1
Department of Medicine, Royal Melbourne Hospital, University of Melbourne, Parkville, VIC, Australia
2
Department of Psychiatry, University of Melbourne, Parkville, VIC, Australia
The pathogenesis of mesial temporal lobe epilepsy (MTLE), the most prevalent form of refractory focal epilepsy in adults, is thought to begin in early life, even though seizures may not commence until adolescence or adulthood. Amongst the range of early life factors implicated in MTLE causation (febrile seizures, traumatic brain injury, etc.), stress may be one important contributor. Early life stress is an a priori agent deserving study because of the large amount of neuroscientific data showing enduring effects on structure and function in hippocampus and amygdala, the key structures involved in MTLE. An emerging body of evidence directly tests hypotheses concerning early life stress and limbic epilepsy: early life stressors, such as maternal separation, have been shown to aggravate epileptogenesis in both status epilepticus and kindling models of limbic epilepsy. In addition to elucidating its influence on limbic epileptogenesis itself, the study of early life stress has the potential to shed light on the psychiatric disorder that accompanies MTLE. For many years, psychiatric comorbidity was viewed as an effect of epilepsy, mediated psychologically and/or neurobiologically. An alternative – or complementary – perspective is that of shared causation. Early life stress, implicated in the pathogenesis of several psychiatric disorders, may be one such causal factor. This paper aims to critically review the body of experimental evidence linking early life stress and epilepsy; to discuss the direct studies examining early life stress effects in current models of limbic seizures/epilepsy; and to suggest priorities for future research.
Mesial temporal lobe epilepsy (MTLE), a disabling, often treatment-resistant form of focal epilepsy, arises from brain structures known to be affected by stress. Stressors at all developmental stages – prenatal, postnatal and in adult life – have neuroplastic effects on hippocampus and amygdala in particular (Herbert et al., 2006
). This raises the possibility that stress contributes to the causation of MTLE. In fact, the causation of MTLE is thought to be a multi-stage process commencing in early life, so stress could be relevant at several stages of pathogenesis. Certainly, patients think stress is relevant, citing it as one of the most common precipitants of epilepsy onset and of seizures (Antebi and Bird, 1993
; Frucht et al., 2000
; Spector et al., 2000
; Nakken et al., 2005
; Haut et al., 2007
; Sperling et al., 2008
). Also, epidemiological data implicate stress in the causation of epilepsy and seizures in adults (Temkin and Davis, 1984
; Swinkels et al., 1998
) and children (Bosnjak et al., 2002
). Furthermore, a considerable body of studies in animal models of MTLE provides strong evidence for a role of stress in aggravation of limbic epilepsy in adult animals (Joels, 2009
).
However, there are good a priori reasons to focus – as this review does – on stress in early life and the earliest developmental stages of limbic epileptogenesis. First, the evidence from adult animals, showing effects of stress on limbic neuroplasticity, on electrophysiology and on epileptogenesis itself may be pertinent at younger ages too. In addition, a body of evidence already exists directly implicating stress mediators, notably corticotropin-releasing hormone (CRH), in seizures of early life (Baram and Hatalski, 1998
).
Secondly, early life stress may be a shared causal factor for both MTLE and the psychiatric comorbidity that often accompanies it. Early life stress has been extensively implicated in the causation of depressive and anxiety disorders and of schizophrenia in the general (nonepileptic) community (e.g., Caspi et al., 2003
; Van Praag et al., 2004
; Malaspina et al., 2008
), and may be relevant also to the psychiatric comorbidity of MTLE. In addition, psychiatric comorbidity may not simply be a neurobiological and/or psychosocial consequence of epilepsy as there is increasing evidence that psychiatric comorbidity, particularly depression, may be causal for MTLE (Hesdorffer et al., 2000
, 2006
).
Thirdly, whereas early life stressors generally have adverse effects on epilepsy in experimental models (see Direct Studies Examining the Influence of Early Life Stress on Limbic Excitability and Epileptogenesis), other early life exposures have positive effects. For example, environmental enrichment repeatedly has been demonstrated to provide neuroprotection in animal models of various neurological disorders (Nithianantharajah and Hannan, 2006
), including limbic epilepsy (Auvergne et al., 2002
; Young et al., 2004
; Korbey et al., 2008
). Much is to be gained by comparing and contrasting the effects and neurobiological underpinnings of these different early life exposures. Finally, the study of early life stress may afford insights into approaches to prevention (McEwen, 2008b
), a much under-developed aspect of epilepsy research (Dichter, 2009
).
This review commences by briefly defining and describing human MTLE and current views of its causation. We then review relevant experimental evidence concerning the role of stress: first, indirect evidence linking stress in early life to neurobiological intermediaries known to be relevant to limbic epileptogenesis, notably effects on neuroplasticity, on neuroendocrine and neurochemical systems and on electrophysiology. We then review the small body of direct evidence, i.e. studies testing hypotheses about various forms of stressor in various models of limbic seizures or epilepsy. We conclude with a critique of these bodies of evidence and suggestions for future research directions.
In reviewing this literature, we do not seek to definitively prove a case that stress in early life is involved in causation of human MTLE; we simply aim to convince readers that it is a compelling general hypothesis and that there are numerous specific aspects of the general hypothesis which are testable, both in animals and ultimately humans.
Mesial temporal lobe epilepsy is one of the most common forms of focal epilepsy in humans and is often treatment refractory (Engel et al., 2007b
). It is a cause of serious disability, with significant mortality (due to trauma, SUDEP and suicide). It also is associated with considerable cognitive and psychiatric comorbidity, which add greatly to disability and impaired quality of life (Hermann et al., 2008
). Treatment resistance is common and an indication for temporal lobe epilepsy surgery, the most common form of epilepsy surgery (Duchowny et al., 2008
).
Its clinical features include limbic seizures, often with secondary generalisation. Seizure onset can be at any age, but typically is in late childhood-early adolescence. There is often a history of early life cerebral insult, such as febrile seizures or head trauma, followed by a latent period before emergence of recurrent seizures. The most common underlying neuropathology is hippocampal sclerosis (HS) defined by loss of neurons in CA1 and CA3 (with preservation of CA2) (Engel et al., 2007b
; Scharfman and Pedley, 2007
). The extent varies, in some cases limited, in others encompassing the entire hippocampus. Other common features are mossy fibre sprouting, dispersion of dentate granule cells, loss of dentate hilar neurons and extrahippocampal pathology, most commonly in amygdala but also parahippocampal region, thalamus, cerebellum and other structures. Mesial temporal sclerosis refers to HS combined with involvement of amygdala and mesial temporal cortex (Engel et al., 2007b
; Scharfman and Pedley, 2007
). In many cases, other accompanying neuropathology is to be found in the temporal lobe and elsewhere, pointing to the heterogeneity that is a feature of MTLE and that has been the focus of considerable debate (Wieser, 2004
; Engel et al., 2007b
).
The most parsimonious framework able to accommodate the known facts about causation posits a multi-stage process of epileptogenesis and gene–environment interaction (Scharfman and Pedley, 2007
). The ‘three-stage hypothesis’ proposes: (1) an initial event (a neural insult perinatally or in early childhood, such as cerebral infection, head trauma or febrile seizures); (2) a latent period, during which many structural and functional changes take place at the levels of circuits, synaptic transmission, and ion channels. Also in this phase neuronal injury and gliosis progress and epileptiform discharges begin; (3) a chronic stage, featuring spontaneous, recurrent seizures. This is not an end point however, as brain changes continue to progress, perhaps in part due to the occurrence of seizures themselves. For extended discussions of MTLE causation, see Scharfman and Pedley (2007)
(p357ff), Walker et al. (2007)
(p774ff), and Engel et al. (2007b)
, upon which this brief account is largely based.
All known neural insults, such as severe prolonged febrile seizures, act as risk factors in that they raise the likelihood of subsequent MTLE, but do not invariably cause it. The variation in susceptibility may stem from genetic variation or from early neurodevelopmental abnormalities, which themselves may stem from genetic or environmental factors or interactions between these. A number of familial forms of temporal lobe epilepsy have been identified, some leading to hippocampal atrophy, others featuring febrile seizures in childhood and heightened risk of temporal lobe epilepsy in later life. Microarray analyses of resected human temporal lobe tissue and of limbic tissue from animals experiencing spontaneous seizures following experimental status epilepticus (SE) implicate genes relevant to all stages of temporal lobe epileptogenesis (Gu et al., 2004
; Gorter et al., 2006
; Jamali et al., 2006
; Gorter et al., 2007
; Hatazaki et al., 2007
). This encompasses genes relevant to cell death and survival, inflammation, gliosis, neurotransmission and neuroplasticity.
How these diverse changes result in the hyper-excitability and hyper-synchrony that characterise the epileptic state is not well understood (Engel et al., 2007a
). Research, both human and animal, has attacked this problem at multiple biological levels, from ion channels, to cells, tissues and circuits, both in vivo and in vitro and using combinations of neurochemical, histological, imaging, genetic, electrophysiological and computer modelling approaches amongst others (Jeffreys, 2007
; Lukasiuk et al., 2007
; Scharfman and Schwarcz, 2007
; Swann et al., 2007
; Lytton, 2008
). Despite the accumulation of many insights, key aspects of epileptogenesis remain elusive.
Nevertheless, a great deal of evidence has accrued that stress biology may be highly relevant to all three stages of epileptogenesis and at several biological levels. Most evidence stems from animal models, mostly in adult animals. Some evidence is direct, demonstrating associations between an experimental stressor and epilepsy end-points; other data are indirect, showing effects of experimental stress on diverse biological processes implicated in epileptogenesis. For stress in early life – prenatal and early postnatal – an extensive body of indirect evidence contrasts with a slender literature of direct studies. Many studies of early life stress show robust effects on intermediary processes relevant to limbic epileptogenesis, such as neuroplasticity, while a small number directly test the hypothesis that early life stress can influence the development of limbic epilepsy.
Early Life Stress Effects on Brain and Neuroendocrine Regulation: Potential Intermediary Processes Connecting Early Life Stress and Limbic Epilepsy
Enduring and delayed effects on brain of early life stress are presently being researched in both humans and animals in relation to a wide array of physical and mental health outcomes. This includes studies of prenatal and early postnatal stress, as well as stressors in childhood and adolescence. It also includes studies of many psychiatric disorders, neurological disorders (including neurodegenerative, dementing disorders of later life) (Gunnar and Vazquez, 2006
; Gunnar and Quevedo, 2007
) and physical disorders, notably obesity, metabolic syndrome and cardiovascular disease (McEwen, 2008b
). More recently, evidence has emerged for transgenerational stress effects, including pre-conception stress (Yehuda and Bierer, 2008
; Shachar-Dadon et al., 2009
).
One issue is the definition of ‘early’, as studies have reported enduring effects from exposures in adolescence (e.g., Wright et al., 2008
). Rather than debate definitions of ‘early’, it is preferable to take a life-span perspective, to work from the fact that neurodevelopment is a prolonged, multiphasic process in mammals and that critical time windows occur during which various effects are or are not possible (Andersen et al., 2009
; Lupien et al., 2009
).
This section selectively reviews evidence addressing whether early life stress affects neurobiological ‘intermediaries’ known to be heavily implicated in temporal lobe epileptogenesis. In doing this, we aim to describe relevant candidate ways in which stress may impact disease process, rather than to categorically define the mechanism by which this occurs; this would be premature given the state of the current literature. Using a crime metaphor, we present a ‘line-up’ of suspected offenders, but whether one committed the crime, or several, or all, we do not yet know. The four key ‘intermediaries’ are: macroscopic and microscopic brain structure especially in limbic areas; electrophysiology; neurotransmission; and neuroendocrine function (notably hypothalamic–pituitary–adrenal (HPA) axis function, the CRH system, and neurosteroids). As well as direct effects on brain, we discuss effects on HPA axis regulation as glucocorticoids have extensive effects within the brain, including effects on limbic structures, thus constituting an indirect pathway for stress effects on brain.
Human evidence bearing on these intermediaries is sparse, in contrast to the very extensive animal experimental evidence. Although the enduring and delayed effects of early life stress on brain have been well-reviewed, the great relevance to limbic epilepsy has only occasionally been considered, with some important exceptions which will be noted.
Early Life Stress and Neuroplasticity
The term ‘neuroplasticity’ refers to ‘the ability of the nervous system to adapt its structural and functional organisation to altered circumstances arising from developmental or environmental (including traumatic) changes’ (Gould, 2007
; Rutter, 2009
). Several forms of neuronal reorganisation have been implicated in medial temporal lobe epileptogenesis, including sprouting and reorganisation of mossy fibre axons from granule cells of dentate gyrus; diminished branching of dendrites and reduced dendritic spine density in CA3; dispersion of the granule cell layer and presence of ectopic DG granule cells in hilus (Engel et al., 2007b
; Scharfman and Pedley, 2007
; Walker et al., 2007
). Also, many of the genes identified in microarray analysis of human and animal epileptic limbic tissue are involved in neuronal plasticity (Lukasiuk et al., 2007
). Such neuroplastic changes may help explain in part the common latent period between neural insult and onset of MTLE.
One suggestion is that such insults reactivate developmental programmes, upregulating growth factors involved in normal limbic system development. Seizures themselves, especially in the infant brain, are one such insult (Ben-Ari and Holmes, 2006
). Tissue of adults with severe epilepsy show a reversal of GABAergic signalling to depolarising γ-aminobutyric acid (GABA), a characteristic of the immature brain (Dulac et al., 2007
), which supports the notion of reactivation of developmental programmes. Seizures possibly slow down or accelerate processes unique to the developmental period essential for correct circuit formation. For example, a re-initiation of axonal growth programmes may result in the aberrant axonal contact of DG cells responsible for the generation of synchrony (Cohen et al., 2003
). In addition, following induced seizures, increased expression of mRNAs encoding neonatal splice variants of sodium channel subunits was detected in the hippocampus of the adult rat brain (Gastaldi et al., 1997
; Aronica et al., 2001
). The required modification during development of the pattern and sequence of expression of receptor proteins, such as AMPA receptors, are activity dependent, and may be disturbed by seizures (Holmes and Ben-Ari, 2001
). In the same way, synapse formation in the developing brain can also be altered by early life insults, resulting in consequences for seizure susceptibility. Hence, the neural reorganisation seen in MTLE and relevant animal models may be both cause and effect of seizures (Parent et al., 2007
; Swann et al., 2007
).
Stress biology has been implicated extensively in several of these aspects of limbic neuroplasticity (McEwen, 2008a
), most notably dendritic structure of CA3 pyramidal and amygdala neurons (Vyas et al., 2002
, 2003
) and dentate gyrus neurogenesis (Joels et al., 2007
; Morris, 2007
; McEwen, 2008a
). Both acute and chronic stress can suppress dentate gyrus neurogenesis and/or cell survival and can induce dendritic remodelling in CA3 (reviewed in McEwen, 2008a
). Corticosterone elevation has a key role in these phenomena, but its effects are complex, vary with time and other concurrent circumstances (such as social isolation, physical activity), and involve interactions with other neurochemical systems, including serotonin, GABA and excitatory amino acids (mainly glutamate).
Stress in early life can have short-, medium- and long-term effects on hippocampal structure (Brunson et al., 2001
, 2005
; Andersen and Teicher, 2004
; Mirescu et al., 2004
; Champagne et al., 2008
; Aisa et al., 2009
; Oomen et al., 2009
), as can normal variations in maternal care (Bagot et al., 2009
) (for reviews see Kaufman et al., 2000
; Sanchez et al., 2001
; Fenoglio et al., 2006
; Teicher et al., 2006
; Weinstock, 2008
; Cirulli et al., 2009a
). In a striking demonstration of the delayed onset of such sequelae even after a transient early-life exposure, rat mothers were stressed on PND2–9 by having limited access to nesting/bedding material (Brunson et al., 2005
). Their offspring showed various impairments of hippocampal function (impaired spatial memory and object recognition) and, at 12 months, exhibited increased mossy fibre sprouting, abnormal abundance of mossy fibre terminals in CA3 and marked dendritic atrophy of CA1 pyramidal cells. There is evidence that early CRH elevation is particularly important in generating such enduring effects (Brunson et al., 2001
).
Such effects of early life stress could be relevant to limbic epileptogenesis in several ways. First, they may play a role in seizures and epilepsies with onset in early life (Baram and Hatalski, 1998
). Second, they may contribute to the first stages of evolution of MTLE, long before clinical emergence of seizures. Third, they may create an enduring vulnerability to limbic epilepsy that emerges only with onset of neurodegenerative disorders or other cerebral insults or pathology in later life.
It is important to note that extratemporal structures have been implicated in MTLE pathophysiology (Schwarcz et al., 2002
). Schwarcz et al. (2002)
cite human and animal evidence implicating ventral limbic cortex, perirhinal and piriform cortices, cerebellum, cerebral cortex and thalamus. Whether the abnormalities noted in these areas are epiphenomena or important to MTLE pathophysiology is not clear. Stress effects in these areas have not been extensively studied, but evidence exists for changes in rat infralimbic medial prefrontal cortex (Izquierdo et al., 2006
; Shansky et al., 2009
) and anterior cingulate cortex (Cerqueira et al., 2005
), areas not considered central to MTLE pathogenesis, but which arguably could influence it or its associated psychopathology.
Early Life Stress and Endocrine Responses in Later Life
Given the important role of glucocorticoids (corticosterone in rodents, cortisol in humans) in normal regulation of hippocampal and amygdala electrophysiology and neuroplasticity (Joels, 2009
), there is ample reason to consider their role in limbic epileptogenesis, especially in the immature brain. Indeed, for mature animals there exists strong and consistent evidence that chronically elevated corticosteroids aggravate epileptogenesis (Karst et al., 1999
; Taher et al., 2005
; Kumar et al., 2007
; Mazarati et al., 2009
).
Early life stressors of various kinds, both prenatal and early postnatal, can ‘program’ HPA axis responsivity well into adult life, perhaps for the lifetime of the organism, as well as having a range of neurobiological and behavioural effects (Sanchez et al., 2001
; Levine, 2005
; Phillips, 2007
; Weinstock, 2008
). These ‘programming’ effects may be forms of ‘predictive adaptive responses’, found in many organisms (Gluckman et al., 2007
) and often epigenetically mediated (Gluckman et al., 2008
). An epigenetic basis has been reported for the findings that rat mothers ‘high’ or ‘low’ on maternal behaviour produce offspring with ‘low’ or ‘high’ stress responsivity, respectively. Significantly, the mechanism underlying hyper-reactivity appears to be methylation of the offsprings’ glucocorticoid receptor (GR) (Weaver et al., 2004
), resulting in diminished capacity to terminate corticosterone responses to acute stressors (a mechanism that may apply also to the enduring effects of prenatal restraint stress on rat offspring, Darnaudery and Maccari, 2008
). Evidence suggesting similar processes in humans has emerged recently (Oberlander et al., 2008
; McGowan et al., 2009
). The direction of HPA axis ‘programming’ – whether hyper- or hypo-responsivity – varies considerably by timing and type of stressor and by species (Gunnar and Vazquez, 2006
; Gunnar and Quevedo, 2008
). For example, ‘early handling’ in infant rats results in diminished HPA axis responses to acute stressors in later life, whereas daily periods of more extended maternal separation (3–4 h/day) result in exaggerated responses (reviewed in Sanchez et al., 2001
, p424 ff, Levine, 2005
).
Responses in nonhuman primates can be quite different: repeated prolonged separations typically result in HPA axis hypo-reactivity (discussed in Gunnar and Vazquez, 2006
; Gunnar and Quevedo, 2008,
p138), despite heightened fearfulness and a range of other neurobiological changes (e.g., elevated CSF CRH levels, increased CRH receptor densities in amygdala and prefrontal cortex, and other ‘adverse’ neurochemical and structural changes in limbic areas). The small body of human research that has addressed this question has also commonly found HPA axis hypo-reactivity (Carpenter et al., 2004
, 2007
, 2009
; Tyrka et al., 2008
). Apart from shifts in hormone levels, neuroendocrine regulation of brain function depends on a range of factors, including functionality of receptors (Pariante and Lightman, 2008
). For example, maternal deprivation in rats resulted in a hypersensitivity to normal levels of glucocorticoids, responsible for suppression of adult neurogenesis and plasticity (Mirescu et al., 2004
).
Other hormones have been implicated in the modulation of seizures and epilepsy, notably neurosteroids (Joels, 1997
; Reddy and Rogawski, 2002
; Rhodes et al., 2004
; Belelli et al., 2006
; Biagini et al., 2009
) and sex hormones (Maguire et al., 2005
; Maguire and Mody, 2007
), and have been extensively studied in regard to catamenial epilepsy (e.g., Penovich and Helmers, 2008
). Although the actions of neurosteroids at GABAergic receptors have been extensively studied (Belelli et al., 2006
), neurosteroids modulate glutamatergic neurotransmission also (Sedlacek et al., 2008
), thus having the potential to influence both the inhibitory and excitatory aspects of epilepsy and seizures. Neurosteroid disturbance early in life may result in enduring changes in brain morphology and behaviour (Gizerian et al., 2006
; Grobin et al., 2006
). Conversely, perinatal exposures may result in enduring alterations in neurosteroid levels, e.g., early postnatal handling is followed by lowered hippocampal 3α,5α-THP (5α-pregnane-3α-ol-20-one) in adulthood (Frye et al., 2006
); and perinatal exposure to low-level alcohol results, in adulthood, in elevations of both 3α,5α-THP and progesterone in hippocampus (Barbaccia et al., 2007
). Further, a recent study reported ‘programming’ effects of rat maternal behaviour (‘high’ and ‘low’ mothering, as discussed above) on the hypothalamic–pituitary–gonadal axis of adult female offspring (Cameron et al., 2008
). Thus, like corticosterone, neurosteroids and sex steroids are also extensively implicated in both psychiatric disorder and epilepsy and may have a role in shared causation. Combined, these data from animal models demonstrate the influence of early life stress on several hormonal systems, influences which may be relevant to limbic epilepsy.
Effects of Early Life Stress on GABA Neurotransmission
The spontaneous recurrent seizures that are the defining characteristic of epilepsy are generally thought to involve altered brain electrophysiology and a disruption in the balance between excitation and inhibition in neuronal networks. This disruption could be caused by excessive excitatory glutamate activity, reduced GABA inhibitory activity, or a combination of both, consequently promoting a hyperexcitable environment. While the effects of stress and stress mediators have profound effects on limbic excitability in adult life (see Joels, 2009
for review), limited studies have investigated the enduring effects of early life stress on limbic system electrophysiology. Such studies however, are in general consensus that exposure to stress in early life leads to hyperexcitable limbic circuitry.
The most illuminating study to investigate electrophysiological alterations in adult rats previously exposed to early life stress examined GABA responses in ex vivo dentate granule hippocampal neurons (Hsu et al., 2003
), the dentate gyrus being a key structure within the hippocampus (see below). The results demonstrated a reduction in the efficacy of GABA-evoked inhibitory currents, and a potentiation of the effects of exogenous zinc and zolpidem, two GABA receptor subunit selective ligands which act to reverse GABA-mediated inhibitory function (Hsu et al., 2003
). This shift to a dampened GABA inhibitory system was accompanied by a decrease in the ratio of α1/α2 GABA receptor subunit mRNA levels, suggesting that early life stress promotes maintenance of an immature (excitatory) GABA receptor phenotype. Similar functional and molecular alterations in GABA receptor phenotype have been described in animal models of epilepsy (Brooks-Kayal et al., 1998
; Lauren et al., 2003
) and see also Coulter (2001)
, and in resected temporal lobe from MTLE patients (Brooks-Kayal et al., 1999
), promoting these effects of early life stress on the GABA system as good candidates to influence vulnerability to epilepsy. Findings of changes in GABAA receptor densities, subunit composition and function following a variety of neonatal stressors have also been consistently observed, providing further support of an altered GABA inhibitory system (Caldji et al., 2000
; Jacobson-Pick et al., 2008
; Stevenson et al., 2008
).
Effects of Early Life Stress on Neurochemistry and Other Neurotransmitter Systems
In addition to GABA, a range of other neurotransmitter systems are implicated in the epileptic state, some known to be affected, in enduring fashion, by stress in early life. The excitation-inhibition imbalance that is thought to underlie the epileptic state involves excitatory neurotransmission via glutamate and aspartate receptors (Wilcox et al., 2007
), and is modulated directly or indirectly by a diversity of neuropeptides, calcium binding proteins and growth factors, amongst others (Scharfman and Schwarcz, 2007
; Walker et al., 2007
, p778). In addition, monoaminergic neurotransmitters – notably noradrenaline and serotonin – affect these processes (Jobe and Browning, 2005
, 2007
).
Early life stressors have been shown to have enduring effects on brain serotonergic function in rats (Andersen et al., 1999
; van Riel et al., 2004
; Vicentic et al., 2006
; Arborelius and Eklund, 2007
; Mesquita et al., 2007
; Lambas-Senas et al., 2009
) and nonhuman primates (Ichise et al., 2006
; Maestripieri et al., 2006
), with some preliminary evidence that this may also be the case in humans (Miller et al., 2009
). A further reason to consider serotonergic function with regard to epilepsy generally is that modern antidepressants, particularly SSRIs (serotonin specific reuptake inhibitors), widely used to treat depression comorbid with epilepsy, appear to have an anti-seizure action (Alper et al., 2007
). This anti-seizure action may be mediated via the 5-HT1A receptor (Gariboldi et al., 1996
; Lu and Gean, 1998
), and may also be relevant to epileptogenesis. In MTLE patients, PET studies have shown reduced hippocampal 5-HT1A receptor binding, reduced further with comorbid depression (Hasler et al., 2007
), and see also Ito et al. (2007)
. SSRIs moderate HPA activity in depressed patients and in animal models (Schule, 2007
); and they have well-established effects on hippocampal neurogenesis which may underlie their antidepressant effect (Duman and Monteggia, 2006
) and which also are mediated via the 5-HT1A receptor (Martinowich and Lu, 2008
). An SSRI, fluoxetine has been reported to reverse some of the adverse hippocampal effects of maternal separation stress (Lee et al., 2001
). A further reason to consider serotonergic neurotransmission generally and the 5-HT1A receptor in particular is that this receptor is itself strongly downregulated by corticosterone elevation (Joels and Van Riel, 2004
; Lanfumey et al., 2008
, p1179); and, when downregulated, disinhibits glutamatergic neurons, potentially aggravating excitotoxicity and epileptogenesis (Yuen et al., 2008
). However, at this stage, the data are complex and difficult to reconcile; for example, in one study of electrical amygdala kindling of adult rats, raised levels of 5-HT1A receptor binding and mRNA expression were observed in dentate gyrus (Kalynchuk et al., 2006
).
In contrast to a sizeable number of studies both linking early life stress to serotonergic function, and serotonergic function to epilepsy (including temporal lobe epilepsy), the corresponding literature for other neurotransmitter systems is limited. For example, a small body of evidence exists connecting early life stressors to alterations in glutamate receptor expression in hippocampus (Roceri et al., 2002
; Pickering et al., 2006
); however, to our knowledge the possible causal pathway from early life stress to glutamatergic dysfunction to epilepsy has not been studied. Similar points could be made about CRH and about noradrenergic function. CRH has been implicated in epilepsies of childhood (Baram and Hatalski, 1998
) and, in experimental animals, is altered in various limbic structures by early life stress (e.g., Plotsky et al., 2005
), but to our knowledge has been studied in relation to limbic epilepsy in adult animals to only a limited extent (Ehlers et al., 1983
; Siggins et al., 1985
; Weiss et al., 1986
; Smith and Dudek, 1994
). Similarly, there is considerable evidence for early life ‘programming’ of sympathetic nervous system activity (Young, 2002
) and, given the modulatory role of noradrenergic function on seizures, this may constitute a plausible complementary pathway from early life experience to epilepsy (see discussions in Jobe and Browning, 2005
, 2007
).
In addition, it should be noted that many other ion channels, such as calcium, potassium, and sodium channel subunits have been heavily implicated in epilepsy research (Armijo et al., 2005
; Avanzini et al., 2007
), and in particular anti-epileptic drug action (Rogawski and Loscher, 2004
), but have so far not been extensively investigated from a stress perspective.
Many, if not all, of the neurotransmitter systems discussed here can have important effects on cellular excitability and limbic electrophysiology. As epilepsy is a disease often associated with aberrant electrical discharges and circuits, the next section describes alterations in limbic electrophysiology caused by early life stress.
Effects of Early Life Stress on Cellular Electrophysiology
Modelling of focal seizures describes the propagation of neuronal activity through the limbic system. Using slice preparations bathed in low Mg2+ to induce seizure-like bursts, or inducing seizures in live animals with chemoconvulsants or electrical stimulation, seizure activity has been shown to propagate from the entorhinal cortex to the dentate gyrus via the perforant path, onto the CA3 and CA1 subfields of the hippocampus, and then to the subiculum (e.g., Collins et al., 1983
; Behr et al., 1996
, 1998
), and then onto thalamic and neocortical areas. The dentate gyrus may act as a brake against seizure propagation through this limbic circuitry (Hsu, 2007
), a brake which is compromised in models of seizures and epilepsy (Behr et al., 1998
; Nadler, 2003
). Electrophysiological properties of many of these limbic pathways and hippocampal substructures have been demonstrated to be influenced by exposure to early life stress, influences which may confer vulnerability to seizure propagation, and also the development of epilepsy in later life.
For example, alterations in dentate gyrus in vivo field potentials in anaesthetized animals have been described following early life stress (Stewart et al., 2004
). Rats experiencing chronic stress as adults in addition to early life maternal deprivation demonstrated greater spike amplitudes in dentate gyrus compared with rats undergoing either of the two stressors alone. This is relevant to the three-hit hypothesis of MTLE (see Mesial Temporal Lobe Epilepsy and its Causation), whereby the early life stress primes the system, and the response to the second hit is consequently magnified.
Increased hyperpolarising GABA-mediated currents occur in CA1 pyramidal cells of neonatal rats after maternal separation (Galanopoulou, 2008
), further suggestive of an aberrant GABAergic system as discussed above in Section ‘Effects of Early Life Stress on GABA Neurotransmission’ While this directional change in GABA currents suggests reduced membrane excitability rather than enhanced, previous research examining the h-current (Ih) in models of epilepsy suggests that both up- and down-regulation of this current can lead to neuronal and circuit hyperexcitability (Dyhrfjeld-Johnsen et al., 2009
). Neuronal circuits are complex systems with multiple components held in intricate homeostatic balance. It is difficult to predict the network response of altering a single parameter (for example, a GABA-mediated current). What is clear from the study by Galanopoulou (2008)
is that key electrophysiological properties of the GABAergic system are altered following early life stress.
With regards to effects of early life stress on limbic circuitry, alterations have been demonstrated in limbic long-term potentiation (LTP) (Cui et al., 2006
; Blaise et al., 2008
; Champagne et al., 2008
) and long-term depression (Blaise et al., 2008
), effects consistent with enduring electrophysiological consequences of neonatal stress. From these limited studies, it seems the region investigated and the stress-state of the animal can produce differing directional effects of early life stress. LTP is impaired in CA1 following early life stress (Cui et al., 2006
), but enhanced when recorded from the DG-amygdala pathway (Blaise et al., 2008
). Additionally, work done by Champagne et al. (2008)
examining CA1 LTP responses in animals who had experienced either high or low maternal care showed that the differing responses to high-frequency stimulation were reversed in stressful states (discussed below) (Champagne et al., 2008
). Further, rodents exposed to stress as adults exhibit detrimental effects on LTP – chronic stress reduces LTP recorded in both DG and CA3 (Pavlides et al., 2002
). Intriguingly, the high-frequency stimulation in this study, delivered as part of the LTP protocol, resulted in epileptic afterdischarges in 56% of acutely stressed rats and 29% of chronically stressed rats, but only in 9% of control animals (Pavlides et al., 2002
). The synergies and commonalities between LTP and epilepsy have been discussed previously (McEachern and Shaw, 1996
, 1999
). At this stage, it is unclear whether enhanced or diminished LTP would be predicted to result in a higher or lower susceptibility to develop epilepsy: the contrasting results here, generated by recordings at different sites, suggest that the role of stress, particularly in early life, can have complex effects on limbic system electrophysiology, some of which may be relevant to epilepsy.
An important and difficult question is whether any particular ‘programming’ effect of early life stress – such as electrophysiological responses – is adaptation or pathology. In a recent study, offspring of ‘low-mothering’ dams in adulthood showed reduced dendritic length and spine density in CA1, as well as reduced LTP (Champagne et al., 2008
). A single pulse of corticosterone, simulating an acute stress, reduced LTP in offspring of ‘high-mothering’ dams, but increased LTP in hippocampal slices from offspring of ‘low-mothering’ dams. Consonant with the LTP findings, ‘low mothering’ offspring performed better on a learning task under a high stress condition than did offspring of ‘high mothering’ dams; the latter performed better under low-moderate stress conditions. The enduring effects on offspring of ‘low mothering’ observed here may therefore be adaptive rather than pathological. Clearly, such data have many implications for experimental design, for intervention strategies, and for extrapolation from animal models to humans.
Electrophysiological effects of early life stress are not exclusive to rodents: studies in monkeys reared in isolation demonstrated profound adverse effects relevant to epilepsy (Heath, 1972
). EEG recordings were obtained in adult life from animals experiencing this form of early deprivation, and abnormalities were observed in four out of five subjects, compared with none from the control rearing group. These abnormalities consisted of consistent high amplitude spiking from limbic and other subcortical recording sites (Heath, 1972
). The effects of early life stress noted here demonstrate the potential of early life stressors to influence limbic system electrophysiology, promoting a hyperexcitable system and potentially rendering the organism vulnerable to epilepsy in later life.
Interim Summary
These four key neurobiological processes are known both to be affected by early life stressors and to be involved in limbic epileptogenesis. Our brief account is by no means comprehensive and indeed omits other neurobiological pathways, such as inflammation (Vezzani et al., 2007
), that also might be considered relevant in the same way (inflammation in adult life, for example, has recently been reported to be increased by childhood maltreatment, Danese et al., 2007
). However, taken together, even this limited review providing correlative data between stress and epilepsy makes an adequate a priori case for the potential relevance of early life stress to limbic epilepsy.
Direct Studies Examining the Influence of Early Life Stress on Limbic Excitability and Epileptogenesis
Given the effects of early life stress on structure and function of the limbic system, and the several experimental studies demonstrating the influence of stress hormones such as glucocorticoids and CRH on epileptogenesis and seizure susceptibility, it is compelling to posit that early life stress may be an important contributor to temporal lobe epileptogenesis. Currently, research directly addressing this relationship is mostly limited to rodent studies (summarised in Table 1
), which we review separately as pre- and postnatal stress studies.
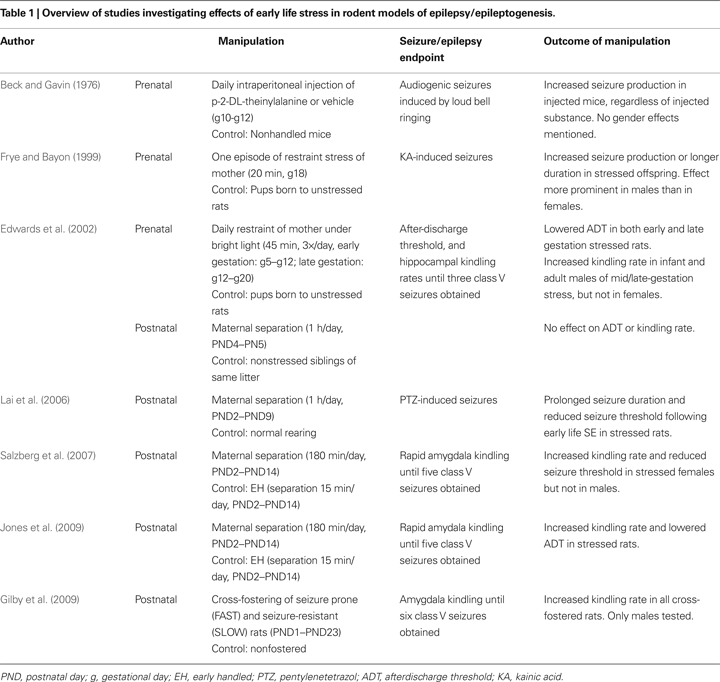
Effects of Prenatal Stress on the Development of Epilepsy
To our knowledge, a single study has examined the contribution of early life stress to epilepsy in later life in humans. This population-based cohort study involved children born to women who had lost a close relative during pregnancy or 1 year before pregnancy, with hospitalisation due to epilepsy as the outcome measure (Li et al., 2008
). In this study, no association was found between this particular form of prenatal stress and risk of epilepsy. However, this study is a weak test of the general hypothesis being considered in this review in that the study population included patients diagnosed with all types of epilepsies, not just MTLE; it included only hospitalised individuals, unrepresentative of the general epilepsy population; and bereavement has a wide array of potential and complex consequences – apart from the grief reaction – some of which may alleviate stress (e.g., the stress of caring for a disabled elderly relative) rather than causing it.
Edwards et al. (2002)
examined how stress exposure at different times during gestation might affect later limbic system excitability and the propensity to develop epilepsy (Edwards et al., 2002
). Pregnant dams were restrained under bright light for 45 min, three times a day during either early gestation (gestational day 5–12) or mid-late gestation (gestational day 12–20). Offspring of the stressed dams were then tested as either infants at postnatal day 10, or as adults, and compared with offspring from nonstressed dams. Outcome measures assessed were the electrically-induced seizure threshold (after-discharge threshold, ADT) and rate of seizure development using electrical hippocampal kindling. Both prenatal stressors significantly lowered ADT in pups, but this effect appeared to diminish by adulthood in the early gestational stress group. In addition, mid-late gestational stress accelerated kindling rates in all infant offspring and also in adult males, but had no effect in adult female rats. This study also incorporated cohorts of rats experiencing postnatal stress (see below).
Similar proconvulsant effects were observed in a study that investigated the effects of prenatal stress on kainic acid (KA)-induced seizures (Frye and Bayon, 1999
). Adult rats born to dams subjected to 20 min restraint stress on gestational day 18 experienced significantly more KA-induced partial and tonic-clonic seizures, and these were of longer duration, compared to rats born to nonstressed dams. These effects seemed more prominent in males rather than females.
In a third early study that initially set out to determine the effects of early life exposure to β-2-thienylalanine on susceptibility to audiogenic seizures later in life, pregnant mice received stressful intraperitoneal injections of β-2-thienylalanine or vehicle on gestational days 10, 11 and 12, or were not handled (Beck and Gavin, 1976
). On postnatal day 23, offspring were exposed to 60 s of loud bell ringing, which promotes seizures in some rodent strains. Offspring mice previously exposed to prenatal stress of injections, regardless of the injected substance, demonstrated increased susceptibility to the audiogenic stimulus, experiencing significantly more seizures than offspring of nonhandled pregnant mice. No mention was made in this manuscript of gender effects.
Effects of Postnatal Stress on the Development of Epilepsy
A recent study performed in our laboratory examined the effects of maternal separation on limbic excitability and the development of amygdala kindling (Salzberg et al., 2007
). Postnatal stress was induced by separating pups from their mothers for 180 min daily from postnatal day 2–14 (PND2–14). The comparison condition was mother/pup separation for 15 min/day over the same period, an exposure referred to as ‘early handling’. At 8 weeks of age, equivalent to young adult life, rats were tested for the afterdischarge threshold (ADT) and subjected to rapid amygdala kindling. Rats exposed to early life stress exhibited significantly lower seizure thresholds and an accelerated rate of kindling, compared to early handled rats. These effects on limbic excitability and epileptogenesis were specifically observed in female rats, whereas males, despite demonstrating increases in anxiety-like behaviour, did not demonstrate changes in epilepsy outcomes. This work has since been replicated (Jones et al., 2009
), this time with larger effect sizes and, although not clearly stated as such, in this study males also exhibited pro-epileptic effects of postnatal stress.
Another study investigating the effects of maternal separation on kindling epileptogenesis utilized a relatively benign separation protocol – 60 min of separation from mothers on PND4 and PND5 (Edwards et al., 2002
). The comparison group in this case were the other littermates which were briefly handled but not removed from the mother. This postnatal manipulation had no effect on ADT or rapid hippocampal kindling rates when assessed at 2 weeks of age. Several factors may be responsible for this negative result and the contrasting findings between this study and that of Salzberg et al., including the shorter follow-up and the milder form of early life stress.
Raising rat pups by foster mothers probes the influence of maternal care and the stress of cross-fostering itself on seizure susceptibility (Gilby et al., 2009
). Rats specifically bred to be seizure-prone (so-called ‘FAST’ rats) or seizure-resistant (‘SLOW’) were raised by a foster mother from their own strain, a foster mother from the opposite strain, or by their own mother, to investigate whether altering postnatal environments influences seizure disposition. Fostering began on the day of birth and persisted until weaning. Amygdala kindling rates were then assessed in adult males raised by the different parents. All cross-fostered animals, regardless of strain, demonstrated faster kindling rates than their nonfostered controls, intimating that the stress of fostering promoted a vulnerability to kindling epileptogenesis. It should be noted that these effects of cross-fostering were not related to any differences in postnatal maternal care (Gilby et al., 2007
).
Effects of postnatal stress on seizure threshold have also been investigated using chemoconvulsants to induce seizures (Lai et al., 2006
). Rats exposed to neonatal isolation from PND2–9 for 1 h/day, neonatal isolation plus lithium-pilocarpine induced SE on PND10, or neonatal isolation plus administration of metyrapone, a corticosteroid synthesis inhibitor prior to SE on PND10 were compared with nonhandled rats and SE-alone rats. Rats exposed to neonatal isolation experienced a longer duration of SE than those not exposed to separation stress, and this effect was reversed in metyrapone-treated rats, suggesting that corticosterone mediates this effect of the stress (Lai et al., 2006
). In adult life (∼PND100), rats were assessed for pentylenetetrazol (PTZ)-induced seizure duration and seizure susceptibility. Rats exposed to neonatal isolation and SE demonstrated a reduced PTZ threshold for seizure induction than nonhandled rats or rats exposed to isolation or SE alone. Metyrapone treatment prior to SE did not reverse this enhanced excitability.
Collectively, the studies described above examining effects of early life stress on various seizure- and epilepsy-related outcomes converge, supporting the proposal that early life stress in rodents promotes vulnerability to limbic epilepsy. These studies encompass a variety of paradigms of both pre- and postnatal stress, as well as diverse models of seizures and/or epilepsy. Publication bias (against ‘negative’ studies) cannot be ruled out, but based on this literature the data consistently point to a detrimental effect of early life stress in this disease.
Some studies report sex-specific effects of stress. Coupled with findings of increased rates of cryptogenic localization-related epilepsy in women, much of which would be MTLE (Christensen et al., 2005
), this is interesting given the higher rates of some mental health disorders (notably depression) in females (Kornstein and Sloan, 2006
), the evidence describing relevant sex-specific effects of early life stress in rodents (Darnaudery and Maccari, 2008
; Galanopoulou, 2008
; Mueller and Bale, 2008
), and the influence of sex hormones on seizures (Maguire et al., 2005
; Maguire and Mody, 2007
) and see discussion in Salzberg et al. (2007
).
Few of the studies we have reviewed linking early life stress to epilepsy are able to shed light on intervening mechanisms. As we have suggested – while there are several candidates, including limbic structural modifications, perturbed neuroendocrine regulation, and neurochemical and electrophysiological alterations – research tackling the mechanism(s) by which these effects are mediated remain rare. Rather than suggest priority research areas, a daunting task given the exceptionally heterogeneous and rapidly evolving worldwide research effort into unravelling epilepsy, we point to several issues which need to be taken into account in future research. These issues are: (1) the validity of animal models of human early life stress; (2) the issue of ‘time windows’ for stress effects; (3) nonhuman primate research; (4) genetic and epigenetic approaches; (5) interventions that ameliorate early life stress effects; and (7) translation to human research.
Prevalence and Types of Early Life Stress in Humans, and the Validity of Animal Models
Stressful experiences, both acute and chronic, are all too common in the lives of children in all societies (Glaser, 2008
; Hodes, 2008
; Jones, 2008
; Sandberg and Rutter, 2008
; Stein et al., 2008
). In industrialised societies these include childhood maltreatment (emotional, physical and sexual abuse) (Jones, 2008
); traumatic experiences such as assault (Sandberg and Rutter, 2008
); parental divorce, instability of caregivers, adoption or foster care; and bereavement. In many poorer societies stressors also include war, severe poverty, child labour, high rates of children orphaned as a result of parental HIV-AIDS and the stress of seeking refuge and asylum (Hodes, 2008
). To these can be added the stress on children of living with parental mental illnesses, such schizophrenia (Stein et al., 2008
), and the stress of childhood mental illness itself. In children with pre-existing chronic disabilities, such as intellectual disability, rates of abuse and neglect are two- to threefold higher than in other children (Jones, 2008
).
In the ‘Dunedin Study’ birth cohort of ∼1000 children, followed prospectively for 32 years (Caspi et al., 2002
), rates of maltreatment were: severe physical punishment (4% of participants); sexual abuse before age 11 (12%); disruptive caregiver changes before age 11 (6%). Twenty-seven percentage of children experienced at least one form of maltreatment; 9% experienced two or more.
The psychological consequences, short and long term, of early life stress in humans have been studied extensively; a smaller, but rapidly growing body of evidence links early stress and trauma to physical health outcomes in adulthood (McEwen, 2008b
). In seeking to understand the biological pathways connecting early stress to later physical illness, animal models are invaluable, but obviously have strengths and weaknesses. Strengths include the short life-times of laboratory animals, which allow testing of life-span hypotheses, e.g., about the enduring effects of perinatal stress.
A key weakness is the generalisability of an animal stressor to humans. For example, maternal separation stress in rats can be used in different ways. It can be employed in a ‘proof of principle’ experiment, simply to establish whether a well-characterised, severe, transient early postnatal stressor is at all capable of affecting some health outcome in adulthood. Alternatively, it can be employed as a model, albeit a limited one, of disruption of human maternal-infant attachment; but clearly, although the rat dam-pup relationship shares some common biological elements with the human mother-infant relationship (Insel et al., 2008
), the differences are extensive and generalisation from the animal model to humans is fraught (Pryce et al., 2005
). Primate models are much more convincing, but their validity too is unclear or limited (Cirulli et al., 2009a
).
Other methodological issues confront the use of these models. For one, the issue regarding the appropriate ‘control’ group in maternal separation studies in rodents has received ongoing debate (for detailed reviews, see Pryce and Feldon, 2003
; Macri and Wurbel, 2006
). Presently, published work employs a variation of comparison groups; some studies utilise nonhandled littermates, while others use briefly handled littermates, the latter an exposure which has been shown to result in ‘stress inoculation’ effects (Levine, 2005
). Finally, rather than studying the effects of early life stress at a single time point later in life – as most studies do to date – it is important to look at various intermediate time points to gain further insight into the order and causality of events leading up to the development of epilepsy.
‘Time Windows’ for Early Life Stress Effects
Neurobiological, behavioural and endocrine effects of early life stressors and of exposure to stress intermediaries (such as glucocorticoids) vary by timing of exposure. There is some human evidence to show this (Andersen et al., 2009
) and considerable animal evidence (Mueller and Bale, 2006
, 2007
, 2008
; Bock et al., 2008
; Gos et al., 2008
), see also Weinstock (2008)
. This has been shown for effects on several brain regions, including hippocampus and amygdala. In the various rodent models of early life stress, differential effects have been reported for stress in early, middle or late pregnancy (Weinstock, 2008
); and for stress before, during or after the stress hyporesponsive period (Vazquez et al., 2006
). For example, a recent study employed chronic variable stress applied to pregnant mice during either first, second or third week of pregnancy (Mueller and Bale, 2008
): pervasive effects were observed in adult (10–16 weeks old) offspring following early – but not middle or late – pregnancy stress exposure and, interestingly, this was apparent in males but not females. These effects included ‘depressive’ behaviours, greater corticosterone response to brief restraint stress, decreased GR levels in CA3 and dentate gyrus, and greater CRH expression in the central nucleus of amygdala. As the foetal brain is not yet formed in the first week of gestation, the authors examined the hypothesis that the sex-specific effects were mediated by actions on the placenta and report alterations in several placental genes and in DNA methyltransferase 1 (DNMT1), an enzyme involved in maintaining DNA methylation; such changes very plausibly could affect ensuing development of the foetal brain in the second and third weeks, although this remains to be tested directly. Thus, their study illustrates marked effects of the timing of prenatal stress exposure on neurodevelopment and, as well, sheds light on potential mechanisms underlying sex differences in stress effects on earliest neurodevelopment (in this case, possible epigenetic alterations in key placental genes).
A major challenge is how to extrapolate animal data to humans, given the different developmental trajectories and different relationship of neurodevelopmental stages to pregnancy and birth (Avishai-Eliner et al., 2002
). There is a general consensus that, with regard to neurodevelopment, the early neonatal period in the rat (PND0–10) corresponds to the third trimester of human foetal development. Thus for example, dentate granule cells begin to form at prenatal day ∼18 in the rat and gestation week 13–14 in the human (Avishai-Eliner et al., 2002
). Despite many commonalities, there are also many differences in development of the HPA axis and of distribution of central MR and GR in rats, nonhuman primates and humans (Gunnar and Quevedo, 2008
). If there is a stress-hyporesponsive period in humans, it is not present in the early postnatal period, when the HPA axis is normally very reactive, but develops later in the first year (Champagne et al., 2009
).
Research Involving Nonhuman Primates
The barriers of cost and expertise to studies using nonhuman primates are well known, however a strong case can be made that such research is justifiable and necessary to understand the linkages between early life stress and limbic epilepsy. Rodent studies are exceptionally valuable, shedding light on many aspects of epileptogenesis and providing innumerable leads to studies in humans, but are seriously limited when it comes to exploring the interplay of psychosocial development, neurodevelopment and epilepsy. In part this is because of the differences in central regulation of the HPA axis and stress systems in primates versus rodents, in part due to the very different nature of parenting and other aspects of social behaviour (Sanchez, 2006
; Nelson and Winslow, 2009
).
Nonhuman primate studies of psychopathology provide an excellent model: they adopt a life-span developmental perspective, examine the interplay of psychosocial processes with neurodevelopment and of genes with environment (Barr et al., 2003
; Suomi, 2006
; Nelson and Winslow, 2009
; Stevens et al., 2009
), a perspective which increasingly is seen to be relevant also to epilepsy and its neurobehavioural comorbidities (Hermann et al., 2008
). Deprivation studies in nonhuman primates are the closest animal model of early neglect in humans, and have been shown to produce effects similar to those seen in children with mental, behavioural and emotional disorders (Sanchez et al., 2001
; Gilmer and McKinney, 2003
; Pryce et al., 2005
; Sanchez, 2006
; Stevens et al., 2009
).
Some studies are of direct relevance to MTLE. For example, utilising positron emission tomography (PET), Ichise et al. (2006)
reported decreased serotonin transporter binding in right medial temporal structures (amongst others) as a result of early life stress in rhesus macaques (Ichise et al., 2006
). Other research in nonhuman primates suggests that early life stress affects neurotrophin levels (BDNF and NGF), factors highly involved in brain development and function (Cirulli et al., 2009a
,b
) and in the neuroplastic processes thought to underlie limbic epileptogenesis. Apart from one early study (Heath, 1972
), it appears stress has not been explored in nonhuman primate epilepsy research.
Genetic and Epigenetic Approaches
Abundant studies now describe altered expression of many different genes in human and experimental models of epilepsy. These include, but are not limited to studies of neurotrophins, neuropeptides, plasticity-related genes and those associated with neurotransmission, particularly those directly influencing neuronal excitability (see Elliott and Lowenstein, 2004
for review). Whether these alterations in gene expression are cause or consequence of the disease process or simply epiphenomena remains, in several cases, to be elucidated. Nevertheless, a greater understanding of the expression profiles of causative genes, and those also influenced by exposure to early life stress, has immense potential to provide understanding of the disease process of epileptogenesis, and also to provide novel approaches for gene therapy. Particular focus should be paid to genetic aspects of the CRH system, which is strongly implicated in the pathophysiology of epilepsy (Baram and Hatalski, 1998
), and in the enduring effects of early life stress (Brunson et al., 2001
). Genes for many epilepsy syndromes have been identified in recent years (Steinlein, 2008
), however it is likely that the genetic contribution to most of the burden of the epilepsies involves multiple genes of small effect, as is the case for other neural disorders such as Alzheimer’s disease and schizophrenia. In addition, as with many other neural disorders, it’s likely that various forms of gene-environment interaction are important and, here too, various forms of life stress may well play a role (Casey et al., 2009
).
Epigenetic mechanisms may underlie many of the gene changes involved in epilepsy. Studies exploring chromatin structural alterations following seizures, including DNA methylation and histone modification, report effects on genes relevant to the development of epilepsy, including those coding for glutamate receptors and BDNF (Palm et al., 1998
; Huang et al., 2002
; Tsankova et al., 2004
; Sng et al., 2006
; Casey et al., 2009
). Additionally, valproic acid, a commonly used antiepileptic drug, is a recognised inhibitor of histone deacetylases (Phiel et al., 2001
), a mechanism by which this drug may exert its therapeutic effects (Jessberger et al., 2007
). Early life stress, too, induces enduring epigenetic alterations of the promoter regions of relevant genes (Weaver et al., 2004
; Mueller and Bale, 2008
; Roth et al., 2009
), and so a synergy between these chromatin modifications may result in alterations in gene transcription, leading to accelerated rates of epilepsy. The field of epigenetics is largely unexplored with respect to both epilepsy and stress biology, but warrants further study – both separately and combined.
Interventions in Pathways Between Early Life Stress and Limbic Epilepsy
Interventions are of interest both as ways to probe causal mechanism and as leads to potential treatments and prevention. Experimentally, in studies of stress in early life, a wide range of interventions have been deployed to manipulate endocrine or autonomic components of the stress response, including CRH antagonists, glucocorticoid synthesis inhibitors (metyrapone) and glucocorticoid receptor antagonists. Some of these have been trialled as psychotropic agents, e.g., CRH antagonists and mifepristone; however most drugs used in experimental stress research are not clinically useful (with the possible exception of mifepristone, which may have utility in severe depression DeBattista and Belanoff, 2006
) and, when considering interventions for stress in early life in particular, would raise serious concerns about long-term adverse consequences.
Other experimental interventions have been deployed to interfere with aspects of epileptogenesis. As well as antiepileptic medications, these include viral delivery of gene-modifying agents (Raol et al., 2006
) and agents that manipulate neurogenesis, such as NMDA receptor antagonists, mitosis inhibitors and irradiation (Scharfman and Gray, 2007
). An interesting question is whether antiepileptic drugs modify stress-related effects in limbic epileptogenesis. Recently, ethosuximide has been shown to suppress the development of spike-wave epilepsy in WAG/Rij rats, a genetic model of human absence epilepsy (Blumenfeld et al., 2008
). Additionally, treated rats failed to show the usual abnormal elevations of ion channels in somatosensory cortex (Nav1.1, Nav1.5 and HCN1) that are part of the WAG/Rij phenotype. This study demonstrates that anti-epileptic drugs can modify epileptogenesis for nonlimbic epilepsy, but whether this is also achievable for limbic epilepsy has not yet been established. Interestingly, the evolution of seizures in WAG/Rij rats is exacerbated by both early handling and by maternal separation in early life (Schridde et al., 2006
). Similar dysregulation of ion channels is certainly a feature of limbic epileptogenesis, and also a feature of febrile seizures (Dube et al., 2009
), but whether early life stress modifies ion channels in this way is not yet known.
‘Second generation’ antidepressants, such as serotonin specific reuptake inhibitors (SSRIs) are known to affect processes involved in limbic epileptogenesis, such as dentate gyrus neurogenesis (Dranovsky and Hen, 2006
; Sahay and Hen, 2007
; Balu and Lucki, 2009
); to mitigate effects of early life stress in experimental studies (e.g., Lee et al., 2001
); to moderate the HPA activation that is often a part of the depressed state (Pericic et al., 2005
; Pariante and Lightman, 2008
); and to reduce seizure activity in humans (Alper et al., 2007
), as well as experimentally in vivo and in vitro (Jobe and Browning, 2005
; Pericic et al., 2005
; Alper et al., 2007
; Jobe and Browning, 2007
). However, the actions of antidepressants on limbic epileptogenesis – both before and after the onset of overt seizures – are not yet known.
As well as these pharmacological approaches, some evidence suggests environmental enrichment is protective against epilepsy and its behavioural and affective accompaniments (Auvergne et al., 2002
; Koh et al., 2005
, 2007
; Dhanushkodi and Shetty, 2008
; Korbey et al., 2008
). This is an important line of research both because of the light it may shed on mechanism and because of the opportunities it suggests for prevention of epilepsy in humans (Hernandez-Reif et al., 2007
; Maguire et al., 2008
). Unfortunately, this is a neglected area of research: psychosocial interventions in early life are known to have neurobehavioural developmental benefits in children at risk; it’s plausible that such interventions also ameliorate epileptogenesis through some of the mechanisms already discussed, but to our knowledge this has not been studied with the exception of a small and encouraging study in adolescents with epilepsy, showing a time-limited cognitive-behavioural intervention reduced both depression and seizure frequency (Martinovic et al., 2006
).
Human Studies
Definitive tests of hypotheses linking early life stress causally to MTLE (and its psychiatric comorbidities) require cohort studies; given the relatively low incidence of MTLE, such studies, as stand-alone projects, are most unlikely to be mounted. Excellent models exist of highly productive cohort studies of early life stress and psychopathology – such as the Dunedin study (Caspi et al., 2003
; Caspi and Moffitt, 2006
) – addressing high-prevalence psychiatric comorbidity, and others of low prevalence disorders, such as schizophrenia (Malaspina et al., 2008
). Other strategies are to nest studies of epilepsy within large cohort studies mounted for other purposes or to employ case-control studies. In our view, the strength of the experimental evidence, both indirect and direct, is such as to justify such research.
In addition, it is important that genetic epidemiological studies of epilepsy examine both the psychiatric and epilepsy phenotypes and histories of the subjects (and families) they recruit. As in the case of psychiatric disorder, much of the burden of epilepsy is likely to involve the actions of multiple genes of small effect interacting in complex ways with environmental exposures over the lifespan (Crino, 2007
; Steinlein, 2008
) and probably modified epigenetically. Arguably, one of the key exposures to be considered is life stress, including – and perhaps especially – stress in early life.
We have presented both direct evidence and a supporting rationale for the general proposition that early life stress may have detrimental effects on the development of limbic epilepsy. Although publication bias against ‘negative’ studies cannot be ruled out, the consistency of the small body of studies that directly address the hypothesis is striking. Several enduring neurobiological consequences of early life stress, including structural modifications of limbic regions, electrophysiological and neurotransmitter perturbations, and HPA axis modifications may underlie these effects, although at this stage, little has been comprehensively proven regarding the responsible mechanisms. Study of early life stress may also shed light on the relationships between MTLE and its common psychiatric comorbidities, such as depression. Taken together, these diverse lines of evidence and argument support the proposition that early life stress may contribute causally to MTLE; this is an hypothesis whose time has arrived.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
N.C. Jones is supported by an NHMRC project grant #566544. M.R. Salzberg is supported by an NHMRC project grant #566843.
Barbaccia, M. L., Scaccianoce, S., Del Bianco, P., Campolongo, P., Trezza, V., Tattoli, M., Cuomo, V., and Steardo, L. (2007). Cognitive impairment and increased brain neurosteroids in adult rats perinatally exposed to low millimolar blood alcohol concentrations. Psychoneuroendocrinology 32, 931–942.
Blumenfeld, H., Klein, J. P., Schridde, U., Vestal, M., Rice, T., Khera, D. S., Bashyal, C., Giblin, K., Paul-Laughinghouse, C., Wang, F., Phadke, A., Mission, J., Agarwal, R. K., Englot, D. J., Motelow, J., Nersesyan, H., Waxman, S. G., and Levin, A. R. (2008). Early treatment suppresses the development of spike-wave epilepsy in a rat model. Epilepsia 49, 400–409.
Brunson, K. L., Eghbal-Ahmadi, M., Bender, R., Chen, Y., and Baram, T. Z. (2001). Long-term, progressive hippocampal cell loss and dysfunction induced by early-life administration of corticotropin-releasing hormone reproduce the effects of early-life stress. Proc. Natl. Acad. Sci. U.S.A. 98, 8856–8861.
Carpenter, L. L., Carvalho, J. P., Tyrka, A. R., Wier, L. M., Mello, A. F., Mello, M. F., Anderson, G. M., Wilkinson, C. W., and Price, L. H. (2007). Decreased adrenocorticotropic hormone and cortisol responses to stress in healthy adults reporting significant childhood maltreatment. Biol. Psychiatry 62, 1080–1087.
Casey, B. J., Glatt, C. E., Tottenham, N., Soliman, F., Bath, K., Amso, D., Altemus, M., Pattwell, S., Jones, R., Levita, L., McEwen, B., Magarinos, A. M., Gunnar, M., Thomas, K. M., Mezey, J., Clark, A. G., Hempstead, B. L., and Lee, F. S. (2009). Brain-derived neurotrophic factor as a model system for examining gene by environment interactions across development. Neuroscience. doi: 10.1016/j.neuroscience.2009.03.081.
Champagne, D. L., Bagot, R. C., van Hasselt, F., Ramakers, G., Meaney, M. J., de Kloet, E. R., Joels, M., and Krugers, H. (2008). Maternal care and hippocampal plasticity: evidence for experience-dependent structural plasticity, altered synaptic functioning, and differential responsiveness to glucocorticoids and stress. J. Neurosci. 28, 6037–6045.
Gizerian, S. S., Moy, S. S., Lieberman, J. A., and Grobin, A. C. (2006). Neonatal neurosteroid administration results in development-specific alterations in prepulse inhibition and locomotor activity: neurosteroids alter prepulse inhibition and locomotor activity. Psychopharmacology (Berl.) 186, 334–342.
Gorter, J. A., Van Vliet, E. A., Rauwerda, H., Breit, T., Stad, R., van Schaik, L., Vreugdenhil, E., Redeker, S., Hendriksen, E., Aronica, E., Lopes da Silva, F. H., and Wadman, W. J. (2007). Dynamic changes of proteases and protease inhibitors revealed by microarray analysis in CA3 and entorhinal cortex during epileptogenesis in the rat. Epilepsia 48(Suppl. 5), 53–64.
Hatazaki, S., Bellver-Estelles, C., Jimenez-Mateos, E. M., Meller, R., Bonner, C., Murphy, N., Matsushima, S., Taki, W., Prehn, J. H., Simon, R. P., and Henshall, D. C. (2007). Microarray profile of seizure damage-refractory hippocampal CA3 in a mouse model of epileptic preconditioning. Neuroscience 150, 467–477.
Jamali, S., Bartolomei, F., Robaglia-Schlupp, A., Massacrier, A., Peragut, J. C., Regis, J., Dufour, H., Ravid, R., Roll, P., Pereira, S., Royer, B., Roeckel–Trevisiol, N., Fontaine, M., Guye, M., Boucraut, J., Chauvel, P., Cau, P., and Szepetowski, P. (2006). Large-scale expression study of human mesial temporal lobe epilepsy: evidence for dysregulation of the neurotransmission and complement systems in the entorhinal cortex. Brain 129, 625–641.
Raol, Y. H., Lund, I. V., Bandyopadhyay, S., Zhang, G., Roberts, D. S., Wolfe, J. H., Russek, S. J., and Brooks-Kayal, A. R. (2006). Enhancing GABA(A) receptor alpha 1 subunit levels in hippocampal dentate gyrus inhibits epilepsy development in an animal model of temporal lobe epilepsy. J. Neurosci. 26, 11342–11346.
Schwarcz, R., Scharfman, H., and Bertram, E. (2002). Temporal lobe epilepsy: renewed emphasis on extrahippocampal areas. In Neuropsychopharmacology: The Fifth Generation of Progress, K. Davis, D. S. Charney, J. T. Coyle, and C. B. Nemeroff, eds (Philadelphia, PA: Wolters Kluwer Health/Lippincott Williams and Wilkins), pp. 1843–1856.