 Rafael Pereira Lemos
Rafael Pereira Lemos Diego Mariano
Diego Mariano Sabrina De Azevedo Silveira
Sabrina De Azevedo Silveira Raquel C. de Melo-Minardi
Raquel C. de Melo-Minardi- 1Laboratory of Bioinformatics and Systems, Department of Computer Science, Federal University of Minas Gerais, Belo Horizonte, Brazil
- 2Laboratory of Bioinformatics, Visualization and Systems, Department of Informatics, Federal University of Viçosa, Viçosa, Brazil
Protein interatomic contacts, defined by spatial proximity and physicochemical complementarity at atomic resolution, are fundamental to characterizing molecular interactions and bonding. Methods for calculating contacts are generally categorized as cutoff-dependent, which rely on Euclidean distances, or cutoff-independent, which utilize Delaunay and Voronoi tessellations. While cutoff-dependent methods are recognized for their simplicity, completeness, and reliability, traditional implementations remain computationally expensive, posing significant scalability challenges in the current Big Data era of bioinformatics. Here, we introduce COC
1 Introduction
Proteins are essential biological macromolecules, composed of amino acid residues linked by covalent peptide bonds. Their final three-dimensional structure is shaped not only by these covalent connections but also by weaker interactions such as hydrogen bonds, electrostatic forces, and hydrophobic effects (Smetana and Misra, 2017). The correct folding and stability of proteins are critical for their biological functions, making structural analysis fundamental for understanding cellular mechanisms, identifying therapeutic targets, and guiding the development of new drugs.
Since the first experimental resolution of a protein structure in 1958 (Kendrew et al., 1958), the field of structural biology has seen tremendous advances. Initiatives such as the Protein Data Bank (PDB, (Berman et al., 2000)) and, more recently, the AlphaFold Protein Structure Database (AFDB, (Varadi et al., 2024)) have centralized experimentally resolved and computationally predicted protein structures, making them widely accessible. The rapid growth of these repositories reflects not only advances in experimental techniques, such as X-ray crystallography, NMR spectroscopy, and cryo-electron microscopy, but also the impact of computational modeling approaches, including deep learning-based tools such as AlphaFold2 (Jumper et al., 2021) and AlphaFold3 (Abramson et al., 2024). These advances are part of the “Big Data era in Bioinformatics”, characterized by challenges related to data storage, processing, and interpretation at scale (Mura et al., 2018; Pal et al., 2020; Mariano et al., 2023).
The PDB currently holds 238,922 entries, with approximately 92% corresponding to protein structures1. The archive continues to grow at an annual rate of around 6.5%2, driven by both experimental and computational contributions (Kovalevskiy et al., 2024). This exponential expansion highlights the urgent need for computational strategies that can efficiently organize, validate, and analyze structural data at large scale. In particular, it is crucial to develop tools capable of supporting fundamental research, as well as applications in biomedical and biotechnological fields.
One key aspect of protein structure analysis is the characterization of interatomic contacts. Contacts are defined as spatial relationships between atoms or residues either within a molecule or between molecules, and are crucial for understanding protein-protein interactions, structural stability, and ligand binding mechanisms (da Silveira et al., 2009; Pires et al., 2011). In this context, it is important to distinguish between “contacts”, defined purely by spatial proximity, and “interactions”, which imply energetic contributions such as hydrophobic or electrostatic forces (Godzik et al., 1992; da Silveira et al., 2009). While not every contact results in a functional interaction, the presence of contacts is often a prerequisite for biologically relevant interactions. Therefore, in the remainder of this paper, the terms contact and interaction may be used interchangeably where appropriate, with “contact” referring primarily to spatial proximity and “interaction” to biochemical context.
Computational methods for contact identification offer an efficient alternative to labor-intensive experimental approaches, facilitating large-scale analyses across protein families and databases (Ding and Kihara, 2018). Traditionally, contacts are identified using Euclidean distance thresholds or cutoff-independent methods such as Voronoi (Voronoi, 1908) or Delaunay tessellations (Delaunay, 1934). Although cutoff-independent approaches are more sophisticated in theory, distance-based methods are often preferred for their simplicity, efficiency, and interpretability (da Silveira et al., 2009; Pires et al., 2011). Recent refinements incorporate physicochemical characteristics such as polarity or charge alongside spatial proximity, improving the biological relevance of computational predictions and reducing the incidence of false positives.
Several tools and databases have been developed to identify and analyze protein contacts (Wallace et al., 1995; Mancini et al., 2004; Schreyer and Blundell, 2009; Laskowski and Swindells, 2011; Bickerton et al., 2011; Pires et al., 2011; Schreyer and Blundell, 2013; Jubb et al., 2017; Fassio et al., 2020; Pimentel et al., 2021). However, existing solutions often present one or more limitations: they may be static, based on predefined datasets; computationally expensive, hindering large-scale use; restricted by server bottlenecks; limited to specific contact types such as residue-residue or protein-ligand; based on cutoff-independent methods; unsupported for modern file formats such as mmCIF; or discontinued altogether.
While these algorithms are well-established in the literature and typically can perform well for single structures, their computational cost becomes a bottleneck in large-scale analyses. Although our current study is based on experimentally determined structures from the PDB, the underlying method is designed with scalability in mind. The landscape of available protein structures has been further expanded by ultra-large-scale prediction initiatives. Notably, the AFDB now provides access to millions of high-confidence predicted models, vastly increasing the volume of structural data available for analysis. This shift underscores the growing importance of methods that combine accuracy with computational efficiency, as the feasibility of analyzing such extensive datasets hinges on scalable algorithms. In addition, time-resolved techniques such as molecular dynamics (MD) simulations introduce another dimension of complexity. These simulations generate thousands of frames per trajectory, each representing a unique protein conformation. Performing contact calculations across such datasets requires algorithms that can process structural information repeatedly and efficiently.
In response to these challenges, we propose COC
To validate and benchmark COC
2 Methodology
Figure 1 outlines the methodology for developing and benchmarking COC
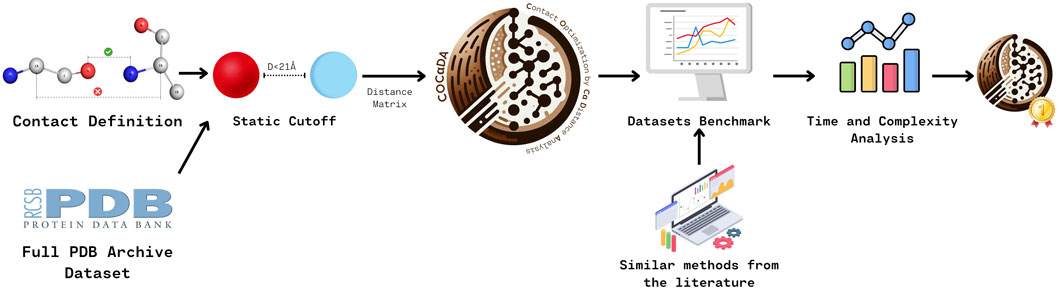
Figure 1. Overview of the methodology used to create and benchmark COC
2.1 Contact definition
To store the contact types and their conditions, we used a dictionary containing all heavy atoms from the 20 standard amino acids, as defined in (Sobolev et al., 1999; Silva et al., 2019; Fassio et al., 2020; Barroso et al., 2020; Pimentel et al., 2021; Dos Santos et al., 2022). All 20 standard amino acids had their heavy atoms classified by the following characteristics, in binary form (Table 1): tendency to contribute to hydrophobic effects, belonging to aromatic groups, having positive charge, having negative charge, capability of donating or accepting electrons. The full atom classification table is available in the Supplementary Table S1.
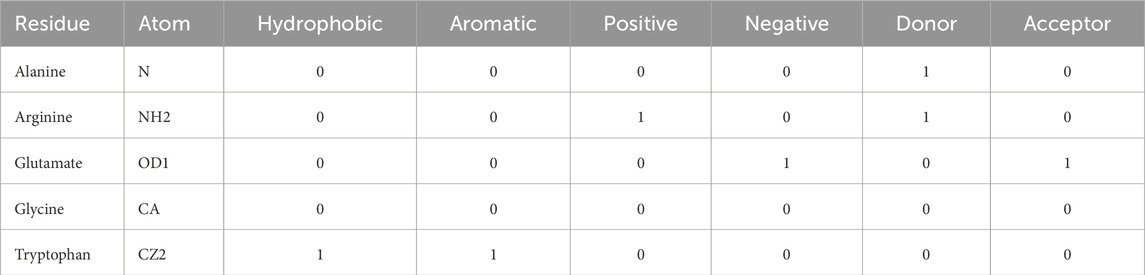
Table 1. Example of the binary classification of heavy atoms, according to their characteristics. For each amino acid residue, all their heavy atoms were classified in a binary manner, according to the following characteristics (hydrophobic, aromatic, positive, negative, donor, acceptor). Atom names follow the PDB nomenclature.
The possible contact types are: hydrogen and disulfide bonds; hydrophobic effects; attractive, repulsive, and salt bridge interactions; and aromatic stackings. This dictionary also contains the conditions needed for the contact (e.g., to form an attractive interaction, the atoms must be differently charged), and the range of Euclidean distances, in angstroms, for the contact to occur (Table 2).
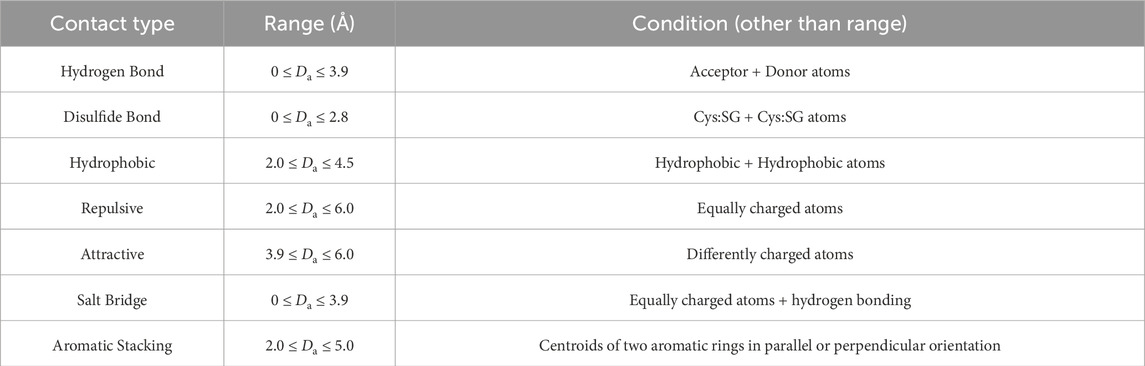
Table 2. Summary of Types, Range and Conditions for contacts to occur.
2.2 Protein Data Bank archive
The full PDB protein archive, in ‘.cif’ format, was obtained using in-house scripts to query and download entries directly from the PDB API. First, a script was used to query the API for entries containing “Protein” as an exact match from the parameter “entity_poly.rcsb_entity_polymer_type”.
To avoid rate limits and overwhelming the server, queries had a 1 s delay from one another, and only 25,000 IDs were obtained at a time. Then, a second script was used, together with the Biopython Bio. PDB module (Cock et al., 2009), to download all files that matched the IDs gathered in the first step. All files were downloaded between July 4th and 10 July 2024.
2.3 Neighbor search implementation using biopython
To serve as a comparison to our method, the Biopython package (Cock et al., 2009), largely used in bioinformatics, was used. The Bio. PDB module contains tools to parse a. pdb or. cif file, as well as the NeighborSearch (NS) class, which is useful in interatomic contact determination.
We used an in-house Python script to perform an all-atom neighbor search of 6Å radius, the maximum distance for contacts defined in our dictionary. Then, the neighbors were filtered based on their distance and physicochemical properties relative to the parent atom. Redundant comparisons were excluded; for example, if atom “
The contacts obtained contained the following information: chain, residue number, and parent atom name; chain, residue number, and neighbor atom name (i.e., the atomic pair making the contact); type of contact; and distance between the two atoms.
2.4 General implementation
To analyze the PDB protein archive and obtain the maximum distances matrix used in the rest of this work, we first devised a Static Cutoff (SC) implementation, where the C
After parsing, the protein object is passed to a contact calculation script, where the C
The atoms from the residues that are in range to interact are then compared to the dictionary previously described, based on their distance to each other, and their physicochemical properties. Finally, the contacts are returned in a custom object containing all their information, similar to the NS method.
2.5 Distance matrix
Throughout the processing of the complete PDB archive using the SC method, the maximum distances (across all proteins in the PDB) between the C
The distance matrix
where each
A total of 210 distance values were obtained, representing each possible residue pair and excluding redundancies (e.g., Ala-Val is the same as Val-Ala) (Equation 2).
where
2.6 COC
The COC
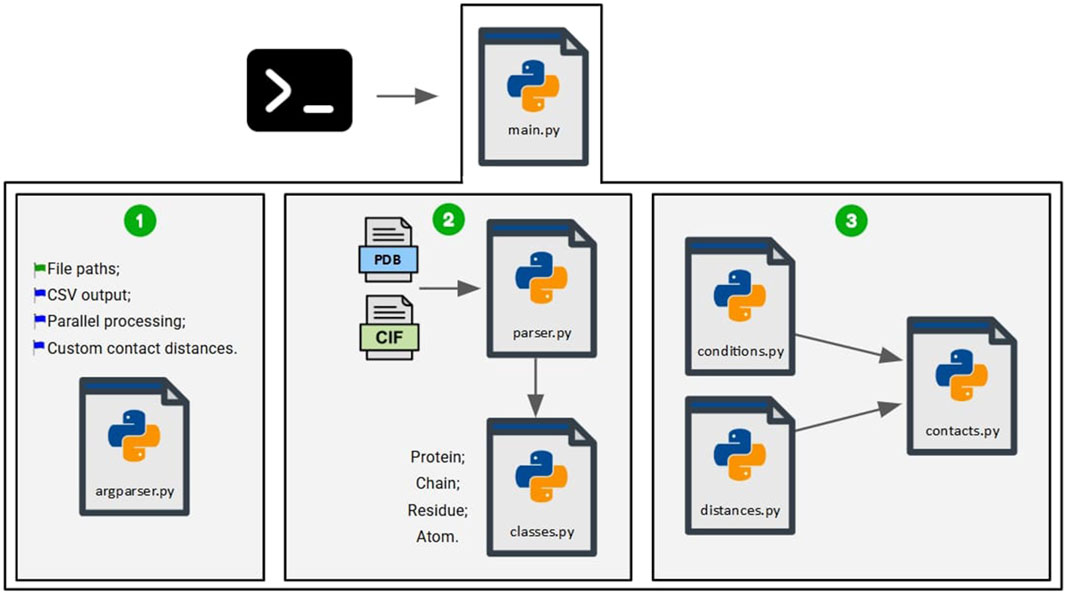
Figure 2. Scripts used for the COC
First, the script “main.py” is executed via the command line, along with the required parameters, which are processed by the script “argparser.py” (1). The parameters include the mandatory file paths (wildcards are accepted), an optional binary flag for generating an output file in. csv format (default = no), an optional parameter to parallelize file processing in batches across any combination of available CPU cores, and an optional parameter to use custom contact distances defined by the user instead of those defined in Table 2 (using the “contact_distances.json” configuration file). All parameters are fully explained using the ‘-h’ or ‘–help’ flags.
When users specify custom contact cutoffs greater than the default (6Å), the static C
The files are then processed by the script “parser.py” (2), which utilizes the previously defined classes to create objects representing proteins, their chains, residues, and atoms. Finally, the script “contacts.py” receives the objects generated by the parser (3). The scripts “conditions.py”, which stores the conditions required for contact detection, and “distances.py”, which stores the values obtained from the distance matrix, are used to compute the contacts.
If the user does not specify the. csv output file parameter, only a summary is displayed in the terminal, containing the protein name, residue count, number of contacts, and processing time in seconds. If the parameter is used, in addition to the summary, a. csv file is generated with detailed information about each detected protein contact. The columns in the output file are organized as follows: Chain 1, Residue Number 1, Residue Name 1, Atom Name 1, Chain 2, Residue Number 2, Residue Name 2, Atom Name 2, Distance, Contact Type. An example output file for PDB ID 101M, as well as a PyMOL (Schrödinger, LLC) visualization script to help users quickly explore the results, are available at the Supplementary GitHub repository.
2.7 Datasets
Two datasets were selected to benchmark our results and compare them to other competitors (
2.8 Benchmarks
To ensure fairness and eliminate biases, all benchmarks were conducted simultaneously on a server with the following specifications: NVIDIA A100 GPU, 768 GB RAM, and a 128-thread AMD Ryzen Threadripper 5995WX processor. To prevent memory overload and parallelization issues, each process was executed on an individual core. Due to its size, dataset D2 was divided into nine batches of approximately 25,000 files each, with each batch being processed independently on separate cores.
Although multithreading and batch processing is available for all implementations (NS, SC, and COC
3 Results and discussion
3.1 Maximum distance matrix
In total, 217,454 PDB entries were downloaded in. cif format, totaling approximately 450 GB (The full ID list is available on the Supplementary GitHub repository). Proteins ranged from three (PDB IDs: 1Q7O, 8DDG, 8DDH) to 503,221 (PDB ID: 8GLV) modeled amino acid residues. To obtain the values for the distance matrix, we processed all the downloaded files using a fixed C
Using the SC implementation and the 217,454 entries downloaded from the PDB, over 211 million amino acid residues and 819 million contacts were processed and identified. Along this process, we stored the maximum C
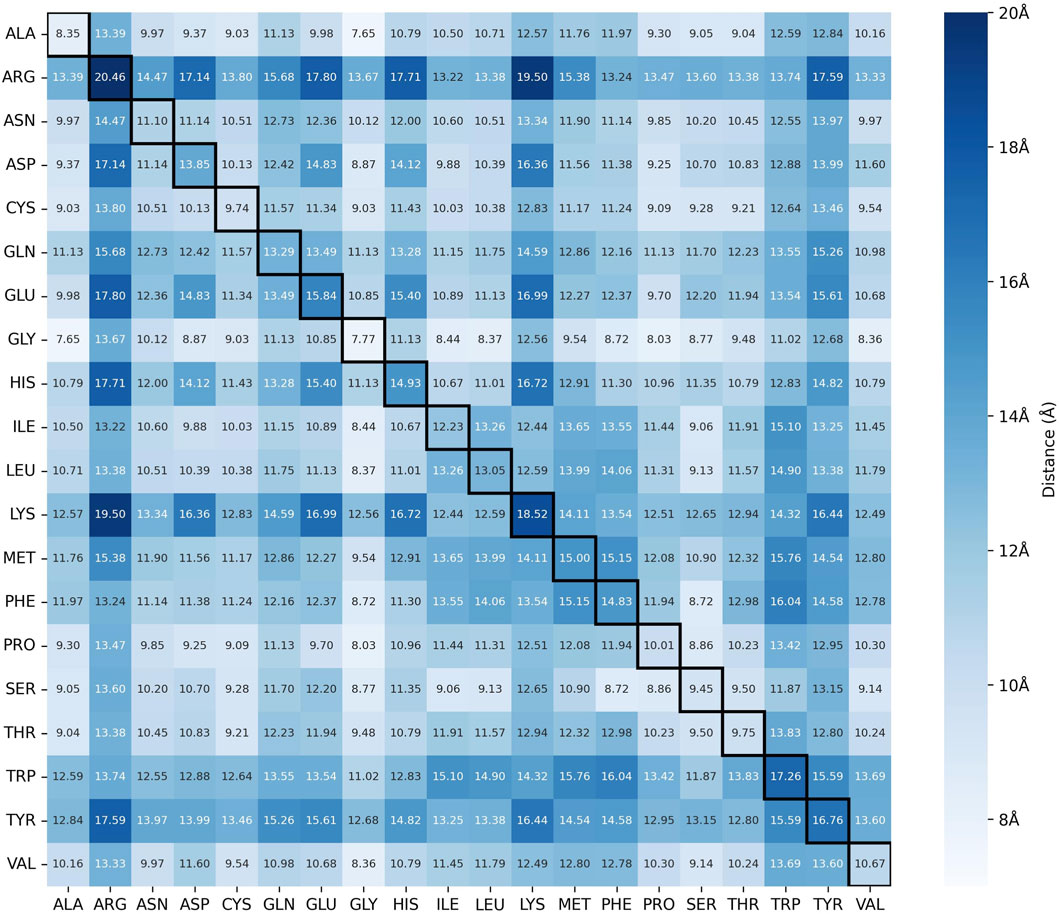
Figure 3. Distance Matrix between the C
As the distance matrix is color-coded based on the value of the maximum C
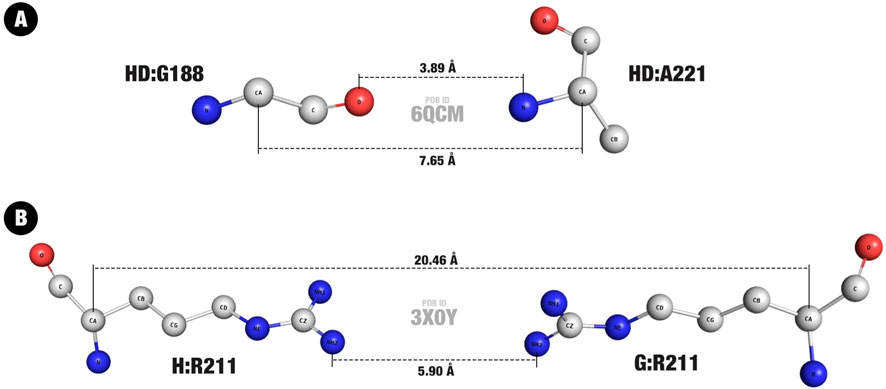
Figure 4. Minimum and maximum entries in the Distance Matrix. (A) Minimum value, in a hydrogen bond between a glycine and an alanine residue. (B) Maximum value, in a repulsive interaction between two arginine residues. The higher number (larger dotted line) represents the distance between C
On the other hand, a pair of two arginine residues, from chains G and H of PDB ID 3X0Y, represents the highest C
If the user wishes to apply custom contact distances instead of those defined in Table 2, the optional ‘-d’ flag can be used, specifying the desired values in the “contact_distances.json” configuration file. This flag extends the C
Use-case scenarios for modifying the default cutoff distances include adopting alternative minimum or maximum values reported in the literature, such as 6Å for aromatic stacking (Fassio et al., 2022), or 5Å for hydrophobic effects (Bickerton et al., 2011), instead of the default 5Å and 4.5Å, respectively. Another common scenario involves exploratory analyses using step-wise cutoff variations (da Silveira et al., 2009; Vangone and Bonvin, 2015).
3.2 COC
With the maximum possible C
By using tightly-defined, pair-specific cutoff distances, we can further enhance the search space pruning compared to using a single fixed distance threshold. Analysis of the distribution of maximum C
To improve efficiency, COC
The residue-pair-specific distance matrix used in COC
While the method is robust to structural variability at the backbone level, the accuracy of contact detection may be influenced by uncertainty in side-chain atom positions, particularly in lower-resolution or flexible regions. In such cases, performance near cutoff boundaries may be affected, which is an inherent limitation of any deterministic cutoff-dependent method.
3.3 Benchmarks
To benchmark COC
Both Arpeggio versions were too slow to process even small proteins, as our tests showed processing times of approximately 5 and 23 min for a single 1,000 residue protein (PDB ID 6RTH) for Arpeggio CLI and Arpeggio Web, respectively. For comparison, the same protein was processed in 0.62s using COC
For the AllAtoms approach, a new implementation was developed in Python, in order to incorporate all the previously defined definitions and constraints, and for the NS method, a custom implementation was created using Biopython (Cock et al., 2009), since the PICCOLO tool is currently unavailable. Thus, for D1, the following methods were compared: AllAtoms, NS, SC and COC
The first dataset contains 896 entries, ranging from one to 994 residues. The initial choice of a smaller dataset was made to include the AllAtoms approach, which is significantly slower than the other three (but still considerably faster than both versions of Arpeggio), which would make a large-scale analysis unfeasible.
In Figure 5, it is possible to see that the AllAtoms approach (orange) rapidly explodes in a quadratic curve compared to the three others, which maintain rather linear calculation times up to 1,000 residues. Once again, no contacts of any type were missed in any of the approaches (Supplementary Table S2), but the AllAtoms approach was removed from further analysis because of its performance. Comparing the faster approaches, SC (yellow) obtained calculation times 1.5x faster on average than NS (magenta), while COC
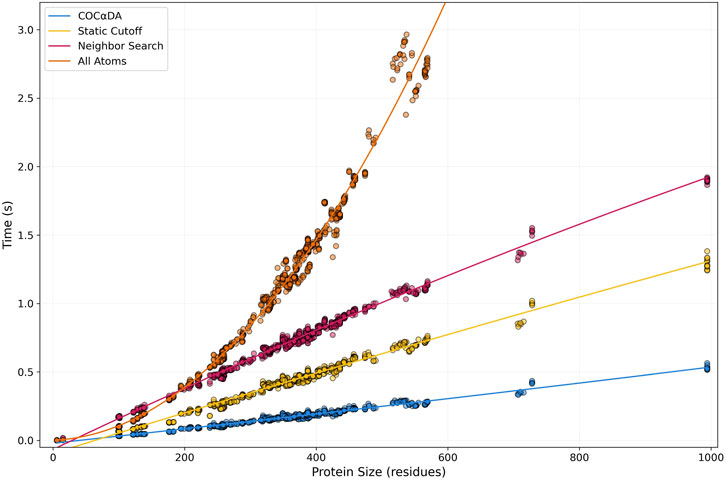
Figure 5. Protein Size vs. Computation Time plot of Benchmark 1. In the first benchmark, 896 files ranging from 1 to 994 residues were analyzed. COC
As the results from the first, small dataset showed a significant difference in processing times between the 3 fastest approaches, we then moved to D2, which contains 215,716 unique entries, ranging from three to 10,000 residues, making approximately 99.2% of the PDB protein archive. The choice to remove entries above 10,000 residues was made due to the nature of those entries, which are mostly protein complexes, containing several copies of each unique chain. This makes them not suitable for contact analysis directly, requiring some kind of pre-processing, like splitting only the unique chains or working with each individual protein present in the complex separately. This can also be true for entries below 10,000 residues, but we believe that this slice correctly represents the diversity of experimentally resolved protein structures.
Figure 6 shows the results for D2 when comparing the COC
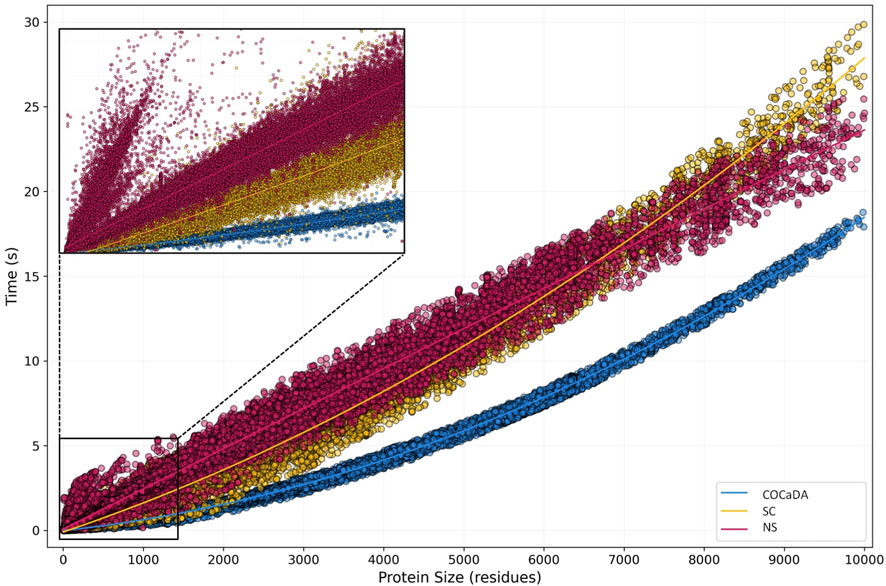
Figure 6. Protein Size vs. Computation Time plot of Benchmark 2. In the second benchmark, 215,716 files were analyzed, ranging from 3 to 10,000 residues. COC
Outliers were considered as entries that had a processing time
The protein size range of 0–1,000 residues is noteworthy, as shown in detail in the upper left corner of Figure 6. At this range, we can see the distribution pattern observed in D1, while also identifying several divergent entries in the NS approach. The divergent spike is composed exclusively of Nuclear Magnetic Resonance (NMR) resolved entries, which are usually deposited as several individual models of the same protein. Due to the nature of Biopython native parsing, all the models need to be parsed even if only the first one is of interest, unless the user creates specific functions for this purpose, thus deviating from the original implementation of the library. As these NMR entries are small, the parsing time of several NMR models outpaces the contact calculation time of the first one, leading to a spike in processing time. This does not occur in the COC
3.4 Empirical complexity analysis
Computing interatomic contacts is inherently a quadratic problem (O
In the case of small entries, like most of the protein structures, the memory overhead associated with the allocation and creation of the tree usually does not outweigh the computational gains in search, insertion, and deletion operations. Thus, in addition to the high implementation complexity of
Our approach in COC
In this study, we chose to evaluate the complexity of various algorithms empirically, by comparing standard methods commonly used in the structural bioinformatics community with the COC
The curve fittings of the three approaches against the second dataset demonstrate that both COC
where
However, in the case of the NS approach, the memory overhead in tree construction is evident, especially for small inputs. It is possible to observe, in Figure 5, that for proteins of up to 200 residues even the most computationally expensive approach (AllAtoms) achieves lower processing times than NS. This can also be seen in the range that includes small entries in D2, although with less detail due to the massive number of points (Figure 6, detail).
The analysis of the coefficients from the obtained equations is another way to explain the poorer performance of NS, even though it grows linearly compared to the quadratic growth of the other approaches. Starting with the quadratic coefficients
As for the linear coefficients
Finally, regarding the constant coefficients
As previously shown, the NS approach (using k-d trees) has a linear time complexity in the worst case. Meanwhile, COC
where
However, only slightly more than 2,000 entries in the PDB have a size greater than 14,000 residues, representing less than 1% of all protein structures. Furthermore, all of these entries correspond to protein complexes or repetitions of the same protein, as previously discussed. Therefore, for all practical purposes of contact detection in proteins, the COC
4 Conclusion
In a context where the influx of biological data is greater than ever, there is an increasing need for solutions that are efficient, robust, and scalable. In response to this demand, we developed COC
Contact calculation between residues provides essential information on protein structure, function, stability, evolution, and molecular interactions. Although existing algorithms perform well for individual structures, their computational cost can become limiting in large-scale applications, such as analysis of structural databases like the PDB and AFDB, and molecular dynamics simulations where contacts must be recalculated for thousands of frames. More efficient methods, like the one we present here, enable faster processing and broader analyses across large datasets.
By leveraging structural and physicochemical knowledge of amino acids, we derived optimal main-chain alpha-carbon cutoff values for each amino acid pair, which significantly reduces the computational cost of detecting interatomic contacts. Advanced data structures such as
This approach led to a stable and efficient method tailored to real-world structural datasets. This strategic simplicity not only outperforms more complex alternatives in practice but also simplifies implementation by requiring neither external libraries nor advanced programming skills. Its scalable performance makes COC
The current version of COC
Data availability statement
The datasets presented in this study can be found in online repositories. This data can be found here: https://github.com/lbs-ufmg/cocada_supplementary.
Author contributions
RL: Writing – review and editing, Software, Methodology, Writing – original draft, Formal Analysis, Visualization, Data curation, Validation, Investigation, Conceptualization. DM: Validation, Visualization, Formal Analysis, Writing – original draft, Investigation, Data curation, Methodology, Writing – review and editing. SS: Supervision, Writing – review and editing, Methodology, Conceptualization, Writing – original draft. Raquel CM-M: Writing – original draft, Methodology, Resources, Project administration, Validation, Conceptualization, Supervision, Funding acquisition, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Finance Code 001; Fundação de Amparo à Pesquisa do Estado de Minas Gerais - Brasil (FAPEMIG) - Finance Codes APQ-01834-21, APQ-02690-22, APQ-01838-24; and Conselho Nacional de Desenvolvimento Científico e Tecnológico - Brasil (CNPq) - Finance Codes 310406/2023-4, 440307/2022-8.
Acknowledgments
The authors thank the funding agencies: Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbinf.2025.1630078/full#supplementary-material
Footnotes
1Available at https://www.rcsb.org/stats/explore/polymer_entity_type. Accessed 23 August 2024.
2Available at https://www.rcsb.org/stats/growth/growth-protein. Accessed 23 August 2024.
3Available at https://github.com/PDBeurope/arpeggio/.
References
Abramson, J., Adler, J., Dunger, J., Evans, R., Green, T., Pritzel, A., et al. (2024). Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500. doi:10.1038/s41586-024-07487-w
Barroso, J. R. M. S., Mariano, D., Dias, S. R., Rocha, R. E. O., Santos, L. H., Nagem, R. A. P., et al. (2020). Proteus: an algorithm for proposing stabilizing mutation pairs based on interactions observed in known protein 3D structures. BMC Bioinforma. 21, 275. doi:10.1186/s12859-020-03575-6
Bentley, J. L. (1975). Multidimensional binary search trees used for associative searching. Commun. ACM 18, 509–517. doi:10.1145/361002.361007
Berg, M., Cheong, O., Kreveld, M., and Overmars, M. (2008). “Orthogonal range searching,” in Computational geometry (Berlin, Heidelberg: Springer), 95–120.
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The protein data bank. Nucleic Acids Res. 28, 235–242. doi:10.1093/nar/28.1.235
Bickerton, G. R., Higueruelo, A. P., and Blundell, T. L. (2011). Comprehensive, atomic-level characterization of structurally characterized protein-protein interactions: the PICCOLO database. BMC Bioinforma. 12, 313. doi:10.1186/1471-2105-12-313
Brown, R. A. (2015). Building a balanced k-d tree in o(kn log n) time. J. Comput. Graph. Tech. (JCGT) 4, 50–68. doi:10.48550/arXiv.1410.5420
Brown, S. D., Gerlt, J. A., Seffernick, J. L., and Babbitt, P. C. (2006). A gold standard set of mechanistically diverse enzyme superfamilies. Genome Biol. 7, R8. doi:10.1186/gb-2006-7-1-r8
Cock, P. J. A., Antao, T., Chang, J. T., Chapman, B. A., Cox, C. J., Dalke, A., et al. (2009). Biopython: freely available python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. doi:10.1093/bioinformatics/btp163
da Silveira, C. H., Pires, D. E. V., Minardi, R. C., Ribeiro, C., Veloso, C. J. M., Lopes, J. C. D., et al. (2009). Protein cutoff scanning: a comparative analysis of cutoff dependent and cutoff free methods for prospecting contacts in proteins. Proteins 74, 727–743. doi:10.1002/prot.22187
Delaunay, B. (1934). “Sur la sphère vide. À la mémoire de georges voronoï,” in Bulletin de l’Académie des Sciences de l’URSS. Classe des sciences mathématiques et naturelles VII, 793–800.
Ding, Z., and Kihara, D. (2018). Computational methods for predicting protein-protein interactions using various protein features. Curr. Protoc. Protein Sci. 93, e62. doi:10.1002/cpps.62
Dos Santos, V. P., Rodrigues, A., Dutra, G., Bastos, L., Mariano, D., Mendonça, J. G., et al. (2022). E-Volve: understanding the impact of mutations in SARS-CoV-2 variants spike protein on antibodies and ACE2 affinity through patterns of chemical interactions at protein interfaces. PeerJ 10, e13099. doi:10.7717/peerj.13099
Fassio, A. V., Santos, L. H., Silveira, S. A., Ferreira, R. S., and de Melo-Minardi, R. C. (2020). Napoli: a graph-based strategy to detect and visualize conserved protein-ligand interactions in large-scale. IEEE/ACM Trans. Comp. Biol. Bioinf. 17, 1317–1328. doi:10.1109/tcbb.2019.2892099
Fassio, A. V., Shub, L., Ponzoni, L., McKinley, J., O’Meara, M. J., Ferreira, R. S., et al. (2022). Prioritizing virtual screening with interpretable interaction fingerprints. J. Chem. Inf. Model. 62, 4300–4318. doi:10.1021/acs.jcim.2c00695
Godzik, A., Kolinski, A., and Skolnick, J. (1992). Topology fingerprint approach to the inverse protein folding problem. J. Mol. Biol. 227, 227–238. doi:10.1016/0022-2836(92)90693-e
Jubb, H. C., Higueruelo, A. P., Ochoa-Montaño, B., Pitt, W. R., Ascher, D. B., and Blundell, T. L. (2017). Arpeggio: a web server for calculating and visualising interatomic interactions in protein structures. J. Mol. Biol. 429, 365–371. doi:10.1016/j.jmb.2016.12.004
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi:10.1038/s41586-021-03819-2
Kendrew, J. C., Bodo, G., Dintzis, H. M., Parrish, R. G., Wyckoff, H., and Phillips, D. C. (1958). A three-dimensional model of the myoglobin molecule obtained by x-ray analysis. Nature 181, 662–666. doi:10.1038/181662a0
Kovalevskiy, O., Mateos-Garcia, J., and Tunyasuvunakool, K. (2024). AlphaFold two years on: validation and impact. Proc. Natl. Acad. Sci. U. S. A. 121, e2315002121. doi:10.1073/pnas.2315002121
Laskowski, R. A., and Swindells, M. B. (2011). LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786. doi:10.1021/ci200227u
Mancini, A. L., Higa, R. H., Oliveira, A., Dominiquini, F., Kuser, P. R., Yamagishi, M. E. B., et al. (2004). STING contacts: a web-based application for identification and analysis of amino acid contacts within protein structure and across protein interfaces. Bioinformatics 20, 2145–2147. doi:10.1093/bioinformatics/bth203
Mariano, D., Da Fonseca Júnior, N. J., Santos, L. H., and de Melo-Minardi, R. C. (2023). Editorial: bioinformatics in the age of data science: algorithms, methods, and tools applied from omics to structural data. Front. Bioinforma. 3, 1246859. doi:10.3389/fbinf.2023.1246859
Mura, C., Draizen, E. J., and Bourne, P. E. (2018). Structural biology meets data science: does anything change? Curr. Opin. Struct. Biol. 52, 95–102. doi:10.1016/j.sbi.2018.09.003
Pal, S., Mondal, S., Das, G., Khatua, S., and Ghosh, Z. (2020). Big data in biology: the hope and present-day challenges in it. Gene Rep. 21, 100869. doi:10.1016/j.genrep.2020.100869
Pimentel, V., Mariano, D., Cantão, L. X. S., Bastos, L. L., Fischer, P., de Lima, L. H. F., et al. (2021). VTR: a web tool for identifying analogous contacts on protein structures and their complexes. F. Bioinf. 1, 730350. doi:10.3389/fbinf.2021.730350
Pires, D. E. V., de Melo-Minardi, R. C., dos Santos, M. A., da Silveira, C. H., Santoro, M. M., and Meira, W. (2011). Cutoff scanning matrix: structural classification and function prediction by protein inter-residue distance patterns. BMC Gen. 12, S12. doi:10.1186/1471-2164-12-S4-S12
Schreyer, A., and Blundell, T. (2009). CREDO: a protein-ligand interaction database for drug discovery. Chem. Biol. Drug Des. 73, 157–167. doi:10.1111/j.1747-0285.2008.00762.x
Schreyer, A. M., and Blundell, T. L. (2013). CREDO: a structural interactomics database for drug discovery. Database (Oxford) 2013, bat049. doi:10.1093/database/bat049
Silva, M. F. M., Martins, P. M., Mariano, D. C. B., Santos, L. H., Pastorini, I., Pantuza, N., et al. (2019). Proteingo: motivation, user experience, and learning of molecular interactions in biological complexes. Entertain. Comput. 29, 31–42. doi:10.1016/j.entcom.2018.11.001
Smetana, J. H. C., and Misra, G. (2017). “Principles of protein structure and function,” in Intro. to biomol. Struct. and biophys. (Singapore: Springer), 1–32.
Sobolev, V., Sorokine, A., Prilusky, J., Abola, E. E., and Edelman, M. (1999). Automated analysis of interatomic contacts in proteins. Bioinformatics 15, 327–332. doi:10.1093/bioinformatics/15.4.327
Vangone, A., and Bonvin, A. M. (2015). Contacts-based prediction of binding affinity in protein-protein complexes. Elife 4, e07454. doi:10.7554/elife.07454
Varadi, M., Bertoni, D., Magana, P., Paramval, U., Pidruchna, I., Radhakrishnan, M., et al. (2024). AlphaFold protein structure database in 2024: providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 52, D368–D375. doi:10.1093/nar/gkad1011
Voronoi, G. (1908). Nouvelles applications des paramètres continus à la théorie des formes quadratiques. deuxième mémoire. recherches sur les parallélloèdres primitifs. J. für die reine und angewandte Math. 134, 198–287. doi:10.1515/crll.1908.133.97
Keywords: COCαDA, protein interactions, contacts, structural bioinformatics, command-line tool
Citation: Lemos RP, Mariano D, Silveira SDA and de Melo-Minardi RC (2025) COCαDA - a fast and scalable algorithm for interatomic contact detection in proteins using Cα distance matrices. Front. Bioinform. 5:1630078. doi: 10.3389/fbinf.2025.1630078
Received: 16 May 2025; Accepted: 11 August 2025;
Published: 01 September 2025.
Edited by:
Fabricio Martins Lopes, Universidade Tecnológica Federal do Paraná (UTFPR), BrazilReviewed by:
Yuki Kagaya, Purdue University, United StatesGaihua Zhang, Hunan Normal University, China
Copyright © 2025 Lemos, Mariano, Silveira and de Melo-Minardi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rafael Pereira Lemos, cmFmYWVsbGVtb3NAdWZtZy5icg==