 Ling-bing Meng
Ling-bing Meng Yuan-meng Zhang3
Yuan-meng Zhang3 Tao Gong
Tao Gong- 1Department of Cardiology, National Center of Gerontology, Institute of Geriatric Medicine, Beijing Hospital, Chinese Academy of Medical Sciences, Beijing, China
- 2Graduate School of Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing, China
- 3Department of Internal Medicine, The Third Medical Centre of Chinese People's Liberation Army (PLA) General Hospital, The Training Site for Postgraduate of Jinzhou Medical University, Beijing, China
- 4Department of Respiratory, The First Affiliated Hospital of Jinzhou Medical University, Jinzhou, China
- 5Department of Neurology, National Center of Gerontology, National Center of Gerontology, Institute of Geriatric Medicine, Beijing Hospital, Chinese Academy of Medical Sciences, Beijing, China
Atherosclerosis (AS) is a chronic vascular inflammatory disease, in which the lipid accumulation in the intima of the arteries shows yellow atheromatous appearance, which is the pathological basis of many diseases, such as coronary artery disease, peripheral artery disease and cerebrovascular disease. In recent years, it has become the main cause of death in the global aging society, which seriously endangers human health. As a result, research on AS is increasing. Lesions of atherosclerosis contain macrophages, T cells and other cells of the immune response, together with cholesterol that infiltrates from the blood. Recent studies have shown that chronic stress plays an important role in the occurrence and development of AS. From the etiology of disease, social, environmental and genetic factors jointly determine the occurrence of disease. Atherosclerotic cardio-cerebrovascular disease (ASCVD) is often caused by chronic stress (CS). If it cannot be effectively prevented, there will be biological changes in the body environment successively, and then the morphological changes of the corresponding organs. If the patient has a genetic predisposition and a combination of environmental factors triggers the pathogenesis, then chronic stress can eventually lead to AS. Therefore, this paper discusses the influence of chronic stress on AS in the aspects of inflammation, lipid metabolism, endothelial dysfunction, hemodynamics and blood pressure, plaque stability, autophagy, ferroptosis, and cholesterol efflux.
Introduction
Atherosclerosis (AS) is considered as a non-specific inflammatory disease, mainly involving the intima and medial layer of the arterial wall, which is the pathological basis of various cardiovascular and cerebrovascular diseases (1, 2). Cardiovascular disease is still the leading cause of death worldwide, with an increasing prevalence in developing countries (3). In recent years, the rapid economic development in China has led to the change of lifestyle and the aggravation of population aging (4). The incidence and prevalence of chronic non-communicable diseases, such as hypertension, hyperlipidemia, diabetes, hyperuricemia, and chronic psychological stress, are increasing year by year, and the AS caused by these diseases is also becoming more and more serious (5–9). AS is the main cause of atherosclerotic cardiovascular disease (ASCVD), which is the leading cause of disability and death among urban and rural residents in China (10). In China, cardiovascular diseases account for about 45% of the deaths of the population, causing a serious medical burden and becoming a major public health problem (11). What's more, the incidence of ASCVD in China continues to rise. As is the main cause of ASCVD (12). The pathophysiological development of AS is closely related to the mutation and abnormal expression of genes, including fms-like tyrosine kinase-1 (Flt-1), tumor necrosis factor-α (TNF-α), apolipoprotein A-I (apo A-I), Vascular Endothelial Growth Factor (VEGF), and Angiogenin (ANG). Previous studies have shown that low expression of Flt-1 could predict the development of endothelial injury, which leads to the development of AS (13). In addition, the stronger the proliferative ability of endothelial progenitor cells (EPCs), the lower the vulnerability of vascular endothelium. Thus, TNF-α overexpression damages the vascular endothelium by disrupting the proliferation process of EPCs (14). The mutation of the anti-atherosclerosis gene, apo A-I, could accelerate the apoptosis of vascular endothelial cells by down-regulating the levels of endothelial nitric oxide synthase (eNOS) and heme oxygenase-1, and eventually lead to the formation of atherosclerotic plaque (15). The expression of VEGF and ANG could promote the regeneration of vascular endothelial cells (16, 17). Therefore, the abnormal expression of VEGF and ANG might play an important role in the occurrence and development of AS (18–20). The up-regulation of “VEGF and ANG” plays a significant role in the development of AS. Compared with the normal artery tissues, the expression of “VEGF and ANG” were higher in the AS tissues. The main biological function of ANG is to promote angiogenesis, which promotes plaque instability (21). VEGF is the strongest known factor promoting angiogenesis, which could promote endothelial cell mitosis and proliferation, increase vascular permeability and promote endothelial cell migration (22). Furthermore, VEGF could promote intimal hyperplasia and aggravate AS by promoting monocyte activation, adhesion, and migration and increasing permeability of endothelial cells (23). However, one of the important reasons for the current inability to effectively control the occurrence and recurrence of ASCVD is that the occurrence and progress of atherosclerotic stenosis and vulnerable plaques cannot be detected in time, dynamically monitored, and effectively controlled, which is also the main research field for ASCVD in China and abroad (24–26). So it is imperative to explore the risk factors for the occurrence and development of atherosclerosis for the early diagnosis and precise treatment of ASCVD (27).
Chronic stress induces changes in organisms that increase the risk of atherosclerotic diseases, including heart disease, stroke, and transient ischemic attack (8, 28). The report shows that stress increases the risk of cardiovascular disease (29). A large amount of evidence confirms that chronic stress plays a significant role in the occurrence and development of AS, but the specific mechanism is still unclear (30–33). The purpose of this paper is to provide a comprehensive review of studies on the effects of chronic stress in healthy individuals and patients with cardiovascular disease (CVD). This study focuses on the research progress of the relationship between chronic stress and AS in the aspects of inflammation, lipid metabolism, endothelial dysfunction, hemodynamics and blood pressure, plaque stability, autophagy, ferroptosis, and cholesterol efflux.
Methods
Our systematic review is a new method of literature synthesis. It systematically and comprehensively collects the published and unpublished studies on a specific clinical problem, and uses the principles and methods of strict evaluation of clinical epidemiology to select the literatures that meet the quality standards for qualitative combination, so as to draw reliable comprehensive conclusions. This systematic review was conducted in accordance with the Preferred Reporting Items for Systematic Reviews statement guidelines. A protocol was developed prior to commencing this review on PROSPERO. The procedure of searching the references in the databases was manifested in the flow diagram (Figure 1; Table 1).
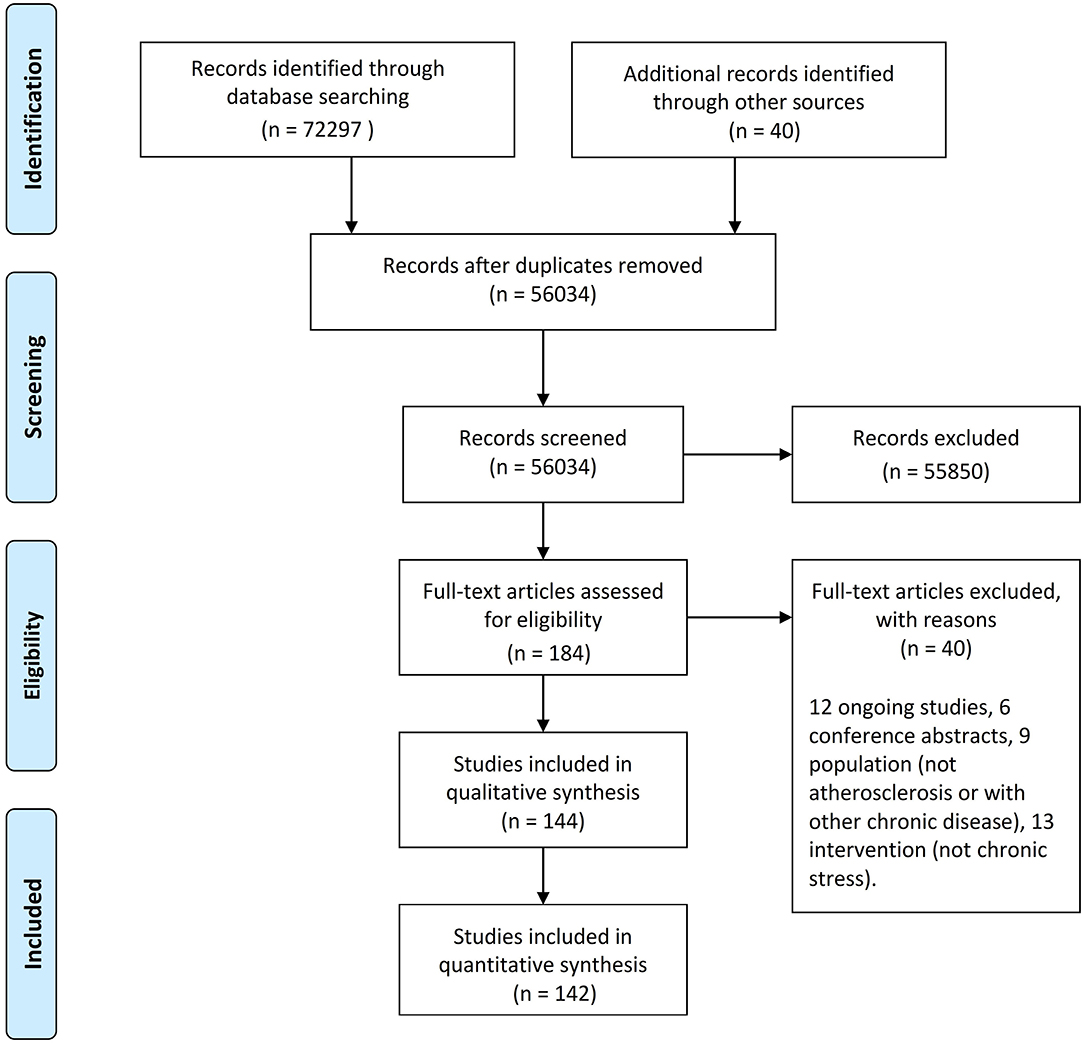
Figure 1. Flow diagram showing the procedure of searching the references in the databases.
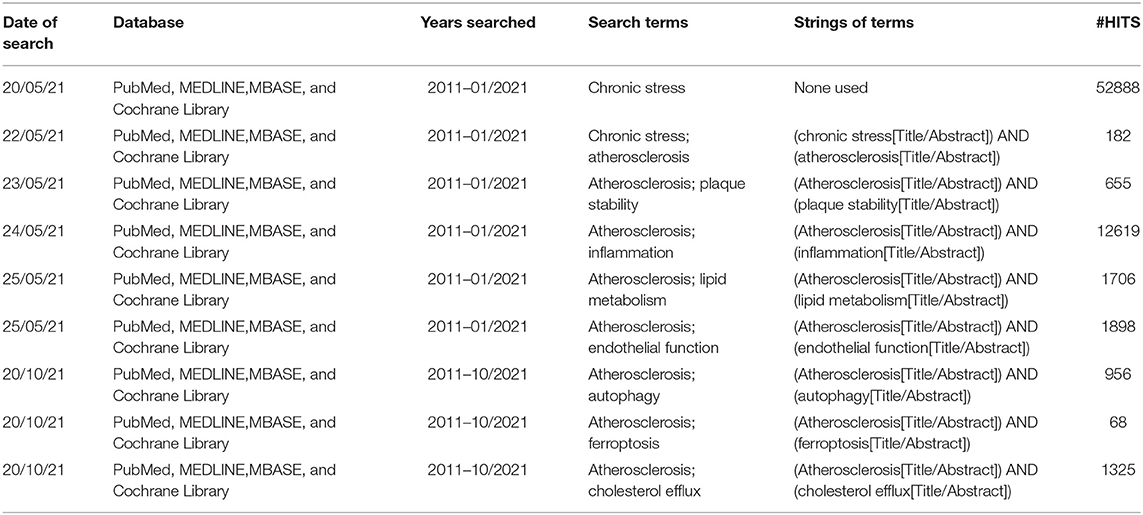
Table 1. Literature search tracking sheet.
Search Strategy
This systematic review focused on the period 2011–2021. Main focus of this paper is on basic medical research about the AS and chronic stress. Researches included in this paper were screened by keyword searches in PubMed, MEDLINE, EMBASE, and Cochrane Library databases. These databases were searched using a combination of subject headings (such as MeSH) and filters (such as Time) when available. We reviewed references of included studies to identify pertinent studies. We imposed no language restriction. The keywords included “chronic stress,” “atherosclerosis,” “inflammation,” “lipid metabolism,” “endothelial function,” and “plaque stability.” And the strings of terms were “(chronic stress[Title/Abstract]) AND (atherosclerosis[Title/Abstract]),” “(Atherosclerosis[Title/Abstract]) AND (plaque stability[Title/Abstract]),” “(Atherosclerosis[Title/Abstract]) AND(inflammation[Title/Abstract]),” “(Atherosclerosis[Title/Abstract]) AND (lipidmetabolism[Title/Abstract]),” “(Atherosclerosis[Title/Abstract]) AND (endothelialfunction[Title/Abstract]),” “(Atherosclerosis[Title/Abstract]) AND (autophagy [Title/Abstract]),” “(Atherosclerosis[Title/Abstract]) AND (ferroptosis [Title/Abstract]),” “(Atherosclerosis[Title/Abstract]) AND (cholesterol efflux [Title/Abstract]).”
Inclusion and Exclusion Criteria
Two reviewers independently assessed records identified from the search for eligibility. Any discrepancies were resolved by consensus. We included any studies referring to chronic stress and atherosclerosis. The researches mainly included the basic medical study with molecular exploration. Outcomes must be objectively measured “atherosclerosis.” We accepted 2011–2021 duration of intervention.
We excluded studies with confounding chronic conditions such as “ventricular remodeling, arrhythmia, Peripheral hemangitis.”
Study Quality
Study quality was assessed by two reviewers based on the seven domains defined by the Cochrane Collaboration's tool for assessing risk of bias. Namely, (1) random sequence generation; (2) allocation concealment; (3) blinding of participants and personnel; (4) blinding of outcome assessment; (5) incomplete outcome data; (6) selective reporting; and (7) other biases, including baseline imbalance, early stopping and bias due to vested financial interest or academic bias.
Potential publication bias across studies was assessed using a funnel plot.
Data Extraction
One author extracted all the data, and two authors reviewed the data for accuracy. The following data was collected: all papers about the association between “chronic stress” and “atherosclerosis.”
Results and Discussion
Chronic Stress Accelerating Atherosclerosis via Inflammation
Although the specific biological mechanisms by which chronic stress increases cardiovascular disease risk remain unclear (34). However, chronic low-grade inflammatory load appears as a possible link because chronic stress exacerbates this load and leads to early progression of atherosclerosis and thrombotic complications (35–37). Inflammation plays a key role in the overall atherosclerotic step, involving the accumulation of foam cells, the formation of fatty stripe tissue and fibrous plaques, the rupture of acute plaques, and the formation of thrombus (38–40). Persistence of inflammation is necessary for plaque development and instability, and plays a decisive role in the pathogenesis and progression of coronary artery disease (41–44). Animal experiments have shown that the levels of intercellular adhesion molecule-1 (ICAM-1), the reactant C-reactive protein (CRP) in the acute phase, and the pro-inflammatory cytokine are significantly increased in apolipoprotein E (ApoE) knockout mice preconditioned by chronic stress (45, 46). Plenty of evidence shows that chronic stress could activate inflammation in the brain and surrounding areas (47, 48). Some researchers believe that stress might activate the Sympathetic Nervous System (SNS) to release noradrenaline (NE) and Nerve Peptide Y (NPY), and these two stress hormones further promote the phosphorylation of mitogen activated protein kinases (MAPKs) or the release of High Mobility Group Box 1 (HMGB1), thereby inducing systemic inflammation to accelerate the development of CVD (49). Chronic stress alters the dynamic balance of the sympathetic and vagal nervous systems. Decrease of vagal tone could promote inflammation. It has been found that chronic stress could enhance the activity of dipeptidyl peptidase-4 (DPP4) in plasma and reduce plasma glucagon-like peptide-1 (GLP-1) and adiponectin (APN) concentrations, thus promoting the development of inflammation (50–52). However, whether it is possible to reduce the promoting effect of chronic stress on atherosclerosis through the targeted inhibition of some cellular inflammatory factors remains to be further studied.
It has been proved that chronic stress and its related diseases anxiety and depression interact with inflammatory response (53). IL-6 is an important inflammatory factor, and its changes represent the body's defense response to chronic stress and help the body adapt to the environment (54). And IL-6 is a kind of polypeptide cytokines with immunomodulatory effects, mainly produced by mononuclear macrophages and T lymphocytes. In the central nervous system, both neurons and glial cells produce this factor (55). Study has shown that IL-6 is involved in the occurrence and development of atherosclerosis in hypertensive patients, and the size of cerebral infarction is positively correlated with the level of serum IL-6 (56). At the same time, IL-6 might promote the progression of atherosclerosis. Studies have shown that IL-6, a pro-inflammatory factor, is elevated in the serum of patients with chronic stress (57). Serum IL-6 increased significantly after chronic stress, and the increase was more obvious in the high-fat diet group. Cortisol acts as an anti-inflammatory, and as IL-6 levels rise in the body, so do cortisol levels. Studies have shown that chronic stress can promote the development of AS through high levels of cortisol mediated by IL-6 (58). IL-6 could promote platelet activation, accelerate the coagulation process, cause endothelial and smooth muscle cell necrosis, and accelerate the formation of AS. IL-6 could damage the vascular endothelium and interfere with uptake of low-density lipoprotein (LDL) by macrophages, resulting in lipid accumulation in the vascular wall and leading to AS (59).
Disorder of Lipid Metabolism in Atherosclerosis Under Chronic Stress
Studies have shown that chronic stress-induced hyperlipidemia and oxidative damage can contribute to the development of atherosclerosis (60). Although atherosclerosis is a chronic inflammatory disease, currently, more and more evidence manifested that atherosclerosis is a complex systematic pathology, and hyperlipidemia is a major risk factor for changes in intimal and media thickness during atherosclerosis (41). Experiments have found that compared with the control group, the high concentrations of serum total cholesterol, triglyceride, low density lipoprotein cholesterol (LDLC), and very low density lipoprotein cholesterol (VLDLC) could increase the atherosclerosis index in the chronic stress group, while the concentration of high density lipoprotein cholesterol did not change significantly (61–65). Chronic stress caused by long-term social pressure leads to obesity to some extent. Obesity is the result of excessive accumulation of fat (66). Scientific studies have shown that obesity can increase the incidence of cardiovascular and cerebrovascular diseases (67, 68). However, the accumulation of subcutaneous fat was not associated with an increased risk of cardiovascular disease. One study found that chronic stress promoted the accumulation of visceral fat, which in turn led to atherosclerosis and cardiovascular events, rather than the accumulation of subcutaneous fat (69). The chronic stress might stimulate the production of glucocorticoid, which can promote visceral obesity, and accompanied by a series of metabolic disorders, including dyslipidemia, impaired glucose tolerance and insulin resistance, unstable or elevated blood pressure (70–73). These factors will be harmful to the arteries, and promote the development of atherosclerosis (67, 74). Other studies have found that Neuropeptide Y (NPY) is a mediator of vascular lipid metabolism disorder under chronic stress and a risk factor for stress-induced lipid metabolic syndrome and atherosclerosis (75–78). Understanding how neuropeptide Y and its homologous receptors regulate lipid metabolism may provide new ideas for the study of the mechanism and treatment of atherosclerosis (79, 80). A large number of studies have shown that hyperlipidemia, induced by chronic stress, is closely related to atherosclerosis (60, 81–83). Therefore, the understanding of lipid metabolism under stress state has important guiding significance for the study of the relationship between chronic stress and atherosclerosis.
Effect of Chronic Stress on Endothelial Dysfunction in Atherosclerosis
Studies have shown that stress is a risk factor for cardiovascular disease (CVD) (84–86). However, the underlying mechanism is not clear. Studies have shown that mental stress activates the sympathetic nervous system (87), which might cause a range of adverse cardiovascular effects, including increased blood pressure, increased heart rate, and endothelial dysfunction. The endothelial dysfunction represent an important link between chronic stress and cardiovascular disease (CVD) risk (46, 88). Recent data from human and animal stress model studies highlight the critical role of endothelial dysfunction in stress-induced cardiovascular disease (89). It was found that under chronic stress, thoracic aortic rings exhibited high sensitivity to vasoconstrictors by inhibiting nitric oxide synthase activity or removing endothelial cells (90–92). Chronic stress could reduce NO production and induce physiological and biological changes of blood vessels, leading to endothelial dysfunction and the progression of atherosclerotic plaques (93, 94). One study examined the effect of vascular endothelial dysfunction on subclinical atherosclerotic plaques by measuring arterial elasticity by observing changes in the percentage of intima-media. The results showed that the loss of endothelial cells could affect the percentage of intima-media and induce atherosclerosis. It has also been found that poor vascular endothelial function will increase the incidence of atherosclerosis (95, 96). Endothelial dysfunction is an important cause of atherosclerosis. Stress can directly inhibit the vasodilator function of endothelial cells. Patients with long-term chronic psychological stress may develop impaired vascular endothelial function. Maintaining homeostasis is a new way to prevent and treat atherosclerosis.
Variation of Hemodynamics and Blood Pressure Under Chronic Stress
Chronic stress is associated with increased cardiovascular risk, including increased incidence of atherosclerosis, myocardial ischemia, coronary heart disease, and death. The association between stress and cardiovascular dysfunction represents an important node for therapeutic interventions for cardiovascular disease, especially in the aging population, where hypertension is a well-known risk factor (97). Chronic stress plays a very important role in the development of hypertension, and its mechanisms are known to involve long-term abnormal neurological and endocrine activity, such as significantly elevated levels of corticosteroids, cortisol, epinephrine, norepinephrine, and angiotensin. Initially, the sympathetic nerve-adrenal medulla system is an important factor in the development of hypertension. Under chronic stress, plasma adrenaline, norepinephrine, and dopamine increase rapidly (98). It is now clear that in hypertension, the sympathetic nervous system activity is increased, and sympathetic excitation causes small arteriovenous contractions, leading to an increase in diastolic/systolic blood pressure (99–101). Catecholamine is an important humoral factor in the sympathetic adrenal myeloid system, which can cause constriction of peripheral blood vessels and increase diastolic pressure. The renin-angiotensin-aldosterone system also plays an important role in chronic stress by inducing increased angiotensin levels, regulating catecholamine secretion, and increasing blood pressure (102–105). Sympathetic excitation is known to increase angiotensin II production by stimulating proximal cells and beta receptors in local tissues to promote renin secretion. Finally, on the hypothalamic-pituitary-adrenal axis (106, 107), chronic psychological stress stimulates the secretion of corticosteroid releasinghormone (CRH) and vasopressin (AVP) in the hypothalamus, which promotes the secretion of corticotropic hormone. Glucocorticoids are important factors in maintaining the normal response of the circulatory system to catecholamines. Glucocorticoid deficiency was associated with significantly reduced response, decreased myocardial contractibility, decreased output, and decreased blood pressure (108). In addition, endothelin (ET) was also an important factor regulating cardiovascular function, and plays an important role in maintaining vascular tension and cardiovascular system homeostasis. As endodermal vascular active factors, endothelin has the strongest and most lasting effect among the endogenous vasoconstrictor peptides. The endothelium could contract vessels and promote endothelial cell proliferation by releasing endothelin. Hypertension and diabetes could lead to endothelial dysfunction and promote release of endothelin (109). The levels of endothelin in patients with diabetes and coronary heart disease were higher than those in control group. The level of endothelin increased significantly in diabetic patients with coronary heart disease. These results demonstrate that endothelin is a good response to vascular endothelial disease regardless of the primary etiology. One study suggests that plasma endothelin levels in atherosclerotic patients are proportional to the severity of atherosclerotic vascular lesions. The more damaged vessels, the higher the endothelin level (110). Endothelin might be an independent risk factor for atherosclerosis. Endothelin causes coronary artery dysfunction, promotes coronary artery wall remodeling, platelet activation, and aggregation (111).
Reduced Plaque Stability by Chronic Stress
Chronic stress could reduce the intimal mediators of atherosclerosis and accelerate plaque instability by promoting apoptosis and neovascularization (28). In our current study, chronic stress increased plaque vulnerability, characterized by thinning of the fibrous cap, larger lipid nuclei, increased macrophages and neovascularization, but fewer smooth muscle cells and elastic fibers (112–114). Thus, chronic stress may not induce larger plaque areas, but rather lead to advanced atherosclerotic lesions. So, how does chronic stress affect the stability of atherosclerotic plaque? Levels of inflammation and oxidative stress, which can be exacerbated by chronic stress, have been shown to be associated with atherosclerotic plaque instability (115, 116).
The Effect of Chronic Stress on Atherosclerosis via Autophagy
Autophagy is a self-protective cellular catabolic pathway involved in protein and organelle degradation (117, 118). Autophagy plays an important role in inhibiting inflammation and apoptosis, and in promoting efferocytosis and cholesterol efflux, and in maintaining cellular metabolic homeostasis. Autophagy is related to oxidative stress, inflammation, and foam cell formation, further promoting atherosclerosis. Therefore, autophagic homeostasis is essential for the development and outcome of atherosclerosis (119). Atherosclerotic lesions are continuously challenged by stressful insults such as DNA damaging molecules, ROS, oxidized lipids, inflammatory cytokines, hypoxia, etc. and will respond in three different ways: either fight (autophagy), adapt (senescence), or die (apoptosis/necrosis). All the three pathways are interconnected and negatively control each other. Atherosclerosis is the progressive buildup of plaque in the arterial wall ultimately resulting in rupture and thrombosis manifesting (120). Moderate activation of autophagy prevents macrophages and vascular smooth muscle cells (VSMCs) from forming foam cells and preventing the progression of atherosclerotic plaques (121, 122). Stimulation of autophagy suppresses vascular smooth muscle cell senescence, whereas inhibition of autophagy promotes it (123). Autophagy is an evolutionarily conserved process in eukaryotes that processes the turnover of intracellular substances. In patients, excessive autophagy activation leads to cell death, plaque instability, or even plaque rupture (119, 124). Abnormal autophagy regulation may lead to atherosclerosis (125).
The Relationship Between Atherosclerosis and Ferroptosis
Ferroptosis is a newly identified form of regulated cell death characterized by the iron-dependent accumulation of lipid hydroperoxides to lethal levels (126), this type of cell death was found to have molecular characteristics distinct from other forms of regulated cell death (127), which exhibits distinct features from apoptosis, necrosis and autophagy in morphology, biochemistry, and genetics (128, 129). Ferroptosis is a type of autophagy-dependent cell death (130). Emerging mechanisms of ferroptosis is related to disease (131). Ferroptosis is closely related to atherosclerosis, and might occur during the initiation and development of AS (129). Apoptosis, necrosis and autophagy-dependent cell death are the three major types of cell death. Traditionally, necrosis is thought as a passive and unregulated form of cell death. However, certain necrosis can also occur in a highly regulated manner, referring to regulated necrosis. Depending on the signaling pathways, regulated necrosis can be further classified as necroptosis, pyroptosis, ferroptosis, parthanatos, and CypD-mediated necrosis. endothelial progenitor cell (EPC)-EVs transferred miR-199a-3p to inhibit Sp1 Transcription Factor (SP1), thus repressing ferroptosis of endothelial cells and retarding the occurrence of AS (132). Inhibition of ferroptosis could alleviate AS through attenuating lipid peroxidation and endothelial dysfunction in AECs (129, 133). Therefore, ferroptosis as a central gene in human coronary atherosclerosis (134).
Aggregating Atherosclerosis via Cholesterol Efflux Under the Chronic Stress
Cholesterol is an important lipid for maintaining cell membrane fluidity and generation of various hormones and bile acids. Thus, it is critical to maintain cholesterol homeostasis including absorption, trafficking, biosynthesis, and efflux. Dysregulation of cholesterol homeostasis may lead to human disorders (135). The phenomena of lipid accumulation, inflammation, oxidative stress, hypoxia, and insulin resistance commonly associated with AS lesions can regulate the expression of cholesterol transporter, and then regulate intracellular cholesterol efflux, affecting the occurrence, and development of As. Cholesterol efflux is a key step in cholesterol reverse transport (136). The reverse cholesterol transport, a process that removes excess cholesterol from peripheral tissues/cells including macrophages to circulating HDL, is one of the main mechanisms responsible for anti-atherogenic properties of HDL. Reverse cholesterol transport (RCT) may counteract the pathogenic events leading to the formation and development of atheroma, by promoting the high-density lipoprotein (HDL)-mediated removal of cholesterol from the artery wall (137, 138). The key proteins of reverse cholesterol transport-ATP-binding cassette transporters A1 (ABCA1) and G1 (ABCG1)-mediate the cholesterol efflux from macrophages and prevent their transformation into foam cells (139). The formation of foam cells is a typical pathological feature of early atherosclerosis, the imbalance of cholesterol metabolism homeostasis of macrophages runs through the whole process of foam cell formation.
Atherosclerosis is characterized by significant aggregation of macrophage foam cells in atherosclerotic plaques and associated pro-inflammatory responses in pathological cells. Results from animal and human studies suggest that in these cells, especially in diseased macrophages, dyshomeostasis plays a key role in the pro-inflammatory response. The cholesterol efflux pathway also inhibits the accumulation of cholesterol esters in macrophages, namely the formation of macrophage foam cells (140). Cholesterol efflux is a key link in regulating the cholesterol dynamic balance of macrophages, which is of great significance in reducing intracellular cholesterol accumulation, preventing the formation of foam cells and the occurrence of As. Cholesterol efflux pathways exert anti-inflammatory and anti-atherogenic effects by suppressing proliferation of hematopoietic stem and progenitor cells, and inflammation and inflammasome activation in macrophages. Therefore, atherosclerosis can be prevented by promoting cholesterol efflux from macrophages (141, 142).
In summary, the overview map presented the effect of chronic stress on atherosclerosis (Figure 2).
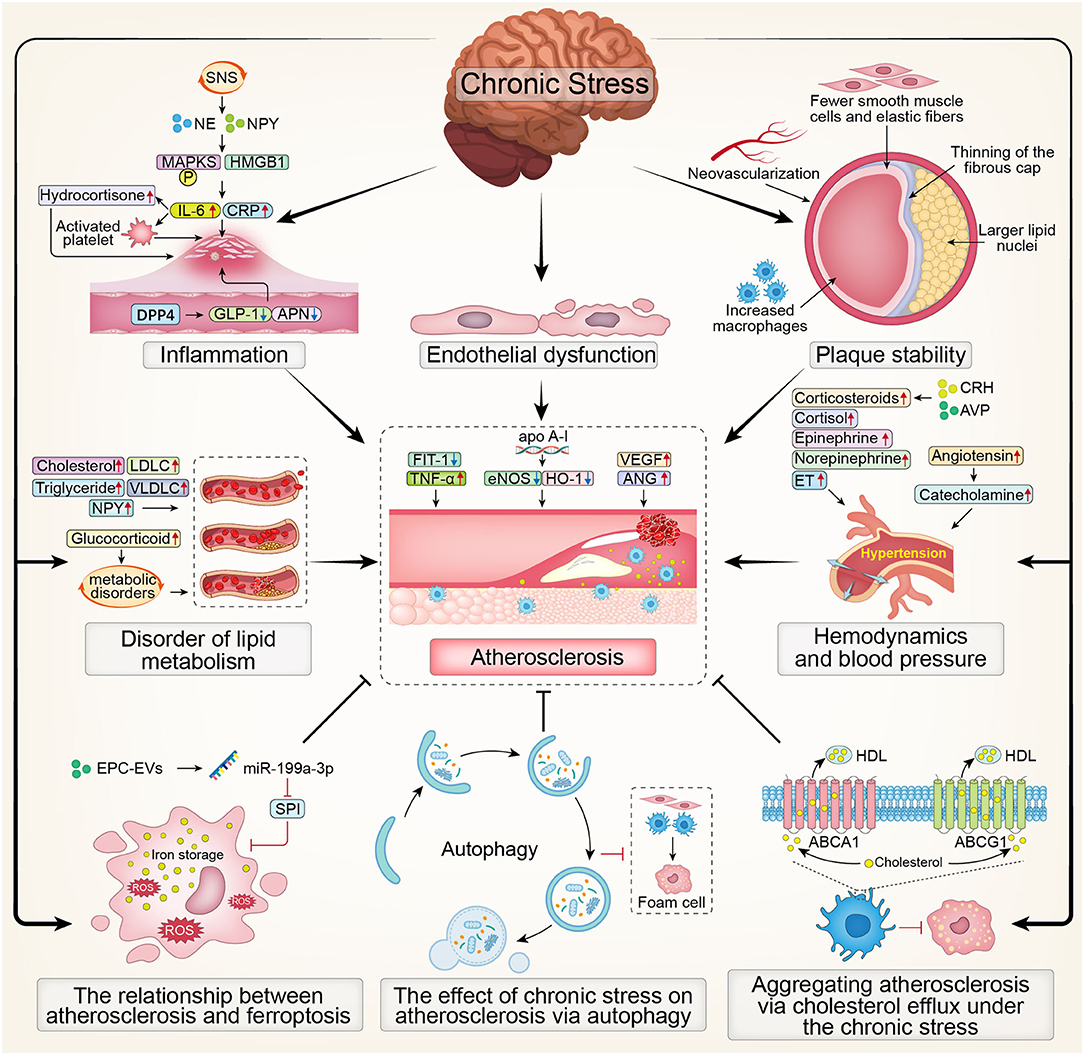
Figure 2. The overview map presenting the effect of chronic stress on atherosclerosis. SNS, Sympathetic Nervous System; NE, noradrenaline; NPY, Nerve Peptide Y; MAPKS, mitogen activated protein kinases; HMGB1, High Mobility Group Box 1; CRP, C-reactive protein; IL-6, interleukin; DPP4, dipeptidyl peptidase-4; GLP-1, glucagon-like peptide-1; APN, adiponectin; LDLC, low density lipoprotein cholesterol; VLDLC, very low density lipoprotein cholesterol; Flt-1, fms-like tyrosine kinase-1; TNF-α, tumor necrosis factor-α; eNOS, endothelial nitric oxide synthase; HO-1, Hemeoxygenase-1; VEGF, Vascular Endothelial Growth Factor; ANG, Angiogenin; CRH, corticosteroid releasinghormone; AVP, vasopressin; ET, endothelin; ROS, Reactive oxygen species; EPC, endothelial progenitor cell; SP1, Sp1 Transcription Factor; HDL, high-density lipoprotein; ABCA1, ATP-binding cassette transporters A1; ABCG1, ATP-binding cassette transporters G1.
Conclusion
Chronic stress is the cause of atherosclerotic cardiovascular and cerebrovascular diseases. If it cannot be effectively prevented, biological changes in the body environment will occur successively, such as inflammation, lipid metabolism, endothelial function, hemodynamics and other changes, and then morphologic changes of the corresponding organs will appear. If the patient has a genetic predisposition, and at the same time the environmental factors work together to activate the pathogenic mechanism, then the chronic stress factors will eventually lead to the development of atherosclerotic cardiovascular disease.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Author Contributions
L-bM was major contributor in writing and was involved in critically revising manuscript for important intellectual content. TG and D-pL made substantial contributions to research conception and designed the draft of the research process. YL and Y-mZ were major contributors in submitting the manuscript and they gave the technical support in the methods. All authors read and approved the final manuscript.
Funding
The present study was funded by the National Key R&D Program of China (Grant Nos. 2020YFC2003000 and 2020YFC2003001), Chinese Academy of Medical Sciences, CAMS Innovation Fund for Medical Sciences (Grant No. 2018-I2M-1-002), and National Natural Science Foundation of China (Grant Nos. 31271097 and 51672030).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank Xiao-Meng Yu for his assistance and suggestions during the submitting process.
Abbreviations
AS, Atherosclerosis; ASCVD, Atherosclerotic cardio-cerebrovascular disease; Flt-1, fms-like tyrosine kinase-1; TNF-α, tumor necrosis factor-α; apo A-I, apolipoprotein A-I; VEGF, Vascular Endothelial Growth Factor; ANG, Angiogenin; EPCs, endothelial progenitor cells; eNOS, endothelial nitric oxide synthase; CVD, cardiovascular disease; ICAM-1, intercellular adhesion molecule-1; CRP, C-reactive protein; ApoE, apolipoprotein E; SNS, Sympathetic Nervous System; NE, noradrenaline; NPY, Nerve Peptide Y; ET, endothelin; MAPKs, mitogen activated protein kinases; HMGB1, High Mobility Group Box 1; DPP4, dipeptidyl peptidase-4; GLP-1, glucagon-like peptide-1; APN, adiponectin; LDLC, low density lipoprotein cholesterol; VLDLC, very low density lipoprotein cholesterol; NPY, Neuropeptide Y; CRH, corticosteroid releasinghormone; AVP, vasopressin; RCT, Reverse cholesterol transport; HDL, high-density lipoprotein; ROS, Reactive oxygen species; EPC, endothelial progenitor cell; SP1, Sp1 Transcription Factor; HDL, high-density lipoprotein; ABCA1, ATP-binding cassette transporters A1; ABCG1, ATP-binding cassette transporters G1.
References
1. Allahverdian S, Ortega C, Francis GA. Smooth muscle cell-proteoglycan-lipoprotein interactions as drivers of atherosclerosis. Handb Exp Pharmacol. (2020). doi: 10.1007/164_2020_364
2. Yin K, Liang S, Tang X, Li M, Yuan J, Wu M, et al. The relationship between intracranial arterial dolichoectasia and intracranial atherosclerosis. Clin Neurol Neurosurg. (2020) 200:106408. doi: 10.1016/j.clineuro.2020.106408
3. Cainzos-Achirica M, Glassner K, Zawahir HS, Dey AK, Agrawal T, Quigley E, et al. Inflammatory bowel disease and atherosclerotic cardiovascular disease: JACC review topic of the week. J Am Coll Cardiol. (2020) 76:2895–905. doi: 10.1016/j.jacc.2020.10.027
4. De Winter G. Aging as disease. Med Health Care Philos. (2015) 18:237–43. doi: 10.1007/s11019-014-9600-y
5. Song P, Rudan D, Zhu Y, Fowkes F, Rahimi K, Fowkes F, et al. Global, regional, and national prevalence and risk factors for peripheral artery disease in 2015: an updated systematic review and analysis. Lancet Glob Health. (2019) 7:e1020–30. doi: 10.1016/S2214-109X(19)30255-4
6. Sharif K, Watad A, Coplan L, Amital H, Shoenfeld Y, Afek A. Psychological stress and type 1 diabetes mellitus: what is the link. Expert Rev Clin Immunol. (2018) 14:1081–8. doi: 10.1080/1744666X.2018.1538787
7. Bernstein CN. Psychological stress and depression: risk factors for IBD. Dig Dis. (2016) 34:58–63. doi: 10.1159/000442929
8. Marin MF, Lord C, Andrews J, Juster RP, Sindi S, Arsenault-Lapierre G, et al. Chronic stress, cognitive functioning and mental health. Neurobiol Learn Mem. (2011) 96:583–95. doi: 10.1016/j.nlm.2011.02.016
9. Ioachimescu AG. Diabetes and atherosclerotic cardiovascular disease. Endocrinol Metab Clin North Am. (2018) 47:xiii–xiv. doi: 10.1016/j.ecl.2017.12.002
10. Tang X, Zhang D, He L, Wu N, Si Y, Cao Y, et al. Performance of atherosclerotic cardiovascular risk prediction models in a rural Northern Chinese population: results from the Fangshan Cohort Study. Am Heart J. (2019) 211:34–44. doi: 10.1016/j.ahj.2019.01.009
11. Low Wang CC, Hess CN, Hiatt WR, Goldfine AB. Clinical update: cardiovascular disease in diabetes mellitus: atherosclerotic cardiovascular disease and heart failure in type 2 diabetes mellitus - mechanisms, management, and clinical considerations. Circulation. (2016) 133:2459–502. doi: 10.1161/CIRCULATIONAHA.116.022194
12. Yang X, Li J, Hu D, Chen J, Li Y, Huang J, et al. Predicting the 10-Year risks of atherosclerotic cardiovascular disease in Chinese population: the China-PAR project (prediction for ASCVD risk in China). Circulation. (2016) 134:1430–40. doi: 10.1161/CIRCULATIONAHA.116.022367
13. Mitsides N, Cornelis T, Broers N, Diederen N, Brenchley P, Heitink-Ter Braak N, et al. Inflammatory and angiogenic factors linked to longitudinal microvascular changes in hemodialysis patients irrespective of treatment dose intensity. Kidney Blood Press Res. (2017) 42:905–18. doi: 10.1159/000485048
14. Zhang Y, Liu H, Tang W, Qiu Q, Peng J. Resveratrol prevents TNF-α-induced VCAM-1 and ICAM-1 upregulation in endothelial progenitor cells via reduction of NF-κB activation. J Int Med Res. (2020) 48:300060520945131. doi: 10.1177/0300060520945131
15. Xu W, Qian M, Huang C, Cui P, Li W, Du Q, et al. Comparison of mechanisms of endothelial cell protections between high-density lipoprotein and apolipoprotein A-I mimetic peptide. Front Pharmacol. (2019) 10:817. doi: 10.3389/fphar.2019.00817
16. Apte RS, Chen DS, Ferrara N. VEGF in signaling and disease: beyond discovery and development. Cell. (2019) 176:1248–64. doi: 10.1016/j.cell.2019.01.021
17. Fagiani E, Christofori G. Angiopoietins in angiogenesis. Cancer Lett. (2013) 328:18–26. doi: 10.1016/j.canlet.2012.08.018
18. Jaipersad AS, Lip GY, Silverman S, Shantsila E. The role of monocytes in angiogenesis and atherosclerosis. J Am Coll Cardiol. (2014) 63:1–11. doi: 10.1016/j.jacc.2013.09.019
19. Braile M, Marcella S, Cristinziano L, Galdiero MR, Modestino L, Ferrara AL, et al. VEGF-A in cardiomyocytes and heart diseases. Int J Mol Sci. (2020) 21:5294. doi: 10.3390/ijms21155294
20. Marino M, Del Bo' C, Tucci M, Klimis-Zacas D, Riso P, Porrini M. Modulation of adhesion process, E-selectin and VEGF production by anthocyanins and their metabolites in an in vitro model of atherosclerosis. Nutrients. (2020) 12:655. doi: 10.20944/preprints202001.0113.v1
21. Gürses KM, Yalçin MU, Koçyigit D, Beşler MS, Canpinar H, Evranos B, et al. The association between serum angiogenin and osteopontin levels and coronary collateral circulation in patients with chronic total occlusion. Anatol J Cardiol. (2019) 22:77–84. doi: 10.14744/AnatolJCardiol.2019.88555
22. Melincovici CS, Boşca AB, Suşman S, Mărginean M, Mihu C, Istrate M, Moldovan IM, et al. Vascular endothelial growth factor (VEGF) - key factor in normal and pathological angiogenesis. Rom J Morphol Embryol. (2018) 59:455–67. Available online at: https://pubmed.ncbi.nlm.nih.gov/30173249/
23. Ahmed S, Ahmed A, Säleby J, Bouzina H, Lundgren J, Rådegran G. Elevated plasma tyrosine kinases VEGF-D and HER4 in heart failure patients decrease after heart transplantation in association with improved haemodynamics. Heart Vessels. (2020) 35:786–99. doi: 10.1007/s00380-019-01548-1
24. Song P, Rudan D, Wang M, Chang X, Rudan I. National and subnational estimation of the prevalence of peripheral artery disease (PAD) in China: a systematic review and meta-analysis. J Glob Health. (2019) 9:010601. doi: 10.7189/jogh.09.010601
25. Rosenblit PD. Extreme atherosclerotic cardiovascular disease (ASCVD) risk recognition. Curr Diab Rep. (2019) 19:61. doi: 10.1007/s11892-019-1178-6
26. Hussain A, Ballantyne CM, Saeed A, Virani SS. Triglycerides and ASCVD risk reduction: recent insights and future directions. Curr Atheroscler Rep. (2020) 22:25. doi: 10.1007/s11883-020-00846-8
27. Sun GZ, Ye N, Wu SJ, Zhou Y, Sun YX. 10-year ASCVD risk is positively correlated with depressive symptoms in a large general population. BMC Psychiatry. (2019) 19:125. doi: 10.1186/s12888-019-2114-7
28. Yu ZM, Deng XT, Qi RM, Xiao LY, Yang CQ, Gong T. Mechanism of chronic stress-induced reduced atherosclerotic medial area and increased plaque instability in rabbit models of chronic stress. Chin Med J. (2018) 131:161–70. doi: 10.4103/0366-6999.222322
29. Iob E, Steptoe A. Cardiovascular disease and hair cortisol: a novel biomarker of chronic stress. Curr Cardiol Rep. (2019) 21:116. doi: 10.1007/s11886-019-1208-7
30. Kovaleva M, Spangler S, Clevenger C, Hepburn K. Chronic stress, social isolation, and perceived loneliness in dementia caregivers. J Psychosoc Nurs Ment Health Serv. (2018) 56:36–43. doi: 10.3928/02793695-20180329-04
31. Kattoor AJ, Pothineni N, Palagiri D, Mehta JL. Oxidative stress in atherosclerosis. Curr Atheroscler Rep. (2017) 19:42. doi: 10.1007/s11883-017-0678-6
32. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primers. (2019) 5:56. doi: 10.1038/s41572-019-0106-z
33. Esler M. Mental stress and human cardiovascular disease. Neurosci Biobehav Rev. (2017) 74:269–76. doi: 10.1016/j.neubiorev.2016.10.011
34. Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. (2018) 15:505–22. doi: 10.1038/s41569-018-0064-2
35. Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. (2019) 25:1822–32. doi: 10.1038/s41591-019-0675-0
36. Raggi P, Genest J, Giles JT, Rayner KJ, Dwivedi G, Beanlands RS, et al. Role of inflammation in the pathogenesis of atherosclerosis and therapeutic interventions. Atherosclerosis. (2018) 276:98–108. doi: 10.1016/j.atherosclerosis.2018.07.014
37. Pant S, Deshmukh A, Gurumurthy GS, Pothineni NV, Watts TE, Romeo F, et al. Inflammation and atherosclerosis–revisited. J Cardiovasc Pharmacol Ther. (2014) 19:170–8. doi: 10.1177/1074248413504994
38. Wolf D, Ley K. Immunity and inflammation in atherosclerosis. Circ Res. (2019) 124:315–27. doi: 10.1161/CIRCRESAHA.118.313591
39. Geovanini GR, Libby P. Atherosclerosis and inflammation: overview and updates. Clin Sci. (2018) 132:1243–52. doi: 10.1042/CS20180306
40. Zhu Y, Xian X, Wang Z, Bi Y, Chen Q, Han X, et al. Research progress on the relationship between atherosclerosis and inflammation. Biomolecules. (2018) 8:80. doi: 10.3390/biom8030080
41. Fioranelli M, Bottaccioli AG, Bottaccioli F, Bianchi M, Rovesti M, Roccia MG. Stress and inflammation in coronary artery disease: a review psychoneuroendocrineimmunology-based. Front Immunol. (2018) 9:2031. doi: 10.3389/fimmu.2018.02031
42. Bäck M, Yurdagul A Jr, Tabas I, Öörni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol. (2019) 16:389–406. doi: 10.1038/s41569-019-0169-2
43. Shah PK. Inflammation, infection and atherosclerosis. Trends Cardiovasc Med. (2019) 29:468–72. doi: 10.1016/j.tcm.2019.01.004
44. Ruparelia N, Choudhury R. Inflammation and atherosclerosis: what is on the horizon. Heart. (2020) 106:80–5. doi: 10.1136/heartjnl-2018-314230
45. Chumaeva N, Hintsanen M, Pulkki-Råback L, Jokela M, Juonala M, Lehtimäki T, et al. Interleukin-6 gene polymorphism, chronic stress and atherosclerosis: interleukin-6-174G>C polymorphism, chronic stress and risk of early atherosclerosis in the Cardiovascular Risk in Young Finns Study. J Psychosom Res. (2014) 76:333–8. doi: 10.1016/j.jpsychores.2014.01.007
46. Kershaw KN, Lane-Cordova AD, Carnethon MR, Tindle HA, Liu K. Chronic stress and endothelial dysfunction: the multi-ethnic study of atherosclerosis (MESA). Am J Hypertens. (2017) 30:75–80. doi: 10.1093/ajh/hpw103
47. Yao BC, Meng LB, Hao ML, Zhang YM, Gong T, Guo ZG. Chronic stress: a critical risk factor for atherosclerosis. J Int Med Res. (2019) 47:1429–40. doi: 10.1177/0300060519826820
48. Rohleder N. Stimulation of systemic low-grade inflammation by psychosocial stress. Psychosom Med. (2014) 76:181–9. doi: 10.1097/PSY.0000000000000049
49. Liu YZ, Wang YX, Jiang CL. Inflammation: the common pathway of stress-related diseases. Front Hum Neurosci. (2017) 11:316. doi: 10.3389/fnhum.2017.00316
50. Lei Y, Yang G, Hu L, Piao L, Inoue A, Jiang H, et al. Increased dipeptidyl peptidase-4 accelerates diet-related vascular aging and atherosclerosis in ApoE-deficient mice under chronic stress. Int J Cardiol. (2017) 243:413–20. doi: 10.1016/j.ijcard.2017.05.062
51. Yang G, Lei Y, Inoue A, Piao L, Hu L, Jiang H, et al. Exenatide mitigated diet-induced vascular aging and atherosclerotic plaque growth in ApoE-deficient mice under chronic stress. Atherosclerosis. (2017) 264:1–10. doi: 10.1016/j.atherosclerosis.2017.07.014
52. Halaris A. Co-morbidity between cardiovascular pathology and depression: role of inflammation. Mod Trends Pharmacopsychiatry. (2013) 28:144–61. doi: 10.1159/000343981
53. Bauer ME, Teixeira AL. Inflammation in psychiatric disorders: what comes first. Ann NY Acad Sci. (2019) 1437:57–67. doi: 10.1111/nyas.13712
54. Wang YL, Han QQ, Gong WQ, Pan DH, Wang LZ, Hu W, et al. Microglial activation mediates chronic mild stress-induced depressive- and anxiety-like behavior in adult rats. J Neuroinflammation. (2018) 15:21. doi: 10.1186/s12974-018-1054-3
55. Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. (2015) 16:448–57. doi: 10.1038/ni.3153
56. Tyrrell DJ, Goldstein DR. Ageing and atherosclerosis: vascular intrinsic and extrinsic factors and potential role of IL-6. Nat Rev Cardiol. (2021) 18:58–68. doi: 10.1038/s41569-020-0431-7
57. Molnar DS, Moore J, O'Leary DD, MacNeil AJ, Wade TJ. Perfectionistic cognitions, Interleukin-6, and C-Reactive protein: a test of the perfectionism diathesis stress model. Brain Behav Immun Health. (2021) 13:100211. doi: 10.1016/j.bbih.2021.100211
58. Okutsu M, Lira VA, Higashida K, Peake J, Higuchi M, Suzuki K. Corticosterone accelerates atherosclerosis in the apolipoprotein E-deficient mouse. Atherosclerosis. (2014) 232:414–9. doi: 10.1016/j.atherosclerosis.2013.11.076
59. Luo P, Shi W, Wang Y, Ma H, Liu T, Yan D, et al. Raloxifene inhibits IL-6/STAT3 signaling pathway and protects against high-fat-induced atherosclerosis in ApoE(-/-) mice. Life Sci. (2020) 261:118304. doi: 10.1016/j.lfs.2020.118304
60. Devaki M, Nirupama R, Yajurvedi HN. Chronic stress-induced oxidative damage and hyperlipidemia are accompanied by atherosclerotic development in rats. Stress. (2013) 16:233–43. doi: 10.3109/10253890.2012.719052
61. Neves VJ, Moura MJ, Tamascia ML, Ferreira R, Silva NS, Costa R, et al. Proatherosclerotic effects of chronic stress in male rats: altered phenylephrine sensitivity and nitric oxide synthase activity of aorta and circulating lipids. Stress. (2009) 12:320–7. doi: 10.1080/10253890802437779
62. Poznyak A, Grechko AV, Poggio P, Myasoedova VA, Alfieri V, Orekhov AN. The diabetes mellitus-atherosclerosis connection: the role of lipid and glucose metabolism and chronic inflammation. Int J Mol Sci. (2020) 21:1835. doi: 10.3390/ijms21051835
63. Meng LB, Qi R, Xu L, Chen Y, Yu Z, Guo P, et al. The more critical murderer of atherosclerosis than lipid metabolism: chronic stress. Lipids Health Dis. (2018) 17:143. doi: 10.1186/s12944-018-0795-4
64. Çimen I, Kocatürk B, Koyuncu S, Tufanli Ö, Onat UI, Yildirim AD, et al. Prevention of atherosclerosis by bioactive palmitoleate through suppression of organelle stress and inflammasome activation. Sci Transl Med. (2016) 8:358ra126. doi: 10.1126/scitranslmed.aaf9087
65. Ou L, Li X, Chen B, Ge Z, Zhang J, Zhang Y, et al. Recombinant human cytoglobin prevents atherosclerosis by regulating lipid metabolism and oxidative stress. J Cardiovasc Pharmacol Ther. (2018) 23:162–73. doi: 10.1177/1074248417724870
66. Bullon P, Newman HN, Battino M. Obesity, diabetes mellitus, atherosclerosis and chronic periodontitis: a shared pathology via oxidative stress and mitochondrial dysfunction. Periodontology. (2014) 64:139–53. doi: 10.1111/j.1600-0757.2012.00455.x
67. Shively CA, Register TC, Clarkson TB. Social stress, visceral obesity, and coronary artery atherosclerosis: product of a primate adaptation. Am J Primatol. (2009) 71:742–51. doi: 10.1002/ajp.20706
68. Ortega-Montiel J, Posadas-Romero C, Ocampo-Arcos W, Medina-Urrutia A, Cardoso-Saldaña G, Jorge-Galarza E, et al. Self-perceived stress is associated with adiposity and atherosclerosis. The GEA Study. BMC Public Health. (2015) 15:780. doi: 10.1186/s12889-015-2112-8
69. Peters A, McEwen BS. Stress habituation, body shape and cardiovascular mortality. Neurosci Biobehav Rev. (2015) 56:139–50. doi: 10.1016/j.neubiorev.2015.07.001
70. Palumbo ML, Prochnik A, Wald MR, Genaro AM. Chronic stress and glucocorticoid receptor resistance in asthma. Clin Ther. (2020) 42:993–1006. doi: 10.1016/j.clinthera.2020.03.002
71. Chiba S, Numakawa T, Ninomiya M, Richards MC, Wakabayashi C, Kunugi H. Chronic restraint stress causes anxiety- and depression-like behaviors, downregulates glucocorticoid receptor expression, and attenuates glutamate release induced by brain-derived neurotrophic factor in the prefrontal cortex. Prog Neuropsychopharmacol Biol Psychiatry. (2012) 39:112–9. doi: 10.1016/j.pnpbp.2012.05.018
72. Vyas S, Rodrigues AJ, Silva JM, Tronche F, Almeida OF, Sousa N, et al. Chronic stress and glucocorticoids: from neuronal plasticity to neurodegeneration. Neural Plast. (2016) 2016:6391686. doi: 10.1155/2016/6391686
73. Horchar MJ, Wohleb ES. Glucocorticoid receptor antagonism prevents microglia-mediated neuronal remodeling and behavioral despair following chronic unpredictable stress. Brain Behav Immun. (2019) 81:329–40. doi: 10.1016/j.bbi.2019.06.030
74. Hui N, Morris MJ, Allison MA, Tsai MY, Rye KA, Tabet F, et al. Lipoprotein (a) and the risk of elevated depressive symptoms: The Multi-Ethnic Study of Atherosclerosis. J Psychiatr Res. (2020) 133:119–24. doi: 10.1016/j.jpsychires.2020.12.022
75. Sun WW, Zhu P, Shi YC, Zhang CL, Huang XF, Liang SY, et al. Current views on neuropeptide Y and diabetes-related atherosclerosis. Diab Vasc Dis Res. (2017) 14:277–84. doi: 10.1177/1479164117704380
76. Tan C, Green P, Tapoulal N, Lewandowski AJ, Leeson P, Herring N. The role of neuropeptide Y in cardiovascular health and disease. Front Physiol. (2018) 9:1281. doi: 10.3389/fphys.2018.01281
77. Zheng YL, Wang WD, Li MM, Lin S, Lin HL. Updated role of neuropeptide Y in nicotine-induced endothelial dysfunction and atherosclerosis. Front Cardiovasc Med. (2021) 8:630968. doi: 10.3389/fcvm.2021.630968
78. Wu WQ, Peng S, Wan XQ, Lin S, Li LY, Song ZY. Physical exercise inhibits atherosclerosis development by regulating the expression of neuropeptide Y in apolipoprotein E-deficient mice. Life Sci. (2019) 237:116896. doi: 10.1016/j.lfs.2019.116896
79. Abe K, Kuo L, Zukowska Z. Neuropeptide Y is a mediator of chronic vascular and metabolic maladaptations to stress and hypernutrition. Exp Biol Med. (2010) 235:1179–84. doi: 10.1258/ebm.2010.009136
80. Vähätalo LH, Ruohonen ST, Ailanen L, Savontaus E. Neuropeptide Y in noradrenergic neurons induces obesity in transgenic mouse models. Neuropeptides. (2016) 55:31–7. doi: 10.1016/j.npep.2015.11.088
81. Park K, Li Q, Evcimen ND, Rask-Madsen C, Maeda Y, Maddaloni E, et al. Exogenous insulin infusion can decrease atherosclerosis in diabetic rodents by improving lipids, inflammation, and endothelial function. Arterioscler Thromb Vasc Biol. (2018) 38:92–101. doi: 10.1161/ATVBAHA.117.310291
82. Han Y, Lin M, Wang X, Guo K, Wang S, Sun M, et al. Basis of aggravated hepatic lipid metabolism by chronic stress in high-fat diet-fed rat. Endocrine. (2015) 48:483–92. doi: 10.1007/s12020-014-0307-x
83. Takahashi K, Yamada T, Tsukita S, Kaneko K, Shirai Y, Munakata Y, et al. Chronic mild stress alters circadian expressions of molecular clock genes in the liver. Am J Physiol Endocrinol Metab. (2013) 304:E301–9. doi: 10.1152/ajpendo.00388.2012
84. Aboa-Eboulé C, Brisson C, Maunsell E, Mâsse B, Bourbonnais R, Vézina M, et al. Job strain and risk of acute recurrent coronary heart disease events. JAMA. (2007) 298:1652–60. doi: 10.1001/jama.298.14.1652
85. Bairey Merz CN, Dwyer J, Nordstrom CK, Walton KG, Salerno JW, Schneider RH. Psychosocial stress and cardiovascular disease: pathophysiological links. Behav Med. (2002) 27:141–7. doi: 10.1080/08964280209596039
86. De Bacquer D, Pelfrene E, Clays E, Mak R, Moreau M, de Smet P, et al. Perceived job stress and incidence of coronary events: 3-year follow-up of the Belgian Job Stress Project cohort. Am J Epidemiol. (2005) 161:434–41. doi: 10.1093/aje/kwi040
87. Hjemdahl P, Fagius J, Freyschuss U, Wallin BG, Daleskog M, Bohlin G, et al. Muscle sympathetic activity and norepinephrine release during mental challenge in humans. Am J Physiol. (1989) 257:E654–4. doi: 10.1152/ajpendo.1989.257.5.E654
88. Ross R. The pathogenesis of atherosclerosis: a perspective for the (1990s). Nature. (1993) 362:801–9. doi: 10.1038/362801a0
89. Golbidi S, Frisbee JC, Laher I. Chronic stress impacts the cardiovascular system: animal models and clinical outcomes. Am J Physiol Heart Circ Physiol. (2015) 308:H1476–98. doi: 10.1152/ajpheart.00859.2014
90. Ghiadoni L, Bruno RM. The endothelium as a target for chronic stress. Am J Hypertens. (2017) 30:19–20. doi: 10.1093/ajh/hpw116
91. Stanley SC, Brooks SD, Butcher JT, d'Audiffret AC, Frisbee SJ, Frisbee JC. Protective effect of sex on chronic stress- and depressive behavior-induced vascular dysfunction in BALB/cJ mice. J Appl Physiol. (2014) 117:959–70. doi: 10.1152/japplphysiol.00537.2014
92. Balkaya M, Prinz V, Custodis F, Gertz K, Kronenberg G, Kroeber J, et al. Stress worsens endothelial function and ischemic stroke via glucocorticoids. Stroke. (2011) 42:3258–64. doi: 10.1161/STROKEAHA.110.607705
93. Dikalov SI, Mayorov VI, Panov AV. Physiological levels of nitric oxide diminish mitochondrial superoxide. Potential role of mitochondrial dinitrosyl iron complexes and nitrosothiols. Front Physiol. (2017) 8:907. doi: 10.3389/fphys.2017.00907
94. Yang HJ, Kim KY, Kang P, Lee HS, Seol GH. Effects of Salvia sclarea on chronic immobilization stress induced endothelial dysfunction in rats. BMC Complement Altern Med. (2014) 14:396. doi: 10.1186/1472-6882-14-396
95. Mausbach BT, Roepke SK, Ziegler MG, Milic M, von Känel R, Dimsdale JE, et al. Association between chronic caregiving stress and impaired endothelial function in the elderly. J Am Coll Cardiol. (2010) 55:2599–606. doi: 10.1016/j.jacc.2009.11.093
96. Chumaeva N, Hintsanen M, Ravaja N, Juonala M, Raitakari OT, Keltikangas-Järvinen L. Chronic stress and the development of early atherosclerosis: moderating effect of endothelial dysfunction and impaired arterial elasticity. Int J Environ Res Public Health. (2009) 6:2934–49. doi: 10.3390/ijerph6122934
97. Goodson ML, Packard A, Buesing DR, Maney M, Myers B, Fang Y, et al. Chronic stress and Rosiglitazone increase indices of vascular stiffness in male rats. Physiol Behav. (2017) 172:16–23. doi: 10.1016/j.physbeh.2016.03.031
98. Imperatore R, Palomba L, Cristino L. Role of orexin-A in hypertension and obesity. Curr Hypertens Rep. (2017) 19:34. doi: 10.1007/s11906-017-0729-y
99. Bohuslavova R, Cerychova R, Papousek F, Olejnickova V, Bartos M, Görlach A, et al. HIF-1α is required for development of the sympathetic nervous system. Proc Natl Acad Sci USA. (2019) 116:13414–23. doi: 10.1073/pnas.1903510116
100. DeLalio LJ, Sved AF, Stocker SD. Sympathetic nervous system contributions to hypertension: updates and therapeutic relevance. Can J Cardiol. (2020) 36:712–20. doi: 10.1016/j.cjca.2020.03.003
101. Esler M. The sympathetic nervous system in hypertension: back to the future. Curr Hypertens Rep. (2015) 17:11. doi: 10.1007/s11906-014-0519-8
102. Lezama-Martínez D, Valencia-Hernández I, Flores-Monroy J, Martínez-Aguilar L. Combination of β adrenergic receptor block and renin-angiotensin system inhibition diminished the angiotensin ii-induced vasoconstriction and increased bradykinin-induced vasodilation in hypertension. Dose Response. (2017) 15:1559325817737932. doi: 10.1177/1559325817737932
103. Wang C, Luo Z, Carter G, Wellstein A, Jose PA, Tomlinson J, et al. NRF2 prevents hypertension, increased ADMA, microvascular oxidative stress, and dysfunction in mice with two weeks of ANG II infusion. Am J Physiol Regul Integr Comp Physiol. (2018) 314:R399–406. doi: 10.1152/ajpregu.00122.2017
104. Holbein WW, Blackburn MB, Andrade MA, Toney GM. Burst patterning of hypothalamic paraventricular nucleus-driven sympathetic nerve activity in ANG II-salt hypertension. Am J Physiol Heart Circ Physiol. (2018) 314:H530–41. doi: 10.1152/ajpheart.00560.2017
105. Ishigaki S, Ohashi N, Matsuyama T, Isobe S, Tsuji N, Iwakura T, et al. Melatonin ameliorates intrarenal renin-angiotensin system in a 5/6 nephrectomy rat model. Clin Exp Nephrol. (2018) 22:539–49. doi: 10.1007/s10157-017-1505-7
106. O'Connor DB, Green JA, Ferguson E, O'Carroll RE, O'Connor RC. Effects of childhood trauma on cortisol levels in suicide attempters and ideators. Psychoneuroendocrinology. (2018) 88:9–16. doi: 10.1016/j.psyneuen.2017.11.004
107. Tang AR, Rabi DM, Lavoie KL, Bacon SL, Pilote L, Kline GA. Prolonged hypothalamic-pituitary-adrenal axis activation after acute coronary syndrome in the GENESIS-PRAXY cohort. Eur J Prev Cardiol. (2018) 25:65–72. doi: 10.1177/2047487317734323
108. Ewart CK, Elder GJ, Jorgensen RS, Fitzgerald ST. The role of agonistic striving in the association between cortisol and high blood pressure. Psychosom Med. (2017) 79:416–25. doi: 10.1097/PSY.0000000000000412
109. Barton M, Yanagisawa M. Endothelin: 30 years from discovery to therapy. Hypertension. (2019) 74:1232–65. doi: 10.1161/HYPERTENSIONAHA.119.12105
110. Pernow J, Shemyakin A, Böhm F. New perspectives on endothelin-1 in atherosclerosis and diabetes mellitus. Life Sci. (2012) 91:507–16. doi: 10.1016/j.lfs.2012.03.029
111. Li MW, Mian MO, Barhoumi T, Rehman A, Mann K, Paradis P, et al. Endothelin-1 overexpression exacerbates atherosclerosis and induces aortic aneurysms in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. (2013) 33:2306–15. doi: 10.1161/ATVBAHA.113.302028
112. Roth L, Rombouts M, Schrijvers DM, Lemmens K, De Keulenaer GW, Martinet W, et al. Chronic intermittent mental stress promotes atherosclerotic plaque vulnerability, myocardial infarction and sudden death in mice. Atherosclerosis. (2015) 242:288–94. doi: 10.1016/j.atherosclerosis.2015.07.025
113. Giannarelli C, Rodriguez DT, Zafar MU, Christoffel D, Vialou V, Peña C, et al. Susceptibility to chronic social stress increases plaque progression, vulnerability and platelet activation. Thromb Haemost. (2017) 117:816–8. doi: 10.1160/TH16-10-0817
114. Mury P, Chirico EN, Mura M, Millon A, Canet-Soulas E, Pialoux V. Oxidative stress and inflammation, key targets of atherosclerotic plaque progression and vulnerability: potential impact of physical activity. Sports Med. (2018) 48:2725–41. doi: 10.1007/s40279-018-0996-z
115. Nation DA, Gonzales JA, Mendez AJ, Zaias J, Szeto A, Brooks LG, et al. The effect of social environment on markers of vascular oxidative stress and inflammation in the Watanabe heritable hyperlipidemic rabbit. Psychosom Med. (2008) 70:269–75. doi: 10.1097/PSY.0b013e3181646753
116. Legein B, Temmerman L, Biessen EA, Lutgens E. Inflammation and immune system interactions in atherosclerosis. Cell Mol Life Sci. (2013) 70:3847–69. doi: 10.1007/s00018-013-1289-1
117. Shao BZ, Han BZ, Zeng YX, Su DF, Liu C. The roles of macrophage autophagy in atherosclerosis. Acta Pharmacol Sin. (2016) 37:150–6. doi: 10.1038/aps.2015.87
118. Kraft C, Martens S. Mechanisms and regulation of autophagosome formation. Curr Opin Cell Biol. (2012) 24:496–501. doi: 10.1016/j.ceb.2012.05.001
119. Xu J, Kitada M, Ogura Y, Koya D. Relationship between autophagy and metabolic syndrome characteristics in the pathogenesis of atherosclerosis. Front Cell Dev Biol. (2021) 9:641852. doi: 10.3389/fcell.2021.641852
120. Evans TD, Jeong SJ, Zhang X, Sergin I, Razani B. TFEB and trehalose drive the macrophage autophagy-lysosome system to protect against atherosclerosis. Autophagy. (2018) 14:724–6. doi: 10.1080/15548627.2018.1434373
121. Grootaert MO, da Costa Martins PA, Bitsch N, Pintelon I, De Meyer GR, Martinet W, et al. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy. (2015) 11:2014–32. doi: 10.1080/15548627.2015.1096485
122. Li BH, Yin YW, Liu Y, Pi Y, Guo L, Cao XJ, et al. TRPV1 activation impedes foam cell formation by inducing autophagy in oxLDL-treated vascular smooth muscle cells. Cell Death Dis. (2014) 5:e1182. doi: 10.1038/cddis.2014.146
123. Grootaert M, Moulis M, Roth L, Martinet W, Vindis C, Bennett MR, et al. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc Res. (2018) 114:622–34. doi: 10.1093/cvr/cvy007
124. Miao J, Zang X, Cui X, Zhang J. Autophagy, hyperlipidemia, and atherosclerosis. Adv Exp Med Biol. (2020) 1207:237–64. doi: 10.1007/978-981-15-4272-5_18
125. Kim KH, Lee MS. Autophagy–a key player in cellular and body metabolism. Nat Rev Endocrinol. (2014) 10:322–37. doi: 10.1038/nrendo.2014.35
126. Weiland A, Wang Y, Wu W, Lan X, Han X, Li Q, et al. Ferroptosis and its role in diverse brain diseases. Mol Neurobiol. (2019) 56:4880–93. doi: 10.1007/s12035-018-1403-3
127. Hirschhorn T, Stockwell BR. The development of the concept of ferroptosis. Free Radic Biol Med. (2019) 133:130–43. doi: 10.1016/j.freeradbiomed.2018.09.043
128. Hu H, Chen Y, Jing L, Zhai C, Shen L. The link between ferroptosis and cardiovascular diseases: a novel target for treatment. Front Cardiovasc Med. (2021) 8:710963. doi: 10.3389/fcvm.2021.710963
129. Bai T, Li M, Liu Y, Qiao Z, Wang Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic Biol Med. (2020) 160:92–102. doi: 10.1016/j.freeradbiomed.2020.07.026
130. Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. (2020) 66:89–100. doi: 10.1016/j.semcancer.2019.03.002
131. Stockwell BR, Jiang X, Gu W. Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol. (2020) 30:478–90. doi: 10.1016/j.tcb.2020.02.009
132. Li L, Wang H, Zhang J, Chen X, Zhang Z, Li Q. Effect of endothelial progenitor cell-derived extracellular vesicles on endothelial cell ferroptosis and atherosclerotic vascular endothelial injury. Cell Death Discov. (2021) 7:235. doi: 10.1038/s41420-021-00610-0
133. Ouyang S, You J, Zhi C, Li P, Lin X, Tan X, et al. Ferroptosis: the potential value target in atherosclerosis. Cell Death Dis. (2021) 12:782. doi: 10.1038/s41419-021-04054-3
134. Zhou Y, Zhou H, Hua L, Hou C, Jia Q, Chen J, et al. Verification of ferroptosis and pyroptosis and identification of PTGS2 as the hub gene in human coronary artery atherosclerosis. Free Radic Biol Med. (2021) 171:55–68. doi: 10.1016/j.freeradbiomed.2021.05.009
135. Guo S, Li L, Yin H. Cholesterol homeostasis and liver X receptor (LXR) in atherosclerosis. Cardiovasc Hematol Disord Drug Targets. (2018) 18:27–33. doi: 10.2174/1871529X18666180302113713
136. Hafiane A, Gasbarrino K, Daskalopoulou SS. The role of adiponectin in cholesterol efflux and HDL biogenesis and metabolism. Metab Clin Exp. (2019) 100:153953. doi: 10.1016/j.metabol.2019.153953
137. Favari E, Chroni A, Tietge UJ, Zanotti I, Escolà-Gil JC, Bernini F. Cholesterol efflux and reverse cholesterol transport. Handb Exp Pharmacol. (2015) 224:181–206. doi: 10.1007/978-3-319-09665-0_4
138. Annema W, Tietge UJ. Role of hepatic lipase and endothelial lipase in high-density lipoprotein-mediated reverse cholesterol transport. Curr Atheroscler Rep. (2011) 13:257–65. doi: 10.1007/s11883-011-0175-2
139. Demina EP, Miroshnikova VV, Schwarzman AL. Role of the ABC transporters A1 and G1, key reverse cholesterol transport proteins, in atherosclerosis. Mol Biol. (2016) 50:223–30. doi: 10.1134/S0026893316020047
140. Groenen AG, Halmos B, Tall AR, Westerterp M. Cholesterol efflux pathways, inflammation, and atherosclerosis. Crit Rev Biochem Mol Biol. (2021) 56:426–39. doi: 10.1080/10409238.2021.1925217
141. Ren K, Li H, Zhou HF, Liang Y, Tong M, Chen L, et al. Mangiferin promotes macrophage cholesterol efflux and protects against atherosclerosis by augmenting the expression of ABCA1 and ABCG1. Aging. (2019) 11:10992–1009. doi: 10.18632/aging.102498
Keywords: chronic stress, atherosclerosis, inflammation, lipid metabolism, endothelial function, plaque stability
Citation: Meng L-b, Zhang Y-m, Luo Y, Gong T and Liu D-p (2021) Chronic Stress A Potential Suspect Zero of Atherosclerosis: A Systematic Review. Front. Cardiovasc. Med. 8:738654. doi: 10.3389/fcvm.2021.738654
Received: 09 July 2021; Accepted: 29 November 2021;
Published: 20 December 2021.
Edited by:
Yi Yang, First Affiliated Hospital of Jilin University, ChinaReviewed by:
Achuthan Raghavamenon, Amala Cancer Research Centre, IndiaFu-Liang Zhang, First Affiliated Hospital of Jilin University, China
Copyright © 2021 Meng, Zhang, Luo, Gong and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: De-ping Liu, bGxpdWRlcGluZ0AyNjMubmV0; Tao Gong, bWFjMDg1MkAxNjMuY29t