Abstract
Atrial fibrillation (AF) is the most common arrhythmia attacking 1. 5–2.0% of general population worldwide. It has a significant impact on morbidity and mortality globally and its prevalence increases exponentially with age. Therapies like catheter ablation or conventional antiarrhythmic drugs have not provided effective solution to the recurrence for AF over the past decades. Over 100 genetic loci have been discovered to be associated with AF by Genome-wide association studies (GWAS) but none has led to a therapy. Recently potential involvement of epigenetics (DNA methylation, histone modification, and non-coding RNAs) in the initiation and maintenance of AF has partly emerged as proof-of-concept in the mechanism and management of AF. Here we reviewed the epigenetic features involved in AF pathophysiology and provided an update of their implications in AF therapy.
Introduction
The most common arrhythmia is atrial fibrillation (AF). It has an estimated worldwide prevalence of 1.5–2.0% in the general population (1). Currently, over 33 million individuals are suffering from AF worldwide, and the prevalence is anticipated to more than double in the next 40 years (2). AF has substantially increased hospitalization rates, stroke occurrence, and social medical burden and impaired quality of life.
There is a growing understanding of the mechanisms underlying the onset and maintenance of AF. The pathophysiology of atrial fibrillation focuses on ectopic firing promotion and reentrant mechanisms, including ion channel dysfunction, Ca2+-signaling abnormalities, structural remodeling, and autonomic neural dysregulation (3). The potential implications of the pathophysiology of AF for its management have been improved rhythm control pharmacotherapy, rate control therapy, AF ablation, and the prevention of thromboembolic events (4). But these treatments have shown limited improvement in AF patients.
Genome-wide association studies (GWAS) have uncovered over 100 AF associated genetic loci (5). But none has been identified to be a potential therapeutic target, indicating additional candidates to AF pathophysiology. High blood pressure (HBP), diabetes mellitus (DM), and heart failure (HF) are the common risk factors of AF. The oxidative stress and inflammation in HBP, DM, or HF probably contribute to the initiation of AF through calcium signaling related-structural remodeing, electrical reentry mechanism, and autonomic nerve activation. Recently, the possible mechanism linking epigenetics and AF are reactive oxygen species (ROS) levels. One study found that ROS levels promotes AF via increased intracellular Ca2+ release by oxidized RyR2 (6). A recent research found that long non-coding RNA ZNF593-AS facilitated RyR2 mRNA stability (7). The changes in epigenetic state induced by ROS levels could provide the basis for uncovering the pathophysiology and management of AF. The functional characterization of regulatory mechanisms, involving DNA methylation, histone modification, and non-coding RNAs, may be related to AF epigenetics (Figure 1) (8). Here we will review the epigenetic features involved in AF pathophysiology and provide a latest functional implications in AF therapy.
Figure 1
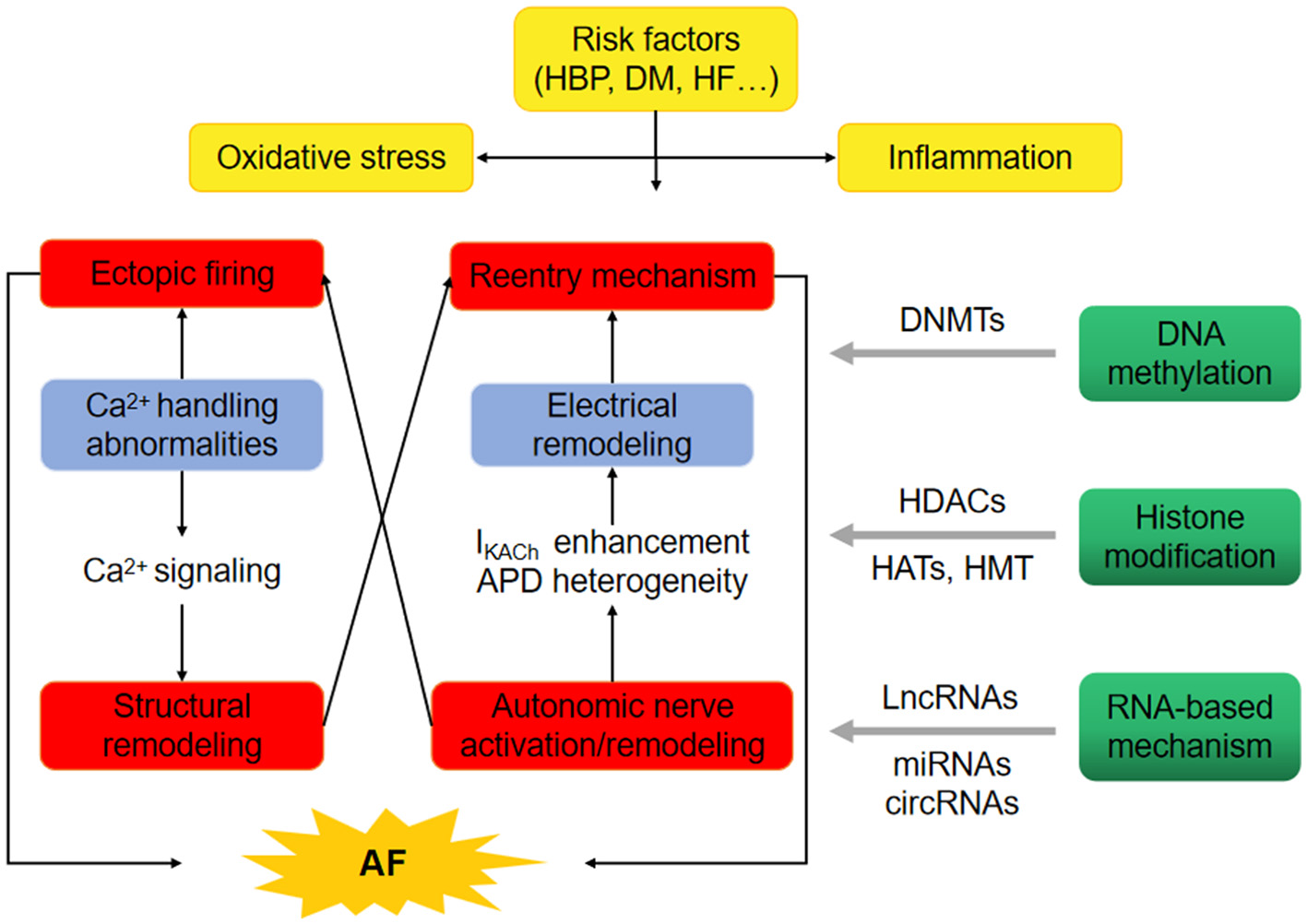
Epigenetics provide the basis for uncovering the pathophysiology of atrial fibrillation (AF). The oxidative stress and inflammation in high blood pressure (HBP), diabetes mellitus (DM) and heart failure (HF) probably promote the occurrence of AF through ectopic firing and reentry mechanism. Epigenetic regulatory mechanisms, involving DNA methylation through DNA methyltransferases (DNMTs), histone modification through histone acetyltransferases (HATs), histone methyltransferases (HMT) and/or histone deacetylases (HDACs), non-coding RNAs such as long-ncRNA (LncRNAs), microRNAs (miRNAs) and circular RNAs (circRNAs) may comtribute to the initiation of AF.
AF Associated Epigenetic Regulation
AF is a highly heterogeneous genetic disease, which consists of distinct subtypes characterized by a few specific chromosomal abnormalities or gene mutations. Although candidate genes and GWAS have identified a number of genetic variants of AF, and elucidated the heritability of AF, a large proportion of AF cases cannot be interpreted by genetic variants alone. Epigenetic regulation including DNA methylation, histone modification, and non-coding RNAs has been explored in the patients and animal/cell modes of AF (Tables 1, 2).
Table 1
| Species | Model/tissue | Discovery | References |
|---|---|---|---|
| DNA methylation | |||
| Human | AF/peripheral blood | 2 CpG site associated with prevalent AF (n = 183) 5 CpG site associated with incident AF (n = 220) |
(11) |
| Human | PeAF/LA | 417 differentially methylated CpG sites AF susceptible loci hypermethylation |
(12) |
| Human | valvular AF/RA | DNMT3B → NPR-A promoter hypermethylation | (13) |
| Rats | ISO treatment/primary cell culture from SD rats | DNMT3A → RAASSF1A-ERK1/2 → cardiac fibrosis | (14) |
| Human Rats Cell line |
VHD/LA ISO treatment/SHR AngII/HL-1 cell |
HF → Pitx2 promoter hypermethylation | (15, 17) |
| Mice Cell line |
C57BI6/J mice HL-1 atrial CM |
SUR2 promoter hypermethylation | (16) |
| Histone modifications and chromatin remodeling | |||
| Human | AF/Heart | (20) | |
| Human | PeAF/RA, AFB | Elevated expression of EZH2, an HMT specific for H3K27me | (21) |
| Human Cell line |
PeAF/RAA,LAA HL-1 atrial CM |
HDAC5 activation and subsequent MEF2-related fetal gene expression | (22) |
| Human Canine Cell line |
PeAF,PAF/RAA,LAA HL-1 atrial CM |
HDAC6 activation and subsequent derailment of alpha-tubulin proteostasis and disruption of the cardiomyocyte microtubule structure | (23) |
| Mice | HopXTg/heart | HDAC inhibition reverses myocardial fibrosis and reduces atrial arrhythmia independent of angiotensin | (24) |
| Rabbits | PVs and SANs | HDAC6,HDAC8 (≥70% inhibition) HDAC4,HDAC5,HDAC7,HDAC9 (≤50% inhibition) Reduces PVs arrhythmogenesis through calcium regulation |
(25) |
| Human Mice Canine |
Sustained AF/HopXTg Atrial tachypacing dogs |
Class I HDAC (HDAC1, HDAC2, HDAC3, HDAC8) inhibition Reduce the total time of fibrillation, atrial fibrosis, intra-atrial adipocytes and immune cell infiltration |
(26) |
| Mice | CREM-IbΔC-XTg | HDAC inhibitor VPA Attenuates atrial remodeling and delays the onset of AF |
(31) |
| Non-coding RNAs | |||
| Canine | RAP/atrium | miR-133, miR-590 downregulation → atrial fibrosis | (34) |
| Human Mice |
PeAF/LAA | miR-21 upregulation → atrial fibrosis | (35) |
| Human Canine Mice |
Persistent AF/ Blood,RAA |
miR-29b downregulation → ECM protein → atrial fibrosis | (37) |
| Human | Persistent AF/ LA |
miR-1 downregulation → inward rectifier potassium currents upregulation → AF | (39) |
| Human Canine Mice |
AF/RAA | miR-26 downregulation → inward rectifier potassium currents(IK1) → KIR2.1/KCNJ2 → fibroblast remodeling | (40) |
| Rabit | Atrial tachypacing model/atrial myocyte | miR-1 → KCNE1,KCNB2 → AERP shortening | (41) |
| Human Mice |
PAF/RAA | miR-106b-25 cluster → RyR2 → calcium leak | (43) |
| Human Canine |
AF/ atrium, LA | miR-223, miR-328, miR-664 upregulation miR-101, miR-320, miR-499 downregulation |
(44) |
| Mice | heart | miR-17-92 and miR-106b-25 downregulation | (45) |
| Rabbits | AF/atrium | LncRNA(TCONS-00106987) upregulation By sponging miR-26 in electrical remodeling |
(47) |
| Rabbits | AF/RA | TCONS_00075467 sponging miR-328 → electrical remodeling | (48) |
| Human | AF/atrium | NEAT1-miR320-NPAS2 axis | (49) |
| Human | AF/EAT | lncRNA differentially expressed in AF and SR | (50, 51) |
| Human | VHD(50%PeAF)/LAA | circRNA-miRNA-mRNA network DEcircRNA between AF group and SR group (circ 255-ITGA7, circ 418-KCNN2, circ 13913-MIB1, circ 44670-BARD1, circ 44782-LAMA2, circ 81906-RYR2, circ 35880-ANO5, circ 22249-TNNI3K, circ 3136-TNNI3K, circ 56186-TNNI3K) |
(52) |
| Canine | RAP | circRNA-miRNA interaction | (53) |
| Human | AF/Heart | circRNA-miRNA interaction (e.g., has_circRNA_100612 and has_miR_133b) |
(54) |
| Human | VHD/AA | 147 DEcircRNA between AF group and SR group circRNA-miRNA interaction (e.g., has-miRNA-2215p and has_circRNA_0005643) (e.g., has-miRNA-2215p and has_circRNA_0077334) |
(55) |
| Human | AF/peripheral blood | DEcircRNA-DEmiRNA-DEmRNA network (e.g., hsa-circRNA-100053-hsa-mirR-455-5p-TRPV1) (e.g., hsa-circRNA-005843-hsa-mirR-188-5p-SPON1) |
(56, 57) |
| Human | AF/LA | circRNAs sponging activities in PeAF | (59) |
| Human | RNA sequencing data | Dysregulation of circRNAs in inflammation response in AF | (60, 61) |
Epigenetic modification in AF.
AF, atrial fibrillation; PAF, paroxysmal AF; PeAF, permanent AF/persistent AF; LA, left atrium; VHD, valvular heart disease; RA, right atrium; DNMTs, DNA methyltransferases; NPR-A, natriuretic peptide receptor-A; ISO, isoproterenol; SHR, spontaneously hypertensive rats; SD, Sprague-Dawley; AngII,Angiotensin II; RAASSF1A, Ras association domain family 1 isoform A; ERK, extracellular regulated protein kinases; Pitx2, paired-like homeodomain 2; CM, cardiomyocytes; SUR, sulfonylurea recepor; AFB, atrial fibroblast; EZH2, zeste homolog2; HMT, histone methyltransferase; RAA, right atrial appendage; LAA, left atrial appendage; HDAC, histone deacetylase; VPA, Valproic Acid; ECM, extracellular matrix; MEF2, myocyte enhancer factor-2; AERP, atrial effective refractory period; RyR2, ryanodine receptor; HopX, homeo-domain-only protein; PV, pulmonary vein; SAN, sinoatrial node; RAP, rapid atrial pacing; NEAT1, Nuclear-Enriched Abundant Transcript 1; EAT, epicardial adipose tissue; SR, sinus rhythm; VHD, valvular heart diseases; DEcircRNA, differentially expressed circRNA.
Table 2
| Epigenetic modification | Target | Biological function | Ref |
|---|---|---|---|
| DNA methylation | |||
| Hypomethylation | BMP6, BMP8B | TGF-β signaling pathway | (12) |
| MSRA, CLU, DUOX2 | Response to oxidative stress | (12) | |
| SLC12A7, SLC38A7, SLC9A9, SCN1A | Sodium ion transport | (12) | |
| KCNS3, KCNA4, ATP12A, SUR2 | Potassium ion transport | (12, 16) | |
| ITGA5, ITGAE, DOCK1 | Integrin-mediated signaling pathway | (12) | |
| PSMB9, OPRD1, HLA-G, HLA-C, BMP6, HLA-DMB, IL16, MR1, HLA-DRB1, TAPBP, TGFBR3, HLA-DQA1, HLA-DRA | Immune response, Antigen processing and presentation | (12) | |
| GMCL1L, PPP1R9A, RNASE4, BMP8B, PAX8, CYFIP1, HLX, MGP | Cell differentiation | (12) | |
| STEAP3, CSRNP1, LGALS7, CLU, DOCK1, UBE4B | Apoptosis | (12) | |
| Hypermethylation | APOL6, OSBPL3, APOA5 | Lipid transport, Lipid transport | (12) |
| SPDEF, HOXC4, NXN, MSX1, MEIS1, HDAC4, ARHGAP22, DUSP22, HOXA3, EBF4, EBF3, PRM1, DMBX1, PITX2 | Development | (12, 15, 17) | |
| F3, MIB2 | Notch signaling pathway | (12) | |
| NPR-A | Cardiac hypertrophy | (13) | |
| RASSF1A | Cardiac fibrosis | (14) | |
| Histone modifications and chromatin remodeling | |||
| H3K27ac, H3K4me1 | GATA4, MYH6, NKX2-5, PITX2, TBX5 CFL2, MYH7, PKP2, RBM20, SGCG, SSPN | Serious heart defects Striated muscle function and integrity |
(20) |
| H3K27me3 | EZH2, ACTA2 | Atrial fibrosis | (21) |
| HDAC5 phosphorylation | MEF2-related fetal gene e.g., β-MHC, α-MHC, BNP |
cardiomyocyte remodeling | (22) |
| HDAC6 activation | α-Tubulin | disruption of the cardiomyocyte microtubule structure | (23) |
| HDAC inhibition | Connexin 40 | Atrial structural remodelin | (24) |
| HDAC inhibition | Ca2+transient amplitudes, sodium-calcium exchanger currents, and ryanodine receptor | Calcium homeostasis | (25) |
| Class I HDAC inhibition | CD19. CD4, CD163 TNF-α, IL-1β, IL-6, IL-8, leptin, and adiponectin |
Angiotensin II signaling in atrial remodeling | (26) |
| HDAC inhibition | RhoA | Oxidative phosphorylation | (31) |
| Non-coding RNAs | |||
| miR-133,miR-590 | TGF-β1, TGF-βRII | Nicotine-induced atrial fibrosis | (34) |
| miR-21 | Spry 1, CTGF | AngII-induced atrial fibrosis | (35) |
| miR-133, miR30 | CTGF | Hypertension-induced LVH TAC-induced LVH |
(36) |
| miR-29b | COL1A1, COL3A1 | CHF related-atrial fibrosis | (37, 38) |
| miR1, miR-26, LncRNA(TCONS-00106987) | KCNJ2 | Regulation of Kir2.1(subunit of Ik1) | (39, 40, 47) |
| miR-1 | KCNE1, KCNB2 | Regulation of subunit of Iks | (41) |
| miR-106b-25 cluster | RyR2 | calcium leak | (43) |
| miR-328 TCONS_00075467 |
CACNA1C, CACNB1 | L-type calcium channel regulation | (44, 48) |
| miR-17-92, miR-106b-25 | Shox2, Tbx3 | Sinoatrial node dysfunction | (45) |
| miR320 | NPAS2 | AngII-induced atrial fibrosis | (49) |
Target of epigenetic modification in AF.
Ref, reference; AF, atrial fibrillation; TGF-β1, transforming growth factor-beta1; TGF-βR1, transforming growth factor-beta R II; NPR-A, natriuretic peptide receptor-A; RAASSF1A, Ras association domain family 1 isoform A; AngII, Angiotensin II; Pitx2, paired-like homeodomain 2; SUR, sulfonylurea recepor; MEF2, myocyte enhancer factor-2; RhoA, Ras homolog gene family, member A; CTGF, connective tissue growth factor; H3K27AC, acetylation of histone H3 lysine 27; H3K4me1, methylation of histone H3 lysine 4; H3K27me3, trimethylation of lysine 27 on histone 3; Spry 1, Sprouty 1; ECM, extracelluar matrix; TAC, transverse aortic constriction; LVH, left ventricular hypertrophy; CHF, congestive heart failure; AngII, angiotensin II; RyR2, ryanodine receptor; NPAS2, neuronal per arnt sim domain protein2.
AF Associated DNA Methylation
DNA methylation is well-characterized as a heritable regulation of gene expression. A methyl group is catalyzed by DNA methyltransferases (DNMTs) to shift from the S-adenosyl-L-methionine to the 5' carbon of cytosine which mostly located in cytosine-phosphate-guanine (CpG) islands (9). The gene promoter hypermethylation correlates with transcriptional silencing, whereas hypomethylation leads to increased expression of the gene (10). DNA methylation regulation may serve an important role in AF pathogenesis.
It has been identified the methylation of CpG sites in prevalent, permanent, and paroxysmal AF. Differential methylation of CpG sites were significantly related to prevalent AF (two CpG sites) and incident AF (five other CpGs) by GWAS of the participants' peripheral blood in the Framingham Heart Study (11). The majority of 417 differentially methylated CpG sites discovered in the fibrillating atrium were located in intergenic regions outside of CpG islands (12).
DNMTs dysregulations likely plays an important role in the pathogenesis of AF. Significantly higher level of the whole DNA methylation was found in the AF group than SR group (13). DNMT3b likely contributes to the DNA methylation dysregulations in valvular AF (13). DNMT3A was shown to be involved in Ras association domain family 1 isoform A (RASSF1A)-mediated upregulation of extracellular signal regulated kinases 1/2 (ERK1/2) in cardiac fibrosis (14).
DNA hypermethylation also participated in the association of transcription factor, fibrosis and potassium ion transport with pathophysiology of AF (15, 16). Hypermethylation of paired-like homeodomain 2 (Pitx2) promoter was shown to be associated with AF in humans and aging spontaneously hypertensive rats (SHR) (15). DNA methylation inhibitor 5-Aza-2'-deoxycitidine treatment reduced the left ventricular fibrosis in SHR (13). The CpG hypermethylation of SUR2, a subunit of the ATP-sensitive potassium channel, leads to its silencing expression in the HL-1 atrial cardiomyocyte cell line (16).
Heart failure (HF), a risk factor of AF, may induce Pitx2c promoter hypermethylation in vivo and in vitro (17) Pitx2c promoter methylation increased and Pitx2c protein level decreased with the increase of DNMT 1 in isoproterenol-induced HF atria compared with normal atria (17). The same trend was shown in Angiotensin II (AngII)-treated HL-1 cells compared with control cells (17). The methylation inhibitor 5-AZa-2 '-deoxycytitine and the AngII receptor blocker Losartan attenuated these effects (17). However, isoproterenol did not alter the expression of Pitx2c and DNMT1 (17).
AF Associated Histone Modifications and Chromatin Remodeling
Chromatin is the state in which DNA is wrapped in the cell. The nucleosome, the basic unit of chromatin, is an octamer composed of four core histones (H3, H4, H2A, H2B) surrounded by 147 DNA base pairs. A primary component of chromatin that plays an essential role in this regulation is the modification of histones. The core histones are mainly globular except for their unstructured N-terminal “tails.” A distinctive feature of histones, especially their tails, is the large number and type of modified residues they possess.
The gene expression regulation in euchromatin requires the delivery of chromatin-modifying enzymes by DNA-bound transcription factors (TFs). Following the external cues, TFs bind to the specific genes' promoter and promote the gene expression or silencing. So there will be activation-related and repression-related modification for the purposes of transcription. Histone modifications are involved in affecting gene expression. The mono-methylations of H3K27, H3K9, H4K20, H3K79, and H2BK5 are related to gene activation, whereas trimethylations of H3K27, H3K9, and H3K79 are linked to repression.
More and more evidences show that N-terminal tail of histone is subjected to covalent and reversible post-translational modifications, such as methylation and acetylation (18, 19). These modifications are used to modify chromatin compress or create anchoring sites for other transcriptional regulators (18). The methylation and acetylation status of chromatin is regulated by writers and erasers. The most studied representative of writers and erases are histone methyltransferases (HMT) and histone deacetylases (HDACs) (19).
Emerging evidences reveal a role for histone acetylation or methylation to modulate pathogenic gene expression in AF patients. GWAS of AF patients (n = 60,620) and controls (n = 970,216) indicated active enhancers as indicated by acetylation of histone H3 lysine 27 (H3K27ac) in right atrium, which has demonstrated that AF-associated risk variants fell near the genes acting via structural cardiac remodeling (20). Enhancer of zeste homolog2 (EZH2), a histone-lysine N-methyltransferase enzyme encoded by the EZH2 gene, is participating in histone methylation by binding to H3K27me3 (trimethylation of lysine 27 on histone 3). The expression of EZH2 and H3K27me3 is upregulated in permanent AF patients with atrial fibrosis (21). Compared to the people with sinus rhythm (SR), HDAC3 protein expression and activity levels were increased in paroxysmal AF (PAF), persistent AF (PeAF) (n = 5), and long-standing PeAF (n = 7) (22). The phosphorylated-HDAC5 levels were correlated with significantly increased BNP gene expression in PeAF patients (22). Similarly, the remarkable elevated expression and activity of HDAC6 were correlated with PeAF duration (23). However, whether the overall acetylation level of protein or the total HDAC activity had no dramatically difference between patients with PeAF/PAF and subjects in SR (23).
Furthermore, the mechanisms in epigenetic regulation of AF, especially HDACs inhibition, have been investigated in vivo and in vitro. The use of HDAC6 inhibitor tubastatin A in vivo can protect dogs with atrial tachycardia pacing from electrical remodeling, while the dominant negative HDAC6 mutant can completely rescue the systolic dysfunction induced by tachycardia pacing (23). HDACs may regulate transcriptional reprogramming in AF. The homeo-domain-only protein (hopx) transgenic mice were recruited into chromatin to induce serum response factor (SRF)—dependent transcription and myocardial hypertrophy. They were administered with or without pan HDAC inhibitor trichostatin A (TSA). TSA treated mice have protective effects on atrial arrhythmia and fibrosis induced by rapid pacing (24). Pan-HDAC inhibitor TSA and class I HDAC inhibitor MPT0E014 reduce the onset of AF by reducing calcium spark through normalize the expression of NCX1 and ryanodine receptors in rabbit pulmonary vein cardiomyocytes (25). Hopx transgenic mice with atrial remodeling and dogs with atrial tachypacing treated with HDAC inhibitor (CI-994) showed no significant effects on cardiac function but decrease of the total time of fibrillation and atrial fibrosis in atrial tachypacing-induced sustained AF (26).
So various HDACs inhibitors are emerging as interesting druggable targets for AF (27, 28). RGFP966, a specific inhibitor of HDAC3, could prevent systolic dysfunction in cardiomyocyte for AF (29). ACY-1215 (ricolinostat), one of HDAC6 inhibitors, will be a potential candidate drug for trials in patients with AF. Because it is undergoing Phase I and II clinical trials for the treatment of multiple myeloma at present with no reports of serious side effects so far (30). Valproic acid (VPA), an HDAC class I/IIA inhibitor, alleviated atrial remodeling in transgenic mice, animal AF models, and human AF (31). And the researchers used ChIP to identify 9 VPA-downregulated genes (Atp5l, Ces1d, Myl7, Ndufa12, Ndufa8, Ndufs7, Pdha1, Tnni3, Uqcr10) (31).
AF Associated Non-coding RNAs
Most AF-associated GWAS variants reside in the non-coding genome. The non-coding RNAs (ncRNAs) can be classified as short-chain ncRNAs (<200 nucleotides) and long-ncRNA (lncRNAs, >200 nucleotides) (32, 33). The short-chain ncRNAs include microRNAs (miRNAs), transfer RNAs, small nuclearRNAs, small nucleolar RNAs, piwi-interacting RNAs, telomerase RNAs, and other endogenous RNA species (32). CircularRNAs (circRNAs) emerge as novel non-coding RNAs that differ from traditional linear RNAs (33).
The transition from paroxysmal to permanent AF is characterized by a pattern of dysregulated miRNAs, which involve in atrial remodeling or fibrosis, electrical remodeling, calcium signaling, and dysregulation of transcription factors (TFs) (34–42). Mir-133, mir-590, mir-21, miR30, and mir-29b regulated the genes related to arial fibrosis or fibroblast remodeling in atrial fibrillation (34–37). There was upregulation of transforming growth factor (TGF) TGF-β1 and TGFβRII at the protein level and downregulation of miR-133 and miR-590 in the levels of miRNAs in nicotine stimulated atrial fibrosis of AF in dog (34). The angiotensin II (AngII)-induced upregulation of miR-21 and repression of Spry1 was showed in neonatal cardiac fibroblasts (35). Connective tissue growth factor (CTGF) is a secreted protein as a powerful inducer of extracellular matrix (ECM) synthesis. Its levels are substantially increased in hypertension-induced left ventricular hypertrophy (LVH) and transverse aortic constriction (TAC)-induced LVH rodent models (36). The expression of miR29b decreased and the expression of miR29b ECM target-genes collagen-1A1 (COL1A1), collagen-3A1 (COL3A1) increased significantly in congestive heart failure (CHF) atrial fibroblasts (37, 38). The regulation of miR-1, miR-26, miR-208a, miR-328, and miR-499 on genes associated with electrical remodeling promotes reentry circuits by shortening of the action potential duration and effective refractory period (37–40). The increased inward-rectifier K+ current (IK1), along with increased expression of the principal underlying subunit KCNJ2 mRNA and its encoded Kir2.1 protein, has been demonstrate to exhibit a pro-AF-related atrial electrical remodeling. Girmatsion et al. reported that miR-1 levels decreased significantly in human AF and led to increased IK1 possibly by up-regulating of KCNJ2 (39), miR-26 was found to be downregulated accompanied by upregulation of IK1/Kir2.1 protein in atrial samples of AF patients (40). The slowly activating delayed rectifier potassium currents (IKs) and atrial effecitve refractory period (AERP) shortening play an important role in the electronic remodeling of AF. The expression of miR-1 was upregulated to induce AERP shortening by targeting IK1 channel genes (KCNE1, KCNB2) in right atrial tachypacing in New Zealand white rabbits (41). MicroRNAs have also been shown to control the expression of genes encoding important Ca2+ processing and signaling proteins in AF (42). Studies have shown that the atrial level of ryanodine receptor type-2 (RyR2) protein is elevated in paroxysmal AF (pAF) patients, suggesting that post-transcriptional regulation of RyR2 might contribute to the pathogenesis of AF (43). Members of the miR-106b-25 cluster, such as miR-106b and miR-93, could suppress the translation of RyR2 by binding to its 3'-untransted region (UTR) (43). The downregulation of the miR-106b-25 cluster and upregulation of RyR2-mediated sarcoplasmic reticulum Ca2+ leak were demonstratedin atria of pAF patients (44). The miR-328 level was elevated by more than 3-fold in AF patients and AF dogs compared to non-AF subjects. Overexpression of miR-328 diminished the L-type Ca2+ currents by targeting CACNA1C and CACNB1, which encode its α1c and β1subunits (44). In addtion, miRNA loss-of-function could develop sinoatrial node dysfunction to increase susceptibility of AF (45). miR-17-92 and miR-106b-25 directly supress the genes required for sinoatrial node function, such as Shox2 and Tbx3 (45). Both miR-17-92 and miR-106b-25 inactivation exhibited pacing-induced AF in mice (45).
Several studies have shown that lncRNAs are participanting in basic mechanism of AF development, including electrical remodeling, atrial remodeling, and metabolic remodeling (46). IncRNA TCONS-00106987 was confirmed to promote electrical remodeling of increasing inward-rectifier K+ current (IK1) through endogenous competition with microRNA-26 (miR-26) by luciferase reporter assays and whole-cell patch-clamp recording in AF rabbit model (47). Silencing of TCONS_00075467 can shorten the atrial effective refractory period in vivo and reduce duration of the L-type calcium current and action potential in vitro, which may also play important roles in electrical remodeling regulation during AF (48). The expression of lncRNAs nuclear-enriched abundant transcript 1 (NEAT1) in atrial tissue of AF patients was up-regulated, and NEAT1 knockdown could improve Ang II-induced atrial fibrosis via the miR-320-NPAS2 axis in mice (49). Recent evidence suggested that lncRNAs in epicardial adipose tissue (EAT) may modulate atrial remodeling (50, 51). Seventeen upregulated lncRNAs and 40 downregulated lncRNAs were differentially expressed in EAT samples collected from persistent non-valvular AF and sinus rhythm (SR) (P < 0.05; fold change>1.5) (50). These differentially expressed lncRNAs were mainly related with stress response and metabolic remodeling, which indicating pathogenesis of AF (50). The biological function predictions for the RNA sequencing data collecting from atrial EAT samples of AF and SR revealed that TNF signaling pathway was the most frequent pathway that the lncRNAs might involve in (51).
Recently, more attention has been paid on circRNAs and their association with miRNA and lncRNAs in AF intiation and perpetuation. The differentially expression of circRNAs (Table 1) were verified between persistent AF patients and SR people (52). One hundred and forty-six different circRNAs were found between control and rapid atrial pacing (RAP) dogs (53). And analysis showed that the differentially expressed circRNAs may be involved in the process of “cytoskeleton structural composition and ion channel activity” as well as extensive interaction among different circRNAs and AF related miRNAs and mRNAs (53–55). Integrated analysis speculated that circRNA-microRNA interaction pairs and intricate cross-talk may be involved in AF (Table 1) (56, 57).
Circular RNAs show higher stability than other RNAs. They also exhibit more functional patterns, such as sponging microRNAs (58). The sponging activities of the circRNAs could be responsible for the down-regulation of specific miRNAs in establishment of a permanent AF condition (59).
In addtion, the dysregulated circRNAs may be enriched in participation of inflammatory response in AF (60, 61). 250 up- and 126 down-regulated circRNAs were differentially expressed between AF subjects and healthy donors. It should be noted that the enrichment analysis identified five circRNAs showing the highest significance. Among them, four were enriched in cytokine-cytokine receptor interaction (60). A well-recognized participant for atrial fibrosis association with AF was TGF-beta signaling pathway (61). Differential expression of 14,215 circRNAs were detected in AF patients and healthy controls. Among them, hsa_circ_0000075 and hsa_circ_0082096 was exhibited to be invovled in TGF-beta signaling pathway of the AF pathogenesis (61).
All these researches make ncRNAs promising candidates for the development of latent diagnostic or prognostic biomarkers and even therapeutic targets for AF.
Epigenetic Modifications in Paroxysmal, Persistent, and Permanent AF
Although differential methylation of CpG sites was found between persistent AF (PeAF) and paroxysmal AF (PAF) in the Framingham Heart study (11), there was no significant difference of the total level of acetylation or the HDAC activity between PeAF and PAF (23). Previous study, whether Genome-wide DNA methylation profiling, HDAC inhibition or role of miR-21, had been performed in permant AF or sustained AF (12, 26, 35). Recent research has been more and more focused on AF promotion (22, 59). Converse role of class I and IIa HDACs was showed in the progression of AF (22). The down-regulation miRNA sponged by circRNA is a characteristics in the transition from PAF to PeAF (59).
Chromatin Immunoprecipitation (ChIP) and ChIP Sequencing (ChIP-Seq) Study in AF
Chromatin immunoprecipitation (ChIP) is a powful tool to analyze protein-DNA interactions in vivo. ChIP-seq, which combines ChIP with second-generation sequencing technology, can efficiently detect genome-wide DNA segments that interact with histones or transcription factors. A ChIP assay showed increased specific binding of EZH2 protein to the DNA of α-SMA promoter, indicating EZH2 was responsible for the atrial fibroblast activation through α-SMA (21). Valproic acid (VPA), an HDAC class I/IIA inhibitor, alleviated atrial remodeling in transgenic mice, animal AF models, and human AF (31). The researchers used ChIP to identify 9 VPA-downregulated genes (Atp5l, Ces1d, Myl7, Ndufa12, Ndufa8, Ndufs7, Pdha1, Tnni3, Uqcr10) (31). miR-26 was found to be downregulated accompanied by upregulation of IK1/Kir2.1 protein in atrial samples of AF patients (40). The ChIP results displayed the binding of nuclear factor of activated T cells (NFAT) to the 3 cis-acting elements in the 5' flanking regions of miR-26 (40). ChIP-Seq analysis revealed that Pitx2 directly bound to conserved chromatin upstream of miR-17-92 and miR-106b-25 (45). All these ChIP and ChIP-seq results suggested the possible signaling pathway in the pathogenesis of epigenetics of AF.
Epigenetic Therapeutic Implications in AF
Therapies like radiofrequency ablation or pharmacotherapy targeting dysfunction of ectopic firing promotion and reentrant mechanisms have not provided effective solution to the recurrence for AF over the past decades (3–6). RNA-therapy has been paved the way for the translation of experimental studies to human clinical trial. Antisense oligonucleotides (ASO), small interfering RNAs (siRNAs), and microRNAs could consist a potential target in RNA-therapy. However, the most challenging setback is off-target effects. ISIS-CRPRx, a second generation ASO complementary to the coding region of the human c-reactive protein (CRP) mRNA, could reduce the CRP levels substantially but not AF burden in a phase 2 clinical trial in patients with PAF (62). The identification of the epigenetic mechanism and proof-of-concept in preclinical models of AF suggest epigenetic therapies, the so-called epidrugs, will be potential gene therapy approaches for AF.
The epidrugs target and reprogram the epigenome. First- and second-generation epidrugs are small-molecule inhibitors of DNMTs or histone-modifying enzyme. Transcriptional procedures that inhibit DNMTs to reverse DNA hypermethylation and restore sinus rhythm have been proposed as a therapy in AF animals (14, 15). The chronic administration of decitabine (one of the inhibitors of DNMTs) improved ECG arrhythmias and reduced left ventricular fibrosis in a SHR model of AF (15). Although there is poor exploration of inhibitory effect of histone modifying enzymes on AF, they have attracted attention as potential targets for AF therapeutic strategy. Cardiac hypertrophy was reversed by simultaneous treatment of HDAC inhibitors (valproic acid, trichostatin A, and SK-7041) in rodents (63). Administration with tubastatin A (an isoform selective inhibitor of HDAC6) to atrial tachypaced dogs can protects them from atrial remodeling (23). CI-994, second-generation HDAC inhibitors, did not have significant effects on cardiac function in sustained AF model of canine, while the time of fibrillation and atrial fibrosis was reduced (26).
Epigenome plays an important role in regulating cardiac fibroblast activation based on cardiac inflammation, indicating that epidrugs can provide an innovative method to reverse AF related atrial fibrosis (64). EZH2 inhibitor GSK126 can inhibit Ang II induced atrial fibroblast differentiation and ECM production in a mouse model (21).
The epidrugs have been approved for clinical treatment of cancer. Notable examples are the inhibitors of DNMT and HDAC for hematological cancer (65, 66). Azacytidine and decitabine, the first generation of DNMT inhibitors, have been efficacious with poor bioavailability, toxic side effects, and lack of locus-specificity. Second-generation DNMT inhibitors, such as zebularine and guadecitabine, have been developed to make up for the deficiency (65). Some HDAC inhibitors have been approved by FDA for the therapy of hematological malignancies—Romidepsin or Vorinostat for cutaneous T cell lymphoma, Panobinostat for multiple myeloma, and Belinostat for peripheral T cell lymphoma (66). While some HDAC inhibitors are still in clinical trials: Givinostat for haemological/muscular cancer, Entinostat for breast/ovarial cancer, and Resminostat for hepatocellular/colorectal carcinoma. BET inhibitors and HMT inhibitors emerged as the latest generation of epidrugs for cancer, such as OTX-015 or I-BET762, are now in clinical trials (67). However, due to the inadequacy of selectivity and efficacy, these drugs have not been used in the clinical application of AF.
Recently, proteolysis targeting chimeric (PROTAC) has emerged as a promising and potential epidrug. The PROTACs are heteobifunctional molecules with two ligands and a linker: a ligand for binding protein of interest, a ligand for recruiting E3 ligase and a liner connecting the two (68). It can promote the degradation of targeted proteins rather than inhibit the degradation of proteins (68). PROTACs have been used to target specific genetic defects of epigenome modifying enzymes or to develop synthetic lethality in clinic trial of cancer subsets (69). Ongoing studies aimed to selectively use PROTACs to degrade HDAC6 (70). We believe that PROTACs will emerge as a target for AF Implications.
Conclusions and Future Directions in AF
Epigenetics is a promising and potential tool to reveal unknown mechanisms of AF and for patient-specific pharmacotherapy. Here, we have provided proof-of-concept of epigenetic regulation of histone modifications and non-coding RNA in AF. However, evidences are lacking about the epigenetic regulation of risk factors, such as aging, diabetes mellitus, or ROS, on AF. For example, aging is an important risk factor of AF and aging is associated with global DNA hypomethylation and promoter hypermethylation. But the relationship between aging and DNA methylation in AF remains to be explored.
How to treat AF through correcting associated genetic variants or to modify the epigenome is a challenge and future direction. Recently the emergence of patient-specific human induced pluripotent stem cell-derived CMs (iPSC-CMs) has been as a pharmacologic testing for AF–both for toxicology and efficacy on a patient-specific background (71). CRISPR/Cas9 (epi)genome editing is used in iPSC and animal models to modulate gene function and induce epigenetic changes (72). However, the off-target effect remains these methods to be controversial ethically.
Integrative analysis using computational model and machine learning to better associate epigenetic changes with AF will bring new insights (73). Performing these analyses (PROTAC, iPSC-CMs, and the integrative analysis using computational modeling and machine learning) in a patient-specific mode will further improve strategies for AF prediction, diagnosis, therapeutic intervention, and prognosis (Figure 2).
Figure 2
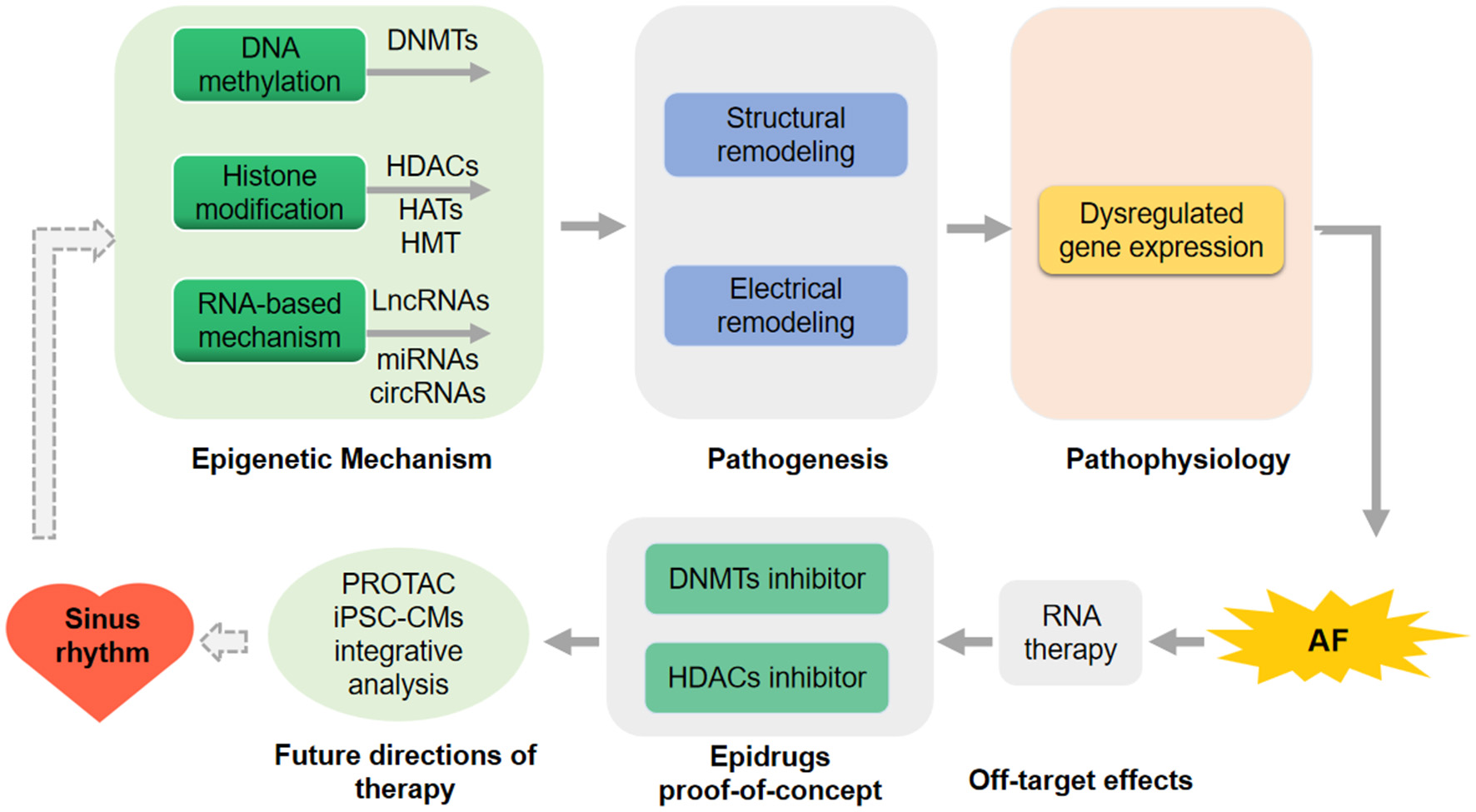
Future direction for atrial fibrillation (AF) prediction and therapeutic intervention. RNA-therapy has not applied in the therapy of human being because of the off-target effects in the human clinical trial. DNA methyltransferases (DNMTs) inhibitors and histone deacetylases (HDACs) inhibitors as the epidrugs have been proof-of-concept in preclinical models of AF. proteolysis targeting chimeric (PROTAC), human induced pluripotent stem cell-derived CMs (iPSC-CMs) and the integrative analysis using computational modeling or machine learning in a patient-specific mode will be performed to further improve strategies for AF intervention.
Funding
This work is supported by National Natural Science Foundation of China (Nos. 82070354 and 81470519) and Program for Huazhong University of Science and Technology Academic Frontier Youth Team (No. 2019QYTD08).
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Author contributions
LN design the study. DL take the responsibility of literature search and manuscript writing. All authors critically revised the manuscript for important intellectual content and gave final approval for the version to be published.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
Richter S Biase LD Hindricks G . Atrial fibrillation ablation in heart failure. Eur Heart J. (2019) 40:663–71. 10.1093/eurheartj/ehy778
2.
Wijesurendra RS Casadei B . Mechanisms of atrial fibrillation. Heart. (2019) 105:1860–67. 10.1136/heartjnl-2018-314267
3.
Andrade J Khairy P Dobrev D Nattel S . The clinical profile and pathophysiology of atrial fibrillation: relationships among clinical features, epidemiology, and mechanisms. Circ Res. (2014) 114:1453–68. 10.1161/CIRCRESAHA.114.303211
4.
Iwasaki YK Nishida K Kato T Nattel S . Atrial fibrillation pathophysiology: implications for management. Circulation. (2011) 124:2264–74. 10.1161/CIRCULATIONAHA.111.019893
5.
Lozano-Velasco E Franco D Aranega A Daimi H . Genetics and epigenetics of atrial fibrillation. Int J Mol Sci. (2020) 21:5717. 10.3390/ijms21165717
6.
Xie W Santulli G Reiken SR Yuan Q Osborne BW Chen BX et al . Mitochondrial oxidative stress promotes atrial fibrillation. Sci Rep. (2015) 5:11427. 10.1038/srep11427
7.
Fan J Li H Xie R Zhang X Nie X Shi X et al . LncRNA ZNF593-AS alleviates contractile dysfunction in dilated cardiomyopathy. Circ Res. (2021) 128:1708–23. 10.1161/CIRCRESAHA.120.318437
8.
Doñate Puertas R Arora R Rome S Asatryan B Roderick HL Chevalier P . Epigenetics in atrial fibrillation: a reappraisal. Heart Rhythm. (2021) 18:824–32. 10.1016/j.hrthm.2021.01.007
9.
Mahmood N Rabbani SA . DNA methylation readers and cancer: mechanistic and therapeutic applications. Front Oncol. (2019) 9:489. 10.3389/fonc.2019.00489
10.
Whayne TF . Epigenetics in the development, modification, and prevention of cardiovascular disease. Mol Biol Rep. (2015) 42:765–76. 10.1007/s11033-014-3727-z
11.
Lin H Yin X Xie Z Lunetta KL Lubitz SA Larson MG et al . Methylome-wide association study of atrial fibrillation in framingham heart study. Sci Rep. (2017) 7:40377. 10.1038/srep40377
12.
Zhao G Zhou J Gao J Liu Y Gu S Zhang X et al . Genome-wide DNA methylation analysis in permanent atrial fibrillation. Mol Med Rep. (2017) 16:5505–14. 10.3892/mmr.2017.7221
13.
Shen K Tu T Yuan Z Yi J Zhou Y Liao X et al . DNA methylation dysregulations in valvular atrial fibrillation. Clin Cardiol. (2017) 40:686–91. 10.1002/clc.22715
14.
Tao H Yang JJ Chen ZW Xu SS Zhou X Zhan HY et al . DNMT3A silencing RASSF1A promotes cardiac fibrosis through upregulation of ERK1/2. Toxicology. (2014) 323:42–50. 10.1016/j.tox.2014.06.006
15.
Puertas RD Meugnier E Romestaing C Rey C Morel E Lachuer J et al . Atrial fibrillation is associated with hypermethylation in human left atrium, and treatment with decitabine reduces atrial tachyarrhythmias in spontaneously hypertensive rats. Transl Res. (2017) 184:57–67.e5. 10.1016/j.trsl.2017.03.004
16.
Fatima N Schooley JF Jr Claycomb WC Flagg TP . Promoter DNA methylation regulates murine SUR1 (Abcc8) and SUR2 (Abcc9) expression in HL-1 cardiomyocytes. PLoS ONE. (2012) 7:e41533. 10.1371/journal.pone.0041533
17.
Kao YH Chen YC Chung CC Lien GS Chen SA Kuo CC et al . Heart failure and angiotensin II modulate atrial Pitx2c promotor methylation. Clin Exp Pharmacol Physiol. (2013) 40:379–84. 10.1111/1440-1681.12089
18.
Kouzarides T . Chromatin modifications and their function. Cell. (2007) 128:693–705. 10.1016/j.cell.2007.02.005
19.
Gillette TG Hill JA . Readers, writers, and erasers: chromatin as the whiteboard of heart disease. Circ Res. (2015) 116:1245-53. 10.1161/CIRCRESAHA.116.303630
20.
Nielsen JB Thorolfsdottir RB Fritsche LG Zhou W Skov MW Graham SE et al . Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat Genet. (2018) 50:1234–39. 10.1038/s41588-018-0171-3
21.
Song S Zhang R Mo B Chen L Liu L Yu Y et al . EZH2 as a novel therapeutic target for atrial fibrosis and atrial fibrillation. J Mol Cell Cardiol. (2019) 135:119–33. 10.1016/j.yjmcc.2019.08.003
22.
Zhang D Hu X Li J Hoogstra-Berends F Zhuang Q Esteban MA et al . Converse role of class I and class IIa HDACs in the progression of atrial fibrillation. J Mol Cell Cardiol. (2018) 125:39–49. 10.1016/j.yjmcc.2018.09.010
23.
Zhang D Wu CT Qi X Meijering RA Hoogstra-Berends F Tadevosyan A et al . Activation of histone deacetylase-6 induces contractile dysfunction through derailment of alpha-tubulin proteostasis in experimental and human atrial fibrillation. Circulation. (2014) 129:346–58. 10.1161/CIRCULATIONAHA.113.005300
24.
Liu F Levin MD Petrenko NB Lu MM Wang T Yuan LJ et al . Histone-deacetylase inhibition reverses atrial arrhythmia inducibility and fifibrosis in cardiac hypertrophy independent of angiotensin. J Mol Cell Cardiol. (2008) 45:715–23. 10.1016/j.yjmcc.2008.08.015
25.
Lkhagva B Chang SL Chen YC Kao YH Lin YK Chiu CT et al . Histone deacetylase inhibition reduces pulmonary vein arrhythmogenesis through calcium regulation. Int J Cardiol. (2014) 177:982–9. 10.1016/j.ijcard.2014.09.175
26.
Seki M LaCanna R Powers JC Vrakas C Liu F Berretta R et al . Class I Histone deacetylase inhibition for the treatment of sustained atrial fibrillation. J Pharmacol Exp Ther. (2016) 358:441–9. 10.1124/jpet.116.234591
27.
Brundel B Li J Zhang D . Role of HDACs in cardiac electropathology: therapeutic implications for atrial fibrillation. Biochim Biophys Acta Mol Cell Res. (2020) 1867:118459. 10.1016/j.bbamcr.2019.03.006
28.
Zhang D Hu X Henning RH Brundel BJ . Keeping up the balance: role of HDACs in cardiac proteostasis and therapeutic implications for atrial fibrillation. Cardiovasc Res. (2016) 109:519–26. 10.1093/cvr/cvv265
29.
van Marion DM Lanters EA Wiersma M Allessie MA Brundel BB de Groot NM . Diagnosis and therapy of atrial fibrillation: the past, the present and the future. J Atr Fibrillation. (2015) 8:1216. 10.4022/jafib.1216
30.
Ashjian E Redic K . Multiple myeloma: updates for pharmacists in the treatment of relapsed and refractory disease. J Oncol Pharm Pract. (2016) 22:289–302. 10.1177/1078155215572036
31.
Scholz B Schulte JS Hamer S Himmler K Pluteanu F Seidl MD et al . HDAC (Histone Deacetylase) Inhibitor valproic acid attenuates atrial remodeling and delays the onset of atrial fibrillation in mice. Circ Arrhythm Electrophysiol. (2019) 12:e007071. 10.1161/CIRCEP.118.007071
32.
Beermann J Piccoli MT Viereck J Thum T . Non-coding RNAs in development and disease: background, mechanisms, and therapeutic approaches. Physiol Rev. (2016) 96:1297–325. 10.1152/physrev.00041.2015
33.
Zhou D Yang K Hu J Shi J Hu X . Noncoding RNAs in atrial fibrillation: current status and prospect. J Cardiovasc Pharmacol. (2020) 75:10–7. 10.1097/FJC.0000000000000762
34.
Shan H Zhang Y Lu Y Zhang Y Pan Z Cai B et al . Downregulation of miR-133 and miR-590 contributes to nicotine-induced atrial remodelling in canines. Cardiovasc Res. (2009) 83:465–72. 10.1093/cvr/cvp130
35.
Adam O Löhfelm B Thum T Gupta SK Puhl SL Schäfers HJ et al . Role of miR-21 in the pathogenesis of atrial fibrosis. Basic Res Cardiol. (2012) 107:278. 10.1007/s00395-012-0278-0
36.
Duisters RF Tijsen AJ Schroen B Leenders JJ Lentink V van der Made I et al . miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res. (2009) 104:170–8. 10.1161/CIRCRESAHA.108.182535
37.
Dawson K Wakili R Ordög B Clauss S Chen Y Iwasaki Y et al . MicroRNA29: a mechanistic contributor and potential biomarker in atrial fibrillation. Circulation. (2013) 127:1466–75:1475e1–28. 10.1161/CIRCULATIONAHA.112.001207
38.
Chen Y Wakili R Xiao J Wu CT Luo X Clauss S et al . Detailed characterization of microRNA changes in a canine heart failure model: Relationship to arrhythmogenic structural remodeling. J Mol Cell Cardiol. (2014) 77:113–24. 10.1016/j.yjmcc.2014.10.001
39.
Girmatsion Z Biliczki P Bonauer A Wimmer-Greinecker G Scherer M Moritz A et al . Changes in microRNA-1 expression and IK1 up-regulation in human atrial fibrillation. Heart Rhythm. (2009) 6:1802–9. 10.1016/j.hrthm.2009.08.035
40.
Luo X Pan Z Shan H Xiao J Sun X Wang N et al . MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. J Clin Invest. (2013) 123:1939–51. 10.1172/JCI62185
41.
Jia X Zheng S Xie X Zhang Y Wang W Wang Z et al . MicroRNA-1 accelerates the shortening of atrial effective refractory period by regulating KCNE1 and KCNB2 expression: an atrial tachypacing rabbit model. PLoS One. (2013) 8:e85639. 10.1371/journal.pone.0085639
42.
Harada M Luo X Murohara T Yang B Dobrev D Nattel S . MicroRNA regulation and cardiac calcium signaling: role in cardiac disease and therapeutic potential. Circ Res. (2014) 114:689–705. 10.1161/CIRCRESAHA.114.301798
43.
Chiang DY Kongchan N Beavers DL Alsina KM Voigt N Neilson JR et al . Loss of microRNA-106b-25 cluster promotes atrial fibrillation by enhancing ryanodine receptor type-2 expression and calcium release. Circ Arrhythm Electrophysiol. (2014) 7:1214–22. 10.1161/CIRCEP.114.001973
44.
Lu Y Zhang Y Wang N Pan Z Gao X Zhang F et al . MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation. (2010) 122:2378–87. 10.1161/CIRCULATIONAHA.110.958967
45.
Wang J Bai Y Li N Ye W Zhang M Greene SB et al . Pitx2-microRNA pathway that delimits sinoatrial node development and inhibits predisposition to atrial fibrillation. Proc Natl Acad Sci USA. (2014) 111:9181–6. 10.1073/pnas.1405411111
46.
Babapoor-Farrokhran S Gill D Rasekhi RT . The role of long noncoding RNAs in atrial fibrillation. Heart Rhythm. (2020) 17:1043–9. 10.1016/j.hrthm.2020.01.015
47.
Du J Li Z Wang X Li J Liu D Wang X et al . Long noncoding RNA TCONS-00106987 promotes atrial electrical remodelling during atrial fibrillation by sponging miR-26 to regulate KCNJ2. J Cell Mol Med. (2020) 24:12777–88. 10.1111/jcmm.15869
48.
Li Z Wang X Wang W Du J Wei J Zhang Y et al . Altered long non-coding RNA expression profile in rabbit atria with atrial fibrillation: TCONS_00075467 modulates atrial electrical remodeling by sponging miR-328 to regulate CACNA1C. J Mol Cell Cardiol. (2017) 108:73–85. 10.1016/j.yjmcc.2017.05.009
49.
Dai H Zhao N Liu H Zheng Y Zhao L . LncRNA nuclear-enriched abundant transcript 1 regulates atrial fibrosis via the miR-320/NPAS2 axis in atrial fibrillation. Front Pharmacol. (2021) 12:647124. 10.3389/fphar.2021.647124
50.
Zhao L Ma Z Guo Z Zheng M Li K Yang X . Analysis of long non-coding RNA and mRNA profiles in epicardial adipose tissue of patients with atrial fibrillation. Biomed Pharmacother. (2020) 121:109634. 10.1016/j.biopha.2019.109634
51.
Shi X Shao X Liu B Lv M Pandey P Guo C et al . Genome-wide screening of functional long noncoding RNAs in the epicardial adipose tissues of atrial fibrillation. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165757. 10.1016/j.bbadis.2020.165757
52.
Zhu X Tang X Chong H Cao H Fan F Pan J et al . Expression profiles of circular rna in human atrial fibrillation with valvular heart diseases. Front Cardiovasc Med. (2020) 7:597932. 10.3389/fcvm.2020.597932
53.
Shangguan W Liang X Shi W Liu T Wang M Li G . Identification and characterization of circular RNAs in rapid atrial pacing dog atrial tissue. Biochem Biophys Res Commun. (2018) 506:1–6. 10.1016/j.bbrc.2018.05.082
54.
Jiang S Guo C Zhang W Che W Zhang J Zhuang S et al . The integrative regulatory network of circRNA, microRNA, and mRNA in atrial fibrillation. Front Genet. (2019) 10:526. 10.3389/fgene.2019.00526
55.
Zhang Y Shen H Wang P Min J Yu Y Wang Q et al . Identification and characterization of circular RNAs in atrial appendage of patients with atrial fibrillation. Exp Cell Res. (2020) 389: 111821. 10.1016/j.yexcr.2020.111821
56.
Liu T Zhang G Wang Y Rao M Zhang Y Guo A et al . Identificaiton of circular RNA-MicroRNA-Messenger RNA regulatory network in atrial fibrillation by integrated analysis. Biomed Res Int. (2020) 2020:8037273. 10.1155/2020/8037273
57.
Ruan ZB Wang F Yu QP Chen GC Zhu L . Integrative analysis of the circRNA-miRNA regulatory network in atrial fibrillation. Sci Rep. (2020) 10:20451. 10.1038/s41598-020-77485-1
58.
Wang Y Liu B . Circular RNA in diseased heart. Cells. (2020) 9:1240. 10.3390/cells9051240
59.
Costa MC Cortez-Dias N Gabriel A de Sousa J Fiúza M Gallego J et al . circRNA-miRNA cross-talk in the transition from paroxysmal to permanent atrial fibrillation. Int J Cardiol. (2019) 290:134–7. 10.1016/j.ijcard.2019.04.072
60.
Hu X Chen L Wu S Xu K Jiang W Qin M et al . Integrative analysis reveals key circular rna in atrial fibrillation. Front Genet. (2019) 10:108. 10.3389/fgene.2019.00108
61.
Zhang PP Sun J Li W . Genome-wide profiling reveals atrial fibrillation-related circular RNAs in atrial appendages. Gene. (2020) 728:144286. 10.1016/j.gene.2019.144286
62.
Laina A Gatsiou A Georgiopoulos G Stamatelopoulos K Stellos K . RNA therapeutics in cardiovascular precision medicine. Front Physiol. (2018) 9:953. 10.3389/fphys.2018.00953
63.
Kee HJ Sohn IlS Nam KIl Park JE Qian YR Yin Z et al . Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation. (2006) 113:51–9. 10.1161/CIRCULATIONAHA.105.559724
64.
Felisbino MB McKinsey TA . Epigenetics in cardiac fibrosis: emphasis on inflammation and fibroblast activation. JACC Basic Transl Sci. (2018) 3:704–15. 10.1016/j.jacbts.2018.05.003
65.
Pechalrieu D Etievant C Arimondo PB . DNA methyltransferase inhibitors in cancer: From pharmacology to translational studies. Biochem Pharmacol. (2017)129:1–13. 10.1016/j.bcp.2016.12.004
66.
Sun Y Sun Y Yue S Wang Y Lu F . Histone deacetylase inhibitors in cancer therapy. Curr Top Med Chem. (2018) 18:2420–8. 10.2174/1568026619666181210152115
67.
Alqahtani A Choucair K Ashraf M Hammouda DM Alloghbi A Khan T et al . Bromodomain and extra-terminal motif inhibitors: a review of preclinical and clinical advances in cancer therapy. Future Sci OA. (2019) 5:FSO372. 10.4155/fsoa-2018-0115
68.
Sun X Gao H Yang Y He M Wu Y Song Y et al . PROTACs: great opportunities for academia and industry. Signal Transduct Target Ther. (2019) 4:64. 10.1038/s41392-019-0101-6
69.
Jones PA Issa JP Baylin S . Targeting the cancer epigenome for therapy. Nat Rev Genet. (2016) 17:630–41. 10.1038/nrg.2016.93
70.
An Z Lv W Su S Wu W Rao Y . Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein Cell. (2019) 10:606–9. 10.1007/s13238-018-0602-z
71.
Benzoni P Campostrini G Landi S Bertini V Marchina E . Iascone M,et al. Human iPSC modeling of a familial form of atrial fibrillation reveals a gain of function of If and ICaL in patient-derived cardiomyocytes. Cardiovasc Res. (2020) 116:1147–60. 10.1093/cvr/cvz217
72.
van Kampen SJ van Rooij E . CRISPR craze to transform cardiac biology. Trends Mol Med. (2019) 25:791–802. 10.1016/j.molmed.2019.06.008
73.
Zhao J Aslanidi O Kuklik P Lee G Tse G Niederer S et al . Editorial: recent advances in understanding the basic mechanisms of atrial fibrillation using novel computational approaches. Front Physiol. (2019) 10:1065. 10.3389/fphys.2019.01065
Summary
Keywords
atrial fibrillation, epigenetic regulation, DNA methylation, histone modification, non-coding RNAs
Citation
Li D, Nie J, Han Y and Ni L (2022) Epigenetic Mechanism and Therapeutic Implications of Atrial Fibrillation. Front. Cardiovasc. Med. 8:763824. doi: 10.3389/fcvm.2021.763824
Received
24 August 2021
Accepted
08 December 2021
Published
21 January 2022
Volume
8 - 2021
Edited by
Yong Xia, The Ohio State University, United States
Reviewed by
Junco S. Warren, Virginia Tech Carilion, United States; Amelia Eva Aranega, University of Jaén, Spain
Updates
Copyright
© 2022 Li, Nie, Han and Ni.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Ni nili@tjh.tjmu.edu.cn
This article was submitted to Cardiac Rhythmology, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.