1
Departments of Psychology, The Ohio State University, Columbus, OH, USA
2
Department of Neuroscience, The Ohio State University, Columbus, OH, USA
3
The Institute for Behavioral Medicine Research, The Ohio State University, Columbus, OH, USA
Chronic stress is capable of exacerbating each major, modifiable, endogenous risk factor for cerebrovascular and cardiovascular disease. Indeed, exposure to stress can increase both the incidence and severity of stroke, presumably through activation of the hypothalamic-pituitary-adrenal (HPA) axis. Now that characterization of the mechanisms underlying epigenetic programming of the HPA axis is well underway, there has been renewed interest in examining the role of early environment on the evolution of health conditions across the entire lifespan. Indeed, neonatal manipulations in rodents that reduce stress responsivity, and subsequent life-time exposure to glucocorticoids, are associated with a reduction in the development of neuroendocrine, neuroanatomical, and cognitive dysfunctions that typically progress with age. Although improved day to day regulation of the HPA axis also may be accompanied by a decrease in stroke risk, evidence from rodent studies suggest that an associated cost could be increased susceptibility to inflammation and neuronal death in the event that a stroke does occur and the individual is exposed to persistently elevated corticosteroids. Given its importance in regulation of health and disease states, any long-term modulation of the HPA axis is likely to be associated with both benefits and potential risks. The goals of this review article are to examine (1) the clinical and experimental data suggesting that neonatal experiences can shape HPA axis regulation, (2) the influence of stress and the HPA axis on stroke incidence and severity, and (3) the potential for neonatal programming of the HPA axis to impact adult cerebrovascular health.
Activation of the hypothalamic-pituitary-adrenal (HPA) axis following exposure to a stressor is part of an adaptive response that enables an organism to respond appropriately to changes in the environment (McEwen, 2000
). The hormonal cascade initiated by stress involves the release of corticotropin-releasing hormone (CRH) from the paraventricular nucleus of the hypothalamus into hypothalamo-hypophyseal portal blood vessels, through which it travels to the anterior pituitary gland, where it stimulates the release of adrenocorticotrophic hormone (ACTH). ACTH then enters the general circulation, and stimulates the adrenal cortex to produce and secrete glucocorticoids (GCs). There are two main, structurally similar, GCs among mammals: corticosterone (predominant in most rodents) and cortisol (predominant in humans and most other mammals). Norepinephrine and epinephrine, two additional hormones important in coordinating the physiological and behavioral responses to stress, are released by the adrenal medulla within seconds of exposure to stress. Although the physiological necessity of GC and catecholamine secretion is apparent when responding to an acute stressor, repeated and prolonged exposure to these hormones over the course of a life-time can increase vulnerability to a wide array of physical and psychological pathologies (Korte et al., 2005
; McEwen, 2008
).
Prior to disease onset, chronic stress or increased exposure to endogenous or exogenous corticosteroids can precipitate physiological and psychological changes that increase risk for chronic disease; the effects of stress have been particularly well-documented in the context of contributing to the development of cardiovascular and cerebrovascular disease. Several of the important endogenous risk factors for human vascular disease can be modified by stress or are associated with HPA axis dysregulation, including high blood pressure, elevated serum cholesterol, glucose intolerance, high body mass index, and inflammation. For example, systemic hypertension is associated with blunted morning cortisol and impaired negative feedback of the HPA axis (Wirtz et al., 2007
), as well as increased cortisol responses to stress (Nyklicek et al., 2005
). Serum cholesterol is elevated immediately following an acute laboratory stressor and remains elevated among those experiencing chronic stress (Stoney et al., 1999a
,b
); in turn, elevated cholesterol can facilitate atherosclerosis (reviewed in Insull, 2009
). Likewise, extended release of HPA axis hormones that are responsible for increasing blood glucose to meet energy demands during an acute stressor, can result in the development of myopathy (Tomas et al., 1979
; Mitsui et al., 2002
) and steroid diabetes (Homo-Delarche et al., 1991
) over time. Exposure to stress also can alter eating habits (Zellner et al., 2006
, 2007
; Adam and Epel, 2007
) and the distribution of body fat; specifically, increased corticosteroid and psychological responses to stress are associated with increased abdominal accumulation of fat (Epel et al., 2000
), which in turn is associated with elevated cardiovascular and cerebrovascular risk (Mathieu et al., 2009
). Stress also can affect vascular health via alterations in the immune system. For example, chronic care-giver stress is associated with overproduction of serum IL-6 (Kiecolt-Glaser et al., 2003
), a pro-inflammatory cytokine that is associated with increased risk for heart disease and stroke (Cesari et al., 2003
; Kaplan et al., 2008b
). Furthermore, in correlational analyses, established risk factors for cardiovascular disease and stroke typically form anthropometric, metabolic, and haemodynamic clusters that are not intercorrelated (Rosmond and Bjorntorp, 2000
). However, when subjects exhibiting signs of HPA axis dysregulation are analyzed separately, these clusters become strongly intercorrelated, such that one cluster, including all anthropometric, metabolic and haemodynamic factors (except high density lipoprotein), is apparent rather than three separate clusters (Rosmond and Bjorntorp, 2000
). Thus, HPA axis function may have an intervening influence on nearly all other major risk factors for vascular disease in humans. This relationship between HPA axis function and disease factors provides a mechanism through which genetic influences (for example, see Rosmond et al., 2000
; Koeijvoets et al., 2008
), aging (Ferrari and Magri, 2008
), and life-time exposure to stress (Korte et al., 2005
) can modify vulnerability to vascular disease.
Individual differences in stress responsivity play an important role in determining both the acute and cumulative effects of GC exposure over the course of a life-time. There are significant differences in how individuals perceive and interpret stressors, and the encoding of memories of past stressful events influences one’s response to future stressors (Buchanan and Lovallo, 2001
; Marti et al., 2001
; Wolf, 2003
). Genetic factors also can contribute to individual differences in HPA function and stress responsivity (Kuningas et al., 2007
; Ising et al., 2008
; Peeters et al., 2008
; Raef et al., 2008
; Duan et al., 2009
). Recognizing the potential for an interaction between glucocorticoid gene polymorphisms and environment in influencing physiological and behavioral phenotypes is also important (Bet et al., 2008
). However, maternal influences during the prenatal and early post-natal developmental periods may be another important source of variance in HPA axis function, allostatic load, and the onset of psychopathology and age-related disease (Matthews, 2000
; Barrington, 2001
; McEwen, 2002
; Gluckman et al., 2005
; Patin et al., 2005
), including stroke and heart disease (Barker, 1997
; Lamireau et al., 2002
; Sinclair et al., 2007
).
The quality of the uterine environment and maternal-infant interactions in early life varies widely among humans. A plethora of studies over the past two decades have suggested that these factors can contribute to early programming of the HPA axis (reviewed in Barker, 1997
; Seckl, 2000
; Mesquita et al., 2009
). Although there is some evidence that the human fetal adrenal cortex is capable of synthesizing cortisol as early as the first trimester of gestation (Goto et al., 2006
), expression of intracellular glucocorticoid receptors does not become apparent in the hippocampus and other major systems and tissues until mid to late gestation (Noorlander et al., 2006
). The onset of GR expression is important for normal fetal development, but it also increases vulnerability of the fetus to elevated circulating GCs originating from the mother’s adrenal gland. The fetus is partially protected by placental 11beta-hydroxysteroid dehydrogenase, which enables GCs from the mother to be converted into an inactive form when crossing the placenta (Edwards et al., 1993
; Benediktsson et al., 1997
; reviewed in O’Donnell et al., 2009
); indeed a negative correlation between cortisol concentrations in umbilical cord and placental 11beta-HSD activity has been observed in full-term human pregnancies (Mericq et al., 2009
). Furthermore, mutations in the human 11beta-hydroxysteroid dehydrogenase gene are associated with low birth weight (Mune et al., 1995
), which is presumably related to increased in utero exposure to GCs (Seckl and Holmes, 2007
). However, even in the absence of such a mutation, a precipitous rise in maternal glucocorticoid concentrations in response to extreme stress or administration of exogenous hormones, could overwhelm the placental 11beta-hydroxysteroid dehydrogenase, and increase fetal exposure to glucorticoids, which could conceivably alter the structure and function of endocrine and insulin-sensitive target tissues in the fetus (Holness et al., 2000
). For example, there is a stronger positive correlation between maternal and amniotic cortisol concentrations among pregnant women who report elevated anxiety during pregnancy than those who are not anxious (Glover et al., 2009
). Indeed, there are a plethora of studies in humans linking perinatal measures, such as birth weight, with adult metabolic disorders and vascular disease; however, much of the evidence supporting a causal relationship between early exposure to GCs and health measures in adulthood have been gleaned from experimental studies utilizing non-human primates and rodents. These studies also aid in identifying and confirming underlying mechanisms, such as the effects of maternal stress on placental 11beta-hydroxysteroid dehydrogenase type 2 expression and activity, organ weights, and the expression of a variety of genes related to HPA axis function and metabolism in rats (Mairesse et al., 2007
). Thus, there are several human and experimental studies supporting the conclusion that the relatively protective environment of the fetus can be compromised by stress, which in turn can alter fetal development, and cause potental long-term health consequences.
To explain such consequences of fetal environment on long-term health, the fetal programming hypothesis was developed. The fetal programming hypothesis proposes that the physiological, neuroendocrine, or metabolic adaptations that enable the fetus to adequately respond to changes in the early life environment can result in semi-permanent phenotypic changes in the fetus (reviewed in Barker, 1998
). The altered programming of the developmental pattern of proliferation and differentiation events within key tissue and organ systems can permanently alter the set points in the metabolic, cardiovascular, and immune systems of the individual (Barker, 1997
, 2002
; Coe et al., 1999
; Fagoaga and Nehlsen-Cannarella, 2002
; Louey and Thornburg, 2005
; Phillips, 2006
). One of the primary areas of interest in fetal programming research has been the correlation between low birth weight and adult cardiovascular disease (Seckl, 2001
; Barker et al., 2009
). In the past, the majority of investigations of the long-term consequences of low birth weight have focused on fetal nutrition. It was suggested that malnourishment resulted in fetal metabolic adaptations, such as the development of tissue insulin resistance, that reduce glucose metabolism and cell growth. Thus, by decelerating energy metabolism and cell growth, the fetus would be able to survive the under-nutritional status and conserve glucose and nutrients for the more critical process of brain development (Phillips, 1996
). However, if such mechanisms are semi-permanently programmed in the fetus, then the insulin resistance and decreased metabolism could eventually lead to type 2 diabetes, obesity, hypertension, and cardiovascular disease as the individual ages (reviewed in Sallout and Walker, 2003
; Barker et al., 2009
). Certainly, shortcomings in the fetal programming hypothesis exist; critics have argued since its inception that many of the original studies suffered design limitations, such as not adequately describing the study population, high subject dropout rates, and inadequate control of socioeconomic status (Klebanoff et al., 1999
). Furthermore, as is often the case when human populations are examined, neonatal programming studies could not effectively establish causation nor thoroughly explore mechanisms under consistent conditions. Most of the clinical studies also can not distinguish between prenatal and early post-natal influences on health. In contrast, animal studies have been crucial for rigorously testing hypotheses regarding the mechanisms underlying neonatal programming of the HPA axis and adult health outcomes, but are limited by the uncertainty as to how closely the neonatal manipulation used in the animal models and the physiological responses approximate the human condition. So far, however, the concurrence between human and non-human tests of the neonatal programming hypothesis is promising.
Recently, the connection between low birth weight and adult cardiovascular health in humans has focused increasingly on concomitant changes in HPA axis function (reviewed in Van den Bergh and Marcoen, 2004
). Exposing the fetus to stress-induced concentrations of GCs may permanently alter HPA axis development, stress responsivity, life-time glucocorticoid exposure, and associated disease risks (Welberg and Seckl, 2001
; Rieger et al., 2004
; Austin et al., 2005
). For example, babies that were exposed to dexamethasone, a synthetic glucocorticoid, to promote lung growth in utero were found to have significantly higher arterial blood pressure at birth than babies of mothers who received the control treatment only (Nathanielsz et al., 2003
). High arterial blood pressure, in turn, has been linked to an increased risk of stroke and heart disease in adulthood (MacMahon et al., 1990
). Prenatal treatment with corticosteroids also has been associated with persistent behavioral problems, including increased aggression, attentional deficits, and cognitive deficits (Trautman et al., 1995
; French et al., 2004
).
There also are indications that fetal stress responsivity can be influenced by the mother. For example, fetuses of mothers with depression or high levels of anxiety have greater heart rate increases as the mother undergoes a psychological stressor than fetuses of healthy, low anxiety, non-depressed mothers; fetal heart rate responses may serve as a predictor for childhood stress responsivity (Monk et al., 2004
). Furthermore, high levels of maternal anxiety during critical periods of fetal brain development may result in increased susceptibility of the offspring to develop childhood affective and behavioral disorders, such as symptoms of attention deficit hyperactivity disorder, externalization of problems, and heightened anxiety in response to stressors (Van den Bergh and Marcoen, 2004
). Though the impact of early mother-infant interactions cannot be completely separated from genetics and prenatal factors in these studies, they do suggest that stress exposure during early development can have long-lasting effects on the psychiatric and physiological health of the offspring. To address the issue of inherited versus pre- and post-natal environmental factors on adverse health and behavioral outcomes in the offspring, a clinical study observed children born to related or unrelated mothers as a result of in vitro fertilization. The conclusion was that regardless of genetic relatedness, maternal reports of elevated prenatal stress were associated with lower infant birth weight and gestational age, and increased anxiety and reported antisocial behavior among children ranging in age from 4- to 10-years old (Rice et al., 2009
). However, when current symptoms of maternal anxiety/depression were used as an additional covariate, it was found to significantly mediate the association between prenatal stress and increased childhood anxiety (Rice et al., 2009
). Furthermore, neonates born to mothers with prenatal diagnoses of anxiety or depression had increased basal cortisol concentrations if the mother provided insensitive or unresponsive care following birth, but were indistinguishable from neonates born to prenatally healthy mothers if provided with more sensitive post-natal care (Kaplan et al., 2008a
). Thus, the effect of prenatal conditions on offspring behavioral outcomes may rely, in part, on the post-natal environment that the neonate experiences.
In common with prenatal stress and glucocorticoid exposure, neonatal and early childhood environment can have pervasive effects on HPA axis functioning and stress responsivity (reviewed in Repetti et al., 2002
). Though the critical period of limbic development, and subsequent HPA axis functioning, is thought to be complete by birth in human neonates, over-stimulation of the stress response in infants and children can permanently alter stress responsivity and risk for disease. For example, young boys of non-supportive parents were found to have heightened sympathetic-adrenomedullary reactivity to laboratory stressors, as measured my increased heart rate responses, in comparison to boys of supportive families (Woodall and Matthews, 1989
). Cardiovascular reactivity also correlates with increased left ventricular mass, a major risk factor for chronic heart disease (Allen et al., 1997
). Furthermore, the biological dysregulations that occur in response to poor family relationships and negative rearing environments can persist into adulthood; poor parental relationships, as determined by college students rating their early family environment, are associated with elevated cortisol responses to a laboratory stressor (Luecken, 1998
; Pruessner et al., 2004
). In addition, women with a history of childhood abuse show greater responses to staged challenges than women without an abusive childhood; responses are further exaggerated among women with a history of abuse as a child and current symptoms of anxiety and depression (Heim et al., 2000
, 2008
). Although not directly correlated with altered HPA axis functioning, childhood abuse or household dysfunction have been associated with increased incidence of adult diseases, such as ischemic heart disease, cancer, chronic lung disease, skeletal fractures, and liver disease (Felitti et al., 1998
). The Harvard Mastery of Stress Study, for example, indicated a strong correlation between low perceived childhood parental care and midlife onset of stress-related illnesses, such as coronary heart disease, hypertension, duodenal ulceration, and alcoholism (Russek and Schwartz, 1997a
,b
). Furthermore, the incidence of overall disease was significantly heightened in individuals who responded to stressors with severe anxiety (Russek et al., 1990
). Thus, there appears to be an emerging association in human literature among negative post-natal environment and negative consequences on adult HPA axis responsivity and stress-related disease.
Though the human correlational data provide convincing evidence for the relationship among fetal programming of the HPA axis, early parental care, and adult health, it is not possible to establish a causal relationships among early life experiences, HPA axis dysregulation, and compromised health from the clinical datasets; it is in this regard that rodent studies on the topic of prenatal and post-natal programming of the HPA axis are critically important (reviewed in Weinstock, 2008
; Champagne, 2009
). Studies in rats have established that one of the molecular mechanisms underlying perinatal programming of the HPA axis involves epigenetic changes in glucocorticoid receptor expression due to changes in histone acetlyation and DNA demethylation (reviewed in Weaver, 2009
). These findings suggest that maternal care can produce semi-permanent changes in gene expression in brain regions vital to stress responsivity, thereby providing a potential mechanism for fetal and neonatal programming of adult, stress-induced disease in humans.
Cerebral ischemia is the loss of blood flow and oxygen to cells in the brain. When the reduction in blood flow occurs throughout the entire brain, for example during cardiac arrest, it is termed global cerebral ischemia. In contrast, if only a region of the brain experiences reduced blood flow, for example during stroke, it is termed focal ischemia. At the cellular level, ischemia initiates neuronal death pathways and immunological trafficking (Doyle et al., 2008
; Ribe et al., 2008
; Amantea et al., 2009
). Clinical and experimental data suggest that the HPA axis may have important influences on these pathophysiological responses to ischemia.
Activation of the HPA axis is among the first, easily measurable, physiological responses to cerebral ischemia (Fassbender et al., 1994
; Johansson et al., 1997
; Slowik et al., 2002
). There is a sustained surge in GCs that persists for days after the ischemic attack. Though post-ischemic GC-release was initially postulated to be beneficial (i.e. by reducing edema), the functional significance of this response remains largely speculative and, in fact, may adversely affect neuronal survival (Anne et al., 2007
). Until recently, treatment of stroke patients with exogenous GCs upon admission to the hospital was standard protocol, but it is no longer recommended in light of the relationship between elevated endogenous cortisol and poor stroke outcome, and the potential for GCs to alter other physiological processes, such as glycemic balance, that are associated with poor stroke prognosis (Gomes et al., 2005
). Indeed, among stroke patients, high acute phase endogenous cortisol is associated with increased infarct volume, greater functional disability, and increased mortality (Murros et al., 1993
; Fassbender et al., 1994
; Christensen et al., 2004
; Marklund et al., 2004
; Anne et al., 2007
). Thus, environmental and genetic factors that modify HPA axis activity could, in turn, affect susceptibility to ischemic disease.
The literature provides a convincing association between stress and stroke outcome, with evidence presented from both clinical and experimental studies (Table 1
). Pre-ischemic stress and glucocorticoid exposure can influence stroke onset and outcome in human stroke patients and animal models (Carasso et al., 1981
; Harmsen et al., 1990
; House et al., 1990
; DeVries et al., 2001
; Madrigal et al., 2003
; Kivimaki et al., 2008
; Surtees et al., 2008
; Tsutsumi et al., 2009
). For example, self and family reports of stroke patients indicate that stressful life events often precede the onset of stroke, with stroke victims reporting a higher incidence of stress occurring days, or even months, prior to stroke onset, in comparison to controls (House et al., 1990
). Two prospective studies confirm that chronic occupational stress also is associated with a substantial increase in stroke risk compared to individuals with low reported job stress (Kivimaki et al., 2008
; Tsutsumi et al., 2009
). Similarly, experimental data indicate that rodents that are exposed to restraint or social stress prior to experimental stroke develop significantly larger infarcts than non-stressed cohorts (DeVries et al., 2001
; Sugo et al., 2002
; Madrigal et al., 2003
; Caso et al., 2007
, 2008
, 2009
; McDonald et al., 2008
). Furthermore, the treatment of mice with a glucocorticoid receptor antagonist immediately prior to stress prevents the negative effects of stress on ischemic outcome, while injection of exogenous corticosterone mimics the effects of stress on infarct size and cognitive function after stroke (Sugo et al., 2002
). Together, the clinical and experimental data suggest that exposure to stress or elevated corticosteroids increases both the risk for, and the severity of, stroke.
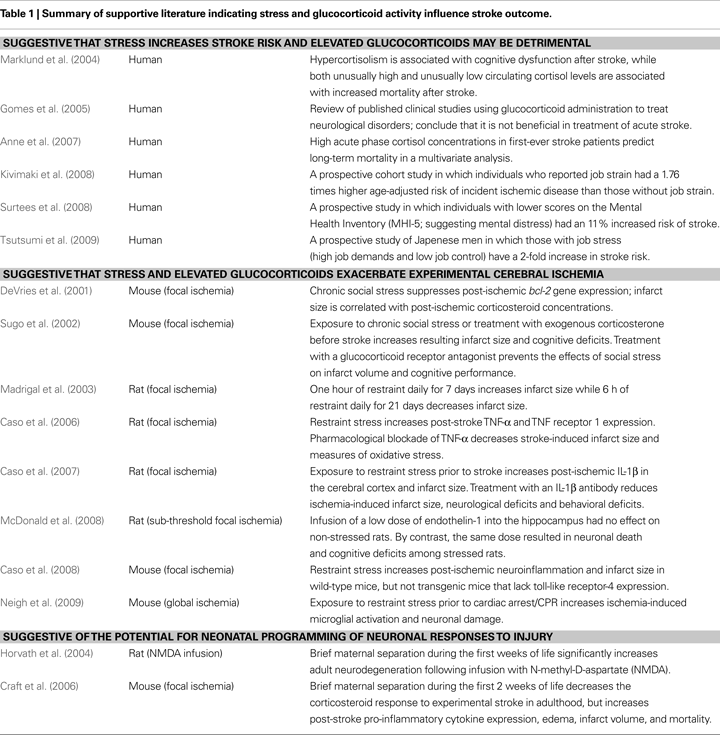
There are equally convincing data concerning the role of post-ischemic corticosteroids on outcome. As stated above, several clinical studies have documented that elevated post-stroke serum concentrations of cortisol are strongly correlated with increased physical and mental deficits (Murros et al., 1993
; Marklund et al., 2004
), and are associated with increased morbidity and mortality (Feibel et al., 1977
; Olsson, 1990
; Marklund et al., 2004
). Similarly, post-ischemic cortisol concentrations are negatively correlated with scores on the Scandinavian Stroke Scale, suggesting that high post-stroke cortisol concentrations are associated with exaggerated functional and cognitive deficits in patients (Christensen et al., 2004
). Experimental evidence also suggests that elevated post-ischemic corticosterone concentrations increase infarct size in mice and rats (Rami et al., 1998
; DeVries et al., 2001
; May et al., 2002
; Neigh et al., 2009
). In contrast, adrenalectomy and peri-ischemic treatment with compounds that decrease circulating corticosterone concentrations or glucocorticoid receptor activation in turn lead to a decrease in neuronal damage following both focal and global cerebral ischemia in rodents (Sapolsky and Pulsinelli, 1985
; Smith-Swintosky et al., 1996
; Antonawich et al., 1999
; Risedal et al., 1999
; Krugers et al., 2000
; Cheng et al., 2009
). Taken together, these studies suggest that glucocorticoid exposure may be a critical determinant of survival and functional outcome following cerebral ischemia.
Extensive exploration of the basic mechanisms of GC action in the brain after ischemia has only recently begun, and several hypotheses have been proposed to explain how stress and GC-release exacerbate ischemic cell death. A direct effect of GCs on survival in otherwise healthy neurons is possible (Crochemore et al., 2005
), but probably relatively rare in vivo. It is more likely that GCs induce a physiological state that renders neurons more susceptible to other mechanisms of cell death following a neurological insult, such as stroke (Sapolsky, 1985
). Indeed, the pattern of neuronal death following global cerebral ischemia is similar to the general pattern of GC receptor concentration; cell death is greatest in the hippocampus, where GR are most abundant, despite the fact that the entire brain is experiencing a similar level of ischemia (Sapolsky, 1985
). However, exposure to stress or exogenous GCs also increases cell death in brain regions with low to moderate GR concentration, such as the cortex, striatum and caudate putamen, so either GR expression is low, but sufficient to promote cell death in neurons already compromised by ischemia, or GC may be acting via multiple mechanism to compromise neuronal survival (Sapolsky and Pulsinelli, 1985
; Sapolsky, 1999
).
Three of the most thoroughly established mechanisms of GC-mediated ischemic cell death involve increased glutamatergic excitotoxicity, suppression of bcl-2 expression, and increased neuroinflammation. Glutamate accumulates in the core infarct region during ischemia (Sapolsky, 2001
; Smith, 2004
), then spreads within hours to tissue surrounding the infarct (Woitzik et al., 2009
). The role of glutamate in ischemic cell death is confirmed by several studies that show glutamate receptor antagonists (Rothman, 1984
; Simon et al., 1984
; Kundrotiene et al., 2004
; cf. Hoyte et al., 2004
) and lesions of glutamatergic inputs (Johansen et al., 1986
; Onodera et al., 1986
; Kimura and Saji, 1997
) reduce ischemic injury. Ischemia raises extracellular glutamate concentrations to toxic levels (Globus et al., 1988
; Arundine and Tymianski, 2004
) which are further amplified by an increase in circulating GCs. For example, stress levels of corticosterone drastically increase glutamate accumulation (3–4 fold increase over basal concentrations) with or without a coincident neurological insult (Stein-Behrens et al., 1992
); thus, the elevation of GCs can exacerbate glutamatergic excitotoxicity following stroke. Furthermore, the ischemia-induced deficits in glutamate transporter function are exaggerated by stress; glutamate transport is impaired by the further depletion of ATP supply, and transporter gene expression is altered in neuronal tissue (Madrigal et al., 2003
). The combination of a stress-induced increase in extracellular glutamate and decrease in glutamate transport has the potential to escalate glutamate’s neurotoxic effect during cerebral ischemia.
Stress-released hormones also have a profound effect on expression of the bcl-2 family of genes, which are important regulators of cell death (Merry and Korsmeyer, 1997
). The bcl-2 family consists of both pro-apoptotic proteins, such as bax, and anti-apoptotic proteins, such as bcl-2. Over-expression of bcl-2 has been found to protect against both apoptosis and necrosis following ischemia (Linnik et al., 1995
; Alkayed et al., 2001
; reviewed in Kuschinsky and Gillardon, 2000
). It has been proposed that bcl-2 protects neurons from apoptosis through regulation of intracellular CA2+ concentrations (McConkey and Orrenius, 1997
). Though the exact mechanism of bcl-2 neuroprotection remains to be determined, there is a large body of experimental literature demonstrating how its expression can be altered. For example, socially stressed mice have larger infarcts than controls due to GC-mediated suppression of stroke-induced bcl-2 expression (DeVries et al., 2001
). Furthermore, transgenic mice that have enhanced bcl-2 expression are protected from the deleterious effects of stress on stroke outcome (DeVries et al., 2001
). Aged animals may be particularly sensitive to a stress-induced shift in the ratio of bax:bcl-2 expression, that would increase the apopototic vulnerability of neurons (Almeida et al., 2000
).
Neuroinflammation is a third mechanism through which stress appears to modify ischemic outcome. A growing body of literature has provided evidence that brain injury is also accompanied by marked inflammatory reaction, characterized by infiltration of granulocytes, monocytes and macrophages into the respective brain parenchyma (Perry and Gordon, 1991
), activation of microglia and astrocytes, and expression of pro-inflammatory cytokines, adhesion molecules and other inflammatory mediators (Feuerstein et al., 1998
; Dirnagl et al., 1999
; Dinkel et al., 2003
). Cytokines, in particular, have a marked upregulation following stroke (reviewed in DeGraba, 1998
). Experimental models of stroke have demonstrated increased concentrations of interleukin mRNA in ischemic tissue, including IL-1α, IL-1β, IL-1ra, and IL-6 (Zhai et al., 1997
; Hill et al., 1999
; Legos et al., 2000
). Similar evidence exists in clinical studies, with increased interleukin concentrations reported in both cerebrospinal fluid (CSF) and serum following stroke (Tarkowski et al., 1995
; Ferrarese et al., 1999
; Rodriguez-Yanez and Castillo, 2008
). Furthermore, post-ischemic concentrations of pro-inflammatory cytokines are positively correlated with infarct size (Tarkowski et al., 1995
; Yang et al., 1999
; Vila et al., 2000
; Boutin et al., 2001
; Touzani et al., 2002
; Allan et al., 2005
), whereas over-expression of, or treatment with, anti-inflammatory cytokines (i.e. IL-1ra; Yang et al., 1999
; Emsley et al., 2005
) or growth factor (i.e. neuregulin-1β; Xu et al., 2005
) that down-regulate pro-inflammatory cytokine expression result in reduced ischemic injury. Thus, cytokine regulation is vital to ischemic outcome, and is becoming a key target for therapeutic and preventive treatment of stroke (Jordan et al., 2008
).
The inflammatory cytokine cascade that occurs in the brain following stroke has been most thoroughly characterized in rats and mice. The primary response to ischemia is mediated by IL-1β and TNF-α, both of which orchestrate the migration of inflammatory cells to the site of ischemic injury (DeGraba, 1998
; Zhang and Stanimirovic, 2002
). IL-1β and TNF-α are detected post-stroke as early as 1–3 h and 3–6 h, respectively. Though both reach their peak concentration at 12 h, the actual functional outcome of IL-1β protein expression does not peak until 72 h (Legos et al., 2000
). At approximately 6-h post-ischemia, just prior to the down-regulation of IL-1β and TNF-α, a surge in the expression of IL-10 is observed. IL-10 is an anti-inflammatory cytokine that may control the duration and extent of the inflammatory response to ischemia by inhibiting the production of TNF-α (Zhai et al., 1997
). The secondary response involves IL-6 and IL-8 (DeGraba, 1998
). The large influx of inflammatory cells initiated by IL-1β and TNF-α may be responsible for the synthesis of IL-6 (Legos et al., 2000
), which peaks at 24-h post-stroke. IL-6 has recently been found to have important anti-inflammatory and immunosuppressive effects after stroke (Dziedzic et al., 2002
; Karelina et al., 2009
). IL-6 regulates concentrations of pro-inflammatory cytokines via a negative feedback function initiated by its upregulation by TNF-α (DeGraba, 1998
). IL-8, on the other hand, remains elevated for 7 days following stroke and is thought to have neurodegenerative effects; indeed, a decrease in infarct size and edema are observed when IL-8 is neutralized or its receptors blocked during cerebral ischemia (Villa et al., 2007
). Thus, the careful balance of pro- and anti-inflammatory cytokines can have critical implications in neuronal survival following stroke.
Though the HPA axis and immune system both have significant roles in the pathophysiology of stroke, they are often considered as separate entities rather than interacting physiological processes. Indeed, GCs have often been used to treat inflammatory responses within the central nervous system, such as to suppress tumor-induced edema in the brain, to treat viral or bacterial encephalitis, and to improve acute onset of multiple sclerosis (Salaki et al., 1984
; Barnes and Adcock, 1993
; Coyle, 1999
; Filippini et al., 2000
); however, there is no evidence that exogenous GCs improve clinical stroke outcome by suppressing inflammation (Gomes et al., 2005
). Dose may be important in determining whether exogenous GCs have an inflammatory or anti-inflammatory effect in the brain during certain disease processes; for example, in primary hippocampal cultures, low doses of corticosteroids suppress the expression of inflammatory cytokines while, higher doses of corticosteroids result in a significant increase in IL-1β and TNF-α expression (MacPherson et al., 2005
). In vivo, kainic acid induces more TNF-α and IL-1β expression and neuronal damage if the rats have been chronically treated with GCs at a level that approximates stress (Dinkel et al., 2003
). Likewise, peri-ischemic exposure to stress or exogenous glucorticoids increases neuroinflammation, including the expression of pro-inflammatory cytokines such as TNF-α and IL-1β that are associated with exacerbation of ischemia-induced neuronal damage (Caso et al., 2006
, 2007
, 2008
, 2009
). Thus, the experimental studies suggest that there is a causal relationship between elevated corticosteroids and increased neuroinflammation after cerebral ischemia.
As reviewed above, the HPA axis is an important modulator of ischemic outcome. In light of the association between peri-ischemic glucocorticoid exposure and stroke outcome, one might logically predict that vulnerability to stroke would be increased by perinatal manipulations that increase exposure to GCs and decreased by perinatal manipulations that decrease exposure to GCs. However, a review of the literature concerning neonatal manipulations and their effects on immune function suggests that the relationship may not be so simple; one consequence of chronically dampened HPA axis reactivity may be increased susceptibility to inflammation and inflammatory disorders (Shanks and Lightman, 2001
), which in the case of stroke could be lethal. To examine the effects of early programming of the HPA axis on stroke, we used the brief maternal separation (BMS)/handling paradigm that has been most thoroughly characterized in rats, but successfully applied to several species with similar results, including mice (Parfitt et al., 2004
). Brief periods of maternal separation (approximately 15 min per day) during the first several weeks of development leads to increased expression of GR in the hippocampus and frontal cortex (Meaney et al., 1985
; O’Donnell et al., 1994
; McCormick et al., 2000
; Ladd et al., 2004
) and dampened physiological and behavioral responses to stressors (Meaney et al., 1991b
; Liu et al., 1997
; Vallee et al., 1997
; Francis et al., 1999
), presumably due to increased sensitivity of the HPA axis to negative feedback regulation (Meaney et al., 1989
). This neonatal programming of the HPA axis also is protective against the decline in hippocampal neural density and cognitive function that is typically observed among aged rats (Meaney et al., 1988
, 1991a
). As predicted based on these studies, adult mice that were exposed to BMS as neonates had significantly lower corticosteroid responses to the induction of experimental stroke than mice that had not been manipulated during development (Craft et al., 2006
). However, despite the lower endogenous corticosteroid response during ischemia, BMS mice sustained significantly more stroke-induced neuroinflammation, neuronal damage and behavioral deficits than mice that were raised under typical colony conditions (Craft et al., 2006
). Interestingly, 24 h after stroke the size of the core infarct was similar between the BMS and control mice, and while infarct size remained steady among controls over the subsequent 48 h, it nearly tripled among the BMS mice. Furthermore, if the mice were adrenalectomized prior to stroke and implanted with pellets that produced circulating corticosterone concentrations that were similar to those observed among intact mice 24 h after stroke, then the same infarct pattern emerged. In contrast, if the adrenalectomized mice were implanted with pellets that produced relatively low circulating corticosterone concentrations (half the typical post-stroke concentration), then there was no difference in infarct volume between BMS and control mice at 72 h (Craft et al., 2006
). Together these data suggest that the BMS mice were more sensitive than unmanipulated mice to the deleterious effects of persistently high corticosterone concentrations following stroke, possibly due to increased GR expression among BMS mice (reviewed above). In addition, the increase in edema, IL-1β expression, and TNF-α expression also may have contributed to the large infarcts among the BMS mice. Interestingly, BMS also is associated with increased N-methyl-D-aspartate (NMDA)-induced neurodegeneration in otherwise healthy rats (Horvath et al., 2004
), and it is possible that alterations in glutamate signaling among BMS mice could have contributed to the exacerbation of neuronal damage after experimental stoke, as well. Thus, there are several mechanisms through which BMS could be exacerbating stroke outcome, including increased sensitivity to GR-mediated effects on neuronal damage during a neurological insult, increased neuroinflammation, and altered glutamate signaling. Whether these are independent or interrelated pathways affecting stroke outcome remains to be determined.
Recent clinical data suggest that prenatal and early post-natal programming of the HPA axis may influence vulnerability to an array of stress-related health conditions, including cerebrovascular disease. Indeed, the HPA axis plays an important role in determining stroke outcome. Exposure to stress appears to increase both the incidence and severity of stroke, and activation of the HPA axis is among the first, easily measurable, physiological responses to stroke. Peri-ischemic glucocorticoid concentrations are positively correlated with stroke severity in both humans and rodents, and converging evidence from several labs using different species and experimental stroke models provides strong support for a causal relationship between elevated per-ischemic glucocorticoid concentrations and increased neuronal death following ischemia. A large body of work in rats and mice suggest that it is also possible to program the HPA axis in such a way as to decrease life-time exposure to glucocorticoids, which in turn can lead to improved neurological and cognitive aging. By pairing these data with the prospective clinical studies reporting increased risk of stroke among individuals who report elevated personal and work-related stress, one could propose that a reduction in stress responsivity and life-time exposure to glucocorticoids could lead to reduced incidence of stroke. However, when we used a well-developed handling and brief maternal separation model in mice to examine how reduced HPA activity affected adult ischemic outcome, we were surprised by the results. The neonatally manipulated mice had a reduced initial corticosteroid response to stroke, as expected, but ultimately developed more neuroinflammation, edema, and neuronal damage than unmanipulated cohorts. Also, post-stroke survival was reduced in the neonatally manipulated mice. From these data we surmise that one potential cost associated with more efficient day to day regulation of the HPA axis may be increased susceptibility to neuroinflammation and neurological damage if a stroke occurs. An independent study in rats confirms that neonatal handling also increases susceptibility to NMDA-induced neuronal damage. Thus, increased neuronal sensitivity to certain types of injury could be a cost associated with neonatal dampening of HPA axis activity. Given the importance of the HPA axis in the regulation of health and disease states, it seems reasonable to expect that any long-term modulation of the HPA axis is likely to be associated with both benefits and potential risks.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Almeida, O. F. X., Conde, G. L., Crochemore, C., Demeneix, B. A., Fischer, D., Hassan, A. H. S., Meyer, M., Holsboer, F., and Michaelidis, T. M. (2000). Subtle shifts in the ratio between pro- and antiapoptotic molecules after activation of corticosteroid receptors decide neuronal fate. FASEB J. 14, 779–790.
Bet, P. M., Penninx, B. W., Bochdanovits, Z., Uitterlinden, A. G., Beekman, A. T., van Schoor, N. M., Deeg, D. J., and Hoogendijk, W. J. (2008). Glucocorticoid receptor gene polymorphisms and childhood adversity are associated with depression: New evidence for a gene-environment interaction. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B, 660–669.
Caso, J. R, Hurtado, O., Pereira, M. P., Garcia-Bueno, B., Menchen, L., Alou, L., Gomez-Lus, M. L., Moro, M. A., Lizasoain, I., and Leza, J. C. (2009). Colonic bacterial translocation as a possible factor in stress-worsening experimental stroke outcome. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296, R979–R985.
Felitti, V. J., Anda, R. F., Nordenberg, D., Williamson, D. F., Spitz, A. M., Edwards, V., Koss, M. P., and Marks, J. S. (1998). Relationship of childhood abuse and household dysfunction to many of the leading causes of death in adults. The Adverse Childhood Experiences (ACE) Study. Am. J. Prev. Med. 14, 245–258.
Kaplan, R. C., McGinn, A. P., Baird, A. E., Hendrix, S. L., Kooperberg, C., Lynch, J., Rosenbaum, D. M., Johnson, K. C., Strickler, H. D., and Wassertheil-Smoller, S. (2008b). Inflammation and hemostasis biomarkers for predicting stroke in postmenopausal women: the Women’s Health Initiative Observational Study. J. Stroke Cerebrovasc. Dis. 17, 344–355.
Koeijvoets, K. C., van der Net, J. B., van Rossum, E. F., Steyerberg, E. W., Defesche, J. C, Kastelein, J. J., Lamberts, S. W., and Sijbrands, E. J. (2008). Two common haplotypes of the glucocorticoid receptor gene are associated with increased susceptibility to cardiovascular disease in men with familial hypercholesterolemia. J. Clin. Endocrinol. Metab. 93, 4902–4908.
Kundrotiene, J., Cebers, G., Wagner, A., and Liljequist, S. (2004). The NMDA NR2B subunit-selective receptor antagonist, CP-101,606, enhances the functional recovery the NMDA NR2B subunit-selective receptor and reduces brain damage after cortical compression-induced brain ischemia. J. Neurotrauma 21, 83–93.
Ladd, C. O., Huot, R. L., Thrivikraman, K. V., Nemeroff, C. B., and Plotsky, P. M. (2004). Long-term adaptations in glucocorticoid receptor and mineralocorticoid receptor mRNA and negative feedback on the hypothalamo-pituitary-adrenal axis following neonatal maternal separation. Biol. Psychiatry 55, 367–375.
MacMahon, S., Peto, R., Cutler, J., Collins, R., Sorlie, P., Neaton, J., Abbott, R., Godwin, J., Dyer, A., and Stamler, J. (1990). Blood pressure, stroke, and coronary heart disease. Part 1, Prolonged differences in blood pressure: prospective observational studies corrected for the regression dilution bias. Lancet 335, 765–774.
McCormick, J. A., Lyons, V., Jacobson, M. D., Noble, J., Diorio, J., Nyirenda, M., Weaver, S., Ester, W., Yau, J. L., Meaney, M. J., Seckl, J. R., and Chapman, K. E. (2000). 5’-heterogeneity of glucocorticoid receptor messenger RNA is tissue specific: differential regulation of variant transcripts by early-life events. Mol. Endocrinol. 14, 506–517.
Rosmond, R., Chagnon, Y. C., Holm, G., Chagnon, M., Pérusse, L., Lindell, K., Carlsson, B., Bouchard, C., and Björntorp, P. (2000). A glucocorticoid receptor gene marker is associated with abdominal obesity, leptin, and dysregulation of the hypothalamic-pituitary-adrenal axis. Obes. Res. 8, 211–218.
Villa, P., Triulzi, S., Cavalieri, B., Di Bitondo, R., Bertini, R., Barbera, S., Bigini, P., Mennini, T., Gelosa, P., Tremoli, E., Sironi, L., and Ghezzi, P. (2007). The interleukin-8 (IL-8/CXCL8) receptor inhibitor reparixin improves neurological deficits and reduces long-term inflammation in permanent and transient cerebral ischemia in rats. Mol. Med. 13, 125–133.
Wirtz, P. H., von Kanel, R., Emini, L., Ruedisueli, K., Groessbauer, S., Maercker, A., and Ehlert, U. (2007). Evidence for altered hypothalamus-pituitary-adrenal axis functioning in systemic hypertension: blunted cortisol response to awakening and lower negative feedback sensitivity. Psychoneuroendocrinology 32, 430–436.