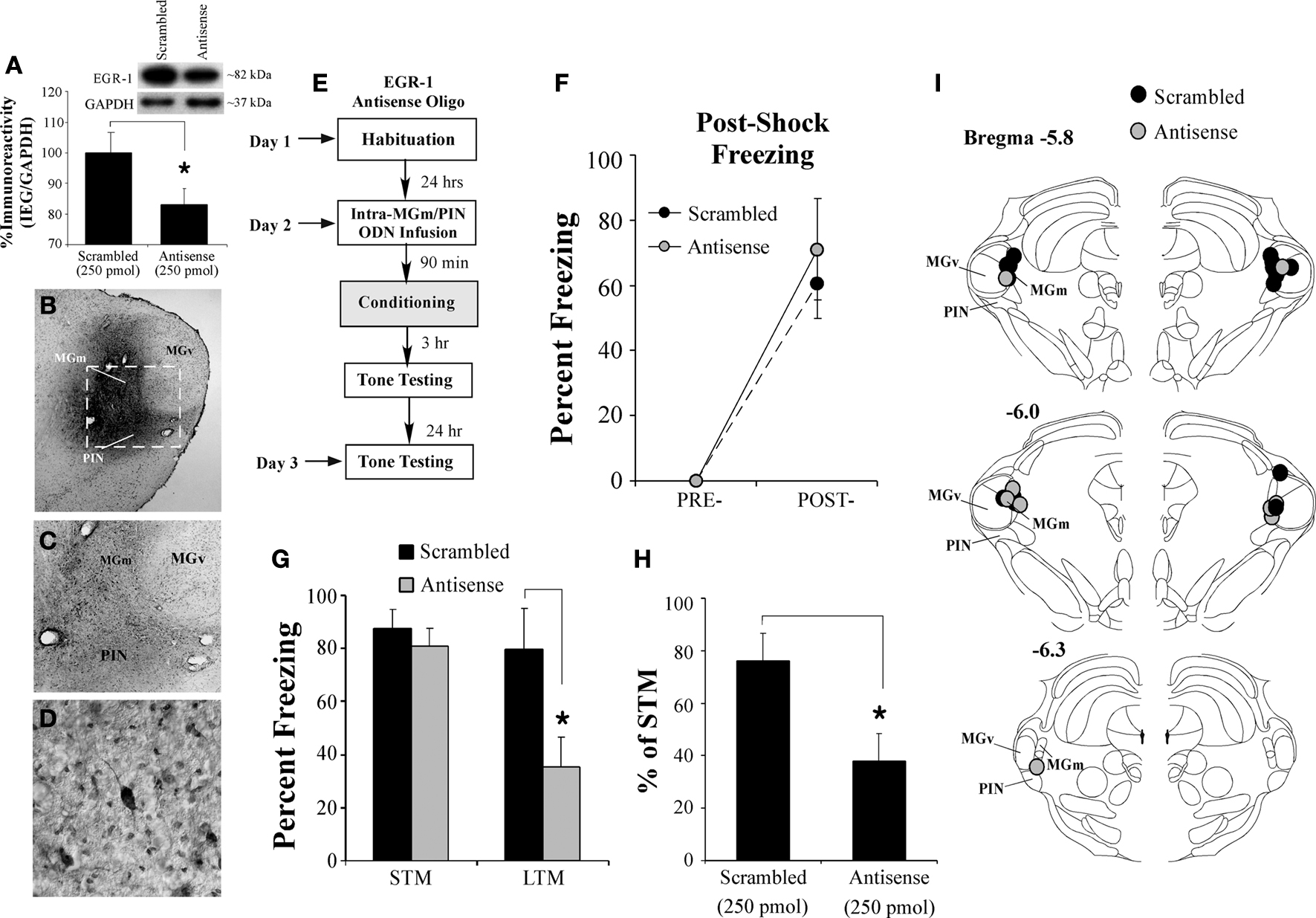
Figure 5. Intra-MGm/PIN infusion of an EGR-1 antisense ODN impairs fear memory consolidation. (A) Western blot analysis of EGR-1 protein in the MGm/PIN of rats given intra-MGm/PIN infusion of scrambled ODN (n = 10) or EGR-1 antisense ODN (n = 10). Representative blots can be seen in the inset. *p < 0.05 relative to the scrambled ODN group. (B) Representative 4X photomicrograph of an animal given intra-MGm/PIN infusion of a biotinylated EGR-1 ODN (0.5 µl; 250 pmol) and sacrificed 30 min later. Note that the ODN diffusion is largely restricted to the MGm and PIN, and spares the MGv. (C–D) Higher level (20 and 40X, respectively) magnifications of MGm/PIN neurons containing biotinylated EGR-1 ODN label from the box in (B). Note the large number of cells exhibiting uptake of the ODN. (E) Schematic of the behavioral protocol. (F) Post-shock freezing scores in rats infused with either scrambled ODN (250 pmol, n = 9) or EGR-1 antisense ODN (250 pmol, n = 5) immediately after the conditioning trial. (G) Auditory fear memory assessed at both 3 and 24 h after fear conditioning in each group. The black bars represent the scrambled ODN-infused groups, while the gray bars represent the EGR-1 antisense ODN-infused groups. *p < 0.05 relative to the scrambled ODN-infused groups. (H) Data depicting LTM as a percentage of STM for each rat in each group. *p < 0.05 relative to the scrambled ODN-infused groups. (I) Cannula placements for rats infused with scrambled ODN (black circles) or EGR-1 antisense ODN (gray circles). Panels adapted from Paxinos and Watson (1997)
.
Next, we used a biotinylated EGR-1 antisense ODN to assay the extent of EGR-1 antisense diffusion throughout the MGm/PIN. Figures 5
B–D depicts the diffusion of biotinylated EGR-1 antisense ODN in the MGm/PIN 30 min after infusion (250 pmol; 0.5 µl). The ODN remained remarkably confined to the borders of the MGm and PIN, largely sparing the adjacent MGv. At higher magnification, EGR-1 antisense ODN was observed to be localized to cell bodies and dendrites in the MGm/PIN, indicating that the ODN was actively taken up by cells (Figures 5
C,D).
In our behavioral experiments, we gave rats intra-MGm/PIN infusion of either scrambled ODN or EGR-1 antisense ODN 90 min prior to fear conditioning (250 pmol; 0.5 µl) and then tested each rat for retention of auditory fear conditioning 3 h (STM) and 24 h (LTM) later (Figure 5
E). Rats infused with either scrambled or EGR-1 antisense ODN had intact post-shock freezing after training (Figure 5
F). The ANOVA revealed only a significant effect of time (pre vs. post-training), F(1,12) = 42.39, p < 0.05. The effect of group [F(1,12) = 0.77, p > 0.05] and the time by group interaction [F(1,12) = 0.77, p > 0.05] were not significant.
When tested for retention of auditory fear conditioning 3 h after training, both scrambled and EGR-1 antisense-infused groups had intact STM (Figure 5
G). In contrast, during the 24-h retention test, the group infused with EGR-1 antisense ODN had impaired LTM relative to scrambled controls (Figure 5
G). The ANOVA (group by test) revealed a significant effect of group [F(1,24) = 5.17, p < 0.04], test [F(1,24) = 20.71, p < 0.01], and the group by test interaction [F(1,24) = 4.37, p < 0.05]. Duncan’s post hoc t-tests revealed that the scrambled and EGR-1 antisense-infused groups differed significantly on the LTM test (p < 0.05), but failed to differ on the STM test (p > 0.05). To further analyze this effect, we expressed each animal’s LTM score as a percentage of its STM score (Figure 5
H). As before, a significant difference emerged between the scrambled and EGR-1 antisense ODN-infused groups, t(12) = 2.52, p < 0.03. Thus, while knockdown of EGR-1 protein in the MGm/PIN has no effect on acquisition or STM formation of auditory fear conditioning, LTM is significantly impaired.
Cannula placements for rats infused with either scrambled or EGR-1 antisense ODNs are depicted in Figure 5
I.
Thalamic EGR-1 Expression Regulates Training-Induced Elevations in Presynaptically Localized Proteins in the LA
In this study, we have hypothesized that the functional significance of ERK-driven transcription in MGm/PIN neurons is to promote presynaptic aspects of plasticity and memory consolidation at thalamo-LA synapses. In support of the idea that fear conditioning is accompanied by presynaptic alterations, a recent report indicated that auditory fear conditioning is accompanied by enhanced expression of the presynaptically localized protein synaptophysin the LA (Nithianantharajah and Murphy, 2008
). In the present experiment, we have asked whether fear conditioning-induced alterations in the presynaptically localized proteins synaptophysin and synapsin in the LA are driven by ERK-driven gene expression at the level of the MGm/PIN. Of particular importance to the present paper, the synapsin family of genes, which consist of a group of at least five related members (Thiel, 1993
), has been shown to be regulated by EGR-1 (Thiel et al., 1994
; Petersohn et al., 1995
).
In the first experiment, we examined whether fear conditioning regulates the expression of both synapsin and synaptophysin proteins in LA homogenates (Figure 6
B). Relative to Naïve, Tone Alone, and Immediate Shock controls, we observed a significant increase in the expression of both synapsin and synaptophysin proteins in the LA in rats that received fear conditioning. The ANOVAs revealed a significant effect for each protein [synapsin (bottom band): F(3,25) = 5.90, p < 0.01; synapsin (top band): F(3,25) = 3.39, p < 0.05; synaptophysin: F(3,25) = 4.82, p < 0.01]. Duncan’s post-hoc t-tests revealed that Paired rats exhibited significantly higher expression relative to each of the other groups for each protein (p < 0.05). Further, no differences were observed between Naïve, Tone Alone, and Immediate Shock groups for either protein (p > 0.05).
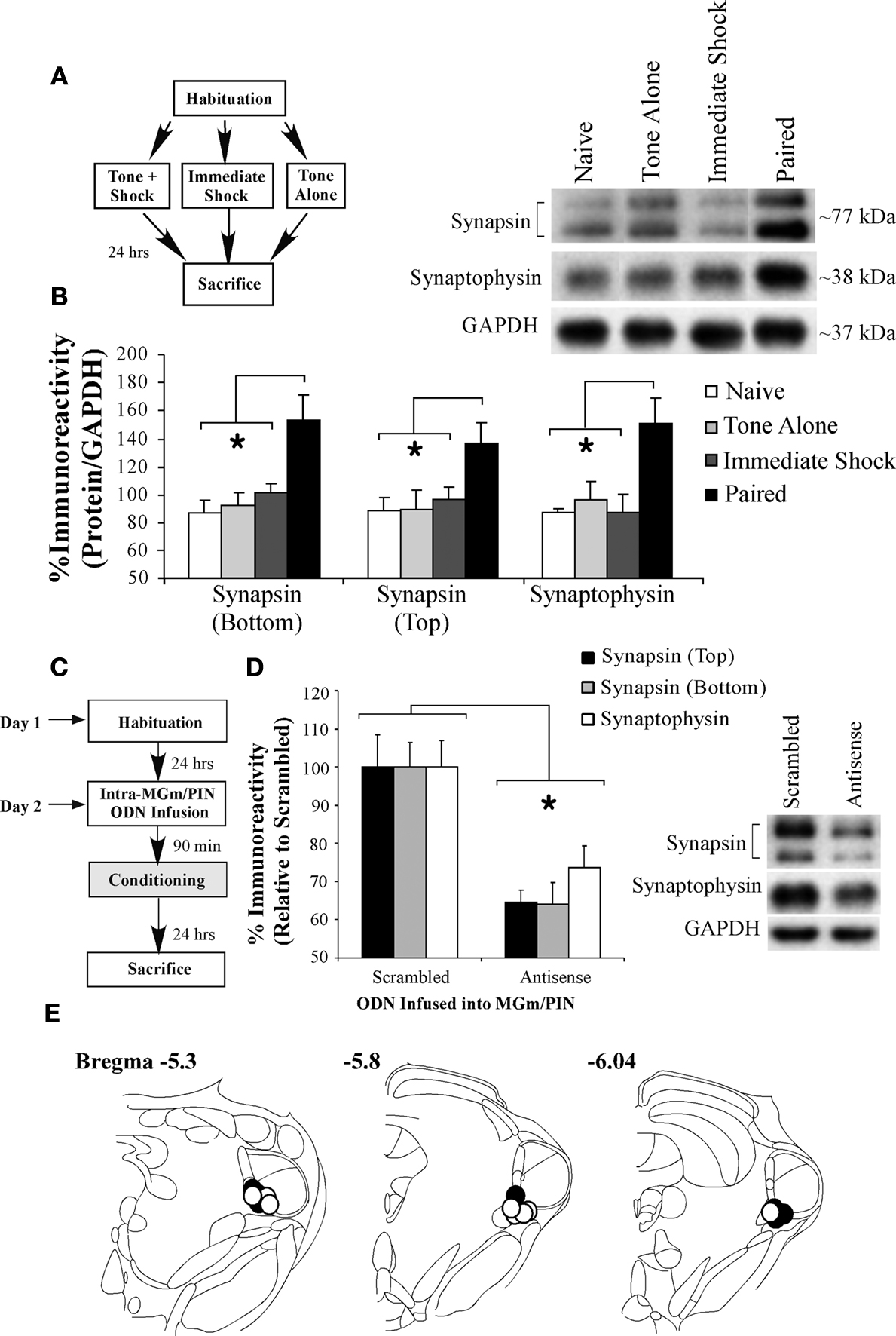
Figure 6. Intra-MGm/PIN infusion of an EGR-1 antisense ODN impairs training-induced elevation of presynaptically localized proteins in the LA. (A) Schematic of the behavioral protocol. (B) Western blot analysis of synapsin (top and bottom bands) and synaptophysin proteins in the LA of Naïve (n = 6), Tone Alone (n = 8), Imm. Shock (n = 8), and Paired (n = 7) groups. *p < 0.05 relative to the Paired group. Representative blots can be seen in the inset. (C) Schematic of the behavioral protocol. (D) Western blot analysis of synapsin (top band, black bars; bottom band, grey bars) and synaptophysin proteins (white bars) in the LA of rats that received either scrambled (n = 8) or EGR-1 antisense ODN (n = 8). *p < 0.05 relative to the scrambled group. Representative blots can be seen in the inset. (E) Cannula placements for rats infused with scrambled ODN (black circles) or EGR-1 antisense ODN (white circles). Panels adapted from Paxinos and Watson (1997)
.
In the second experiment, we asked whether training-induced expression of EGR-1 protein in the MGm/PIN is obligatory for the increase in presynaptically localized proteins in the LA that is observed 24 h after training. Rats were given intra-MGm/PIN infusion of either scrambled ODN or EGR-1 antisense ODN (250 pmol; 0.5 µl) 90 min prior to fear conditioning consisting of three tone-shock pairings. Twenty-four hours later, rats were sacrificed and LA homogenates were processed for synapsin and synaptophysin protein expression using Western blotting (Figure 6
C). Relative to scrambled ODN controls, we observed a significant decrease in the expression of both synapsin and synaptophysin proteins in the LA in the group that received intra-MGm/PIN infusions of the EGR-1 antisense ODN (Figure 6
D). The ANOVA (group by protein with repeated measure for protein) revealed a significant effect of group, F(1,14) = 16.93, p < 0.01. The effect of protein [F(2,28) = 0.30, p > 0.05] and the group by protein interaction [F(2,28) = 0.30, p > 0.05] were not significant. Further, this reduction in presynaptically localized proteins in the LA following intra-MGm/PIN knockdown of EGR-1 was not observed in naïve animals that did not receive fear conditioning (data not shown). Relative to naïve rats infused with scrambled ODN, naïve rats infused with EGR-1 antisense ODN exhibited intact expression of synapsin [top band: 83.2 ± 26.75%; bottom band: 94.6 ± 29.98%] and synaptophysin [98.32 ± 21.39%]. The ANOVA (group by protein with repeated measure for protein) revealed no significant effect of group [F(1,11) = 0.06, p > 0.05], protein [F(2,22) = 0.41, p > 0.05] or the group by protein interaction [F(2,22) = 0.41, p > 0.05].
Cannula placements for rats infused with either scrambled or EGR-1 antisense ODNs are depicted in Figure 6
E.
While studies employing in vitro preparations in both invertebrate and vertebrate models of synaptic plasticity have consistently implicated the NO-cGMP-PKG signaling pathway in coordinating plastic changes at pre- and post-synaptic sites, a convincing role of NO as a “retrograde signal” in mammalian memory formation has been difficult to establish. Using auditory fear conditioning, we have shown that NMDAR-mediated synaptic plasticity and NO signaling in the LA at the time of training drive changes in the expression of the ERK-driven immediate early gene EGR-1 in auditory thalamic projection neurons within the first several hours following fear conditioning. Further, functional knockdown of training-induced EGR-1 in the MGm/PIN impairs in parallel the consolidation of auditory fear conditioning as well as training-induced alterations in the presynaptically localized proteins synapsin and synaptophysin in the LA. Collectively, our findings support the hypothesis that NMDAR-driven synaptic plasticity and NO signaling in the LA drive changes in gene expression in thalamic projection neurons that are critical for fear memory formation by promoting presynaptic aspects of plasticity at LA synapses, and provide compelling support for a role of NO as a “retrograde signal” in associative fear memory formation.
Most recent studies in the fear conditioning literature have pointed to a critical role for NMDAR-driven regulation of protein kinase signaling pathways in LA neurons that are thought to promote fear memory consolidation, in part, by engaging activators of transcription in the nucleus (Rodrigues et al., 2004
). Other recent work, however, has suggested that intracellular signaling and transcriptional regulation in the MGm/PIN are equally important for fear memory formation (Apergis-Schoute et al., 2005
; Parsons et al., 2006
; Han et al., 2008
). The findings of the present study provide a means for reconciling these two findings. Specifically, our findings are consistent with a revised model of fear learning in which synaptic plasticity and NO signaling in the LA promote presynaptic aspects of fear memory formation at thalamo-LA synapses via regulation of ERK-driven gene expression in thalamic neurons (Figure 7
). Convergence of CS and US inputs in the LA during fear conditioning appears to engage not only NMDAR-mediated alterations in ERK-driven transcription and translation in LA principal neurons (Step 1) (Bailey et al., 1999
; Schafe and LeDoux, 2000
; Schafe et al., 2000
; Josselyn et al., 2001
), but also ERK-driven transcription in thalamic projection neurons (Apergis-Schoute et al., 2005
; Han et al., 2008
) via retrograde signaling by NO (Step 2) (Schafe et al., 2005
; Ota et al., 2008
). ERK-driven transcription in LA neurons is widely believed to lead to transcriptional changes in LA cells that are necessary to promote structural and/or functional changes on the postsynaptic side of LA synapses (Rodrigues et al., 2004
). Concurrently, downstream activation of the ERK/MAPK signaling pathway in presynaptic thalamic targets of LA neurons may lead to transcriptional changes in MGm/PIN cells (Steps 3, 4) that are necessary to promote structural and/or functional changes on the presynaptic side of LA synapses (Step 5). Together with the postsynaptic modifications driven by ERK signaling in the LA, these presynaptic modifications act to strengthen the connectivity of thalamo-LA synapses, which is reflected neurophysiologically in an enhanced response to the CS in the LA after training (Step 6). Interestingly, while in the present study pharmacological inhibition of synaptic plasticity (via ifenprodil) or NO signaling (via 7-Ni, c-PTIO or Rp-8-Br-PET-cGMPS) in the LA was shown to impair EGR-1 expression in the MGm/PIN, inhibition of ERK/MAPK (via U0126) in the LA had no effect (Figure 3
K). This intriguing pattern of findings suggests that a fairly circumscribed series of cellular events in LA neurons at the time of fear learning engage ERK-driven transcription in the MGm/PIN. This also suggests that there are two functionally distinct pools of ERK/MAPK, one in LA neurons and another in MGm/PIN neurons, that are required for fear memory consolidation and linked by a common upstream mechanism in the LA. While the experimental findings outlined in the present manuscript are restricted to an analysis of thalamo-LA synapses, it will also be of interest in future studies to examine the contribution of auditory cortical area TE3 to fear memory consolidation, as previous studies have suggested that cortical inputs to the LA may also undergo presynaptic alterations with fear conditioning (Huang and Kandel, 1998
; Tsvetkov et al., 2002
).
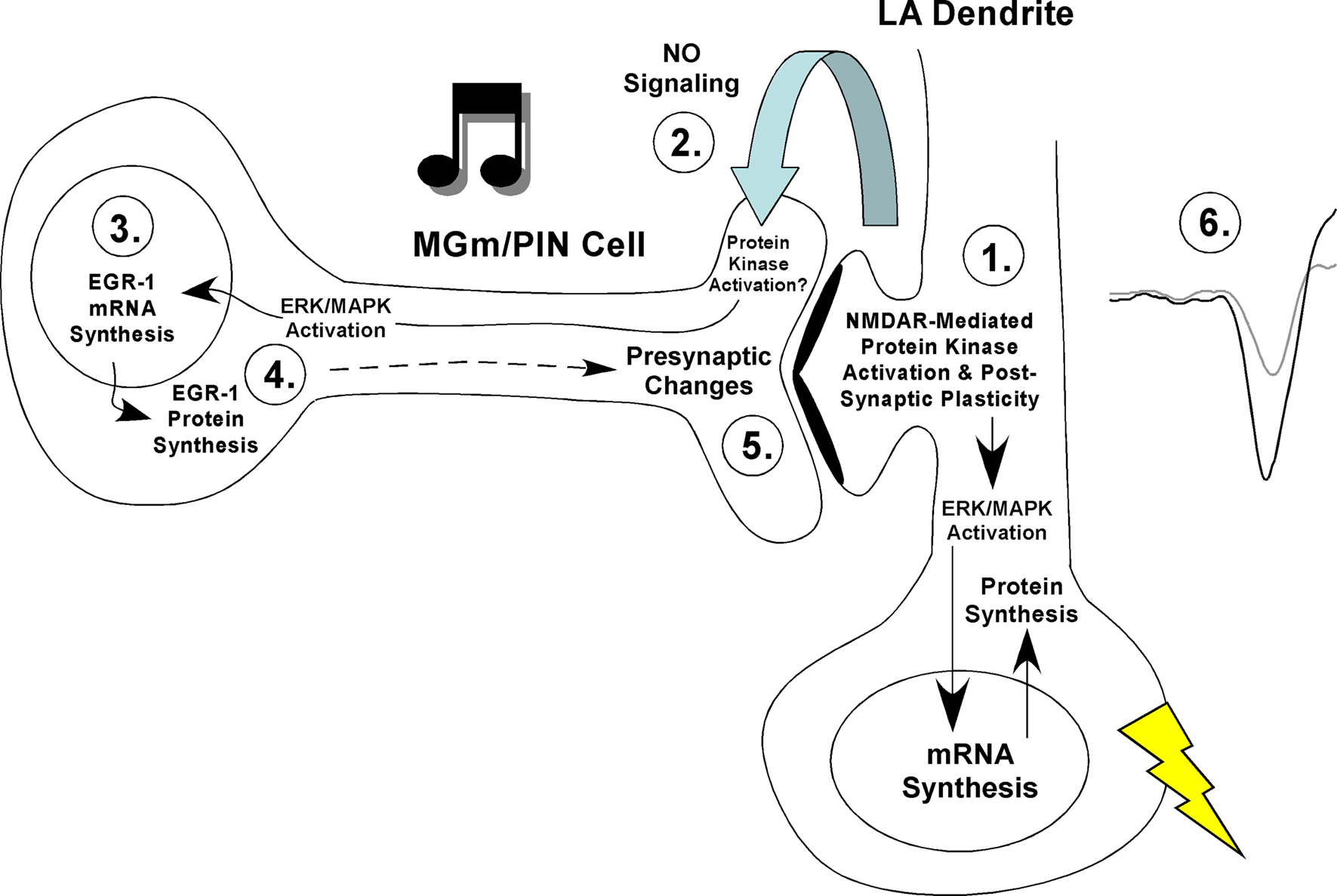
Figure 7. A model of fear memory consolidation. Fear memory consolidation is hypothesized to involve both pre- and postsynaptic modifications at thalamo-LA synapses. These modifications are first triggered by NMDAR-mediated alterations in protein kinase signaling pathways in LA neurons that ultimately promote postsynaptic functional and/or structural changes that contribute to the formation of the memory (Step 1). Second, NMDAR-driven synaptic plasticity in LA neurons is hypothesized to lead to the activation of nNOS in LA neurons and the release of nitric oxide (NO; Step 2), which can in turn engage ERK-dependent signaling and transcription in MGm/PIN neurons (Step 3). ERK-driven gene expression in MGm/PIN neurons (Step 4) is, in turn, hypothesized to promote presynaptic functional and/or structural changes at thalamo-LA synapses (Step 5). Together with the postsynaptic modifications driven by ERK signaling in the LA, these presynaptic modifications act to strengthen the connectivity of thalamo-LA synapses, which is reflected neurophysiologically in an enhanced response to the CS in the LA after training (Step 6).
Our findings are consistent with a large body of in vitro evidence which suggests that synaptic plasticity in vertebrate models of memory formation involves both pre- and postsynaptic alterations coordinated by extracellular signaling. Of particular relevance to the present manuscript, LTP induced by glutamate application in hippocampal cell cultures has been observed to lead to an increase in GluR1, synaptophysin, and synapsin I labeled puncta, as well as a corresponding increase in the number of sites where GluR1 and synaptophysin/synapsin I are co-localized (Antonova et al., 2001
). This increase in clusters of pre- and post-synaptically localized proteins is blocked by bath application of the NMDAR antagonists APV or MK-801 (Antonova et al., 2001
), and also by inhibitors of NO signaling (Wang et al., 2005
). Conversely, application of exogenous NO or cGMP analogs alone leads to an increase in clusters of both GluR1 and synaptophysin/synapsin I puncta (Wang et al., 2005
). Remarkably, these LTP-induced alterations in pre- and post-synaptic proteins occur very rapidly in culture (within 5 min), and are independent of protein synthesis (Antonova et al., 2001
). They are, however, blocked by inhibitors of actin polymerization (Antonova et al., 2001
) and associated with increases in the phosphorylation of the actin cytoskeleton regulators VASP and RhoA (Wang et al., 2005
). In our own experiments, we observed increases in both synapsin and synaptophysin in the LA at 24 h following fear conditioning; we did not examine the regulation of these proteins at very short intervals following fear learning. However, when considered collectively with the in vitro findings it is tempting to speculate that there may be two phases of structural plasticity following fear learning – one leading to rapid, protein synthesis-independent increases in clusters of pre- and post-synaptic proteins, and another leading to more permanent, protein synthesis-dependent modifications involving ERK-driven transcription at both sides of the thalamo-LA synapse. Interestingly, previous studies have reported that both stimulation and BDNF-induced LTP at dentate gyrus synapses leads to a robust activation of CREB and the protein synthesis-dependent expression of synaptophysin in the entorhinal cortex, events which have been hypothesized to be critical for promoting presynaptic morphological changes at entorhinal-granule cell synapses (Mullany and Lynch, 1997
; Gooney et al., 2004
).
One outstanding question in our revised model of fear learning is the critical locus of protein translation within MGm/PIN neurons during fear learning. Previous studies have shown that inhibition of ERK/MAPK or mRNA synthesis in the MGm/PIN impairs memory consolidation of auditory fear learning (Apergis-Schoute et al., 2005
). Conversely, over-expression of CREB, a downstream target of ERK signaling and a regulator of transcription, enhances auditory fear memory formation in the MGm/PIN (Han et al., 2008
). In the present study, we now show that antisense knockdown of the CREB-driven IEG EGR-1 in MGm/PIN neurons impairs fear memory consolidation. Paradoxically, however, most studies have failed to find an effect of protein synthesis inhibition in the MGm/PIN on fear memory formation (Maren et al., 2003
; Apergis-Schoute et al., 2005
), but see (Parsons et al., 2006
). One likely explanation of these negative findings may be that the timing of infusion in the previous studies (e.g. immediately after training) may not have been optimal (Maren et al., 2003
; Apergis-Schoute et al., 2005
). For example, it is quite possible that two rounds of translation are critical for fear memory formation in the LA; one that occurs shortly after training (via local translation at synapses) and another that is driven by de novo transcription hours later (Paul et al., 2007
). In the MGm/PIN, however, only the latter round of translation may be critical for fear memory formation. As a consequence, immediate post-training infusion of a protein synthesis inhibitor into the LA would be expected to impair fear memory formation (Schafe and LeDoux, 2000
), while that in the MGm/PIN would not (Maren et al., 2003
; Apergis-Schoute et al., 2005
). Additional experiments using multiple training-to-infusion intervals will be required to more thoroughly explore the role of protein synthesis in the MGm/PIN during fear memory formation.
A second outstanding question is the nature of the presynaptic target of NO signaling at thalamo-LA synapses in our revised model of fear learning. Our findings suggest that it cannot be ERK itself; intra-LA infusion of the MEK inhibitor U0126, which would presumably affect ERK activation both pre- and post-synaptically, has no effect on training-induced elevations of EGR-1 in the MGm/PIN (Figure 3
K). Two additional candidates are PKG and αCaMKII. Recent findings suggest that presynaptic αCaMKII is a critical substrate for LTP induced by the NO-cGMP-PKG signaling pathway. Presynaptic injection of an αCaMKII inhibitor peptide blocks both LTP and accompanying presynaptic morphological alterations induced by an NO donor or cGMP analog (Ninan and Arancio, 2004
), suggesting that αCaMKII may act downstream of cGMP-PKG signaling to promote presynaptic aspects of plasticity in CA1. Our lab has recently shown that intra-LA infusion of either an inhibitor or activator of cGMP-PKG signaling impairs or enhances, respectively, LTP at thalamo-LA synapses, fear memory consolidation, and ERK activation in the LA (Ota et al., 2008
). Similarly, fear conditioning has been shown to regulate the autophosphorylation of αCaMKII at postsynaptic sites, while inhibition of CaMKII activity in the LA impairs fear memory formation and synaptic plasticity at thalamo-LA synapses (Rodrigues et al., 2004
). While these findings suggest that signaling via PKG and αCaMKII are critical regulators of postsynaptic signaling in LA neurons (Rodrigues et al., 2004
; Ota et al., 2008
), these same pathways may also be critical for regulating ERK-driven gene expression and associated presynaptic aspects of plasticity in MGm/PIN neurons following fear learning.
While the findings of the present manuscript point to a role for ERK-driven gene expression at the level of the MGm/PIN in contributing to presynaptic aspects of plasticity back at the level of the LA, they may also shed light on the cellular mechanisms underlying the distinct yet potentially related phenomenon of neurophysiological alterations in MGm/PIN neurons that accompany fear learning. It has long been appreciated, for example, that auditory fear conditioning induces neurophysiological alterations not only in the LA (Quirk et al., 1995
; Rogan et al., 1997
; Maren, 2000
; Maren and Quirk, 2004
), but also in regions of the auditory thalamus (Gabriel et al., 1975
; Edeline and Weinberger, 1992
; Weinberger, 1993
); that is, neurons in each of these regions exhibit enhanced responding to a tone following fear conditioning. In the present study, we have proposed that the neurophysiological changes that are observed within the LA rely on coordinated pre- and post-synaptic alterations at thalamo-LA synapses induced by NMDAR-driven synaptic plasticity and NO signaling within the LA at the time of training. However, one intriguing possibility is that synaptic plasticity and NO signaling in the LA may also promote the acquisition and/or retention of neurophysiological changes in MGm/PIN neurons by altering their intrinsic excitability. Studies in Aplysia, for example, have shown that training-induced alterations in the excitability of (presynaptic) sensory neurons are blocked by injection of BAPTA into the postsynaptic neuron (Antonov et al., 2003
), suggesting that a postsynaptically-generated transynaptic signal may be sufficient to drive intrinsic changes in presynaptic excitability. While it remains an open question whether a similar mechanism underlies training-induced changes in the MGm/PIN following fear learning, it is noteworthy that previous studies have reported that either lesions or functional inactivation of the LA disrupt training-induced neurophysiological changes in the MGm/PIN (Maren et al., 2001
; Poremba and Gabriel, 2001
). Future experiments employing pharmacological manipulations combined with neurophysiological recordings at both sides of the thalamo-LA synapse in awake-behaving animals will be required to further examine this important question.
In summary, the findings of the present study clearly suggest that NMDAR-driven synaptic plasticity and NO signaling in the LA drive ERK-mediated transcription in the MGm/PIN that is required for both fear memory consolidation as well as presynaptic correlates of memory formation in the LA. These findings reveal a potential biochemical mechanism whereby intracellular signaling pathways in the LA at the time of fear learning may engage presynaptic aspects of plasticity at thalamo-LA synapses. These findings make an additional contribution towards understanding the cellular and molecular processes underlying emotional memory formation in the mammalian brain, and also provide support for the hypothesis that NO signaling serves as a “retrograde messenger” in mammalian memory formation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Kathie A. Overeem and Kristie T. Ota contributed equally to this work. This research was supported by the National Science Foundation (NSF 0444632 to Glenn E. Schafe), the National Institutes of Health (MH 073949 to Glenn E. Schafe), and by Yale University. Kristie T. Ota is supported by a National Science Foundation Graduate Research Fellowship. Kathie A. Overeem is supported by the New Zealand Tertiary Education Commission T.A.D Scholarship.
Apergis-Schoute, A. M., Debiec, J., Doyere, V., LeDoux, J. E., and Schafe, G. E. (2005). Auditory fear conditioning and long-term potentiation in the lateral amygdala require ERK/MAP kinase signaling in the auditory thalamus: a role for presynaptic plasticity in the fear system. J. Neurosci. 25, 5730–5739.
Paul, S., Olausson, P., Venkitaramani, D. V., Ruchkina, I., Moran, T. D., Tronson, N., Mills, E., Hakim, S., Salter, M. W., Taylor, J. R., and Lombroso, P. J. (2007). The striatal-enriched protein tyrosine phosphatase gates long-term potentiation and fear memory in the lateral amygdala. Biol. Psychiatry 61, 1049–1061.