 Konstantin Kochergin-Nikitsky
Konstantin Kochergin-Nikitsky Alexander Lavrov
Alexander Lavrov Svetlana Smirnikhina
Svetlana Smirnikhina- Laboratory of Genome Editing, Research Centre for Medical Genetics, Moscow, Russia
Detailed mapping of viral vector integration sites, including retroviral and particularly lentiviral vectors, is critical for assessing their safety in preclinical and clinical studies. Although integration into the host genome follows certain virus-specific patterns, it remains a stochastic event and can cause insertional mutagenesis with diverse consequences, such as oncogene activation. In this review, we trace the evolution of ISA methods applied to retroviruses and derived vectors, from early labor-intensive approaches with limited coverage—such as combined strategies involving restriction analysis, Southern blotting, and subcloning—to modern high-throughput strategies. We discuss key methodologies that shaped the field, including inverse PCR (iPCR), ligation-mediated PCR (LM-PCR), and linear amplification-mediated PCR (LAM-PCR), highlighting their contributions to more comprehensive and unbiased mapping, along with limitations associated with systematic errors stemming from dependence on restriction endonuclease digestion and amplification biases. We also examine recent approaches designed to overcome these limitations, independent of PCR and restriction analysis, which enable a more accurate and undistorted representation of retroviral vector integration profiles. Despite the emergence of new techniques, classical methods—particularly LAM-PCR and its modifications, such as nrLAM-PCR—remain widely used and continue to serve as the standard in many commercial platforms.
1 Introduction
Personalized medicine is a key field in the development of modern medicine. In turn, controlled modification of the genetic context provides the basis for many of its approaches. While delivering transgenes into cell lines using plasmids and methods such as lipofection or electroporation poses no major challenge, recombinant viral vectors (VVs) are valuable tools for delivering genetic constructs into cells of multicellular organisms due to their evolutionarily developed adaptations facilitating infection of specific cells and tissues, including viral tropism and host cell entry mechanisms. Among different types of recombinant VVs, retroviral VVs have the intrinsic ability to maintain prolonged expression of exogenous genetic agents, integrating the viral genome into the genome of the host cell (proviruses). At the same time, the aforementioned capability is obviously potentially dangerous. For retroviruses, including lentiviruses, integration, while showing some preference for certain types of genomic regions, is not strictly targeted. It is considered that lentiviruses tend to prefer intragenic loci, whereas other commonly used retroviral vectors show a preference for 5′ transcriptional regulatory regions (Vijaya et al., 1986; Scherdin et al., 1990; Schröder et al., 2002; Hanai et al., 2004; Kok et al., 2024; Daniel and Smith, 2008; Yoder et al., 2021; Shao et al., 2022). Although gene knockout caused by lentivector insertion is usually not the goal of the manipulation, it can be less detrimental than the occasional random activation of oncogenes resulting from retroviral vector integration in regulatory regions (Corcoran et al., 1984; Hacein-Bey-Abina et al., 2003a; Ye et al., 2023). This observed tendency makes integrating lentiviral vectors (LVVs) more acceptable for drug development without the need for exhaustive genome sequencing of every transduced cell, while not alleviating the necessity of stringent control measures. Notably, early SCID-X1 gene therapy trials using γ-retroviral vectors aimed to restore functional T cells by ex vivo transduction of hematopoietic stem cells, followed by autologous transplantation, which led to LMO2-associated clonal T-cell proliferation in several patients. This revealed the severe oncogenic risks of untargeted retroviral integrations. Detailed analysis of these cases showed that the insertional activation of proto-oncogenes could drive clonal dominance even when only a small fraction of transduced cells were affected. This critical observation directly motivated the development of comprehensive, unbiased integration-site analysis (ISA) methods, enabling systematic monitoring of clonal expansion and identification of potentially oncogenic integration events, which is considered essential for all types of integrating vectors, particularly in preclinical studies, and in the case of retrovirus-based vectors, it often requires genome-wide analysis (Hacein-Bey-Abina et al., 2003a; Hacein-Bey-Abina et al., 2008). In most cases, the seemingly straightforward approach to IS identification—whole-genome sequencing (WGS) of transduced cells—proves impractical as achieving the required sensitivity when DNA from pooled cell samples is being sequenced demands extremely high coverage (by WGS standards), making the analysis prohibitively expensive and laborious. Moreover, in most use-case scenarios involving integrating vectors—whether during in vivo administration or ex vivo cell modification followed by autologous transplantation—sequencing the entire population of transduced cells would imply sacrificing the treated organism or the transplant itself. Therefore, at the preclinical stage, methods designed to ensure a maximally representative description of integration events are usually applied, focusing on the general profiling of integrations and the identification of their hot spots specific to the viral vector under investigation.
2 Early approaches to identification of insertions
Before the advent of automated sequencing and, later, WGS (late 1970s–1980s), the analysis of proviral integration relied mainly on restriction endonuclease analysis (REA) combined with Southern blot (SB), which enabled the detection of specific sequences up to several tens of kb and the estimation of the proviral load (Chan and Martin, 1980). Using probes in provirus-flanking regions, integrations could be mapped within preselected loci (Vijaya et al., 1986; Scherdin et al., 1990; Selten et al., 1984; King et al., 1985; Lavu and Reddy, 1986; Robinson and Gagnon, 1986; Isfort et al., 1987; Lobel et al., 1989; Ben-David et al., 1990), leading to the description of structural features such as LTR redundancy (Hughes et al., 1978; Varmus et al., 1981) and the identification of oncogenic hotspots such as INT1/WNT1 (Nusse and Varmus, 1982). Molecular cloning and local sequencing of host–virus junctions (Lobel et al., 1989; van Ooyen and Nusse, 1984; Frankel et al., 1985; Shih et al., 1984; Wolf and Rotter, 1984; Swift et al., 1987; van Lohuizen et al., 1989; Clurman and Hayward, 1989) increased the resolution, revealing both dispersed and recurrent insertions, including those activating c-Myc (Corcoran et al., 1984) or disrupting B2M, Mov-13, and INT1 (van Ooyen and Nusse, 1984; Frankel et al., 1985; Harbers et al., 1984).
In the 1990s, the methodological toolkit expanded with S1-mapping, shuttle vector systems (Cepko et al., 1984), and hybridization approaches such as IS-phage libraries, which demonstrated RSV preference for AT-rich, DNase I-hypersensitive regions (Shih et al., 1988). Genetic strategies (backcrosses and co-segregation tracking) identified new common integration sites (EVI-5 and NOTCH1) (Tsichlis et al., 1990; Buchberg et al., 1999; Liao et al., 1995; Girard et al., 1996; Tam et al., 1997; Blaydes et al., 2001), while expression analyses (Northern blot and transcript sequencing) confirmed the functional impact of insertions, e.g., in NOTCH1 (Lee et al., 1999; Yanagawa et al., 2000), and the tendency of M-MuLV to integrate into actively transcribed regions (Mooslehner et al., 1990). Dideoxy sequencing of distorted DNA regions provided early evidence of integration bias toward curved DNA (Pryciak and Varmus, 1992; Müller and Varmus, 1994; Pruss et al., 1994), signaling the gradual methodological shift of the 1990s toward PCR-based and more systematic sequencing approaches (Pryciak and Varmus, 1992; Müller and Varmus, 1994; Pruss et al., 1994; Benkel et al., 1992; Gong et al., 1998).
By the early 2000s, classical methods were increasingly becoming insufficient and lacked sensitivity and throughput, particularly for polyclonal samples or large-scale safety assessment of vector integrations (Tsichlis et al., 1990; Buchberg et al., 1999; Liao et al., 1995).
3 PCR-based recovery of non-predicted IS with downstream sequencing
Non-targeted strategies for isolating proviruses together with their flanking host sequences, enabling unbiased mapping of integration sites and subsequent sequencing, began to emerge in the 1990s, reflecting a methodological shift driven by the need for more comprehensive and detailed analyses of retroviral integrations. Various PCR-based techniques were utilized, such as inverse PCR (iPCR), ligation-mediated PCR (LM-PCR), splinkerette-PCR, vectorette-PCR, LAM-PCR, and nrLAM-PCR
IPCR. This PCR variation was developed as a “genome-walking” approach, allowing investigation of the genome “terra incognita” starting from regions with known sequences. It was presumably invented independently and simultaneously by several groups. Triglia et al. (1988) and Ochman et al. (1988) proposed essentially the same method, enabling amplification of sequences “that lie outside the boundaries of known sequences” (c), named by the latter as “inverse” PCR. One year later, Silver and Keerikatte (1989) proposed a similar approach, allowing the isolation of provirus-adjacent regions and having the potential to significantly accelerate the analysis of proviral integration sites.
The method is based on a simple yet practical idea. Standard PCR requires known flanking regions for primers to anneal. If there is a known sequence (provirus in the context of ISA) surrounded by uncharted DNA, this known island can be turned “inside out” so that the unknown flanking sequences (or sequence, if the second cut site is within the provirus) become flanked by parts of the known region, enabling simple two-primer exponential PCR. To do this, the unknown sequences are cut at some distance and circularized using DNA-ligase. Then, primers placed in the known region with their 5′ ends facing each other and 3′ ends directed into the terra incognita allow conventional PCR on the circular template (Figure 1A). To improve the efficiency of such PCR, it is also recommended to cut this circular DNA somewhere between the 5′ ends of primers. In the absence of sequencing methods distinguishing individual reads, the natural approach was to subclone single amplicons and perform Sanger sequencing on each clone, thereby obtaining two deciphered flanks for each known cutout isle. A notable complication (and prolongation) in using this method arises from the fact that achieving efficient monomeric ligation rather than concatemers requires careful empirical adjustment of fragment concentrations (despite the theoretical rationale described) and is complicated by the widely varying fragment sizes (Green and Sambrook, 2019).
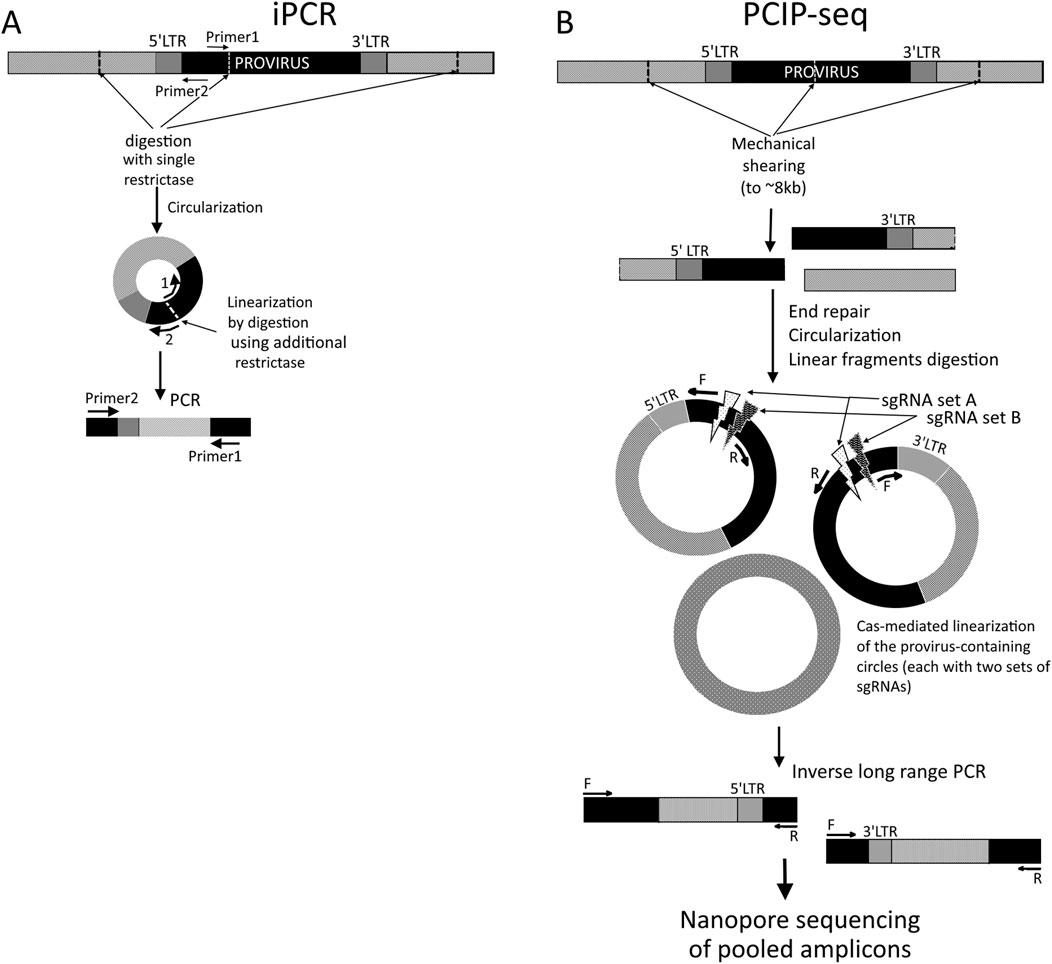
Figure 1. Schematic representation of the iPCR (A) and PCIP-seq (B) methods. Black boxes represent known sequences (proviral), while light-gray boxes represent genomic DNA. (A) 5′ LTR-containing circles are shown. Optional linearization is performed using another restriction enzyme with a cut site between 5′ ends of primers.
Being less time- and labor-intensive, more sensitive, and allowing far more comprehensive ISA than earlier locus-limited methods, iPCR was widely applied in the late 1990s and 2000s to study clonal properties of host cells infected with integrating viruses such as HTLV-1, STLV-1, HIV-1, MMLV, and even HBV or mobile elements, enabling the detection of clones at <1% frequency (Hanai et al., 2004; Takemoto et al., 1994; Cavrois et al., 1995; Ohshima et al., 1997; Kikuchi et al., 1997; Gabet et al., 2003; d’Offay et al., 2013). Precise mapping of insertion sites (ISs) revealed non-random integration patterns influenced by chromatin, gene proximity, or sequence motifs, with biases such as avoidance of centromeric repeats (HIV-1), targeting of proto-oncogenes (MMLV and HTLV-1), or conserved insertion hot spots (IncJ in E. coli), supporting adaptive or context-dependent integration mechanisms (Hanai et al., 2004; Cartea et al., 1998; Li et al., 1999; Mack et al., 2003; McGrath and Pembroke, 2004; Zhu et al., 2012).
3.1 LM-PCR/LAM-PCR/nrLAM-PCR
3.1.1 LM-PCR
A real breakthrough came with the LM-PCR method, introduced in the last 1980s to early 1990s (Pfeifer et al., 1989; Garrity and Wold, 2025; Mueller and Wold, 2025), which became widely adopted by the early 2000s as a core technique for mapping proviruses. In the context of mapping proviruses, this approach involves “blind” ligation of short adapters to restriction-digested host genomic DNA, including fragments containing viral insertions. PCR primers are typically designed so that one anneals to the viral sequences and the other anneals to the ligated adapters, enabling simultaneous amplification of many distinct DNA fragments in a single reaction with a common primer pair or two pairs in the nested PCR variant. This allows potential amplification of a large number of fragments. Without additional cloning, this method is suitable for creating libraries for high-throughput sequencing (next-generation sequencing, NGS) as it allows the observation of individual reads. However, NGS was not yet available, and researchers typically relied on automated Sanger capillary sequencers, which limited the method’s capacity and required cloning of amplicons to separate products amplified from different loci.
The LM-PCR-based ISA approach enabled, in one of the early foundational studies by Schröder et al. (2002), the detailed characterization of HIV-1 and recombinant viral vector integration specificity, resulting in 524 mapped integration sites in SupT1 cells and 111 in vitro control sites, revealing a preferential integration into active gene bodies. Subsequently, similar approaches were applied in clinical gene-marking studies (Wehnert et al., 2004), which revealed the leukemogenic potential of SIN vectors with strong enhancers integrating near critical targets such as EVI1 or PRDM16 (Correa de Freitas et al., 2014) and others. Since the mid-2000s, some NGS techniques, starting with pyrosequencing, began to be incorporated into LM-PCR-based ISA pipelines, allowing the avoidance of amplicon subcloning and significantly increasing the throughput of ISA (Hacein-Bey-Abina et al., 2008; Biffi et al., 2011; Dawes et al., 2020). For example, recently, Welles et al. (2022) showed ongoing clonal expansion of infected cells despite antiretroviral therapy (ART) (Rozera et al., 2022).
With the implementation of NGS techniques, restriction endonuclease digestion (RED) in some ISA protocols has been replaced by random DNA shearing via sonication. This alleviated RED-dependent limitations such as limited restriction site availability, causing incomplete or biased coverage, site-dependent blind spots where integrations distant from restriction sites remain undetected, and the overall loss of proviral insertions. Simultaneously, it simplified and expedited the workflow, rendering the methods more versatile, sensitive, and better suited for high-throughput applications (Hacein-Bey-Abina et al., 2008; Gillet et al., 2011; Berry et al., 2012; Serrao et al., 2016; Wells et al., 2020). A representative example is the approach used by Justice et al. (2015), who applied sonication-based fragmentation followed by adapter ligation and nested PCR with virus-specific and adapter-specific primers to selectively amplify virus–host junctions, preparing libraries for Illumina high-throughput sequencing, which enabled mapping of 32,050 unique ALV integration sites in vivo and the analysis of integration clusters and clonal expansion.
Non-perfect conversion of fragments processed in a single-primer reaction into the linker-ligated product should be kept in mind. There are mostly inevitable losses during the repair of DNA ends with incompatible overhangs, nicks, or missing phosphates (generated by sonication) and, in protocols such as Illumina library preparation, during A-tailing (Serrao et al., 2016). Nevertheless, an LM-PCR-based approach has gained widespread adoption. It has been applied, for example, to detect T-DNA insertions in the genome of Arabidopsis thaliana arising from agrobacterium-mediated transformation (O’Malley et al., 2007). Moreover, there are commercially available kits for IS identification, in which libraries are prepared using LM-PCR, such as the Retro-X Retrovirus Integration Site Analysis Kit, which is available online at https://www.takarabio.com/about (TaKaRa Bio, Japan).
3.1.2 Important LM-PCR derivatives
One recent LM-PCR modification is cassette-ligation PCR. Here, the conventional adapter/linker is replaced with a cassette of two partially complementary strands: a long strand (∼27 nt) serving as a primer template and a short strand (∼14 nt) with a 3′-end mismatch, preventing nonspecific priming. The 3′-ends of genomic fragments are blocked with ddCTP before ligation to reduce the background. This design provides more uniform and accurate amplification of integration sites. Zhang et al. (2020) combined this method with nanopore sequencing, using RED-based digestion with NcoI and BspHI (cutting throughout the provirus but not within LTRs), significantly reducing amplification bias compared to that with inverse PCR and classical LM-PCR, and enabling representative capture of polyclonal sites after T-cell gene therapy. Coupling with nanopore sequencing allows high-throughput, sensitive detection of thousands of unique integration sites with low background (Zhang et al., 2020).
Vectorette-PCR and splinkerette-PCR. Vectorette- and splinkerette-PCR are LM-PCR variants with modified adapters, originally developed for genome walking. Vectorette-PCR was developed in the early 1990s (Arnold and Hodgson, 1991), and splinkerette-PCR was developed around 1994 (Qureshi et al., 1994). Both use complex adapters, which are designed so that primers cannot anneal directly to the adapter itself, ensuring that amplification occurs only from newly synthesized strands containing viral and flanking genomic sequences. In vectorette adapters, a central non-complementary region reduces the background, while splinkerette adapters form a polymerase-blocking hairpin on one strand at all stages except at denaturation, due to a self-complementary region. In splinkerette-PCR, only one adapter strand can cause nonspecific priming, whereas in vectorette-PCR, both strands may still allow erroneous priming.
Apparently, due to the greater specificity and structural stability of the adapter, the splinkerette-PCR variant has largely replaced the vectorette-PCR over time. In particular, in the field of viral integration-site detection, splinkerette-PCR became relatively widespread in the 2010s (Dambrot et al., 2014; Uren et al., 2009; Yin, 2011; Šenigl et al., 2012; Shao and Lok, 2014; Jong et al., 2014; Šenigl et al., 2017; Potter and Luo, 2010; Lenvi et al., 2002; Lund et al., 2002), while the number of studies using vectorette-PCR remains limited (Cartea et al., 1998; Allen et al., 1994; Sfanos et al., 2011).
3.1.3 LAM-PCR
Schmidt et al. (2001) presented the first demonstrated modification of LM-PCR, which later evolved into the so-named LAM-PCR, specifically adapted for high-sensitivity mapping of retroviral integrations. They developed a method called EPTS/LM-PCR (magnetic extension primer tag selection/ligation-mediated PCR), which significantly expanded the capabilities for finding integration sites (Schmidt et al., 2001). The key modifications involved the inclusion of an IS surrounding-independent linear amplification stage and an enrichment stage—EPTS. A biotinylated virus-specific primer complementary to the LTR region was used for single-primer extension. The products, containing a biotin tag, could then be selectively captured by streptavidin-coated magnetic beads after the removal of unused primers, with the washing away of non-target genomic DNA and restriction digestion of the extension products. The approach provided high sensitivity, allowing the detection of proviral sequences at low copy numbers: down to one copy per 100–1,000 cells in complex biological samples. In this case, nucleotide sequences were obtained for individual identified amplicons via cycle sequencing. With minimal modification, the approach originally proposed by Schmidt et al. (2001) was adopted by other groups (Laufs et al., 2003; Varas et al., 2009).
The method and name LAM-PCR were first introduced by Schmidt M et al in 2003 and later in 2007 and 2009 (Woods et al., 2003; Schmidt et al., 2007; Schmidt et al., 2009) by building on earlier strategies to develop a highly sensitive technique for identifying proviral insertions in the host genome. Like earlier EPTS/LM-PCR, LAM-PCR involves restriction digestion, single-primer extension using a biotinylated primer, and ligation of a known adapter to unknown genomic DNA fragments flanking the provirus–host junctions. However, the sequence of steps in LAM-PCR differs. After multicycle LA with a biotinylated primer complementary to the viral sequence and solid-phase enrichment of target sequences on streptavidin-coated beads, the immobilized single-stranded DNA is enzymatically converted to double-stranded DNA. This double-stranded DNA is then restriction-digested, ligated to a double-stranded linker of known sequence, and subjected to nested PCR using primers that are specific to the viral region and the linker (Figure 2). The subsequent steps are similar to those of the standard LM-PCR approaches: either cloning into plasmids for Sanger sequencing or direct sequencing using NGS-based methods.
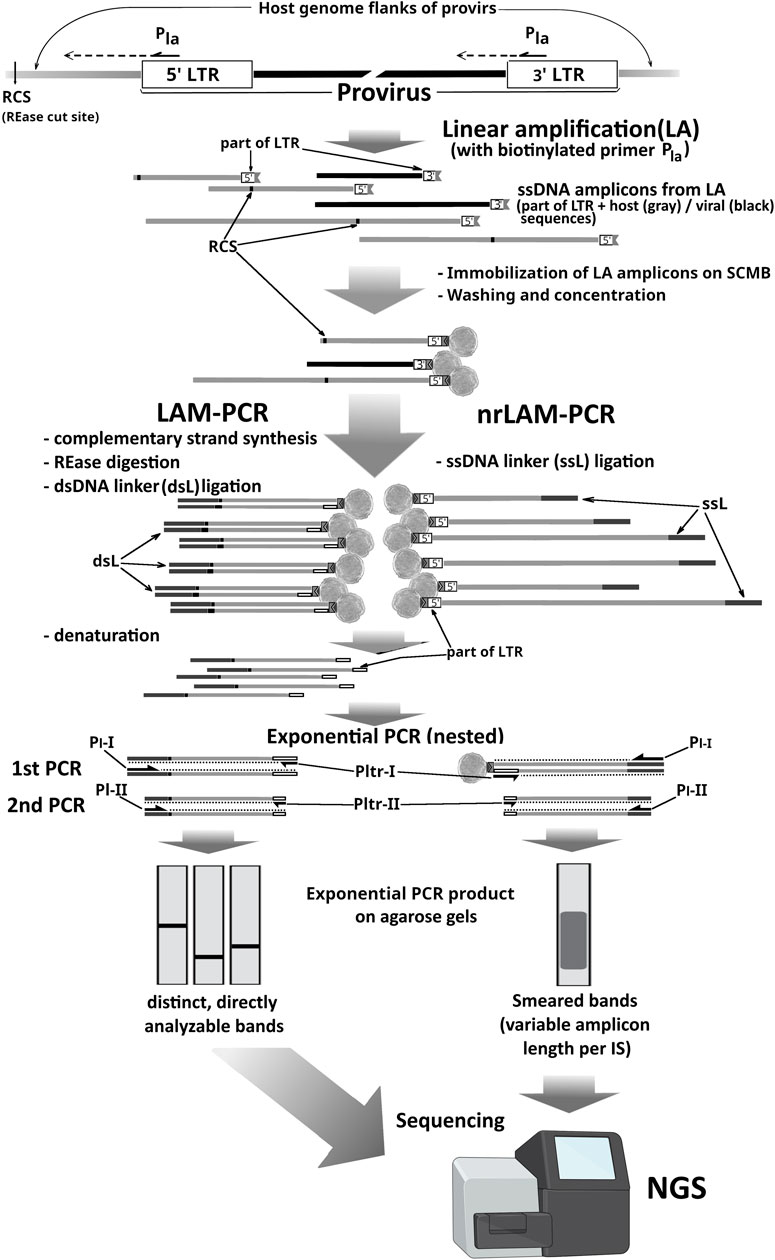
Figure 2. Comparison of LAM-PCR and nrLAM-PCR. LA, linear amplification reaction; Pla, biotinylated primer used in LA; RCS, digestion sites for the chosen REase; Pltr-I/II, LTR-complementary primers for exponential PCR (expPCR); Pl-I/II, linker-complementary expPCR primers; ssL/dsL, linker cassettes (dark gray lines); gray lines, host genome sequences; black lines, internal viral sequences; black-bordered white boxes, 3′LTR or 5′ LTR part of amplicons.
Since the early 2000s, the LAM-PCR method has been implemented in numerous studies without significant modifications. It has been widely applied to identify retroviral integration sites in various experimental and clinical contexts, including the transplantation of transduced hematopoietic stem cells (Gonzalez-Murillo et al., 2008; Hayakawa et al., 2009; Montini et al., 2009; Mitsuhashi et al., 2010; Rong et al., 2017). As a relevant method, LAM-PCR was subsequently adapted for use with high-throughput sequencing, starting with pyrosequencing and extending to modern platforms such as Illumina (Cattoglio et al., 2010; Cornils et al., 2013; Zhang et al., 2013). An improved pipeline for bioinformatical analysis of the NGS results, acquired using LAM-PCR, was proposed by Rosewick et al. (2020). This pipeline provided faster, more accurate, and reproducible identification of integration sites by replacing BLAST with Bowtie2 for read mapping, automating event filtering and clustering, and compensating for underrepresentation of GC-rich regions typically caused by PCR and NGS-related GC bias (Rosewick et al., 2020).
Despite its high sensitivity and specificity, LAM-PCR remains methodologically complex, requiring primer biotinylation, capture steps, and adapter ligation. Its performance depends on the genomic distribution of restriction sites, which limits its universality. These limitations prompted the development of new approaches that overcome the drawbacks of LAM-PCR and offer greater flexibility in application. As early as 2007, criticism of the LAM-PCR method was expressed by Harkey et al. (2007), who noted that standard LAM-PCR fails to detect 30%–40% of clones even after thorough analysis and that the relative abundance of specific clones in a mixture can be distorted by up to 60-fold, which severely limits quantitative assessment and reliable clonal tracking. Additional limitations included the labor-intensive nature of the method, the need for large sequence databases that are difficult to generate using the existing approach, and the dependence on restriction-site availability, which can suppress the detection of certain integration sites. As an alternative, the authors proposed a modification involving the use of multiple restriction enzymes to account for random clustering and systematic bias, along with an extra digestion step targeting the recognition sites within the provirus, followed by the removal of cleaved fragments to eliminate non-informative internal amplicons. According to the authors, these improvements increased the global detection capacity to over 90%, reduced systematic errors, and improved the accuracy of clonal quantification.
3.1.4 nrLAM-PCR
With the expanding use of recombinant retroviral vectors in gene therapy and, particularly, following reports of insertional activation of proto-oncogenes leading to clonal outgrowth and leukemogenesis in clinical trials (Hacein-Bey-Abina et al., 2003a; Hacein-Bey-Abina et al., 2003b; Ott et al., 2006; Howe et al., 2008), the need for comprehensive, genome-wide mapping of IS has gained significant demand. The identification of integration loci with high sensitivity and minimal bias is essential for understanding vector–host interactions and ensuring the safety of integrating vector systems.
By 2009–2010, all three widely used ISA PCR-based approaches were inverse PCR (iPCR), ligation-mediated PCR (LM-PCR), and linear amplification-mediated PCR (LAM-PCR), which shared a fundamental limitation determined by their RED-dependency for the extraction of the provirus–genome junctions. The positioning of some integration sites relative to the recognition sites of the selected restriction enzymes could be suboptimal; the recognition sites themselves are unevenly distributed throughout the genome (especially in heterochromatic regions), and some integration sites might be located in regions less accessible to restriction enzymes. All of this introduced a method-specific bias in the extraction of integration sites, leading to the underrepresentation of many integration events (Dawes et al., 2020; Schmidt et al., 2007; Harkey et al., 2007). Later, with the growing implementation of NGS, several of these methods were adapted to use mechanical DNA fragmentation—typically via sonication—as a more random and restriction-independent strategy; the inherent limitations of restriction-based fragmentation remained a concern during this transition period.
Thus, to address some of these concerns, the inventors of the LAM-PCR method proposed an improved alternative in 2009–2010: nonrestrictive linear amplification-mediated PCR (nrLAM-PCR) (an advancement over classical LAM-PCR) (Gabriel et al., 2009; Paruzynski et al., 2010), thus enabling genome-wide identification of IS without the need for restriction digestion. By removing the dependency on restriction enzymes, nrLAM-PCR directly addresses the site-specific bias of earlier methods, allowing a more uniform and representative recovery of integration sites across the genome. This improvement enhances the reliability of clonal analysis in gene-modified cells.
The nrLAM-PCR protocol starts from LA on genomic DNA with a single biotinylated primer annealing to the vector’s LTR. Amplification of the 3′ junction requires the primer to be positioned within the U5 region of the 3′ LTR, as close as possible to the host genome. Conversely, for the 5′ junction, it should be placed in the U3 region, since the LTR sequence—more than 600 nucleotides in the HIV-1 case—reduces the effective length of informative reads during sequencing. Similarly to LAM-PCR, biotinylated ssDNA products are enriched using streptavidin-coated beads. The key distinction lies in the unnecessary RED shearing of host DNA. Specific ssDNA-linkers (5′-phosphorylated and bearing a 3′-terminal dideoxycytidine) are ligated to the ssDNA products of linear amplification that serve as priming sites for the universal primer in the subsequent exponential PCR amplification (usually in nested variants) (Figure 2). Modification of the linkers prevents undesired ligation and ensures unidirectional attachment. The variable length of products from the LA stage prevents the resolution of individual junctions by AGE after PCR, so the sequencing is essential for clonality analysis, and an additional stage introducing platform-specific adapters is required (Wang et al., 2016). Another drawback of nrLAM-PCR-based ISA approaches is the lower sensitivity than LAM-PCR at low amounts of input DNA, while still being comparable to that of the LM-PCR-based approaches (Gabriel et al., 2009; Paruzynski et al., 2010; Wang et al., 2016; Paruzynski et al., 2012; Gabriel et al., 2014).
This method, proposed as a comprehensive methodological approach for highly sensitive and accurate analysis of the clonal repertoire of gene-corrected cells, including hematopoietic stem/progenitor cells (HSPCs) (Paruzynski et al., 2012; Giordano et al., 2015), has been in use since the 2010s. It was implemented for the ISA of integration events in transgenesis techniques using integrating vectors, such as piggyBac-based vectors (Marh et al., 2012), in studies of natural integrating viruses, including HIV-1 in the context of ART (Kok et al., 2024; Hauber et al., 2013; Einkauf et al., 2025), along with the safety assessment of retroviral (lenti- and γ-retroviral) vectors in autologous (Carbonaro et al., 2014; Zhang et al., 2022) or heterologous HSPC transplantation (Shi et al., 2014) in differentiating cell lines (Karumbayaram et al., 2012; Chin et al., 2016; Saito et al., 2020).
3.1.5 Approaches aiming to improve uniformity in PCR-based IS recovery
In addition to the above-mentioned nrLAM-PCR, the bias introduced by the use of RED-dependent DNA shearing in standard ISA methods such as iPCR, LM-PCR, and LAM-PCR, related to the uneven distribution of recognition sites across the host genome and variable cleavage efficiency and, thus, the incomplete recovery of integration events, has been addressed in different approaches. For example, s-EPTS/LM-PCR (van Haasteren et al., 2021) and MGS-PCR were proposed as mechanical host DNA shearing-adapted variations of EPTS/PCR and LAM-PCR methods, respectively. Both methods implied NGS sequencing. In MGS-PCR (Beard et al., 2014), the key differences are the replacement of the standard ligatable adapter with a modified splinkerette bearing a 3′-T overhang complementary to the A overhang of the APE-PCR product, which enhances specificity and reduces nonspecific amplification, and the use of nanosized magnetic beads instead of standard magnetic beads (Xu et al., 2013).
MIP-seq. In 2019, in a study focused on intact HIV-1 proviruses accumulating at distinct chromosomal positions during prolonged antiretroviral therapy, Einkauf et al. (2025) proposed a comprehensive approach called MIP-seq, enabling the simultaneous identification of integrated proviral sequences and precise IS localization from genetic material derived from as few as a single cell. The main innovation was the addition to widely used pipelines based on LM-PCR and nrLAM-PCR of whole-genome nonspecific amplification of all available DNA in samples diluted to approximately one IS per reaction (i.e., down to single cells in some cases) via multiple displacement amplification (MDA) with phi29 DNA polymerase, characterized by high processivity, 3′→5′ exonuclease proofreading activity, and strand displacement capability. MDA is an isothermal amplification method wherein phi29 polymerase synthesizes multiple new strands while simultaneously displacing previously synthesized fragments (strand displacement) from random hexamer primers, resulting in branched and exponential DNA amplification without denaturation cycles. MDA-amplified genetic material was then split into two workflows (Vijaya et al., 1986): amplification of near full-length (∼8–9 kb) HIV-1 proviral sequences using specific primers and (Scherdin et al., 1990) analysis of integration sites using LM-PCR, nrLAM-PCR, or ISLA. Since both reactions were performed from the same MDA product derived from a single viral copy, it was possible to reliably link each sequenced proviral genome to its chromosomal integration site—a task previously achievable only with more laborious and less scalable methods. The method also proved useful for detecting and confirming clonal origins of individual integrated proviruses.
Another approach relying on MDA to improve sensitivity is the individual proviral sequencing assay (IPSA), which was proposed in 2022 by Joseph et al. (2022) as a method aimed at simultaneously determining the near-full-length structure of individual HIV-1 proviruses and their precise chromosomal integration sites in patients receiving antiretroviral therapy. Its workflow starts from MDA of genomic DNA using human genome-biased random decamers at 40 °C with trehalose to suppress nonspecific amplification. MDA products are purified with magnetic SPRI beads and split into two branches. In the first, nested PCR amplifies nearly full-length proviruses from the gag leader to the 3′ LTR. In the second, integration sites are identified using an LM-PCR-like NGS-adapted strategy: MDA products are digested with four restriction enzymes, end-repaired, A-tailed, ligated to adapters containing nullomers, and subjected to nested PCR using primers specific to both the adapter and the 5′ LTR, selectively amplifying host–provirus junctions. Amplicons from both branches are sequenced on the Illumina MiSeq platform. A key step is limiting the dilution of genomic DNA to a concentration at which fewer than 30% of the reactions are PCR-positive, ensuring that most wells contain no more than one provirus and allowing unambiguous linking of its structure to a specific integration site.
The method demonstrated high specificity, with an average of 41% of wells yielding informative results, thus linking individual provirus structures to their genomic integration sites. Limitations include low MDA efficiency due to endpoint dilution (a large fraction of reactions remains empty), complications related to deletions in primer-binding sites, a limited 49 bp overlap between NFL and IS amplicons, and absence of the final 69 bp of the 3′ LTR. The workflow is labor-intensive, requires manual screening, and is poorly scalable. When multiple proviruses occur in a single reaction (10%–15% of cases), data may become ambiguous, causing chimeric assemblies and misinterpretations.
Among other methods aimed at improving ISA uniformity, FLEA-PCR (Pule et al., 2008) strongly resembles nrLAM-PCR, with the main difference lying in the stage of the so-called “anchor primer” attachment to the products of linear amplification, which, in this approach, could include either a sequence external to the provirus (when the LA primer anneals within the 5′ LTR) or a section of the provirus sequence (within the 3′ LTR). This anchor primer includes a predefined 5′ part (used as a target for universal primers in the downstream PCR) and a random 3′ part, which is able to anneal to the unknown 3′ ends of LA stage products to prime dsDNA synthesis by either DNA polymerase I Klenow fragment or T7 DNA polymerase.
Approximately 23% of the sequenced clones contain only internal retroviral sequences and have to be excluded from analysis. In NGS workflows, bioinformatics pipelines can automatically filter such reads. The use of anchored primers with randomized 3′ tails may generate multiple amplicons from the same integration site, complicating data interpretation and requiring careful PCR optimization. FLEA-PCR was not widely adopted but has been used in a few studies relevant to this review, typically with NGS and protocol modifications (Henssen et al., 2015; Xu et al., 2023).
Recently developed RAISING, introduced in 2022 by a Japanese research team (Wada et al., 2022), is also a conceptual advancement of the nrLAM-PCR method. Unlike nrLAM-PCR, RAISING eliminates adapter ligation and the use of biotinylated primers, significantly reducing the assay time to approximately 3.5 h and lowering costs. A key feature of the method is the use of polynucleotide tailing (polyAG-tailing) combined with thermomodulation, enabling selective hybridization of a complementary oligo-dT adapter without the need for high-temperature denaturation and with controlled temperature ramping during annealing. This reduces the amplification of non-target genomic DNA regions, thereby increasing specificity and sensitivity. RAISING can detect integrations at a proviral load as low as approximately 0.03%, which is five times more sensitive than its predecessor RAIS, which itself was reported to be approximately 100 times more sensitive than nrLAM-PCR. The method is suitable for analyzing both monoclonal and polyclonal integrations and is compatible with Sanger sequencing and high-throughput sequencing (NGS). For Sanger data analysis, a specialized R package, CLOVA, with a web interface, is used to assess the clonality of integrated fragments.
Limitations of the methods include sensitivity to the quality of input DNA, requiring an optimal number of ssDNA synthesis cycles (25 cycles), and the use of the highly specific Q5 polymerase to ensure uniform and specific double-strand synthesis. Additionally, the method requires a column-based ssDNA purification step, which may affect the overall efficiency. Occasionally, nonspecific products may appear, necessitating careful PCR optimization.
In recent years, the iPCR methodology has advanced through a novel comprehensive approach called pooled CRISPR inverse PCR sequencing (PCIP-seq). Artesi et al. (2021) proposed it for the simultaneous determination of integration sites, proviral sequences, and clonal composition of infected cells in a single protocol using Oxford Nanopore long reads. This approach helps overcome the limitations of traditional iPCR-based methods with short-read sequencing as PCIP-seq enables the simultaneous identification of integration sites and near-complete provirus sequencing, along with RED-dependent bias. Genomic DNA from infected cells is mechanically sheared to ∼8 kb, typically producing two fragments per provirus (on average): one containing the 5′ end and upstream host DNA and the other containing the 3′ end and downstream host DNA. Fragments are then circularized using T4 ligase, and linear DNA is removed by a plasmid-safe ATP-dependent procedure. Selective linear cleavage of circular molecules is performed with Cas9 guided by pools of sgRNAs targeting multiple sites near the 5′ and 3′ LTRs. Two sets of sgRNAs and corresponding primers are used, designed so that each provirus fragment resulting from shearing contains a binding site for only one sgRNA-binding locus and a primer pair from each duplicate set. Primers are oriented outward from the cleavage site toward unknown sequences, enabling the amplification of regions in iPCR, including flanking host DNA and part of the provirus (Figure 1B). The resulting amplicons are purified, indexed, pooled, and sequenced on the Oxford Nanopore MinION. This design provides overlapping amplification of provirus regions and near-complete coverage.
The method demonstrated high sensitivity for detecting integration sites due to long reads, enabling precise mapping in complex repetitive regions and simultaneous retrieval of proviral sequence, clonality, and epigenetic information, but it is mostly limited by the need for large input DNA (3 µg), reducing its applicability for samples with low proviral copy numbers. MDA reduces this to 10 ng–100 ng, albeit at the cost of completeness in site detection.
Mechanical shearing (sonication) is not the sole approach that allows escaping the RED-dependent bias in PCR-based techniques. One example is the tagmentation-assisted PCR (tag-PCR). Tag-PCR is a method for preparing amplification-based libraries for DNA sequencing based on simultaneous fragmentation and adapter attachment using Tn5 transposase. Originally introduced by Adey et al. (2010), it was designed to simplify and accelerate the construction of shotgun NGS libraries by reducing the protocol steps and processing time. The method utilizes a dimeric Tn5 transposase pre-loaded with a pair of double-stranded adapters containing short 19-bp mosaic ends (ME) derived from IS50 sequences, which are essential for transposition. The loaded enzyme (“transposome”) introduces double-stranded breaks across genomic DNA with near-random distribution while simultaneously attaching sequencing adapters. The initial tagmentation step is universal, while the downstream steps depend on the specific application and sequencing platform. In its original form, limited-cycle PCR is performed using primers that incorporate flow cell adapters and sample barcodes, allowing the preparation of full NGS libraries from as little as 10 pg input DNA in under 30 min.
Tag-PCR provides high coverage and efficiency, though some sequence bias may occur due to Tn5 preferences. Because of its speed and low input requirement, Tag-PCR has been widely used in genomics, including for ISA, transcriptomics, and profiling of rare samples (Hesselberg Løvestad et al., 2023; Kim et al., 2023; Hamada et al., 2018). In the ISA context Tag-PCR protocols typically use one primer targeting the adapter (e.g., Nextera R1) and another primer that is specific to the transgene (or proviral sequence). Several modifications of this method have been developed to reduce nonspecific background amplification.
One such modification is saTag-PCR, which uses a customized Tn5 transposase loaded with two identical adapters (usually Nextera R1). This prevents the amplification of fragments flanked by R1 and R2 (i.e., without the transgene), and the missing adapter is introduced during a second PCR round. Another approach, esTag-PCR, developed by Ryu et al. (2022), utilizes standard commercial Tn5 with dual adapters but modifies the amplification strategy: in the first PCR, only one adapter-specific primer and one transgene-specific primer are used, and no full-length adapter is restored. In the second, nested PCR round, a new internal transgene-specific primer is used, which also carries a TruSeq-compatible adapter. As both PCR rounds depend on gene-specific priming, background amplification is minimized. The method supports multiplexed primer pools targeting both 5′ and 3′ ends of the transgene, allowing the bidirectional mapping of integration sites. esTag-PCR yielded 8–13 times more on-target reads than Tag-PCR and saTag-PCR and successfully validated all tested integration sites by Sanger sequencing. It enables the detection of integration events from as little as 0.2 ng to 0.5 ng of genomic DNA, corresponding to ∼30–150 cells, demonstrating high sensitivity and accuracy.
Another variant of the classic tagmentation-assisted PCR is DIStinct-seq, developed by Kim et al. (2023), which was designed for rapid and efficient genome-wide ISA of lentiviral vectors. DIStinct-seq uses a bead-linked Tn5 transposome (Illumina DNA Prep) that allows up to 500 ng input DNA and reduces sample loss and handling time. The method achieves high sensitivity and reproducibility, detecting 5,000–6,000 unique integration sites per sample with minimal background. DIStinct-seq was applied to study lentiviral integration in CAR-T cells from multiple donors, accurately recapitulating known lentiviral integration biases (e.g., enrichment near TSS, oncogenes, and CpG islands). The method supports the quantitative assessment of clonal composition and longitudinal dynamics of CAR-T cells both ex vivo and in vivo, confirming associations between integration site location and clonal expansion. A custom bioinformatics pipeline filters out artifacts such as chimeric reads, PCR recombination products, and improper orientations, enabling reliable high-throughput ISA.
Alongside DNA sequence analysis-based methods for proviral integration site mapping, RT-PCR-based techniques emerged to specifically capture transcriptionally active insertions. Valk et al. (1997) developed a targeted approach exploiting the typical outcome of retroviral mutagenesis: when proviruses integrate near proto-oncogenes, their 5′ LTRs often drive aberrant chimeric transcripts fused to host gene exons. By designing an oligo (dT)-adapter primer for cDNA synthesis (anchored to the poly (A) tails of these transcripts) paired with an LTR-specific PCR primer, they selectively amplified only integration-derived chimeric cDNAs and excluded transcriptionally silent insertions. This bypassed the need for genomic DNA processing steps such as restriction digestion and adapter ligation that are inherent to LM-PCR/iPCR. The resulting amplicons, representing LTR–host gene fusions, were directly cloned and sequenced, enabling the rapid identification of oncogenic drivers such as EVI1 in murine leukemias (Valk et al., 1997).
3.2 Modern methods with no reliance on PCR
Amplification artifacts and variable efficiency of PCR-based methods can distort quantitative assessment of clonal characteristics of infected cells, complicating the accurate determination of clone prevalence and size. Since these characteristics are often as important as precise IS mapping (sometimes even more so), many recent approaches avoid PCR altogether, aiming to overcome its limitations and provide more reliable quantitative analysis of integration profiles. Several such RED- and PCR-independent pipelines have now been developed.
3.2.1 AFIS-seq
Since 2013, the CRISPR/Cas system has gained broad popularity and widespread application in laboratory practice due to its relatively easy and inexpensive targeting of specific loci. The CRISPR/Cas system has also found application in the ISA field. In particular, van Haasteren et al. (2021) proposed a method called amplification-free integration-site sequencing (AFIS-Seq), which is based on the excision of internal proviral regions using Cas9 RNPs delivered into cells along with guide RNAs targeting sites located within the viral (or vector) genome, approximately 500 base pairs from the LTRs. Host genomic DNA is first isolated using high molecular weight (HMW) DNA extraction to ensure acquired fragment size >50 kb and protected from ligation by dephosphorylation. After Cas9-mediated cleavage, new exposed DNA ends are A-tailed and ligated with Nanopore adapters (in the authors’ study, a 9.4.1 flow cell on a MinION Mk1B sequencer was used), and sequencing proceeds with in silico filtering of informative reads containing both host genome and provirus sequences.
When applied to genomic DNA from HEK293T cells transduced with rHIV.VSV-G and rSIV.F/HN, the authors obtained approximately 200,000 reads per vector, each approximately 12 kb in length, indicating up to 285- and 1,612-fold enrichment (for HIV and SIV, respectively), normalized to the number of integrated vector copies. In a direct comparison, the efficiency of unique IS mapping was 4% (HIV) and 3% (SIV) for S-EPTS/LM-PCR and 0.4% and 2%, respectively, for AFIS-Seq, reflecting lower enrichment efficiency of the amplification-free method. However, AFIS-Seq demonstrated higher mapping quality: long reads (∼11 kb) enabled confident identification of IS even in difficult-to-map genomic regions (including repetitive elements), where S-EPTS/LM-PCR failed. Moreover, AFIS-Seq provided simultaneous information on the proviral sequence, clonal composition of infected cell populations, and DNA modifications (e.g., CpG methylation). The method is scalable and can be implemented on higher-throughput Oxford Nanopore platforms such as GridION or PromethION.
The main limitation of AFIS-Seq is the requirement for a large amount of input genomic DNA (∼10 μg), which reduces sensitivity when the proviral copy number (VCN) is low, especially in samples with low infection rates or rare transduced cells.
3.2.2 CReVIS-seq
A year later, in 2021, another CRISPR/Cas-related method was proposed by Kim et al. (2021). In their approach, genomic DNA is mechanically fragmented (by sonication) to generate fragments of approximately 300 base pairs. After end-repair and phosphorylation, the fragments are circularized by self-ligation (this requires a low DNA concentration to reduce concatemer formation). The remaining linear fragments are removed by treatment with T5 exonuclease and DNA circles are re-linearized using Cas9 guided to a site within the LTR. As a result, only fragments containing proviral sequences are cleaved, ensuring the selectivity of the method. The linearized DNA fragments undergo A-tailing (addition of an adenine to the 3′ end) and ligation to sequencing adapters. After several cycles of PCR amplification to enrich the libraries, they are sequenced on high-throughput platforms such as Illumina MiSeq or MiniSeq, using paired-end reads that enable the precise identification of the host–virus junction. Since the target fragments have already been selected and linearized independently of PCR, the subsequent PCR amplification for library enrichment does not introduce the same systematic distortions in the representation of integration sites as observed in PCR-based methods. This method allowed the authors to accurately and clonally identify multiple IS of lentiviral vectors throughout the host genome, even in the presence of multiple integrations and heterogeneous cell populations, and detect circular forms of lentiviral DNA.
The method’s potential limitations are the relatively high input DNA requirement (in the microgram range), its dependence on the presence of an intact sgRNA recognition site within the LTR, a technical constraint on fragment length imposed by the chosen sequencing platform, and the need for careful optimization of circularization conditions to avoid concatemer formation.
3.2.3 Targeted sequence capture for lentiviral integration site identification
Ustek et al. (2012) proposed a targeted sequence capture method for analyzing lentiviral vector integration sites in the human genome without PCR amplification in the step of provirus–host junction retrieval. This method combines hybridization capture of fragmented genomic DNA (100 bp–600 bp), containing viral genome regions (using a set of overlapping probes covering the entire provirus), with subsequent high-throughput sequencing (454 GS FLX pyrosequencing). Viral–host junctions were identified by aligning reads first to the vector and then to the human genome (GRCh37/hg19), thus filtering out nonspecific fragments. This avoids PCR and restriction enzyme biases, enabling accurate mapping of integration sites and reconstitution of provirus structure. The authors identified 203 unique integration sites for the HIV-1-based vector, predominantly in introns and away from CpG islands and transcription start sites.
The advantages, in addition to the absence of PCR and restriction digestion, include high sensitivity for detecting novel integration sites, versatility in analyzing various genomic contexts (genes, TSS, CpG islands, and repetitive elements), and cost efficiency. The limitations, common to all amplification-free methods, are high DNA input (∼500 ng) and low capture efficiency (>90% of reads lack viral sequences), which increase sequencing costs.
3.3 Single-cell integration site analysis
Currently, single-cell analysis of integration sites (single-cell ISA) is increasingly being considered useful as it may allow the determination of both the number and precise location of individual proviruses within single cells following transduction with retroviral vectors, providing a potential link between individual integration events and the clonal properties of transduced cells, while overcoming the limitations of bulk analysis, where signals from different cells are averaged. This is particularly relevant in clinical applications of retroviral vectors. In the context of CAR-T cells, a single-cell approach enables the direct correlation of integration sites with the functional state of individual clones as cells differ in survival and proliferative capacity. Similarly, when using integrating vectors in gene therapy, for example in the modification of hematopoietic stem cells or during iPSC reprogramming, single-cell ISA allows linking specific integration events to safe and functionally successful clones and assessing therapeutic gene expression stability and oncogenic risk.
Some of the methods discussed above, such as MIP-seq, have performed reasonably well in this regard, demonstrating sufficient sensitivity to work with individual cells. In this case, the increased sensitivity was achieved through whole-genome nonspecific amplification of DNA from highly diluted samples using MDA.
The EpiVIA method (a modified single-cell ATAC-seq, another variant of tagmentation-based approaches) was proposed by Wang et al. (2020) to simultaneously determine the functional identity of individual CAR-T cells and precisely map lentiviral integration sites, addressing whether integration affects proliferative potential and clonal properties. Tn5 transposase fragments and tags nucleosome-free, accessible regions of chromatin and integrated proviral DNA, generating host–virus chimeric fragments that are sequenced and mapped to a combined reference genome. In an experiment with ∼1,000 cells, 188 integrations were identified in 172 CAR-T cells; in the most favorable conditions—cells containing more than 72,499 unique fragments—integration site detection reached 35%, while proviral reads were detected in 96% of cells, confirming CAR-T identity. In Yan et al. (2023), EpiVIA was applied for additional validation in a limited number of cells from a single patient. The method detected two integration sites within one cell, demonstrating its capability to identify multiple integration events in individual cells (Wang et al., 2020; Yan et al., 2023).
The method has limitations related to the low detection rate of integrations, which is inherent to all droplet-based single-cell protocols, and a strong dependence of sensitivity on sequencing depth and the number of fragments obtained per cell. In addition, PCR amplification of libraries could also potentially introduce bias.
4 Conclusion
Accurate methods for identifying integration sites (ISA) remain crucial for assessing the safety of integrating viral vector application and understanding patterns of integration, i.e., studying clonal dynamics of transduced cells. Random (with a certain previously described bias) integration of retroviruses, including lentiviruses, and retroviral vectors into the host cell genome poses a risk of insertional mutagenesis and oncogene activation. Modern ISA methods enable systematic detection of such events, refinement of integration patterns, and assessment of the clonal dynamics of modified cells in vivo, thus providing critically important data for preclinical and clinical studies.
The history of ISA methods reflects a transition from limited and labor-intensive approaches to high-throughput and more precise strategies. Early methods, involving the combined use of SB, RED, and molecular cloning, detected integrations primarily at preselected loci and exhibited significant locus-dependent variability in sensitivity. Over time, these were supplemented by local sequencing. The emergence of PCR-based methods, such as inverse PCR (iPCR), LM-PCR, and LAM-PCR, allowed for more comprehensive and unbiased mapping of integration sites across the host genome, although biases related to restriction digestion and amplification remained. LM-PCR and LAM-PCR continue to be used; however, more modern variants, including nrLAM-PCR and Tag-PCR, minimize systematic biases associated with restriction digestion and provide a more uniform representation of integration events. For even more accurate analysis, methods free from major sources of distortion—RED and PCR amplification—have been developed, such as AFIS-seq and CReVIS-seq. Although most contemporary methods, including PCR-dependent techniques, are adapted for high-throughput sequencing, the combination of NGS with relatively “bias-free” approaches provides the most accurate and undistorted representation of various IS.
In the commercial and research sectors, LM-PCR, LAM-PCR, and their modifications (nrLAM-PCR) continue to be applied, being offered by companies such as TaKaRa Bio, Azenta GENEWIZ, and ProtaGene, along with some PCR-free approaches utilizing mechanical DNA fragmentation. A summary comparison of ISA methodologies, aiming to assist in method selection, is provided in Table 1.
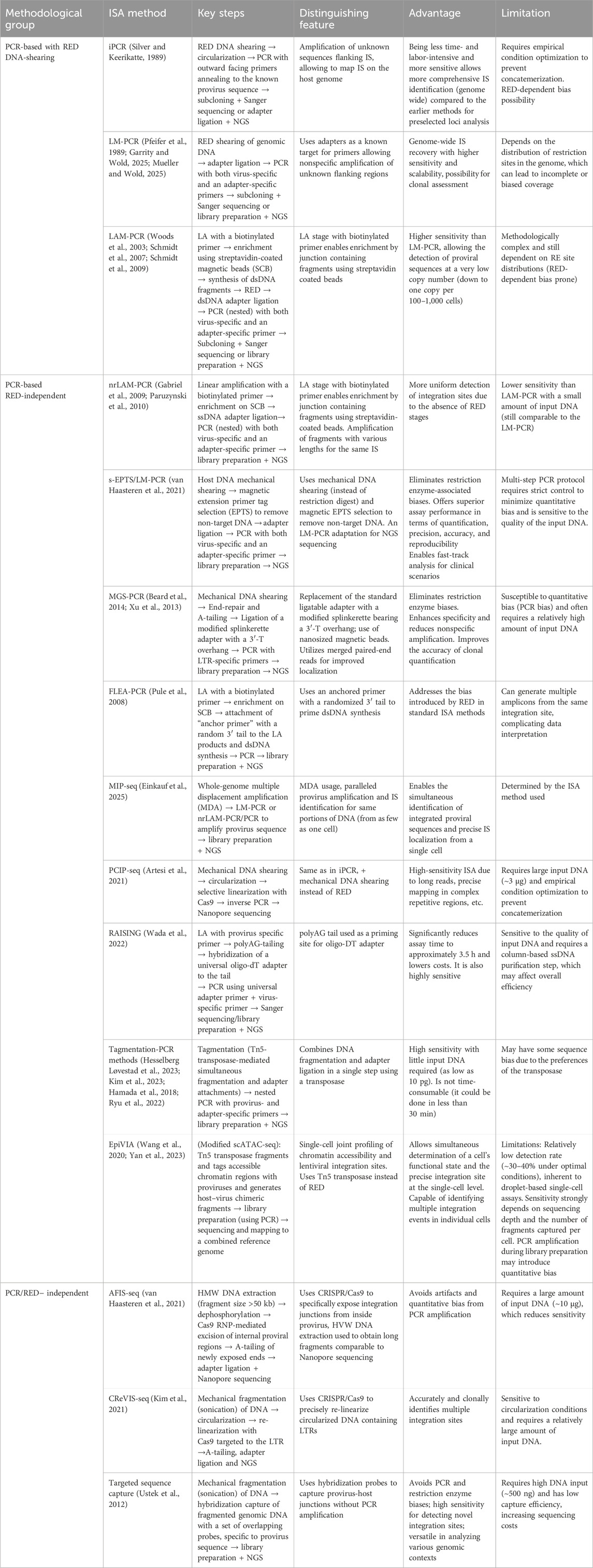
Table 1. Comparison of some modern ISA approaches.
Author contributions
KK-N: Writing – original draft, Conceptualization. AL: Conceptualization, Writing – review and editing. SS: Conceptualization, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was carried out within the state assignment of the Ministry of Science and Higher Education of the Russian Federation for the FSBI Research Centre for Medical Genetics.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. Generative AI was used in some parts of this manuscript to assist with language editing and refinement of phrasing. The authors take full responsibility for the content.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adey, A., Morrison, H. G., Asan, N., Xun, X., Kitzman, J. O., Turner, E. H., et al. (2010). Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol. 11 (12), R119. doi:10.1186/gb-2010-11-12-r119
Allen, M. J., Collick, A., and Jeffreys, A. J. (1994). Use of vectorette and subvectorette PCR to isolate transgene flanking DNA. PCR Methods Appl. 4 (2), 71–75. doi:10.1101/gr.4.2.71
Arnold, C., and Hodgson, I. J. (1991). Vectorette PCR: a novel approach to genomic walking. PCR Methods Appl. 1 (1), 39–42. doi:10.1101/gr.1.1.39
Artesi, M., Hahaut, V., Cole, B., Lambrechts, L., Ashrafi, F., Marçais, A., et al. (2021). PCIP-seq: simultaneous sequencing of integrated viral genomes and their insertion sites with long reads. Genome Biol. 22 (1), 97. doi:10.1186/s13059-021-02307-0
Beard, B. C., Adair, J. E., Trobridge, G. D., and Kiem, H. P. (2014). “High-throughput genomic mapping of vector integration sites in gene therapy studies,” in Hematopoietic stem cell protocols. Editors K. D. Bunting, and C. K. Qu (New York, NY: Springer), 321–344. doi:10.1007/978-1-4939-1133-2_22
Ben-David, Y., Giddens, E. B., and Bernstein, A. (1990). Identification and mapping of a common proviral integration site Fli-1 in erythroleukemia cells induced by friend murine leukemia virus. Proc. Natl. Acad. Sci. U. S. A. 87 (4), 1332–1336. doi:10.1073/pnas.87.4.1332
Benkel, B. F., Mucha, J., and Gavora, J. S. (1992). A new diagnostic method for the detection of endogenous rous-associated virus-type provirus in chickens. Poult. Sci. 71 (9), 1520–1526. doi:10.3382/ps.0711520
Berry, C. C., Gillet, N. A., Melamed, A., Gormley, N., Bangham, C. R. M., and Bushman, F. D. (2012). Estimating abundances of retroviral insertion sites from DNA fragment length data. Bioinformatics 28 (6), 755–762. doi:10.1093/bioinformatics/bts004
Biffi, A., Bartolomae, C. C., Cesana, D., Cartier, N., Aubourg, P., Ranzani, M., et al. (2011). Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection. Blood 117 (20), 5332–5339. doi:10.1182/blood-2010-09-306761
Blaydes, S. M., Kogan, S. C., Truong, B. T. H., Gilbert, D. J., Jenkins, N. A., Copeland, N. G., et al. (2001). Retroviral integration at the Epi1 locus cooperates with Nf1 gene loss in the progression to acute myeloid leukemia. J. Virol. 75 (19), 9427–9434. doi:10.1128/jvi.75.19.9427-9434.2001
Buchberg, A. M., Bedigian, H. G., Jenkins, N. A., and Copeland, N. G. (1999). Evi-2, a common integration site involved in murine myeloid leukemogenesis. Mol. Cell Biol. 10 (9), 4658–4666. doi:10.1128/mcb.10.9.4658
Carbonaro, D. A., Zhang, L., Jin, X., Montiel-Equihua, C., Geiger, S., Carmo, M., et al. (2014). Preclinical demonstration of lentiviral vector-mediated correction of immunological and metabolic abnormalities in models of adenosine deaminase deficiency. Mol. Ther. 22 (3), 607–622. doi:10.1038/mt.2013.265
Carteau, S., Hoffmann, C., and Bushman, F. (1998). Chromosome structure and human immunodeficiency virus type 1 cDNA integration: centromeric alphoid repeats are a disfavored target. J. Virol. 72 (5), 4005–4014. doi:10.1128/jvi.72.5.4005-4014.1998
Cattoglio, C., Maruggi, G., Bartholomae, C., Malani, N., Pellin, D., Cocchiarella, F., et al. (2010). High-definition mapping of retroviral integration sites defines the fate of allogeneic T cells after donor lymphocyte infusion. PLOS ONE 5 (12), e15688. doi:10.1371/journal.pone.0015688
Cavrois, M., Wain-Hobson, S., and Wattel, E. (1995). Stochastic events in the amplification of HTLV-I integration sites by linker-mediated PCR. Res. Virol. 146 (3), 179–184. doi:10.1016/0923-2516(96)80578-4
Cepko, C. L., Roberts, B. E., and Mulligan, R. C. (1984). Construction and applications of a highly transmissible Murine retrovirus shuttle vector. Cell 37 (3), 1053–1062. doi:10.1016/0092-8674(84)90440-9
Chan, H. W., and Martin, M. A. (1980). Identification of ecotropic proviral sequences in twelve inbred mouse strains. Am. J. Trop. Med. Hyg. 29 (5_Part_2), 1107–1110. doi:10.4269/ajtmh.1980.29.1107
Chin, C. J., Cooper, A. R., Lill, G. R., Evseenko, D., Zhu, Y., He, C. B., et al. (2016). Genetic tagging during human mesoderm differentiation reveals tripotent lateral plate mesodermal progenitors. Stem Cells 34 (5), 1239–1250. doi:10.1002/stem.2351
Clurman, B. E., and Hayward, W. S. (1989). Multiple proto-oncogene activations in avian leukosis virus-induced lymphomas: evidence for stage-specific events. Mol. Cell Biol. 9 (6), 2657–2664. doi:10.1128/mcb.9.6.2657-2664.1989
Corcoran, L. M., Adams, J. M., Dunn, A. R., and Cory, S. (1984). Murine T lymphomas in which the cellular myc oncogene has been activated by retroviral insertion. Cell 37 (1), 113–122. doi:10.1016/0092-8674(84)90306-4
Cornils, K., Bartholomae, C. C., Thielecke, L., Lange, C., Arens, A., Glauche, I., et al. (2013). Comparative clonal analysis of reconstitution kinetics after transplantation of hematopoietic stem cells gene marked with a lentiviral SIN or a γ-retroviral LTR vector. Exp. Hematol. 41 (1), 28–38.e3. doi:10.1016/j.exphem.2012.09.003
Correa de Freitas, M. C., Fontes, A. M., de Castilho Fernandes, A., Picanço-Castro, V., de Sousa Russo, E. M., and Covas, D. T. (2014). Murine leukemia virus-derived retroviral vector has differential integration patterns in human cell lines used to produce recombinant factor VIII. Rev. Bras. Hematol. Hemoter. 36 (3), 213–218. doi:10.1016/j.bjhh.2014.03.002
Dambrot, C., Buermans, H. P. J., Varga, E., Kosmidis, G., Langenberg, K., Casini, S., et al. (2014). Strategies for rapidly mapping proviral integration sites and assessing cardiogenic potential of nascent human induced pluripotent stem cell clones. Exp. Cell Res. 327 (2), 297–306. doi:10.1016/j.yexcr.2014.05.001
Daniel, R., and Smith, J. A. (2008). Integration site selection by retroviral vectors: molecular mechanism and clinical consequences. Hum. Gene Ther. 19 (6), 557–568. doi:10.1089/hum.2007.148
Dawes, J. C., Webster, P., Iadarola, B., Garcia-Diaz, C., Dore, M., Bolt, B. J., et al. (2020). LUMI-PCR: an illumina platform ligation-mediated PCR protocol for integration site cloning, provides molecular quantitation of integration sites. Mob. DNA 11 (1), 7. doi:10.1186/s13100-020-0201-4
d’Offay, J. M., Eberle, R., Wolf, R. F., Kosanke, S. D., Doocy, K. R., Ayalew, S., et al. (2013). Simian T-lymphotropic virus-associated lymphoma in 2 naturally infected baboons: T-Cell clonal expansion and immune response during tumor development. Comp. Med. 63 (3), 288–294.
Einkauf, K. B., Lee, G. Q., Gao, C., Sharaf, R., Sun, X., Hua, S., et al. (2025). Intact HIV-1 proviruses accumulate at distinct chromosomal positions during prolonged antiretroviral therapy. J. Clin. Invest 129 (3), 988–998. doi:10.1172/jci124291
Frankel, W., Potter, T. A., Rosenberg, N., Lenz, J., and Rajan, T. V. (1985). Retroviral insertional mutagenesis of a target allele in a heterozygous murine cell line. Proc. Natl. Acad. Sci. 82 (19), 6600–6604. doi:10.1073/pnas.82.19.6600
Garrity, P. A., and Wold, B. J. (2025). Effects of different DNA polymerases in ligation-mediated PCR: enhanced genomic sequencing and in vivo footprinting. Proc Natl Acad Sci U S A. 89 (3):1021–5. doi:10.1073/pnas.89.3.1021
Gabet, A. S., Gessain, A., and Wattel, E. (2003). High simian T-cell leukemia virus type 1 proviral loads combined with genetic stability as a result of cell-associated provirus replication in naturally infected, asymptomatic monkeys. Int. J. Cancer 107 (1), 74–83. doi:10.1002/ijc.11329
Gabriel, R., Eckenberg, R., Paruzynski, A., Bartholomae, C. C., Nowrouzi, A., Arens, A., et al. (2009). Comprehensive genomic access to vector integration in clinical gene therapy. Nat. Med. 15 (12), 1431–1436. doi:10.1038/nm.2057
Gabriel, R., Kutschera, I., Bartholomae, C. C., von Kalle, C., and Schmidt, M. (2014). Linear amplification mediated PCR – localization of genetic elements and characterization of unknown flanking DNA. J. Vis. Exp. (88), 51543. doi:10.3791/51543
Gillet, N. A., Malani, N., Melamed, A., Gormley, N., Carter, R., Bentley, D., et al. (2011). The host genomic environment of the provirus determines the abundance of HTLV-1–infected T-cell clones. Blood 117 (11), 3113–3122. doi:10.1182/blood-2010-10-312926
Giordano, F. A., Appelt, J. U., Link, B., Gerdes, S., Lehrer, C., Scholz, S., et al. (2015). High-throughput monitoring of integration site clonality in preclinical and clinical gene therapy studies. Mol. Ther. - Methods and Clin. Dev. 2, 14061. doi:10.1038/mtm.2014.61
Girard, L., Hanna, Z., Beaulieu, N., Hoemann, C. D., Simard, C., Kozak, C. A., et al. (1996). Frequent provirus insertional mutagenesis of Notch1 in thymomas of MMTVD/myc transgenic mice suggests a collaboration of c-myc and Notch1 for oncogenesis. Genes Dev. 10 (15), 1930–1944. doi:10.1101/gad.10.15.1930
Gong, M., Semus, H. L., Bird, K. J., Stramer, B. J., and Ruddell, A. (1998). Differential selection of cells with proviral c-myc and c-erbB integrations after avian leukosis virus infection. J. Virol. 72 (7), 5517–5525. doi:10.1128/jvi.72.7.5517-5525.1998
Gonzalez-Murillo, A., Lozano, M. L., Montini, E., Bueren, J. A., and Guenechea, G. (2008). Unaltered repopulation properties of mouse hematopoietic stem cells transduced with lentiviral vectors. Blood 112 (8), 3138–3147. doi:10.1182/blood-2008-03-142661
Green, M. R., and Sambrook, J. (2019). Inverse polymerase chain reaction (PCR). Cold Spring Harb. Protoc. 2019, pdb.prot095166. doi:10.1101/pdb.prot095166
Hacein-Bey-Abina, S., Von Kalle, C., Schmidt, M., McCormack, M. P., Wulffraat, N., Leboulch, P., et al. (2003a). LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 302 (5644), 415–419. doi:10.1126/science.1088547
Hacein-Bey-Abina, S., von Kalle, C., Schmidt, M., Le Deist, F., Wulffraat, N., McIntyre, E., et al. (2003b). A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 348 (3), 255–256. doi:10.1056/nejm200301163480314
Hacein-Bey-Abina, S., Garrigue, A., Wang, G. P., Soulier, J., Lim, A., Morillon, E., et al. (2008). Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest 118 (9), 3132–3142. doi:10.1172/jci35700
Hamada, M., Nishio, N., Okuno, Y., Suzuki, S., Kawashima, N., Muramatsu, H., et al. (2018). Integration mapping of piggyBac-Mediated CD19 chimeric antigen receptor T cells analyzed by novel Tagmentation-Assisted PCR. EBioMedicine 34, 18–26. doi:10.1016/j.ebiom.2018.07.008
Hanai, S., Nitta, T., Shoda, M., Tanaka, M., Iso, N., Mizoguchi, I., et al. (2004). Integration of human T-cell leukemia virus type 1 in genes of leukemia cells of patients with adult T-cell leukemia. Cancer Sci. 95 (4), 306–310. doi:10.1111/j.1349-7006.2004.tb03207.x
Harbers, K., Kuehn, M., Delius, H., and Jaenisch, R. (1984). Insertion of retrovirus into the first intron of alpha 1(I) collagen gene to embryonic lethal mutation in mice. Proc. Natl. Acad. Sci. U. S. A. 81 (5), 1504–1508. doi:10.1073/pnas.81.5.1504
Harkey, M. A., Kaul, R., Jacobs, M. A., Kurre, P., Bovee, D., Levy, R., et al. (2007). Multiarm high-throughput integration site detection: limitations of LAM-PCR technology and optimization for clonal analysis. Stem Cells Dev. 16, 381–392. doi:10.1089/scd.2007.0015
Hauber, I., Hofmann-Sieber, H., Chemnitz, J., Dubrau, D., Chusainow, J., Stucka, R., et al. (2013). Highly significant antiviral activity of HIV-1 LTR-specific tre-recombinase in humanized mice. PLoS Pathog. 9 (9), e1003587. doi:10.1371/journal.ppat.1003587
Hayakawa, J., Washington, K., Uchida, N., Phang, O., Kang, E. M., Hsieh, M. M., et al. (2009). Long-term vector integration site analysis following retroviral mediated gene transfer to hematopoietic stem cells for the treatment of HIV infection. PLoS One 4 (1), e4211. doi:10.1371/journal.pone.0004211
Henssen, A. G., Henaff, E., Jiang, E., Eisenberg, A. R., Carson, J. R., Villasante, C. M., et al. (2015). Genomic DNA transposition induced by human PGBD5. Botchan MR. eLife 25 (4), e10565. doi:10.7554/eLife.10565
Hesselberg Løvestad, A., Stosic, M. S., Costanzi, J. M., Christiansen, I. K., Aamot, H. V., Ambur, O. H., et al. (2023). TaME-seq2: tagmentation-assisted multiplex PCR enrichment sequencing for viral genomic profiling. Virol. J. 20 (1), 44. doi:10.1186/s12985-023-02002-5
Howe, S. J., Mansour, M. R., Schwarzwaelder, K., Bartholomae, C., Hubank, M., Kempski, H., et al. (2008). Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest 118 (9), 3143–3150. doi:10.1172/jci35798
Hughes, S. H., Shank, P. R., Spector, D. H., Kung, H. J., Bishop, J. M., Varmus, H. E., et al. (1978). Proviruses of Avian sarcoma virus are terminally redundant, co-extensive with unintegrated linear DNA and integrated at many sites. Cell 15 (4), 1397–1410. doi:10.1016/0092-8674(78)90064-8
Isfort, R., Witter, R. L., and Kung, H. J. (1987). C-myc activation in an unusual retrovirus-induced avian T-lymphoma resembling Marek’s disease: proviral insertion 5’ of exon one enhances the expression of an intron promoter. Oncogene Res. 2 (1), 81–94.
Jong, J., Akhtar, W., Badhai, J., Rust, A. G., Rad, R., Hilkens, J., et al. (2014). Chromatin landscapes of retroviral and transposon integration profiles. PLOS Genet. 10 (4), e1004250. doi:10.1371/journal.pgen.1004250
Joseph, K. W., Halvas, E. K., Brandt, L. D., Patro, S. C., Rausch, J. W., Chopra, A., et al. (2022). Deep sequencing analysis of individual HIV-1 proviruses reveals frequent asymmetric long terminal repeats. J. Virol. 96 (13), e00122-22–22. doi:10.1128/jvi.00122-22
Justice, J. F., Morgan, R. W., and Beemon, K. L. (2015). Common viral integration sites identified in Avian leukosis virus-induced B-Cell lymphomas. mBio 6 (6), e01863-15. doi:10.1128/mbio.01863-15
Karumbayaram, S., Lee, P., Azghadi, S. F., Cooper, A. R., Patterson, M., Kohn, D. B., et al. (2012). From skin biopsy to neurons through a pluripotent intermediate under good manufacturing practice protocols. Stem Cells Transl. Med. 1 (1), 36–43. doi:10.5966/sctm.2011-0001
Kikuchi, A., Ohata, Y., Matsumoto, H., Sugiura, M., and Nishikawa, T. (1997). Anti-HTLV-1 antibody positive cutaneous T-cell lymphoma. Cancer 79 (2), 269–274. doi:10.1002/(sici)1097-0142(19970115)79:2<269::aid-cncr10>3.0.co;2-z
Kim, H. S., Hwang, G. H., Lee, H. K., Bae, T., Park, S. H., Kim, Y. J., et al. (2021). CReVIS-Seq: a highly accurate and multiplexable method for genome-wide mapping of lentiviral integration sites. Mol. Ther. Methods Clin. Dev. 20, 792–800. doi:10.1016/j.omtm.2020.10.012
Kim, J., Park, M., Baek, G., Kim, J. I., Kwon, E., Kang, B. C., et al. (2023). Tagmentation-based analysis reveals the clonal behavior of CAR-T cells in association with lentivector integration sites. Mol. Ther. - Oncolytics 21 (30), 1–13. doi:10.1016/j.omto.2023.05.004
King, W., Patel, M. D., Lobel, L. I., Goff, S. P., and Nguyen-Huu, M. C. (1985). Insertion mutagenesis of embryonal carcinoma cells by retroviruses. Science 228 (4699), 554–558. doi:10.1126/science.3838595
Kok, Y. L., Vongrad, V., Chaudron, S. E., Shilaih, M., Leemann, C., Neumann, K., et al. (2024). HIV-1 integration sites in CD4+ T cells during primary, chronic, and late presentation of HIV-1 infection. JCI Insight 6 (9), e143940. doi:10.1172/jci.insight.143940
Laufs, S., Gentner, B., Nagy, K. Z., Jauch, A., Benner, A., Naundorf, S., et al. (2003). Retroviral vector integration occurs in preferred genomic targets of human bone marrow–repopulating cells. Blood 101 (6), 2191–2198. doi:10.1182/blood-2002-02-0627
Lavu, S., and Reddy, E. P. (1986). Structural organization and nucleotide sequence of mouse c-myb oncogene: activation in ABPL tumors is due to viral integration in an intron which results in the deletion of the 5’ coding sequences. Nucleic Acids Res. 14 (13), 5309–5320. doi:10.1093/nar/14.13.5309
Lee, J. S., Ishimoto, A., Honjo, T., and Yanagawa, S. (1999). Murine leukemia provirus-mediated activation of the Notch1 gene leads to induction of HES-1 in a mouse T lymphoma cell line, DL-3. FEBS Lett. 455 (3), 276–280. doi:10.1016/s0014-5793(99)00901-1
Lenvik, T., Lund, T. C., and Verfaillie, C. M. (2002). Blockerette-ligated capture T7-Amplified RT-PCR, a new method for determining flanking sequences. Mol. Ther. 6 (1), 113–118. doi:10.1006/mthe.2002.0637
Li, J., Shen, H., Himmel, K. L., Dupuy, A. J., Largaespada, D. A., Nakamura, T., et al. (1999). Leukaemia disease genes: large-scale cloning and pathway predictions. Nat. Genet. 23 (3), 348–353. doi:10.1038/15531
Liao, X., Buchberg, A. M., Jenkins, N. A., and Copeland, N. G. (1995). Evi-5, a common site of retroviral integration in AKXD T-cell lymphomas, maps near Gfi-1 on mouse chromosome 5. J. Virol. 69 (11), 7132–7137. doi:10.1128/jvi.69.11.7132-7137.1995
Lobel, L. I., Murphy, J. E., and Goff, S. P. (1989). The palindromic LTR-LTR junction of moloney murine leukemia virus is not an efficient substrate for proviral integration. J. Virol. 63 (6), 2629–2637. doi:10.1128/jvi.63.6.2629-2637.1989
Lund, A. H., Turner, G., Trubetskoy, A., Verhoeven, E., Wientjens, E., Hulsman, D., et al. (2002). Genome-wide retroviral insertional tagging of genes involved in cancer in Cdkn2a-deficient mice. Nat. Genet. 32 (1), 160–165. doi:10.1038/ng956
Mack, K. D., Jin, X., Yu, S., Wei, R., Kapp, L., Green, C., et al. (2003). HIV insertions within and proximal to host cell genes are a common finding in tissues containing high levels of HIV DNA and macrophage-associated p24 antigen expression. J. Acquir Immune Defic. Syndr. 33 (3), 308–320. doi:10.1097/00126334-200307010-00004
Marh, J., Stoytcheva, Z., Urschitz, J., Sugawara, A., Yamashiro, H., Owens, J. B., et al. (2012). Hyperactive self-inactivating piggyBac for transposase-enhanced pronuclear microinjection transgenesis. Proc. Natl. Acad. Sci. 109 (47), 19184–19189. doi:10.1073/pnas.1216473109
McGrath, B. M., and Pembroke, J. T. (2004). Detailed analysis of the insertion site of the mobile elements R997, pMERPH, R392, R705 and R391 in E. coli K12. FEMS Microbiol. Lett. 237 (1), 19–26. doi:10.1016/j.femsle.2004.06.009
Mitsuhashi, J., Hosoyama, H., Tsukahara, S., Katayama, K., Noguchi, K., Ito, Y., et al. (2010). In vivo expansion of MDR1-transduced cells accompanied by a post-transplantation chemotherapy regimen with mitomycin C and methotrexate. J. Gene Med. 12 (7), 596–603. doi:10.1002/jgm.1474
Montini, E., Cesana, D., Schmidt, M., Sanvito, F., Bartholomae, C. C., Ranzani, M., et al. (2009). The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. American Society for Clinical Investigation. Available online at: https://www.jci.org/articles/view/37630/pdf.
Mooslehner, K., Karls, U., and Harbers, K. (1990). Retroviral integration sites in transgenic mov mice frequently map in the vicinity of transcribed DNA regions. J. Virol. 64 (6), 3056–3058. doi:10.1128/jvi.64.6.3056-3058.1990
Müller, H. P., and Varmus, H. E. (1994). DNA bending creates favored sites for retroviral integration: an explanation for preferred insertion sites in nucleosomes. EMBO J. 13 (19), 4704–4714. doi:10.1002/j.1460-2075.1994.tb06794.x
Mueller, P. R., and Wold, B. (2025). Ligation-mediated PCR: applications to genomic footprinting - ScienceDirect. Available online at: https://www.sciencedirect.com/science/article/abs/pii/S1046202305801227
Nusse, R., and Varmus, H. E. (1982). Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 31 (1), 99–109. doi:10.1016/0092-8674(82)90409-3
Ochman, H., Gerber, A. S., and Hartl, D. L. (1988). Genetic applications of an inverse polymerase chain reaction. Genetics 120 (3), 621–623. doi:10.1093/genetics/120.3.621
Ohshima, K., Mukai, Y., Shiraki, H., Suzumiya, J., Tashiro, K., and Kikuchi, M. (1997). Clonal integration and expression of human T-cell lymphotropic virus type I in carriers detected by polymerase chain reaction and inverse PCR. Am. J. Hematol. 54 (4), 306–312. doi:10.1002/(sici)1096-8652(199704)54:4<306::aid-ajh8>3.0.co;2-z
Ott, M. G., Schmidt, M., Schwarzwaelder, K., Stein, S., Siler, U., Koehl, U., et al. (2006). Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 12 (4), 401–409. doi:10.1038/nm1393
O’Malley, R. C., Alonso, J. M., Kim, C. J., Leisse, T. J., and Ecker, J. R. (2007). An adapter ligation-mediated PCR method for high-throughput mapping of T-DNA inserts in the arabidopsis genome. Nat. Protoc. 2 (11), 2910–2917. doi:10.1038/nprot.2007.425
Paruzynski, A., Arens, A., Gabriel, R., Bartholomae, C. C., Scholz, S., Wang, W., et al. (2010). Genome-wide high-throughput integrome analyses by nrLAM-PCR and next-generation sequencing. Nat. Protoc. 5 (8), 1379–1395. doi:10.1038/nprot.2010.87
Paruzynski, A., Glimm, H., Schmidt, M., and von Kalle, C. (2012). “Chapter four - analysis of the clonal repertoire of gene-corrected cells in gene therapy,” in Methods in enzymolog. Editor T. Friedmann (Academic Press), 59–87. Available online at: https://www.sciencedirect.com/science/article/pii/B9780123865090000041.
Pfeifer, G. P., Steigerwald, S. D., Mueller, P. R., Wold, B., and Riggs, A. D. (1989). Genomic sequencing and methylation analysis by ligation mediated PCR. Science 246, 810–813. doi:10.1126/science.2814502
Potter, C. J., and Luo, L. (2010). Splinkerette PCR for mapping transposable elements in Drosophila. PLoS One 5 (4), e10168. doi:10.1371/journal.pone.0010168
Pruss, D., Bushman, F. D., and Wolffe, A. P. (1994). Human immunodeficiency virus integrase directs integration to sites of severe DNA distortion within the nucleosome core. Proc. Natl. Acad. Sci. U. S. A. 91 (13), 5913–5917. doi:10.1073/pnas.91.13.5913
Pryciak, P. M., and Varmus, H. E. (1992). Nucleosomes, DNA-binding proteins, and DNA sequence modulate retroviral integration target site selection. Cell 69 (5), 769–780. doi:10.1016/0092-8674(92)90289-o
Pule, M. A., Rousseau, A., Vera, J., Heslop, H. E., Brenner, M. K., and Vanin, E. F. (2008). Flanking-sequence exponential anchored–polymerase chain reaction amplification: a sensitive and highly specific method for detecting retroviral integrant–host–junction sequences. Cytotherapy 10 (5), 526–539. doi:10.1080/14653240802192636
Qureshi, S. J., Porteous, D. J., and Brookes, A. J. (1994). Alu-based vectorettes and splinkerettes: more efficient and comprehensive polymerase chain reaction amplification of human DNA from complex sources. Genetic analysis. Biomol. Eng. 11 (4), 95–101. doi:10.1016/1050-3862(94)90046-9
Robinson, H. L., and Gagnon, G. C. (1986). Patterns of proviral insertion and deletion in avian leukosis virus-induced lymphomas. J. Virol. 57 (1), 28–36. doi:10.1128/jvi.57.1.28-36.1986
Rong, L., Bian, Y., Liu, S., Liu, X., Li, X., Liu, H., et al. (2017). Identifying tumor promoting genomic alterations in tumor-associated fibroblasts via retrovirus-insertional mutagenesis. Oncotarget 8 (57), 97231–97245. doi:10.18632/oncotarget.21881
Rosewick, N., Hahaut, V., Durkin, K., Artesi, M., Karpe, S., Wayet, J., et al. (2020). An improved sequencing-based bioinformatics pipeline to track the distribution and clonal architecture of proviral integration sites. Front. Microbiol. 11, 587306. doi:10.3389/fmicb.2020.587306
Rozera, G., Sberna, G., Berno, G., Gruber, C. E. M., Giombini, E., Spezia, P. G., et al. (2022). Intact provirus and integration sites analysis in acute HIV-1 infection and changes after one year of early antiviral therapy. J. Virus Erad. 8 (4), 100306. doi:10.1016/j.jve.2022.100306
Ryu, J., Chan, W., Wettengel, J. M., Hanna, C. B., Burwitz, B. J., Hennebold, J. D., et al. (2022). Rapid, accurate mapping of transgene integration in viable rhesus macaque embryos using enhanced-specificity tagmentation-assisted PCR. Mol. Ther. Methods Clin. Dev. 24, 241–254. doi:10.1016/j.omtm.2022.01.009
Saito, M., Hasegawa, H., Yamauchi, S., Nakagawa, S., Sasaki, D., Nao, N., et al. (2020). A high-throughput detection method for the clonality of human T-cell leukemia virus type-1-infected cells in vivo. Int. J. Hematol. 112 (3), 300–306. doi:10.1007/s12185-020-02935-5
Scherdin, U., Rhodes, K., and Breindl, M. (1990). Transcriptionally active genome regions are preferred targets for retrovirus integration. J. Virol. 64 (2), 907–912. doi:10.1128/jvi.64.2.907-912.1990
Schmidt, M., Hoffmann, G., Wissler, M., Lemke, N., Müßig, A., Glimm, H., et al. (2001). Detection and direct genomic sequencing of multiple rare unknown flanking DNA in highly complex samples. Hum. Gene Ther. 12 (7), 743–749. doi:10.1089/104303401750148649
Schmidt, M., Schwarzwaelder, K., Bartholomae, C., Zaoui, K., Ball, C., Pilz, I., et al. (2007). High-resolution insertion-site analysis by linear amplification-mediated PCR (LAM-PCR). Nat. Methods 4 (12), 1051–1057. doi:10.1038/nmeth1103
Schmidt, M., Schwarzwaelder, K., Bartholomae, C. C., Glimm, H., and Kalle, C. (2009). “Detection of retroviral integration sites by linear amplification-mediated PCR and tracking of individual integration clones in different samples,” in Genetic modification of hematopoietic stem cells: methods and protocols. Editor C. Baum (Totowa, NJ: Humana Press), 363–372. doi:10.1007/978-1-59745-409-4_24
Schröder, A. R. W., Shinn, P., Chen, H., Berry, C., Ecker, J. R., and Bushman, F. (2002). HIV-1 integration in the human genome favors active genes and local hotspots. Cell 110 (4), 521–529. doi:10.1016/s0092-8674(02)00864-4
Selten, G., Cuypers, H. T., Zijlstra, M., Melief, C., and Berns, A. (1984). Involvement of c-myc in MuLV-induced T cell lymphomas in mice: frequency and mechanisms of activation. EMBO J. 3 (13), 3215–3222. doi:10.1002/j.1460-2075.1984.tb02281.x
Šenigl, F., Auxt, M., and Hejnar, J. (2012). Transcriptional provirus silencing as a crosstalk of de novo DNA methylation and epigenomic features at the integration site. Nucleic Acids Res. 40 (12), 5298–5312. doi:10.1093/nar/gks197
Šenigl, F., Miklík, D., Auxt, M., and Hejnar, J. (2017). Accumulation of long-term transcriptionally active integrated retroviral vectors in active promoters and enhancers. Nucleic Acids Res. 45 (22), 12752–12765. doi:10.1093/nar/gkx889
Serrao, E., Cherepanov, P., and Engelman, A. N. (2016). Amplification, next-generation sequencing, and genomic DNA mapping of retroviral integration sites. J. Vis. Exp. (109), 53840. doi:10.3791/53840
Sfanos, K. S., Aloia, A. L., Hicks, J. L., Esopi, D. M., Steranka, J. P., Shao, W., et al. (2011). Identification of replication competent murine gammaretroviruses in commonly used prostate cancer cell lines. PLOS ONE 6 (6), e20874. doi:10.1371/journal.pone.0020874
Shao, H., and Lok, J. B. (2014). Detection of piggyBac-mediated transposition by splinkerette PCR in transgenic lines of Strongyloides ratti. Bio Protoc. 4 (1), e1015. doi:10.21769/bioprotoc.1015
Shao, L., Shi, R., Zhao, Y., Liu, H., Lu, A., Ma, J., et al. (2022). Genome-wide profiling of retroviral DNA integration and its effect on clinical pre-infusion CAR T-cell products. J. Transl. Med. 20 (1), 514. doi:10.1186/s12967-022-03729-5
Shi, Q., Kuether, E. L., Chen, Y., Schroeder, J. A., Fahs, S. A., and Montgomery, R. R. (2014). Platelet gene therapy corrects the hemophilic phenotype in immunocompromised hemophilia A mice transplanted with genetically manipulated human cord blood stem cells. Blood 123 (3), 395–403. doi:10.1182/blood-2013-08-520478
Shih, C. K., Linial, M., Goodenow, M. M., and Hayward, W. S. (1984). Nucleotide sequence 5’ of the chicken c-myc coding region: localization of a noncoding exon that is absent from myc transcripts in most avian leukosis virus-induced lymphomas. Proc. Natl. Acad. Sci. U. S. A. 81 (15), 4697–4701. doi:10.1073/pnas.81.15.4697
Shih, C. C., Stoye, J. P., and Coffin, J. M. (1988). Highly preferred targets for retrovirus integration. Cell 53 (4), 531–537. doi:10.1016/0092-8674(88)90569-7
Silver, J., and Keerikatte, V. (1989). Novel use of polymerase chain reaction to amplify cellular DNA adjacent to an integrated provirus. J. Virol. 63 (5), 1924–1928. doi:10.1128/jvi.63.5.1924-1928.1989
Swift, R. A., Boerkoel, C., Ridgway, A., Fujita, D. J., Dodgson, J. B., and Kung, H. J. (1987). B-lymphoma induction by reticuloendotheliosis virus: characterization of a mutated chicken syncytial virus provirus involved in c-myc activation. J. Virol. 61 (7), 2084–2090. doi:10.1128/jvi.61.7.2084-2090.1987
Takemoto, S., Matsuoka, M., Yamaguchi, K., and Takatsuki, K. (1994). A novel diagnostic method of adult T-cell leukemia: monoclonal integration of human T-cell lymphotropic virus type I provirus DNA detected by inverse polymerase chain reaction. Blood 84 (9), 3080–3085. doi:10.1182/blood.v84.9.3080.3080
Tam, W., Ben-Yehuda, D., and Hayward, W. S. (1997). Bic, a novel gene activated by proviral insertions in avian leukosis virus-induced lymphomas, is likely to function through its noncoding RNA. Mol. Cell Biol. 17 (3), 1490–1502. doi:10.1128/mcb.17.3.1490
Triglia, T., Peterson, M. G., and Kemp, D. J. (1988). A procedure for in vitro amplification of DNA segments that lie outside the boundaries of known sequences. Nucleic Acids Res. 16 (16), 8186. doi:10.1093/nar/16.16.8186
Tsichlis, P. N., Lee, J. S., Bear, S. E., Lazo, P. A., Patriotis, C., Gustafson, E., et al. (1990). Activation of multiple genes by provirus integration in the Mlvi-4 locus in T-cell lymphomas induced by moloney murine leukemia virus. J. Virol. 64 (5), 2236–2244. doi:10.1128/jvi.64.5.2236-2244.1990
Uren, A. G., Mikkers, H., Kool, J., van der Weyden, L., Lund, A. H., Wilson, C. H., et al. (2009). A high-throughput splinkerette-PCR method for the isolation and sequencing of retroviral insertion sites. Nat. Protoc. 4 (5), 789–798. doi:10.1038/nprot.2009.64
Ustek, D., Sirma, S., Gumus, E., Arikan, M., Cakiris, A., Abaci, N., et al. (2012). A genome-wide analysis of lentivector integration sites using targeted sequence capture and next generation sequencing technology. Infect. Genet. Evol. 12 (7), 1349–1354. doi:10.1016/j.meegid.2012.05.001
Valk, P. J., Joosten, M., Vankan, Y., Löwenberg, B., and Delwel, R. (1997). A rapid RT-PCR based method to isolate complementary DNA fragments flanking retrovirus integration sites. Nucleic Acids Res. 25 (21), 4419–4421. doi:10.1093/nar/25.21.4419
van Haasteren, J., Munis, A. M., Gill, D. R., and Hyde, S. C. (2021). Genome-wide integration site detection using Cas9 enriched amplification-free long-range sequencing. Nucleic Acids Res. 49 (3), e16. doi:10.1093/nar/gkaa1152
van Lohuizen, M., Breuer, M., and Berns, A. (1989). N-myc is frequently activated by proviral insertion in MuLV-induced T cell lymphomas. EMBO J. 8 (1), 133–136. doi:10.1002/j.1460-2075.1989.tb03357.x
van Ooyen, A., and Nusse, R. (1984). Structure and nucleotide sequence of the putative mammary oncogene int-1; proviral insertions leave the protein-encoding domain intact. Cell 39 (1), 233–240. doi:10.1016/0092-8674(84)90209-5
Varas, F., Stadtfeld, M., de Andres-Aguayo, L., Maherali, N., di Tullio, A., Pantano, L., et al. (2009). Fibroblast-derived induced pluripotent stem cells show no common retroviral vector insertions. Stem Cells 27 (2), 300–306. doi:10.1634/stemcells.2008-0696
Varmus, H. E., Quintrell, N., and Ortiz, S. (1981). Retroviruses as mutagens: insertion and excision of a nontransforming provirus alter expression of a resident transforming provirus. Cell 25 (1), 23–36. doi:10.1016/0092-8674(81)90228-2
Vijaya, S., Steffen, D. L., and Robinson, H. L. (1986). Acceptor sites for retroviral integrations map near DNase I-hypersensitive sites in chromatin. J. Virol. 60 (2), 683–692. doi:10.1128/jvi.60.2.683-692.1986
Wada, Y., Sato, T., Hasegawa, H., Matsudaira, T., Nao, N., Coler-Reilly, A. L. G., et al. (2022). RAISING is a high-performance method for identifying random transgene integration sites. Commun. Biol. 5 (1), 535. doi:10.1038/s42003-022-03467-w
Wang, W., Bartholomae, C. C., Gabriel, R., Deichmann, A., and Schmidt, M. (2016). “The LAM-PCR method to sequence LV integration sites,” in Lentiviral vectors and exosomes as gene and protein delivery tools. Editor M. Federico (New York, NY: Springer), 107–120. doi:10.1007/978-1-4939-3753-0_9
Wang, W., Fasolino, M., Cattau, B., Goldman, N., Kong, W., Frederick, M. A., et al. (2020). Joint profiling of chromatin accessibility and CAR-T integration site analysis at population and single-cell levels. Proc. Natl. Acad. Sci. 117 (10), 5442–5452. doi:10.1073/pnas.1919259117
Wehnert, S., Krüger, T., von Neuhoff, N., Hertenstein, B., Ganser, A., and Weissinger, E. M. (2004). Analysis of vector integration with LM-PCR after retroviral HSV-Tk/ΔLNGFR gene transfer. Blood 104 (11), 1752. doi:10.1182/blood.v104.11.1752.1752
Wells, D. W., Guo, S., Shao, W., Bale, M. J., Coffin, J. M., Hughes, S. H., et al. (2020). An analytical pipeline for identifying and mapping the integration sites of HIV and other retroviruses. BMC Genomics 21 (1), 216. doi:10.1186/s12864-020-6647-4
Wolf, D., and Rotter, V. (1984). Inactivation of p53 gene expression by an insertion of moloney murine leukemia virus-like DNA sequences. Mol. Cell Biol. 4 (7), 1402–1410. doi:10.1128/mcb.4.7.1402
Woods, N. B., Muessig, A., Schmidt, M., Flygare, J., Olsson, K., Salmon, P., et al. (2003). Lentiviral vector transduction of NOD/SCID repopulating cells results in multiple vector integrations per transduced cell: risk of insertional mutagenesis. Blood 101 (4), 1284–1289. doi:10.1182/blood-2002-07-2238
Xu, Z., Li, Y., Mao, Z. J., and Yin, B. (2013). The development of APE-PCR for the cloning of genomic insertion sites of DNA elements. Biologia 68 (4), 766–772. doi:10.2478/s11756-013-0214-2
Xu, D., Tang, L., Zhou, J., Wang, F., Cao, H., Huang, Y., et al. (2023). Evidence for widespread existence of functional novel and non-canonical human transcripts. BMC Biol. 21 (1), 271. doi:10.1186/s12915-023-01753-5
Yan, K. K., Condori, J., Ma, Z., Metais, J. Y., Ju, B., Ding, L., et al. (2023). Integrome signatures of lentiviral gene therapy for SCID-X1 patients. Sci. Adv. 9, eadg9959. doi:10.1126/sciadv.adg9959
Yanagawa, S. ichi, Lee, J. S., Kakimi, K., Matsuda, Y., Honjo, T., and Ishimoto, A. (2000). Identification of Notch1 as a frequent target for provirus insertional mutagenesis in T-Cell lymphomas induced by leukemogenic mutants of mouse mammary tumor virus. J. Virol. 74 (20), 9786–9791. doi:10.1128/jvi.74.20.9786-9791.2000
Ye, R., Wang, A., Bu, B., Luo, P., Deng, W., Zhang, X., et al. (2023). Viral oncogenes, viruses, and cancer: a third-generation sequencing perspective on viral integration into the human genome. Front. Oncol. 13, 1333812. doi:10.3389/fonc.2023.1333812
Yin, B. (2011). Isolation of genomic insertion sites of proviruses using Splinkerette-PCR-based procedures. Methods Mol. Biol. 687, 25–42. doi:10.1007/978-1-60761-944-4_3
Yoder, K. E., Rabe, A. J., Fishel, R., and Larue, R. C. (2021). Strategies for targeting retroviral integration for safer gene therapy: advances and challenges. Front. Mol. Biosci. 8, 662331. doi:10.3389/fmolb.2021.662331
Zhang, W., Muck-Hausl, M., Wang, J., Sun, C., Gebbing, M., Miskey, C., et al. (2013). Integration profile and safety of an adenovirus hybrid-vector utilizing hyperactive sleeping beauty transposase for somatic integration. PLoS One 8 (10), e75344. doi:10.1371/journal.pone.0075344
Zhang, P., Ganesamoorthy, D., Nguyen, S. H., Au, R., Coin, L. J., and Tey, S. K. (2020). Nanopore sequencing as a scalable, cost-effective platform for analyzing polyclonal vector integration sites following clinical T cell therapy. J. Immunother. Cancer 8 (1), e000299. doi:10.1136/jitc-2019-000299
Zhang, S., Gu, C., Huang, L., Wu, H., Shi, J., Zhang, Z., et al. (2022). The third-generation anti-CD30 CAR T-cells specifically homing to the tumor and mediating powerful antitumor activity. Sci. Rep. 12, 10488. doi:10.1038/s41598-022-14523-0
Keywords: retroviral vectors, provirus mapping, integration site, gene therapy, insertional mutagenesis, clonal expansion
Citation: Kochergin-Nikitsky K, Lavrov A and Smirnikhina S (2025) Methodological landscape in the field of integration site identification of retroviruses and retroviral vectors. Front. Bioeng. Biotechnol. 13:1708724. doi: 10.3389/fbioe.2025.1708724
Received: 19 September 2025; Accepted: 16 October 2025;
Published: 11 November 2025.
Edited by:
Segaran P. Pillai, United States Department of Health and Human Services, United StatesReviewed by:
Koon-Kiu Yan, St. Jude Children’s Research Hospital, United StatesLipei Shao, National Institutes of Health (NIH), United States
Copyright © 2025 Kochergin-Nikitsky, Lavrov and Smirnikhina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Konstantin Kochergin-Nikitsky, a29jaG5pay5rc0BnbWFpbC5jb20=