Department of Psychology & Neuroscience, Duke University, Durham, NC, USA
The immune system is well characterized for its critical role in host defense. Far beyond this limited role however, there is mounting evidence for the vital role the immune system plays within the brain, in both normal, “homeostatic” processes (e.g., sleep, metabolism, memory), as well as in pathology, when the dysregulation of immune molecules may occur. This recognition is especially critical in the area of brain development. Microglia and astrocytes, the primary immunocompetent cells of the CNS, are involved in every major aspect of brain development and function, including synaptogenesis, apoptosis, and angiogenesis. Cytokines such as tumor necrosis factor (TNF)α, interleukin [IL]-1β, and IL-6 are produced by glia within the CNS, and are implicated in synaptic formation and scaling, long-term potentiation, and neurogenesis. Importantly, cytokines are involved in both injury and repair, and the conditions underlying these distinct outcomes are under intense investigation and debate. Evidence from both animal and human studies implicates the immune system in a number of disorders with known or suspected developmental origins, including schizophrenia, anxiety/depression, and cognitive dysfunction. We review the evidence that infection during the perinatal period of life acts as a vulnerability factor for later-life alterations in cytokine production, and marked changes in cognitive and affective behaviors throughout the remainder of the lifespan. We also discuss the hypothesis that long-term changes in brain glial cell function underlie this vulnerability.
Developmental or “fetal programming” is an area of research that has received increasing interest in recent years. This field has emerged based on the idea that experience during the perinatal period may modulate or “program” the normal course of development, with the result that adult outcomes, including behavior, are significantly and often permanently altered (Bennet and Gunn, 2006
). Much of this work has been done in relation to the long-term consequences of early-life stress and resulting glucocorticoid exposure, maternal interactions, and fetal growth (Champagne, 2008
; de Boo and Harding, 2006
; Owen et al., 2005
, and see this issue). In contrast, the role of the immune system in neurodevelopment and potential effects on brain and behavior in adulthood remains relatively unexplored. Nonetheless, there is strong evidence that perinatal exposure to infectious agents has a number of influences on later life, including reactivity to stress, disease susceptibility, and notably, increased vulnerability to cognitive and/or neuropsychiatric disorders, including Alzheimer’s, Parkinson’s, schizophrenia, and autism (Hornig et al., 1999
; Nelson and Willoughby, 2000
; Rantakallio et al., 1997
; Shi et al., 2003
).
Beyond its traditional role in host defense, the immune system is now considered a diffuse sensory organ and regulator of metabolism, which works in concert with the nervous system to achieve and maintain homeostasis throughout the body (Husband, 1995
; Vitkovic et al., 2000
). Immunocompetent cells are located throughout virtually every organ of the body, including the brain, and sophisticated interactions occur among these cells, primarily via soluble protein messengers called cytokines. Important for this review, bidirectional communication between the brain and immune system by cytokines has significant consequences for behavior. For instance, sick animals exhibit several well-characterized behavioral changes, including reductions in food and water intake, activity, exploration, and social and sexual interactions (Hart, 1988
; Kent et al., 1992
). Importantly, these so-called “sickness behaviors” are not mediated by the infectious pathogens themselves, but rather are orchestrated by the host via cytokines (Dantzer and Kelley, 2007
). Numerous studies have now demonstrated that these behaviors are organized, adaptive strategies that are critical to host survival (Dantzer and Kelley, 2007
; Nesse and Williams, 1994
). Thus, sickness behavior reflects an overall change in the motivational state of the individual that is organized by both the nervous and the immune systems.
We believe that cytokines are important not only for behavioral changes during acute illness, but may also underlie long-term changes in behavior as a consequence of infection early in life. Thus, the purpose of this review is to: (1) summarize the evidence that infections occurring during the perinatal period can produce effects on brain and subsequent behavior that endure throughout an organism’s life span, and (2) discuss the potential role of cytokines and glia in these long-term changes. Cytokines are produced within the brain during normal brain development, but are expressed at much higher levels during the course of an immune response. In contrast to overt neural damage, we present data indicating that increased cytokine exposure during key periods of brain development may also act as a “vulnerability” factor for later-life pathology, by sensitizing the underlying neural substrates and altering the way that the brain responds to a subsequent immune challenge in adulthood. In turn, this altered immune response has significant and enduring consequences for behavior, including social, cognitive, and affective abilities. We discuss the evidence that one mechanism responsible for enduring cytokine changes is chronic activation of brain microglia, the primary immunocompetent cells of the CNS.
A link between infection and neuropsychiatric disorders has a long history, beginning as early as the establishment of the germ theory of diseases (e.g., “the infectious theory of psychosis”, recently reviewed by Yolken and Torrey, 2008
), a full discussion of which goes beyond the scope of this paper. However, it is worth noting that one of the strongest relationships in the literature for a specific developmental origin of psychopathology is that between infection and the later presentation of schizophrenia (Fruntes and Limosin, 2008
; Meyer et al., 2005
; Yolken and Torrey, 2008
). Such a connection may have first been proposed by Scottish psychiatrist Thomas Clouston in 1891, in which he suggested a developmental component to “adolescent insanity” (Bennet and Gunn, 2006
). This literature has recently been extensively reviewed (Fatemi and Folsom, 2009
). Briefly, the mechanisms by which infections may lead to psychopathology, either alone or in genetically vulnerable individuals, include direct infection of the developing fetus and subsequent abnormal neural development, the generation of auto-antibodies by the mother that subsequently react with fetal neural tissue, and, notably, cytokine production, which may be involved in all of these mechanisms (Pearce, 2001
). There are several reports in humans that influenza infection induces cytokine production by the maternal immune system, the fetal immune system, and the placenta (Hillier et al., 1993
; Urakubo et al., 2001
), and each has been linked to increased risk of schizophrenia in the offspring (Brown et al., 2004
). Similarly, influenza administered to pregnant mice increases cytokine levels in placental tissue and amniotic fluid, and results in abnormal development of the offspring (Shi et al., 2003
). Notably, a replicating virus is not required; rats exposed prenatally to double-stranded RNA, a viral mimic and synthetic cytokine inducer, exhibit hippocampal dysmorphology and an impaired ability to filter irrelevant information, traits characteristic of schizophrenia in humans (Zuckerman and Weiner, 2003
). It should be noted that there remains considerable debate about this topic, as the majority of individuals that suffer from infections early in life do not develop schizophrenia (Bennet and Gunn, 2006
). Nonetheless, it is clear that a link is present in at least a subset of individuals, and these combined data from the schizophrenia literature have in large part set the stage for an “immune origins of neurodevelopmental disorders hypothesis” (Meyer et al., 2005
), which extends more broadly to a number of cognitive and affective disorders.
Normal Brain Development
Researchers continue to identify ways in which the immune system influences normal brain development (Merrill, 1992
). Developmental and region specific regulation of cytokine expression has been demonstrated (Pousset, 1994
), suggesting an important role for individual cytokines in the development of specific brain regions. IL-1β is expressed at high levels in the developing CNS, but at very low (constitutive) levels in the adult brain (Giulian et al., 1988
; Schmitz and Chew, 2008
). Cytokines are critically involved in neuronal and glial cell migration, differentiation, synaptic maturation, and many other important processes of brain development (Nawa and Takei, 2006
). Glial cells also play crucial roles in a wide variety of developmental processes, including apoptosis, axonal growth, and angiogenesis (Rakic and Zecevic, 2000
; Streit, 2001
). Astrocytes are critical for the formation of synapses during development (Ullian et al., 2004
), and microglial development occurs in parallel with neurogenesis in most brain structures (Rice and Barone, 2000
). Finally, we know that proteins of the classical complement cascade, important for innate immunity and pathogen elimination in the periphery, are also required for developmental synapse elimination within the CNS (Stevens et al., 2007
).
Cytokines and Infection
Given their crucial role in brain development, it is not surprising that elevated levels of pro-inflammatory cytokines generated by the maternal or fetal immune system during perinatal infection have been associated with abnormal fetal brain development and increased risk for neurodevelopmental disorders (Cai et al., 2000
; Meyer et al., 2006
; Pang et al., 2003
; Richardson-Burns and Tyler, 2004
; Urakubo et al., 2001
; Yu et al., 2004
). For instance, concentrations of IL-1β, IL-6, and tumour necrosis factor (TNF) are elevated in infants with severe perinatal complications (Miller et al., 1990
), and children with bacterial meningitis have elevated levels of IL-1β that strongly correlate with the occurrence of neurological disorders (Mustafa et al., 1989
). Increased levels of IL-6 in amniotic fluid have also been a clinically useful marker of increased risk for neurological disorders and morbidity (Yoon et al., 1995
). Lipopolysaccharide (LPS), the cell wall component of gram-negative bacteria, has been used to mimic infection in many studies because it initiates a rapid and well-characterized immune response (see Wang et al., 2006
for review). LPS markedly induces cytokine expression in the immature rat brain, which has been linked to white matter injury and cerebral palsy (Cai et al., 2000
; Fidel et al., 1994
; Golan et al., 2005
; Urakubo et al., 2001
). LPS also induces a number of long-term behavioral changes, which we discuss more fully below. Importantly, in each of these cases the infectious agents themselves do not enter the fetal circulation, thus implicating the critical role of the maternal immune response. Furthermore, the strong association between infection and developmental disorders spans a diverse number of bacterial and viral agents, suggesting a common mechanism such as cytokine release.
Bacterial infections represent the number one cause of infection in newborns worldwide, and are a significant cause and consequence of premature birth, which have increased 30% in the past 25 years (Osrin et al., 2004
; Skogstrand et al., 2008
). Human newborns are particularly impaired in their abilities to mount an immune response against the polysaccharide regions of bacteria, which results in significant infant mortality, particularly in developing countries (Marshall-Clarke et al., 2000
; Osrin et al., 2004
). Major recent advances in maternal and perinatal medicine have greatly increased survival rates among these populations in developed countries. However, it remains to be determined what the total impact of infection during the perinatal period may have on subsequent physiology and behavior in individuals (see Adams-Chapman and Stoll, 2006
; Hagberg and Mallard, 2005
for review). We have developed a model of postnatal bacterial infection in order to explore these questions. Rats are injected subcutaneously on postnatal day (P) 4 with phosphate-buffered-saline (PBS) or a non-lethal dose of live Escherichia coli. P4 was originally selected based on models of neonatal LPS challenge in rats (Hodgson et al., 2001
; Shanks et al., 2000
), and represents a time relatively comparable to the third trimester in humans, during which significant brain growth occurs (Dobbing and Sands, 1979
; Rodier, 1980
). Thus, the newborn rat is arguably a good model for human premature infants (Vaccarino and Ment, 2004
). E. coli is the number one cause of infection in low-birth weight premature infants in the US (Stoll et al., 2005
), which is largely resistant to antibiotics (Stoll et al., 2005
).
Our data have demonstrated that neonatal E. coli infection in pups markedly increases circulating cytokines (IL-1β, IL-6, TNF) and the primary stress hormone, corticosterone, for at least 48 h after infection (Bilbo et al., 2005a
). Within the brain, there is a significant and specific increase in IL-1β mRNA and protein (Bilbo et al., 2005a
), but not in other analyzed cytokines (IL-6, TNF), compared to PBS injection. These results suggest that IL-1β may be a key mediator of events occurring within the developing brain in response to infection at this time. These data are intriguing given evidence that IL-1β levels are naturally elevated in the neonatal brain, peaking around P2, and declining thereafter into adulthood (Giulian et al., 1988
). Thus, neonatal infection leads to exaggerated IL-1β production at a time that a natural developmental peak occurs, suggesting that this may be a sensitive period for influences on this particular cytokine. Further support for this idea is evidence that human placenta from full-term infants increases the release of IL-1β receptor antagonist compared to IL-1β in response to LPS, whereas preterm infant placenta increases both equally (and is thus more inflammatory), suggesting the early fetal period is quite distinct from later developmental time points in terms of cytokine expression (Holcberg et al., 2008
).
Cognitive Impairment
What enduring effects does such exposure have on the brain, and ultimately on behavior? Sepsis in infants was first linked to white matter damage in the 1970s (Leviton and Gilles, 1973
), and there is now a strong literature linking perinatal infection to cerebral palsy (see Garnier et al., 2003
; Huleihel et al., 2004
for review). Beyond these major developmental impairments, one of the most common consequences of perinatal infection/inflammation generally is cognitive dysfunction, including learning, memory, and attention disorders (Bauman et al., 1997
). E. coli infections in particular in premature infants are associated with significant neurodevelopmental delays, which occur regardless of infection location (e.g., brain, blood, or intestines; Stoll et al., 2004
). Pro-inflammatory cytokines in the amniotic fluid and/or neonatal blood stream are cited as the primary predictor of adverse outcomes (Deguchi et al., 1996
; Leviton and Dammann, 2002
). Cytokine receptors are distributed throughout the brain, but have one of the highest densities in the hippocampus (Cunningham and De Souza, 1993
; Schneider et al., 1998
). The hippocampus also exhibits marked vulnerability (compared to other brain regions) to events such as severe stress, epilepsy, stroke, and cardiac arrest (Fujioka et al., 2000
; Petito et al., 1987
; Salmenpera et al., 1998
; Sapolsky et al., 1990
). Thus the hippocampus, and thus cognition, may be particularly vulnerable to immune-related alterations (Lynch et al., 2004
; Morrison and Hof, 1997
).
One implication of the previous discussion is that early-life infection might influence the formation of memories that depend on the hippocampus in adulthood. Initially, we considered two possible ways by which neonatal infection may influence memory: (1) it could directly influence the neural pathways supporting memory or, (2) it could alter how the adult animal responds to a subsequent immune challenge, thereby affecting the processes that support memory. Our initial experiments centered on a modified version of contextual fear conditioning known as the context pre-exposure task (Fanselow, 1990
; Rudy et al., 2004
). This paradigm assesses the rat’s memory for a recently explored context. In this task, when a rat is placed into a conditioning context and immediately shocked, he later displays little or no conditioned fear (freezing) to the context. This absence of fear to the context is thought to occur because an immediately shocked rat does not have the opportunity to sample the environment and thus store a representation of its features. However, if a rat is pre-exposed to the context for several minutes the day before, immediate shock conditioning will then produce substantial freezing on a subsequent test day (Fanselow, 1990
; Rudy and O’Reilly, 2001
; Westbrook et al., 1994
). Importantly, each stage of this task is critically dependent on the hippocampus (Rudy and O’Reilly, 2001
; Rudy et al., 2002
, 2004
).
In order to discriminate between the two possible ways an early-life infection might influence memory, we designed our experiments so that we could determine whether the E. coli infection itself affected memory permanently throughout the remainder of the lifespan, or if it affected instead the adult’s immune response to a subsequent infectious challenge, which might then affect memory. In the latter case, the memory impairment would only be “unmasked” by a subsequent immune challenge. Rats were treated on P4 with PBS or E. coli as described previously, and were then as adults pre-exposed to a novel environment on the first day of testing. Immediately after pre-exposure, rats from each neonatal group received no injection, saline, or a low-dose immune challenge (25 μg/kg intraperitoneal bacterial LPS). Thus, if infection on P4 altered the neural pathways that support memory formation, then these rats should display impaired memory regardless of whether they received the LPS immune challenge after pre-exposure. However, if the infection on P4 altered how the adult immune system responds to the second challenge, then only rats that experience the combination of the infection on P4 and LPS after training should display impaired memory. As shown in Figure 1
A, rats that experienced the combination displayed impaired memory for the explored context. In contrast, rats that only experienced the P4 infection were not affected, identical to PBS controls in any condition. These data support option 2 above, i.e., that some aspect of the immune reaction is itself interfering with memory.
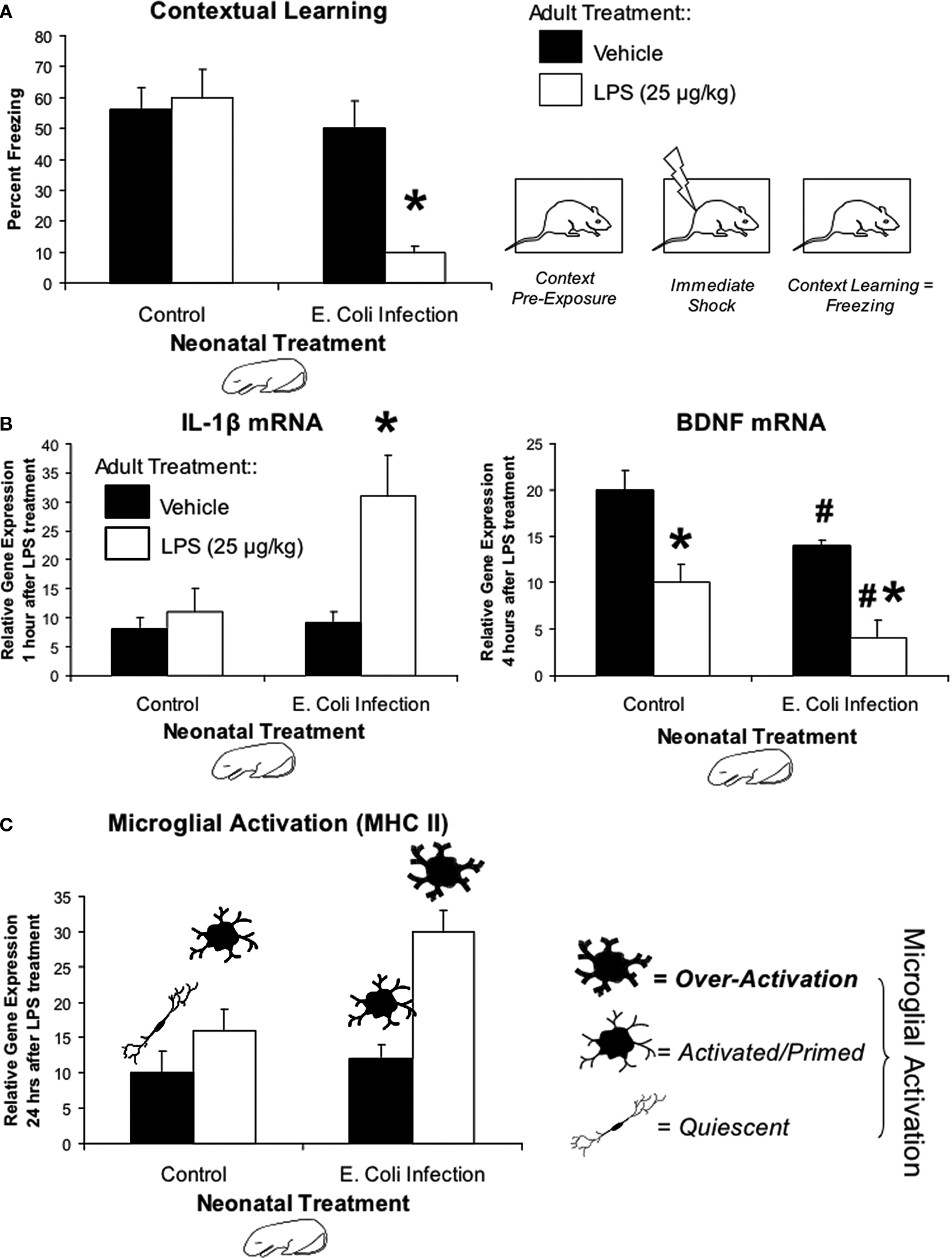
Figure 1. Early-life infection alters memory, cytokine expression, and microglial activation after a subsequent immune challenge in adulthood. (A) Mean (+SEM) percent freezing during the context pre-exposure task. Neonatal E. coli infection impairs memory formation in adulthood only in the presence of a subsequent immune challenge, LPS (white bars; *p < 0.05 from all other groups) (adapted from Bilbo et al., 2005a
). (B) Mean (+SEM) relative gene expression of IL-1β and BDNF in the hippocampus during the context pre-exposure task. Neonatal E. coli infection induces a more than three-fold increase in the transcription of IL-1β only when exposed in adulthood to LPS (white bars). Animals infected neonatally with E. coli also show the greatest decrease in hippocampal BDNF levels 4 h after context pre-exposure and subsequent LPS (*p < 0.05 compared to Vehicle treated animals from same neonatal treatment group; #p < 0.05 compared to Control) (adapted from Bilbo et al., 2008a
). (C) Mean (+4 SEM) relative gene expression of MHC II in the hippocampus, a marker of microglial activation. MHC II activity in response to a vehicle injection (black bars) is low in both groups. Neonatal E. coli infection combined with adult LPS (white bars) significantly increases microglial activation above all other treatment groups (*p < 0.05). These data are consistent with the idea of glial priming, illustrated by the cartoon pictorials, in that neonatal infection can permanently activate or “prime” microglia leading to an exaggerated response to LPS.
Additional behavioral experiments have revealed the selective nature of the impairment. The combination of P4 infection and adult LPS impaired the long-term (48 h) but not the short-term (1 h) memory for a contextual fear experience, indicating that the rats initially learned the association, but that long-term consolidation was impaired or prevented. Moreover, this treatment had no effect on conditioned fear to a tone paired with shock. This finding is important because conditioning to a tone does not depend on the hippocampus (Bilbo et al., 2006
). Collectively these results support the hypothesis that the P4 infection leads to selective impairment of a long-term memory that depends on the hippocampus.
Three additional points should also be mentioned: First (and most critical), an infection later in development, on P30, does not lead to LPS-induced memory impairment later in life (Bilbo et al., 2006
). These data indicate that the early-infection induced vulnerability is specifically a developmental effect, and not a general sensitizing event that can occur at any time. Second, no differences in overall activity, basal anxiety, or exploration have been observed in adulthood as a consequence of the neonatal infection (Bilbo et al., 2006
). Third, we have also observed no differences in spatial learning or memory using a water maze task performed in the absence of an adult LPS injection (Bilbo et al., 2007
). This is consistent with our findings that neonatal infection by itself has no overt effects on hippocampal memory formation in adulthood. However, we did find that P4 E. coli infection prevents a beneficial/augmenting effect of neonatal handling (brief, daily separation of pups from mom) on both anxiety and spatial memory in adulthood, a paradigm that consistently enhances learning and memory and decreases anxiety (Caldji et al., 1998
; Fish et al., 2004
; Liu et al., 2000
). In summary, the collective results yield two important conclusions. First they support the hypothesis that the infection occurring during a sensitive period of life leads to a selective impairment in the consolidation of memories that depend on the hippocampus in adulthood. Second, the early infection produces a latent or hidden change in the immune system that is unmasked by either a second immune challenge or by preventing the effectiveness of the handling intervention. These data are well in accord with much of the human schizophrenia literature, which posits that a combination of early perinatal plus young-adult challenge (e.g., stress, infection) is required for the manifestation of the illness, the so-called “two-hit hypothesis” (Choy et al., 2009
; Keshavan, 1999
; Maynard et al., 2001
; Pantelis et al., 2003
).
A growing body of evidence suggests a role for cytokines, notably IL-1β, in normal, non-pathological, synaptic plasticity mechanisms within the brain, including memory (McAfoose and Baune, 2009
). IL-1β is induced within the hippocampus in response to fear conditioning (Goshen et al., 2007
), and is critical for long-term potentiation (LTP) maintenance during learning (Ross et al., 2003
; Spulber et al., 2009
). Mice lacking endogenous IL-1β or its receptor, or with overexpression of the endogenous IL-1 receptor antagonist (IL-1ra), exhibit markedly impaired hippocampal-dependent learning and memory (Goshen et al., 2007
; Spulber et al., 2009
).
In contrast to these data, exaggerated IL-1β within the brain is also strongly associated with memory impairment. IL-1β is the most consistently induced cytokine within the brain following immune stimulation (Abraham and Williams, 2003
), but is very tightly regulated in the normal brain, suggesting that any dysregulation will result in pathology. Patients with AIDS-related dementia, cancer, chronic inflammatory diseases (e.g., Alzheimer’s), or autoimmune diseases often exhibit exaggerated levels of IL-1β co-occurring with cognitive impairment (Gallo et al., 1989
; Griffin et al., 1989
; Meyers, 2000
; Stanley et al., 1994
). Exogenously applied IL-1β inhibits LTP in vitro within major hippocampal pathways (Cunningham et al., 1996
; Katsuki et al., 1990
). Similarly, peripheral high dose LPS interferes with LTP in vivo, with this effect depending on hippocampal IL-1β production (Vereker et al., 2000
). Finally, rats injected with high levels of IL-1β directly into the dorsal hippocampus display memory impairments (Barrientos et al., 2002
; Pugh et al., 1999
), as do rats that receive other treatments that induce hippocampal IL-1β (Pugh et al., 1998
).
Based on these data, we explored whether IL-1β production in the hippocampus is dysregulated in response to the LPS challenge in adulthood as a consequence of the early-life infection. We find no detectable IL-1β protein within the hippocampus of either group following a saline injection, indicating that neonatal infection does not result in a chronic exaggerated IL-1β response within the brain. However, in response to LPS, early-infected rats exhibit a prolonged expression of IL-1β protein specifically within the hippocampus and adjacent association cortex (Figure 1
B). At the messenger RNA level, the genes encoding for IL-1β, IL-1β converting enzyme (caspase-1), and the IL-1β type I receptor, are all significantly elevated after LPS within the hippocampus of early-infected rats compared to controls. Notably, however, mRNA for the anti-inflammatory IL-1ra is not significantly elevated above controls (Bilbo et al., 2007
). Importantly, neither splenic/circulating cytokines nor corticosterone production to the adult LPS challenge are altered by the neonatal infection, suggesting that these cytokine alterations occurring within the brain are not simply a consequence of an altered cytokine signal to the brain from the periphery (Bilbo et al., 2005a
). Thus, the early-life infection appears to create a shift towards a pro-inflammatory bias in the adult brain. The importance of these findings to the memory impairment is confirmed by preventing the synthesis of IL-1β prior to the LPS in conjunction with learning, which completely prevents the memory impairment (Bilbo et al., 2005a
).
How might exaggerated brain IL-1β expression interfere with memory? IL-1β injected directly into the hippocampus dramatically suppresses the induction of BDNF following contextual learning, a molecule critical for long-term memory formation (Barrientos et al., 2004
). IL-1β also interferes with the neuroprotective actions of BDNF when applied to neuronal cultures (Tong et al., 2008
). We have reported that prior to conditioning, basal BDNF mRNA does not differ in adults as a consequence of early infection. However, following fear conditioning, adult LPS decreases BDNF mRNA induction in all hippocampal regions of neonatally-infected rats compared to controls, and induces a faster decay within CA1. This decrease in E. coli rats is accompanied by a large increase in IL-1β mRNA in CA1 (Bilbo et al., 2008a
).
Taken together, our results suggest that excess IL-1β at the time of learning can set in motion a cascade, involving BDNF and likely other cellular players, that ultimately impairs memory. Importantly, PBS controls did also increase IL-1β protein in response to the low-dose LPS, but this increase did not impair memory. Collectively, these data provide support for an inverted “U” function for IL-1β and memory, with physiological levels being important for memory, and any deviation from this range resulting in pathology and impairment (Goshen et al., 2007
; Spulber et al., 2009
). Early-life infection appears to sensitize the “cytokine thermostat”, so to speak, with profound implications for optimal production within the brain and ultimately, cognitive function.
Our results suggest that a neonatal infection can alter the developmental trajectory of the immune system and the brain. They support the idea that whereas direct neurological injury is often associated with severe neonatal infection (e.g., Borna Disease Virus or high dose LPS), mild-to-moderate infection and subsequent cytokine exposure during key periods of brain development may result in more subtle alterations, such as altering the response or vulnerability to a subsequent insult (Hagberg and Mallard, 2005
). For instance, low-dose LPS administration in the immature brain several days prior to hypoxia/ischemia results in far more damage than ischemia alone (i.e., sensitization) (Cai et al., 2000
; Eklind et al., 2001
). In contrast, pretreatment with pro-inflammatory cytokines has also been shown to protect against subsequent cerebral ischemia or cytotoxic insult, a phenomenon known as preconditioning (Hagberg et al., 2004
). Whether an inflammatory event within the CNS confers either resilience or vulnerability to a later challenge is a key unanswered question. In the next sections we review some of the potential pathways whereby neonatal infection might exert its influence on long-term changes in IL-1β, and subsequent behavioral changes.
Cytokines and Glia
One of the most consistent changes associated with perinatal infection is glial activation (Billiards et al., 2006
). Interestingly, one of the first physicians to suggest a relationship between infection and later psychosis in the late 19th century cited “changes in glia” as one 4 primary anatomical changes within the brain (Kraepelin, 1919
). While virtually ignored for years by the neuroscience community, the literature on glia has now reached a critical mass; to name but two recent reviews, glia are described as critical for “every major aspect of brain development, function, and disease” (Barres, 2008
), as “central in brain homeostasis”, and that, “brain pathology is, to a very great extent, a pathology of glia” (Giaume et al., 2007
). Microglia are the primary immunocompetent cells of the brain, existing under non-pathological conditions in a resting, highly ramified morphology, with low or non-detectable expressions of cell surface “activation” markers (e.g., CD11b [complement 3 receptor/CR3], major histocompatibility complex [MHC] II). However, the term “activation” marker is somewhat of a misnomer, given that resting cells are by no means dormant or inactive. Nonetheless, in response to injury or immune stimulation, microglia become considerably more active, and upregulate a number of the aforementioned surface receptors (Morale et al., 2006
). Once activated, glia produce a plethora of cytokines, chemokines [e.g., monocyte chemoattractant protein (MCP)-1], and other inducible factors [e.g., nitric oxide (NO), superoxide], which in turn influence neural function.
Glia and Early Brain Development
Most researchers believe that microglia derive from mesodermal precursors such as infiltrating blood monocytes during pre- and postnatal life, beginning around embryonic day 14 in rodents (Ling and Wong, 1993
). Notably, the first week of life in rats is a time of extensive brain growth when microglial proliferation, migration, and density peak (Bayer et al., 1993
; Wu et al., 1992
). Furthermore, a number of studies have demonstrated that microglia are largely in an activated or amoeboid state during the early postnatal period, in contrast to the quiescent, ramified microglia found in adults (Fujita et al., 1981
; Ling and Wong, 1993
; Rezaie et al., 1997
). Interestingly, the developmental peak in IL-1β concentrations discussed earlier coincides exactly with the presence of ameboid microglia in the rat brain (Giulian et al., 1988
). For these reasons, we have hypothesized that the early postnatal stage may be particularly vulnerable to insult, with subsequent changes in microglial cell number, function, and/or neural trajectory (i.e. vulnerability or protection) (see Figure 2
). Microglia are an excellent candidate for long-term changes within the brain, because these cells are long-lived and can become and remain chronically activated (Town et al., 2005
).
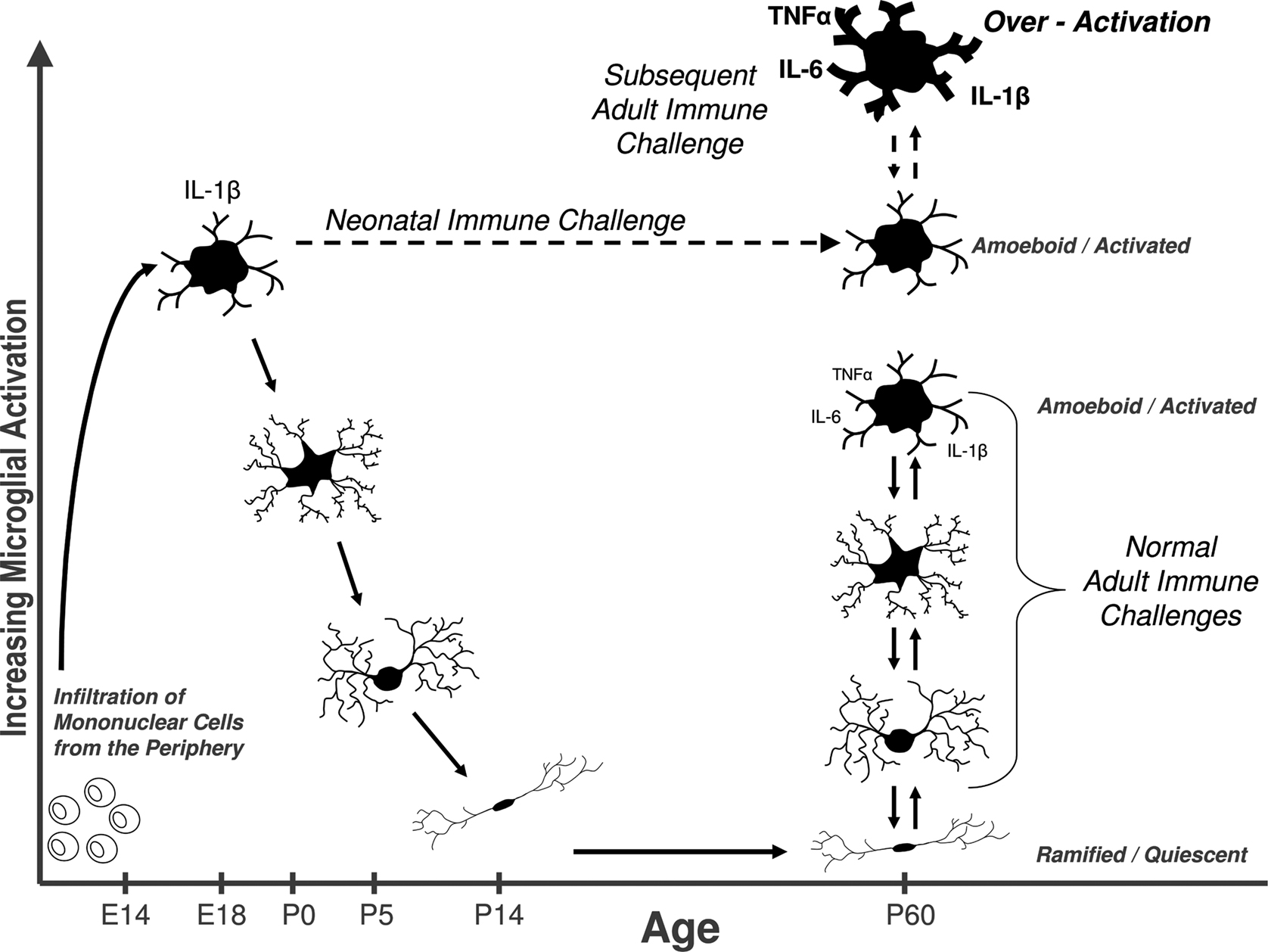
Figure 2. Early brain development is a sensitive period for long-term changes in glial cell function. Bone marrow-derived mononuclear cells infiltrate the brain in rodents beginning around embryonic day (E) 14, where they eventually differentiate into fully mature, ramified microglia by ∼P14. Microglia are critical for early brain development, and exist primarily in an amoeboid/phagocytic state during this time, and can respond vigorously to infection or injury (e.g., cytokine production). In adulthood as well, quiescent microglial can rapidly transition into an activated state in response to infection or injury. We hypothesize that a subset of glia are permanently maintained in an activated or primed state (represented by the dashed line) into adulthood as a consequence of neonatal infection. Thus, when animals are exposed to a subsequent immune challenge in adulthood, primed microglia produce exaggerated levels of cytokines, which in turn impair memory. Early-life priming of microglial function likely has life-long consequences for brain function, homeostasis, and ultimately behavior.
To test this hypothesis, we assessed the impact of early-life infection on glial activation by measuring expression levels for CD11b mRNA in neonate brains immediately following E. coli infection. We found a significant, sustained increase (≥72 h) in the same hippocampal tissue in which we observed increased IL-1β (Bilbo et al., 2005a
). Moreover, both CD11b and IL-1β expression increased with the same time course, peaking 24 h post-infection. Although this remains to be demonstrated directly, these findings suggest that the source of the IL-1β in the neonate brains in response to neonatal infection is activated microglia. In contrast, expression levels of the astrocyte cell marker glial-fibrillary acidic protein (GFAP) did not change, suggesting a selective change in microglial activation after neonatal infection (Bilbo et al., 2005a
).
Double-Edged Sword of Microglia
The long-term consequences of early infection-induced changes in microglial cell number or function in adulthood are largely unknown. Microglia have demonstrated roles in both protection and pathology. For instance, they produce neurotrophic factors that aid in cellular repair, and recruit immune cells into the brain, which aid in clearing infection or cellular debris (Lalancette-Hebert et al., 2007
). Furthermore, the selective ablation of microglia following stroke worsens outcome (Lalancette-Hebert et al., 2007
). In contrast to these benefits, chronic or exaggerated microglial activation is associated with multiple neuroinflammatory and neurodegenerative diseases, including Parkinson’s disease, Multiple Sclerosis, Alzheimer’s, and Huntington’s disease (Perry, 2004
).
Glial Priming
The factors that may cause a switch in glial function from helpful to harmful remain speculative. However, there is increasing support for the concept of “glial priming”, in which cells can become sensitized by an insult, challenge, or injury, such that subsequent responses to a challenge are exaggerated (Perry et al., 2003
). For instance, a systemic inflammatory challenge in an animal with a chronic neurodegenerative disease leads to exaggerated brain inflammation compared to a control animal (Combrinck et al., 2002
). The morphology of primed glial cells is similar to that of “activated” cells (e.g. amoeboid, phagocytic), but primed glial cells do not chronically produce cytokines and other pro-inflammatory mediators typical of cells in an activated state. Upon challenge, however, such as infection or injury in the periphery, these primed cells will over-produce cytokines within the brain compared to cells that were not previously primed or sensitized (Perry et al., 2002
). This overproduction may then lead to cognitive and/or other impairments (Cunningham et al., 2005
; Frank et al., 2006
; Godbout et al., 2005
).
We know that an E. coli infection activates hippocampal microglia in neonatal pups. The finding that glia can be primed long-term raises the critical question, does early-life infection prime glia into adulthood? To answer this question we quantified mRNA levels for two different cell surface activation markers (CD11b and MHC II) in the adult hippocampus of rats treated on P4 with PBS or E. coli, and injected as adults with saline or LPS. CD11b is both constitutively expressed by microglia and somewhat activity-dependent, whereas MHC II is largely expressed only upon activation (Butovsky et al., 2005
). In E. coli-infected rats, we found that CD11b, but not MHC II expression, was elevated following saline compared to controls. Furthermore, both markers were significantly elevated in response to adult LPS treatment in E. coli treated rats compared to controls (Figure 1
C). GFAP was also measured, but again did not differ by neonatal or adult treatment (Bilbo et al., 2005a
, 2007
). The increase in CD11b in early-infected rats regardless of adult treatment suggests an increase in the number of microglial cells (because its expression is relatively constitutive), whereas exaggerated MHC II expression (which is activity-dependent) to the LPS suggests these cells are sensitized or primed in E. coli rats. Finally, we have recent data indicating that the number of cells in adulthood is largely unchanged, whereas their morphology is consistent with a primed phenotype (Bland et al., submitted).
Is there a sensitive period? Does an immune challenge early in life influence brain and behavior in a way that depends on developmental processes? Since 2000 alone, there have been numerous reports in the animal literature of perinatal immune challenges ranging from early gestation to the juvenile period, and their consequences for adult offspring phenotypes (see Table 1
). It is clear that the timing of a challenge is likely a critical factor for later outcomes, impacting the distinct developmental time courses of different brain regions and their underlying mechanisms (e.g., neurotransmitter system development, synapse formation, glial and neural cell genesis, etc; Herlenius and Lagercrantz, 2004
; Stead et al., 2006
). However, the original question of whether these changes depend on development has been surprisingly little addressed. We have demonstrated that infection on P30 does not result in memory impairments later in life (Bilbo et al., 2006
), nor does it induce the long-term changes in glial activation and cytokine expression observed with a P4 infection (Bilbo et al., unpublished data). The factors defining this “sensitive period” are undoubtedly many, as suggested above. However, our working hypothesis is that one primary reason the early postnatal period in rats is a sensitive or critical period for later-life vulnerabilities to immune stimuli, is because the glia themselves are functionally different at this time. Several studies have demonstrated that amoeboid, “macrophage-like”, microglia first appear in the rat brain no earlier than E14, and steadily increase in density until about P7. By P15 they have largely transitioned to a ramified, adult morphology. Thus, the peak in density and amoeboid morphology (and function) occurs within the first postnatal week, with slight variability depending on brain region (Giulian et al., 1988
; Wu et al., 1992
). In the human brain, this same transition occurs, with a shifted time course of late second-to-early third trimester, consistent with comparable neural development to the early postnatal rodent (Rezaie and Male, 2002
). Thus, infectious or traumatic events occurring at this time are hypothesized to have an increased capacity to influence later-life neural function via specific, long-term influences on these long-living and dynamic cells. By extension, we would predict that challenges occurring earlier or later in development would have a reduced impact on glia, and thus on long-term glial function.
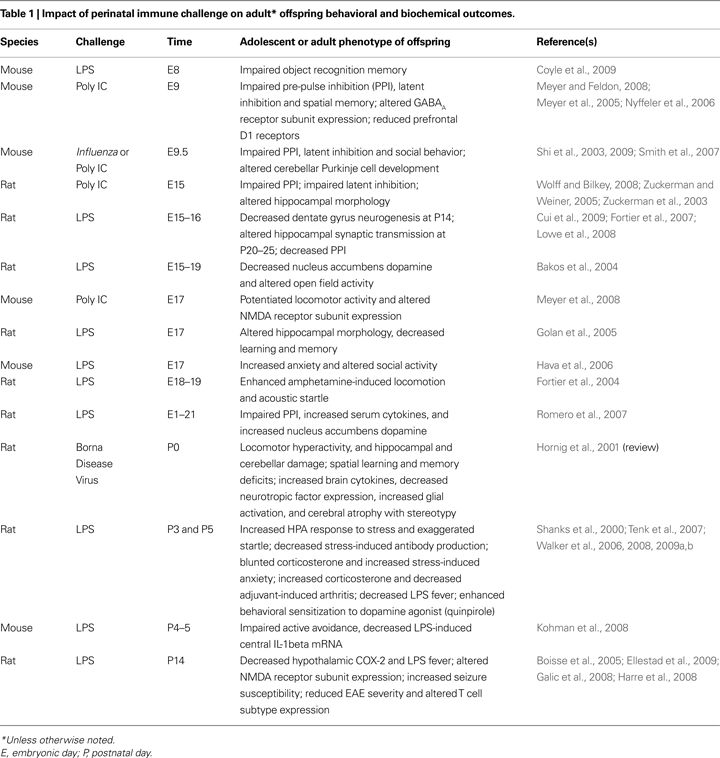
In this review, we have presented evidence that early-life infection with bacteria leads to marked behavioral changes in adulthood. The primary change we have characterized is memory impairment specific to the hippocampus, which is dependent upon the infection occurring early in postnatal life, and is only revealed upon subsequent immune challenge. We have further determined that exaggerated IL-1β production and long-term changes in microglial function/activation are associated with this vulnerability. Several groups have characterized deficits in various aspects of cognition in adulthood following early immune challenge, including pre-pulse inhibition (PPI), latent inhibition, object recognition, and active avoidance (see Table 1
). Unlike our own work, the majority of these deficits occur independent of any adult challenge or precipitating event, although not always (e.g., Walker et al., 2008
). However, several are manifest only in adulthood (e.g., Zuckerman and Weiner, 2003
), suggesting an underlying developmental mechanism is required to unveil the deficits, and together with our own work lend support for the “two hit” hypothesis of developmental pathology. A number of other behavioral changes are also associated with early-life immune activity. It goes beyond the scope of this paper to thoroughly review this literature, but we highlight some of the findings here for comparison to our own work, and their subsequent implications.
HPA Axis Reactivity
The stress axis and the immune system are inextricably linked; during infection, cytokines profoundly activate the HPA axis, and alterations in HPA axis development and function have been characterized by a number of groups following perinatal immune challenge or infection. Unfortunately, this literature is by no means clear, beyond the simple consensus that there are changes. For instance, neonatal LPS challenge (on P3 and again on P5) increases corticosterone within 4 h after exposure (Shanks and Meaney, 1994
), and results in increased anxiety-like behavior in adulthood (Breivik et al., 2002
; Walker et al., 2004
). In contrast, Spencer et al. (2006)
report no significant change in adult anxiety following LPS on P7, similar to our own findings with early E. coli. LPS treatment on P3/P5 also increases corticosterone responses to stress in adulthood (Hodgson et al., 2001
; Shanks et al., 2000
), although this is not always the case (Breivik et al., 2002
; Granger et al., 1996
). Similarly, we have never observed differences in corticosterone in response to adult LPS as a consequence of early infection, although we have observed differences in corticosterone levels in response to an adult (non-immune) stressor, which we discuss more fully below (Bilbo et al., 2005a
,b
, 2008b
). Finally, in humans, early childhood infections are inversely correlated with adult cortisol concentrations, a pattern similar to our own findings (Vedhara et al., 2007
). It should be noted that inconsistencies between our own findings and LPS models are not unexpected given fundamental differences between live E. coli and purified LPS; perhaps most importantly, E. coli is capable of activating the immune system independent of (or in addition to) LPS molecules and toll-like-receptor (TLR) 4 signaling (e.g., via TLR 2/5/9; (Hayashi et al., 2001
; Hemmi et al., 2000
; Steiner et al., 2005
), and results in a more prolonged immune response.
Depression
Exaggerated expression of CNS cytokines, including IL-1β, has been linked to depression in a large number of studies (Dantzer et al., 2008
; Hayley et al., 2005
; Irwin and Miller, 2007
; Maier and Watkins, 1998
; Pollak and Yirmiya, 2002
). Because neonatal E. coli infection in rats leads to exaggerated brain cytokine production to the LPS challenge in adulthood, we predicted that stressor-induced depressive-like symptoms would be more severe in these rats as well, similar to the memory impairment. To test this idea, rats treated on P4 with PBS or E. coli were as adults exposed to an acute bout of inescapable tailshock stress, and then tested for several well-characterized measures of depressive-like behavior in rodents: sucrose preference, social exploration with a juvenile, and overall activity following the stressor. Stress increased corticosterone and decreased all three behaviors in adult rats compared to non-stressed controls, as expected. Surprisingly, however, rats that suffered a bacterial infection early in life had blunted corticosterone responses during and after the stressor, and recovered much more quickly from the depressive symptoms compared to the controls (Bilbo et al., 2008b
).
Thus, in striking contrast to our data on memory, the same early-life infection provides a marked protection against stress-induced depression in adulthood. This divergence in behavioral outcomes is very intriguing, and remains to be more fully explored. Because corticosterone leads to cytokine expression in response to stress, it is possible that rats infected as neonates exhibited a decrease in cytokine production compared to controls, which was ultimately protective. A second hypothesis is that an initial exaggerated immune response in the adult may work to resolve inflammation more quickly. Depressive symptoms are strongly linked to chronic inflammation (Dantzer et al., 2008
). We have evidence, of course, of exaggerated brain IL-1β in these animals, which may be indicative of changes in overall measures of inflammation. If an exaggerated initial response results in a faster resolution, then one would predict a faster recovery from depression in early-infected rats as well, and this is exactly what we observed. From this perspective, memory impairment may simply come as a “cost” to this later benefit.
In summary, there is increasing support for a critical role of CNS immune cells, primarily microglia, and their products such as cytokines, in both health and disease processes within the brain. However, the role of immune factors in homeostatic functions such as synaptic plasticity, and complex behaviors such as cognition, anxiety, and depression remain little understood. We have argued here that the early-life environment of an individual is especially critical in shaping the way that the immune system and hence the brain develop, with significant consequences for brain and behavior throughout the remainder of the lifespan.
The available data indicate a number of common influences of early-life immune activation on later life brain and behavior, namely mechanisms such as cytokine production and glial activation. Thus, while we have been working specifically with a model of bacterial infection in neonatal rats, we believe the results we have reported likely apply more generally to brain-immune interactions. On the other hand, the data also suggest a number of challenge (e.g., LPS vs. E. coli) and gestational time-point specific influences on later outcomes, a further analysis of which will lead to greater understanding of the mechanisms involved. Finally, just as distinct brain regions exhibit markedly different functions and cell populations, immune activity within each region is likely similarly diverse, and should be considered in relation to the microenvironment in which it occurs.
Our data in particular also suggest that the consequences of early-life immune challenge should be considered within a cost/benefit perspective, in which outcomes in adulthood may be differentially protected or impaired (which may indeed be a consequence of distinct functions depending on different brain regions as suggested above). This idea is not without precedence—the “hygiene hypothesis” was proposed nearly 20 years ago as a way to explain the increased incidence of asthma/allergy and other autoimmune disorders in developed countries, and the idea that “too-sterile” environments do not provide sufficient early training for the immune system, with resulting over-reactions in later life (Bufford and Gern, 2005
; Strachan, 1989
). Another elegant example of this concept comes from the field of metabolism in which the “fetal origins of adult disease” was first characterized – Dr. Barker and colleagues observed a striking correlation between low birth weight (due to famine, and/or fetal growth restriction) and risk for obesity/overweight and its associated health problems (e.g., heart disease, diabetes) in adulthood, a causal relationship that has since been overwhelmingly confirmed in animals (Armitage et al., 2008
; de Boo and Harding, 2006
). This hypothesis has been nicknamed the “thrifty phenotype” (Hales and Barker, 1992
), because in the absence of sufficient calories in the fetal environment, the neonatal metabolism prepares or “programs” the individual to conserve calories, even in adulthood. In our modern world of excess food, this becomes maladaptive. Similarly, our data provide support for the hypothesis that early experience with infectious organisms may help to “tune” an appropriate immune responsiveness, and that this has significant consequences, both adaptive and maladaptive, for complex behavioral responses throughout life.
In closing, we believe that the data discussed here are small pieces of a much larger puzzle, in which the immune system is critically involved in normal brain function and, conversely, cognitive/behavioral pathology on a large scale. More research is certainly warranted on the extraordinarily complex influence of immune activity on neural development, and the role that the brain’s immune response plays in adaptive behavior, in both sickness and in health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Jerry Rudy for insightful comments on the manuscript. Supported in part by R01MH083698.
Morale, M. C., Serra, P. A., L’Episcopo, F., Tirolo, C., Caniglia, S., Testa, N., Gennuso, F., Giaquinta, G., Rocchitta, G., Desole, M. S., Miele, E., and Marchetti, B. (2006). Estrogen, neuroinflammation and neuroprotection in Parkinson’s disease: glia dictates resistance versus vulnerability to neurodegeneration. Neuroscience 138, 869–878.
Steiner, A. A., Chakravarty, S., Robbins, J. R., Dragic, A. S., Pan, J., Herkenham, M., and Romanovsky, A. A. (2005). Thermoregulatory responses of rats to conventional preparations of lipopolysaccharide are caused by lipopolysaccharide per se – not by lipoprotein contaminants. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289, R348–R352.
Stevens, B., Allen, N. J., Vazquez, L. E., Howell, G. R., Christopherson, K. S., Nouri, N., Micheva, K. D., Mehalow, A. K., Huberman, A. D., Stafford, B., Sher, A., Litke, A. M., Lambris, J. D., Smith, S. J., John, S. W., and Barres, B. A. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178.
Stoll, B. J., Hansen, N. I., Higgins, R. D., Fanaroff, A. A., Duara, S., Goldberg, R., Laptook, A., Walsh, M., Oh, W., and Hale, E. (2005). Very low birth weight preterm infants with early onset neonatal sepsis: the predominance of gram-negative infections continues in the National Institute of Child Health and Human Development Neonatal Research Network, 2002–2003. Pediatr. Infect. Dis. J. 24, 635–639.
Walker, A. K., Nakamura, T., Byrne, R. J., Naicker, S., Tynan, R. J., Hunter, M., and Hodgson, D. M. (2009a). Neonatal lipopolysaccharide and adult stress exposure predisposes rats to anxiety-like behaviour and blunted corticosterone responses: Implications for the double-hit hypothesis. Psychoneuroendocrinology. (in press).
Zuckerman, L., Rehavi, M., Nachman, R., and Weiner, I. (2003). Immune activation during pregnancy in rats leads to a postpubertal emergence of disrupted latent inhibition, dopaminergic hyperfunction, and altered limbic morphology in the offspring: a novel neurodevelopmental model of schizophrenia. Neuropsychopharmacology 28, 1778–1789.