 Seong-Rae Lee
Seong-Rae Lee Jun Su Kang
Jun Su Kang Pyung Cheon Lee
Pyung Cheon Lee- Department of Molecular Science and Technology and Advanced College of Bio-convergence Engineering, Ajou University, Suwon, Republic of Korea
Wickerhamomyces ciferrii: a non-conventional yeast with significant industrial potential for tetraacetyl phytosphingosine (TAPS), remains underutilized due to the lack of a comprehensive genetic toolbox. In this study, we developed a modular genetic system tailored for Wickerhamomyces ciferrii to enable strain engineering and metabolic pathway optimization. This toolkit includes episomal plasmids incorporating multiple selectable markers, replication origins, and fluorescent reporters. Systematic evaluation of four antibiotic resistance markers demonstrated that nourseothricin, geneticin, and zeocin effectively confer resistance, whereas hygromycin B did not support selection in this host. Among three tested replication origins, 2μ and CEN6/ARS4 enabled stable episomal maintenance, whereas panARS failed to replicate. Expression analysis of six fluorescent proteins under the endogenous PGK1 promoter revealed significant variability in transcript levels, which correlated with codon adaptation index values, emphasizing the importance of codon optimization for heterologous expression. Additionally, characterization of the endogenous TDH3, PGK1, and PDA1 promoters using two highly expressed fluorescent proteins confirmed that promoter strength is largely independent of the downstream coding sequence. To demonstrate the functional application of this toolkit, we overexpressed a phosphorylation-insensitive mutant of acetyl-CoA carboxylase (ACC1S26A-S1161A), resulting in a 2.4-fold increase in TAPS production. Collectively, this study establishes a versatile genetic platform for W. ciferrii, providing a robust foundation for future synthetic biology and metabolic engineering applications.
Introduction
The increasing demand for sustainable and scalable bioproduction systems has driven interest in microbial platforms as alternatives to traditional chemical synthesis (Patra et al., 2021; Ploessl et al., 2023). Yeasts are particularly attractive due to their eukaryotic cellular machinery, metabolic versatility, and ability to perform post-translational modifications, making them well-suited for industrial biotechnology (Nielsen, 2013). While Saccharomyces cerevisiae is the most extensively studied yeast, its industrial applicability is often limited by stress sensitivity, inefficient precursor accumulation, and rigid metabolic constraints (Fraczek et al., 2018; Liang et al., 2024; Singh et al., 2022; Utomo et al., 2021). Consequently, non-conventional yeasts with enhanced stress tolerance and unique biosynthetic capabilities are increasingly being explored as alternative chassis organisms (Deparis et al., 2017; Lacerda et al., 2020; Walker and Basso, 2020; Zhou et al., 2025; Baptista and Domingues, 2022; Cao et al., 2022; Duman-Özdamar and Binay, 2021; Liu et al., 2023; Ma et al., 2022; Manfrão-Netto et al., 2019; Park and Ledesma-Amaro, 2023; Qiao et al., 2017; Sun et al., 2020; Tran et al., 2019).
Among these, W. ciferrii (formerly Hansenula ciferri and later Pichia ciferrii) is notable for its native ability to synthesize tetraacetyl phytosphingosine (TAPS), a high-value sphingolipid precursor with applications in cosmetics and pharmaceuticals (Wickerham and Stodola, 1960; Barenholz et al., 1971). Since TAPS shares biosynthetic pathways with other sphingolipids, including sphinganine and sphingosine, Wickerhamomyces ciferrii presents a promising platform for engineering sphingolipid biosynthesis (Eun and Lee, 2023). Early metabolic engineering strategies have focused on the overexpression of LCB2, disruption of negative regulatory genes (ORM12, LCB4), and optimization of serine metabolism, leading to significant improvements in TAPS yield (Bae et al., 2003; Börgel et al., 2012; Schorsch et al., 2012). More recently, genome sequencing, adaptive laboratory evolution, and gamma-ray mutagenesis have facilitated further strain improvements, culminating in the highest reported TAPS titer of 20.2 g/L in fed-batch fermentation (Schneider et al., 2012; Choi et al., 2021; Yoo et al., 2023).
In addition to its native sphingolipid biosynthetic capacity, W. ciferrii possesses several desirable traits as a chassis for synthetic biology. It is among the fast-growing yeast species, with a reported specific growth rate of 0.27–0.64 h-1 under defined conditions (Wronska et al., 2020). Indeed, W. ciferrii is already used for commercial-scale production of ceramide-related compounds for skincare (Yoo et al., 2023; Schorsch et al., 2012; Pérez-Rivero and López-Gómez, 2023). These features, along with its demonstrated tractability in fermentation processes, support its potential as a safe and efficient microbial host for biomanufacturing.
Despite these advancements, the lack of a well-developed genetic toolkit remains a major barrier to W. ciferrii’s broader adoption in synthetic biology (Schorsch et al., 2009). Currently, available tools are limited to a small set of antibiotic selection markers and replication origins, restricting the flexibility of plasmid-based genetic manipulation. Expanding the repertoire of replication elements, fluorescent reporters, and tunable promoters would greatly improve strain engineering efforts, allowing for more precise pathway optimization and metabolic control.
To address these limitations, this study aims to systematically characterize and expand the genetic toolkit for W. ciferrii by evaluating four antibiotic selection markers, three replication origins, and six fluorescent proteins. Additionally, three endogenous promoters—pTDH3, pPGK1, and pPDA1—were assessed for their strength and expression variability (Figure 1). Finally, to validate the functionality of this toolkit, we overexpressed a phosphorylation-insensitive mutant of acetyl-CoA carboxylase (ACC1S26A−S1161A), demonstrating its impact on TAPS biosynthesis. Together, these findings provide a versatile molecular platform for future strain engineering, metabolic optimization, and synthetic biology applications in W. ciferrii.
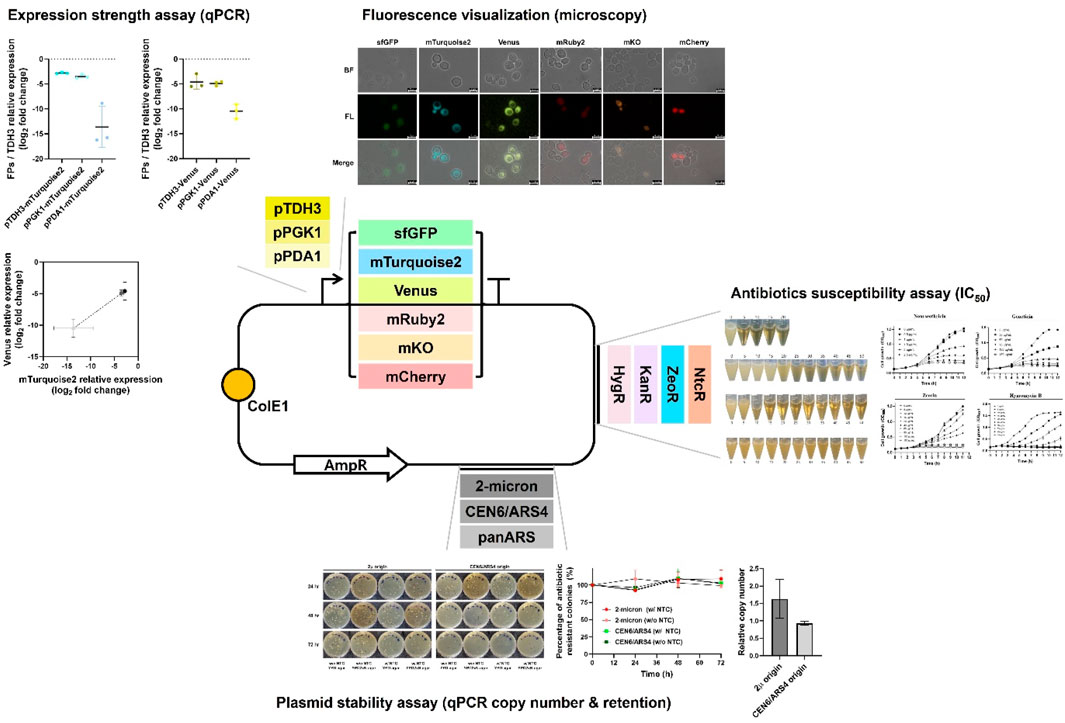
Figure 1. Overview of the genetic toolkit construction and functional characterization in Wickerhamomyces ciferrii. This schematic illustrates the modular plasmid system developed in this study, highlighting the key genetic elements and their experimental validation. The base plasmid contains a bacterial backbone (ColE1 origin and ampicillin resistance marker) for propagation in Escherichia coli. Functional modules evaluated in Wickerhamomyces ciferrii include: (i) three replication origins (2μ, CEN6/ARS4, panARS), compared in terms of plasmid maintenance and copy number; (ii) four antibiotic resistance markers (NtcR, ZeoR, KanR, HygR), tested for selection efficiency on both solid and liquid media; (iii) six fluorescent protein genes (sfGFP, mTurquoise2, Venus, mRuby2, mKO, mCherry), evaluated by fluorescence microscopy and transcript analysis; (iv) three endogenous promoters (pTDH3, pPGK1, pPDA1), assessed for transcriptional strength via quantitative real-time PCR (qPCR). Representative experimental results for each functional module are summarized adjacent to the schematic.
Material and methods
Strains, media, and growth conditions
Wickerhamomyces ciferrii (F-60-10A NRRL 1031) wild-type strain was used for the overall study. Single colonies were obtained by streaking the cell stock on YPD agar (10 g/L yeast extract, 20 g/L peptone, 20 g/L glucose, and 1.5% agar) and incubating at 25°C for 2 days. For 96-well plate cultivation in an antibiotic IC50 test, single colonies were inoculated into 200 μL of YPD medium (10 g/L yeast extract, 20 g/L peptone, and 20 g/L glucose) and aerobically cultured at 25°C with shaking at 800 rpm. For short-term cultivation prior to genomic DNA extraction, fluorescence analysis, plasmid copy number determination, transcriptional expression analysis, and main culture inoculation, single colonies were inoculated into test tubes containing 2 mL of YPD medium (with or without appropriate antibiotics) and incubated at 25°C with shaking at 250 rpm for 12–24 h. Cells were subsequently harvested and processed for each respective assay. For long-term cultivation, test tube-scale pre-cultured cells were inoculated into 250 mL Erlenmeyer flasks containing 50 mL of YMglSC medium at an initial OD600 of 0.1 and cultured at 25°C, 250 rpm until the carbon source was depleted. Appropriate antibiotics were added to the culture medium at concentrations to be determined in this study. Cell growth was monitored at a wavelength of 600 nm (OD600) with a SPECTRAmax PLUS384 spectrophotometer (Molecular Devices, United States). For plasmid cloning and propagation, Escherichia coli NEB 10-beta (New England Biolabs Korea, Republic of Korea) was aerobically grown overnight in Luria-Bertani (LB) medium (10 g/L tryptone, 5 g/L yeast extract, and 5 g/L NaCl) at 37°C with shaking at 250 rpm. Solid medium was prepared by adding 1.5% agar to the recipe. For selection, 100 μg/mL ampicillin or 50 μg/mL chloramphenicol was added to the solid agar plates and culture tubes containing 4 mL of LB medium.
Genomic DNA extraction
Genomic DNA of W. ciferrii was extracted following the protocol of Dymond et al., with slight modifications (Dymond, 2013). Briefly, a single colony generated from YPD agar was inoculated into 2 mL of YPD medium in a test tube and incubated overnight at 25°C with shaking at 250 rpm. One milliliter of the culture was transferred to a 1.7 mL microcentrifuge tube and centrifuged at 14,000 rpm for 1 min at room temperature. After removing the supernatant, the cell pellet was resuspended in 200 μL of yeast lysis buffer [10 mM Tris-HCl (pH 8.0), 1 mM EDTA (pH 8.0), 100 mM NaCl, 2% Triton X-100, 1% sodium dodecyl sulfate (SDS)]. Next, 150 μL of glass beads and 200 μL of phenol/chloroform/isoamyl alcohol (25:24:1, v/v/v) were added to the suspension, followed by vigorous vortexing for 10 min. Subsequently, 400 μL of TE buffer [10 mM Tris-HCl (pH 8.0), 1 mM EDTA (pH 8.0)] was added, and the mixture was vortexed for one additional minute. After centrifugation at 4°C and 13,000 rpm for 10 min, the upper aqueous layer was transferred to a new 1.7 mL microcentrifuge tube. An equal volume of isopropyl alcohol was added and gently inverted to mix. After centrifugation at 4°C and 13,000 rpm for 10 min, a pellet was observed. The pellet was washed with 1 mL of chilled 70% ethanol. After discarding the supernatant by centrifugation at 4°C and 13,000 rpm for 5 min, the pellet was air-dried at room temperature for approximately 5 min. Finally, the dried pellet was resuspended in 100 μL of TE buffer and used for subsequent polymerase chain reaction (PCR).
Plasmid construction and sequencing
Plasmids and strains used in this study are listed in Table 1, and primers used for PCR are provided in Table 2. Plasmid were constructed using Golden Gate assembly or Gibson assembly. DNA fragments used for assembly were amplified by PCR, with detailed information on the fragments and their corresponding primers listed in Table 3. Oligonucleotides for PCR were obtained from Macrogen (Seoul, Republic of Korea), and PCR was performed using Phusion High-Fidelity DNA polymerase (New England Biolabs Korea, Republic of Korea) following the manufacturer’s manual. The DNA fragments used for assembly were gel-purified with a gel extraction kit (GeneAll, Republic of Korea).

Table 1. Plasmids and strains used in this study.
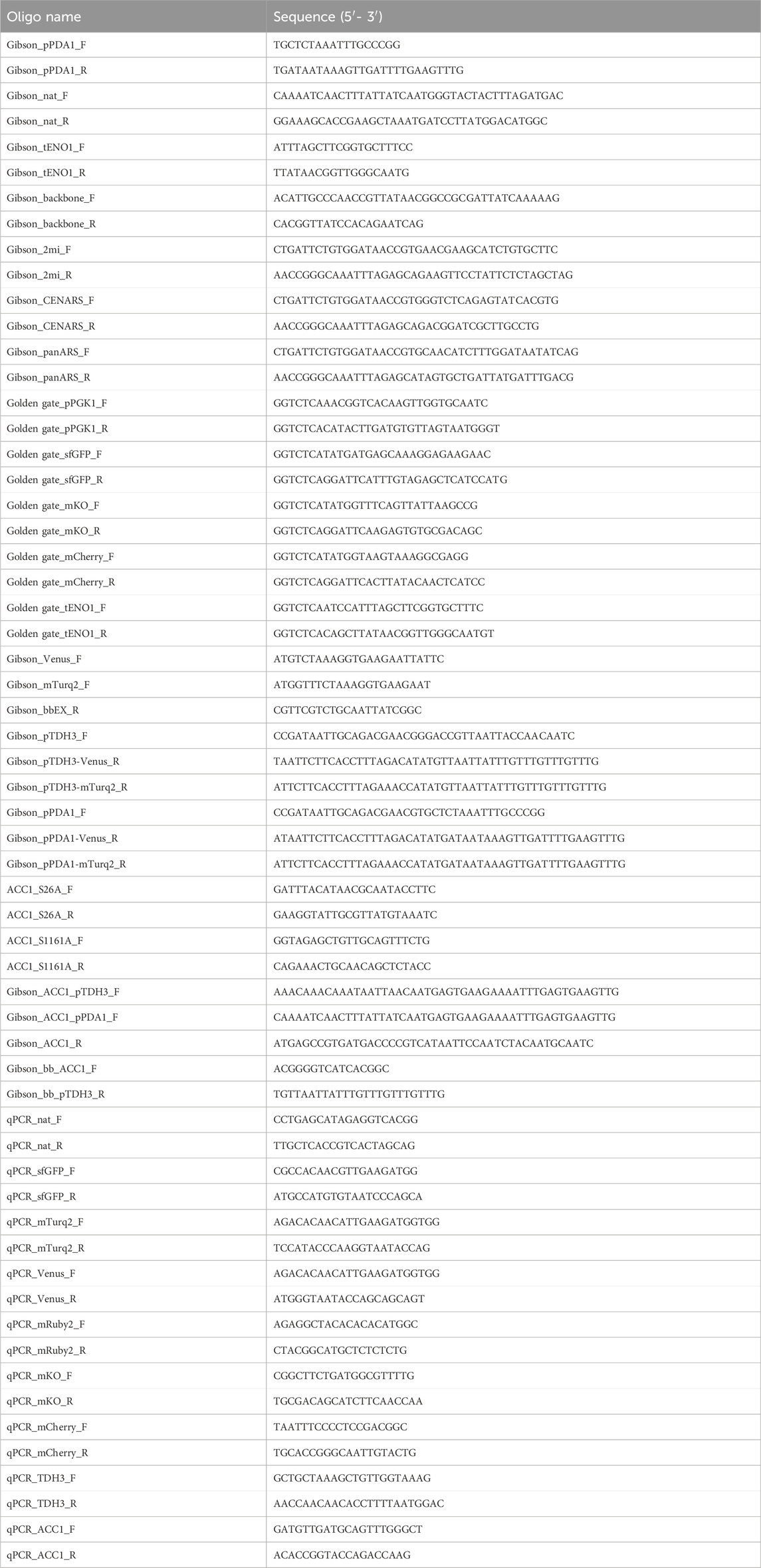
Table 2. Primers used in this study.
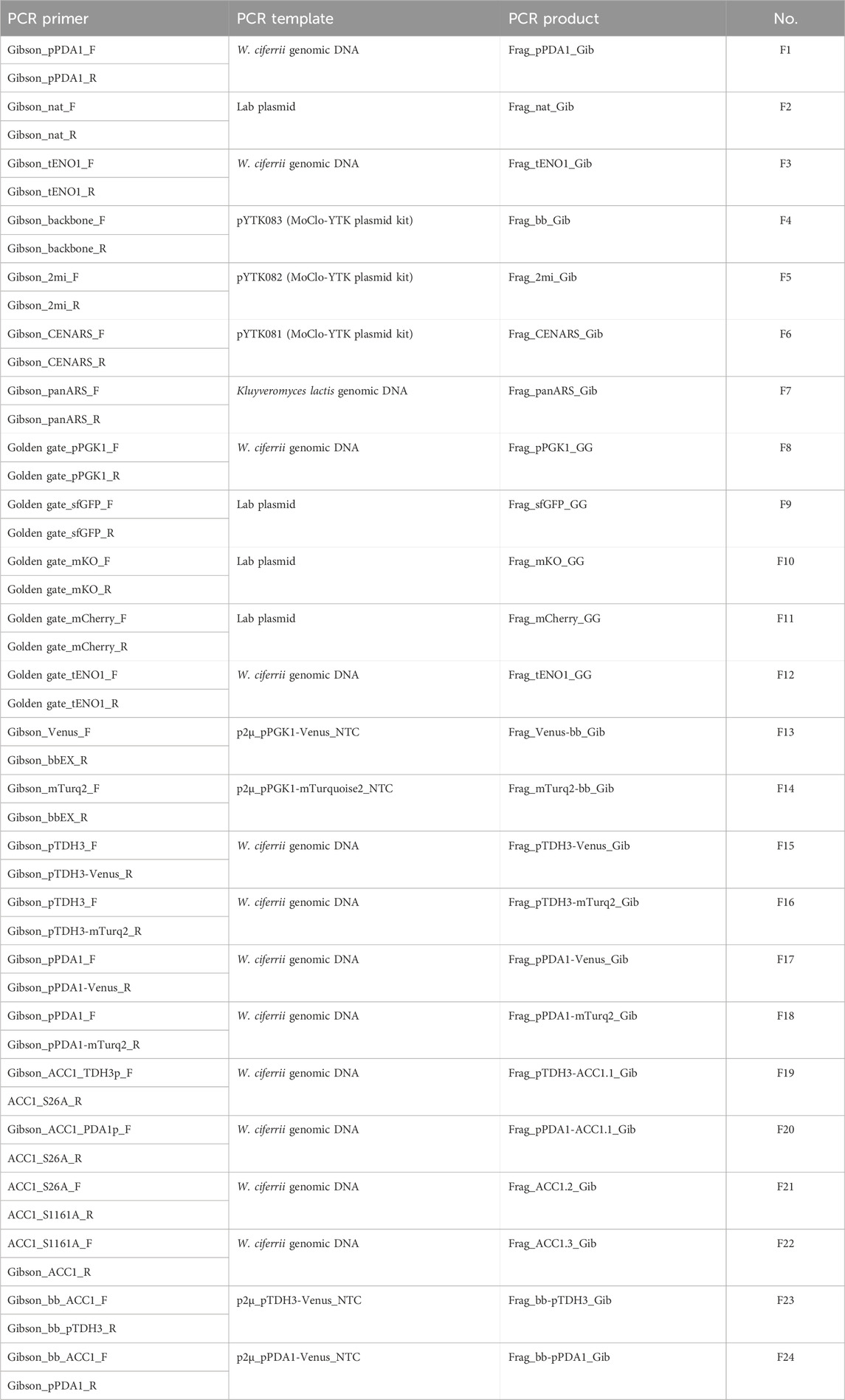
Table 3. PCR fragment used for assembly in this study.
The MoClo-YTK plasmid kit was used to obtain four antibiotic resistance marker sequences (nourseothricin, geneticin, zeocin, and hygromycin B), each expressed under the Ashbya gossypii TEF promoter and TEF terminator, three fluorescent protein coding genes (mTurquoise2, Venus, and mRuby2), two origins of replication commonly used in Saccharomyces cerevisiae (2μ and CEN6/ARS4), and a bacterial vector backbone containing the ampicillin resistance marker and ColE1 origin of replication for selection in E. coli (Lee et al., 2015). The MoClo-YTK plasmid kit was a gift from John Dueber (Addgene kit # 1000000061).
To identify a replication origin sequence capable of supporting stable plasmid replication in W. ciferrii, plasmids p2μ_WcNAT, pCEN_WcNAT, and ppan_WcNAT were constructed by assembling one of three replication origin sequences—2μ and CEN6/ARS4 from S. cerevisiae, and panARS from Kluyveromyces lactis—with a nat marker cassette and a bacterial vector backbone. The 2μ and CEN6/ARS4 sequences were obtained from pYTK082 and pYTK081 of MoClo-YTK plasmid kit, and panARS was obtained from genomic DNA of K. lactis. This nat marker cassette consisted of the W. ciferrii endogenous pyruvate dehydrogenase subunit E1 alpha (PDA1) promoter, a codon-optimized nat gene, and the W. ciferrii endogenous enolase (ENO1) terminator. The assembly was performed using Gibson Assembly Master Mix (New England Biolabs Korea, Republic of Korea). The PDA1 promoter and the ENO1 terminator sequence were identified using the reference genome sequence available in the NCBI database. Specifically, the upstream 675 bp region of the start codon was extracted from the annotated reference sequence (Gene ID: BN7_3959a, RefSeq: NW_011887617.1) for the PDA1 promoter, and the downstream 332 bp region of the stop codon was extracted from the reference sequence (Gene ID: BN7_149, RefSeq: NW_011887739.1) for the ENO1 terminator.
Next, for screening of selectable markers applicable to W. ciferrii, four additional plasmids—p2μ_NTC, p2μ_G418, p2μ_ZEO, and p2μ_HYG—were constructed. Four marker cassettes expressed under the A. gossypii TEF promoter and TEF terminator were amplified by PCR from the MoClo-YTK plasmid kit (pYTK078, pYTK077, pYTK080, and pYTK079, respectively). Each amplified cassette was then cloned via Gibson assembly, together with the 2μ origin of replication sequence amplified from pYTK082 and the bacterial vector backbone from pYTK083.
For the construction of fluorescent protein expression vectors used in functional reporter protein screening, Golden Gate cloning was performed following the protocol by Michael E. Lee et al. to generate twelve plasmids carrying either 2μ or CEN6/ARS4 replication origin: p2μ_pPGK1-sfGFP_NTC, p2μ_pPGK1-mTurquoise2_NTC, p2μ_pPGK1-Venus_NTC, p2μ_pPGK1-mRuby2_NTC, p2μ_pPGK1-mKO_NTC, p2μ_pPGK1-mCherry_NTC, pCEN_pPGK1-sfGFP_NTC, pCEN_pPGK1-mTurquoise2_NTC, pCEN_pPGK1-Venus_NTC, pCEN_pPGK1-mRuby2_NTC, pCEN_pPGK1-mKO_NTC, and pCEN_pPGK1-mCherry_NTC (Lee et al., 2015). The plasmid assembly required eight parts: (1) linker, (2) promoter, (3) gene, (4) terminator, (5) linker, (6) yeast selectable marker, (7) yeast origin of replication, and (8) bacterial vector backbone. To incorporate the W. ciferrii endogenous phosphoglycerate 1 (PGK1) promoter and ENO1 terminator into positions (2) and (4), respectively, PCR amplification was carried out using W. ciferrii genomic DNA as the template with primers containing BsaⅠ restriction sites along with appropriate flanking sequences to ensure compatibility with adjacent parts. The PGK1 gene sequence was identified from the NCBI database (Gene ID: BN7_3735, RefSeq: NW_011887629.1), and the 497 bp upstream region of the start codon was used as the promoter. To further expand the fluorescent protein library beyond mTurquoise2, Venus, and mRuby2 from the MoClo-YTK plasmid kit, additional fluorescent proteins, including superfolder GFP (sfGFP), mKO, and mCherry, were amplified using the same primer design strategy and incorporated into part (3). For the assembly, PCR-amplified parts: (2) PGK1 promoter, (3) sfGFP, mKO, and mCherry genes, and (4) ENO1 terminator were used, while the remaining parts were directly obtained from the MoClo-YTK plasmid kit. The specific part numbers used in the assembly are listed in Table 3. Each part was mixed equimolarly (20 fmol) in a single tube together with 20 U of BsaⅠ, 400 U of T4 DNA ligase, 2 μL of T4 DNA ligase buffer (all from New England Biolabs Korea, Republic of Korea), and deionized water to a final volume of 20 μL. The reaction was carried out under the following conditions [37°C for 5 min, 16°C for 5 min] × 30 cycles, followed by 60°C for 5 min, and an infinite hold at 4°C.
To compare fluorescence intensity based on promoter strength, three representative endogenous promoters commonly used in yeast research were selected: the glyceraldehyde 3-phosphate dehydrogenase promoter (pTDH3), the phosphoglycerate kinase 1 promoter (pPGK1), and the pyruvate dehydrogenase subunit E1 alpha promoter (pPDA1). Using these promoters, four additional plasmids—p2μ_pPDA1-mTurquoise2_NTC, p2μ_pTDH3-mTurquoise2_NTC, p2μ_pPDA1-Venus_NTC, and p2μ_pTDH3-Venus_NTC—were constructed in addition to the two previously generated plasmids carrying the PGK1 promoter, p2μ_pPGK1-mTurquoise2_NTC and p2μ_pPGK1-Venus_NTC. The promoter sequences were identified based on the reference genome of W. ciferrii available in the NCBI database: TDH3 gene (Gene ID: BN7_5327, RefSeq: NW_011887532.1), PGK1 gene (Gene ID: BN7_3735, RefSeq: NW_011887629.1), PDA1 gene (Gene ID: BN7_3959a, RefSeq: NW_011887617.1). The upstream 418 bp (pTDH3), 497 bp (pPGK1), and 675 bp (pPDA1) regions of the start codon were extracted and used as the endogenous promoter parts. These three promoters, amplified from W. ciferrii genomic DNA by PCR, were individually cloned with both mTurquoise2 and Venus, which were the most highly expressed fluorescent proteins in W. ciferrii, using Gibson assembly together with the endogenous ENO1 terminator, the nourseothricin resistance marker, the 2μ origin of replication, and the bacterial vector backbone.
To construct an ACC1 mutant expression vector for practical application in enhancing tetraacetyl phytosphingosine (TAPS) production, the W. ciferrii ACC1 sequence was identified through a BLASTp search, using the amino acid sequence of Rattus norvegicus (rat) ACC1, retrieved from UniProtKB/Swiss-Prot (Accession: P11497.1) as the query. To enhance enzyme activity, site-directed mutagenesis was performed on the W. ciferrii ACC1 gene sequence to introduce the S26A and S1161A mutations, as previously reported. For vector construction, the ACC1S26A−S1161A gene, including the coding sequence and the terminator region (from the start codon to 225 bp downstream of the stop codon), was amplified by PCR. The resulting fragment was then cloned using Gibson assembly, incorporating two different promoters (pTDH3 for strong expression and pPDA1 for weak expression), the ACC1S26A−S1161A coding sequence and terminator, the nourseothricin resistance marker, the 2μ origin of replication, and the bacterial vector backbone parts in the designated order, resulting in the p2μ_pTDH3-ACC1(mut)_NTC and p2μ_pPDA1-ACC1(mut)_NTC plasmids.
The sequences of all constructed vectors were confirmed by Sanger sequencing (Macrogen, Republic of Korea).
Transformation of Wickerhamomyces ciferrii and Escherichia coli
The Golden Gate and Gibson assembly mixtures, prepared as described above, were transformed into E. coli 10-beta competent cells (New England Biolabs Korea, Republic of Korea) via electroporation following the manufacturer’s protocol. The cloned plasmids were then introduced into W. ciferrii using a slightly modified version of a previously published protocol (Bae et al., 2003).
A single W. ciferrii colony from YPD agar was inoculated into 2 mL of YPD medium and cultured until reaching an OD600 of 0.8–1.2. The cells were harvested by centrifugation and resuspended in 0.1 culture volume of 50 mM phosphate buffer (pH 7.5) supplemented with 25 mM dithiothreitol (DTT). After shaking incubation at 37°C for 15 min, the cells were washed twice with one culture volume of STM solution (10 mM Tris-HCl [pH 7.5], 270 mM sucrose, 1 mM MgCl2) and finally resuspended in 0.01 culture volume of STM solution. The prepared competent cells were aliquoted into 50-μL portions and stored at −80°C until use.
For transformation, 50 μL of frozen W. ciferrii competent cells were mixed with ∼1 μg of plasmid DNA and kept on ice until fully thawed. The mixture was transferred to a 0.2-cm electroporation cuvette (Bio-Rad, United States), and electroporation was performed using a GenePulser Xcell™ (Bio-Rad, United States) at 500 V, 50 μF, and 700 Ω. Following electroporation, cells were immediately resuspended in 500 μL of STM solution and transferred to a test tube containing 2 mL of YPD medium. After incubation at 25°C and 250 rpm for 6–12 h, the cells were plated onto YPD agar containing the appropriate antibiotic. Colonies were observed after 3 days of incubation at 25°C.
Screening of functional origin of replication
To assess whether plasmids could be maintained and confer resistance in W. ciferrii, three plasmids, p2μ_WcNAT, pCEN_WcNAT, and ppan_WcNAT, each carrying a replication origin—2μ and CEN6/ARS4 from S. cerevisiae and panARS from K. lactis—along with the nourseothricin resistance marker and the bacterial vector backbone, were transformed into W. ciferrii competent cells, which were then plated onto YPD2xN agar (YPD medium containing 100 μg/mL nourseothricin). Colony formation was assessed after incubation at 25°C for 3 days.
To evaluate plasmid stability over culture time, transformants obtained from YPD2xN agar were inoculated into 2 mL of YPDN liquid medium (YPD containing 50 μg/mL nourseothricin) and cultured at 25°C, 250 rpm for 12 h. The overnight culture was then transferred into 50 mL of YMglSCN medium (YMglSC medium containing 50 μg/mL nourseothricin) in a 250 mL baffled flask at an initial OD600 = 0.1 and incubated for 72 h. Every 24 h, equal numbers of cells were plated onto YPD agar (without antibiotics) and YPD2xN agar (with antibiotics), and the percentage of antibiotic-resistant colonies was calculated.
Antibiotic and selectable marker screening
To screen for antibiotics and selectable markers applicable to the wild-type diploid W. ciferrii strain, four commonly used antibiotics in yeast research were tested at various concentrations to assess cell viability. Cells were pre-cultured in 2 mL of YPD medium in test tubes and incubated at 25°C, 250 rpm for 12 h. After incubation, cultures were inoculated at 1% (v/v) into 200 µL of YPD medium containing different antibiotic concentrations in a 96-well plate. Since W. ciferrii exhibited varying susceptibility to each antibiotic, multiple trials were conducted to determine the appropriate concentration ranges: nourseothricin (0–2.5 μg/mL), geneticin (0–45 μg/mL), zeocin (0–45 μg/mL), and hygromycin B (0–45 μg/mL). Cell growth was monitored by measuring OD600 at 1-h intervals, and statistical analysis was performed using biological triplicates. The specific growth rate of W. ciferrii was calculated using the formula “ln (OD2/OD1)/(t2-t1)”. The obtained values were normalized to the untreated antibiotic set, and concentrations of antibiotics inhibiting 50% of the growth were determined.
In order to identify selectable markers that confer effective resistance, four plasmids—p2μ_NTC, p2μ_G418, p2μ_ZEO, and p2μ_HYG—carrying each antibiotic resistance marker sequence (nourseothricin, geneticin, zeocin, and hygromycin B) from the MoClo-YTK plasmid kit, along with the previously constructed p2μ_WcNAT plasmid were transformed into W. ciferrii competent cells and spread onto YPD agar containing the respective antibiotics to assess colony formation. Antibiotic concentrations were systematically optimized by incrementally increasing from previously reported values for various yeast species—nourseothricin (50 μg/mL), geneticin (200 μg/mL), zeocin (100 μg/mL), and hygromycin B (100 μg/mL) (Bai et al., 2021; Chen et al., 2016; Schorsch et al., 2009; Zhao et al., 2020)—to minimize false-positive colonies while maintaining effective selection.
Relative plasmid copy number determination
To determine the relative plasmid copy number in W. ciferrii, a quantitative real-time PCR (qPCR) was performed using the TDH3 gene as a reference and the nourseothricin resistance marker (nat) gene as the target. Transformants were generated by transforming W. ciferrii competent cells with p2u_NTC or pCEN_NTC plasmids, each carrying the nourseothricin resistance marker and either the 2μ origin or CEN6/ARS4 origin. Single colonies were isolated from YPD2xN agar plates, and five single colonies from each plate were inoculated into test tubes containing 2 mL of YPDN medium. Cultures were incubated at 25°C and 250 rpm overnight. Following incubation, cells were harvested by centrifugation, and genomic DNA was extracted. qPCR was performed on genomic DNA extracted from a total of 10 transformant samples (five from each plasmid type) using the SensiFAST™ SYBR No-ROX One-Step Kit (Bioline, United States). For each sample, both the TDH3 reference gene and the nat target gene were amplified, and Ct values were obtained using a Rotor-Gene system (Qiagen, Germany). Relative plasmid copy number was determined using the ΔCt method, where ΔCt = Ct (nat) - Ct (TDH3). Since W. ciferrii is diploid, the TDH3 gene copy number was set to 2, and relative plasmid copy number was calculated as 2−ΔCt × 2. The sequences of primers used—qPCR_nat_F, qPCR_nat_R, qPCR_TDH3_F, and qPCR_TDH3_R—are listed in Table 2.
Fluorescence analysis
For fluorescence observation in W. ciferrii, plasmids expressing six different fluorescent proteins—sfGFP, mTurquoise2, Venus, mRuby2, mKO, and mCherry—were introduced into W. ciferrii competent cells via transformation. Single colonies selected on YPD2xN agar plates were inoculated into test tubes containing 2 mL of YPDN medium, followed by incubation at 25°C and 250 rpm overnight. Cultures typically reached OD600 ≈ 4 after overnight incubation, which corresponds to the mid-logarithmic phase of W. ciferrii growth. After cultivation, cells were harvested by centrifugation and washed twice with 1× phosphate-buffered saline (PBS) to remove residual medium. Fluorescence microscopy was performed using a LEICA DM2500 microscope (LEICA, Germany) equipped with a LEICA DFC450 C digital camera. To visualize the fluorescence signals, two different filter sets were used depending on the excitation and emission spectra of each fluorescent protein. The FITC filter set (LEICA L5 filter) was used for sfGFP (488/510 nm), mTurquoise2 (434/474 nm), and Venus (515/528 nm), while the TRITC filter set (LEICA N2.1 filter) was used for mRuby2 (559/600 nm), mKO (548/559 nm), and mCherry (587/610 nm).
Transcriptional gene expression analysis
To assess the transcriptional expression levels of heterologous genes in W. ciferrii transformants, cells were cultivated at 25°C and 250 rpm until reaching the logarithmic growth phase. At this stage, a portion of the culture corresponding to an OD600 of approximately 10 was harvested by centrifugation. The supernatant was completely removed, and the cell pellet was resuspended in 1 mL of RNAlater stabilization solution (Thermo Fisher, United States) to preserve RNA integrity. Total RNA was extracted from the harvested cells using the easy-BLUE™ Total RNA Extraction Kit (iNtRON Biotechnology, Republic of Korea) following the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized using the ReverTraAce™ qPCR RT Kit (Toyobo, Japan). Quantitative reverse transcription PCR (qRT-PCR) was subsequently performed using the SensiFAST™ No-ROX One-Step Kit (Bioline, United States) on a Rotor-Gene (Qiagen, Germany). For each cDNA sample, two separate qRT-PCR reactions were conducted: one for the gene of interest and one for the reference gene TDH3. Relative transcriptional expression levels were analyzed using both the ΔCt and ΔΔCt methods. In the ΔCt method, ΔCt was calculated as Ct (target) - Ct (TDH3), with TDH3 serving as the reference gene. For comparative expression analysis across strains, the ΔΔCt method was applied, where the ΔCt values of the target gene in overexpression strains were normalized to its ΔCt value in the designated negative control strain. Primer sequences used for amplification are listed in Table 2, specifically those designated as ‘qPCR_gene_F’ and ‘qPCR_gene_R’.
Extraction and analysis of TAPS
Recombinant W. ciferrii strains were cultivated in 50 mL of YMglSCN medium in 250 mL baffled flasks. Cultures were incubated at 25°C and 250 rpm until glycerol depletion in triplicate. For TAPS quantification, 1 mL of each culture was mixed with 4 mL of methanol, followed by vigorous vortexing for 30 min. After extraction, the mixture was centrifuged, and the resulting supernatant was filtered through a 0.2 μm PTFE syringe membrane filter. The filtrate was then analyzed using high-performance liquid chromatography (HPLC) on an Agilent 1,260 system (Agilent Technology, United States) equipped with a photodiode array detector and a ZORBAX Eclipse XDB-C18 column (150 mm × 4.6 mm, 5 μm, Agilent Technology) at 20°C. Gradient elution was performed at a flow rate of 1 mL/min with mobile phase A (100% acetonitrile) and mobile phase B (100% deionized distilled water, DDW) under the following conditions: 45%–80% A (0–18 min), 80%–90% A (18–20 min), 90%–100% A (20–23 min), 100% A (23–25 min), 100%–45% A (25–28 min), and 45% A (28–30 min). TAPS was quantified at 200 nm based on peak area values, using a standard curve established in this study.
Codon adaptation index (CAI) calculation and correlation analysis
To evaluate the potential impact of codon usage on gene expression in W. ciferrii, the codon adaptation index (CAI) was calculated using the online tool CAIcal (http://genomes.urv.es/CAIcal/) with codon usage tables generated via the Countcodon program (https://www.kazusa.or.jp/codon/) (Nakamura et al., 2000; Puigbò et al., 2008). The reference gene set for CAI calculation consisted of ten genes involved in the sphingoid base biosynthetic pathway of W. ciferrii: ATF2 (NCBI Gene ID: BN7_5921, RefSeq: NW_011887500.1), LCB1 (NCBI Gene ID: BN7_169, RefSeq: NW_011887738.1), LCB2 (NCBI Gene ID: BN7_5771, RefSeq: NW_011887508.1), LCB3 (NCBI Gene ID: BN7_3163, RefSeq: NW_011887655.1), LCB4 (NCBI Gene ID: BN7_4658, RefSeq: NW_011887561.1), ORM1 (NCBI Gene ID: BN7_3097, RefSeq: NW_011887657.1), SLI1 (NCBI Gene ID: BN7_6322, RefSeq: NW_011887476.1), SYR2 (NCBI Gene ID: BN7_5855, RefSeq: NW_011887504.1), TSC3 (NCBI Gene ID: BN7_3864, RefSeq: NW_011887620.1), and TSC10 (NCBI Gene ID: BN7_3057, RefSeq: NW_011887660.1).
CAI values were computed for six fluorescent proteins (sfGFP, mTurquoise2, Venus, mRuby2, mKO, and mCherry) and four antibiotic resistance markers (nourseothricin, zeocin, geneticin, and hygromycin B) obtained from the MoClo-YTK plasmid kit. To examine the relationship between codon adaptation and transcript levels, linear regression analysis was performed using CAI values as the independent variable (x-axis) and the relative transcriptional expression levels of the fluorescent proteins (log2 fold change of FP/TDH3) as the dependent variable (y-axis). Each data point represents biological triplicates of transcript levels for each fluorescent protein. The mKO dataset was excluded from this analysis due to its undetectable transcript levels. The correlation between the relative transcriptional expression levels of mTurquoise2 and Venus under different promoter strengths was also analyzed, with mTurquoise2 plotted on the x-axis and Venus on the y-axis. The correlation coefficient (R2) and statistical significance (p-value) were determined.
Statistical analysis
All data are presented as mean ± standard deviation (SD) from 3 or 5 biological replicates. Linear regression analysis and unpaired t-test with Welch’s correction were performed using GraphPad Prism (version 8.0.2). Statistical significance was defined as p < 0.05 (ns: p ≥ 0.05, *p < 0.05, **p < 0.01, ***p < 0.001).
Results
Screening and verification of replication origins for stable episomal maintenance in Wickerhamomyces ciferrii
The availability of autonomously replicating plasmids in W. ciferrii remains largely unexplored. To date, the only reported sequence enabling episomal maintenance in W. ciferrii is the S. cerevisiae CEN/ARS origin (Yoo et al., 2023). However, no additional replication origins have been systematically characterized, and most genetic studies have relied on integrative vectors for gene expression and genome modifications (Bae et al., 2003; Börgel et al., 2012; Schorsch et al., 2009; 2012). To address this limitation, we aimed to establish a versatile plasmid system incorporating alternative replication origins for efficient gene expression in W. ciferrii.
We selected three replication origins: the 2μ origin and CEN6/ARS4 sequence, both widely used in S. cerevisiae, and the panARS sequence from K. lactis, suggested as a broad-host-range origin (Camattari et al., 2016; Liachko and Dunham, 2014) (Figure 2A). Plasmids carrying these origins—p2μ_WcNAT (2μ origin), pCEN_WcNAT (CEN6/ARS4 origin), and ppan_WcNAT (panARS origin)—were transformed into W. ciferrii and plated onto selective YPD2xN agar. After 3 days at 25°C, colonies formed for W. c_p2μ_WcNAT and W. c_pCEN_WcNAT, whereas no growth was observed for W. c_ppan_WcNAT, indicating that panARS does not support episomal replication in W. ciferrii.
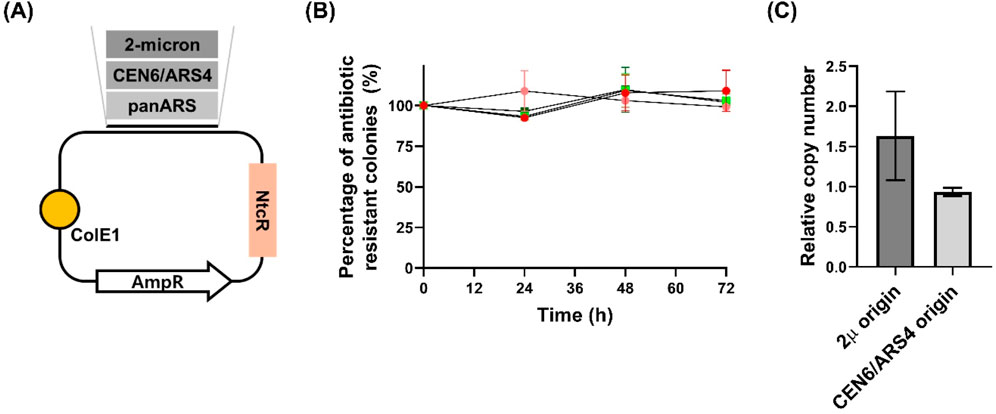
Figure 2. Stability and copy number analysis of plasmids with different origins of replication in Wickerhamomyces ciferrii (A) Schematic representation of the three constructed plasmids. Each plasmid harbors a distinct candidate origin of replication (2μ, CEN6/ARS4, or panARS) and includes a nourseothricin resistance (NtcR) marker functional in Wickerhamomyces ciferrii (B) Long-term segregational stability assay. Transformants were cultivated in both selective and non-selective media, and at designated time points (0, 24, 48, and 72 h), equal numbers of cells were plated onto both antibiotic-containing and antibiotic-free agar plates. The percentage of antibiotic-resistant colonies was calculated to assess plasmid retention. Data are presented as mean ± standard deviation (SD) from three biological replicates (n = 3) (C) Quantification of relative plasmid copy number. The relative copy numbers of plasmids carrying the 2μ and CEN6/ARS4 origins were determined via quantitative PCR. Values were normalized to the genomic TDH3 gene, which was used as a diploid reference (copy number = 2). Data represent the mean ± SD from three biological replicates (n = 3).
To confirm plasmid stability, single colonies of W. c_p2μ_WcNAT and W. c_pCEN_WcNAT were grown in YPDN liquid medium (YPD supplemented with 50 μg/mL nourseothricin) for 12 h, showing normal growth. Long-term plasmid stability was further assessed by cultivating both strains for 72 h in YMglSC medium (without selection) and YMglSCN medium (with 50 μg/mL nourseothricin). At 24-h intervals, equal numbers of cells were plated onto selective and non-selective YPD agar. After 3 days at 25°C, CFU counts showed no significant differences across time points, indicating stable plasmid retention even after 72 h without selection (Figure 2B). Additionally, quantitative PCR (qPCR) analysis revealed that plasmids with the 2μ origin exhibited a higher copy number than those with the CEN6/ARS4 origin (Figure 2C), reinforcing the difference in replication properties.
Collectively, these findings establish that 2μ and CEN6/ARS4 support stable episomal replication in W. ciferrii, enabling long-term plasmid maintenance without selection. This work expands the genetic toolkit for W. ciferrii, providing foundational resources for strain engineering and synthetic biology applications.
Evaluation of antibiotic susceptibility and selectable markers in Wickerhamomyces ciferrii
The range of antibiotic selection markers available for W. ciferrii remains limited, with previous studies primarily utilizing nourseothricin, geneticin, cycloheximide (via ribosomal protein L41 mutation), syringomycin E (for SYR2 mutant screening), and 5-fluoroorotic acid (for URA3-based counter-selection) (Bae et al., 2003; Börgel et al., 2012; Schorsch et al., 2009; Yoo et al., 2023). However, no studies have systematically evaluated the half-maximal inhibitory concentrations (IC50) for these antibiotics in W. ciferrii. To address this gap, we assessed the susceptibility of W. ciferrii to four widely used antibiotics—nourseothricin, geneticin, zeocin, and hygromycin B—by culturing wild-type cells in YPD medium with increasing antibiotic concentrations and monitoring growth inhibition. The IC50 values were determined as follows: nourseothricin, 1.48 ± 0.12 μg/mL; geneticin, 16.52 ± 2.07 μg/mL; zeocin, 14.96 ± 1.43 μg/mL; and hygromycin B, 9.72 ± 0.65 μg/mL, indicating differential susceptibility. To evaluate whether W. ciferrii could acquire resistance through heterologous expression of selection markers, four plasmids—p2μ_NTC, p2μ_G418, p2μ_ZEO, and p2μ_HYG—encoding resistance genes for nourseothricin, geneticin, zeocin, and hygromycin B, respectively, were transformed into W. ciferrii. Transformants were plated on YPD agar supplemented with standard antibiotic concentrations (50 μg/mL nourseothricin, 200 μg/mL geneticin, 100 μg/mL zeocin, and 100 μg/mL hygromycin B). Despite strong growth inhibition in liquid culture, high levels of false positives and lawn formation were observed on three solid media (supplemented with nourseothricin, geneticin, and zeocin), suggesting that standard antibiotic concentrations were insufficient for effective selection. To improve selection efficiency, antibiotic concentrations were optimized, establishing effective selection thresholds of 100 μg/mL (solid) and 50 μg/mL (liquid) for nourseothricin, 300 μg/mL (solid) and 50 μg/mL (liquid) for geneticin, and 200 μg/mL (solid) and 100 μg/mL (liquid) for zeocin, except for hygromycin B. Under these conditions, resistant colonies were consistently recovered for nourseothricin, geneticin, and zeocin, whereas no viable hygromycin B-resistant colonies were obtained. This observed differences in selection efficiency between solid and liquid media, with lower antibiotic concentrations required for effective selection in liquid culture may be attributed to differences in antibiotic diffusion rates, microbial cell density effects, or metabolic adaptations in response to solid-phase growth. Similar phenomena have been reported previously, where structured environments such as agar surfaces reduced antibiotic efficacy due to metabolic dormancy or decreased penetration (Stokes et al., 2019). In addition, bacterial cell debris has been shown to bind and sequester antibiotics, further reducing their bioavailability in solid media (Yuan et al., 2022), thus requiring higher concentrations for effective selection.
Determination of copy number and structural integrity of episomal plasmids in Wickerhamomyces ciferrii
While the 2 μ and CEN6/ARS4 origins supported stable plasmid replication in W. ciferrii, we quantified their retention and relative copy numbers. qPCR analysis revealed that plasmids carrying the 2 μ origin maintained a higher copy number (1.6 ± 0.59 copies per cell) than those with the CEN6/ARS4 origin (0.9 ± 0.05 copies per cell) (Figure 2C). These findings confirm that while both origins enable episomal maintenance, the 2 μ origin provides a greater plasmid retention advantage in W. ciferrii.
To verify sequence integrity, episomal plasmids were extracted from W. ciferrii and subsequently transformed into E. coli for propagation and high-quality plasmid preparation suitable for Sanger sequencing. This approach enabled confirmation that the plasmids maintained during episomal replication in W. ciferrii did not undergo structural rearrangements or point mutations. Both p2μ_NTC and pCEN_NTC were confirmed to be intact, reinforcing the genetic stability of episomal vectors in W. ciferrii and their reliability for engineering applications.
Characterization of functional reporter fluorescent proteins in Wickerhamomyces ciferrii
Fluorescent reporter proteins (FPs) are essential tools for monitoring gene expression, protein localization, and cellular dynamics in genetic engineering. To expand the FP toolkit for W. ciferrii, we evaluated six distinct FPs—sfGFP, mTurquoise2, Venus, mRuby2, mKO, and mCherry—for their expression efficiency and functionality (Figure 3A). The endogenous PGK1 promoter (pPGK1) was selected for FP expression based on its stronger transcriptional activity compared to PDA1 promoter (pPDA1), while avoiding overexpression artifacts (Peng et al., 2015). Twelve expression plasmids, carrying either the 2μ or CEN6/ARS4 replication origin, were generated via Golden Gate assembly and transformed into W. ciferrii, resulting in 12 FP-expressing strains: W. c_2μ-sfGFP-mod, W. c_2μ-mTurq2-mod, W. c_2μ-Venus-mod, W. c_2μ-mRuby2-mod, W. c_2μ-mKO-mod, W. c_2μ-mCherry-mod, W. c_CEN-sfGFP-mod, W. c_CEN-mTurq2-mod, W. c_CEN-Venus-mod, W. c_CEN-mRuby2-mod, W. c_CEN-mKO-mod, and W. c_CEN-mCherry-mod.
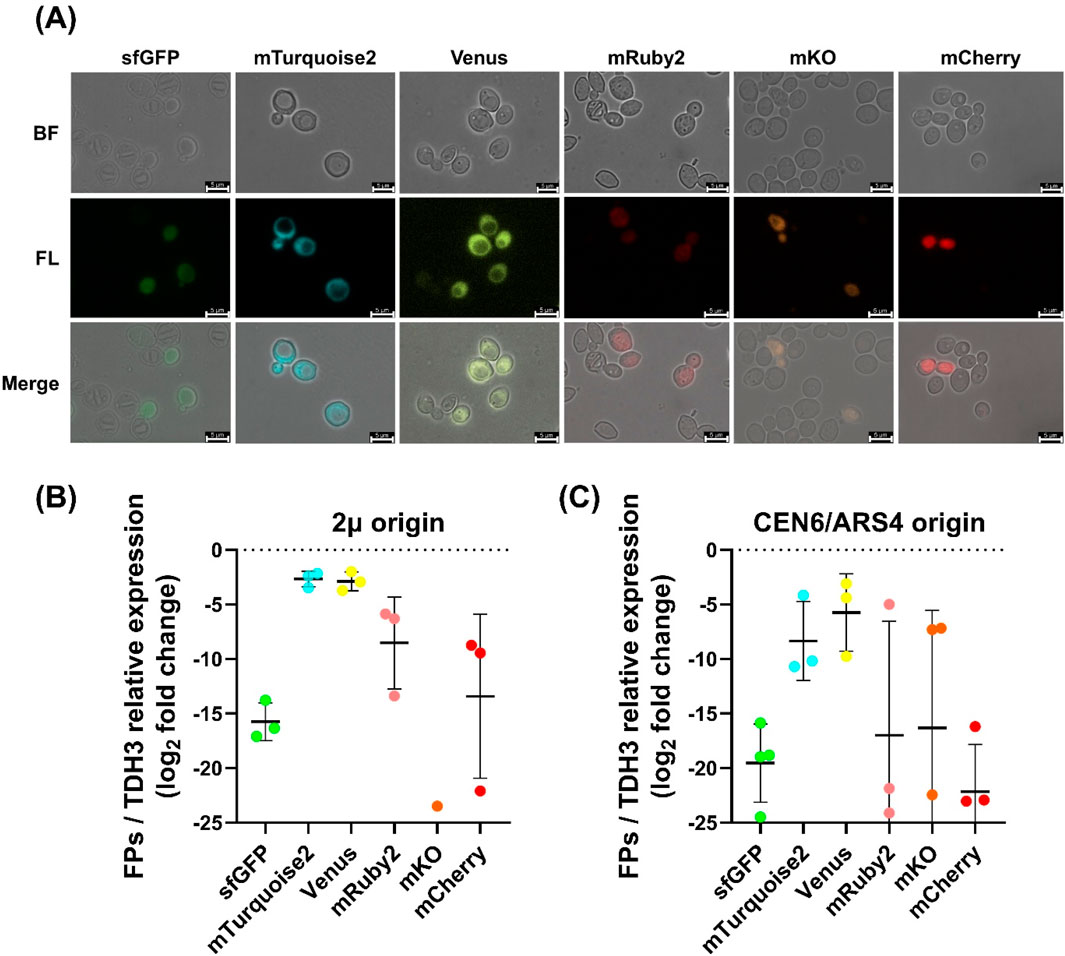
Figure 3. Fluorescence imaging and transcriptional analysis of fluorescent protein expression in Wickerhamomyces ciferrii (A) Fluorescence microscopy images of Wickerhamomyces ciferrii transformants expressing different fluorescent proteins from 2μ-based vectors. Cells harboring plasmids encoding sfGFP, mTurquoise2, Venus, mRuby2, mKO, or mCherry were imaged under bright-field (BF) and fluorescence (FL) microscopy, with merged images provided for comparison. Representative single-cell fluorescence images are shown to visualize protein-level expression from 2μ-based vectors. Fluorescence images of strains carrying CEN6/ARS4-based vectors are provided in Supplementary Data S2. Scale bars, 5 μm (B) Relative transcriptional expression of fluorescent protein genes in 2μ-based plasmids. Quantitative PCR (qPCR) analysis was conducted to determine mRNA levels, normalized to TDH3 gene and presented as log2 fold changes. Data represent the mean ± standard deviation (SD) from three biological replicates (n = 3). Only detectable clones were analyzed for mKO (C) Relative transcriptional expression of fluorescent protein genes in CEN6/ARS4-based plasmids. qPCR analysis was performed as in (B) but for CEN6/ARS4-based vectors. Data represent the mean ± SD from three biological replicates (n = 3).
Cells for fluorescence microscopy were harvested after overnight cultivation in YPDN medium containing 50 μg/mL nourseothricin at 25°C and 250 rpm, reaching OD600 ≈ 4, indicative of mid-log phase of W. ciferrii. The fluorescence microscopy images in Figure 3A were obtained from strains carrying 2μ-based vectors and are shown as representative examples of protein-level expression. Fluorescence images of strains harboring CEN6/ARS4-based vectors are provided in Supplementary Data S2.
Following plasmid construction and transformation, we compared the performance of vectors carrying different origins of replication. Overall, 2μ-based vectors conferred more stable FP expression than CEN6/ARS4-based vectors, as evidenced by lower colony-to-colony variability in transcriptional expression levels (Figures 3B,C). Among the tested FPs, mTurquoise2 and Venus displayed the highest fluorescence intensity and expression stability, whereas sfGFP showed lower but uniform fluorescence. In contrast, mRuby2 and mCherry exhibited substantial variability, with weak fluorescence in most cells, and mKO displayed almost no detectable fluorescence. qPCR analysis corroborated these observations (Figures 3B,C). Transformants carrying 2μ-based vectors showed lower expression variability, while mTurquoise2 and Venus had the highest transcript levels, approximately 212-fold and 211-fold higher than sfGFP, respectively. mTurquoise2 was expressed ∼1.25-fold higher than Venus, with both showing similar trends. In contrast, mKO exhibited negligible transcript levels, consistent with its undetectable fluorescence.
These findings establish mTurquoise2 and Venus as optimal FPs for gene expression studies in W. ciferrii, providing valuable tools for metabolic engineering and synthetic biology applications.
Reporter protein expression using three endogenous promoters in Wickerhamomyces ciferrii
The availability of stable endogenous promoters is essential for expanding the genetic toolkit in W. ciferrii, particularly for optimizing heterologous protein expression. To characterize promoter strength, we evaluated three native promoters—PDA1 promoter (pPDA1), PGK1 promoter (pPGK1), and TDH3 promoter (pTDH3)— in a plasmid-based expression system. While pPDA1 and pPGK1 have been previously utilized, pTDH3 was selected for its strong constitutive activity in yeast (Lee et al., 2015; Mumberg et al., 1995; Redden and Alper, 2015; Xiong et al., 2018). We hypothesized that these promoters would exhibit differential expression levels (pTDH3 > pPGK1 > pPDA1) and tested their ability to drive fluorescent protein (FP) expression. Additionally, to assess promoter strength independent of the coding sequence effects, two highly transcribed FPs, mTurquoise2 and Venus, were expressed under each promoter using 2μ origin-based vectors (Figures 4A,B).
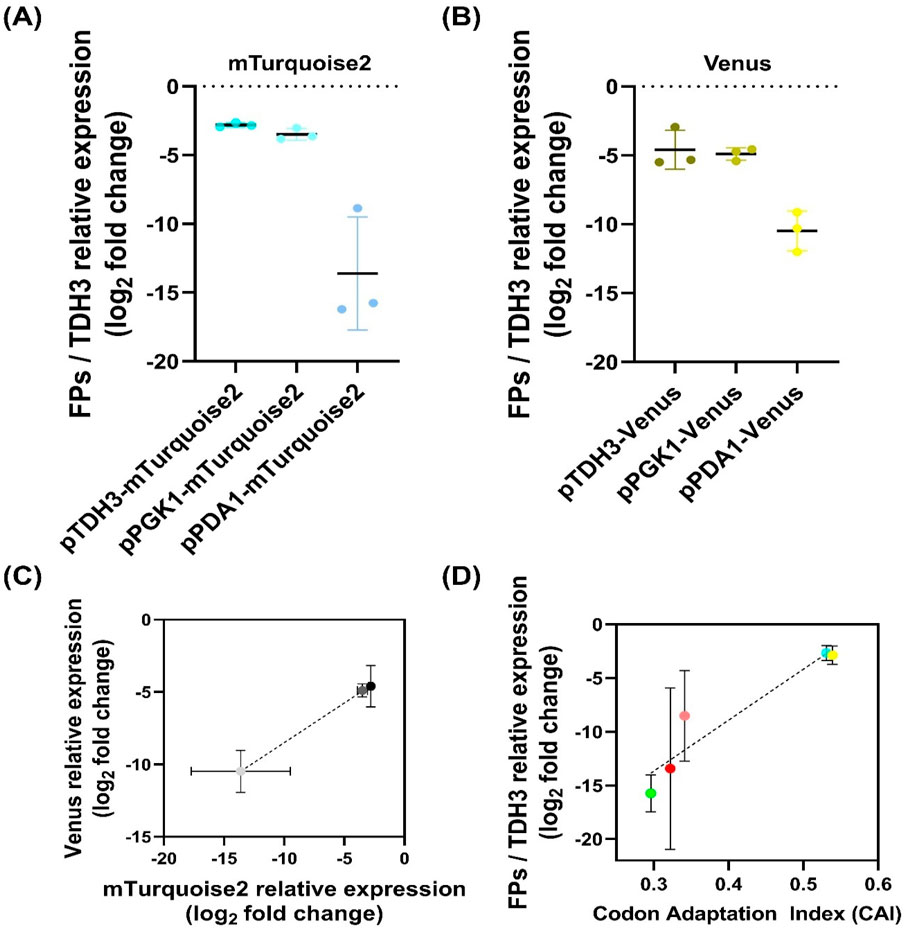
Figure 4. Promoter-dependent transcriptional expression and correlation analysis in Wickerhamomyces ciferrii (A, B) Promoter-dependent transcriptional expression of mTurquoise2 (A) and Venus (B) in Wickerhamomyces ciferrii. Expression levels were quantified by qPCR, normalized to TDH3, and presented as log2 fold change values. Data represent biological triplicates. (C) Correlation between promoter strength and transcriptional expression. The log2 fold change values of mTurquoise2 and Venus from (A, B) were plotted and analyzed by linear regression, confirming that promoter strength is independent of the downstream coding sequence (D) Correlation between transcriptional expression and codon adaptation index (CAI). The relative transcriptional expression levels of five fluorescent proteins were plotted against their CAI values and analyzed by linear regression to assess the influence of codon usage on gene expression.
Expression analysis confirmed that pTDH3 drove the highest transcriptional levels, followed by pPGK1, while pPDA1 exhibited significantly lower transcriptional expression with higher colony-to-colony variation (Figures 4A,B). For mTurquoise2, pTDH3 expression was 1.6-fold higher than pPGK1 and 1,791-fold higher than pPDA1, while pPGK1 expression was 1,107-fold higher than pPDA1. Similar trends were observed for Venus, where pTDH3 was 1.2-fold higher than pPGK1 and 59.4-fold higher than pPDA1, while pPGK1 was 48.1-fold higher than pPDA1 (Figures 4A,B). To confirm that expression differences were due to promoter strength rather than coding sequence variation, a correlation analysis between mTurquoise2 and Venus transcription levels was performed (Figure 4C). Linear regression analysis revealed a strong correlation (R2 = 0.8841, p = 0.0002), indicating that promoter strength was the primary determinant of transcript abundance.
Correlation between CAI values and transcriptional expression levels
To assess the impact of codon usage on transcript stability in W. ciferrii, we calculated codon adaptation index (CAI) values for the FPs and antibiotic resistance genes used in this study. The CAI values for the FPs were as follows: sfGFP (0.296), mTurquoise2 (0.531), Venus (0.539), mRuby2 (0.341), mKO (0.322), and mCherry (0.322). For antibiotic resistance genes, nourseothricin had a CAI of 0.463, zeocin 0.546, geneticin 0.293, and hygromycin B 0.140.
A positive correlation was observed between CAI values and transcriptional expression levels of FPs (Figure 4D). Consistent with qPCR results, mTurquoise2 and Venus exhibited the highest expression, while mRuby2 and mCherry showed moderate levels, and sfGFP displayed the lowest. Due to undetectable transcripts, mKO data were excluded.
Linear regression analysis confirmed a strong correlation between CAI values and FP/TDH3 expression (R2 = 0.6551, p = 0.0003), suggesting that codon optimization significantly influences transcript abundance. Notably, W. ciferrii exhibited resistance to nourseothricin, zeocin, and geneticin but failed to grow under hygromycin B selection. The low CAI value of the hygromycin B resistance gene (0.140) suggests that poor codon adaptation may contribute to its inefficacy in W. ciferrii.
Overexpression of ACC1 using the developed genetic toolbox in Wickerhamomyces ciferrii for enhanced TAPS production
To apply the developed genetic toolkit for improving tetraacetyl phytosphingosine (TAPS) production, we targeted ACC1, a key gene in fatty acid biosynthesis that may influence TAPS biosynthesis (Choi and Da Silva, 2014). The endogenous ACC1 gene in W. ciferrii was identified via BLASTp using R. norvegicus ACC1 as a query, revealing 46.5% overall amino acid identity. Conserved domain analysis confirmed the presence of essential catalytic regions, including the biotin carboxylation, biotin-binding, and carboxyltransferase domains, aligning with known ACC1 orthologs (Blum et al., 2025) (Supplementary Data S1). Since ACC1 activity is regulated by phosphorylation, we examined conserved inhibitory sites in Rattus norvegicus ACC1 (Davies et al., 1990; Ha et al., 1994) and identified two functionally conserved serine residues (S26 and S1161) in W. ciferrii. To generate a constitutively active variant, we introduced S26 A and S1161 A mutations via site-directed mutagenesis, producing ACC1S26A-S1161A.
This mutant was cloned into a 2μ based plasmid carrying a nourseothricin resistance marker under either the strong pTDH3 promoter or the weak pPDA1 promoter, generating p2μ_pTDH3-ACC1(mut)_NTC and p2μ_pPDA1-ACC1(mut)_NTC. The resulting plasmids were transformed into W. ciferrii, generating three recombinant strains: W. c_2μ-ACC1m-str (with pTDH3), W. c_2μ-ACC1m-weak (with pPDA1), and W. c_2μ-ev (empty vector control) (Figure 5A). These strains were cultured in YMglSCN medium at 25°C and 250 rpm for 96 h to assess growth and TAPS production.
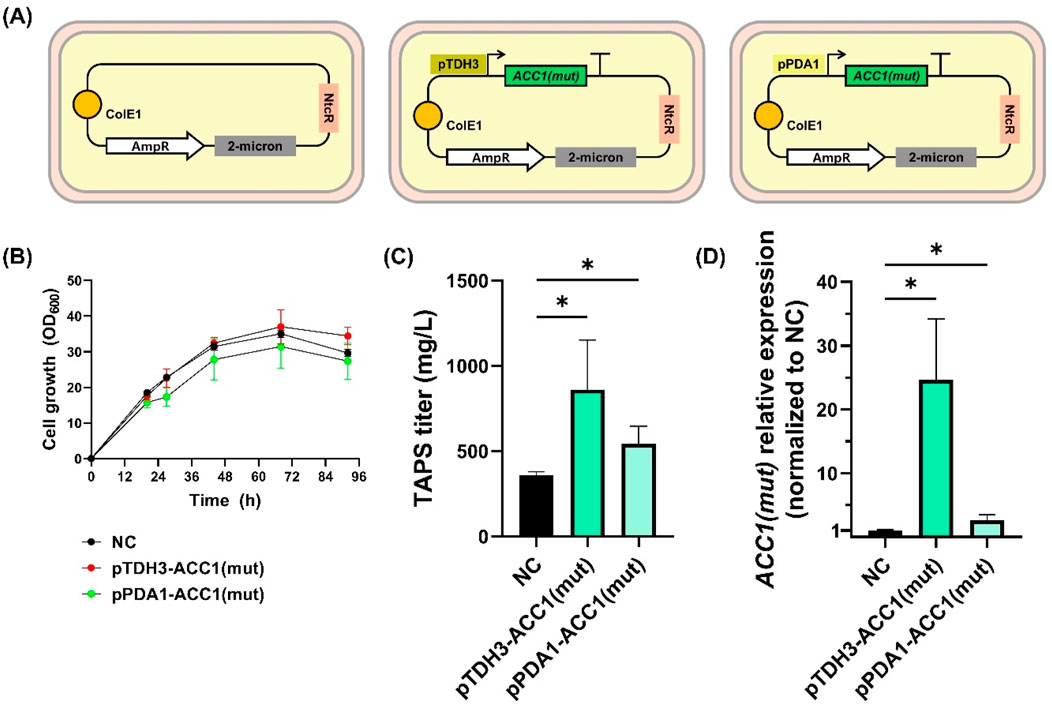
Figure 5. Growth and TAPS production of Wickerhamomyces ciferrii expressing mutant ACC1S26A-S1161A (A) Schematic representation of plasmid constructs used in this study. Wickerhamomyces ciferrii strains were transformed with either an empty vector (W.c_2μ-ev), a plasmid expressing ACC1S26A-S1161A under the strong TDH3 promoter (W.c_2μ-ACC1m-str), or a plasmid expressing ACC1S26A-S1161A under the weak PDA1 promoter (W.c_2μ-ACC1m-weak). (B) Growth curves of the three strains cultured in YMglSCN medium at 25°C with shaking at 250 rpm. Optical density at 600 nm (OD600) was measured at regular intervals. (C) TAPS titers after 96 h of shake-flask cultivation (D) Relative transcriptional expression of mutant ACC1S26A-S1161A in each strain, normalized to TDH3 expression and subsequently to the empty vector control (W.c_2μ-ev). Statistical significance was determined using an unpaired t-test with Welch’s correction (p < 0.05). Error bars represent standard deviations from biological replicates.
Growth analysis revealed that W. c_2μ-ev strain reached an OD600 of 29.6, while W. c_2μ-ACC1m-str and W. c_2μ-ACC1m-weak reached 34.4 and 27.4, respectively (Figure 5B). Notably, overexpression of ACC1S26A-S1161A significantly enhanced TAPS production compared to the control (Figure 5C). Average TAPS titers were 359.0 ± 20.3 mg/L in W. c_2μ-ev, 860.8 ± 291.5 mg/L in W. c_2μ-ACC1m-str (2.4-fold increase, p < 0.05), and 542.4 ± 104.5 mg/L in W. c_2μ-ACC1m-weak (1.5-fold increase, p < 0.05). qPCR analysis confirmed that ACC1S26A-S1161A expression correlated with TAPS titers, with pTDH3-driven expression yielding a 24.6-fold increase, whereas pPDA1 resulted in a 2.6-fold increase, both significantly higher than the control (p < 0.05) (Figure 5D).
Overall, these results demonstrate that ACC1S26A-S1161A overexpression enhances TAPS production in W. ciferrii, with the strong pTDH3-driven expression yielding the highest titers. These findings validate the developed genetic tools for strain engineering and establish ACC1 engineering as a viable strategy for spingoid base biosynthetic pathway optimization.
Discussion
This study established a stable episomal plasmid system for W. ciferrii, making a significant advancement in enabling efficient genetic modifications in this non-model yeast. We demonstrated that S. cerevisiae-derived replication origins, 2μ and CEN6/ARS4, support plasmid replication and maintenance in W. ciferrii, with the 2μ origin yielding a higher copy number and lower colony-to-colony variation. These findings provide a versatile platform for genetic engineering applications, facilitating rapid strain development for metabolic engineering and synthetic biology.
Despite successful episomal plasmid maintenance, the measured 2μ plasmid copy number in W. ciferrii was significantly lower than in S. cerevisiae, averaging only 1–2 copies per cell. In S. cerevisiae, native 2μ plasmids are typically maintained at 40–80 copies per haploid cell, though copy numbers drop to 8–14 copies when carrying selective markers (Clark-Walker and Miklos, 1974; Futcher, 1986; Futcher and Cox, 1984; Gerbaud and Guérineau, 1980; Gnügge and Rudolf, 2017; Karim et al., 2013). This discrepancy is likely due to the absence of the Flp/FRT recombination system in W. ciferrii, which amplifies 2μ plasmid copy number in S. cerevisiae through site-specific recombination (Chan et al., 2013; Gronostajski and Sadowski, 1985). Without this amplification mechanism, W. ciferrii may rely solely on passive plasmid replication, leading to a lower plasmid copy number. Future studies could explore W. ciferrii-specific centromere sequences to develop native CEN-based replication systems for stable plasmid inheritance without selection pressure (Cao et al., 2017).
Our evaluation of selectable markers identified nourseothricin, zeocin, and geneticin as reliable selection agents, whereas hygromycin B failed to confer resistance. The failure is likely due to hygromycin B resistance gene’s low codon adaptation index (CAI) (0.140), suggesting inefficient expression in W. ciferrii. These findings underscore the importance of codon optimization when designing selection markers for non-model yeasts. Previous studies have demonstrated that codon optimization can drastically enhance gene expression in yeasts. For example, Kaishima et al. showed that codon-optimized GFP variants exhibited over 100-fold higher fluorescence in S. cerevisiae compared to non-optimized versions (Kaishima et al., 2016). Similarly, Gordon et al. reported that codon adaptation of selectable markers was essential for functional expression in non-model yeasts such as Metschnikowia borealis, where non-optimized versions failed to produce transformants (Gordon et al., 2019). These findings suggest that codon-optimized versions of fluorescent proteins and resistance markers may also yield improved performance in W. ciferrii.
Additionally, we observed that higher antibiotic concentrations were required for selection on solid media compared to liquid cultures, likely due to differences in antibiotic diffusion and agar binding, reducing bioavailability. Similar challenges in antibiotic efficacy have been observed in structured environments, where factors such as metabolic dormancy and limited diffusion reduce the effectiveness of antibiotics (Stokes et al., 2019). Furthermore, cell debris has been shown to bind and sequester antibiotics, further diminishing their activity in solid-phase conditions (Yuan et al., 2022). These findings underscore the importance of media-specific antibiotic optimization for robust selection in W. ciferrii genetic engineering.
Expanding the fluorescent protein (FP) toolkit for W. ciferrii, we validated the expression of mTurquoise2, Venus, mRuby2, and mCherry, in addition to the previously reported GFP. These tools enable multi-gene expression tracking, subcellular localization studies, and high-throughput screening (HTS). Fluorescent reporters facilitate pathway engineering by allowing real-time monitoring of enzyme expression and metabolic flux adjustments. Additionally, fluorescence-activated cell sorting (FACS) can enhance strain selection and single-cell heterogeneity analysis, improving metabolic pathway refinement. Interestingly, significant transcriptional variability was observed among different FPs expressed under the same promoter. While promoter strength is a key determinant of transcript abundance, codon adaptation showed a strong correlation with expression levels (R2 = 0.6551, p = 0.0003), suggesting that codon bias affects both mRNA stability and translational efficiency (Brule and Grayhack, 2017; Presnyak et al., 2015). This effect may be exacerbated by the low genomic G + C content (30.4%) of W. ciferrii, which influences mRNA folding, ribosome binding, and protein synthesis efficiency (Schneider et al., 2012). The failure of the hygromycin B resistance gene further supports this hypothesis, emphasizing the necessity of codon optimization for heterologous gene expression in W. ciferrii.
Overexpression of ACC1S26A−S1161A significantly increased TAPS production, confirming that acetyl-CoA availability is a key limiting factor in sphingoid base biosynthesis. However, despite higher ACC1 transcription levels compared to control strains, the increase in TAPS production was relatively modest between strong (pTDH3) and weak (pPDA1) promoter-driven strains, suggesting the presence of additional metabolic bottlenecks. Previous studies have shown that overexpression of LCB1, LCB2, and SYR2 enhances sphingoid base production (Schorsch et al., 2012), indicating that a combinatorial approach integrating ACC1 activation with pathway gene co-expression may be more effective. Future research should focus on optimizing metabolic flux balancing strategies to further enhance TAPS yields.
Despite successful episomal expression, colony-to-colony variation remained a challenge, with substantial heterogeneity in FP expression across individual cells. This variability likely results from differences in plasmid copy number or transcriptional stability, necessitating rigorous clone selection before downstream experiments. Genome integration may provide a more stable alternative, ensuring consistent gene expression (Zhao et al., 2020). The higher transformation efficiency further supports genome integration as a viable alternative for stable gene expression. Advances in CRISPR-based genome editing could enable precise targeted gene integration, reducing variability and improving genetic stability in engineered W. ciferrii strains (Cai et al., 2019; Raschmanová et al., 2018).
Beyond its use in TAPS biosynthesis, W. ciferrii and its genetic components also hold promise for broader synthetic biology applications. Notably, the W. ciferrii α-mating factor secretion signal has demonstrated superior efficiency compared to the S. cerevisiae α-factor when used in Komagataella phaffii, enhancing heterologous protein secretion (Zou et al., 2022). This enhanced secretion observed in K. phaffii suggests that W. ciferrii may possess an inherently strong secretory system. Accordingly, W. ciferrii itself could be engineered as a chassis for industrial protein production, particularly for lipid-associated or membrane proteins that may benefit from its native metabolic context. Moreover, the high-level production of TAPS implies a robust acetyl-CoA and fatty acid precursor pool. This metabolic capacity suggests potential for the biosynthesis of a broader range of lipid-derived compounds, including ceramides, long-chain fatty alcohols, and other hydrophobic molecules. The combination of strong lipid-handling capacity and the versatile genetic tools developed in this study positions W. ciferrii as a valuable yet underutilized host for industrial lipid biotechnology.
In conclusion, this study established a versatile genetic toolkit for W. ciferrii, including a robust episomal plasmid system, optimized selection markers, and an expanded set of functional fluorescent proteins. Our findings highlight the importance of codon adaptation in heterologous gene expression and validate ACC1S26A-S1161A overexpression as a strategy to enhance TAPS biosynthesis. Future efforts should focus on refining genome editing strategies using CRISPR-based targeted gene manipulation, optimizing metabolic pathways through combinatorial gene expression approaches, and enhancing episomal plasmid retention by leveraging native replication elements in W. ciferrii. Together, these advancements not only position W. ciferrii as a genetically accessible non-conventional yeast but also reinforce its industrial relevance as a platform for scalable bioproduction and cross-system synthetic biology applications.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
S-RL: Data curation, Formal Analysis, Visualization, Writing – original draft, Writing – review and editing. JS: Formal Analysis, Writing – original draft, Writing – review and editing. PL: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Research Foundation of Korea (NRF) (2022M3A9I3082366) and the Korea Institute of Marine Science and Technology Promotion (KIMST) funded by the Ministry of Oceans and Fisheries (RS-2022-KS221581).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2025.1586218/full#supplementary-material
References
Bae, J.-H., Sohn, J.-H., Park, C.-S., Rhee, J.-S., and Choi, E.-S. (2003). Integrative transformation system for the metabolic engineering of the sphingoid base-producing yeast Pichia ciferrii. Appl. Environ. Microbiol. 69, 812–819. doi:10.1128/AEM.69.2.812-819.2003
Bai, Q., Cheng, S., Zhang, J., Li, M., Cao, Y., and Yuan, Y. (2021). Establishment of genomic library technology mediated by non-homologous end joining mechanism in Yarrowia lipolytica. Sci. China Life Sci. 64, 2114–2128. doi:10.1007/s11427-020-1885-x
Baptista, M., and Domingues, L. (2022). Kluyveromyces marxianus as a microbial cell factory for lignocellulosic biomass valorisation. Biotechnol. Adv. 60, 108027. doi:10.1016/j.biotechadv.2022.108027
Barenholz, Y., Edelman, I., and Gatt, S. (1971). The metabolic basis for the accumulation of acetylated sphingosine bases in the yeast Hansenula ciferri. Biochim. Biophys. Acta Lipids Lipid Metab. 248, 458–465. doi:10.1016/0005-2760(71)90235-9
Blum, M., Andreeva, A., Florentino, L. C., Chuguransky, S. R., Grego, T., Hobbs, E., et al. (2025). InterPro: the protein sequence classification resource in 2025. Nucleic Acids Res. 53, D444–D456. doi:10.1093/nar/gkae1082
Börgel, D., Van Den Berg, M., Hüller, T., Andrea, H., Liebisch, G., Boles, E., et al. (2012). Metabolic engineering of the non-conventional yeast Pichia ciferrii for production of rare sphingoid bases. Metab. Eng. 14, 412–426. doi:10.1016/j.ymben.2012.03.003
Brule, C. E., and Grayhack, E. J. (2017). Synonymous codons: choose wisely for expression. Trends Genet. 33, 283–297. doi:10.1016/j.tig.2017.02.001
Cai, P., Gao, J., and Zhou, Y. (2019). CRISPR-mediated genome editing in non-conventional yeasts for biotechnological applications. Microb. Cell Factories 18, 63. doi:10.1186/s12934-019-1112-2
Camattari, A., Goh, A., Yip, L. Y., Tan, A. H. M., Ng, S. W., Tran, A., et al. (2016). Characterization of a panARS-based episomal vector in the methylotrophic yeast Pichia pastoris for recombinant protein production and synthetic biology applications. Microb. Cell Fact. 15, 139. doi:10.1186/s12934-016-0540-5
Cao, M., Gao, M., Lopez-Garcia, C. L., Wu, Y., Seetharam, A. S., Severin, A. J., et al. (2017). Centromeric DNA facilitates nonconventional yeast genetic engineering. ACS Synth. Biol. 6, 1545–1553. doi:10.1021/acssynbio.7b00046
Cao, M., Tran, V. G., Qin, J., Olson, A., Mishra, S., Schultz, J. C., et al. (2022). Metabolic engineering of oleaginous yeast Rhodotorula toruloides for overproduction of triacetic acid lactone. Biotechnol. Bioeng. 119, 2529–2540. doi:10.1002/bit.28159
Chan, K.-M., Liu, Y.-T., Ma, C.-H., Jayaram, M., and Sau, S. (2013). The 2 micron plasmid of Saccharomyces cerevisiae: a miniaturized selfish genome with optimized functional competence. Plasmid, Special Issue based Int. Soc. Plasmid Biol. Meet. Santander 2012 70, 2–17. doi:10.1016/j.plasmid.2013.03.001
Chen, Y., Xiao, W., Wang, Y., Liu, H., Li, X., and Yuan, Y. (2016). Lycopene overproduction in Saccharomyces cerevisiae through combining pathway engineering with host engineering. Microb. Cell Fact. 15, 113. doi:10.1186/s12934-016-0509-4
Choi, J. W., and Da Silva, N. A. (2014). Improving polyketide and fatty acid synthesis by engineering of the yeast acetyl-CoA carboxylase. J. Biotechnol. 187, 56–59. doi:10.1016/j.jbiotec.2014.07.430
Choi, J. Y., Hwang, H. J., Cho, W. Y., Choi, J., and Lee, P. C. (2021). Differences in the fatty acid profile, morphology, and tetraacetylphytosphingosine-forming capability between wild-type and mutant Wickerhamomyces ciferrii. Front. Bioeng. Biotechnol. 9, 662979. doi:10.3389/fbioe.2021.662979
Clark-Walker, G. D., and Miklos, G. L. (1974). Localization and quantification of circular DNA in yeast. Eur. J. Biochem. 41, 359–365. doi:10.1111/j.1432-1033.1974.tb03278.x
Davies, S. P., Sim, A. T. R., and Hardie, D. G. (1990). Location and function of three sites phosphorylated on rat acetyl-CoA carboxylase by the AMP-activated protein kinase. Eur. J. Biochem. 187, 183–190. doi:10.1111/j.1432-1033.1990.tb15293.x
Deparis, Q., Claes, A., Foulquié-Moreno, M. R., and Thevelein, J. M. (2017). Engineering tolerance to industrially relevant stress factors in yeast cell factories. FEMS Yeast Res. 17, fox036. doi:10.1093/femsyr/fox036
Duman-Özdamar, Z. E., and Binay, B. (2021). Production of industrial enzymes via Pichia pastoris as a cell factory in bioreactor: current status and future aspects. Protein J. 40, 367–376. doi:10.1007/s10930-021-09968-7
Dymond, J. S. (2013). “Preparation of genomic DNA from Saccharomyces cerevisiae,” in Methods in enzymology, laboratory methods in enzymology: DNA. Editor J. Lorsch (Academic Press), 153–160. doi:10.1016/B978-0-12-418687-3.00012-4
Eun, S. W., and Lee, P. C. (2023). Investigation of the effects of culture conditions on cell growth and tetraacetylphytosphingosine production by mutant Wickerhamomyces ciferrii. Process Biochem. 130, 203–210. doi:10.1016/j.procbio.2023.04.017
Fraczek, M. G., Naseeb, S., and Delneri, D. (2018). History of genome editing in yeast. Yeast 35, 361–368. doi:10.1002/yea.3308
Futcher, A. B. (1986). Copy number amplification of the 2 μm circle plasmid of Saccharomyces cerevisiae. J. Theor. Biol. 119, 197–204. doi:10.1016/S0022-5193(86)80074-1
Futcher, A. B., and Cox, B. S. (1984). Copy number and the stability of 2-micron circle-based artificial plasmids of Saccharomyces cerevisiae. J. Bacteriol. 157, 283–290. doi:10.1128/jb.157.1.283-290.1984
Gerbaud, C., and Guérineau, M. (1980). 2 μm plasmid copy number in different yeast strains and repartition of endogenous and 2 μm chimeric plasmids in transformed strains. Curr. Genet. 1, 219–228. doi:10.1007/BF00390947
Gnügge, R., and Rudolf, F. (2017). Saccharomyces cerevisiae Shuttle vectors. Yeast 34, 205–221. doi:10.1002/yea.3228
Gordon, Z. B., Soltysiak, M. P. M., Leichthammer, C., Therrien, J. A., Meaney, R. S., Lauzon, C., et al. (2019). Development of a transformation method for Metschnikowia borealis and other CUG-serine yeasts. Genes (Basel) 10, 78. doi:10.3390/genes10020078
Gronostajski, R. M., and Sadowski, P. D. (1985). The FLP recombinase of the Saccharomyces cerevisiae 2 microns plasmid attaches covalently to DNA via a phosphotyrosyl linkage. Mol. Cell Biol. 5, 3274–3279. doi:10.1128/mcb.5.11.3274
Ha, J., Daniel, S., Broyles, S. S., and Kim, K. H. (1994). Critical phosphorylation sites for acetyl-CoA carboxylase activity. J. Biol. Chem. 269, 22162–22168. doi:10.1016/S0021-9258(17)31770-2
Kaishima, M., Ishii, J., Matsuno, T., Fukuda, N., and Kondo, A. (2016). Expression of varied GFPs in Saccharomyces cerevisiae: codon optimization yields stronger than expected expression and fluorescence intensity. Sci. Rep. 6, 35932. doi:10.1038/srep35932
Karim, A. S., Curran, K. A., and Alper, H. S. (2013). Characterization of plasmid burden and copy number in Saccharomyces cerevisiae for optimization of metabolic engineering applications. FEMS Yeast Res. 13, 107–116. doi:10.1111/1567-1364.12016
Lacerda, M. P., Oh, E. J., and Eckert, C. (2020). The model system Saccharomyces cerevisiae versus emerging non-model yeasts for the production of biofuels. Life (Basel) 10, 299. doi:10.3390/life10110299
Lee, M. E., DeLoache, W. C., Cervantes, B., and Dueber, J. E. (2015). A highly characterized yeast toolkit for modular, multipart assembly. ACS Synth. Biol. 4, 975–986. doi:10.1021/sb500366v
Liachko, I., and Dunham, M. J. (2014). An autonomously replicating sequence for use in a wide range of budding yeasts. FEMS Yeast Res. 14, 364–367. doi:10.1111/1567-1364.12123
Liang, Y., Gao, S., Qi, X., Valentovich, L. N., and An, Y. (2024). Progress in gene editing and metabolic regulation of Saccharomyces cerevisiae with CRISPR/Cas9 tools. ACS Synth. Biol. 13, 428–448. doi:10.1021/acssynbio.3c00685
Liu, D., Hwang, H. J., Otoupal, P. B., Geiselman, G. M., Kim, J., Pomraning, K. R., et al. (2023). Engineering Rhodosporidium toruloides for production of 3-hydroxypropionic acid from lignocellulosic hydrolysate. Metab. Eng. 78, 72–83. doi:10.1016/j.ymben.2023.05.001
Ma, Y., Liu, N., Greisen, P., Li, J., Qiao, K., Huang, S., et al. (2022). Removal of lycopene substrate inhibition enables high carotenoid productivity in Yarrowia lipolytica. Nat. Commun. 13, 572. doi:10.1038/s41467-022-28277-w
Manfrão-Netto, J. H. C., Gomes, A. M. V., and Parachin, N. S. (2019). Advances in using Hansenula polymorpha as chassis for recombinant protein production. Front. Bioeng. Biotechnol. 7, 94. doi:10.3389/fbioe.2019.00094
Mumberg, D., Müller, R., and Funk, M. (1995). Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156, 119–122. doi:10.1016/0378-1119(95)00037-7
Nakamura, Y., Gojobori, T., and Ikemura, T. (2000). Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Res. 28, 292. doi:10.1093/nar/28.1.292
Nielsen, J. (2013). Production of biopharmaceutical proteins by yeast: advances through metabolic engineering. Bioengineered 4, 207–211. doi:10.4161/bioe.22856
Park, Y.-K., and Ledesma-Amaro, R. (2023). What makes Yarrowia lipolytica well suited for industry? Trends Biotechnol. 41, 242–254. doi:10.1016/j.tibtech.2022.07.006
Patra, P., Das, M., Kundu, P., and Ghosh, A. (2021). Recent advances in systems and synthetic biology approaches for developing novel cell-factories in non-conventional yeasts. Biotechnol. Adv. 47, 107695. doi:10.1016/j.biotechadv.2021.107695
Peng, B., Williams, T. C., Henry, M., Nielsen, L. K., and Vickers, C. E. (2015). Controlling heterologous gene expression in yeast cell factories on different carbon substrates and across the diauxic shift: a comparison of yeast promoter activities. Microb. Cell Fact. 14, 91. doi:10.1186/s12934-015-0278-5
Pérez-Rivero, C., and López-Gómez, J. P. (2023). Unlocking the potential of fermentation in cosmetics: a review. Fermentation 9, 463. doi:10.3390/fermentation9050463
Ploessl, D., Zhao, Y., and Shao, Z. (2023). Engineering of non-model eukaryotes for bioenergy and biochemical production. Curr. Opin. Biotechnol. 79, 102869. doi:10.1016/j.copbio.2022.102869
Presnyak, V., Alhusaini, N., Chen, Y.-H., Martin, S., Morris, N., Kline, N., et al. (2015). Codon optimality is a major determinant of mRNA stability. Cell 160, 1111–1124. doi:10.1016/j.cell.2015.02.029
Puigbò, P., Bravo, I. G., and Garcia-Vallve, S. (2008). CAIcal: a combined set of tools to assess codon usage adaptation. Biol. Direct 3, 38. doi:10.1186/1745-6150-3-38
Qiao, K., Wasylenko, T. M., Zhou, K., Xu, P., and Stephanopoulos, G. (2017). Lipid production in Yarrowia lipolytica is maximized by engineering cytosolic redox metabolism. Nat. Biotechnol. 35, 173–177. doi:10.1038/nbt.3763
Raschmanová, H., Weninger, A., Glieder, A., Kovar, K., and Vogl, T. (2018). Implementing CRISPR-Cas technologies in conventional and non-conventional yeasts: current state and future prospects. Biotechnol. Adv. 36, 641–665. doi:10.1016/j.biotechadv.2018.01.006
Redden, H., and Alper, H. S. (2015). The development and characterization of synthetic minimal yeast promoters. Nat. Commun. 6, 7810. doi:10.1038/ncomms8810
Schneider, J., Andrea, H., Blom, J., Jaenicke, S., Rückert, C., Schorsch, C., et al. (2012). Draft genome sequence of Wickerhamomyces ciferrii NRRL Y-1031 F-60-10. Eukaryot. Cell 11, 1582–1583. doi:10.1128/EC.00258-12
Schorsch, C., Köhler, T., Andrea, H., and Boles, E. (2012). High-level production of tetraacetyl phytosphingosine (TAPS) by combined genetic engineering of sphingoid base biosynthesis and L-serine availability in the non-conventional yeast Pichia ciferrii. Metab. Eng. 14, 172–184. doi:10.1016/j.ymben.2011.12.002
Schorsch, C., Köhler, T., and Boles, E. (2009). Knockout of the DNA ligase IV homolog gene in the sphingoid base producing yeast Pichia ciferrii significantly increases gene targeting efficiency. Curr. Genet. 55, 381–389. doi:10.1007/s00294-009-0252-z
Singh, R., Chandel, S., Ghosh, A., Gautam, A., Huson, D. H., Ravichandiran, V., et al. (2022). Easy efficient HDR-based targeted knock-in in Saccharomyces cerevisiae genome using CRISPR-Cas9 system. Bioengineered 13, 14857–14871. doi:10.1080/21655979.2022.2162667
Stokes, J. M., Lopatkin, A. J., Lobritz, M. A., and Collins, J. J. (2019). Bacterial metabolism and antibiotic efficacy. Cell Metab. 30, 251–259. doi:10.1016/j.cmet.2019.06.009
Sun, W., Vila-Santa, A., Liu, N., Prozorov, T., Xie, D., Faria, N. T., et al. (2020). Metabolic engineering of an acid-tolerant yeast strain Pichia kudriavzevii for itaconic acid production. Metab. Eng. Commun. 10, e00124. doi:10.1016/j.mec.2020.e00124
Tran, V. G., Cao, M., Fatma, Z., Song, X., and Zhao, H. (2019). Development of a CRISPR/Cas9-Based tool for gene deletion in Issatchenkia orientalis. mSphere 4, 1–11. doi:10.1128/mSphere.00345-19
Utomo, J. C., Hodgins, C. L., and Ro, D.-K. (2021). Multiplex genome editing in yeast by CRISPR/cas9 – a potent and agile tool to reconstruct complex metabolic pathways. Front. Plant Sci. 12, 719148. doi:10.3389/fpls.2021.719148
Walker, G. M., and Basso, T. O. (2020). Mitigating stress in industrial yeasts. Fungal Biol. Fungal Adapt. Stress 124, 387–397. doi:10.1016/j.funbio.2019.10.010
Wickerham, L. J., and Stodola, F. H. (1960). Formation of extracellular sphingolipides by microorganisms: I. Tetraacetylphytosphingosine from Hansenula ciferri. J. Bacteriol. 80, 484–491. doi:10.1128/jb.80.4.484-491.1960
Wronska, A. K., Haak, M. P., Geraats, E., Bruins Slot, E., van den Broek, M., Pronk, J. T., et al. (2020). Exploiting the diversity of saccharomycotina yeasts to engineer biotin-independent growth of Saccharomyces cerevisiae. Appl. Environ. Microbiol. 86, 002700–e320. doi:10.1128/AEM.00270-20
Xiong, L., Zeng, Y., Tang, R.-Q., Alper, H. S., Bai, F.-W., and Zhao, X.-Q. (2018). Condition-specific promoter activities in Saccharomyces cerevisiae. Microb. Cell Factories 17, 58. doi:10.1186/s12934-018-0899-6
Yoo, S. W., Kim, M., Park, B.-S., Yoon, J., and Oh, M.-K. (2023). Wickerhamomyces ciferrii auxotroph and expression vector for improved production of tetraacetyl phytosphingosine. Biotechnol. Bioproc E 28, 804–812. doi:10.1007/s12257-023-0128-y
Yuan, Q., Yu, P., Cheng, Y., Zuo, P., Xu, Y., Cui, Y., et al. (2022). Chlorination (but not UV disinfection) generates cell debris that increases extracellular antibiotic resistance gene transfer via proximal adsorption to recipients and upregulated transformation genes. Environ. Sci. Technol. 56, 17166–17176. doi:10.1021/acs.est.2c06158
Zhao, Y., Yao, Z., Ploessl, D., Ghosh, S., Monti, M., Schindler, D., et al. (2020). Leveraging the hermes transposon to accelerate the development of nonconventional yeast-based microbial cell factories. ACS Synth. Biol. 9, 1736–1752. doi:10.1021/acssynbio.0c00123
Zhou, T., Park, Y.-K., Fu, J., Hapeta, P., Klemm, C., and Ledesma-Amaro, R. (2025). Metabolic engineering of Yarrowia lipolytica for the production and secretion of the saffron ingredient crocetin. Biotechnol. Biofuels Bioprod. 18, 1. doi:10.1186/s13068-024-02598-y
Keywords: genetic toolkit, non-model yeast, Wickerhamomyces ciferrii, episomal plasmid system, tetraacetyl phytosphingosine
Citation: Lee S-R, Kang JS and Lee PC (2025) A versatile genetic toolkit for engineering Wickerhamomyces ciferrii for tetraacetyl phytosphingosine production. Front. Bioeng. Biotechnol. 13:1586218. doi: 10.3389/fbioe.2025.1586218
Received: 02 March 2025; Accepted: 16 April 2025;
Published: 28 April 2025.
Edited by:
Jung-Kul Lee, Konkuk University, Republic of KoreaReviewed by:
Soon Ho Hong, University of Ulsan, Republic of KoreaJingqi Chen, University of Illinois at Urbana–Champaign, United States
Se Jong Han, Korea Polar Research Institute, Republic of Korea
Copyright © 2025 Lee, Kang and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pyung Cheon Lee, cGNsZWVAYWpvdS5hYy5rcg==