1
Northwest Network VISN 20 Mental Illness Research, Education and Clinical Center, VA Puget Sound Health Care System, Seattle, WA, USA
2
Department of Psychiatry and Behavioral Sciences, University of Washington, Seattle, WA, USA
3
Geriatric Research, Education and Clinical Center, VA Puget Sound Health Care System, Seattle, WA, USA
Early studies of posttraumatic stress disorder (PTSD) reported that abnormal function of the hypothalamic–pituitary–adrenocortical (HPA) system was associated with the disorder. However, subsequent studies attempting to identify a specific aspect of HPA dysfunction that characterizes PTSD have been marked by considerable inconsistency of results. A facet of HPA regulation that has been considered but not definitively investigated is the possibility that the responsiveness of the adrenal cortex to physiological concentrations of adrenocorticotropin (ACTH) is diminished in PTSD. Relationships between PTSD and the adrenal androgen dehydroepiandrosterone (DHEA) have also been postulated. In this study we investigated the magnitude and time course of changes in concentrations of plasma cortisol and DHEA in response to bolus infusions of physiological doses of ACTH1–24 in PTSD patients and control subjects. We found no evidence for PTSD-related alterations in cortisol or DHEA secretion in response to stimulation by low doses of ACTH and conclude that adrenocortical responsiveness is normal in PTSD. Results from this and other studies suggest that the occurrence of defects in HPA function in PTSD may be specific responses to particular combinations of trauma type, genetic susceptibility, and individual history.
Attribution of relationships between posttraumatic stress disorder (PTSD) and hypothalamic–pituitary–adrenocortical (HPA) dysfunction is intuitively apposite because of the importance of the HPA axis in the mediation of stress responses, and several early studies of PTSD reported HPA dysregulation (Smith et al., 1989
; Yehuda et al., 1990
, 1991
, 1993
; Hockings et al., 1993
). However, from the beginning, consistency in the specifics of these findings has been lacking. Low plasma or serum cortisol has been the most frequently observed HPA abnormality in PTSD (Yehuda, 2002
), but a large number of studies have failed to find decreased circulating cortisol (Meewisse et al., 2007
). A recent meta-analysis found that across 37 studies, 828 people with PTSD and 800 controls did not differ in cortisol levels (Meewisse et al., 2007
). However, cortisol levels were lower in PTSD patients than in controls not exposed to trauma. The meta-analysis indicated that an association of PTSD with low cortisol is most likely to be observed when: (a) women are included in the study, (b) cortisol is sampled in the afternoon near its circadian nadir, and (c) the PTSD is the result of physical or sexual abuse. A variety of specific mechanisms and loci of HPA dysregulation have been suggested to explain low cortisol in PTSD (de Kloet et al., 2006
). The currently prevailing model argues for enhanced negative feedback of the HPA axis by cortisol. This viewpoint has been supported by several studies (Yehuda et al., 1995
, 2002
, 2004a
,b
, 2006b
; Newport et al., 2004
) but not by others (Kanter et al., 2001
; Kellner et al., 2004
; Lindley et al., 2004
). A review of the experimental evidence for a specific pathophysiological mechanism for PTSD that surveyed studies of both pharmacological manipulations and non-pharmacological stress challenges concluded that there was insufficient evidence to support any of the models advanced so far to implicate HPA axis dysfunction in PTSD (de Kloet et al., 2006
).
A mechanistic possibility that has received relatively little investigative attention is hyposensitivity to adrenocorticotropin (ACTH) at the level of the adrenal cortex, which could explain both subnormal circulating cortisol concentrations and compensatory upregulation of CNS corticotropin-releasing factor (Kanter et al., 2001
). However, Rasmusson et al. (2001
) found that patients with PTSD produce more cortisol in response to the standard clinical 250 μg ACTH stimulation test than controls. The supraphysiological dose of ACTH used in this test, which was designed to be a clinical indicator of adrenal insufficiency, provides an index of the adrenal’s maximal secretory capability, but is of very limited value in gauging the responsiveness of the adrenal to submaximal, physiological concentrations of ACTH.
Therefore, to examine adrenocortical responsiveness to physiological stimulation in PTSD patients, we performed a pilot study to determine doses of ACTH that would stimulate a low to moderate cortisol response and one that would reflect a high level of stress but remain within the physiological range. On the basis of the pilot study we administered placebo and two doses of ACTH (1.5 and 7.5 ng/kg body weight) to PTSD patients and control participants. We hypothesized that in response to administration of low dose exogenous ACTH, cortisol concentrations of PTSD participants would increase less than the cortisol concentrations of control participants, reflecting adrenal hyposensitivity to ACTH in PTSD.
Circulating levels of dehydroepiandrosterone (DHEA) have also been investigated in connection with PTSD. DHEA, and its sulfated form, DHEA sulfate (DHEAS), are secreted by the human adrenal cortex in concentrations greater than cortisol, and their secretion is also stimulated by ACTH. However, they are secreted from a different adrenocortical zone than cortisol, are differentially regulated, and display biological actions that may oppose those of cortisol (Albertson et al., 1984
; Kimonides et al., 1999
; Muller et al., 2006
; Rainey and Nakamura, 2008
). Discordance among DHEA, DHEAS, and cortisol secretion has often been reported (Griffing et al., 1985
; Baulieu, 1996
; Rainey and Nakamura, 2008
), as well as differences in their relationships to PTSD (Spivak et al., 2000
; Yehuda et al., 2006a
; Bremner et al., 2007
; Gill et al., 2008
). In an earlier study we found plasma DHEA concentrations to be lower in male PTSD patients than in male control subjects (Kanter et al., 2001
). In other studies, DHEA in PTSD has generally been reported to be either unchanged (Pico-Alfonso et al., 2004
; Rasmusson et al., 2006
; Bremner et al., 2007
) or higher than normal (Spivak et al., 2000
; Rasmusson et al., 2004
; Yehuda et al., 2006a
; Gill et al., 2008
). Based on our earlier results, we hypothesized that DHEA concentrations of PTSD participants would increase less than those of control participants in response to administration of physiological doses of ACTH.
Subjects
This study was approved by the institutional review board at the University of Washington, and all subjects provided written informed consent. All subjects underwent a medical history, screening physical examination, 12-lead electrocardiogram, and screening laboratory tests of blood samples for electrolytes, glucose, blood urea nitrogen, creatinine, serum glutamic oxaloacetic transaminase, serum glutamic pyruvic transaminase, thyroid function tests, and complete blood count, and of urine samples for routine urinalysis, urine toxicology screen, and for female subjects of childbearing potential, urine pregnancy test.
Use of any psychoactive medication or any medication known to affect the HPA axis within 2 weeks of the study was exclusionary for all subjects. These medications included antidepressants, antipsychotics, anticonvulsants, tranquilizers, sedative/hypnotics, antihypertensives, and oral or inhaled glucocorticoids. The only medications taken by participants in the study were ibuprofen, the antihistamines cetirizine and loratadine, and glucosamine and chondroitin for treatment of osteoarthritis.
Participants taking medications to treat PTSD were withdrawn from those medications, under medical supervision, for up to 30 days. The period required for withdrawal was dependent on the biological half-life of each medication and management of potential withdrawal symptoms.
Female subjects of childbearing potential underwent urine pregnancy tests on each study day to ensure that no pregnant participants were exposed to ACTH. Female subjects of childbearing potential agreed to use a non-hormonal barrier form of birth control for the period of the study. Hormonal contraceptives were not allowed for at least 3 months prior to the study because of their stimulation of corticosteroid-binding globulin (CBG) that results in elevated levels of total plasma cortisol.
The inclusion criteria for participants in the PTSD group were: (a) meeting DSM-IV diagnostic criteria for PTSD based on administration of the Structured Clinical Interview for DSM-IV (SCID) – Patient Edition (First et al., 1995
); (b) a Clinician-Administered PTSD scale (CAPS) score ≥65 (Blake et al., 1990
, 1995
); (c) a Life Event History (LEH) scale score of ≥1 (Kessler et al., 1995
); (d) a Mississippi Scale for Combat-Related Posttraumatic Stress Disorder score of ≥111 (or ≥107 on the civilian version) (Keane et al., 1988
); (e) an Impact of Event Total Scale score ≥50 (Weiss and Marmar, 1997
); (f) a Posttraumatic Stress Diagnostic Scale score ≥34 (Foa, 1995
); and (g) a PTSD Checklist score ≥50 (Weathers et al., 1993
; Blanchard et al., 1996
; Weathers and Ford, 1996
). Control subjects were administered the SCID – Nonpatient Edition and the LEH. To confirm the participants’ self-report of never having been exposed to trauma, an LEH scale score of 0 was required for inclusion in the control group.
All participants were free of current (past 6 months) acute or chronic Axis I psychiatric disorders (with the exception of PTSD in the PTSD subject group) based on SCID assessment. Two control and one PTSD subject had past histories of depression. Absence of substance abuse within 3 months of the study by subject report and urine toxicology screen was required. One control and seven PTSD subjects had a past history of alcohol abuse, and one individual in each group had a history of alcohol dependence. Two control and one PTSD participant had past histories of substance abuse (cannabis). One subject in each group was a current smoker; no information about past smoking history was requested.
Participants included nine PTSD subjects: eight males with combat trauma PTSD and one female with civilian trauma PTSD, 50.3 ± 2.5 years old [mean ± standard error of the mean (SEM)]. There were nine healthy non-trauma exposed aged-matched controls: six males and three females, 41.2 ± 3.5 years old. The ages of the two groups did not differ significantly. The PTSD participants had moderately severe symptoms according to the CAPS (total CAPS score = 76.7 ± 6.0).
ACTH Infusion
For each participant, the three study infusions were performed on different days, separated by at least 1 week, in the Clinical Research Unit of the Veterans Affairs Puget Sound Health Care System, Seattle Division. Participants were instructed to abstain from alcohol on the evening prior to each study session, to eat a normal breakfast (or at least a snack and liquid) before 10:00 am. Subjects were instructed not to eat or drink and not to smoke after 10:00 am and to arrive at the VA Special Studies Unit by noon. The two smokers were allowed to wear a nicotine patch. The 1.5 and 7.5 ng/kg doses of ACTH1–24 (Cortrosyn®, Organon) and placebo (normal saline) were administered in a random, counterbalanced manner on the three separate study days. The study room was quiet and free of stimuli that might precipitate PTSD symptoms. Subjects were maintained in a supine position and an intravenous (IV) catheter was placed in the antecubital vein of each arm; one catheter was used for infusion of ACTH or placebo, and the other, for blood draws. The IV lines were kept patent with a slow (50 ml/h) infusion of normal saline. We obtained baseline blood samples 30 and 35 min following the placement of the IV catheters. Forty minutes (time 0) after catheter placement, the study drug for the given day was administered via IV bolus push (<30 s). All infusions were begun by 1:00 pm. The afternoon study time was selected because the circadian rhythms of circulating ACTH and cortisol rhythms undergo a rapid concentration decline in the morning. By early afternoon the levels of both hormones provide a relatively low, flat, and stable baseline, particularly in the absence of a mid-day meal, which is typically followed by a postprandial hormonal spike (Van Cauter et al., 1992
).
Subjects, but not investigators, were blind to the study condition. ACTH was diluted in normal saline in order to measure the small doses precisely, and the IV line was flushed thoroughly after ACTH administration.
Blood Collection and Hormonal Measurements
Blood samples were collected every 5 min for 60 min following ACTH administration and then every 15 min for the next 90 min. Blood was collected into chilled tubes containing ethylenediaminetetraacetic acid; the tubes were placed on ice until processed. The samples were cold-centrifuged within 1 h of collection. The plasma was aliquoted and stored at −80°C until assayed. For each hormone, all samples from a given subject were included in a single assay, and each assay contained samples from an equal number of subjects from each group.
Plasma ACTH concentration was measured with DSL-2300 ACTH RIA Kits according to the manufacturer’s protocol (Diagnostic Systems Laboratories, Webster, TX, USA). The method was chosen because the ACTH antibody has 100% cross-reactivity with the infused ACTH1–24, thus allowing an accurate measurement of total circulating ACTH (ACTH1–24 and endogenous ACTH1–39 combined). The intra- and inter-assay coefficients of variation are 6.0 and 7.0%, respectively, and the minimum detection limit is 3.5 pg/ml.
Cortisol was measured by radioimmunoassay in unextracted plasma. Samples were diluted with phosphate buffer and heated for 20 min at 80°C to denature binding globulins. Cortisol antiserum was obtained from ICN Biomedicals (Costa Mesa, CA, USA). The detection limit for cortisol is 0.5 mg/dl. Intra- and inter-assay coefficients of variation are 4.6 and 10.2%, respectively.
Plasma concentrations of DHEA were measured with the DSL-8900 DHEA Radioimmunoassay Kit (Diagnostic Systems Laboratories, Webster, TX, USA) following the manufacturer’s protocol. Cross-reactivity of the antiserum with DHEA-sulfate is 0.02%, and cross-reactivities with other steroids are all <1%. Sensitivity of the assay is 0.03 ng/ml. The intra- and inter-assay coefficients of variation are 2.8 and 5.9%, respectively.
Statistical Analysis
The effects of subject group and ACTH dose on the magnitude and time-course of the cortisol and DHEA responses were determined using the concentration values at each time point minus the baseline values (response delta) by analysis of variance (ANOVA) for repeated measures followed by Fisher’s protected least significant difference (PLSD) post hoc test for paired comparisons. Areas under the response curves (AUCs) of the three groups were compared by ANOVA. Correlations between PTSD symptom severity and cortisol and DHEA measures were determined by linear regression.
ANOVA for repeated measures for the effect of participant diagnosis on the baseline levels of each of the three hormones (two baseline samples at each of three sessions) showed no differences in ACTH or DHEA concentrations between groups, but significantly higher circulating cortisol levels in the PTSD group (F5,52 = 4.2, p = 0.046).
There were no adverse effects of the ACTH infusion. At the peak of ACTH concentration (5 min after infusion in both groups and for both ACTH doses), there was a highly significant effect of dose (F1,48 = 34.7, p < 0.0001) on the change in circulating ACTH relative to baseline (ACTH delta). Paired comparisons of the ACTH deltas of the three treatment conditions with Fisher’s PLSD also indicated highly significant separation between placebo, 1.5 and 7.5 ng/kg conditions (Figure 1
). There was no significant group effect or group–dose interaction, demonstrating that the infusions produced comparable levels of circulating ACTH in both groups. The change in ACTH plasma concentration following the higher dose (115.8 ± 16.6 pg/ml) was more than threefold greater than that following the low dose ACTH infusion (34.8 ± 6.6). Delta ACTH was slightly negative for the placebo condition (−5.1 ± 12). The area under the curve of the plasma ACTH concentration was calculated for the first six sampling times (30 min) after the infusion during which ACTH concentration rapidly rises to a peak and falls to baseline again. ANOVA of the AUC showed a highly significant effect of dose (F2,48 = 21.4, p < 0.0001), but no significant group or interaction effects.
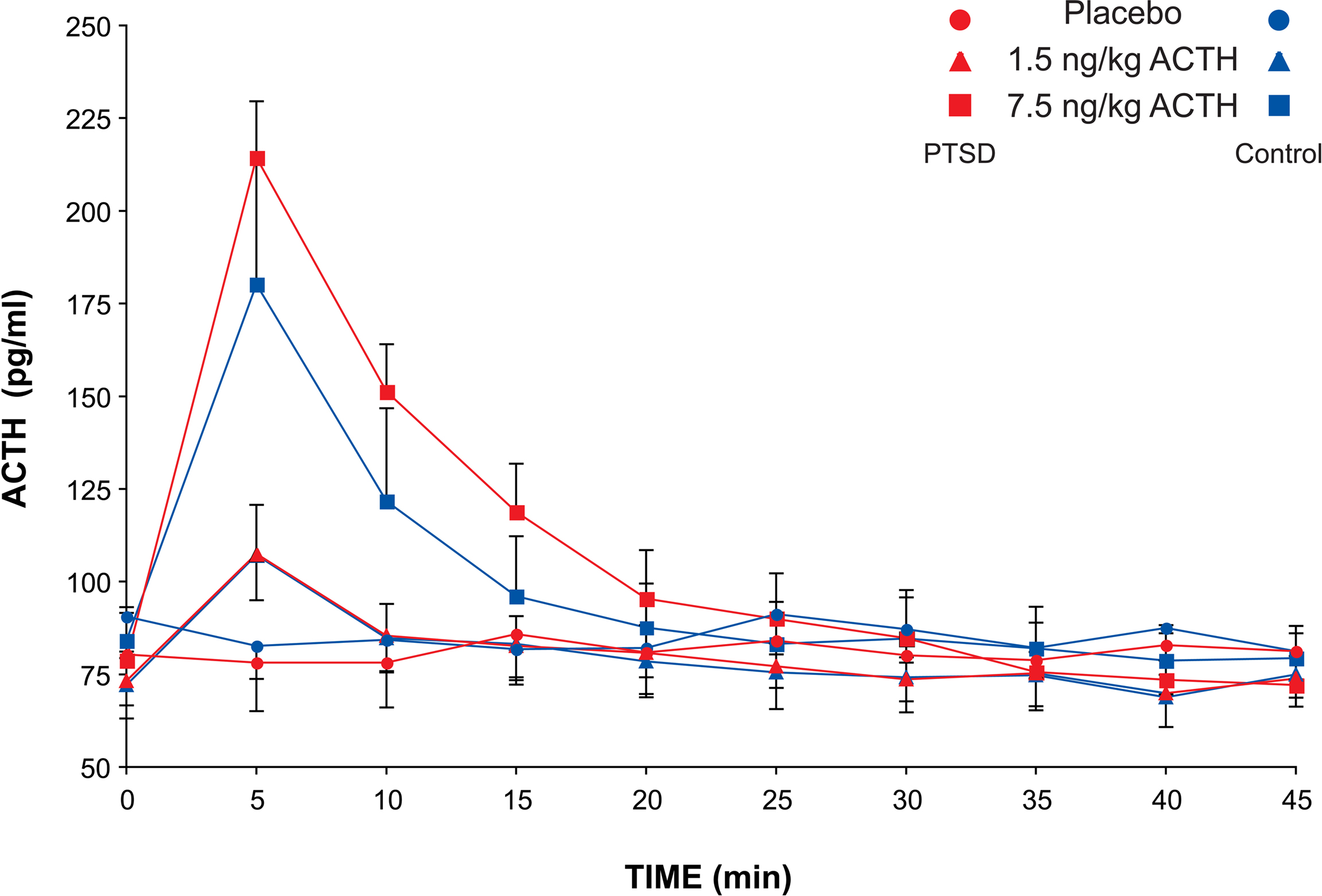
Figure 1. Plasma ACTH concentration in PTSD and control subjects after ACTH infusion. Sampling occurred at 5-min intervals for 60 min and at 15-min intervals for the following 90 min. Mean and SEM values are plotted for each subject group and each dose of ACTH for the first 45 min after infusion during the dynamic portion of the ACTH concentration profile.
There were significant dose-related differences in the magnitude of the cortisol responses to the ACTH injections in both groups of subjects (effect of dose on cortisol AUC: F2,48 = 19.7, p < 0.0001). However, neither the cortisol response delta nor the area under the cortisol response curve differed between subject groups at either dose, and there was no group–dose interaction (Figure 2
).
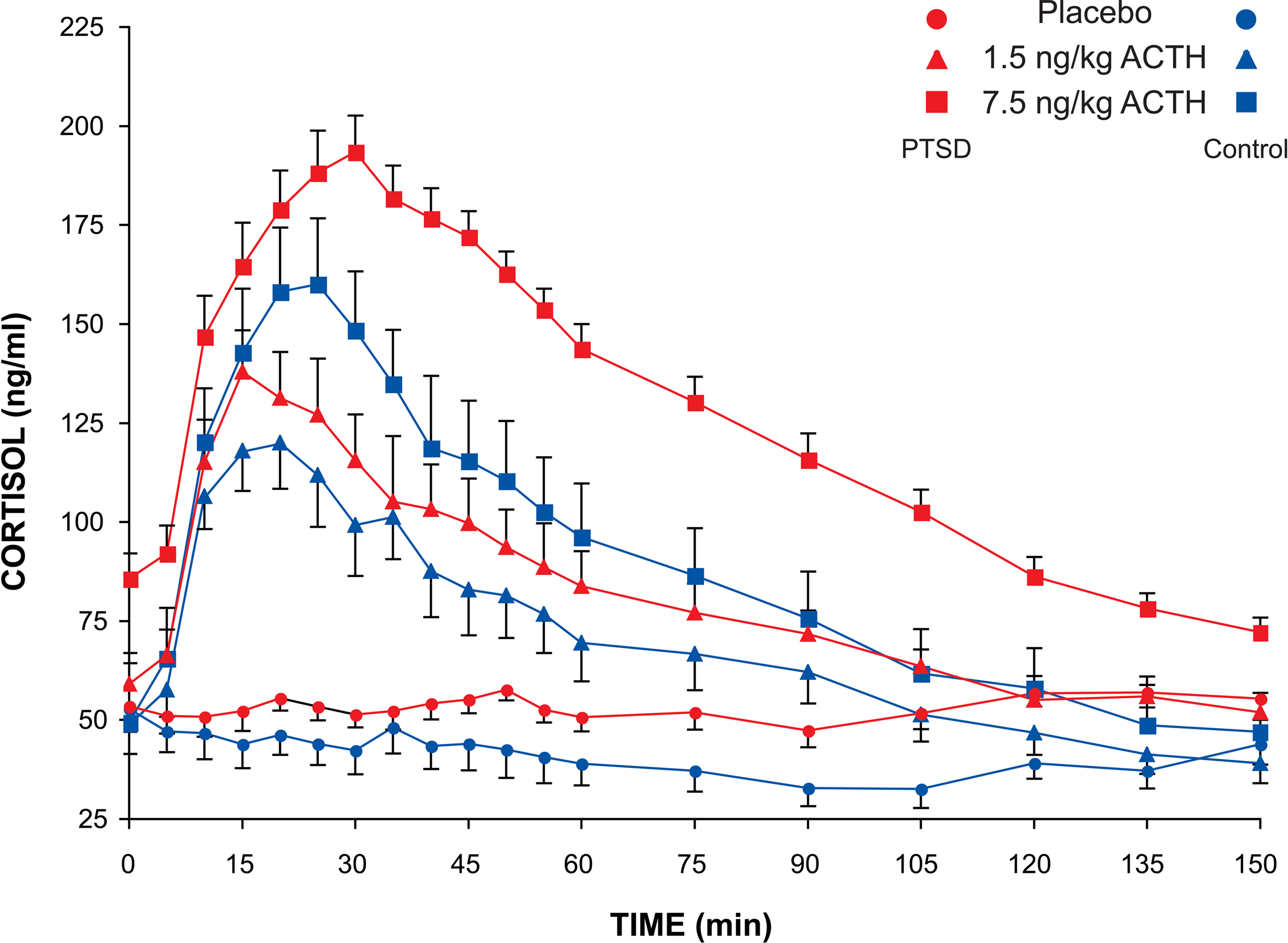
Figure 2. Plasma cortisol concentration in PTSD and control subjects after ACTH infusion. Sampling occurred at 5-min intervals for 60 min and at 15-min intervals for the following 90 min. Mean and SEM values are plotted for each subject group and each dose of ACTH for the entire 150-min sampling period after infusion.
ANOVA for repeated measures for delta DHEA concentration showed no effect of subject group (F1,48 = 0.004, p = 0.95) on delta DHEA, although there were highly significant dose (F2,48 = 6.2, p < 0.005), time and time–dose interaction effects (both p < 0.001) (Figure 3
). The area under the DHEA dose-response curve increased from 11.7 ± 5.8 after the placebo infusion to 15.5 ± 6.4 after the 1.5 ng/kg dose ACTH infusion to 31.8 ± 10.7 after the 7.5 ng/kg dose. However, due to high variability in the DHEA responses to ACTH, there were no significant effects of dose or subject group on the DHEA AUC. The ANOVA of the AUC of the ratio of circulating cortisol to DHEA was highly significant (F2,48 = 12.5, p < 0.0001), consistent with the finding of a pronounced dose effect on cortisol. There was no group or group by dose effect on the cortisol/DHEA ratio.
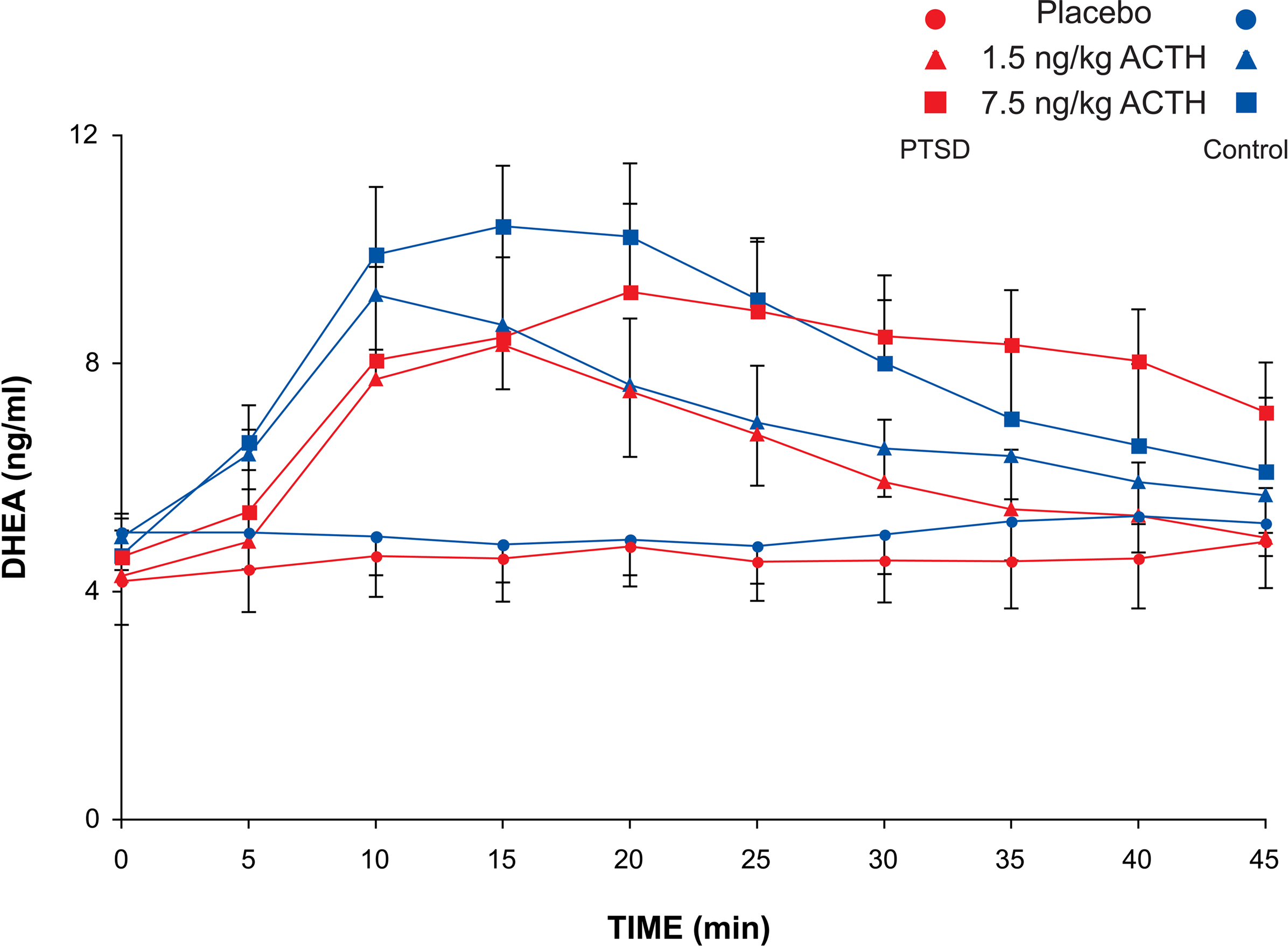
Figure 3. Plasma DHEA concentration of PTSD and control subjects after ACTH infusion. Sampling occurred at 5-min intervals for 60 min and at 15-min intervals for the following 90 min. Mean and SEM values are plotted for each subject group and each dose of ACTH for the first 45 min after infusion.
PTSD symptom severity, as measured by baseline CAPS total scores, did not correlate significantly with the increases in cortisol or DHEA. After the lower dose of ACTH, the correlations of cortisol and DHEA AUCs with symptom severity as measured by CAPS total scores were r = −0.47, p = 0.21 and r = −0.10, p = 0.79, respectively. After infusion with the higher dose, the correlations of CAPS scores with cortisol and DHEA were r = −0.27, p = 0.49 and r = 0.31, p = 0.42, respectively.
Our primary finding was that cortisol and DHEA responses to physiological doses of ACTH did not differ significantly between PTSD and control participants. This result suggests that adrenal function and responsiveness to ACTH are normal in PTSD and that if indeed, HPA dysregulation is a concomitant of PTSD, the locus of the functional deficits is not at the level of the adrenal cortex.
The peak ACTH concentrations achieved after the higher of our two doses (7.5 ng/kg) were approximately 150–250 pg/ml, which are comparable to those reached after administration of the standard insulin tolerance test to control subjects (plasma glucose concentration ≤2.2 mmol/l) (Radikova et al., 2006
) or after exhaustive exercise (Farrell et al., 1983
; Inder et al., 1998
). Extreme stresses such as severe sepsis or multiple trauma (Vermes et al., 1995
) are required to elicit increases in ACTH concentration (∼350–800 pg/ml) similar to the peak levels observed after the clinical 1 μg “low-dose” ACTH test (∼550–800 pg/ml) (Gerritsen and Vermes, 1997
). The ACTH levels (∼4000–5800 pg/ml) resulting from the standard clinical 250 μg test are much higher yet (Raff and Findling, 1989
). The 250 μg dose (∼3,570 ng/kg for a 70-kg subject), is over 475 times greater than the higher of the two doses used in the present study.
Our results differ from those of Rasmusson and colleagues (Rasmusson et al., 2001
, 2004
) who found that cortisol concentrations of PTSD patients rose significantly more than those of control participants following injection of 250 μg of ACTH. The dose of ACTH administered is one of several methodological differences that may explain these conflicting results. Several researchers have noted that standard clinical high-dose ACTH administration, and even the 1 μg “low-dose” (approximately 14.3 ng/kg for a 70-kg individual) ACTH challenge test maximally stimulate ACTH receptors. These doses provide a valid index of maximal adrenocortical capacity to respond to ACTH but do not measure the sensitivity of the adrenal cortex to stimulation by ACTH in the normal physiological range (Thaler and Blevins Jr., 1998
; Arvat et al., 2000
; Tordjman et al., 2000
; Rasmusson et al., 2001
).
There are also multiple differences in the characteristics of the study populations of the Rasmusson studies (Rasmusson et al., 2001
, 2004
, 2006
) and the current study that make comparisons difficult. The participants in the Rasmusson studies were all premenopausal women, whereas the subjects in the current study were predominantly male, but included some female participants. All but one of the participants in the Rasmusson studies had been victims of civilian sexual trauma, whereas all but one of the PTSD patients in the current study had experienced combat trauma. The Rasmusson PTSD groups included some participants with comorbid diagnoses of major depressive disorder, obsessive compulsive disorder, dysthymia, or generalized anxiety disorder. We excluded subjects with Axis I diagnoses other than PTSD. The Rasmusson studies also included subjects taking oral contraceptives, which result in significant elevations in total plasma cortisol responses to ACTH due to increased liver production of CBG (Gozansky et al., 2005
; Šimunková et al., 2008
). In the first study (Rasmusson et al., 2001
), four of the PTSD subjects and none of the controls were taking oral contraceptives, which may have contributed to greater plasma cortisol responses associated with PTSD.
The DHEA results of the present study are consistent with the conclusion drawn from the cortisol data that adrenocortical responses to physiological concentrations of ACTH are normal in PTSD. However, most studies have found elevated DHEA to be associated with PTSD (Maninger et al., 2009
), and injection of 250 μg ACTH elicited significantly higher DHEA concentrations in PTSD than in control subjects (Rasmusson et al., 2004
).
We did not measure DHEAS in the current study and cannot speculate about DHEAS-related changes in PTSD. There is a tendency in some review articles to combine DHEA and DHEAS data as if the concentrations of the two steroids reflected similar reactions to stimuli. However, DHEA and DHEAS are differentially regulated, fluctuations in their circulating levels are discordant (Baulieu, 1996
; Chen and Parker Jr., 2004
), and their concentrations have been shown not to change in tandem in PTSD patients (Bremner et al., 2007
; Olff et al., 2007
). Circulating DHEA concentrations undergo circadian oscillations coincidentally with cortisol, but because of its low metabolic clearance rate (MCR), DHEAS remains at approximately the same concentrations throughout the day and is much less susceptible to stress-related perturbations (Baulieu, 1996
). DHEAS has a half-life of 7–10 h as opposed to the 15–30 min half-life of DHEA, and a MCR of 5–20 L/day compared to approximately 2,000 L/day for DHEA.
The dissimilarity in the observed effects of ACTH administration on DHEA and cortisol between the Rasmusson study and this study may reflect differences in ACTH dose, inclusion/exclusion criteria, gender, and/or the nature of the precipitating trauma. Generalizations from the present study may be limited by the relatively small number of subjects and the inclusion of a mixed gender study population, although gender differences in human adrenal responsiveness to physiological doses of ACTH have not been demonstrated. Serum cortisol responses to eight ACTH doses from 0.01 to 250 μg did not differ significantly between men and women (Arvat et al., 2000
). However, gender differences occur in other aspects of human HPA function, and gender effects in the present study cannot be ruled out. Consistent effects of gender on free cortisol responses to specific stressors have been reported (Seeman et al., 2001
; Kudielka et al., 2009
), and estrogens stimulate hepatic production of CBG, resulting in elevated levels of total plasma cortisol in women (Wilkinson et al., 2001
). In addition, sensitivity to glucocorticoid feedback inhibition differs between men and women in the morning (Wilkinson et al., 1997
), but not in the evening (Wilkinson et al., 2001
). Therefore, to test the possibility that the absence of an effect of PTSD on adrenal responsiveness observed here was the result of an interaction of gender and PTSD, additional statistical analyses were carried out using data from male subjects only (not shown), yielding an n of 8 in the PTSD group and 6 in the control group. There were no significant group differences in plasma ACTH concentration in the dynamic phase during the first 30 min after infusion, and no significant group effects or group–dose interactions on ACTH, cortisol or DHEA AUC as determined by ANOVAs.
The absence of significant differences between PTSD and control participants in cortisol and DHEA responses to physiological levels of ACTH suggests that abnormalities in adrenal responsiveness are unlikely to be a major factor in PTSD-related alterations in HPA axis function. Thus, future research on the neuroendocrinology of PTSD may be more successfully directed toward detailed examination of central mechanisms such as enhanced negative feedback of the HPA axis by cortisol or alteration of glucocorticoid or mineralocorticoid receptor function or number. Studies of glucocorticoid feedback in PTSD have seldom made explicit reference to the three discrete time domains and mechanisms of inhibition that have been demonstrated (Keller-Wood and Dallman, 1984
; Posener et al., 1997
; Posener et al., 1998
). However, the possibility exists that differences in feedback inhibition between PTSD and control subjects may be restricted to, or more pronounced in, a single time domain. To our knowledge only two studies have investigated fast feedback inhibition in PTSD, one finding increased feedback in PTSD (Yehuda et al., 2006b
) and one finding no group differences (Kanter et al., 2001
). Other studies have focused on slow feedback, generally using dexamethasone suppression of ACTH secretion.
It is unlikely that fast feedback affected the magnitude of the cortisol response to ACTH infusion in the present study. Peak circulating cortisol concentrations were reached between 15 and 30 min post-injection. Glucocorticoid fast feedback inhibition results in significant decreases in endogenous ACTH levels within 15–30 min in both rats and humans, but because of the much longer half-life of cortisol and corticosterone, circulating glucocorticoid concentrations are not reduced by fast feedback in either species within 30 min (Liberzon et al., 1997
; Boscaro et al., 1998
).
The diversity of the findings related to HPA dysfunction and the variation in the history and comorbidity of the subject populations in various studies suggests that HPA dysfunction may not be a necessary concomitant of PTSD and that the existence of dysfunction or the specific locus of the dysfunction may vary as a function of differing types or intensities of traumatic experience. Variations in circulating concentrations of adrenocortical steroids, alterations of glucocorticoid feedback inhibition, modifications of adrenal responsiveness, or other changes in HPA axis function, when they occur in conjunction with PTSD, may be characteristic responses to specific constellations of the nature and frequency of the trauma, gender, age, genetic susceptibility, and individual history.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
For their excellent assistance that made this study possible, we thank Jane Hammett RN, and Jennifer Acselrod RN, for nursing care of study participants, Jill Perander, Robert Beckham III, Shawna Feely, and Kristina Purganan for recruitment and study coordination, Libby Colasurdo and Carl Sikkema for hormone assays, Molly Chinn for assistance in preparing ACTH doses, and Nadia Czajkiewicz for graphic design. This material is based upon work supported by the Northwest Network VISN 20 Mental Illness Research, Education and Clinical Center (MIRECC), the VA Puget Sound Geriatric Research, Education and Clinical Center (GRECC), and the Office of Research and Development Medical Research Service, Department of Veterans Affairs.
Albertson, B. D., Hobson, W. C., Burnett, B. S., Turner, P. T., Clark, R. V., Schiebinger, R. J., Loriaux, D. L., and Cutler, G. B. Jr. (1984). Dissociation of cortisol and adrenal androgen secretion in the hypophysectomized, adrenocorticotropin-replaced chimpanzee. J. Clin. Endocrinol. Metab. 59, 13–18.
Arvat, E., Di, V. L., Lanfranco, F., Maccario, M., Baffoni, C., Rossetto, R., Aimaretti, G., Camanni, F., and Ghigo, E. (2000). Stimulatory effect of adrenocorticotropin on cortisol, aldosterone, and dehydroepiandrosterone secretion in normal humans: dose-response study. J. Clin. Endocrinol. Metab. 85, 3141–3146.
Rasmusson, A. M., Vasek, J., Lipschitz, D. S., Vojvoda, D., Mustone, M. E., Shi, Q., Gudmundsen, G., Morgan, C. A., Wolfe, J., and Charney, D. S. (2004). An increased capacity for adrenal DHEA release is associated with decreased avoidance and negative mood symptoms in women with PTSD. Neuropsychopharmacology 29, 1546–1557.
Tordjman, K., Jaffe, A., Trostanetsky, Y., Greenman, Y., Limor, R., and Stern, N. (2000). Low-dose (1 microgram) adrenocorticotrophin (ACTH) stimulation as a screening test for impaired hypothalamo–pituitary–adrenal axis function: sensitivity, specificity and accuracy in comparison with the high-dose (250 microgram) test. Clin. Endocrinol. (Oxf.) 52, 633–640.