 Kirk E. Anderson
Kirk E. Anderson Duan C. Copeland
Duan C. Copeland- Carl Hayden Bee Research Center, USDA-ARS, Tucson, AZ, United States
The microbiome of the honey bee worker hindgut has been explored thoroughly with culturing and next-generation sequencing revealing both composition and function. However, less effort has been devoted to the aerobic social niches associated with the hive environment and colony process. We performed a meta-analysis of 3,800+ publicly available 16S rRNA gene sequence libraries examining the hypothesis of a native aerobic microbiota associated with social interaction and colony resources. We selected high-throughput studies to represent tissue-specific samples, including nine distinct aerobic niches throughout the colony and hive, defined by social nutrient processing. These included queen and worker gut tissues, foregut, midgut, ileum, rectum, mouthparts, worker social glands, developing larvae, and secreted and stored nutrition. We found that the aerobic mouthparts, foregut and midgut niches of queens and workers share a significant portion of their microbiome with that of larval rearing and nutrient secretion and storage, defining the microbiota of the social resource niche. Characterized by species dominance and rapid growth, the social resource microbiota functions primarily in disease prevention at both the individual and colony level and may also function in social communication and gut microbiome resilience. Defining the microbiota of social function contributes to a systems-level understanding of host–microbial interactions in the honey bee.
Introduction
The honey bee (Apis mellifera) is a colonial insect species domesticated worldwide for honey production and pollination services (Gallant et al., 2014; Aslan et al., 2016). The colony and associated built structure consist of a reproductive queen, thousands of cooperative sterile workers, developing larvae, stored food, and a highly predictable hindgut microbiome that populates worker bees (Kwong and Moran, 2016). The honey bee colony has been described as a superorganism because complex social communication and behavioral interactions between individuals result in emergent group properties that benefit the colony as a whole (Fewell, 2003). Herein, we consider the ecology of the holobiont, a unit of selection that includes the genomes of the host and its associated microbiome (Bordenstein and Theis, 2015; Moran and Sloan, 2015). More specifically, we define and discuss the aerobic microbiome associated with healthy social (group) hygiene that occurs on, within, and throughout colony and hive environment of the honey bee host. Strongly allied with the processing and sharing of information and nutrition, the collection of aerobic or microaerophilic niches including stored and secreted nutrition, and host anatomical features we refer to herein as the social resource niche (SRN). While this broad niche space includes “nest materials” like honey and stored pollen, taxonomic similarity across studies suggests that the SRN may extend to the larval gut and the mouthparts, glands, foreguts, and mid-guts of both queens and workers (Figure 1).
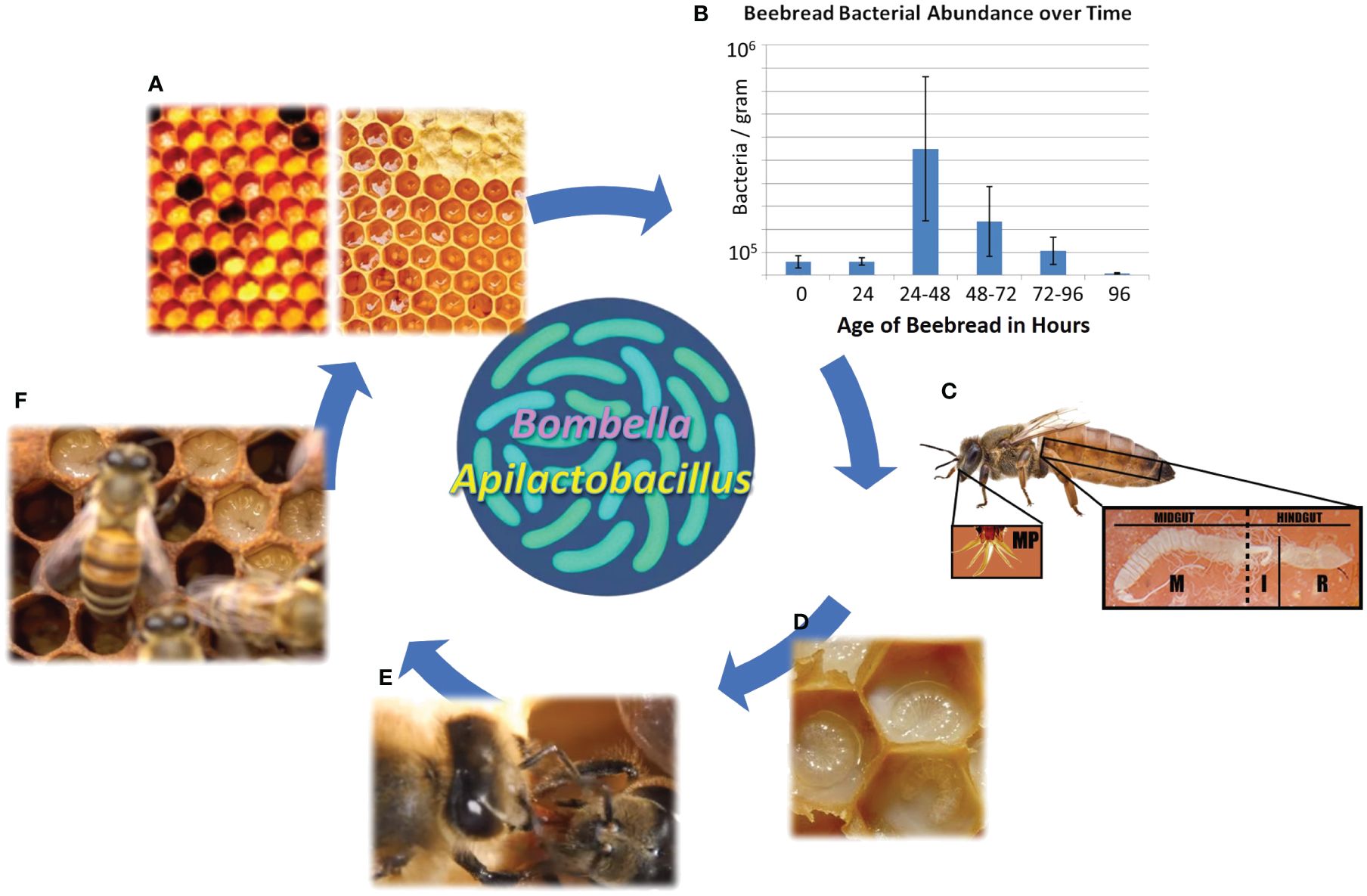
Figure 1 The social resource niche includes the anatomical features and bioactive substances associated with sharing behavior and colony hygiene. (A) Collected nectar is processed into honey, while collected pollen is processed into beebread, both of which are consumed by young adult worker bees as their gut microbiome self-assembles. (B) The storage of beebread fuels a predictable burst of native microbial growth, including sugar tolerant yeasts, that peaks at 24–48 h post-collection before declining precipitously. This is the preferred age at which stored pollen is consumed by nurse bees, and the process aids in the hygienic filtering of bacteria and yeasts vectored from the pollination or floral environment. (C) New adult worker bees become nurse bees, and from a modified head gland (HPG), secrete a highly nutritious and antimicrobial jelly that sustains queen egg-laying and larval development. The queens mouthparts and midgut are often saturated with royal jelly. (D) The highly nutritious jelly contains antimicrobial peptides and other microbial deterrents and can be customized to some degree to meet the immediate social needs of the colony. (E) Social sharing results in the colony-level distribution of antimicrobial substances and associated microbes. (F) The substances and associated microbes provide pathogen protection for developing larvae.
Honey bee life history is amenable to the co-evolution of strict host–microbial relationships. The founding worker population (reproductive swarm) presents a broad and continuous microbial niche for holobiont evolution (Winston, 1987; Engel et al., 2012; Rothman et al., 2018). The honey bee colony shows perpetual worker production, continuous colony fission, and continuously overlapping adult worker generations (Seeley, 1989). The worker bees of the present moment have had continuous and intimate physical contact with their ancestral lineage, as it stretches back through the ages. The depth and continuity of the surviving swarm and lack of a reproductive bottleneck have thus facilitated intimate co-evolution of the total microbiome with the host organism. Here, we suggest that similar to other complex social groups, microbes that contribute to the informational and hygienic function of the nest and colony environment are transmitted across generations with the founding host (Breed et al., 1988; Haeder et al., 2009; Goldstein and Klassen, 2020). With this idea in mind, we review data from 35 NCBI bio-projects to better define the microbiota inhabiting the SRN (Anderson et al., 2013; Vojvodic et al., 2013; Maes et al., 2016; Anderson and Ricigliano, 2017; Anderson et al., 2022). We predict that the social resource microbiota (SRM) is similar across studies and locations and shared throughout the SRN in accordance with colony function. As a null hypothesis, non-native (environmental) microbes vectored from the local pollination environment may typify the SRN.
The SRN is deeply antimicrobial and influenced by colony-level factors like worker activity level and behavioral role. The mechanical processing associated with nutrient storage, honey production, and brood rearing are layered with raw nutritional resources (Anderson et al., 2013). Three substances produced by the honey bee dominate the SRN and are integral to microbial health: honey, jelly, and propolis. All three are uniquely antimicrobial, function throughout distinct but overlapping niche space, and display specialized relationships with native hive microbes (Vojvodic et al., 2013; Corby-Harris et al., 2014b; Dalenberg et al., 2020; Anderson and Maes, 2022). In general, fructophilic lactic acid bacteria (Apilactobacillus) specialize on honey and Bombella species on jelly (Endo et al., 2012; Vojvodic et al., 2013). Produced from collected tree resin, propolis is mixed with host secreted wax and distributed throughout the colony. Propolis promotes microbiome and immune health and provides a form of socialized medicine (Simone et al., 2009; Evans and Spivak, 2010; Dalenberg et al., 2020). Active in both individual and group level immunity (Evans and Spivak, 2010; Anderson and Ricigliano, 2017; Zheng et al., 2018), these three substances and their associated co-evolved microbiotas strongly mitigate microbial growth throughout the SRN. When the activity of worker bees (the colony) is removed from the built structure, resident fungal and bacterial opportunists consume the hive environment. It appears that many of the behavioral processes, substances, and microbes associated with the SRN actively maintain colony hygiene, mitigating disease, and opportunism (Figure 1).
The SRM aids in the rapid sterilization of collected pollen or “beebread” (Figure 1), a process dominated by Bombella, Apilactobacillus, and sugar-tolerant yeasts (Anderson et al., 2014; Anderson and Mott, 2023). Pollen foraging introduces a spectrum of environmental bacteria to the hive environment, but the subsequent treatment of collected pollen and nectar within the hive promotes survival of the SRN microbiota relative to introduced microbes (Anderson et al., 2014; Anderson and Mott, 2023). Half honey by weight, beebread coopts most of its antimicrobial properties from honey (Nicolson, 2011). During the conversion of collected pollen into beebread, the SRM grows fast, producing an extreme acidic environment at the oxygen interface, much like the production of silage in agriculture (Anderson et al., 2014). Apilactobacillus can grow rapidly with exposure to oxygen and can also metabolize p-coumaric acid, a biologically vital monomer abundant in pollen cell walls (Mao et al., 2013; Endo et al., 2018). The SRM peaks in size in 1–2-day-old pollen stores, concurrent with significantly increased beebread consumption by newly emerged worker bees (Anderson et al., 2014; Carroll et al., 2017). Beebread is consumed quickly by the worker bee population to avoid negative host effects associated with long-term pollen storage like increased mortality, delayed development, gut dysbiosis, and increased disease susceptibility (Anderson et al., 2014; Maes et al., 2016; Roessink and van der Steen, 2021). Following the digestion of beebread in the midguts of newly emerged worker bees, hypopharyngeal glands in the head synthesize royal jelly as food for developing larvae and the queen (Feng et al., 2009; Harwood et al., 2021). The HPG and its secreted jelly can contain tailored cocktails of pro-oxidants, antioxidants, and antimicrobial peptides that interface constantly with the SRM and the social information network (Buttstedt et al., 2013; Vojvodic et al., 2015; Anderson and Maes, 2022).
While populations of aerobic bacteria are common in the worker mouthparts, foregut, and midgut, the rectum houses generally anaerobic fermentative metabolism, and the ileum represents a transition from microaerophilic to anaerobic metabolism (Engel et al., 2012; Zheng et al., 2017). However, the hindgut microbiota alters in accord with gut physiology, often supporting aerobic growth in the rectum (Anderson and Ricigliano, 2017; Callegari et al., 2021). In this contribution, we perform a niche- specific meta-analysis of the total honey bee microbiota. We include oxygenated and nutrient-rich niches throughout the SRN including tissue-specific sequencing of aerobic to anaerobic gut niches of workers and queens, mouthparts, foreguts, hypopharyngeal (social) glands and their secretions (royal jelly), developing larvae, beebread, and honey. Using >3,800 libraries from publicly available datasets, we curate, distill, and standardize taxonomy of the total microbiome, testing the hypothesis of a consistent microbiota shared throughout the SRN.
Methods
We selected a broad list of microbiome studies to represent variation of the total honey bee microbiome and emphasize aerobic niches. We normalized these datasets by retrieving the raw data sets then processing them via the same bioinformatics pipeline. The selected studies differ by method (primer sets and extraction protocols) and biological variation: physiological subsets of honey bee life history capturing environments within the gut, colony, and hive, including aerobic, microaerophilic, and anaerobic niches. As our primary goal, we test the hypothesis of a social resource microbiota, using a subset of libraries that have explored a variety of aerobic niches. Supplementary Table S1 describes some of the features (primers, niche, and location) of the 35 NCBI bio-projects included in this meta-analysis (Anderson et al., 2014; Corby-Harris et al., 2014a; Kapheim et al., 2015; Jia et al., 2016; Hubert et al., 2017; Anderson et al., 2018; Jones et al., 2018; Motta et al., 2018; Rothman et al., 2019; Subotic et al., 2019; Taylor et al., 2019; Daisley et al., 2020; Dalenberg et al., 2020; Kešnerová et al., 2020; Sopko et al., 2020; Vernier et al., 2020; Alberoni et al., 2021; Callegari et al., 2021; Damico et al., 2021; Maes et al., 2021; Wang et al., 2021; Anderson and Maes, 2022; Anderson et al., 2022; Liberti et al., 2022; Copeland et al., 2022a, 2022b; Anderson et al., 2023).
Selected studies used 454-amplicon sequencing or Illumina high-throughput sequencing to target 16S rRNA genes producing paired-end reads. Read libraries were processed using mothur v.1.44.3 (Schloss et al., 2009). Paired-end reads were merged and quality filtered using the make.contigs command in mothur v.1.44.3 (Schloss et al., 2009). The command fastq.info was used to create a fasta and quality file. Next, rim.seqs was used to remove sequencing barcodes. The merge.files command was used to combine fasta, count_tables, and group files. The command “screen.seqs” was used to filter sequences with >1 ambiguous bases and a maximum homopolymer length of eight bases.
To assign sequences across different hypervariable regions of the 16S rRNA gene, we employed a closed-reference OTU workflow using the BEExact database (Daisley and Reid, 2021). BEExact allows for species-level taxonomic resolution for honey bee-associated bacteria compared to traditional databases like GreenGenes and Silva (Daisley and Reid, 2021). Chimeras were removed and sequences were clustered into OTUs at 97% similarity using VSEARCH (Rognes et al., 2016). The “merge.otus” command in mothur (Schloss et al., 2009) was used to generate species and genus-level OTUs for further analysis.
Many of the sequenced environments or tissues included in this meta-analysis reflect high exposure to oxygen and highly concentrated sources of nutrition, including early instar larvae, queen and worker mouthparts, foreguts, and midguts. These niches can vary greatly in microbial load (104–108 gene copies per tissue) but average low microbial biomass relative to the worker hindgut (106–109 gene copies per tissue). To determine the likelihood of native aerobic microbiota, we applied a threshold of 1% relative abundance and 70% prevalence across all libraries. The threshold of 70% was chosen to account for the high degree of variability (gut and hive niches) in our dataset, allowing us to capture more biologically relevant signal. Because some of our samples were from low-abundance DNA environments (Salter et al., 2014), we curated sequences to identify and remove sources of contamination. We classified potential contaminants and sparse OTUs based on known culturing results, curated data collections, and documented contaminants specific to reagents or laboratories (Salter et al., 2014; Anderson et al., 2023).
The community composition of samples by niche was assessed with ANOSIM (Bray–Curtis dissimilarity) implemented in mothur (Clarke, 1993). We used a PERMANOVA test to estimate beta diversity variation associated with niche using the ADONIS function from the vegan package performed with 1,000 permutations. We ran a linear discriminant analysis (LDA) of the top 10 OTUs to test the hypothesis of a SRN microbiota shared between larvae, workers, queens, and the hive environment. We used Circos plots (Krzywinski et al., 2009) to display the bacterial relationships by niche.
Results
We analyzed high-throughput 16S rRNA gene sequence archives from 35 NCBI bio-projects representing honey bee alimentary tract tissues, reproductive caste, larval development, secretory glands, and nutritional resources. Overall, VSEARCH assigned 5,773,026 of 7,118,258 (81.10%) of unique sequences and 128M of the 141M sequence reads (90.47%) were at least 97% similar to a representative full-length sequence in the BEExact database (Daisley and Reid, 2021). Sequences that failed to match 97% identity to any reference sequence are examined in a different publication. Our species and genus-level OTU tables provided 1,972 and 669 OTUs, respectively. OTUs occasionally clustered into “x_bxid####”, where x is the first initial of the taxonomic level and the #s are unique identifiers distinguishing group members at each taxonomic rank. These are placeholder names in the BEExact database given to sequences with <98.7% identity to type strain representatives (Supplementary Table S1).
Associated with high‐throughput metagenomics studies investigating low-abundance DNA environments, we identified a number of contaminant OTUs consistent with previous identification from amplicon libraries investigating early instar larvae (Anderson et al., 2023). Our results linking particular OTUs to contamination is further reinforced by previous results sequencing low-abundance queen gut environments and blank controls (Anderson et al., 2018). As determined by previous criteria, we designated the following OTUs as the top 8 contaminants: Ralstonia, Caulobacter, Bradyrhizobium, Pelomonas, Cyanobacteria, Lysinibacillus, Shigella, and Nevskia, and more generally, Chitinophagaceae, Comamonadaceae, Caulobacteraceae, Burkholderiaceae, and Bradyrhizobiaceae. We note that many OTUs confirmed by culturing can also present the character of a contaminant, and the rare biosphere of honey bees remains to be confirmed. Although most prevalent in low-abundance DNA environments, contaminant sequences are found at lower abundance throughout the data set in association with deep sequencing efforts of the worker gut.
A total of 10 genera met a minimum threshold of 70% prevalence with at least 1% relative abundance: Lactobacillus, Gilliamella, Snodgrassella, Bombilactobacillus, Frischella, Bifidobacterium, Bombella, Apilactobacillus, Commensalibacter, and Bartonella (Figure 2). We found that the SRM is comprised of two major genera, Bombella and Apilactobacillus, and various species of Lactobacillus shared across aerobic niches including larvae, worker and queen mouthparts, worker crops, worker hypopharyngeal glands, queen crops and midguts, beebread, royal jelly, and honey. Based on linear discriminant analysis and the resulting feature space, we found a strong taxonomic overlap of nine distinct niches, driven primarily by the frequency and abundance of three major bacterial genera (Figure 3). These niches are dominated by Apilactobacillus, Bombella, and Lactobacillus. Bombella and Apilactobacillus account for >40% of reads throughout the SRN (Figure 4). Fructobacillus accounted for approximately 1% of the reads in this study but did not meet our 70% prevalence criteria. Fructobacillus clustered with Apilatcobacillus and Bombella by study, and it was most prevalent and abundant in larvae, worker mouthparts, and the anterior queen gut.
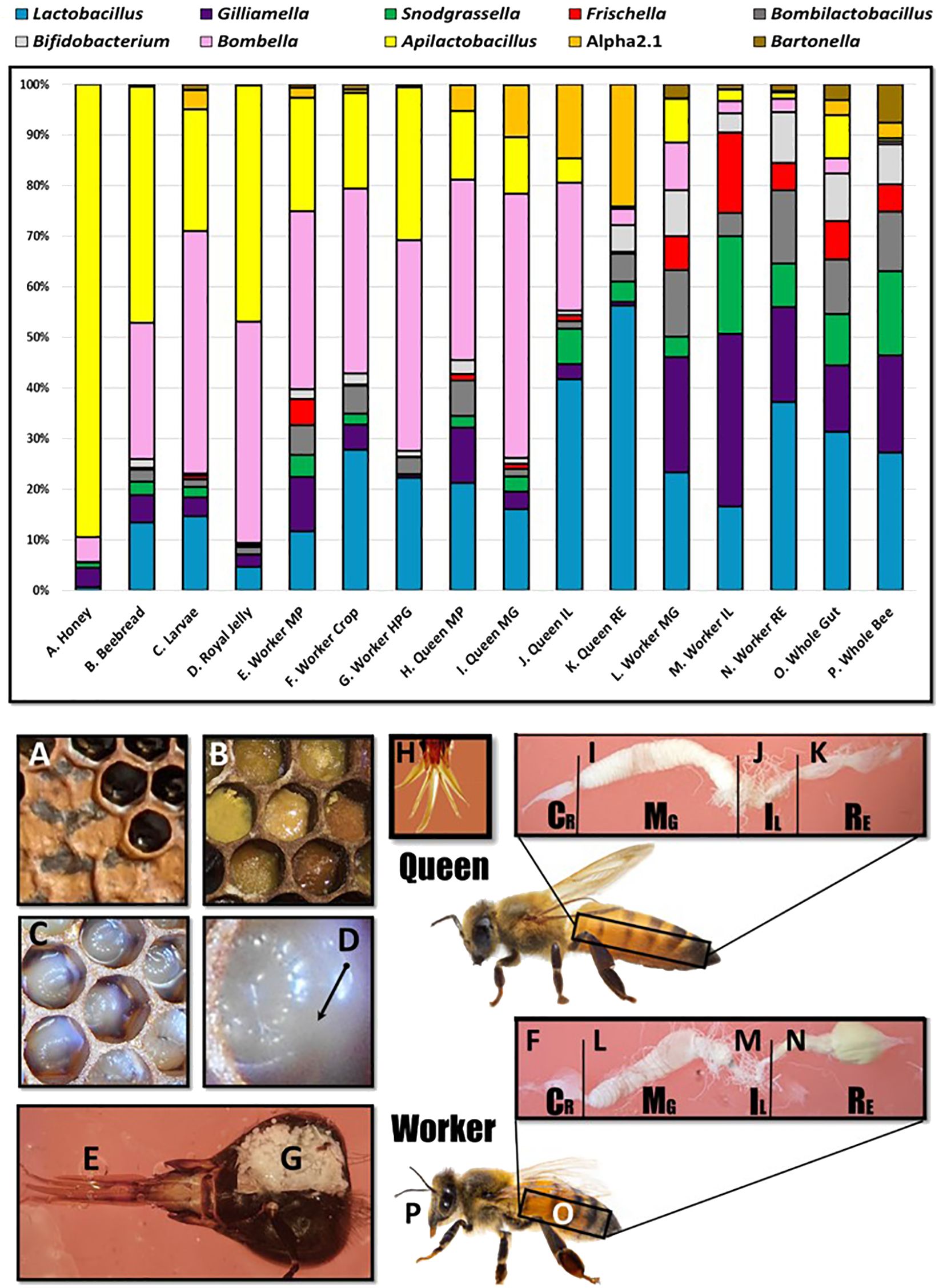
Figure 2 Microbiota of the social resource niche displayed as relative abundance in the upper panel. The niche is saturated with processed and shared nutrition: honey (A), beebread (B), and royal jelly (D). Larvae (C) are fed these substances in varying amounts throughout development. The niche also includes anatomical features associated with producing, processing, consuming, or sharing nutrition including the mouthparts (E), crops (F), and hypopharyngeal (social) glands (G) of workers. The mouthpart microbiota of queens (H) and workers is similar, but beginning at the midgut (I, L), the microbiota diverges significantly in membership and structure by caste. Queens (H–K) contain more Acetobacteraceae, both Bombella and Commensalibacter, while workers (L–P) are typified by the presence/abundance of Snodgrassella, Gilliamella, and Frischella.
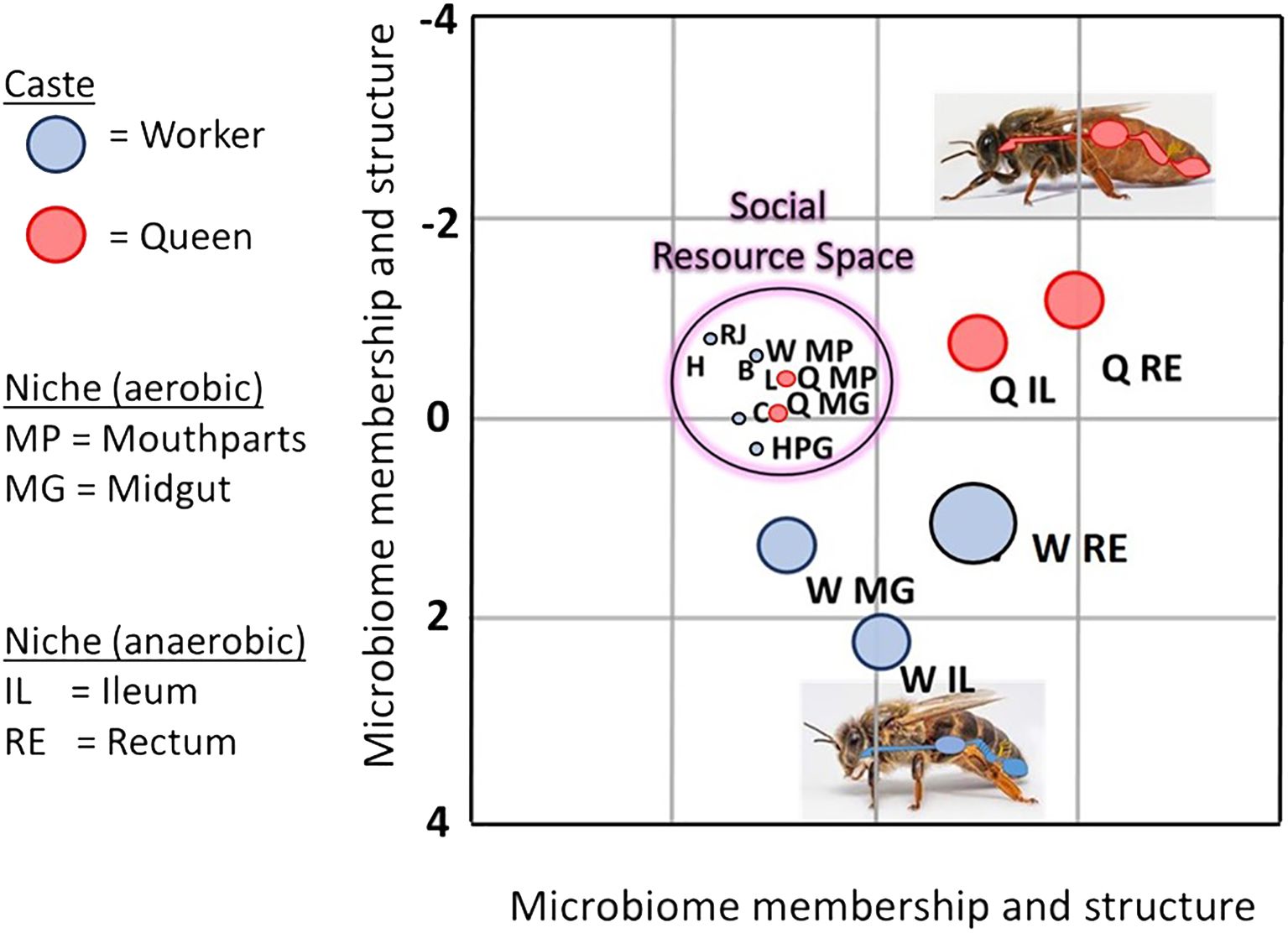
Figure 3 Linear discriminant analysis LDA depicts an overlap of various bacterial species shared by aerobic niches including larvae (L), worker mouthparts (W MP), worker crops (C), worker hypopharyngeal glands (HPG), queen mouthparts (Q MP), queen midguts (Q MG), beebread (B), royal jelly (RJ), and honey (H). Dominated by a few major bacterial species, the social resource niche supports the microbiota of the “hygienesphere” because the associated substances, activities, and microbiota are the demonstrated antagonists of the pathosphere.
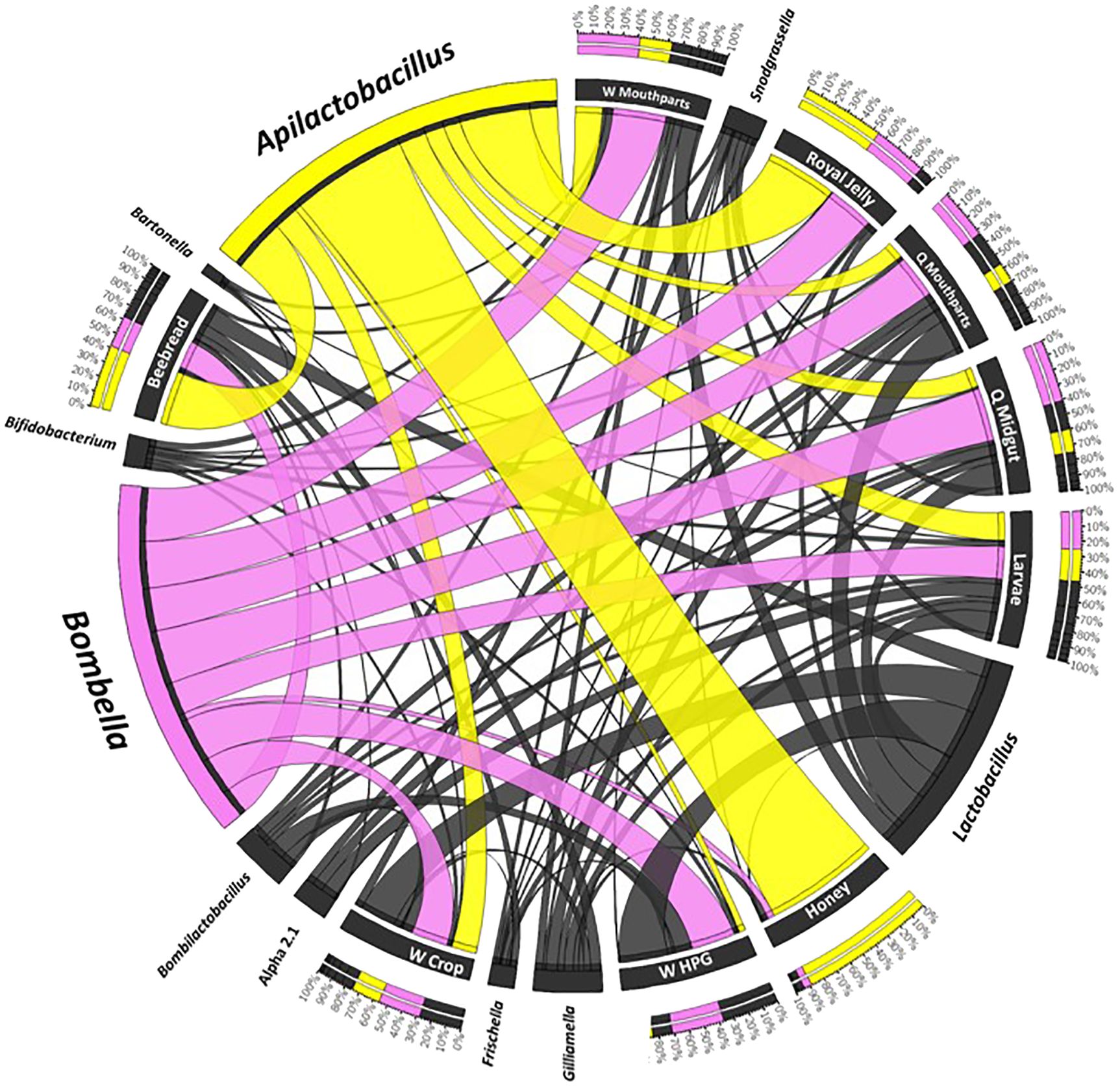
Figure 4 A Circos plot of the social resource niche and genera found therein. Plots were generated from a CSS-normalized OTU table with all singletons removed. Each genus is assigned a specific color, and its abundance is directly proportional to the width of each ribbon connecting bacterial taxa to its respective niche. The outer ring represents the cumulative percent of 16S sequences assigned to a given genus from each sample, while the inner circle represents the number of 16S rRNA sequences assigned to a given taxa in a given sample.
We found significant separation of microbiomes by niche with both ANOSIM (R, 0.145, p = 0.001) and ADONIS (F15 = 28.9, p = 0.001). Linear discriminant analysis reveals an overlap of various bacterial species shared by aerobic niches including larvae, worker mouthparts, worker crops, worker hypopharyngeal glands, queen mouthparts, queen midguts, beebread, royal jelly, and honey. The SRN microbiota is dominated by a few major bacterial species: Bombella apis, Bombella intestini, Bombella spp. (bxid5328), Apilactobacillus kunkeei, Apilactobacillus apinorum, Apilactobacillus spp. (bxid5570), Fructobacillus fructosus, and Fructobacillus spp. (bxid5666).
Discussion
This meta-analysis united >3,800 raw sequence read libraries to normalize bioinformatic methods and evaluate niche specificity by taxonomic group. Our results highlight a native aerophilic, acid-resistant and osmotolerant microbiota shared across a number of socially interconnected niches, referred to herein as the social resource niche, SRN (Endo and Salminen, 2013; Vojvodic et al., 2013; Anderson et al., 2014; Corby-Harris et al., 2014b). Results suggest that the social resource microbiota (SRM) is continuously transmitted to the new budding colony and nesting location in the glands, mouthparts, and guts of workers (Figure 2). Correspondingly, the entire core hindgut microbiota of workers occurs with prevalence and abundance throughout the SRN. Prevalent in the literature, a competing hypothesis had long speculated that these intimate colony niches were dominated by “environmental microbes” introduced from the local foraging environment (Endo et al., 2012; Anderson et al., 2013; Vojvodic et al., 2013; Corby-Harris et al., 2014b; Djukic et al., 2015; Kwong and Moran, 2016). While microbes often originate from the local foraging environment, relatively few microbes have evolved to endure the active hive and colony environment of Apis mellifera. The SRM is closely related to bacteria that populate flowers and solitary bees (McFrederick et al., 2012; Vojvodic et al., 2013; Anderson and Ricigliano, 2017; McFrederick et al., 2017), but has evolved to prosper in the antimicrobial environments of royal jelly, propolis, and honey (Kwakman et al., 2010; Dalenberg et al., 2020; Harwood et al., 2021).
Much like the fermentative hindgut microbiome of workers, the SRM provides a living layer of protection that thwarts the growth and establishment of undesirable microbes, both native and introduced. Simply by surviving on the fringe of the colony activity, within the niche preferred by the pathosphere, the SRN microbiome plays a protective role similar to that of human skin or nasal pharyngeal microbiome. The dynamic SRN connects the behavioral and metabolic activities of a colony and maintains continuous contact with the nutrition, substances, activities, and microbiota that typify colony health and growth. The SRN contains molecular information associated with group nutritional state and microbial threats that inform key behaviors of social immunity and more general colony process (Evans and Spivak, 2010; Spivak et al., 2019). Below, we discuss the SRN and SRM in the context of colony hygiene and social life history. Although many core hindgut OTUs (e.g., Lactobacillus) occur with frequency and abundance throughout the SRN, we limit our discussion to the highly aerobic and fast-growing genera Apilactobacillus and Bombella.
Based on linear discriminant analysis and the resulting feature space, we found a strong taxonomic overlap of nine distinct niches, driven primarily by the prevalence and abundance of two major bacterial genera, Bombella and Apilactobacillus (Figure 3). Throughout this broad and interconnected niche, the lack of moisture, abundance of oxygen, and the combination of host and microbial products result in a somewhat continuous layer of antimicrobial activity that accompanies the colony processes of nutrition processing, queen maintenance, and larval development (Anderson et al., 2014; Anderson and Ricigliano, 2017; Anderson and Maes, 2022; Anderson and Mott, 2023). Anatomically, this niche includes the surface rich and versatile mouthparts and foreguts of queens and workers, queen midguts, worker head glands that produce social secretions, and developing larvae (Figure 2). The niche also includes stored, processed, and secreted nutrition. More generally, the collective worker behaviors and physiology associated with colony process nurture a broad and interconnected niche conducive to the growth of beneficial microbes and inhibitory towards the growth of non-native or pathogenic microbes (Figure 2).
The SRM is the demonstrated antagonist of the honey bee pathosphere (Schwarz et al., 2015) and non-native microbes that populate flowers. A. kunkeei, prevalent in the crop and on the mouthparts, inhibits the growth of 60 different transient flower microorganisms based on in vitro growth experiments (Vásquez et al., 2012). Apilactobacillus also inhibits the major honey bee pathogens Nosema, American foulbrood and European foulbrood (Forsgren et al., 2010; Vásquez et al., 2012; Arredondo et al., 2018; Zendo et al., 2020; Miller et al., 2021). More specifically, A. kunkeei produces Kunkecin, a bacteriocin with specificity for European foulbrood (M. plutonius), a widespread and destructive pathogen (Zendo et al., 2020). Bombella (Parasaccharibacter) treatment at the colony level is associated with significantly lower Varimorpha (Nosema) counts in workers experimentally fed 10,000 Nosema spores (Corby-Harris et al., 2016) and often proliferates in the worker midgut, where Nosema and sugar- tolerant yeast find their reproductive niche (Corby-Harris et al., 2016). The abundance of Bombella shows significant negative associations with general fungal abundance throughout the worker gut (Anderson and Maes, 2022; Anderson et al., 2022). Bombella inhibits the growth of a major fungal pathogen Aspergillus flavus, based on in vitro inhibition assays. This phenotype was confirmed with in vivo larval rearing; bee brood supplemented with Bombella were significantly less likely to be infected by A. flavus. Comparative genome analysis of Bombella suggests that fungal inhibition occurs via the secretion of secondary metabolites (Miller et al., 2021).
The worker mouthparts and foregut work together to generate pathogen protection or “social immunity”, including continuous trophallaxis between worker adults, feeding the larvae and queen and nectar dehydration (Nicolson, 2009; Anderson et al., 2013; Dalenberg et al., 2020). The foregut (crop) is simply an expandable bag in the anterior worker gut used to process, hold, and distribute liquid resources, while the mouthparts are rich with surface area and capable of unfolding, reconfiguring, biting, sucking, and lapping. In a process known as “bubbling”, the mouthparts and the foregut swap nectar loads to dehydrate collected nectar (Nicolson, 2009), a highly oxidative and water- removing process that significantly inhibits non-native microbial growth and selects for the growth of native beneficial species (Corby-Harris et al., 2014a). This antimicrobial effect is transmitted throughout the colony by the continuous sharing of liquid food (Crailsheim, 1998). Our results confirm that only Bombella and Apilactobacillus are primary to the crop niche (Corby-Harris et al., 2014a). While species of Lactobacillus, Bombilactobacillus, and Bifidobacterium are often sampled from the crop and the SRN, these taxonomic groups show much stronger fidelity for the oxygen depleted hindgut (Moran et al., 2012; Vásquez et al., 2012; Corby-Harris et al., 2014a). Similarly, the worker mouthparts were also dominated by Apilactobacillus and Bombella and, to a lesser extent, Fructobacillus fructosus, another aerobe demonstrated to enhance the growth of other beneficial bacteria throughout the system (Rokop et al., 2015; Dalenberg et al., 2020).
Ubiquitous in the worker crop and on the mouthparts, the nutritional resources jelly and honey (often mixed) are associated with distinct but overlapping microbiomes (Vojvodic et al., 2013; Corby-Harris et al., 2014b; Maeno et al., 2016; Anderson and Maes, 2022). Worker brood rearing and feeding activities are reduced during periods of colony stress and disturbance, and the quality of jelly provided by workers may affect the protective powers of the SRN/SRM. The occurrence and abundance of native aerobes throughout the data set suggest that honey, royal jelly, and propolis promote rapid aerobic growth by providing nutrition, decreasing competition, and reducing the cost of using oxygen (Simone-Finstrom and Spivak, 2010). In one study, Bo apis, A. kunkeei, and F. fructosus were all significantly enriched on the mouthparts in colonies with increased propolis collection and deposition (Dalenberg et al., 2020). These same bacteria can also flourish in honey, royal jelly, larvae, and queens (Endo and Salminen, 2013; Vojvodic et al., 2013; Anderson et al., 2023). Although not considered by this study, similar niche-related factors including worker behavior likely mitigate native populations of sugar- tolerant yeast (Detry et al., 2020).
Honey represents one of the most extreme antibiotic environments known to science (Kwakman et al., 2010). Both Apilactobacillus kunkeei and Fructobacillus fructosus are specialized to exploit honey. First recognized by Endo, these genera are obligately fructophilic, preferring D-fructose as a carbon source abundant in honey (Neveling et al., 2012; Endo et al., 2018). Oxygen tolerance and utilization is a primary attribute of the SRM, as O2 is required for rapid growth. A. kunkeei grows exceedingly fast under aerobic lab conditions with a doubling time of approximately 1 h, a trait attributed to a suite of large mystery (unannotated) genes that flank the origin of replication (Tamarit et al., 2015), genes that likely enhance survival in honey. Microbes can survive briefly on the periphery of honey, whereas pure honey quickly kills or inactivates all microbial growth due to severe osmotic conditions and acidic pH, a byproduct of honey bee salivary enzymes and microbial fermentation occurring at the hygroscopic and oxygen- rich surface. Fully processed honey (dehydrated to >82% sugars) is sealed with beeswax, a substance impervious to water and atmospheric gasses.
Abundance measures of Apilactobacillus and Bombella are positively correlated in many studies suggesting resource partitioning. The two major SRN bacteria are specialized to exploit different sugar monomers abundant through the system; Bombella prefers glucose. Highly osmotolerant, gluconic-acid producing strains of Bombella continue growth at 40%–50% sugar concentrations and pH 3, demonstrating their tolerance for honey-rich environments (Ruiz-Argueso and Rodriguez-Navarro, 1975). Bo. apis has lost alternative oxidative pathways and harvests energy almost exclusively from glucose using oxygen as an electron receptor (Bonilla-Rosso and Engel, 2018). Both Apilactobacillus and Bombella are core to the queen’s gut microbiota (Tarpy et al., 2015; Anderson et al., 2018; Copeland et al., 2022a), and Bombella often dominates the queen mouthparts, foregut, ileum, and midgut. The queens gut supports a magnitude less gut bacteria than workers, suggesting a relatively less hospitable microbial environment (Anderson et al., 2018; Copeland et al., 2022b). This may result from continuous royal jelly exposure and constitutive expression of vitellogenin and other antimicrobial molecules throughout the queens system (Salmela and Sundström, 2017; Harwood et al., 2019). In the similar environment of larval guts, Bo. apis is the first bacterium to populate larvae based on culture- dependent and culture- independent data, and Bo. apis and/or A. kunkeei dominate later instars (Vojvodic et al., 2013; Floyd et al., 2020: Anderson et al., 2023).
Genome analysis of Bombella indicates intimate host–microbial evolution. Niche dominance of Bo. apis is facilitated by host- produced glucose oxidase, an enzyme converting glucose into gluconic acid, producing H2O2 as a byproduct (Ohashi et al., 1999). Bo. apis can then further oxidize gluconic acid, fueling its metabolism (Bonilla-Rosso and Engel, 2018; Smith and Newton, 2020). Genome evidence also suggests that Bo. apis has evolved to quickly divert the readily available energy stores in honey or jelly to alleviate omoregulatory and oxidative stress (Smith and Newton, 2020). As a fast growing aerobe, Bo. apis possesses all the conventional mechanisms of oxidative stress management, including superoxide dismutase, catalase, peroxidase, and the suite of enzymes involved in glutathione cycling. Between the inner and outer membrane of Bo. apis cells are extensive networks of periplasmic glucans, providing resistance to acids, enzymes, reactive oxygen species (e.g., H2O2), and rapid changes in osmolarity encountered in food stores and in the queen and larval gut. Not found in its closest relatives, Bo. apis possesses Aquaporin Z, a highly stable transmembrane protein channel that facilitates rapid osmoregulation and resists denaturing due to heat or extremes of pH found throughout the SRN. Consistent with other recent findings (Bonilla-Rosso and Engel, 2018; Smith and Newton, 2020; Härer et al., 2023), our analysis of 16S rRNA sequence variation suggests at least four major Bombella species with fidelity for distinct niche space including a novel species of Bombella that populates the midgut and ileum of queens (Figure 4).
Although their ecology is poorly known, highly osmotolerant and native species of yeast have likely influenced the evolution of the SRN, SRM, and the hindgut microbiota (Gilliam, 1979; Tauber et al., 2019; Detry et al., 2020; Anderson and Mott, 2023). In the human gut, fungi regulate host physiological processes and assembly of the co-residing gut bacterial microbiome (Nash et al., 2017). In honey bees, a general survey throughout the gut using universal fungal primers shows strong abundance relationships of fungi with species of hindgut bacteria (Maes et al., 2021; Anderson and Maes, 2022; Anderson et al., 2022). Based on detailed microscopy, it appears that the vast majority of fungi found throughout the SRN are native osmotolerant yeasts that can attain high numbers where oxygen is readily available (Detry et al., 2020; Anderson and Mott, 2023). Consistently, Apilactobacillus and Bombella show strong negative associations with yeast abundance in the worker gut; fungal load decreases concurrent with increasing bacterial load, attaining greatest values in the midgut and lowest values in the hindgut (Anderson and Maes, 2022; Anderson et al., 2022). However, these yeasts appear unwelcome in some aerobic environments rich in royal jelly including larvae and the queen gut. Queen guts do not tolerate a high fungal load, showing significantly lower fungal load relative to worker guts (Maes et al., 2021; Copeland et al., 2022a). Yeast blooms in times of bacterial gut dysbiosis (Anderson et al., 2022), and Bombella is a demonstrated fungal antagonist, inhibiting the growth of both yeasts and molds ubiquitous throughout the SRN (Corby-Harris et al., 2014b; Anderson et al., 2018; Dalenberg et al., 2020; Miller et al., 2021). Apilactobacillus also inhibits yeast growth (Vásquez et al., 2012; Bisson et al., 2017).
This exploration and review of the SRN uncovered a variety of Enterobacteriaceae with consistent taxonomy across studies, known to participate in gut dysbiosis of workers and invade the hemolymph (Gilliam and Valentine, 1974; Burritt et al., 2016; Raymann et al., 2019; Anderson and Maes, 2022). We suggest that the SRM acts to supplement the function of a compromised worker gut microbiome and discourage the establishment of opportunistic/pathogenic microbes. Following a disturbance, Apilactobacillus and Bombella often replace core hindgut bacteria (Anderson et al., 2022). Typically abundant in the worker mouthparts, foregut, and midgut, the SRM may act in gut microbiome resilience and generally suppress the growth of pathogenic microbes throughout the gut (Corby-Harris et al., 2014b, 2016; Anderson and Ricigliano, 2017; Anderson and Maes, 2022). With a shift in gut physiology, the speed with which these two fast growing and obligate aerobes can dominate available resources and niche space may be important for pathogen protection. Similar to core hindgut bacteria, Bombella and Apilactobacillus expel short- chain fatty acids as a final product of oxidative metabolism. Their presence in guts is tolerated by S. alvi, a core gut bacterium and obligate aerobe with a complete TCA cycle capable of assimilating short- chain fatty acids (Kwong and Moran, 2016). Beyond this, the SRM frequently co-occurs in the worker midgut with Gilliamella, native yeast, and Varimorpha (Nosema). Based on relationships within and among studies, various gut microbiome dynamics are associated with the development of Nosema disease and viral infections in both queens and workers (Ptaszyńska et al., 2016; Tauber et al., 2019; Anderson et al., 2022; Copeland et al., 2024). Understanding the dynamics of the SRM in the context of gut microbiome resilience presents a new perspective on honey bee health.
Conclusion
Research on the honey bee microbiota has focused primarily on six core genera that comprise the worker hindgut microbiome. To entertain the holobiont perspective (Bordenstein and Theis, 2015; Moran and Sloan, 2015), we performed a meta-analysis that included aerobic microbiomes associated with colony maintenance and social interaction, examining both anterior and posterior gut environments by reproductive caste. We identified two genera and a collection of species that are shared among intimate social niches, describing an aerobic “surface-rich” ecosystem maintained by continuous social processing of colony resources. The osmotolerant and acidophilic microbes that evolved to endure the social resource niche have come to play functional roles in disease ecology either by suppressing deleterious microbial growth or through their participation in perturbed hindgut enterotypes. Based on the results presented here and those of others, it is easy to speculate that this native aerobic microbiota contributes many functions at the group level including social communication, preserving stored nutrition, preventing disease and opportunism, and perhaps even gut microbiome resilience.
Author contributions
KA: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. DC: Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the ARS-USDA, research plan 2022–21000-021–00D, and an AFRI-NIFA grant no. 2021–67013-33555 to KA, Meghan O. G. Milbrath, Jay D. Evans, and Brendon M. Mott.
Acknowledgments
The authors would like to acknowledge advisors, lab members, and collaborators: Jay Evans, Kevin Hackett, Brendon Mott, Patrick Maes, Robert Erickson, Nathan Allen, Oliver Kortenkamp, Taylor Rathburn, and Allienna Nezelek. The USDA is an equal opportunity employer and provider.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frbee.2024.1410331/full#supplementary-material
Supplementary Table 1 | Reports the list of NCBI bio-projects and associated publications used for this meta-analysis, and bacterial (OTU) prevalence and abundance by niche.
References
Alberoni D., Favaro R., Baffoni L., Angeli S., Di Gioia D. (2021). Neonicotinoids in the agroecosystem: In-field long-term assessment on honeybee colony strength and microbiome. Sci. Total Environ. 762, 144116. doi: 10.1016/j.scitotenv.2020.144116
Anderson K. E., Carroll M. J., Sheehan T., Mott B. M., Maes P., Corby-Harris V. (2014). Hive-stored pollen of honey bees: Many lines of evidence are consistent with pollen preservation, not nutrient conversion. Mol. Ecol. 23, 5904–5917. doi: 10.1111/mec.12966
Anderson K. E., Copeland D. C., Erickson R. J., Floyd A. S., Maes P. C., Mott B. M. (2023). A high-throughput sequencing survey characterizing European foulbrood disease and Varroosis in honey bees. Sci. Rep. 13, 1162. doi: 10.1038/s41598-023-28085-2
Anderson K. E., Maes P. (2022). Social microbiota and social gland gene expression of worker honey bees by age and climate. Sci. Rep. 12, 10690. doi: 10.1038/s41598-022-14442-0
Anderson K. E., Mott B. M. (2023). Ecology of pollen storage in honey bees: sugar tolerant yeast and the aerobic social microbiota. Insects 14, 265. doi: 10.3390/insects14030265
Anderson K. E., Ricigliano V. A. (2017). Honey bee gut dysbiosis: a novel context of disease ecology. Curr. Opin. Insect Sci. 22, 125–132. doi: 10.1016/j.cois.2017.05.020
Anderson K. E., Ricigliano V. A., Copeland D. C., Mott B. M., Maes P. (2022). Social interaction is unnecessary for hindgut microbiome transmission in honey bees: the effect of diet and social exposure on tissue-specific microbiome assembly. Microbial. Ecol 85, 1498–1513. doi: 10.1007/s00248-022-02025-5
Anderson K. E., Ricigliano V. A., Mott B. M., Copeland D. C., Floyd A. S., Maes P. (2018). The queen’s gut refines with age : longevity phenotypes in a social insect model. Microbiome 6, 1–16. doi: 10.1186/s40168-018-0489-1
Anderson K. E., Sheehan T. H., Mott B. M., Maes P., Snyder L., Schwan M. R., et al. (2013). Microbial ecology of the hive and pollination landscape: Bacterial associates from floral nectar, the alimentary tract and stored food of honey bees (Apis mellifera). PloS One 8, 1–16. doi: 10.1371/journal.pone.0083125
Arredondo D., Castelli L., Porrini M. P., Garrido P. M., Eguaras M. J., Zunino P., et al. (2018). Lactobacillus kunkeei strains decreased the infection by honey bee pathogens Paenibacillus larvae and Nosema ceranae. Benef Microbes 9, 279–290. doi: 10.3920/BM2017.0075
Aslan C. E., Liang C. T., Galindo B., Kimberly H., Topete W. (2016). The role of honey bees as pollinators in natural areas. Natural Areas J. 36, 478–488. doi: 10.3375/043.036.0413
Bisson L. F., Walker G., Ramakrishnan V., Luo Y., Fan Q., Wiemer E., et al. (2017). The two faces of lactobacillus kunkeei: wine spoilage agent and bee probiotic. Am. J. Enol Vitic. 1, 1–11. doi: 10.5344/catalyst.2016.16002
Bonilla-Rosso G., Engel P. (2018). Functional roles and metabolic niches in the honey bee gut microbiota. Curr. Opin. Microbiol. 43, 69–76. doi: 10.1016/j.mib.2017.12.009
Bordenstein S. R., Theis K. R. (2015). Host biology in light of the microbiome: ten principles of holobionts and hologenomes. PloS Biol. 13, e1002226. doi: 10.1371/journal.pbio.1002226
Breed M. D., Williams K. R., Fewell J. H. (1988). Comb wax mediates the acquisition of nest-mate recognition cues in honey bees. Proc. Natl. Acad. Sci. United States America 85, 8766–8769. doi: 10.1073/pnas.85.22.8766
Burritt N. L., Foss N. J., Neeno-Eckwall E. C., Church J. O., Hilger A. M., Hildebrand J. A., et al. (2016). Sepsis and hemocyte loss in honey bees (Apis mellifera) Infected with serratia marcescens strain sicaria. PloS One 11, 1–26. doi: 10.1371/journal.pone.0167752
Buttstedt A., Moritz R. F., Erler S. (2013). More than royal food - Major royal jelly protein genes in sexuals and workers of the honeybee Apis mellifera. Front. Zool 10, 72. doi: 10.1186/1742-9994-10-72
Callegari M., Crotti E., Fusi M., Marasco R., Gonella E., De Noni I., et al. (2021). Compartmentalization of bacterial and fungal microbiomes in the gut of adult honeybees. NPJ Biofilms Microbiomes 7, 42. doi: 10.1038/s41522-021-00212-9
Carroll M. J., Brown N., Goodall C., Downs A. M., Sheenan T. H., Anderson K. E. (2017). Honey bees preferentially consume freshly-stored pollen. PloS One 12, e0175933. doi: 10.1371/journal.pone.0175933
Clarke K. R. (1993). Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 18, 117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x
Copeland D. C., Anderson K. E., Mott B. M. (2022a). Early queen development in honey bees : social context and queen breeder source affect gut microbiota and associated. Microbiol. Spectr. 10, e00383-22. doi: 10.1128/spectrum.00383-22
Copeland D. C., Maes P. W., Mott B. M., Anderson K. E. (2022b). Changes in gut microbiota and metabolism associated with phenotypic plasticity in the honey bee Apis mellifera. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.1059001
Copeland D. C., Ricigliano V. A., Mott B. M., Kortenkamp O. L., Erickson R. J., Gorrochategui-Ortega J., et al. (2024). A longitudinal study of queen health in honey bees reveals tissue specific response to seasonal changes and pathogen pressure. Sci. Rep. 14, 8963. doi: 10.1038/s41598-024-58883-1
Corby-Harris V., Maes P., Anderson K. E. (2014a). The bacterial communities associated with honey bee (Apis mellifera) foragers. PloS One 9, e95056. doi: 10.1371/journal.pone.0095056
Corby-Harris V., Snyder L. A., Meador C. A. D., Naldo R., Mott B. M., Anderson K. E. (2016). Parasaccharibacter apium, gen. nov., sp. nov., Improves Honey Bee (Hymenoptera: Apidae) Resistance to Nosema. J. Econ. Entomol 109, 537–543. doi: 10.1093/jee/tow012
Corby-Harris V., Snyder L. A., Schwan M. R., Maes P., McFrederick Q. S., Anderson K. E. (2014b). Origin and effect of Alpha 2.2 Acetobacteraceae in honey bee larvae and description of Parasaccharibacter apium gen. nov., sp. nov. Appl. Environ. Microbiol. 80, 7460–7472. doi: 10.1128/AEM.02043-14
Crailsheim K. (1998). Trophallactic interactions in the adult honeybee (Apis mellifera L.). Apidologie 29, 97–112. doi: 10.1051/apido:19980106
Daisley B. A., Pitek A. P., Chmiel J. A., Al K. F., Chernyshova A. M., Faragalla K. M., et al. (2020). Novel probiotic approach to counter Paenibacillus larvae infection in honey bees. ISME J. 14, 476–491. doi: 10.1038/s41396-019-0541-6
Daisley B. A., Reid G. (2021). BEExact: a metataxonomic database tool for high-resolution inference of bee-associated microbial communities. mSystems 6, 10.1128/msystems.00082-21 doi: 10.1128/mSystems.00082-21
Dalenberg H., Maes P., Mott B., Anderson K. E., Spivak M. (2020). Propolis envelope promotes beneficial bacteria in the honey bee (Apis mellifera) mouthpart microbiome. Insects 11, 1–12. doi: 10.3390/insects11070453
Damico M. E., Rueppell O., Shaffer Z., Han B., Raymann K. (2021). High royal jelly production does not impact the gut microbiome of honey bees. Anim. Microbiome 3, 60. doi: 10.1186/s42523-021-00124-1
Detry R., Simon-Delso N., Bruneau E., Daniel H. M. (2020). Specialisation of yeast genera in different phases of bee bread maturation. Microorganisms 8, 1–14. doi: 10.3390/microorganisms8111789
Djukic M., Poehlein A., Strauß J., Tann F. J., Leimbach A., Hoppert M., et al. (2015). High quality draft genome of Lactobacillus kunkeei EFB6, isolated from a German European foulbrood outbreak of honeybees. Standards Genom. Sci. 10, 16. doi: 10.1186/1944-3277-10-16
Endo A., Irisawa T., Futagawa-Endo Y., Takano K., du Toit M., Okada S., et al. (2012). Characterization and emended description of Lactobacillus kunkeei as a fructophilic lactic acid bacterium. Int. J. System. Evolution. Microbiol. 62, 500–504. doi: 10.1099/ijs.0.031054-0
Endo A., Maeno S., Tanizawa Y., Kneifel W., Arita M., Dicks L., et al. (2018). Fructophilic lactic acid bacteria, a unique group of fructose-fermenting microbes. Appl. Environ. Microbiol. 84, e01290–e01218. doi: 10.1128/AEM.01290-18
Endo A., Salminen S. (2013). Honeybees and beehives are rich sources for fructophilic lactic acid bacteria. System. Appl. Microbiol. 36, 444–448. doi: 10.1016/j.syapm.2013.06.002
Engel P., Martinson V. G., Moran N. A. (2012). Functional diversity within the simple gut microbiota of the honey bee. Proc. Natl. Acad. Sci. U.S.A. 109, 11002–11007. doi: 10.1073/pnas.1202970109
Evans J. D., Spivak M. (2010). Socialized medicine: Individual and communal disease barriers in honey bees. J. Invertebr. Pathol. 103, S62–S72. doi: 10.1016/j.jip.2009.06.019
Feng M., Fang Y., Li J. (2009). Proteomic analysis of honeybee worker (Apis mellifera) hypopharyngeal gland development. BMC Genomics 10, 645. doi: 10.1186/1471-2164-10-645
Floyd A. S., Mott B. M., Maes P., Copeland D. C., McFrederick Q. S., Anderson K. E. (2020). Microbial ecology of european foul brood disease in the honey bee (Apis mellifera): Towards a microbiome understanding of disease susceptibility. Insects 11, 1–16. doi: 10.3390/insects11090555
Forsgren E., Olofsson T. C., Váasquez A., Fries I. (2010). Novel lactic acid bacteria inhibiting Paenibacillus larvae in honey bee larvae. Apidologie 41, 99–108. doi: 10.1051/apido/2009065
Gallant A. L., Euliss N. H., Browning Z. (2014). Mapping large-area landscape suitability for honey bees to assess the influence of land-use change on sustainability of national pollination services. PloS One 9, e99268. doi: 10.1371/journal.pone.0099268
Gilliam M. (1979). Microbiology of pollen and bee bread: the yeasts. Apidologie 10, 45–53. doi: 10.1051/apido:19790106
Gilliam M., Valentine D. K. (1974). Enterobacteriaceae isolated from foraging worker honey bees, Apis mellifera. J. Invertebr. Pathol. 23, 38–41. doi: 10.1016/0022-2011(74)90069-X
Goldstein S. L., Klassen J. L. (2020). Pseudonocardia symbionts of fungus-growing ants and the evolution of defensive secondary metabolism. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.621041
Haeder S., Wirth R., Herz H., Spiteller D. (2009). Candicidin-producing Streptomyces support leaf-cutting ants to protect their fungus garden against the pathogenic fungus Escovopsis. Proc. Natl. Acad. Sci. United States America 106, 4742–4746. doi: 10.1073/pnas.0812082106
Härer L., Stýblová S., Ehrmann M. A. (2023). Bombella pluederhausensis sp. nov., Bombella pollinis sp. nov., Bombella saccharophila sp. nov. and Bombella dulcis sp. nov., four Bombella species isolated from the environment of the western honey bee Apis mellifera. Int. J. System. Evolution. Microbiol. 73, 005927. doi: 10.1099/ijsem.0.005927
Harwood G., Amdam G., Freitak D. (2019). The role of Vitellogenin in the transfer of immune elicitors from gut to hypopharyngeal glands in honey bees (Apis mellifera). J. Insect Physiol. 112, 90–100. doi: 10.1016/j.jinsphys.2018.12.006
Harwood G., Salmela H., Freitak D., Amdam G. (2021). Social immunity in honey bees: royal jelly as a vehicle in transferring bacterial pathogen fragments between nestmates. J. Exp. Biol. 224. doi: 10.1242/jeb.231076
Hubert J., Bicianova M., Ledvinka O., Kamler M., Lester P. J., Nesvorna M., et al. (2017). Changes in the Bacteriome of Honey Bees Associated with the Parasite Varroa destructor, and Pathogens Nosema and Lotmaria passim. Microbial. Ecol. 73, 685–698. doi: 10.1007/s00248-016-0869-7
Jia H., Geng L., Li Y., Wang Q., Diao Q., Zhou T., et al. (2016). The effects of Bt Cry1Ie toxin on bacterial diversity in the midgut of Apis mellifera ligustica (Hymenoptera: Apidae). Sci. Rep. 6, 24664. doi: 10.1038/srep24664
Jones J. C., Fruciano C., Marchant J., Hildebrand F., Forslund S., Bork P., et al. (2018). The gut microbiome is associated with behavioural task in honey bees. Insectes Sociaux 65, 419–429. doi: 10.1007/s00040-018-0624-9
Kapheim K. M., Rao V. D., Yeoman C. J., Wilson B. A., White B. A., Goldenfeld N., et al. (2015). Caste-specific differences in hindgut microbial communities of honey bees (Apis mellifera). PloS One 10, 1–14. doi: 10.1371/journal.pone.0123911
Kešnerová L., Emery O., Troilo M., Liberti J., Erkosar B., Engel P. (2020). Gut microbiota structure differs between honeybees in winter and summer. ISME J. 14, 801–814. doi: 10.1038/s41396-019-0568-8
Krzywinski M. I., Schein J. E., Birol I., Connors J., Gascoyne R., Horsman D., et al. (2009). Circos: An information aesthetic for comparative genomics. Genome Res 1639–1645. doi: 10.1101/gr.092759.109
Kwakman P. H. S., te Velde A., de Boer L., Speijer D., Vandenbroucke-grauls C. M. J. E., Zaat S. A. J., et al. (2010). How honey kills bacteria. FASEB journal 24, 2576–2582. doi: 10.1096/fj.09-150789
Kwong W. K., Moran N. A. (2016). Gut microbial communities of social bees. Nat. Rev. Microbiol. 14, 374–384. doi: 10.1038/nrmicro.2016.43
Liberti J., Kay T., Quinn A., Kesner L., Frank E. T., Cabirol A., et al. (2022). The gut microbiota affects the social network of honeybees. Nat. Ecol. Evol. 6, 1471–1479. doi: 10.1038/s41559-022-01840-w
Maeno S., Tanizawa Y., Kanesaki Y., Kubota E., Kumar H., Dicks L., et al. (2016). Genomic characterization of a fructophilic bee symbiont Lactobacillus kunkeei reveals its niche-specific adaptation. System. Appl. Microbiol. 39, 516–526. doi: 10.1016/j.syapm.2016.09.006
Maes P. W., Floyd A. S., Mott B. M., Anderson K. E. (2021). Overwintering honey bee colonies: effect of worker age and climate on the hindgut microbiota. Insects 12, 224. doi: 10.3390/insects12030224
Maes P. W., Rodrigues P. A. P., Oliver R., Mott B. M., Anderson K. E. (2016). Diet-related gut bacterial dysbiosis correlates with impaired development, increased mortality and Nosema disease in the honeybee (Apis mellifera). Mol. Ecol. 25, 5439–5450. doi: 10.1111/mec.13862
Mao W., Schuler M., Berenbaum M. R. (2013). Honey constituents up-regulate detoxification and immunity genes in the western honey bee Apis mellifera. Proc. Natl. Acad. Sci. United States America 110, 1–5. doi: 10.1073/pnas.1303884110
McFrederick Q. S., Thomas J. M., Neff J. L., Vuong H. Q., Russell K. A., Hale A. R., et al. (2017). Flowers and wild megachilid bees share microbes. Microb. Ecol. 73, 188–200. doi: 10.1007/s00248-016-0838-1
McFrederick Q. S., Wcislo W. T., Taylor D. R., Ishak H. D., Dowd S. E., Mueller U. G. (2012). Environment or kin: whence do bees obtain acidophilic bacteria? Mol. Ecol. 21, 1754–1768. doi: 10.1111/j.1365-294X.2012.05496.x
Miller D. L., Smith E. A., Newton I. L. G. (2021). A bacterial symbiont protects honey bees from fungal disease. mBio 12, 10.1128/mbio.00503-21. doi: 10.1128/mBio.00503-21
Moran N. A., Hansen A. K., Powell J. E., Sabree Z. L. (2012). Distinctive gut microbiota of honey bees assessed using deep sampling from individual worker bees. PloS One 7, e36393. doi: 10.1371/journal.pone.0036393
Moran N. A., Sloan D. B. (2015). The hologenome concept: helpful or hollow? PloS Biol. 13, 1–10. doi: 10.1371/journal.pbio.1002311
Motta E. V. S., Raymann K., Moran N. A. (2018). Glyphosate perturbs the gut microbiota of honey bees. Proc. Natl. Acad. Sci. 115, 10305–10310. doi: 10.1073/pnas.1803880115
Nash A. K., Auchtung T. A., Wong M. C., Smith D. P., Gesell J. R., Ross M. C., et al. (2017). The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 5, 153. doi: 10.1186/s40168-017-0373-4
Neveling D. P., Endo A., Dicks L. M. T. (2012). Fructophilic Lactobacillus kunkeei and Lactobacillus brevis Isolated from Fresh Flowers, Bees and Bee-hives. Curr. Microbiol. 65, 507–515. doi: 10.1007/s00284-012-0186-4
Nicolson S. W. (2009). Water homeostasis in bees, with the emphasis on sociality. J. Exp. Biol. 212, 429–434. doi: 10.1242/jeb.022343
Nicolson S. W. (2011). Bee food: the chemistry and nutritional value of nectar, pollen and mixtures of the two. African Zoology 46, 197–204. doi: 10.1080/15627020.2011.11407495
Ohashi K., Natori S., Kubo T. (1999). Expression of amylase and glucose oxidase in the hypopharyngeal gland with an age-dependent role change of the worker honeybee (Apis mellifera L.). Eur. J. Biochem. 265, 127–133. doi: 10.1046/j.1432-1327.1999.00696.x
Ptaszyńska A. A., Paleolog J., Borsuk G. (2016). Nosema ceranae infection promotes proliferation of yeasts in honey bee intestines. PloS One 11, 1–15. doi: 10.1371/journal.pone.0164477
Raymann K., Coon K. L., Shaffer Z., Salisbury S., Moran N. A. (2019). Pathogenicity of serratia marcescens strains in honey bees. Microbiol. Spectr. 7, 1–16. doi: 10.1128/microbiolspec.gpp3-0053-2018
Roessink I., van der Steen J. J. M. (2021). Beebread consumption by honey bees is fast: results of a six-week field study. J. Apicult. Res. 0, 1–6. doi: 10.1080/00218839.2021.1915612
Rognes T., Flouri T., Nichols B., Quince C., Mahé F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e258. doi: 10.7717/peerj.2584
Rokop Z. P., Horton M. A., Newton I. L. G. (2015). Interactions between cooccurring lactic acid bacteria in honey bee hives. Appl. Environ. Microbiol. 81, 7261–7270. doi: 10.1128/AEM.01259-15
Rothman J. A., Carroll M. J., Meikle W. G., Anderson K. E., McFrederick Q. S. (2018). Longitudinal effects of supplemental forage on the honey bee (Apis mellifera) microbiota and inter- and intra-colony variability. Microbial. Ecol. 76, 814–824. doi: 10.1007/s00248-018-1151-y
Rothman J. A., Leger L., Kirkwood J. S., McFrederick Q. S. (2019). Cadmium and selenate exposure affects the honey bee microbiome and metabolome, and bee-associated bacteria show potential for bioaccumulation. Appl. Environ. Microbiol. 85. doi: 10.1128/AEM.01411-19
Ruiz-Argueso T., Rodriguez-Navarro A. (1975). Microbiology of ripening honey. Appl. Microbiol. 30, 893–896. doi: 10.1128/am.30.6.893-896.1975
Salmela H., Sundström L. (2017). Vitellogenin in inflammation and immunity in social insects. Inflammation Cell Signaling 5, e1506. doi: 10.14800/ics.1506
Salter S. J., Cox M. J., Turek E. M., Calus S. T., Cookson W. O., Moffatt M. F., et al. (2014). Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12, 87. doi: 10.1186/s12915-014-0087-z
Schloss P. D., Westcott S. L., Ryabin T., Hall J. R., Hartmann M., Hollister E. B., et al. (2009). Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schwarz R. S., Huang Q., Evans J. D. (2015). Hologenome theory and the honey bee pathosphere. Curr. Opin. Insect Sci. 10, 1–7. doi: 10.1016/j.cois.2015.04.006
Simone M., Evans J. D., Spivak M. (2009). RESIN COLLECTION AND SOCIAL IMMUNITY IN HONEY BEES. Evolution 63, 3016–3022. doi: 10.1111/evo.2009.63.issue-11
Simone-Finstrom M., Spivak M. (2010). Propolis and bee health: the natural history and significance of resin use by honey bees. Apidologie 41, 295–311. doi: 10.1051/apido/2010016
Smith E. A., Newton I. L. G. (2020). Genomic signatures of honey bee association in an acetic acid symbiont. Genome Biol. Evol. 12, 1882–1894. doi: 10.1093/gbe/evaa183
Sopko B., Zitek J., Nesvorna M., Markovic M., Kamler M., Titera D., et al. (2020). Detection and quantification of Melissococcus plutonius in honey bee workers exposed to European foulbrood in Czechia through conventional PCR, qPCR, and barcode sequencing. J. Apicult. Res. 59, 503–514. doi: 10.1080/00218839.2019.1685148
Spivak M., Goblirsch M., Simone-Finstrom M. (2019). Social-medication in bees: the line between individual and social regulation. Curr. Opin. Insect Sci. 33, 49–55. doi: 10.1016/j.cois.2019.02.009
Subotic S., Boddicker A. M., Nguyen V. M., Rivers J., Briles C. E., Mosier A. C. (2019). Honey bee microbiome associated with different hive and sample types over a honey production season. PloS One 14, e0223834. doi: 10.1371/journal.pone.0223834
Tamarit D., Ellegaard K. M., Wikander J., Olofsson T., Vásquez A., Andersson S. G. E. (2015). Functionally structured genomes in lactobacillus kunkeei colonizing the honey crop and food products of honeybees and stingless bees. Genome Biol. Evol. 7, 1455–1473. doi: 10.1093/gbe/evv079
Tarpy D. R., Mattila H. R., Newton I. L. G. (2015). Characterization of the honey bee microbiome throughout the queen-rearing process. Appl. Environ. Microbiol., 81. doi: 10.1128/AEM.00307-15
Tauber J. P., Nguyen V., Lopez D., Evans J. D. (2019). Effects of a resident yeast from the honeybee gut on immunity, microbiota, and nosema disease. Insects 10, 296. doi: 10.3390/insects10090296
Taylor M. A., Robertson A. W., Biggs P. J., Richards K. K., Jones D. F., Parkar S. G. (2019). The effect of carbohydrate sources: Sucrose, invert sugar and components of mānuka honey, on core bacteria in the digestive tract of adult honey bees (Apis mellifera). PloS One 14, e0225845. doi: 10.1371/journal.pone.0225845
Vásquez A., Forsgren E., Fries I., Paxton R. J., Flaberg E., Szekely L., et al. (2012). Symbionts as major modulators of insect health: Lactic acid bacteria and honeybees. PloS One 7, 10.1371/annotation/3ac2b867-c013-4504-9e06-bebf3fa039d1. doi: 10.1371/journal.pone.0033188
Vernier C. L., Chin I. M., Adu-Oppong B., Krupp J. J., Levine J., Dantas G., et al. (2020). The gut microbiome defines social group membership in honey bee colonies. Sci. Adv. 6, 1–10. doi: 10.1126/sciadv.abd3431
Vojvodic S., Johnson B. R., Harpur B. A., Kent C. F., Zayed A., Anderson K. E., et al. (2015). The transcriptomic and evolutionary signature of social interactions regulating honey bee caste development. Ecol. Evol. 5, 4795–4807. doi: 10.1002/ece3.1720
Vojvodic S., Rehan S. M., Anderson K. E. (2013). Microbial gut diversity of africanized and european honey bee larval instars. PloS One 8, e72106. doi: 10.1371/journal.pone.0072106
Wang X., Zhong Z., Chen X., Hong Z., Lin W., Mu X., et al. (2021). High-fat diets with differential fatty acids induce obesity and perturb gut microbiota in honey bee. Int. J. Mol. Sci. 22, 834. doi: 10.3390/ijms22020834
Zendo T., Ohashi C., Maeno S., Piao X., Salminen S., Sonomoto K., et al. (2020). Kunkecin A, a new nisin variant bacteriocin produced by the fructophilic lactic acid bacterium, apilactobacillus kunkeei FF30-6 isolated from honey bees. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.571903
Zheng H., Powell J. E., Steele M. I., Dietrich C., Moran N. A. (2017). Honeybee gut microbiota promotes host weight gain via bacterial metabolism and hormonal signaling. Proc. Natl. Acad. Sci. U.S.A. 114, 4775–4780. doi: 10.1073/pnas.1701819114
Keywords: aerobic bacteria, social microbiome, nest microbiome, larvae, queen, disease resistance
Citation: Anderson KE and Copeland DC (2024) The honey bee “hive” microbiota: meta-analysis reveals a native and aerobic microbiota prevalent throughout the social resource niche. Front. Bee Sci. 2:1410331. doi: 10.3389/frbee.2024.1410331
Received: 31 March 2024; Accepted: 19 June 2024;
Published: 05 July 2024.
Edited by:
Susan E. Fahrbach, Wake Forest University, United StatesReviewed by:
Solenn Patalano, Alexander Fleming Biomedical Sciences Research Center, GreeceTomas Erban, Crop Research Institute (CRI), Czechia
Terd Disayathanoowat, Chiang Mai University, Thailand
Copyright © 2024 Anderson and Copeland. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kirk E. Anderson, a2lyay5hbmRlcnNvbkB1c2RhLmdvdg==
†These authors have contributed equally to this work