 Natalie Stahr
Natalie Stahr Elena V. Galkina
Elena V. Galkina- Department of Microbiology and Molecular Cell Biology, Eastern Virginia Medical School, Norfolk, VA, United States
Alzheimer's disease (AD) and cardiovascular disease (CVD) are pathologies that are characterized by common signatures of vascular dysfunction and chronic inflammation that are accelerated with aging. Importantly, epidemiological studies report an independent interaction between AD and CVD and data suggest that chronic inflammation in CVD may accelerate AD development. Atherosclerosis affects most large to medium sized arteries including those supplying the cerebral circulation. Vascular dysfunction caused by atherosclerosis results in blood brain barrier breakdown, inflammation, an impaired clearance of amyloid-beta (Aβ), and finally ends with neurovascular dysfunction. Numerous data indicate that innate and adaptive immune responses shape atherogenesis and increasing evidence suggests an implication of the immune response in AD progression. Currently, mechanisms by which these two diseases are interconnected with each other are not well-defined. In this review, we discuss the recent advances in our understanding of the intertwined role of the immune response in atherosclerosis and AD and the implications of these findings for human health.
Introduction
Atherosclerosis is a multi-factorial disease of the large to medium arteries, characterized by the buildup of plaque and aggregation of immune cells in response to subsequent endothelial damage (1–4). Alzheimer's Disease (AD) is a neurodegenerative disease characterized by deposition of amyloid β (Aβ) and hyperphosphorylated tau in the brain, leading to significant cognitive decline (5–7). Although these diseases may seem entirely disparate at first glance, recent research has indicated that both share commonalities in their activation and progression (8, 9). Atherosclerosis and AD are both aging-related diseases that can be described as having common signatures of chronic, low-grade inflammation. Recent epidemiological data, explored in more detail herein, supports the notion that these diseases are intertwined. Importantly, these diseases are both heavily modulated by the immune system, with new insights into AD pointing toward a role for peripheral immune dysfunction in its etiology (4, 6, 10). Neuroinflammation in isolation has been explored extensively, but researchers are only beginning to uncover how the peripheral immune system, and its dysfunction during atherosclerosis, may affect the development of AD. Atherosclerosis and AD share many features in immune cell activation and cytokine release that could change how we view AD as a disease. The effect of hyperlipidemia on immune cell subset differentiation and activation in atherosclerosis may additionally alter or exacerbate the pathology of AD (11). Although a multitude of features connect AD and atherosclerosis, including oxidative distress, mitochondrial dysfunction, hypertension, endothelial dysfunction, vascular aging, the gut-brain axis and hyperlipidemia, this review will particularly focus on the interconnections between peripheral immune cell subsets and their dysfunction in atherosclerosis and AD. This review will therefore follow commonalities in the roles of different immune cell subsets in these two diseases, identifying possible links that need further exploration.
Atherosclerosis and Associated Immune Response
Atherosclerosis is a complex, inflammatory disease associated with lipid accumulation, vascular dysfunction, and inflammatory immune response in the large and medium sized arteries (1–4). Importantly, atherosclerosis continues to be the leading cause of CVD (1–4). Atherosclerosis affects most large to medium sized arteries, including those supplying the cerebral circulation (12). Vascular dysfunction caused by atherosclerosis results in small vessel disease, a breakdown of the blood brain barrier, inflammation, an impaired clearance of Aβ, and finally in neurovascular dysfunctions (13). Many elements that increase the risk of CVD including aging, hypertension, hyperlipidemia, inflammation, and vascular dysfunction also elevate the chances of getting AD (14, 15). Chronic inflammation in the aorta supports atherogenesis, and both innate and adaptive immune responses are involved in this process. The pathological role of neutrophils, Th1, T follicular helper (Tfh), NK and NKT cells, and the protective role of Tregs is established in atherosclerosis, whereas the role of Th2 and Th17 cells remains under debate (1, 3, 4). The role of B cells in atherosclerosis is complex and B cell-subset-specific (16). Follicular (FO) B cells and innate response activator (IRA) B cells are pro-atherogenic, supporting the Th1 response and the production of pro-inflammatory cytokines and immunoglobulins (Igs) (16). Conversely, protective B1a and B1b cells secrete natural antibodies (Abs) that attenuate oxLDL uptake by macrophages (MΦs). A recent study highlighted an anti-atherogenic role of marginal zone B cells (MZ) via the regulation of Tfh cell development (16). The role of regulatory B cells remains controversial. Overall, the immune system is strongly involved in atherogenesis and generates a unique, complex immune response that might affect development of other atherosclerosis-associated pathologies. In this review, we provide evidence for an implication of atherosclerosis in the promotion of AD and discuss potential mechanisms by which a unique immune response developed during atherogenesis might affect progression of AD.
Alzheimer's Disease and Associated Immune Response
Alzheimer's Disease (AD) is a progressive neurodegenerative disorder characterized by the buildup of amyloid β (Aβ) in the brain, followed by neuronal death, and culminates in significant cognitive decline (17, 18). Although numerous etiological theories exist for AD, the predominant hypothesis, the Amyloid Hypothesis, posits that the accumulation of Aβ itself acts as the initial trigger for neurodegeneration (6, 19). Normally, Aβ is cleared by multiple mechanisms including flow–dependent clearance of the interstitial fluid into the cerebral spinal fluid, Aβ uptake by microglia and astrocytes, transport of Aβ by low-density lipoprotein receptor-related protein 1, very low-density lipoprotein receptor and P-glycoprotein, and via clearance by proteases such as neprilysin, insulin-degrading enzymes, matrix metalloprotease-9 (MMP9), and glutamate carboxypeptidase II (20). There then emerges different mechanisms that can lead to the accumulation of Aβ in the brain: excessive production of Aβ and decreased clearance of Aβ from the brain.
Early onset AD, also known as familial AD, is caused by one or more mutations in the genes of APP or presenilin1/2, subunits of the γ-secretase enzyme, leading to AD symptoms as early as 30–50 years old with homozygous dominant inheritance (7). These mutations cause an increase in the amyloidogenic pathway, resulting in higher production of Aβ. The more prevalent form of AD, late onset AD, also known as sporadic AD, appears to be caused by the intermingling of genetic and environmental risk factors, which may play a role in either Aβ overproduction and reduction in clearance. Multiple genetic factors feeding into AD have been identified, including isoforms of the Apolipoprotein E (APOE) gene and heterozygous mutations in triggering receptor expressed on myeloid cells 2 (TREM2), among many others (7). Of note, mutations in APOE and TREM-2 are also strongly associated with atherogenesis via dysregulation of macrophage autophagy making these targets promising candidates for therapeutic manipulation for both pathologies. Although genetics do appear to play a major role in late onset AD, with some estimating a 70:30 split between genetics and environment in the etiology of the disease (21), multiple vascular risk factors are associated with increased risk for dementia such as smoking, diabetes, hypertension, and atherosclerosis (22).
While the role of Aβ in AD is well-established, increasing evidence also suggests an impact of the immune response and associated inflammation in the induction and persistence of AD pathologies (6). It was known for a long time that AD is strongly associated with increased levels of CRP and pro-inflammatory cytokines such as IL-1β, IL-6, and TNFα and some but not all studies found a correlation between levels of peripheral inflammation, aging (23) and AD in humans (24–28). Additionally, several studies demonstrate that elevated levels of peripheral inflammation in midlife to late life correlates with higher risk of dementia and is associated with future cognitive decline and vascular risks (29, 30). Importantly, elevated TNFα levels and reduced TGFβ production have been detected in the cerebrospinal fluid (CSF) of patients with mild cognitive impairment highlighting that CNS-associated inflammation is an early hallmark in the pathogenesis of AD (31). Increased levels of TNFα are also associated with a 2-fold increase in the rate of cognitive decline over a 6-month period (32). Additional evidence on the impact of inflammation are coming from studies demonstrating systemic inflammation induced by either common acute or chronic infections or critical illness might result in AD-like neuropathology in mice (33) and cognitive decline in aged patients (6, 34, 35). These reports imply that systemic inflammation can precede AD-like pathology and likely participate in its induction/acceleration. To date, several lines of evidence including preclinical and genetic data as well as animal studies support the notion that activated immunity accompanies AD pathology at all stages of disease development (9, 36). The pathological role of infiltrating neutrophils and monocytes has been documented in animal models of AD, and a dominant response of Th1 and Th17 cells is a hallmark of the immune response in AD, while a role of Tregs and B cells is still controversial (6, 10). It remains to be determined how peripheral immunity might affect the activation of AD-specific degenerative processes and it is unclear at which stage(s) of AD progression the immune system and associated inflammation play a critical role.
Epidemiological Perspective of the Atherosclerosis and AD Interconnection
The main risk factor for AD and atherosclerosis is advanced age (5). Aging is accompanied by systemic changes to the innate and adaptive immune systems characterized by chronic inflammation and senescence for vascular and immune cell types. The underlying mechanisms leading to AD have yet to be fully elucidated, but epidemiological data point to connections between aging, AD and atherosclerosis mediated by hyperlipidemia, hypometabolism, hypertension, obesity, and atherosclerotic plaque burden (37). It has long been known that cholesterol and lipid metabolism play a critical role in the development of AD, as seen with the APOE4 isoform of APOE, which increases plasma low-density lipoprotein levels (thereby also increasing risk of atherosclerosis), while also increasing the risk for AD (11). Xiang et al. reported that higher carotid intermedial thickness, as a measure of carotid atherosclerosis, is associated with faster cognitive decline (38). Interestingly, post-mortem studies link intracranial atherosclerosis to increased risk for dementia independent of cerebral infarcts suggesting potentially additional pathways unrelated to AD through which vascular changes may influence dementia risk (39). Cortez-Canteli et al. found that carotid plaque burden was associated with increased risk of hypometabolism independent of CVD risk in middle-aged asymptomatic individuals (40). These important data reveal the role of unmitigated cardiovascular risk factors and subclinical atherosclerosis in the establishment of AD-like symptoms in a relatively young group of subjects. Further strong arguments for an implication of vascular risk factors into AD progression came from a study demonstrating that AD patients that were treated for their vascular risk factors had reduced cognitive decline compared to those that did not receive treatment, despite exclusion of patients with pre-existing cardiovascular disease (41). While epidemiological data strongly suggest an impact of atherosclerosis on AD, there is a limited number of in vivo studies, which recapitulate conditions of aging, atherosclerosis and AD. To fill out this gap, development of new models that will display characteristics of both pathologies is necessary as they might provide an important basis for our understanding mechanisms of atherosclerosis-associated AD development and lead to a successful creating an effective and complex pharmacotherapy for patients with AD and atherosclerosis.
Neutrophils Play a Pathological Role in AD and Atherosclerosis
Neutrophils, although crucial in acute immune assault, can prove to be highly damaging in the context of chronic inflammation presented in AD and atherosclerosis (Figure 1). In the case of AD, neutrophil accumulation and activation have negative consequences (42). Neutrophils from AD patients are highly activated and pro-apoptotic, produce intravascular neutrophil extracellular traps (NETs) and higher levels of ROS that correlate with amyloid burden (43). While precise mechanisms of neutrophil activation remain to be determined, it is likely that peripheral proinflammatory cytokines are instrumental in the activation of peripheral blood neutrophils. Neutrophil hyperactivation and impairments in homeostasis appeared to correlate with faster cognitive decline (43). In two transgenic models of AD (5xFAD and 3xTg-AD mice), neutrophils were found in Aβ-positive areas of the brain, where they released NETs and IL-17 (44). LFA-1-dependent infiltration of neutrophils were peaking at 4 months of age, which coincided with the onset of memory loss in cognitive tests of these animals (44). Neutrophil depletion in those mice improved performance in certain memory tests and reduced microgliosis highlighting a functional role of neutrophils in neuroinflammation. In further support of the negative effects of neutrophil aggregation, Cruz-Hernández et al. found that adhesion of neutrophils in the vasculature of the brain leads to reduced cerebral blood flow in APP/PS1 mice, which was subsequently resolved by treatment with anti-Ly6G antibody (Abs) (45). Another avenue through which neutrophils may further exacerbate AD is NETosis. Although NETosis can be activated through numerous mechanisms, of note, neutrophils interact with Aβ-activated platelets in the vasculature of the brain, an interaction that induces NETosis (46, 47). Increased NETosis in the small vessels of the brain promotes blood clotting through thrombin formation and further platelet aggregation, occluding the microvasculature and possibly furthering Aβ buildup through hypoxia-induced increases in β-secretase activity and reduced Aβ clearance (11, 48). NETosis can also damage the blood-brain barrier, further hampering the brain's clearance of Aβ (47). Proteases in these NETs such as MMP-9 also contribute to blood-brain barrier breakdown. In fact, a recent study by Ringland et al. found that inhibition and/or knockout of MMP-9 resulted in reduced anxiety and improved social recognition memory in an AD mouse model, despite no reduction in Aβ levels (49). Thus, burgeoning data demonstrate that neutrophils are a significant component of the immune response that is associated with neuroinflammation and have an instrumental role in AD pathophysiology.
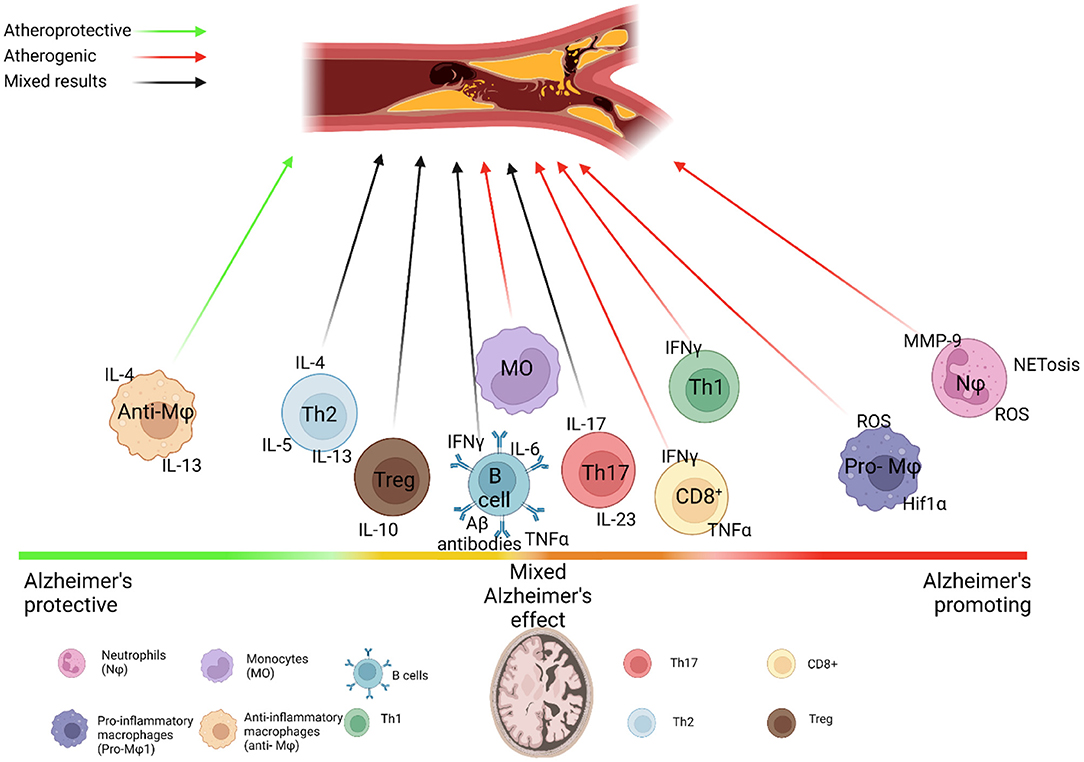
Figure 1. Immune system dysfunction links Alzheimer's and Atherosclerosis. Immune cell subsets vary in their interactions with Alzheimer's disease (AD) and Atherosclerosis. Neutrophils drive both AD and atherosclerosis through their aggregation and production of MMPs, ROS and NETs, which damage the blood brain barrier and heighten hypoxia-induced Aβ production, while the neutrophilia and increased activation of neutrophils seen in atherosclerosis may further feed into this damage. Monocytes phagocytize Aβ, but may also produce proinflammatory cytokines that drive harmful neuroinflammation, while the production of pro-inflammatory monocytes is increased in atherosclerosis, which accelerates atherogenesis. Both diseases drive differential production of classical and non-classical monocytes. TREM2+ macrophages may help to clear Aβ in the brain and mitigate inflammation in atherosclerotic lesions. Similarly, increased proportions of anti-inflammatory macrophage subsets appear to play a protective role in AD, while increased proportions of proinflammatory macrophages drive inflammasome activation and production of pro-inflammatory cytokines in atherosclerosis, causing plaque progression. Adaptive immune cells add further complexity to these diseases. The role of B cells in AD and atherosclerosis appears to be subset specific, with clearance of Aβ via antibodies tempered by the release of pro-inflammatory cytokines and differential effects by follicular B cells and innate response activator B cells on atherosclerosis. This is similar to the differential activity of T cells in these diseases. Controversy over the role of regulatory T cells in AD revolves around whether immune tolerance or immune activation is beneficial for AD. Th1 cells are prominent in progression of atherosclerosis and AD, producing high levels of IFNγ, which increases risk of plaque rupture in atherosclerosis and blood brain barrier breakdown in AD through increased activation of monocytes and macrophages and MMP release. Th17 cells are still largely understudied in AD, but their IL-17 production may contribute to AD possibly through increased migration of neutrophils and monocytes to the brain, although their role in atherosclerosis is still under investigation. Th2 cells do not appear to migrate to the brain, and studies differ on their role in AD, possibly due to the differential role of IL-4, IL-5, and IL-13 that they produce. This is mirrored in atherosclerosis, where IL-4 is atheropromoting, while IL-5 and IL-13 are atheroprotective, making for a complex relationship between Th2 cells and these diseases. CD8+ T cells are associated with AD progression through mechanisms that are still being explored, and may promote plaque instability in atherosclerosis.
The pathological role of neutrophils in atherosclerosis has been firmly established and data indicate that chronic inflammatory environment in atherosclerosis shapes number and phenotype of circulating neutrophils, which has been reviewed elsewhere (50). Pro-atherogenic environment impairs cholesterol efflux, induces inflammasome activation, alters the balance of myeloid and lymphoid hematopoietic stem and progenitor cells in the bone marrow, and results in enhanced myelopoiesis and elevated number of neutrophils and monocytes in the blood (51, 52). It is likely that the increased neutrophil production observed in atherosclerosis may serve to facilitate further neutrophil aggregation in the brain, and future studies using adoptive transfer recruitment experiments could answer this important question.
Not only number, but the activated phenotype of neutrophils in atherosclerosis might result in collateral damage from their effector functions within various tissues including the brain (Figure 1). Several studies demonstrated that increased ROS production by activated neutrophils is implicated in endothelial dysfunction and plaque instability (50). As neutrophils are found in the vasculature of the brain, it is likely that these activated cells may induce vascular cell apoptosis and dysfunction. Neutrophils are also known to contain a large pool of granules such as azurocidin, proteinase 3, α-defensin, myeloperoxidase, LL37 (CRAMP in mouse) and Cathepsin G, which serve as activators of endothelium and some as chemoattractants for monocytes. These active compounds are also known to increase the expression of adhesion molecules and chemokine receptors in endothelial cells and regulate endothelial cell permeability in mice. Taking into an account a pivotal role of neutrophils in regulation of monocyte recruitment, it would be imperative to test the implication of neutrophils in modulation of leukocyte, and particularly, monocyte recruitment into inflammatory brain tissues at different time points of AD development. Neutrophils are found within atherosclerotic lesions and are believed to be responsible for many proinflammatory processes leading to a formation of unstable plaques (2, 50). Once homed to plaques, neutrophils degranulate and release NETs, MPO, proteases such as MMP-9, which contribute to additional ROS production, thinning of the fibrous cap, and ultimately plaque instability and rupture (50). Circulating NET markers have also been identified as risk factors for CVD severity (2). Recently, Silvestre-Roig et al. demonstrated that NET-released histone H4 can bind to and lyse vascular smooth muscle cells (VSMCs), leading to their apoptosis and plaque destabilization, highlighting a novel role of neutrophils in atherosclerotic plaque burden and stability that may additionally be explored in the blood-brain barrier breakdown seen in AD (53). NETs can prime macrophages to produce IL-1β via an inflammasome–dependent mechanism (54) in atherosclerosis and activated neutrophils may pulse microglia to over-reactive activated phenotype as well. Based on the critical functions of neutrophils in AD and their increasing numbers and pathological functions in atherosclerosis, it is clear that neutrophils and particularly neutrophil-derived NETs might be one of the important key players in AD and associated atherosclerosis (Figure 2). To date, several questions require additional studies in order to better understand triggers and mechanisms by which neutrophils can be activated in AD and associated CVD. Neutrophil-derived NETs can interact with monocytes/macrophages, dendritic cells, and platelets, regulate their functions, and accelerate damage of surrounding tissues. What is not well yet known how NETs can regulate neuroinflammation and particularly the impact of NETs on health and functions of microglia as well as vascular cells including pericytes, VSMCs, and endothelial cells and additional studies are warrant in this direction. Taken together, current findings indicate that neutrophils fuel central processes supporting both atherogenesis and AD; thus, targeting pathways of pathological neutrophil activation such as NET formation, may be a promising therapeutic approach that would have a double effect.
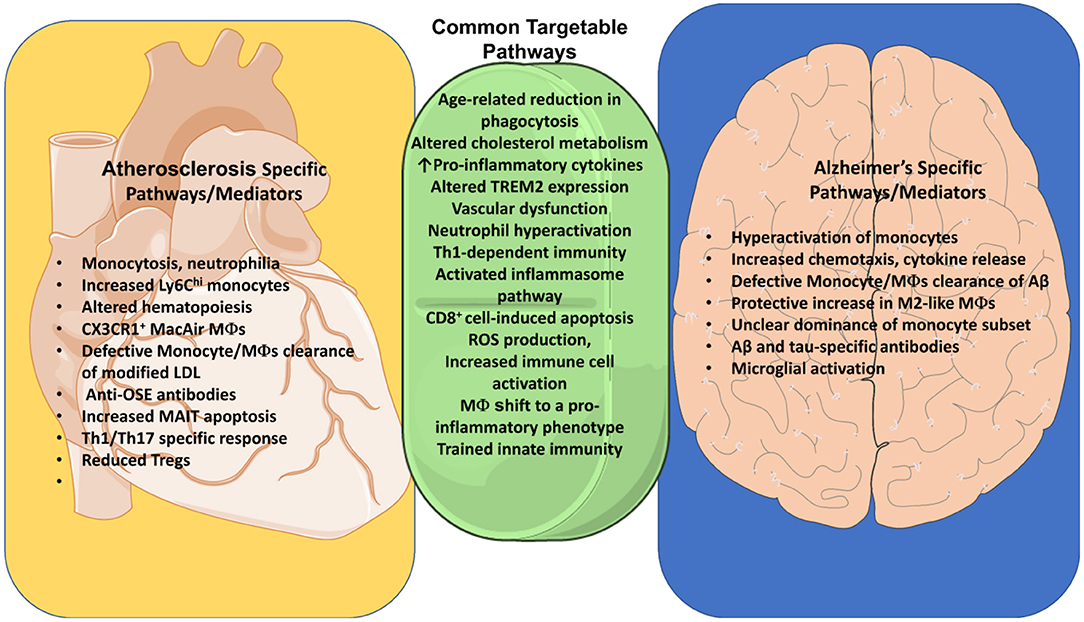
Figure 2. Common pathways of Immune dysregulation between Alzheimer's and Atherosclerosis. Identification of common pathways between AD and atherosclerosis identifies targets for treatment of both diseases. Both diseases are influenced by an age-related reduction in phagocytosis, as well as cholesterol metabolism, vascular dysfunction, activation of the inflammasome pathway and ROS production. Neutrophil hyperactivation and Th1-dependent immunity are both detrimental to these diseases. TREM2 assists myeloid cells in clearance of Aβ and cholesterol in their respective diseases. Both diseases also share an increase in IL-4, as well as apoptosis of endothelial cells by CD8+ T cells. We additionally explore differences between these diseases such as the production of OSE-specific antibodies, increased monocytosis, increased Ly6Chi monocytes, increased apoptosis of MAIT cells, and differentiation of MacAIR macrophages in atherosclerosis. Alzheimer's, in contrast, features a clearance of Aβ by monocytes and macrophages, activation of microglia, as well as Aβ- and tau-specific Abs. Some of these differences may with time be challenged as we uncover more commonalities and ties between these diseases.
Monocytes and Macrophages in AD and Atherosclerosis
The role of monocytes, macrophages and microglia in AD development is a topic of extensive studies (6, 10) and epidemiological and experimental data reveal differential roles of microglia, monocytes and cerebral macrophages in Aβ pathophysiology (55). The significance of microglia and macrophages in systemic inflammation and neurodegenerative diseases have been discussed comprehensively by others (55). In this review, we focus on main points of monocyte/macrophage phenotype in AD that has some commonality with described functions of monocytes/macrophages in atherosclerosis (Figure 2).
There are distinct quantitative and qualitative changes in monocytes as AD progresses with some controversial results in the field (6, 10). Evidence suggests that AD patients demonstrated a decrease in the non-classical CD14+CD16++ monocytes in comparison with patients with mild cognitive impairment or healthy controls (56). In contrast, Munawara et al. reported an increases in the percentage of non-classical and intermediate monocytes and the decreased percentage of classical monocytes in the blood of AD patients vs. healthy controls (57). Importantly, several studies also reported an overall increase in circulating monocytes in AD that exhibit hyperactivated phenotype characterized by increased chemotaxis, ROS production, and cytokine release in response to TLR2 and TLR4 stimulation (57). Importantly, these features of activated phenotype are a common denominator for all reported studies on monocyte phenotype in AD pathology.
Monocytes and monocyte-derived macrophages appear to play a complex role in AD, as these cells help to phagocytize and thereby clear the Aβ in AD patients while simultaneously arousing increased inflammation through release of potentially damaging proinflammatory cytokines such as IL-1α/β, IL-6, TNFα, IL-8 and TGFβ (5). Thus, emigrated monocytes perform dual role within the brain exacerbating or alleviating disease progression. The importance of recruited monocytes in the regulation of neuroinflammation has been elegantly demonstrated in experiments with CCR2-deficient mice (58). Naert et al. showed that an impairment of Ccr2−/− monocyte trafficking to the brain results in increased levels of intracellular Aβ as well as worsening of cognitive impairment in APP/PS1 mice (58), suggesting the overall protective role of CCR2+ monocytes in AD. Additionally, Naert et al. found deficient production of classical CX3CR1lowLy6-ChighGr1+CCR2+ monocytes in the bone marrow of 6-month-old APP/PS1 mice, whose rescue through treatment with M-CSF prevented cognitive decline (59).
As BM-derived macrophages are known to be more efficient than resident microglia in clearing Aβ deposits in AD models (60), it appears that this is a key decisive function of monocyte in the regulation of neuroinflammation. Here, it is important to note that monocyte phagocytic capability becomes compromised with age. Age correlates with reduced phagocytosis of Aβ in both classical CD14+CD16− and non-classical CD14dimCD16+subsets (61). This reduction was concomitant to diminished chemotaxis in response to CCL2 in patients with AD and reduced phagocytic capacity of monocytes in patients with mild cognitive impairment compared to healthy controls (57). Subsequent differentiation into monocyte-derived macrophages using the patients' sera resulted in increased proportions of M2-like vs. M1-like macrophages in AD patients (57). These M2 macrophages may be playing a protective role, as transplantation of M2 macrophages into Aβ-treated rats improved performance in cognitive testing, and in APP/PS1 mice reduced Aβ plaque size (62, 63). Collectively, these data point out the importance of phagocytosis and proper clearance of Aβ and damaged cells at different stages of AD development as a combination of defective phagocytosis and inflammatory insults deadly affects microglia health and functions. Indeed, several studies now demonstrate that the inhibition of key inflammatory mediators improves AD pathology and emphasize a critical role of microglia in this process (10, 64). In these regards, studies investigating a role of microglial surface receptor TREM2 helped to dissect an impact of functional microglia in AD. These reports showed that overexpression of TREM2 improves the cognitive decline in mouse models of AD (65) and TREM2 deletion aggravates pathology via regulation of macrophage and neuronal phenotypes and the Aβ uptake (66). While significant efforts have been directed to examination of microglia and blood-derived macrophage functions in AD, few studies have been devoted to a role of the specific population of macrophages surrounding brain vessels - perivascular macrophages in AD. Depletion of perivascular macrophages significantly increased the number of Aβ+ cortical blood vessels. Conversely, stimulation of perivascular macrophage turnover reduced cerebral amyloid load (67). In contrast, Park et al. reported that a selective depletion of perivascular macrophages abrogates cerebrovascular dysfunction and CD36- and Nox2 expressing by macrophages are responsible for pathological effects of perivascular macrophages (68).
The role of monocytes and macrophages in atherosclerosis have been described in many excellent reviews (4, 69, 70). In this review, we highlight features of monocyte/macrophage responses in atherosclerosis that might have a potential implication in modulation of AD pathophysiology (Figure 1). One of the important factors that is critical for the AD acceleration in atherosclerosis is the impact of pro-atherogenic conditions including hyperlipidemia on circulating monocyte numbers. Hematopoietic stem and progenitor cells (HSPCs) demonstrate reduced regenerative capacities and a shift toward myeloid cell differentiation in atherosclerosis (71) resulting in monocytosis with a dominant increase in pro-inflammatory Ly6Chi monocytes (72–74) that is linked to accelerated atherosclerotic lesion progression in mice as well as increased risk of CVD in humans (72–75).
Macrophages are professional phagocytic cells that uptake lipoproteins and form lipid-enriched foam cells (4). While it was initially thought that lipid uptake results in pro-inflammatory phenotype of macrophages, recent data point out that foam cell formation does not result in a pro-inflammatory phenotype of macrophages (76, 77), but pro-inflammatory conditions reverse the phenotype of these macrophages. The impact of cholesterol on monocyte/macrophage biology in AD and associated atherosclerosis is not well-understood (78). Several studies revealed harmful effect of dyslipidemia on AD. A number of genes involved in cholesterol metabolism or transport are AD susceptibility genes, including apolipoprotein E (APOE), apolipoprotein J (APOJ, CLU), ATP-binding cassette subfamily A member 7(ABCA7), and sortilin-related receptor (SORL1) has been identified as a pivotal components of cholesterol metabolism in the brain (78). Epidemiological studies detected an association between dyslipidemia and AD and clinical trials demonstrated some controversial effects of statin therapy AD (79, 80). As cholesterol loading alters membrane integrity, one of the interesting questions that has to be addressed in the future experiments is how increased cholesterol levels affect ability of monocyte-derived macrophages and microglia response to inflammatory insults and effectively phagocyte accumulating Aβ.
Macrophage polarization is also greatly affected by atherosclerosis and a broad spectrum of macrophages with either proinflammatory or anti-inflammatory features is found within the aortic wall. To date, there are at least five subsets of macrophages detected with the aortic wall (81). Classical inflammatory macrophages display an activated inflammasome pathway and express a myriad of pro-inflammatory cytokines and chemokines. Atherosclerosis-induced subset of macrophages that express various interferon-inducible genes, including Ifit3, Irf7 and Isg15 is also identified in the lesions (81). A subset of pro-inflammatory macrophages also originate from CX3CR1+ precursor cells in the aorta. Characterized by a specific location with the intima, aortic intima-resident macrophage (MacAIR) subset have a pro-atherogenic role at the early lesion development stage. The most interesting population in connection with AD is lipid-loaded macrophages expressing TREM2 that have been found within plaques at different stages of atherogenesis (60). TREM2+ macrophages have differentiated either from circulating monocytes or from embryonic precursors, express cholesterol metabolism-related such as Cd9, Ctsd, Fabp4 and Abcg1, which have roles in lipid metabolism, cholesterol efflux and oxidative phosphorylation. TREM2+ macrophages represent a major population of lipid-filled macrophage-derived foam cells, unexpectedly express low levels of inflammatory genes, and are postulated to have a role in mitigating vascular inflammation (76, 77).
As they are brain-resident cells, microglia in particular are not necessarily of interest in atherosclerosis. However, cells in atherosclerosis do have parallels to disease-associated microglia's (DAMs), and atherosclerosis in general can affect the behavior of microglia (55, 60). The lipid droplets in DAMs are reminiscent of lipid accumulation in foam cells during atherosclerosis and transcriptomic analysis has found enrichment of foam-cell enriched genes in DAMs (82). Aβ-laden DAMs downregulate expression of P2ry12, Cx3xr1, and Tmem119 while upregulating genes such as Apoe and Trem2 (83). These DAMs initially play a protective role, phagocytizing Aβ through upregulated TREM2 without significant inflammation, but in later stages of AD these DAMs become more inflammatory (83, 84). Yeh et al. also found that TREM2 serves as scavenger-like receptor that binds to lipoproteins associated with Aβ, which allows the microglia to uptake Aβ more efficiently (85). It might be possible that TREM2 serves as a key role in the anti-inflammatory properties of TREM2+ microglia and TREM2+ macrophages in the aortas. Since part of TREM2+ aortic macrophages are differentiating from monocytes, it might be beneficial to design studies that would identify pathways leading to a generation of monocyte-derived TREM2+ macrophages and a way how direct this population to migrate to the brain tissues for effective efferocytosis and suppression of neuroinflammation.
It is interesting to note that similar to a shift from initially protective macrophages to pro-inflammatory macrophages in AD, shift from protective/clearing functions of macrophages is also observed in atherosclerosis providing an additional line of similarities and potential therapeutically targeted points for these two diseases (Figure 2). In both pathologies, macrophages (and microglia in AD) play a multidimensional role via clearance of dying cells, processing its metabolites, taken up lipids, secretion of cytokines/chemokines and MMPs and therefore regulation of additional efflux of immune cells and regulation of vascular cell functions. On the other hand, macrophages can also promote resolution of inflammation. Thus, a therapeutic goal may be to find a network that would support “Golden Mean” of macrophages and microglia between their inflammatory functions, proteolytic activities and housekeeping functions of proper cholesterol transport, cellular flux, and effective efferocytosis in both pathologies.
Emerging evidence indicates that innate leukocytes can acquire an innate cell memory that is characterized by enhanced responsiveness to activation triggers. Innate trained memory is formed as a consequence of reprogramming based on gene transcription changes, epigenetic processes, and altered cellular metabolism (86). While more evidence is needed, initial studies demonstrate an existence of innate trained immunity in both atherosclerosis and AD (87, 88). It is likely that common triggers of innate memory such as infections in conjunction with the disease-specific stimuli including oxLDL, LDL and high-mobility group box 1 (HMGB1), can induce trained immunity in atherosclerosis (88). While it is unclear whether endogenous stimuli can induce trained innate immunity in AD, several infections such as Herpes simplex virus type 1, Cyto-megalovirus, Chlamydophila pneumoniae, spirochetes, Helicobacter pylori, and periodontal pathogens are closely associated with AD (89), suggesting these pathogens can serve as triggers of innate memory in AD patients. What are the targets for trained immunity? It has been reported that bone marrow progenitors, mature innate immune cells, microglia as well as NK cells, endothelial and VSCMs can develop memory characteristics. A common signature of innate trained immunity defined as an elevated long-lasting altered inflammatory response is detected in myeloid cells from patients with AD and/or atherosclerosis (87, 88). Indeed, elevated levels of circulating pro-inflammatory cytokines and amplified monocyte/macrophage cell responses have been reported in AD patients (27, 90). Not only AD circulating monocytes/neutrophils but also AD microglia demonstrate an activated phenotype that is consistent with a phenotype of innate memory response with a signature of alterations in cell transcriptome, epigenetic landscape and metabolic phenotype [reviewed in (87)]. Interestingly, in the aging brain and more evidently in AD patients, microglia appear primed: microglia are activated, produce increased amounts of pro-inflammatory mediators and are more susceptible to central damage after peripheral insults (93, 94). Interestingly, circulating pro-inflammatory cytokine levels are also dependent on the stages of disease progression with an overall reduction at the severe AD stages (91), suggesting a dynamic recalibration of the trained innate response likely due to the induction of tolerance.
In line with the trained immunity phenotype in AD, several papers highlighted a signature of trained immunity in atherosclerosis, particularly to various microbial and atherosclerosis-related stimuli in the in vitro system with a macrophage response characterized by elevated levels of TNFα, IL-6, MCP-1, MMP-2 and MMP-9, and increased foam cell formation (92). In vivo mouse studies showed that 4 weeks of LPS-induced TLR4-engagement initiated a reprogramming of monocytes to an inflammatory phenotype and aggravated atherogenesis (93). A similar signature of trained immune response with an increased ex vivo cytokine production and an elevated endothelial cell adhesion and migration was observed in patients with atherosclerosis (94). Not only elevated production of pro-inflammatory cytokines, but also evidence of metabolic reprogramming and epigenetic modifications at the level of histone methylation has been detected in blood monocytes of patients with established atherosclerosis (95). Thus, trained innate immunity is active in both conditions of AD and atherosclerosis and may contribute to the regulation of dysbalanced innate response that has a key role in driving both pathologies. While the majority of studies in both AD and atherosclerosis are focused on the characterization of the innate cell phenotype, very little is known about effects of innate memory responses on other myeloid cell functions such as recruitment, efferocytosis, apoptosis, and DC and macrophage antigen presentation and further studies are necessary in this promising area. It would be also critical to identify key characteristics of a monocyte population, conditions, and mechanisms by which monocytes might be recruited to inflammatory brains at different time points of the AD progression. As AD and atherosclerosis are diseases of aging, it would be further important to develop research programs focused on mechanisms that connect trained immunity and “inflammageing”, particularly in atherosclerosis-associated AD. These studies would help to better understand effects of trained innate memory immune responses in AD and atherosclerosis.
B Cells in Alzheimer's Disease and Atherosclerosis
While the prominent role of the innate immune system is extensively studied in AD, the role of adaptive immune response, and particularly B cells, in this disease is not well-understood, but an increasing body of evidence suggests an implication of B cells in AD (96). Indeed, one of the key discoveries in B cell–related pathophysiology of AD relies on finding that Aβ and tau are immunogenic proteins that induce an Ab response in AD (97). Importantly, these anti-Aβ (98) and anti-tau (99) Abs can bind its antigens in brain tissues of AD patients. In line with the observation of specific Abs against Aβ and tau, the analysis of the BCR repertoire revealed an amplification of high-frequency clonotypes in AD vs. healthy subjects suggesting that BCR-antigen interactions lead to a clone expansion (100). It remains to be determined specific functions of anti-Aβ and anti-tau Abs in vivo as followed up studies provided controversial conclusions for a role of Ab-specific Abs in AD pathology (Figure 1).
Studies in mice suggest that Igs play a dual role in AD since they activate microglia phagocytosis reducing Aβ deposits, but can also induce over-activation of microglia and neuronal neurotoxicity (101). Specifically, in 5 × FAD mice, Rag deficiency exacerbates AD due to loss of non-specific Igs that activate microglial phagocytosis and consequent clearance of Aβ plaques (101). Several studies attempted to use anti-Aβ Abs to reduce progression of AD in humans (102). Intravenous immunoglobulin (IVIG), a mixture of healthy donor-derived polyclonal Abs that contains most of the IgG found in the human immune repertoire, have been used in AD patients and showed some beneficial effects. Not only passive administration of Aβ-specific Ab, but also active immunization with Aβ reduced Aβ deposits in AD patients (103, 104) and mice, ameliorating the severity of disease (105, 106). The immunization with Aβ1−42 reduces Aβ plaque burden and preserves cognitive function in APP transgenic mice. Subsequently, this approach has been used in a clinical trial of AN1792(QS-21) in patients with mild to moderate AD. The study was interrupted because of development of meningoencephalitis in 6% of immunized patients, but initial data on neuropsychological test battery revealed differences favoring Ab responders (103) suggesting a protective role of Igs against Aβ in AD progression.
B cells not only produce Igs but also serve as antigen-presenting cells and cytokine-producing cells (16). Recent study by Kim et al. focused on the overall B cell functions in AD and demonstrated that the loss of mature B cells alone is sufficient to markedly retard the AD progression, improve behavioral and memory capacities and restore TGFβ+ microglia in the transgenic AD mouse models (107). Another function of B cells is to regulate inflammation via production of cytokines and chemokines. In line with a pathological function of B cells, B cells from AD mice acquire an inflammatory phenotype as shown by the upregulation of pro-inflammatory cytokines, including IL-6, TNFα, and IFNγ (42). Thus, B cells might play different roles in AD and associated neuroinflammation via secretion of immunomodulatory molecules, such as cytokines or Abs in periphery or directly affect glial reactivity and induce neuronal damage by secretion of inflammatory proteins at the site of damage via migration through dysfunctional blood-brain barrier.
B cells are active players not only in AD, but also have a prominent subset-specific role in atherosclerosis (16, 108). B1 cells synthesized natural Abs against pathogen-associated molecular patterns and neo-antigens. MZ B cells protect against blood-borne pathogens. FO B cells are activated by antigen stimulation, undergo germinal center reactions, and differentiate into memory B cells or plasma cells. Overall, FO and IRA B cells support atherosclerosis development, while B1 and MZ B cells protect against atherogenesis (45, 109). The role of B cell subsets in AD is not yet investigated, but some evidence suggests that it could be B cell subset specific. For example, several studies showed that neutralizing IgGs against Aβ are protective against AD, suggesting a potential role of FO B cells in AD (110, 111). Interestingly, a wide range of auto-Abs against Aβ, tau, neurotransmitters and related receptors, lipids, as well as microglial markers, presumably produced by B1 cells, are found in AD patients as well as healthy subjects (112). It is possible that activated upon atherogenic conditions in atherosclerosis, FO B cells can provide a further set of Ab-producing cells, while activated B1 cells might release Abs with a specific to neo-antigens including the Aβ protein. Further studies are necessary to determine which Ab isotypes are dominant in AD, characterize antigen-specificity of the response, and finally identify a role for germinal centers in driving AD pathologies. Taking into account some similarities in the nature of the diseases that are characterized by the chronic inflammatory response associated with aging and responses to self-antigens, it might be possible that FO, MZ, and B1 cells play a diverse role in AD. As atherosclerosis shapes number and functions of B cell subsets, it is likely that B cell subsets might display a unique phenotype and functions in atherosclerosis-associated AD and further studies will be necessary in this direction.
T Cells in Alzheimer's Disease and Atherosclerosis
Like B cells, T cells appear to have a complicated relationship with AD that is heavily T cell subset dependent (42). Although the role of T cells in AD is still being explored, there has been extensive effort into identifying changes in T cell subsets with varying degrees of agreement among studies. CD4+ T effector cells vary drastically in their role in AD. Adoptive transfer of Aβ-reactive Th1 or Th17 cells into the APP/PS1 mice led to an increase in Aβ in the brain through downregulation of Tregs (113). Additionally, IFNγ produced by adoptively transferred Aβ-specific Th1 promoted increased Aβ deposition in an APP/PS1 model, alongside cognitive deficits, indicating a detrimental role for Aβ-specific Th1 cells in AD (114). In line with these data, T cell infiltration into the brain is increased in AD compared to steady-state conditions, and studies in humans have found increased levels of Aβ-reactive T cells in AD patients and older adults compared to younger, healthy controls (115).
Circulating Th17 cells are increased in mild cognitive impairment (MCI) compared to cognitively normal controls (116). Neutralization of IL-17 prevents short-term cognitive deficits in an AD mouse model and attenuates deficits in long-term potentiation, although the primary source of IL-17 appeared to be from γδ T cells (117). One of the potential mechanisms of pathological action of infiltrating Th17 cells is upregulation of neuron-associated Fas and Th17-associated FasL, possibly contributing to increased neuronal apoptosis (118). Th2 cells appear to play a vastly different role in the AD-associated adaptive immune response. Adoptive transfer of Aβ-stimulated Th2 cells reversed cognitive impairment, reduced plaque associated microglia, decreased cerebrovascular Aβ deposition, and returned plasma cytokine level to normal levels in a mouse model (119). Specifically, lowered circulating GM-CSF correlated greatly to behavioral improvement, implying Th2 cells may soothe myeloid-induced neuroinflammation (119). In contrast, Baruch et al. found elevation in IL-4 expression in the choroid plexus of older mice coincided with production of CCL11, a chemokine associated with cognitive decline suggesting a pathological role of Th2 in neuroinflammation (120).
CD8+ cells have long been known to infiltrate into the brain during AD (121). Recent studies showed significantly more activated CD8+ T cells, particularly CD8+CD27− Teffector memory CD45RA+, in the blood and cerebrospinal fluid of patients with mild AD compared to healthy controls with a positive association with neuropsychological deficits, suggesting a detrimental role for CD8+ cells in AD (122, 123). In a mouse model of AD, doublecortin+ CD8+ cells clustered at Aβ plaques, and ablation of CD8+ T cells from the blood and brain resulted in changes in neuronal- and synapse-related gene expression (124). This depletion did not improve either cognition or Aβ pathology, although these mice were treated at 12 months of age, and may have already undergone extensive irreparable neurodegeneration. It would be important to understand mechanisms of doublecortin+ CD8+ cells recruitment to the aged brain as these cells can be a key participant in the regulation of synaptic plasticity.
Recent studies have unveiled a significant controversy in the trend for numbers of peripheral Tregs with decrease, increase or unchanged levels of Tregs in AD patients compared to patients with mild cognitive impairment or healthy controls (125, 126). This discrepancy could be due to differences in patient categorization of patient populations or gating strategies in flow cytometry. Mouse studies into the role of Tregs have caused even further confusion, as controversies arise over whether the immune-suppressive nature of Tregs propagates or protects against neurodegeneration. The adoptive transfer of Tregs into 3xTG mice reduced Aβ burden in the brain and improved spatial learning memory while reducing production of IL-2, IL-6, and IFNγ and increasing IL-10 production (127). In contrast, the depletion of Foxp3+ cells reduced Aβ plaque burden in 5xFAD mice (128). In line with pathological role of Tregs, induction of IL-10 in the brain using AAV vectors injected into the cerebral ventricles increased Aβ deposition and worsened fear-conditioned memory (129). Altogether, growing data indicate that T cell subsets are involved in AD-associated pathology and surprisingly demonstrate an altered distributions and activation statuses, thus playing a T cell subset specific role in this pathology (42). The ways in which peripheral T cells in AD showed a high sensitivity to peripheral inflammatory mediators suggest that these cells would be highly susceptible to a Th1/Th17-associated atherogenic response upon atherogenesis.
The overall T cell implication in atherosclerosis and their subset-specific roles have been discussed in several recent reviews (4, 130). Here, we focus on potentially important features of T-cell response in atherosclerosis that could affect the response in AD (Figure 1). Th1 cells are the predominant pathogenic T cell in atherosclerosis, contributing to high levels of IFNγ, which activate macrophages and monocytes and upregulate expression of MMPs, thinning the fibrous cap and increasing risk a plaque rupture. Similar to AD, Th1 cells are associated with worsening of atherosclerosis, as exogenous IFNγ enhances atherosclerosis in Apoe−/− mice (4, 131, 132). An important study using Tbet, a transcription factor responsible for a Th1 differentiation, showed that Tbet deficiency caused a Th2 switch, changed in T-dependent isotypes of oxidized LDL-specific Abs and reduced atherosclerosis (133). How elevated levels of Th1-derived cytokines modulates AD neuroinflammation remains to be determined; however, initial studies, particularly revolving around IFNγ, demonstrated somewhat diametrically opposed functions in the setting of AD-related neurodegeneration (134).
The role of Th17 cells in atherosclerosis is complex, and seems to be dependent on the model of atherosclerosis and timing of measurements of plaque development (135). One of highlighted features of IL-17+ T cells is a capacity to enhance endothelial expression of P-selectin, E-selectin, CXCL1, CXCL2, and CXCL5, potentially increasing migration of neutrophils and monocytes to the tissue specific site (136). This implies that Th1 and Th17 (or other IL-17 producing T cells) could act synergistically to further support recruitment of monocyte and neutrophils in the brain. Tregs are atheroprotective, with their expansion by various means detailed in previous reviews (130) resulting in reduced plaque formation due to the release of anti-inflammatory cytokines IL-10 and TGFβ. In the context of AD and atherosclerosis, modulation of Tregs or their downstream mechanisms could abate the damaging aspects of inflammation seen in both diseases, although more information is needed in regards to the role of Tregs as a whole in AD. Th2 cells are rarely found in atherosclerotic lesions, and are controversial in their role in atherosclerosis, much like in AD (1, 3). Deficiency in IL-4, a Th2 cytokine, in Apoe−/− mice reduced plaque area, implicating it in atherosclerotic progression, while the Th2 cytokines IL-5 and IL-13 appear to be largely atheroprotective (3). CD8+ cells are present within atherosclerotic lesions, with many of these identified as CD45RO- and very late activation antigen-1-expressing memory T cells (137). CD8+ T cells persist in the fibrous caps of plaques, and may contribute to plaque instability and rupture by inducing apoptosis of endothelial cells. In addition to their influence on monocytes and neutrophils, T cells may also be altered in terms of innate immunity. Mucosal-associated invariant T cells (MAITs) are thought to be involved in as well as influenced by immune-mediated chronic diseases, including type 2 diabetes mellitus and coronary artery disease (138, 139). Touch et al. found that individuals with coronary artery disease had decreased circulatory levels of MAITs with increased proapoptotic signaling compared to healthy controls, which may be caused in part by the chronic inflammation associated with the disease (139). Although the role of MAITs in AD has remained relatively unexplored, MAIT cells are known to be present in the brain, where they may interact with microglia and astrocytes via the major histocompatibility complex class Ib molecule called MR1 (140). Circulating levels of MAIT cells have been found to decline in later life (141), an observation whose explanation may further elucidate what role these cells play in atherosclerosis and AD. T cell subsets play an integral role in both AD and atherosclerosis, although whether this role is protective or harmful remains to be fully elucidated in many subsets. Given the fact that T cells are rarely found within the healthy CNS parenchyma, it would be important to investigate when, how and which subpopulation of Th cells can migrate to the brain tissues and regulate the local microenvironment. It is also important to determine whether only CNS antigens- specific Th cells or a broader range of CD4+ effector memory cells might be recruited and modulate the local immune response in the brain under conditions of atherosclerosis. Increasing evidence indicates that homeostasis of peripheral T cell immunity is dysregulated in AD and atherosclerosis and these changes in T cell subset phenotypes and activation status affect the brain and vessel biology and have complex implications in both disease development and persistence. Gaining an understanding as to how proatherogenic Th cells modulate the AD-associated response could elucidate new therapeutic strategies designed to harness the activity of pathological Th subsets in both AD and atherosclerosis.
Hypercholesterolemia in the Crossroad of Atherosclerosis and AD
Hypercholesterolemia has long been agreed upon as a risk for atherosclerosis, but hypercholesterolemia could also play a pivotal role in neurodegeneration in AD (Figure 2). Twenty-five percentage of the body's cholesterol is located in the brain, where it is synthesized de novo due to its inability to cross the brain blood barrier (11). Despite this fact, a meta-analysis of vascular risk factors in AD found that systemic hypercholesterolemia increases the risk of AD (142), and higher levels of LDL have been associated with higher Aβ burden in autopsies of AD patients independent of APOE status, indicating a link between peripheral hypercholesterolemia and AD. This link may in part be driven by the effects of hypercholesterolemia on peripheral immune cells. Hypercholesterolemia induces monocytosis and neutrophilia by increasing expression of CCR2 and CXCR2, promoting atherosclerosis but also potentially contributing to brain blood barrier breakdown and immune infiltration (143). Brain blood barrier breakdown increases the egress of 24S-hydroxy-cholesterol, a metabolite of cholesterol that modulates cholesterol synthesis, which in-turn increases the production of cholesterol in the brain (11). Increased neuronal cholesterol increases amyloidogenic processing of APP through assembly of APP, β-secretases, and γ-secretases in lipid rafts (125). Similarly, hypercholesterolemia increases TCR signaling through increased assembly of lipid rafts, which help in the dimerization of TCR (144). This leads to hyperproliferation in CD4+ T cells, which could serve to exacerbate AD (10). Evidence suggests that lymphatic functions are disregulated under hypercholesterolemic conditions and dysfunctional lymphatics are likely involved in atherogenesis (145). Importantly, meningeal lymphatics have been recently discovered across the species and its role has been implicated in development of several neurodegenerative pathologies including AD (145). Therefore, there is a need in new studies focused on a potential implication of hyperlipidemia in the regulation of lymphatics in AD that might open new strategies for combatting both AD and atherosclerosis. One of the strongest links that ties AD and atherosclerosis is the fact that the APOE4 isotype is a risk factor for both hypercholesterolemia and AD (11). APOE transports cholesterol-carrying lipoproteins from astrocytes to neurons in the brain (11), but it is still unclear how differences in APOE isoforms in the brain mediate increased prevalence of AD. In the periphery, the APOE4 isoform may contribute to AD through increased prevalence of hypercholesterolemia and plasma LDL levels, which may then indirectly modulate AD through changes in immune response. Therefore, although many mechanisms modulate the interconnections interwoven between atherosclerosis and AD, hypercholesterolemia-induced immune perturbation stands as one of a driving force that needs further exploration.
Discussion
Although the brain has previously been considered an immune privileged organ, it is apparent that the peripheral immune system plays a large part in the brain's diseased state. Aging leads to chronic, low-grade inflammation that underlies the pathogenesis of many aging-related diseases, like AD and atherosclerosis, in a process known as inflammageing. Current work in AD focuses mainly on changes in local immune response, while atherosclerosis is known for its unique strong systemic immune response. Based on epidemiological data, AD and atherosclerosis as well as other chronic inflammatory diseases are interlinked, indicating a need for animal models that may elucidate the effects of atherosclerosis-derived systemic immune changes on AD-linked neuroinflammation. More exploration into how chronic peripheral inflammation, such as in atherosclerosis, may alter, exacerbate, or even cause AD may help to further reveal the roles of immune cell subsets in both diseases. Our current understanding of the role of neuroinflammation in AD is very reminiscent of atherosclerosis. Drawing parallels to compare DAMs and Aβ to foam cells and oxLDL has revealed that some of the same underlying mechanisms may be at play, and more similarities may be discovered in future studies. Clarification of the effects of peripheral inflammation on long-term plasticity in the function of the resident brain immune cells, like microglia, is needed to understand the changes in these cells. Additionally, as microglia are long-lived cells, there exists potential for innate trained immunity in the brain that has been explored in recent years (86, 146). Further study into how epigentic changes in atherosclerosis-influenced trained immunity of peripheral immune cells may contribute to AD will additionally shed light into how these two diseases may interact. Although not the central focus of this particular review, both AD and Atherosclerosis share common attributes of endothelial dysfunction and vascular aging, as well as shared risk factors that may contribute to the altered immune landscape. Aging, obesity, type-2 diabetes, and smoking are just a few inflammation-inducing risk factors that AD and atherosclerosis share. Aging of vasculature itself can lead to increased vascular stiffness, increased immune infiltration due to increased expression of leukocyte adhesion molecules on endothelial cells, and increased release of proinflammatory cytokines by immune, endothelial, and VSCMs [further reviewed elsewhere (147, 148)]. Endothelial cells become more senscent-like with age, with increased activation of NF-κB and apoptosis that may play a role in the recruitment of immune cells to the vasculature (149). Therefore, aging of the vaculature and endothelial dysfunction serve as an additional link between these two diseases.
While we have mainly focused in this review on the similarities and intertwined nature of the immune response in AD and atherosclerosis, another important aspect of the discussion should be an implication of gut microbiota and associated immunity in development of both pathologies.
Dysbiosis of gut microbiota has been reported to induce and support the progression of AD reflected by formation of Aβ plaques and neurofibrillary tangles. From the other hand, studies have also demonstrated that alterations in gut microbiota induced by a number of factors can lead to leaky gut, causing an increased inflammation and accelerated atherosclerosis. Thus, gut microbiota and gut-associated immune responses might have a synergistic effect on development of both AD and atherosclerosis. For a more detailed overview of how gut microbiota influences atherosclerosis and AD, we refer to related reviews (150, 151).
Current studies of AD in humans face challenges in unraveling the temporal and spatial aspects of immune infiltration in AD progression. Epidemiological data focuses on either collection of peripheral samples (such as blood and cerebrospinal fluid) in living humans or evaluation of the brain via imaging and/or post-mortem evaluation. New tools may be needed to identify where and when infiltration of the brain by immune cells occurs, perhaps through further development of imaging technology like two-photon microscopy. Although this review has encapsulated many aspects of the immune system in atherosclerosis and AD, it is limited in its discussion of other factors, such as an impact of oxidative stress, hypertension, hyperlipidemia and other metabolic aspects that have led some to refer to AD as type 3 diabetes (152). A detailed knowledge of immune pathways in atherosclerosis and AD and their interconnected molecular networks may also lead to a better understanding and treatment of both pathologies simultaneously, reducing a harmful combinatorial effects of a defective resolution of inflammation in the systemic circulation, aorta, and brain. For example, the function of neutrophils in both diseases leaves them as a promising target population for a more systemic approach to therapy, while the complex interplay between monocyte phagocytosis and release of inflammatory mediators in these two diseases may require a more nuanced approach that still allows for the clearance of oxLDL or Aβ while impeding the oncoming immune assault. With this in mind, the future of these two disparate diseases may not look as different as once thought.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
The authors were supported by the National Institutes of Health under awards R01HL142129, R01HL139000, and HL142129-04S1 (EG).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We apologize to our many colleagues whose important work we were unable to cite due to space limitations. Figure 1 illustrations were created with Biorender. Figure 2 illustrations were created using edited pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
References
1. Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis (*). Annu Rev Immunol. (2009) 27:165–97. doi: 10.1146/annurev.immunol.021908.132620
2. Hansson GK, Libby P, Tabas I. Inflammation and plaque vulnerability. J Intern Med. (2015) 278:483–93. doi: 10.1111/joim.12406
3. Mallat Z, Taleb S, Ait-Oufella H, Tedgui A. The role of adaptive T cell immunity in atherosclerosis. J Lipid Res. (2009) 50(Suppl.):S364–9. doi: 10.1194/jlr.R800092-JLR200
4. Roy P, Orecchioni M, Ley K. How the immune system shapes atherosclerosis: roles of innate and adaptive immunity. Nat Rev Immunol. (2021) 22:251–65. doi: 10.1038/s41577-021-00584-1
5. Feng Y, Li L, Sun X-H. Monocytes and Alzheimer's disease. Neurosci Bull. (2011) 27:115–22. doi: 10.1007/s12264-011-1205-3
6. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer's disease. The Lancet Neurology. (2015) 14:388–405. doi: 10.1016/S1474-4422(15)70016-5
7. Lane CA, Hardy J, Schott JM. Alzheimer's disease. Eur J Neurol. (2018) 25:59–70. doi: 10.1111/ene.13439
8. Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. (2006) 12:1005–15. doi: 10.1038/nm1484
9. Santiago JA, Potashkin JA. The impact of disease comorbidities in Alzheimer's disease. Front Aging Neurosci. (2021) 13:1–13. doi: 10.3389/fnagi.2021.631770
10. Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. (2015) 16:358–72. doi: 10.1038/nrn3880
11. Feringa FM, van der Kant R. Cholesterol and Alzheimer's Disease; From Risk Genes to Pathological Effects. Front Aging Neurosci. (2021) 13:1–17. doi: 10.3389/fnagi.2021.690372
12. Donnan GA, Fisher M, Macleod M, Davis SM. Stroke. Lancet. (2008) 371:1612–23. doi: 10.1016/S0140-6736(08)60694-7
13. Kisler K, Nelson AR, Montagne A, Zlokovic BV. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci. (2017) 18:419–34. doi: 10.1038/nrn.2017.48
14. Samieri C, Perier MC, Gaye B, Proust-Lima C, Helmer C, Dartigues JF, et al. Association of cardiovascular health level in older age with cognitive decline and incident dementia. JAMA. (2018) 320:657–64. doi: 10.1001/jama.2018.11499
15. Shabir O, Berwick J, Francis SE. Neurovascular dysfunction in vascular dementia, Alzheimer's and atherosclerosis. BMC Neurosci. (2018) 19:62. doi: 10.1186/s12868-018-0465-5
16. Ma SD, Mussbacher M, Galkina EV. Functional role of B cells in atherosclerosis. Cells. (2021) 10:270. doi: 10.3390/cells10020270
17. Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer's disease. Nat Rev Dis Prim. (2015) 1:15056. doi: 10.1038/nrdp.2015.56
18. Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. (2019) 179:312–39. doi: 10.1016/j.cell.2019.09.001
19. Chen GF, Xu TH, Yan Y, Zhou YR, Jiang Y, Melcher K, et al. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol Sin. (2017) 38:1205–35. doi: 10.1038/aps.2017.28
20. Yoon SS, Jo SA. Mechanisms of amyloid-β peptide clearance: potential therapeutic targets for Alzheimer's disease. Biomol Ther. (2012) 20:245–55. doi: 10.4062/biomolther.2012.20.3.245
21. König T, Stögmann E. Genetics of Alzheimer's disease. Wiener Med Wochenschrift. (2021) 171:249–56. doi: 10.1007/s10354-021-00819-9
22. Gottesman RF, Albert MS, Alonso A, Coker LH, Coresh J, Davis SM, et al. Associations between midlife vascular risk factors and 25-year incident dementia in the atherosclerosis risk in communities (ARIC) cohort. JAMA Neurol. (2017) 74:1246–54. doi: 10.1001/jamaneurol.2017.1658
23. Jenny NS, French B, Arnold AM, Strotmeyer ES, Cushman M, Chaves PH, et al. Long-term assessment of inflammation and healthy aging in late life: the Cardiovascular Health Study All Stars. J Gerontol Ser A Biol Sci Med Sci. (2012) 67:970–6. doi: 10.1093/gerona/glr261
24. Sundelöf J, Kilander L, Helmersson J, Larsson A, Rönnemaa E, Degerman-Gunnarsson M, et al. Systemic inflammation and the risk of Alzheimer's disease and dementia: a prospective population-based study. J Alzheimers Dis. (2009) 18:79–87. doi: 10.3233/JAD-2009-1126
25. Lanzrein AS, Johnston CM, Perry VH, Jobst KA, King EM, Smith AD. Longitudinal study of inflammatory factors in serum, cerebrospinal fluid, and brain tissue in Alzheimer disease: interleukin-1beta, interleukin-6, interleukin-1 receptor antagonist, tumor necrosis factor-alpha, the soluble tumor necrosis factor receptors I and II, and alpha1-antichymotrypsin. Alzheimer Dis Assoc Disord. (1998) 12:215–27. doi: 10.1097/00002093-199809000-00016
26. Ravaglia G, Forti P, Maioli F, Chiappelli M, Montesi F, Tumini E, et al. Blood inflammatory markers and risk of dementia: The Conselice Study of Brain Aging. Neurobiol Aging. (2007) 28:1810–20. doi: 10.1016/j.neurobiolaging.2006.08.012
27. Swardfager W, Lanctôt K, Rothenburg L, Wong A, Cappell J, Herrmann N, et al. meta-analysis of cytokines in Alzheimer's disease. Biol Psychiatry. (2010) 68:930–41. doi: 10.1016/j.biopsych.2010.06.012
28. Lai KSP, Liu CS, Rau A, Lanctôt KL, Köhler CA, Pakosh M, et al. Peripheral inflammatory markers in Alzheimer's disease: a systematic review and meta-analysis of 175 studies. J Neurol Neurosurg Psychiatry. (2017) 88:876–82. doi: 10.1136/jnnp-2017-316201
29. Singh-Manoux A, Dugravot A, Brunner E, Kumari M, Shipley M, Elbaz A, et al. Interleukin-6 and C-reactive protein as predictors of cognitive decline in late midlife. Neurology. (2014) 83:486–93. doi: 10.1212/WNL.0000000000000665
30. Yaffe K, Kanaya A, Lindquist K, Simonsick EM, Harris T, Shorr RI, et al. The metabolic syndrome, inflammation, and risk of cognitive decline. JAMA. (2004) 292:2237–42. doi: 10.1001/jama.292.18.2237
31. Tarkowski E, Andreasen N, Tarkowski A, Blennow K. Intrathecal inflammation precedes development of Alzheimer's disease. J Neurol Neurosurg Psychiatry. (2003) 74:1200–5. doi: 10.1136/jnnp.74.9.1200
32. Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, et al. Systemic inflammation and disease progression in Alzheimer disease. Neurology. (2009) 73:768–74. doi: 10.1212/WNL.0b013e3181b6bb95
33. Krstic D, Madhusudan A, Doehner J, Vogel P, Notter T, Imhof C, et al. Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J Neuroinflammation. (2012) 9:151. doi: 10.1186/1742-2094-9-151
34. Cunningham C. Systemic inflammation and delirium: important co-factors in the progression of dementia. Biochem Soc Trans. (2011) 39:945–53. doi: 10.1042/BST0390945
35. Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA. (2010) 304:1787–94. doi: 10.1001/jama.2010.1553
36. Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell. (2013) 153:707–20. doi: 10.1016/j.cell.2013.03.030
37. Maciejewska K, Czarnecka K, Szymański PA. review of the mechanisms underlying selected comorbidities in Alzheimer's disease. Pharmacol Rep. (2021) 73:1565–81. doi: 10.1007/s43440-021-00293-5
38. Xiang J. Carotid atherosclerosis promotes the progression of Alzheimer's disease: a three-year prospective study. Exp Ther Med. (2017) 14:1321–6. doi: 10.3892/etm.2017.4661
39. Dolan H, Crain B, Troncoso J, Resnick SM, Zonderman AB, Obrien RJ. Atherosclerosis, dementia, and Alzheimer disease in the Baltimore Longitudinal Study of Aging cohort. Ann Neurol. (2010) 68:231–40. doi: 10.1002/ana.22055
40. Cortes-Canteli M, Gispert JD, Salvadó G, Toribio-Fernandez R, Tristão-Pereira C, Falcon C, et al. Subclinical atherosclerosis and brain metabolism in middle-aged individuals: The PESA Study. J Am Coll Cardiol. (2021) 77:888–98. doi: 10.1016/j.jacc.2020.12.027
41. Deschaintre Y, Richard F, Leys D, Pasquier F. Treatment of vascular risk factors is associated with slower decline in Alzheimer disease. Neurology. (2009) 73:674–80. doi: 10.1212/WNL.0b013e3181b59bf3
42. Bettcher BM, Tansey MG, Dorothée G, Heneka MT. Peripheral and central immune system crosstalk in Alzheimer disease - a research prospectus. Nat Rev Neurol. (2021) 17:689–701. doi: 10.1038/s41582-021-00549-x
43. Dong Y, Lagarde J, Xicota L, Corne H, Chantran Y, Chaigneau T, et al. Neutrophil hyperactivation correlates with Alzheimer's disease progression. Ann Neurol. (2018) 83:387–405. doi: 10.1002/ana.25159
44. Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Marongiu L, Budui S, et al. Neutrophils promote Alzheimer's disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med. (2015) 21:880–6. doi: 10.1038/nm.3913
45. Cruz Hernández JC, Bracko O, Kersbergen CJ, Muse V, Haft-Javaherian M, Berg M, et al. Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer's disease mouse models. Nat Neurosci. (2019) 22:413–20. doi: 10.1038/s41593-018-0329-4
46. Donner L, Fälker K, Gremer L, Klinker S, Pagani G, Ljungberg LU, et al. Platelets contribute to amyloid-β aggregation in cerebral vessels through integrin αIIbβ3-induced outside-in signaling and clusterin release. Sci Signal. (2016) 9:ra52. doi: 10.1126/scisignal.aaf6240
47. Pietronigro EC, Della Bianca V, Zenaro E, Constantin G. NETosis in Alzheimer's Disease. Front Immunol. (2017) 8:211. doi: 10.3389/fimmu.2017.00211
48. Korte N, Nortley R, Attwell D. Cerebral blood flow decrease as an early pathological mechanism in Alzheimer's disease. Acta Neuropathol. (2020) 140:793–810. doi: 10.1007/s00401-020-02215-w
49. Ringland C, Schweig JE, Eisenbaum M, Paris D, Ait-Ghezala G, Mullan M, et al. MMP9 modulation improves specific neurobehavioral deficits in a mouse model of Alzheimer's disease. BMC Neurosci. (2021) 22:39. doi: 10.1186/s12868-021-00643-2
50. Silvestre-Roig C, Braster Q, Ortega-Gomez A, Soehnlein O. Neutrophils as regulators of cardiovascular inflammation. Nat Rev Cardiol. (2020) 17:327–40. doi: 10.1038/s41569-019-0326-7
51. Fotakis P, Kothari V, Thomas DG, Westerterp M, Molusky MM, Altin E, et al. Anti-inflammatory effects of HDL (high-density lipoprotein) in macrophages predominate over proinflammatory effects in atherosclerotic plaques. Arterioscler Thromb Vasc Biol. (2019) 39:e253–72. doi: 10.1161/ATVBAHA.119.313253
52. Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. (2010) 328:1689–93. doi: 10.1126/science.1189731
53. Silvestre-Roig C, Braster Q, Wichapong K, Lee EY, Teulon JM, Berrebeh N, et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature. (2019) 569:236–40. doi: 10.1038/s41586-019-1167-6
54. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. (2015) 349:316–20. doi: 10.1126/science.aaa8064
55. Fani Maleki A, Rivest S. Innate immune cells: monocytes, monocyte-derived macrophages and microglia as therapeutic targets for Alzheimer's disease and multiple sclerosis. Front Cell Neurosci. (2019) 13:355. doi: 10.3389/fncel.2019.00355
56. Saresella M, Marventano I, Calabrese E, Piancone F, Rainone V, Gatti A, et al. A complex proinflammatory role for peripheral monocytes in Alzheimer's disease. J Alzheimers Dis. (2014) 38:403–13. doi: 10.3233/JAD-131160
57. Munawara U, Catanzaro M, Xu W, Tan C, Hirokawa K, Bosco N, et al. Hyperactivation of monocytes and macrophages in MCI patients contributes to the progression of Alzheimer's disease. Immun Ageing. (2021) 18:29. doi: 10.1186/s12979-021-00236-x
58. Naert G, Rivest SCC. Chemokine receptor 2 deficiency aggravates cognitive impairments and amyloid pathology in a transgenic mouse model of Alzheimer's disease. J Neurosci. (2011) 31:6208–20. doi: 10.1523/JNEUROSCI.0299-11.2011
59. Naert G, Rivest S. Age-related changes in synaptic markers and monocyte subsets link the cognitive decline of APPSwe/PS1 mice. Front Cell Neurosci. (2012) 6:51. doi: 10.3389/fncel.2012.00051
60. Thériault P, ElAli A, Rivest S. The dynamics of monocytes and microglia in Alzheimer's disease. Alzheimers Res Therapy. (2015) 7:41. doi: 10.1186/s13195-015-0125-2
61. Chen S-H, Tian D-Y, Shen Y-Y, Cheng Y, Fan D-Y, Sun H-L, et al. Amyloid-beta uptake by blood monocytes is reduced with ageing and Alzheimer's disease. Transl Psychiatry. (2020) 10:423. doi: 10.1038/s41398-020-01113-9
62. Zhu D, Yang N, Liu YY, Zheng J, Ji C, Zuo PP. M2 Macrophage transplantation ameliorates cognitive dysfunction in amyloid-β-treated rats through regulation of microglial polarization. J Alzheimers Dis. (2016) 52:483–95. doi: 10.3233/JAD-151090
63. Costa-Marques L, Arnold K, Pardon M-C, Leovsky C, Swarbrick S, Fabian C, et al. Transplantation of bone marrow derived macrophages reduces markers of neuropathology in an APP/PS1 mouse model. Transl Neurodegener. (2019) 8:33. doi: 10.1186/s40035-019-0173-9
64. Daniels MJD, Rivers-Auty J, Schilling T, Spencer NG, Watremez W, Fasolino V, et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer's disease in rodent models. Nat Commun. (2016) 7:12504. doi: 10.1038/ncomms12504
65. Jiang T, Zhang YD, Chen Q, Gao Q, Zhu XC, Zhou JS, et al. TREM2 modifies microglial phenotype and provides neuroprotection in P301S tau transgenic mice. Neuropharmacology. (2016) 105:196–206. doi: 10.1016/j.neuropharm.2016.01.028
66. Xiang X, Werner G, Bohrmann B, Liesz A, Mazaheri F, Capell A, et al. TREM2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. EMBO Mol Med. (2016) 8:992–1004. doi: 10.15252/emmm.201606370
67. Hawkes CA, McLaurin J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc Natl Acad Sci USA. (2009) 106:1261–6. doi: 10.1073/pnas.0805453106
68. Park L, Uekawa K, Garcia-Bonilla L, Koizumi K, Murphy M, Pistik R, et al. Brain perivascular macrophages initiate the neurovascular dysfunction of Alzheimer Aβ peptides. Circ Res. (2017) 121:258–69. doi: 10.1161/CIRCRESAHA.117.311054
69. Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. (2011) 145:341–55. doi: 10.1016/j.cell.2011.04.005
70. Swirski FK, Nahrendorf M, Libby P. Mechanisms of myeloid cell modulation of atherosclerosis. Microbiol Spectr. (2016) 4. doi: 10.1128/microbiolspec.MCHD-0026-2015. [Epub ahead of print].
71. Poller WC, Nahrendorf M, Swirski FK. Hematopoiesis and cardiovascular disease. Circ Res. (2020) 126:1061–85. doi: 10.1161/CIRCRESAHA.120.315895
72. Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. (2007) 117:195–205. doi: 10.1172/JCI29950
73. Combadière C, Potteaux S, Rodero M, Simon T, Pezard A, Esposito B, et al. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. (2008) 117:1649–57. doi: 10.1161/CIRCULATIONAHA.107.745091
74. Drechsler M, Megens RT, van Zandvoort M, Weber C, Soehnlein O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation. (2010) 122:1837–45. doi: 10.1161/CIRCULATIONAHA.110.961714
75. Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. (2007) 117:185–94. doi: 10.1172/JCI28549
76. Spann NJ, Garmire LX, McDonald JG, Myers DS, Milne SB, Shibata N, et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell. (2012) 151:138–52. doi: 10.1016/j.cell.2012.06.054
77. Kim K, Shim D, Lee JS, Zaitsev K, Williams JW, Kim KW, et al. Transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models. Circ Res. (2018) 123:1127–42. doi: 10.1161/CIRCRESAHA.118.312804
78. Sáiz-Vazquez O, Puente-Martínez A, Ubillos-Landa S, Pacheco-Bonrostro J, Santabárbara J. Cholesterol and Alzheimer's disease risk: a meta-meta-analysis. Brain Sci. (2020) 10:386. doi: 10.3390/brainsci10060386
79. Sparks DL, Kryscio RJ, Sabbagh MN, Connor DJ, Sparks LM, Liebsack C. Reduced risk of incident AD with elective statin use in a clinical trial cohort. Curr Alzheimer Res. (2008) 5:416–21. doi: 10.2174/156720508785132316
80. Sano M, Bell KL, Galasko D, Galvin JE, Thomas RG, van Dyck CH, et al. A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology. (2011) 77:556–63. doi: 10.1212/WNL.0b013e318228bf11
81. Zernecke A, Winkels H, Cochain C, Williams JW, Wolf D, Soehnlein O, et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ Res. (2020) 127:402–26. doi: 10.1161/CIRCRESAHA.120.316903
82. Claes C, Danhash EP, Hasselmann J, Chadarevian JP, Shabestari SK, England WE, et al. Plaque-associated human microglia accumulate lipid droplets in a chimeric model of Alzheimer's disease. Mol Neurodegener. (2021) 16:50. doi: 10.1186/s13024-021-00473-0
83. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer's disease. Cell. (2017) 169:1276–90.e17. doi: 10.1016/j.cell.2017.05.018
84. Parhizkar S, Arzberger T, Brendel M, Kleinberger G, Deussing M, Focke C, et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci. (2019) 22:191–204. doi: 10.1038/s41593-018-0296-9
85. Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron. (2016) 91:328–40. doi: 10.1016/j.neuron.2016.06.015
86. Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol. (2020) 20:375–88. doi: 10.1038/s41577-020-0285-6
87. Salani F, Sterbini V, Sacchinelli E, Garramone M, Bossù P. Is innate memory a double-edge sword in Alzheimer's disease? A reappraisal of new concepts and old data. Front Immunol. (2019) 10:1768. doi: 10.3389/fimmu.2019.01768
88. Flores-Gomez D, Bekkering S, Netea MG, Riksen NP. Trained immunity in atherosclerotic cardiovascular disease. Arterioscler Thromb Vasc Biol. (2021) 41:62–9. doi: 10.1161/ATVBAHA.120.314216
89. Harris SA, Harris EA. Herpes simplex virus type 1 and other pathogens are key causative factors in sporadic Alzheimer's disease. J Alzheimers Dis. (2015) 48:319–53. doi: 10.3233/JAD-142853
90. Guerreiro RJ, Santana I, Brás JM, Santiago B, Paiva A, Oliveira C. Peripheral inflammatory cytokines as biomarkers in Alzheimer's disease and mild cognitive impairment. Neurodegen Dis. (2007) 4:406–12. doi: 10.1159/000107700
91. Motta M, Imbesi R, Di Rosa M, Stivala F, Malaguarnera L. Altered plasma cytokine levels in Alzheimer's disease: correlation with the disease progression. Immunol Lett. (2007) 114:46–51. doi: 10.1016/j.imlet.2007.09.002
92. Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol. (2014) 34:1731–8. doi: 10.1161/ATVBAHA.114.303887
93. Geng S, Chen K, Yuan R, Peng L, Maitra U, Diao N, et al. The persistence of low-grade inflammatory monocytes contributes to aggravated atherosclerosis. Nat Commun. (2016) 7:13436. doi: 10.1038/ncomms13436
94. van der Valk FM, Bekkering S, Kroon J, Yeang C, Van den Bossche J, van Buul JD, et al. Oxidized phospholipids on lipoprotein (a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. (2016) 134:611–24. doi: 10.1161/CIRCULATIONAHA.116.020838
95. Bekkering S, van den Munckhof I, Nielen T, Lamfers E, Dinarello C, Rutten J, et al. Innate immune cell activation and epigenetic remodeling in symptomatic and asymptomatic atherosclerosis in humans in vivo. Atherosclerosis. (2016) 254:228–36. doi: 10.1016/j.atherosclerosis.2016.10.019
96. Sabatino JJ Jr, Pröbstel AK, Zamvil SS. B cells in autoimmune and neurodegenerative central nervous system diseases. Nat Rev Neurosci. (2019) 20:728–45. doi: 10.1038/s41583-019-0233-2
97. Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, et al. Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. (2008) 372:216–23. doi: 10.1016/S0140-6736(08)61075-2
98. Gaskin F, Finley J, Fang Q, Xu S, Fu SM. Human antibodies reactive with beta-amyloid protein in Alzheimer's disease. J Exp Med. (1993) 177:1181–6. doi: 10.1084/jem.177.4.1181
99. Pascual G, Wadia JS, Zhu X, Keogh E, Kükrer B, van Ameijde J, et al. Immunological memory to hyperphosphorylated tau in asymptomatic individuals. Acta Neuropathol. (2017) 133:767–83. doi: 10.1007/s00401-017-1705-y
100. Xu H, Jia J. Single-cell RNA sequencing of peripheral blood reveals immune cell signatures in Alzheimer's disease. Front Immunol. (2021) 12:645666. doi: 10.3389/fimmu.2021.645666
101. Marsh SE, Abud EM, Lakatos A, Karimzadeh A, Yeung ST, Davtyan H, et al. The adaptive immune system restrains Alzheimer's disease pathogenesis by modulating microglial function. Proc Natl Acad Sci USA. (2016) 113:E1316–25. doi: 10.1073/pnas.1525466113
102. Relkin N. Clinical trials of intravenous immunoglobulin for Alzheimer's disease. J Clin Immunol. (2014) 34(Suppl. 1):S74–9. doi: 10.1007/s10875-014-0041-4
103. Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. (2005) 64:1553–62. doi: 10.1212/01.WNL.0000159740.16984.3C
104. Bombois S, Maurage CA, Gompel M, Deramecourt V, Mackowiak-Cordoliani MA, Black RS, et al. Absence of beta-amyloid deposits after immunization in Alzheimer disease with Lewy body dementia. Arch Neurol. (2007) 64:583–7. doi: 10.1001/archneur.64.4.583
105. Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, et al. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. (2000) 408:982–5. doi: 10.1038/35050116
106. Olkhanud PB, Mughal M, Ayukawa K, Malchinkhuu E, Bodogai M, Feldman N, et al. DNA immunization with HBsAg-based particles expressing a B cell epitope of amyloid beta-peptide attenuates disease progression and prolongs survival in a mouse model of Alzheimer's disease. Vaccine. (2012) 30:1650–8. doi: 10.1016/j.vaccine.2011.12.136
107. Kim K, Wang X, Ragonnaud E, Bodogai M, Illouz T, DeLuca M, et al. Therapeutic B-cell depletion reverses progression of Alzheimer's disease. Nat Commun. (2021) 12:2185. doi: 10.1038/s41467-021-22479-4
108. Srikakulapu P, McNamara CA. B lymphocytes and adipose tissue inflammation. Arterioscler Thromb Vasc Biol. (2020) 40:1110–22. doi: 10.1161/ATVBAHA.119.312467
109. Tsiantoulas D, Sage AP, Mallat Z, Binder CJ. Targeting B cells in atherosclerosis: closing the gap from bench to bedside. Arterioscler Thromb Vasc Biol. (2015) 35:296–302. doi: 10.1161/ATVBAHA.114.303569
110. Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, et al. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. (2010) 330:1774. doi: 10.1126/science.1197623
111. Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. (2015) 11:457–70. doi: 10.1038/nrneurol.2015.119
112. Wu J, Li L. Autoantibodies in Alzheimer's disease: potential biomarkers, pathogenic roles, and therapeutic implications. J Biomed Res. (2016) 30:361–72. doi: 10.7555/JBR.30.20150131
113. Machhi J, Yeapuri P, Lu Y, Foster E, Chikhale R, Herskovitz J, et al. CD4+ effector T cells accelerate Alzheimer's disease in mice. J Neuroinflammation. (2021) 18:272. doi: 10.1186/s12974-021-02308-7
114. Browne TC, McQuillan K, McManus RM, O'Reilly J-A, Mills KHG, Lynch MA. IFN-γ production by amyloid β-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer's disease. J Immunol. (2013) 190:2241–51. doi: 10.4049/jimmunol.1200947
115. Monsonego A, Zota V, Karni A, Krieger JI, Bar-Or A, Bitan G, et al. Increased T cell reactivity to amyloid beta protein in older humans and patients with Alzheimer disease. J Clin Invest. (2003) 112:415–22. doi: 10.1172/JCI200318104
116. Oberstein TJ, Taha L, Spitzer P, Hellstern J, Herrmann M, Kornhuber J, et al. Imbalance of circulating Th17 and regulatory T cells in Alzheimer's disease: a case control study. Front Immunol. (2018) 9:1213. doi: 10.3389/fimmu.2018.01213
117. Brigas HC, Ribeiro M, Coelho JE, Gomes R, Gomez-Murcia V, Carvalho K, et al. IL-17 triggers the onset of cognitive and synaptic deficits in early stages of Alzheimer's disease. Cell Rep. (2021) 36:109574. doi: 10.1016/j.celrep.2021.109574
118. Zhang J, Ke KF, Liu Z, Qiu YH, Peng YP. Th17 cell-mediated neuroinflammation is involved in neurodegeneration of aβ1-42-induced Alzheimer's disease model rats. PLoS ONE. (2013) 8:e75786. doi: 10.1371/journal.pone.0075786
119. Cao C, Arendash GW, Dickson A, Mamcarz MB, Lin X, Ethell DW. Aβ-specific Th2 cells provide cognitive and pathological benefits to Alzheimer's mice without infiltrating the CNS. Neurobiol Dis. (2009) 34:63–70. doi: 10.1016/j.nbd.2008.12.015
120. Baruch K, Ron-Harel N, Gal H, Deczkowska A, Shifrut E, Ndifon W, et al. CNS-specific immunity at the choroid plexus shifts toward destructive Th2 inflammation in brain aging. Proc Natl Acad Sci USA. (2013) 110:2264–9. doi: 10.1073/pnas.1211270110
121. Itagaki S, McGeer PL, Akiyama H. Presence of T-cytotoxic suppressor and leucocyte common antigen positive cells in Alzheimer's disease brain tissue. Neurosci Lett. (1988) 91:259–64. doi: 10.1016/0304-3940(88)90690-8
122. Lueg G, Gross CC, Lohmann H, Johnen A, Kemmling A, Deppe M, et al. Clinical relevance of specific T-cell activation in the blood and cerebrospinal fluid of patients with mild Alzheimer's disease. Neurobiol Aging. (2015) 36:81–9. doi: 10.1016/j.neurobiolaging.2014.08.008
123. Gate D, Saligrama N, Leventhal O, Yang AC, Unger MS, Middeldorp J, et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer's disease. Nature. (2020) 577:399–404. doi: 10.1038/s41586-019-1895-7
124. Unger MS Li E, Scharnagl L, Poupardin R, Altendorfer B, Mrowetz H, et al. CD8+ T-cells infiltrate Alzheimer's disease brains and regulate neuronal- and synapse-related gene expression in APP-PS1 transgenic mice. Brain Behav Immun. (2020) 89:67–86. doi: 10.1016/j.bbi.2020.05.070
125. Fu J, Duan J, Mo J, Xiao H, Huang Y, Chen W, et al. Mild cognitive impairment patients have higher regulatory T-cell proportions compared with Alzheimer's disease-related dementia patients. Front Aging Neurosci. (2021) 12:624304. doi: 10.3389/fnagi.2020.624304
126. Faridar A, Thome AD, Zhao W, Thonhoff JR, Beers DR, Pascual B, et al. Restoring regulatory T-cell dysfunction in Alzheimer's disease through ex vivo expansion. Brain Commun. (2020) 2:fcaa112. doi: 10.1093/braincomms/fcaa112
127. Baek H, Ye M, Kang G-H, Lee C, Lee G, Choi DB, et al. Neuroprotective effects of CD4+CD25+Foxp3+ regulatory T cells in a 3xTg-AD Alzheimer's disease model. Oncotarget. (2016) 7:69347–57. doi: 10.18632/oncotarget.12469
128. Baruch K, Rosenzweig N, Kertser A, Deczkowska A, Sharif AM, Spinrad A, et al. Breaking immune tolerance by targeting Foxp3+ regulatory T cells mitigates Alzheimer's disease pathology. Nat Commun. (2015) 6:7967. doi: 10.1038/ncomms8967
129. Chakrabarty P, Li A, Ceballos-Diaz C, Eddy James A, Funk Cory C, Moore B, et al. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron. (2015) 85:519–33. doi: 10.1016/j.neuron.2014.11.020
130. Ketelhuth DF, Hansson GK. Adaptive response of T and B cells in atherosclerosis. Circ Res. (2016) 118:668–78. doi: 10.1161/CIRCRESAHA.115.306427
131. Gupta S, Pablo AM, Jiang X, Wang N, Tall AR, Schindler C. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J Clin Invest. (1997) 99:2752–61. doi: 10.1172/JCI119465
132. Whitman SC, Ravisankar P, Elam H, Daugherty A. Exogenous interferon-gamma enhances atherosclerosis in apolipoprotein E-/- mice. Am J Pathol. (2000) 157:1819–24. doi: 10.1016/S0002-9440(10)64820-1
133. Buono C, Binder CJ, Stavrakis G, Witztum JL, Glimcher LH, Lichtman AH. T-bet deficiency reduces atherosclerosis and alters plaque antigen-specific immune responses. Proc Natl Acad Sci USA. (2005) 102:1596–601. doi: 10.1073/pnas.0409015102
134. Gorlé N, Vandenbroucke RE. Interferons: a molecular switch between damage and repair in ageing and Alzheimer's disease. Mech Ageing Dev. (2019) 183:111148. doi: 10.1016/j.mad.2019.111148
135. Butcher M, Galkina E. Current views on the functions of interleukin-17A-producing cells in atherosclerosis. Thromb Haemost. (2011) 106:787–95. doi: 10.1160/TH11-05-0342
136. Griffin GK, Newton G, Tarrio ML, Bu D-x, Maganto-Garcia E, Azcutia V, et al. IL-17 and TNF-α sustain neutrophil recruitment during inflammation through synergistic effects on endothelial activation. J Immunol. (2012) 188:6287–99. doi: 10.4049/jimmunol.1200385
137. Schäfer S, Zernecke A. CD8(+) T cells in atherosclerosis. Cells. (2020) 10:37. doi: 10.3390/cells10010037
138. Toubal A, Nel I, Lotersztajn S, Lehuen A. Mucosal-associated invariant T cells and disease. Nat Rev Immunol. (2019) 19:643–57. doi: 10.1038/s41577-019-0191-y
139. Touch S, Assmann KE, Aron-Wisnewsky J, Marquet F, Rouault C, Fradet M, et al. Mucosal-associated invariant T (MAIT) cells are depleted and prone to apoptosis in cardiometabolic disorders. FASEB J. (2018) 32:fj201800052RR. doi: 10.1096/fj.201800052RR
140. Priya R, Brutkiewicz RR. Brain astrocytes and microglia express functional MR1 molecules that present microbial antigens to mucosal-associated invariant T (MAIT) cells. J Neuroimmunol. (2020) 349:577428. doi: 10.1016/j.jneuroim.2020.577428
141. Loh L, Gherardin NA, Sant S, Grzelak L, Crawford JC, Bird NL, et al. Human mucosal-associated invariant T cells in older individuals display expanded TCRalphabeta clonotypes with potent antimicrobial responses. J Immunol. (2020) 204:1119–33. doi: 10.4049/jimmunol.1900774
142. Meng XF Yu JT, Wang HF, Tan MS, Wang C, Tan CC, et al. Midlife vascular risk factors and the risk of Alzheimer's disease: a systematic review and meta-analysis. J Alzheimers Dis. (2014) 42:1295–310. doi: 10.3233/JAD-140954
143. Soehnlein O, Swirski FK. Hypercholesterolemia links hematopoiesis with atherosclerosis. Trends Endocrinol Metab. (2013) 24:129–36. doi: 10.1016/j.tem.2012.10.008
144. Mailer RKW, Gisterå A, Polyzos KA, Ketelhuth DFJ, Hansson GK. Hypercholesterolemia enhances T cell receptor signaling and increases the regulatory T cell population. Sci Rep. (2017) 7:15655. doi: 10.1038/s41598-017-15546-8
145. Oliver G, Kipnis J, Randolph GJ, Harvey NL. The lymphatic vasculature in the 21(st) century: novel functional roles in homeostasis and disease. Cell. (2020) 182:270–96. doi: 10.1016/j.cell.2020.06.039
146. Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. (2018) 556:332–8. doi: 10.1038/s41586-018-0023-4
147. Cortes-Canteli M, Iadecola C. Alzheimer's disease and vascular aging: JACC focus seminar. J Am Coll Cardiol. (2020) 75:942–51. doi: 10.1016/j.jacc.2019.10.062
148. Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. (2012) 111:245–59. doi: 10.1161/CIRCRESAHA.111.261388
149. Lacolley P, Regnault V, Laurent S. Mechanisms of arterial stiffening: from mechanotransduction to epigenetics. Arterioscler Thromb Vasc Biol. (2020) 40:1055–62. doi: 10.1161/ATVBAHA.119.313129
150. Kurilenko N, Fatkhullina AR, Mazitova A, Koltsova EK. Act locally, act globally-microbiota, barriers, and cytokines in atherosclerosis. Cells. (2021) 10:348. doi: 10.3390/cells10020348
151. Zhu S, Jiang Y, Xu K, Cui M, Ye W, Zhao G, et al. The progress of gut microbiome research related to brain disorders. J Neuroinflammation. (2020) 17:25. doi: 10.1186/s12974-020-1705-z
Keywords: atherosclerosis, Alzheimer's disease, immune response, inflammation, aging
Citation: Stahr N and Galkina EV (2022) Immune Response at the Crossroads of Atherosclerosis and Alzheimer's Disease. Front. Cardiovasc. Med. 9:870144. doi: 10.3389/fcvm.2022.870144
Received: 06 February 2022; Accepted: 02 June 2022;
Published: 06 July 2022.
Edited by:
Gabrielle Fredman, Albany Medical College, United StatesReviewed by:
Alexander Akhmedov, University of Zurich, SwitzerlandEsther Lutgens, Academic Medical Center, Netherlands
Carmela Rita Balistreri, University of Palermo, Italy
Copyright © 2022 Stahr and Galkina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena V. Galkina, Z2Fsa2luZXZAZXZtcy5lZHU=