Abstract
Drug-induced cardiotoxicity (DICT) is an important concern of drug safety in both drug development and clinical application. The clinical manifestations of DICT include cardiomyopathy, arrhythmia, myocardial ischemia, heart failure, and a series of cardiac structural and functional changes. The occurrence of DICT has negative impacts on the life quality of the patients, brings additional social and economic burden. It is important to identify the potential factors and explore the mechanisms of DICT. Traditional cardiovascular risk factors can only partially explain the risk of DICT. Pharmacogenomic studies show accumulated evidence of genetics in DICT and suggest the potential to guide precision therapy to reduce risk of cardiotoxicity. The comprehensive application of technologies such as third-generation sequencing, human induced pluripotent stem (iPS) cells and genome editing has promoted the in-depth understanding of the functional role of susceptible genes in DICT. This paper reviewed drugs that cause DICT, the clinical manifestations and laboratory tests, as well as the related content of genetic variations associated with the risk of DICT, and further discussed the implication of new technologies in pharmacogenomics of DICT.
Introduction
Drug induced cardiotoxicity (DICT) is a serious adverse drug reaction, which interferes with the normal physiological function of the cardiovascular system. The clinical manifestations of DICT are diverse and mainly include cardiomyopathy, arrhythmia, valve injury, myocarditis, pericarditis, cardiac insufficiency, and myocardial ischemia (1). Drug-induced prolongation of the QT interval can even lead to fatal severe ventricular tachycardia and sudden death (2). Cardiotoxicity has become an important issue in drug development and public health. Although the safety evaluations of all listed drugs have been obtained in clinical trials, cardiotoxicity is still very common in clinical practice. Some cardiac injuries, such as acquired long QT syndrome (aLQTS), are the causes of relabeling and drug withdrawal. In recent years, cardiovascular complications of cancer therapeutics have even given rise to the new and unique interdisciplinary field of cardio-oncology (2).
The cardiotoxicity of drug is multifactorial. Traditional cardiovascular risk factors, such as gender, age, renal failure, iron overload, drug-drug interactions, and pre-existing cardiovascular diseases, can not fully explain the occurrence of DICT (3). Routine clinical monitoring indicators usually lack the specificity for the diagnosis of DICT. More and more evidence supports the importance of genetic components, which make some individuals more susceptible to DICT. Many risk genes have been identified, and some have been translated into clinic to optimize drug regimens (4). The integration of new technologies in life science and pharmacogenomics, a field focuses on exploring the genetic basis of interindividual difference in drug responses, has paved the way for the discovery and functional analysis of genetic biomarkers associated with risk of DICT.
Drugs with cardiotoxicity and the clinical evaluation
Cardiotoxic drugs and manifestations
Cardiotoxicity occurs in therapies with many of the drugs including the antineoplastic drugs (such as cancer chemotherapeutics, targeted therapies, cancer immunotherapies), anti-infective drugs, antiarrhythmics, and other non-cardiac drugs (such as antihistamines, bronchodilating, the lipid regulating agent, etc.) (Table 1). The manifestations are varied, which include arrhythmia (sinus bradycardia, atrial fibrillation, atrial flutter, ventricular arrhythmia, QT interval prolongation, even torsades de pointes ventricular tachycardia), cardiomyopathy, myocarditis, myocardial ischemia/myocardial infarction, heart dysfunction/heart failure, cardiogenic shock, and even sudden death (Table 1).
Table 1
| Types | Common drugs | Cardiotoxic manifestations |
|---|---|---|
| Antineoplastic drugs (1, 5) | Anthracyclines: adriamycin, aunorubicin, epirubicin | Arrhythmia, cardiomyopathy, heart failure |
| Alkylating agent: cyclophosphamide | Hemorrhagic necrotizing pericardial myocarditis, heart failure, arrhythmia | |
| Anti microtubule: paclitaxel | Myocardial ischemia, sinus bradycardia, heart failure | |
| Antimetabolic drugs: 5-fluorouracil and capecitabine | Coronary spasm, heart failure | |
| Monoclonal antibody drug: trastuzumab, bevacizumab | Heart failure | |
| Small molecule protein kinase inhibitors: imatinib, sunitinib | Atrial fibrillation, heart failure | |
| Proteasome inhibitor: kafezomib | Heart failure | |
| Immunosuppressants: PD-1/PD-1L inhibitors | Myocarditis | |
| Anti-infection drugs (6–8) | Macrolides: clarithromycin, azithromycin | Torsade de pointe, QT interval prolongation |
| β Lactams: penicillin | Arrhythmia, myocarditis, heart failure | |
| Lincomrades: lincomycin and clindamycin | QT interval prolongation, ventricular tachycardia | |
| Quinolone: ciprofloxacin, levofloxacin, moxifloxacin, etc. | QT interval prolongation, ventricular tachycardia, occasionally develops to severe arrhythmias such as torsade de pointe | |
| Antifungal: imidazole antifungal agents (Itraconazole) | QT interval prolongation, ventricular tachycardia | |
| Antiparasitic: chloroquine | Heart block, congestive heart failure, cardiomyopathy | |
| Antiviral: α-Interferon (IFN-α) | Myocarditis, atrioventricular block, bradycardia | |
| Antiarrhythmic drugs (9, 10) | Amiodarone | QT interval prolongation |
| Digitalis | Atrioventricular block, ventricular arrhythmia | |
| Antihistamines (9, 10) | Benamin, cetirizine, loratadine, desloratadine, levocetirizine, | Q-T interval prolongation, palpitations, arrhythmias, sinus bradycardia, supraventricular tachycardia, ventricular tachycardia, torsade de pointe, atrial fibrillation, etc. |
| Lipid lowering drugs (11) | Probucol | QT interval prolongation, ventricular tachycardia |
| Psychotropic drugs (12) | Thiazide antipsychotics, tricyclic antidepressants | Arrhythmia |
| Gastrointestinal motility promoting drugs (13) | Domperidone, cisapride | Q-T interval prolongation and arrhythmia |
| Bronchodilators (14) | Salbutamol | Arrhythmia (atrial fibrillation, sinus tachycardia, etc.) |
Drugs that induced cardiotoxicity and the manifestations.
Indicators for clinical evaluation of cardiac toxicity
DICT is usually comprehensively evaluated by medication history, clinical manifestations, electrocardiogram (ECG), cardiac imaging, laboratory tests for cardiac biomarkers, and pathological examination with endomyocardial biopsy. Consensus definition of cardiac toxicities of cancer therapies has recently been coined by International Cardio-Oncology Society (IC-OS) (1). The cardiotoxicity of most drugs is cumulative, especially in high doses. Of course, cumulative use of low doses can also cause abnormal cardiac function on some occasions. For example, anthracyclines can cause cardiotoxicity at low-dose. During long-term follow-up, cardiac dysfunction was observed in patients received low-dose adriamycin, indicating “no safe” dose for anthracyclines (5, 6). Medication history with potential cardiotoxic drugs is an essential prerequisite for the diagnosis. Symptoms such as chest tightness, palpitation, exertional dyspnea, and in severe cases, upright breathing and syncope may occur (7). Physical examination may show signs of cardiac enlargement, tachycardia, galloping rhythm of the third heart sound, cardiac murmur, etc. ECG and echocardiography are routine non-invasive examinations for clinical monitoring of cardiac structural and/or function changes. The recovery of ECG, especially for QT intervals after drug withdrawal, is helpful for the diagnosis. Left ventricular ejection fraction (LVEF) is a commonly used cardiotoxicity monitoring index, but is insensitive and lacks specificity for early changes in systolic function. Noteworthy, a decrease in LVEF usually indicates more severe myocardial injury.
Serum cardiac biomarkers are also used in the diagnosis of DICT. For example, high sensitive troponin I (HS TnI) can be used to predict early cardiac injury. Troponin is a sensitive marker for detecting anthracycline-induced myocardial damage. Troponin I may be elevated in patients with myositis (8). The increase of Troponin above the 99th percentile limit supports the diagnosis of cardiac injury. B-type natriuretic peptide (BNP) and N-terminal pro-B-type natriuretic peptide (NT proBNP) are also commonly used to establish the diagnosis of heart failure (8). It is reported that serum NT proBNP correlated positively with cardiotoxicity, but the pre-treatment levels should be compared to confirm a drug-specific effect.
Endomyocardial biopsies provides direct histological evidence of cardiac injury and is the most sensitive to cardiotoxicity. However, due to its high risk and great trauma, it cannot be used as a routine examination for DICT. The pathological manifestations of DICT are complex, which requires comprehensive evaluation according to the drug used and clinical manifestations. It is worth noting that though many of the above mentioned clinical indicators are used, most of them lack specificity for drug toxicity.
Pharmacogenomics in DICT
There are significant individual differences in susceptibility to DICT. Genetics can partly account for this difference. Pharmacogenetic studies have shown that genetic variations represented by single nucleotide polymorphisms (SNPs) in drug disposition and response related genes can modify the risk of DICT through both pharmacokinetics (PK) and pharmacodynamics mechanisms [(9, 10, 15), Figure 1, Supplementary Table 1]. In recent decades, genetic polymorphisms of genes encoding drug transporters, drug metabolizing enzymes and drug targets have been extensively studies. Better understanding of the pharmacogenomics of DICT will help optimize the current treatment selection and dosing regimens, and minimize risk of DICT as well.
Figure 1
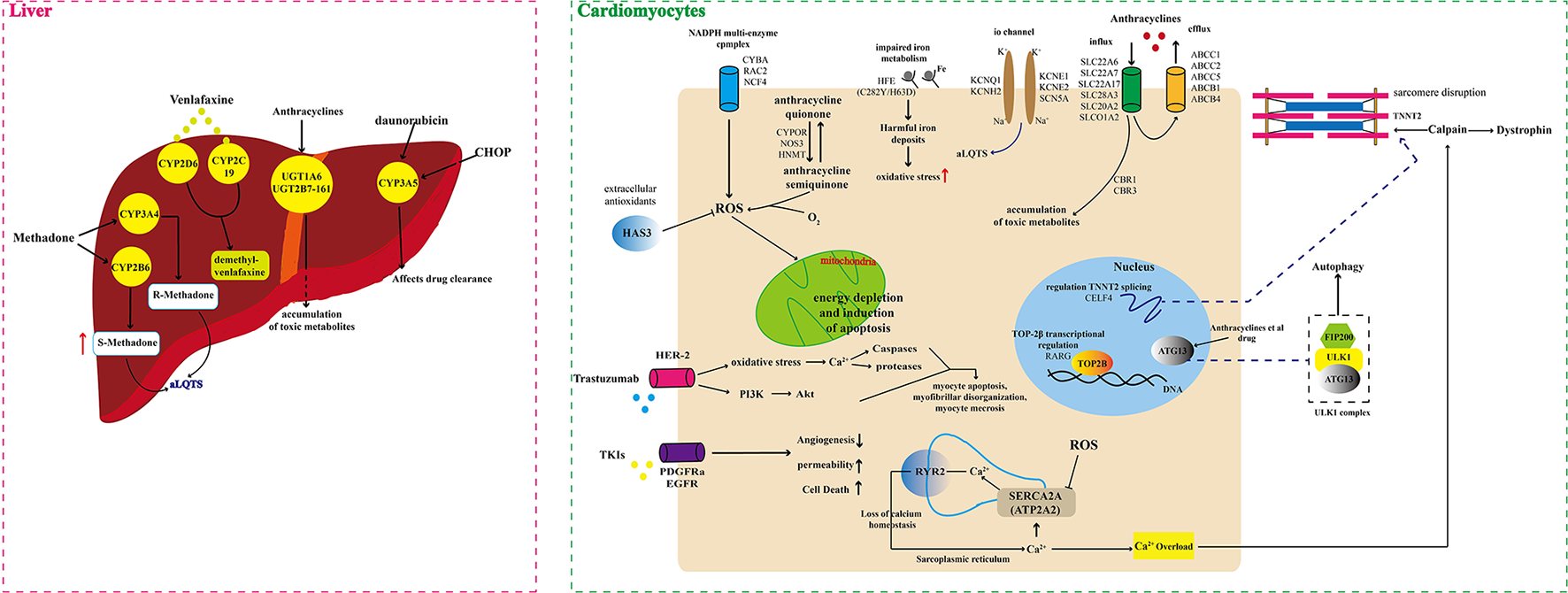
Schematic illustration of multiple genomic mechanisms of drug cardiotoxicity.
Pharmacokinetics (PK) gene polymorphisms
ATP-binding cassette (ABC) family transporters
The ABC family drug transporters play key roles in the transmembrane efflux of many cardiotoxic drugs, such as anthracyclines. For anthracyclines, association of genetic polymorphisms in ABC family members and risk of drug resistance or cardiovascular toxicity have been widely studied (16). Variations in family members including ABCC1 (rs246221, rs4148350, rs45511401), ABCC2 (rs8187710, rs8187694, rs3740066), ABCB4 (rs1149222 and rs4148808), and ABCC5 rs7627754 are associated with increased risk of persistent anthracycline cardiotoxicity in adults and children suffered from hematology and malignant tumors (17–24). SNP variants in these ABC genes that reduce or interfere with expression will lead to the accumulation of detrimental metabolites of anthracyclines in cardiomyocytes, thereby increasing the risk of DICT. ABCB1 (also known as P-glycoprotein) is a component of the heart endothelial blood barrier. Sissung et al. found that the ABCB1 SNPs 1236C>T (rs1128503), 2677G>T/A (rs2032582), and 3435C>T (rs1045642) that alter protein folding can reduce intracardiac concentration of the ABCB1 model substrate romidepsin and drug-induced QT interval prolongation as well, which indicates cardioprotective of these SNPs toward ABCB1 substrates (25).
Transporters of the soluble carrier family (SLCs)
SLCs are the second largest membrane proteins family in human and play important roles in the absorption, distribution and excretion of drugs. Functional genetic variations in SLCs genes are also identified. In a study with 5–10-year follow-up in patients taking anthracyclines, the SLC22A6 rs6591722 AA genotype showed increased risk of decreased left ventricular cardiac function (21). SLC28A3 rs7853758 and rs4877847, SLC10A2 rs9514091, SLC22A7 rs4149178, SLCO1A2 rs2857468, and SLC22A17 rs4982753 were reported to be protective for anthracycline induced cardiotoxicity (22, 23, 26). These variants that lead to reduced gene expression can reduce cellular uptake of anthracyclines, reduce the production of harmful metabolites in cells, and protect cardiomyocytes. SLC28A3 plays a role in the influx of anthracyclines into cancer cells. The SLC28A3 rs7853758 (G>A, Leu461Leu) polymorphism A allele can decrease its mRNA expression and is protective for anthracycline induced cardiotoxicity (21). Pharmacogenetic test for SLC28A3 rs7853758 is suggested before treatment with anthracyclines in pediatric cancer patients (21). By using nanopore-based fine-mapping and base editing technologies, Magdy et al. identified the SNP rs11140490 at the SLC28A3 locus was cardioprotective by regulating the expression of an antisense long non-coding RNA (SLC28A3AS1) that overlaps with SLC28A3 (26).
Cytochrome P450 family of enzymes
Cytochrome P450 enzymes have the highest content in human liver and are responsible for the oxidative metabolism of 50% of clinical drugs. The genetic polymorphisms of CYP450 family members lead to huge individual differences in enzyme activities and drug metabolism, which eventually leads to adverse drug reactions (ADR) or unsatisfactory therapeutic efficacy. In past two decades, many of the interests have been focused on CYP450 genetic polymorphisms and individualized medicine.
Methadone is a racemic mixture of R- and S-methadone, which is considered to be the best antidote. CYP3A4 and CYP2B6 are the major CYP enzymes responsible for the metabolism of R- and S-methadone, respectively (26). High plasma concentration of S-methadone can lead to cardiotoxicity by prolonging the QT interval. A study of 125 death in Caucasians showed that CYP2B6*9 (rs3745274, c516G>T), CYP2B6*5 (rs3211371, c1459C >T), and CYP2B6 rs8192719 (21563 C>T) were associated with the risk of fatal methadone cardiotoxicity (27). In order to identify genetic polymorphisms potentially associated with the risk of acquired long QT syndrome (aLQTS) in 153 cases with 216 QT-prolonging culprit drugs, Gray et al. recently observed that 22.2% and 7.8% of patients bearing rare variations in the LQTS genes and the CYP genes, respectively (28). Rare variant association studies indicated significantly higher burden of rare non-synonymous variants in CYP genes in the aLQTS cases. CYP2B6 c.499C>G, c.1172T>A, c.415A>G, c.445G>A, and CYP3A4 c.1000G>T might lead to increased risk for methadone-induced aLQTS (28). Some aLQTS cases can be explained by drug interactions in metabolism or pharmacodynamic synergy.
Venlafaxine (VEN) is a serotonin-norepinephrine-dopamine reuptake inhibitor. Excessive intake of VEN usually leads to mild cardiotoxicity. VEN is mainly metabolized by CYP2D6 and CYP2C19 to form demethylvenlafaxine (ODV). Patients with lower CYP2D6 or CYP2C19 activity are more likely to suffer from ADR events and have increased risk of developing cardiotoxicity during VEN treatment (29, 30). In patients developed VEN-related cardiotoxicity, CYP2D6 poor metabolizer genotype *4/*9 and CYP2C19 intermediate metabolizer genotype *1/*2 are observed to show higher serum VEN levels (31).
CYP3A5 is involved in the clearance of daunorubicin (DNR) in vivo. CYP3A5*3 is a common loss of function allele that results in the loss of CYP3A5 expression in adult liver. In children with acute lymphoblastic leukemia, an increase in area under the curve (AUC) for DNR plasma concentration was observed in CYP3A5*3/*3 homozygotes (32). The CYP3A5*3 polymorphism is also associated with increased risk of cardiotoxicity in patients with diffuse large B-cell lymphoma adopted combined therapy with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) (33). In addition, the CYP3A5 rs4646450 polymorphism was observed to be a risk factor for doxorubicin induced cardiotoxicity, especially in males (21).
Carbonyl reductases (CBRs)
The CBRs oxido-reductase enzymes CBR1 and CBR3 are involved in the reduction of anthracyclines to the cardiotoxic ethanol metabolites, which play key roles in the induction of cardiovascular toxicity by anthracyclines. Genetic polymorphisms in CBR1 and CBR3 genes are reported to affect the production of the ethanol metabolites. Cancer patients with Down syndrome (DS) are prone to anthracyclines related cardiotoxicity, which could be explained by increased expression of CBR1 and increased metabolism of daunorubicin to the ethanol metabolites in the heart (34). CBR1 1096G>A (rs9024) is a 3′-UTR SNP that interferes with the inhibitory effects of hsa-miR-574-5p and hsa-miR-921 on its mRNA expression. The mutant 1096A allele was initially observed to increase the expression and activity of CBR1 (35). However, following studies with both liver cytosols and lymphoblastoid cell lines indicated that the CBR1 1096 G/A genotype showed a lower maximum rate of doxorubicinol synthesis than the GG genotype in the Whites (35). In support, a DS patient with the trisomic for the rs9024 A allele (A/A/A) exhibited low CBR1 enzymatic activity (34). In addition, among child cancer survivors receiving anthracyclines, the blacks showed relatively higher incidence of cardiotoxicity and lower frequency of the CBR1 rs9024 A allele than the whites. CBR1 rs9024 polymorphism may thus explain racial difference in susceptibility to anthracyclines induced cardiotoxicity (36). These findings suggest protective role of CBR1 rs9024 A variant to anthracyclines induced cardiotoxicity. However, there are conflicting reports. For example, the rs9024 AA genotype is associated with increased risk of cardiotoxicity in children with acute lymphoblastic leukemia treated with UKALL 2003 protocol, and higher systemic doxorubicin exposure in this genotype is assumed (37, 38). Therefore, further studies are required to validate these associations.
CBR3 Val244Met (rs1056892) polymorphism can also affect the risk of anthracyclines induced cardiomyopathy in child cancer survivors and adult breast cancer patients (24). In child cancer survivors, CBR3 Val244Met was dose-dependently associated with the risk of anthracyclines induced cardiomyopathy (39). Compared with CBR3 Met244 carriers, rs1056892 GG (Val244) homozygotes showed 3.3-fold increased risk of cardiomyopathy in adults breast cancer patients when treated with low-dose anthracyclines (<250 mg/m2). Functional study showed that the Val244 (rs1056892 G allele) catalyzed the synthesis of the cardiotoxic doxorubicinol with a rate 2.6-fold higher than the Met24 (40). However, in patients receiving high-dose anthracyclines (≥250 mg/m2), no obvious association between the SNP and cardiotoxicity was observed (41). In a prospective single arm observational pharmacogenetic study with 155 breast cancer patients receiving doxorubicin, carriers of the CBR3 Val244 allele showed significant reduction in LVEF at 6 months following initiation of doxorubicin, and Val244 homozygotes showed a further reduction (42). The CBR3 Val244Met polymorphism was also associated with cardiotoxicity in breast cancer patients treated with trastuzumab (17).
Other drug metabolism genes
Some other drug metabolism genes can also influence the risk of DICT. For example, Uridine diphosphate-glucuronosyltransferase 1A6 (UGT1A6) rs6759892, UGT2B7-161 rs7668258, Histamine N methyltransferase (HNMT) rs17583889, P450 oxidoreductase (POR) rs13240755 have been reported to be associated with increased risk of persistent cardiotoxicity after anthracycline therapy (22, 43, 44). In epidermal growth factor 2 (HER-2)-positive breast cancer patients treated with trastuzumab, the incidence of myocardial injury was reduced in carriers of the UGT2B7-161 (rs7668258) T allele (45). This finding suggests UGT2B7-161 rs7668258 a potential predictor of cardiotoxicity in patients treated with trastuzumab therapy.
Pharmacodynamics (PD) gene polymorphisms
Human epidermal growth factor receptor type 2 (HER-2)
HER-2 is an important target of cancer targeted therapy. About 25–30% of breast cancer patients show HER-2 overexpression or HER-2 gene amplification. The preferred treatment regimen for HER-2-positive breast cancer is based on trastuzumab and anthracycline/cyclophosphamide, which significantly improves the overall survival rate. However, at least 10–15% of the patients experienced anthracycline-induced cardiotoxicity, and 20–33% of patients also suffered from trastuzumab-induced cardiotoxicity (46). A genome wide association study (GWAS) carried out in 481 patients with (11 cases) and without (257 controls) trastuzumab-induced cardiotoxicity in Japanese showed that five SNPs including rs9316695, rs28415722, rs7406710, rs11932853, and rs8032978 were independent predictors of trastuzumab cardiotoxicity (Pcombined = 7.82 × 10−15, OR = 40.0) (47). The HER-2 SNPs Ile654Val (rs1801201), Ile655/Val (rs1136201), and Pro1170Ala (rs1058808) were also associated with susceptibility to cardiotoxicity (48). In a meta-analysis of 344 patients with 43 developed drug induced cardiotoxicity, 67% of the patients carried the Ile/Val genotype, resulting in an OR of 5.35. HER-2 Ile655Val is an independent predictor of cancer-therapy related cardiotoxicity (24, 48). By comparing HER-2 genotype distribution in 29 cases with cardiotoxicity and 111 controls underwent trastuzumab treatment, Stanton et al., observed association between Pro1170Ala polymorphism and increased risk of trastuzumab induced cardiomyopathy (49). The frequency of HER-2 Pro/Pro genotype in cases with cardiotoxicity (10/29, 34.5%) was higher than the controls (19/111, 17.1%) (49).
HER-2 SNPs inhibit HER4/HER4 homodimerization or HER4/HER2 heterodimerization through the HER-2 gene, thereby inhibiting a series of downstream signaling pathways, including PI3K-Akt. Blockade of the PI3K-Akt pathway will lead to the accumulation of ROS in cardiomyocytes, thereby triggering cardiomyocyte apoptosis (50). In addition, blockade of HER-2 signaling induces oxidative stress, leading to NO production and impairment of mitochondrial function, ultimately leading to myocyte apoptosis, myofibrillar disorder, and myocyte necrosis (50).
Tyrosine kinase receptor gene polymorphisms
Tyrosine kinases inhibitors (TKIs) are competitive inhibitors of the enzymes by binding to the adenosine triphosphate (ATP) binding pocket of the enzymes. TKIs are multi-target anticancer drugs with low specificity. Cardiovascular toxicity is one of the common ADR of TKIs. Using several publicly available datasets including drug-gene interaction database and GWAS database of heart failure, Li et al. found a group of overlap genes induced by TKIs and affect HF susceptibility (51). Comprehensive integrated analysis indicated that several SNPs potentially affect RNA binding protein-mediated regulation have the potential to affect cardiotoxicity of TKIs, among which the PDGFRα rs191188930 and EGFR rs142136033 have the potential to affect cardiotoxicity of multiple drugs including sunitinib, pazopanib, sorafenib, dasatinib and nilotinib (51). Of course, these findings require verification in clinic, and functional analysis of the suggested SNPs are also needed. The main mechanisms of TKIs-induced cardiotoxicity include inhibition of VEGF and PDGFR coronary microvascular dysfunction (52), through up-regulation of cardioprotective insulin and insulin-like growth factor (IGF) signaling and down-regulation of the phosphorylation of AKT and ERK affecting cardiomyocyte survival pathways, ultimately leading to cardiomyocyte death (53).
Others
In addition to genetic variants in PK and PD genes, some other genes that may modify risk of DICT have also been explored, such as genes related to regulation of oxidative stress, iron metabolism, autophagy, and myocardial sarcomere structure.
Oxidative stress related genes
Reactive oxygen species (ROS) produced by oxidative stress act as links between underlying cardiovascular disease and drug induced cardiotoxicity. NADPH oxidase (NOX) is the main endogenous source of ROS and a key mediator of cardiac oxidative damage. On the contrary, Hyaluronan synthase 3 (HAS3) is an enzyme that produces low molecular weight hyaluronic acid, which has antioxidant activity and protects the heart by reducing ROS-mediated cardiac damage (54). Several studies have focused on association of genetic variations in genes encoding enzymes involved ROS formation or clearance and risk of DICT.
NOX consists of five subunits, including two membrane-bound subunits (p22phox and gp91phox), three cytoplasmic subunits (p67phox, p47phox, p40phox), and a small G-protein Rac (52). The four NOX subunits are encoded by different autosomal genes: CYBA for p22phox, NCF1 for p47phox, NCF2 for p67phox, and NCF4 for p40phox. Ras-related C3 Botulinum Toxin Substrate 2 gene (RAC2) is a small cytoplasmic GTPase that is required for NOX activation and regulation of ROS production (55, 56). Common polymorphisms in NOX subunit genes, such as CYBA rs4673, NCF4 rs1883112, and RAC2 rs13058338, are identified (57). Kopeva et al. divided 176 breast cancer patients who received anthracycline chemotherapy for 12 months into two groups: the anthracycline-induced cardiotoxicity (AIC) group (52 cases) and non-AIC group (124 cases), and observed that the CYBA rs4673 polymorphism was a risk factor for the occurrence of AIC (58). Alteration in RAC2 can also lead to mitochondrial dysfunction and increased ROS production, and ultimately lead to cardiomyocyte damage (59). In a study aimed at identification of key genes affecting the risk of anthracycline-related congestive heart failure (CHF) in long-term survivors after haematopoietic cell transplantation (HCT), Armenia et al. observed that the odds of developing CHF after HCT was increased nearly 3 times in patients with the RAC2 rs13058338 (7508 T>A) variants (4). Another study also supported association of RAC2 rs13058338 variant with AIC in AML patients (44, 57).
By analyzing 2100 SNPs in genes associated with de novo cardiovascular disease in individuals exposed to high-dose anthracyclines (>250 mg/m2), Wang et al. found that the HAS3 rs2232228 AA genotype was associated with a 8.9-fold increased risk of cardiomyopathy compared with the rs2232228 GG genotype (60). In addition, HAS3 mRNA expression in heart samples of patients with the rs2232228 AA genotype was significantly lower than that of the GA heterozyotes (60). It is assumed that the HAS3 AA genotype may increase the sensitivity of cardiomyocytes to ROS in the presence of high-dose anthracycline, and thereby increases the risk of AIC (60).
Iron homeostasis genes
Iron homeostasis is important for maintaining normal cardiac function. Iron-overload can lead to cardiomyopathy and heart failure. The HFe (high iron) gene on chromosome 6p encodes a protein that regulates iron transport and metabolism. HFE binds to transferrin receptors on the cell surface and promotes the uptake of transferrin bound iron. During anthracycline therapy, individuals with high HFE gene mutations may cause harmful iron deposition in the heart, causing more serious damage to cardiomyocytes (19). C282Y (rs1800562) and H63D (rs1799945) are two main functional SNPs of HFE. The rs1800562 polymorphism results in the substitution of tyrosine to cysteine at position 282 (C282Y), and the rs1799945 is a substitution of aspartate to histidine at position 63 (H63D) (61). A prospective association study of genetic mutations with anthracyclines induced cardiotoxicity in 184 child leukemia survivors observed positive associations for C282Y and H63D, with the H63D rs1799945 shows more prominent heart damage and cardiotoxicity (62).
Cardiac ion channel genes
KCNE1 encodes the β-auxiliary subunit of the voltage-gated slow cardiac potassium IKs current, whose dysfunction leads to cardiac arrhythmia. In a study of 153 aLQTS patients to explore possible rare variations related to TdP, four cases were observed to bear the KCNE1-c.253G>A (rs1805128) variant that is associated with increased risk of drug-induced TdP (28). Other variants in iron channel genes, such as KCNE1-c.253G>A, KCNE2 c.22A>G, SCN5A (c.1715C>A, c.569G>A), KCNQ1 (c.733G>A, c.727C>T), KCNH2-c.3163C>T are also found to induce aLQTS by causing QT prolongation (28).
Other pathways involved in myocardial function
Factors that affect myocardial sarcomere structure or transcriptional regulation may also modify risk of anthracycline induced cardiotoxicity. The cardiac sarcomere protein troponin T2 (TNNT2) is regulated by mRNA splicing, and different isoforms of TNNT2 (the fetal isoform, the adult cTnT3 isoform, for example) have different Ca2+ sensitivity (63, 64). Evidence shows that over-expression of Dual Specificity Tyrosine Phosphorylation Regulated Kinase 1A(DYRK1A) ameliorates the impact of daunorubicin on beating frequency in cardiomyocytes via increasing phosphorylation of the splicing factor Serine/arginine-rich splicing factor 6 (SRSF6), the latter plays a role in TNNT2 mRNA splicing (65). CUGBP ELAV-like family member 4 (CELF4) is a mRNA binding protein that is also involved in regulating TNNT2 mRNA splicing. The gene TNNT2 also encodes cardiac troponin T (cTnT), an established biomarker of myocardial injury in the serum. cTnT is also important in Ca2+ signaling in the myocardium. GWAS in 430 childrens with (162 cases) and without (268 controls) cardiomyopathy after anthracycline therapy found that CELF4 rs1786814 polymorphism was associated with risk of cardiomyopathy. In children exposed to >300 mg/m2 of anthracyclines, the rs1786814 GG homozygotes showed a 10.2-fold increased risk of cardiomyopathy as compared with the G/A or A/A genotypes, while carriers of the CELF4 rs1786814 A allele showed no change in risk of cardiomyopathy regardless of cumulative anthracycline exposure (66). This indicates that CELF4 rs1786814 GG genotype is a risk factor of cardiovascular toxicity of anthracycline therapy (66).
The rs2229774 polymorphismin retinoic acid receptor gamma (RARG) gene (S427L) was associated with increased anthracycline cardiotoxicity with an OR of 4.7 (95% CI: 2.7–8.3) (67). In induced pluripotent stem cell-cardiomyocytes (iPSC-CMs), RARG rs2229774 variant was observed to increase double-strand DNA breaks, ROS production, and cell death, thereby increased susceptibility of the cells to doxorubicin-induced cardiotoxicity (68). Subsequent studies further revealed that RARG may increase DICT susceptibility by reducing mitochondrial numbers and attenuating DNA repair (69). Magdy et al. also observed that the RARG variation can function through disruption of RARG mediated inhibition on topoisomerase 2β (TOP2B) expression and activation of the extracellular regulated kinase (ERK) signaling upon doxorubicin treatment, emphasizing multiple pathways and mechanisms for the protective role of RARG in DICT (15).
Autophagy imbalance is also involved in the mechanism of DICT. To explore whether genetic polymorphisms in autophagy-related genes are associated with risk of DICT, 25 SNPs in genes related to autophagy regulation were genotyped in 147 triple-negative breast cancer (TNBC) patients with relatively complete ECG records during the chemotherapy cycles (70). The results showed that the rs10838611 G allele in autophagy-related 13 (ATG13) was significantly associated with abnormal ECG (OR: 2.258, 95% CI: 1.318–3.869), suggesting an increased risk of cardiac events (70).
New technologies in pharmacogenomics study of DICT
Candidate gene association studies (CGAS) and genome-wide association studies (GWAS) are two main methods in identifying drug response susceptible genes (71, 72). Although these studies have found genetic polymorphisms that may lead to DICT, exploration of the the causal relationship and mechanism between the SNPs and DICT is difficult. Technologies such as human induced pluripotent stem cells (HiPSCs) and genome editing bring opportunities to make the functional analysis of the causative variants more feasible.
Whole genome or whole exon sequencing
In the 1990s, the implication of first-generation sequencing (FGS) technology pushed the completion of the sequencing of the first human genome (73). Subsequently, the rapid development of second-generation sequencing (SGS) promoted studies in genomics (73) as well as mapping of genetic polymorphisms in the human genome (74). Third-generation sequencing (TGS), also known as single-molecule sequencing, was also developed, such as single-molecule real-time (SMRT™) from Pacific Biosciences (PacBio), true single-molecule sequencing (tSMS™) from Helicos, and single-molecule nanopore DNA sequencing from Oxford Nanopore (75). TGS can not only adapt to the reading of longer genomes, identify complex structural changes in DNA samples, and accurately locate the position of sequence changes, but also can recognize DNA/RNA methyltransferase modifications, a successful step toward understanding the biology that occurs between DNA and proteins. Most importantly, TGS sequencing are as accurate as FGS and SGS for assembling complete genomes (76).
TGS has been applied in disease genomic and pharmacogenetics studies. For example, Wang et al. used single-molecule nanopore DNA sequencing to detect the serine/threonine protein kinase gene BRAF V600E mutation in thyroid cancer patient tissues with high sensitivity (77). Magdy et al. used nanopore DNA sequencing to pinpoint the association of GWAS-positive SLC28A3 SNP with doxorubicin-induced cardiotoxicity. The results showed that single-molecule nanopore DNA sequencing can not only provide comprehensive and accurate information on precisely mapped GWAS-positive sites, but also help identify causal SNP/haplotype (78). It can be expected that in the future, TGS technology will be more widely used in the study of DICT.
Other omics-based technologies
In addition to genomics, emerging technologies for biomarker discovery include transcriptomics, metabolomics, proteomics, and gut microbiomics are developed. Application of these new methods can faciliated the identification of both predictive and diagnostic biomarkers for DICT and are prosperous.
RNA sequencing is a kind of high-throughput sequencing that is used to identify differentially expressed genes by detecting samples from different backgrounds (different species, tissues, and periods, etc.), discover potential biomarkers, and reveal the underlying molecular mechanisms for diseases. Combination of single cell RNA-seq (scRNA-seq) and spatial transcriptomics can bring RNA-seq technology into single-cell resolution and tissue-level transcriptomics, providing new insights for disease diagnosis, treatment, and prevention (79–82).
Metabolomics is a collection of small-molecule chemical metabolites that identify biomarkers primarily by methods such as nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry. Both targeted and non-targeted metabolomics have been used to identify circulating metabolites related to drug induced cardiotoxicity. For example, Asnani et al. evaluated metabolite changes in 38 breast cancer female patients treated with anthracyclines and trastuzumab, and found that in patients with cardiotoxicity, citrate levels were reduced, while purine and pyrimidine metabolites were significantly increased, suggesting that metabolomics changes may also contribute to the development of DICT (83).
Proteomics has traditionally been dominated by methods of liquid chromatography-mass spectrometry (LC-MC), which is mainly used to identify and detect diagnostic markers, understand pathogenic mechanisms, and explain functional protein pathways in human diseases (84). Pilot study also used high-throughput proteomic analysis to the identify potential biomarkers associated with doxorubicin and trastuzumab-induced cardiac insufficiency in plasma (85–87). In an cohort of 35 patients treated with doxorubicin and trastuzumab, high baseline immunoglobulin E (lgE) levels was observed to be associated reduced risk of drug-induced cardiac dysfunction (85). These studies suggest that proteomics shed new light on the identification of novel molecular pathways and biomarkers for DICT.
The gut microbiome is another emerging field that attracts much interest in recent years. Liu et al. used 16rRNA gene and metagenomic sequencing to analyze the composition and function of the gut microbiota in mice with doxorubicin-induced cardiotoxicity (88). They observed that depletion of gut microbiota could alleviate adriamycin-induced myocardial injury and cardiomyocyte apoptosis, suggesting important role of gut microbiota in the pathogenesis of adriamycin-induced cardiotoxicity (88). It is also reported that intestinal flora butyric acid (BUT) derivative phenylalanine butyramide (FBA) can prevent anthracyclines induced left ventricular dilation, fibrosis and cardiomyocyte apoptosis in mice (89). FBA reduces anthracyclines induced damage of human cells and is protective against experimental doxorubicin cardiotoxicity by improving mitochondrial function and reducing oxidative stress (89). In conclusion, the gut microbiota may become a new target for the prevention of drug cardiotoxicity and cardiovascular disease.
Human induced pluripotent stem cells (HiPSCs) and 3D HiPSC-derived heart model
As the primary site of DITC, the human cardiac tissue is largely inaccessible and cannot be maintained in tissue culture. HiPSCs are a regenerative cell type that can be obtained by non-invasive manipulations. HiPSCs are also genetically identical to the patients from which they were obtained, which makes manipulating HiPSCs in vitro and comparing them with clinical phenotypes become possible. HiPSC can be used to determine the potential toxicity of drugs, study the mechanism of drug toxicity, verify the determinants of genetic variants in drug toxicity, and provide target information for new drug development. The use HiPSCs is emerging in pharmacogenomics study of DICT in recent years (90).
Patient-specific HiPSC-cardiomyocytes (HiPSC-CMs) develop similar characteristics to the human heart in genomics, transcriptomics, electrophysiology, biochemistry, contraction, and beating. Therefore, HiPSC-CMs have the advantage of reproducing human cardiac tissue in in vitro studies over other models such as animal models, non-human primary cells and immortalized cell lines (91, 92). Burridge and colleagues demonstrated that HiPSC-CMs recapitulate the susceptibility of individuals to doxorubicin-induced cardiotoxicity at the cellular level. They recruited 12 female breast cancer patients who had been treated with doxorubicin or equivalent, with 4 patients without clinical cardiotoxicity, 4 patients with established clinical cardiotoxicity, and 4 age-sex-matched healthy volunteers not received any medication (93). They observed that HiPSC-CMs from patients developed doxorubicin cardiotoxicity were consistently more sensitive to doxorubicin toxicity, suggesting HiPSC-CMs as a suitable cellular model to identify and characterize the genetic basis and molecular mechanisms of doxorubicin cardiotoxicity (93). Using HiPSC-CMs from patients treated with trastuzumab, Kitani et al. identified changes in metabolic pathways to be key important in cardiac dysfunction following trastuzumab treatment (94). The study also supports the use of in vitro HiPSC-CMs assays to investigate drug cardiotoxicity for antibody therapies (94). Non-specific HiPSCs are also used to study the mechanism of drug cardiotoxicity. For example, Sharma et al. using HiPSC-CMs, endothelial HiPSC-ECs, and cardiac fibroblasts HiPSC-CFs to detect the potention of 21 TKIs in inducing cardiotoxicity by high-throughput screening (95). HiPSCs have also been used in the study of cardiovascular toxicity of etoposide (96), arsenic trioxide (97, 98), lapatinib (99), and histone deacetylase inhibitors (100, 101).
Stem cell-derived cardiomyocytes are also used to develop in vitro 3-dimensional (3D) models, namely cardiac micro-tissues and organoids. These models can synergize with genetic engineering to provide tissue-level models for study drug cardiotoxicity. Richards et al. designed an in vitro organotypic disease model of cardiovascular disease based on the principle of tissue engineering (102), and observed that the human cardiac organoids can reproduce drug-induced or aggravate cardiac fibrosis at the tissue level (102). Truitt also used a 3D cardiac microtissue (CMT) model to study the cardiotoxicity of sunitinib. Compared with the 2D model HiPS-CMs, the CMT model is particularly preferable to evaluate the combined effects of drug treatment and afterload (103). Organ-on-a-chip models are micro-microfluidic-controlled 3D organoid models that not only accurately reproduce the physiological parameters of their in vivo counterparts, but also can be linked together by microfluidics in a manner similar to their arrangement in vivo, which make the study of multi-organ interactions possible. Compared with traditional 2D models, multi-organ models can predict human drug responses more accurately. Automated modular design platform based on multi-organ models has also been developed in mimicking the microenvironment in real time and in situ, which provide basis for in-depth study of the mechanism of drug cardiotoxicity (104).
CRIPSR/Cas9 genome editing
To exclude the influence of complex genetic backgrounds, the use of HiPSC-CMs from healthy volunteers or treated patients alone still does not meet the needs of current study. CRIPSR/Cas9 genome editing provides a solution to create HiPSCs genes by introducing targeted mutations in cell lines. The genome editing technology can provide precise control of genome conditions and make HiPSCs a powerful tool for studying drug-induced cardiovascular toxicity (105). Maillet et al. also performed CRISPR/Cas9 genome editing to disrupt the TOP2B gene in HiPSC-CMs to evaluate doxorubicin toxicity on the cells. It was found that disruption of TOP2B reduced the susceptibility of HiPSC-CMs to doxorubicin-induced cell death, and conformed that doxorubicin-induced double-strand DNA breaks (DSB) in HiPSC-CMs was TOP2B-dependent (106).
Expectation
DICT is a common ADR for diversity of drugs, especially in cancer therapies. Current studies have identified some genetic variants associated with risk of DICT through studies based on candidate genes or GWAS. Most variants mentioned above need further replication in different populations and clinical conditions. The availability of high throughput technologies such as whole genome/exon sequencing will facilitate the identification of additional genetic biomarkers potentially affecting DICT risk. Emerging of new models such as patient-derived HiPSC-CMs and genome editing cells or animals makes mechanism study of the variants more approximate to the human myocardium. Of note, many potential pharmacogenomics biomarkers associated with risk of DICT are identified through case-control studies and few are translated into clinic practice in guiding drug therapies or individualized prevention of DICT. More studies, including well designed randomized clinical trials (RCTs), are required to confirm the utility of the genetic variants in future clinic practice. Translation of the pharmacogenomics findings into genotype-guided drug therapy is supposed to maximize drug efficacy and minimize cardiotoxicity for related drugs.
Funding
This project was supported by Scientific Research Program of Hunan Provincial Health Commission of China (No. 202203015026).
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Author contributions
X-PC contributed to conception and design of the study. M-YL wrote the first draft of the manuscript. L-MP wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.966261/full#supplementary-material
References
1.
Herrmann J Lenihan D Armenian S Barac A Blaes A Cardinale D et al . Defining cardiovascular toxicities of cancer therapies: an International Cardio-Oncology Society (IC-OS) consensus statement. Eur Heart J. (2022) 43:280–99. 10.1093/eurheartj/ehab674
2.
Porta-Sánchez A Gilbert C Spears D Amir E Chan J Nanthakumar K et al . Incidence. diagnosis, and management of QT prolongation induced by cancer therapies: a systematic review. J Am Heart Assoc. (2017) 6:e007724. 10.1161/JAHA.117.007724
3.
Osanlou O Pirmohamed M Daly AK . Pharmacogenetics of adverse drug reactions. Adv Pharmacol. (2018) 83:155–90. 10.1016/bs.apha.2018.03.002
4.
Pereira NL Weinshilboum RM . Cardiovascular pharmacogenomics and individualized drug therapy. Nat Rev Cardiol. (2009) 6:632–38. 10.1038/nrcardio.2009.154
5.
Franco VI Lipshultz SE . Cardiac complications in childhood cancer survivors treated with anthracyclines. Cardiol Young. (2015) 25:107–16. 10.1017/S1047951115000906
6.
Salvatorelli E Menna P Chello M Covino E Minotti GJ . Modeling human myocardium exposure to doxorubicin defines the risk of heart failure from low-dose doxorubicin. Pharmacol Exp Ther. (2017) 362:263–70. 10.1124/jpet.117.242388
7.
Mladěnka P Applová L Patočka J Costa VM Remiao F Pourová J et al . Comprehensive review of cardiovascular toxicity of drugs and related agents. Med Res Rev. (2018) 38:1332–403. 10.1002/med.21476
8.
Michel L Mincu RI Mahabadi AA Settelmeier S Al-Rashid F Rassaf T et al . Troponins and brain natriuretic peptides for the prediction of cardiotoxicity in cancer patients: a meta-analysis. Eur J Heart Fail. (2020) 22:350–61. 10.1002/ejhf.1631
9.
Chang VY Wang JJ . Pharmacogenetics of chemotherapy-induced cardiotoxicity. Curr Oncol Rep. (2018) 20:52. 10.1007/s11912-018-0696-8
10.
Castrillon JA Eng C Cheng F . Pharmacogenomics for immunotherapy and immune-related cardiotoxicity. Hum Mol Genet. (2020) 29:R186–96. 10.1093/hmg/ddaa137
11.
Cubeddu LX . Drug-induced inhibition and trafficking disruption of ion channels: pathogenesis of QT abnormalities and drug-induced fatal arrhythmias. Curr Cardiol Rev. (2016) 12:141–54. 10.2174/1573403X12666160301120217
12.
Beach SR Celano CM Noseworthy PA Januzzi JL Huffman JC . QTc prolongation, torsades de pointes, and psychotropic medications. Psychosomatics. (2013) 54:1–13. 10.1016/j.psym.2012.11.001
13.
Yu Z Liu J van Veldhoven JP IJzerman AP Schalij MJ Pijnappels DA et al . Allosteric modulation of Kv11.1 (hERG) channels protects against drug-induced ventricular arrhythmias. Circ Arrhythm Electrophysiol. (2016) 9:e003439. 10.1161/CIRCEP.115.003439
14.
Carroll CL Coro M Cowl A Sala KA Schramm CM . Transient occult cardiotoxicity in children receiving continuous beta-agonist therapy. World J Pediatr. (2014) 10:324–9. 10.1007/s12519-014-0467-z
15.
Magdy T Jiang Z Jouni M Fonoudi H Lyra-Leite D Jung G et al . RARG variant predictive of doxorubicin-induced cardiotoxicity identifies a cardioprotective therapy. Cell Stem Cell. (2021) 28:2076–89.e7. 10.1016/j.stem.2021.08.006
16.
Januchowski R Wojtowicz K Andrzejewska M Zabel M . Expression of MDR1 and MDR3 gene products in paclitaxel-, doxorubicin- and vincristine-resistant cell lines. Biomed Pharmacother. (2014) 68:111–7. 10.1016/j.biopha.2013.09.004
17.
Kim Y Seidman JG Seidman CE . Genetics of cancer therapy-associated cardiotoxicity. J Mol Cell Cardiol. (2022) 167:85–91. 10.1016/j.yjmcc.2022.03.010
18.
Vulsteke C Pfeil AM Maggen C Schwenkglenks M Pettengell R Szucs TD et al . Clinical and genetic risk factors for epirubicin-induced cardiac toxicity in early breast cancer patients. Breast Cancer Res Treat. (2015) 152:67–76. 10.1007/s10549-015-3437-9
19.
Armenian SH Ding Y Mills G Sun C Venkataraman K Wong FL et al . Genetic susceptibility to anthracycline-related congestive heart failure in survivors of haematopoietic cell transplantation. Br J Haematol. (2013) 163:205–13. 10.1111/bjh.12516
20.
Wojnowski L Kulle B Schirmer M Schlüter G Schmidt A Rosenberger A et al . NAD(P)H oxidase and multidrug resistance protein genetic polymorphisms are associated with doxorubicin-induced cardiotoxicity. Circulation. (2005) 112:3754–62. 10.1161/CIRCULATIONAHA.105.576850
21.
Sági JC Egyed B Kelemen A Kutszegi N Hegyi M Gézsi A et al . Possible roles of genetic variations in chemotherapy related cardiotoxicity in pediatric acute lymphoblastic leukemia and osteosarcoma. BMC Cancer. (2018) 18:704. 10.1186/s12885-018-4629-6
22.
Visscher H Ross CJ Rassekh SR Barhdadi A Dubé MP Al-Saloos H et al . Pharmacogenomic prediction of anthracycline-induced cardiotoxicity in children. J Clin Oncol. (2012) 30:1422–8. 10.1200/JCO.2010.34.3467
23.
Magdy T Burmeister BT Burridge PW . Validating the pharmacogenomics of chemotherapy-induced cardiotoxicity: what is missing?. Pharmacol Ther. (2016) 168:113–25. 10.1016/j.pharmthera.2016.09.009
24.
Cartas-Espinel I Telechea-Fernández M Manterola Delgado C Ávila Barrera A Saavedra Cuevas N Riffo-Campos AL . Novel molecular biomarkers of cancer therapy-induced cardiotoxicity in adult population: a scoping review. ESC Heart Fail. (2022) 9:1651–65. 10.1002/ehf2.13735
25.
Sissung TM Gardner ER Piekarz RL Howden R Chen X Woo S et al . Impact of ABCB1 allelic variants on QTc interval prolongation. Clin Cancer Res. (2011) 17:937–46. 10.1158/1078-0432.CCR-10-0925
26.
Magdy T Jouni M Kuo HH Weddle CJ Lyra-Leite D Fonoudi H et al . Identification of drug transporter genomic variants and inhibitors that protect against doxorubicin-induced cardiotoxicity. Circulation. (2022) 145:279–94. 10.1161/CIRCULATIONAHA.121.055801
27.
Ahmad T Sabet S Primerano DA Richards-Waugh LL Rankin GO . Tell-Tale SNPs: the role of CYP2B6 in methadone fatalities. J Anal Toxicol. (2017) 41:325–33. 10.1093/jat/bkw135
28.
Gray B Baruteau AE Antolin AA Pittman A Sarganas G Molokhia M . Rare variation in drug metabolism and long QT genes and the genetic susceptibility to acquired long QT syndrome. Circ Genom Precis Med. (2022) 15:e003391. 10.1161/CIRCGEN.121.003391
29.
Martinez-Matilla M Blanco-Verea A Santori M Ansede-Bermejo J Ramos-Luis E Gil R et al . Genetic susceptibility in pharmacodynamic and pharmacokinetic pathways underlying drug-induced arrhythmia and sudden unexplained deaths. Forensic Sci Int Genet. (2019) 42:203–12. 10.1016/j.fsigen.2019.07.010
30.
McAlpine DE Biernacka JM Mrazek DA O'Kane DJ Stevens SR Langman LJ et al . Effect of cytochrome P450 enzyme polymorphisms on pharmacokinetics of venlafaxine. Ther Drug Monit. (2011) 33:14–20. 10.1097/FTD.0b013e3181fcf94d
31.
Garcia S Schuh M Cheema A Atwal H Atwal PS . Palpitations and asthenia associated with venlafaxine in a CYP2D6 poor metabolizer and CYP2C19 intermediate metabolizer. Case Rep Genet. (2017) 2017:6236714. 10.1155/2017/6236714
32.
Huang Z Wang J Qian J Li Y Xu Z Chen M et al . Effects of cytochrome P450 family 3 subfamily A member 5 gene polymorphisms on daunorubicin metabolism and adverse reactions in patients with acute leukemia. Mol Med Rep. (2017) 15:3493–8. 10.3892/mmr.2017.6470
33.
Rossi D Rasi S Franceschetti S Capello D Castelli A De Paol L et al . Analysis of the host pharmacogenetic background for prediction of outcome and toxicity in diffuse large B-cell lymphoma treated with R-CHOP21. Leukemia. (2009) 23:1118–26. 10.1038/leu.2008.398
34.
Kalabus JL Sanborn CC Jamil RG Cheng Q Blanco JG . Expression of the anthracycline-metabolizing enzyme carbonyl reductase 1 in hearts from donors with Down syndrome. Drug Metab Dispos. (2010) 38:2096–9. 10.1124/dmd.110.035550
35.
Kalabus JL Cheng Q Blanco JG . MicroRNAs differentially regulate carbonyl reductase 1 (CBR1) gene expression dependent on the allele status of the common polymorphic variant rs9024. PLoS ONE. (2012) 7:e48622. 10.1371/journal.pone.0048622
36.
Gonzalez-Covarrubias V Zhang J Kalabus JL Relling MV Blanco JG . Pharmacogenetics of human carbonyl reductase 1 (CBR1) in livers from black and white donors. Drug Metab Dispos. (2009) 37:400–7. 10.1124/dmd.108.024547
37.
Aslam S Ameer S Shabana NA Ahmed M . Pharmacogenetics of induction therapy-related toxicities in childhood acute lymphoblastic leukemia patients treated with UKALL 2003 protocol. Sci Rep. (2021) 11:23757. 10.1038/s41598-021-03208-9
38.
Lal S Sandanaraj E Wong ZW Ang PC Wong NS Lee EJ et al . CBR1 and CBR3 pharmacogenetics and their influence on doxorubicin disposition in Asian breast cancer patients. Cancer Sci. (2008) 99:2045–54. 10.1111/j.1349-7006.2008.00903.x
39.
Blanco JG Sun CL Landier W Chen L Esparza-Duran D Leisenring W et al . Anthracycline-related cardiomyopathy after childhood cancer: role of polymorphisms in carbonyl reductase genes-a report from the Children's oncology group. J Clin Oncol. (2012) 30:1415–21. 10.1200/JCO.2011.34.8987
40.
Blanco JG Leisenring WM Gonzalez-Covarrubias VM Kawashima TI Davies SM Relling MV et al . Genetic polymorphisms in the carbonyl reductase 3 gene CBR3 and the NAD(P)H:quinone oxidoreductase 1 gene NQO1 in patients who developed anthracycline-related congestive heart failure after childhood cancer. Cancer. (2008) 112:2789–95. 10.1002/cncr.23534
41.
Serie DJ Crook JE Necela BM Dockter TJ Wang X Asmann YW et al . Genome-wide association study of cardiotoxicity in the NCCTG N9831 (Alliance) adjuvant trastuzumab trial. Pharmacogenet Genomics. (2017) 27:378–85. 10.1097/FPC.0000000000000302
42.
Lang JK Karthikeyan B Quiñones-Lombraña A Rachael HB Early AP Levine EG et al . CBR3 V244M is associated with LVEF reduction in breast cancer patients treated with doxorubicin. Cardiooncology. (2021) 7:17. 10.1186/s40959-021-00103-0
43.
Li H Hu B Guo Z Jiang X Su X Zhang X . Correlation of UGT2B7 polymorphism with cardiotoxicity in breast cancer patients undergoing epirubicin/cyclophosphamide-docetaxel adjuvant chemotherapy. Yonsei Med J. (2019) 60:30–7. 10.3349/ymj.2019.60.1.30
44.
Linschoten M Teske AJ Cramer MJ van der Wall E Asselbergs FW . Chemotherapy-related cardiac dysfunction: a systematic review of genetic variants modulating individual risk. Circ Genom Precis Med. (2018) 11:e001753. 10.1161/CIRCGEN.117.001753
45.
Li J Luo H Liu YY Chen LX Zhu MQ Deng QT et al . Value of UGT2B7-161 single nucleotide polymorphism in predicting the risk of cardiotoxicity in HER-2 positive breast cancer patients who underwent pertuzumab combined with trastuzumab therapy by PSL. Pharmgenomics Pers Med. (2022) 15:215–25. 10.2147/PGPM.S351718
46.
Tan L Su X Li X Li H Hu B . Correlation of HER2 codon 655 polymorphism with cardiotoxicity risk in Chinese HER2-positive breast cancer patients undergoing epirubicin/cyclophosphamide followed by docetaxel plus trastuzumab adjuvant chemotherapy. Int J Clin Exp Pathol. (2020) 13:286–94.
47.
Nakano MH Udagawa C Shimo A Kojima Y Yoshie R Zaha H et al . A Genome-Wide Association Study identifies five novel genetic markers for trastuzumab-induced cardiotoxicity in Japanese population. Biol Pharm Bull. (2019) 42:2045–53. 10.1248/bpb.b19-00527
48.
Lunardi M Al-Habbaa A Abdelshafy M Davey MG Elkoumy A Ganly S et al . Genetic and RNA-related molecular markers of trastuzumab-chemotherapy-associated cardiotoxicity in HER2 positive breast cancer: a systematic review. BMC Cancer. (2022) 22:396. 10.1186/s12885-022-09437-z
49.
Stanton SE Ward MM Christos P Sanford R Lam C Cobham MV et al . Pro1170 Ala polymorphism in HER2-neu is associated with risk of trastuzumab cardiotoxicity. BMC Cancer. (2015) 15:267. 10.1186/s12885-015-1298-6
50.
Lin M Xiong W Wang S Li Y Hou C Li C et al . The research progress of trastuzumab-induced cardiotoxicity in HER-2-positive breast cancer treatment. Front Cardiovasc Med. (2022) 8:821663. 10.3389/fcvm.2021.821663
51.
Li Y Wang W Gao R Xu X Zhang Y . Genome-wide prioritization reveals novel gene signatures associated with cardiotoxic effects of tyrosine kinase inhibitors. Oncol Lett. (2021) 21:94. 10.3892/ol.2020.12355
52.
Yang Y Bu P . Progress on the cardiotoxicity of sunitinib: prognostic significance, mechanism and protective therapies. Chem Biol Interact. (2016) 257:125–31. 10.1016/j.cbi.2016.08.006
53.
Singh AP Umbarkar P Tousif S Lal H . Cardiotoxicity of the BCR-ABL1 tyrosine kinase inhibitors: emphasis on ponatinib. Int J Cardiol. (2020) 316:214–21. 10.1016/j.ijcard.2020.05.077
54.
Jiang D Liang J Noble PW . Hyaluronan as an immune regulator in human diseases. Physiol Rev. (2011) 91:221–64. 10.1152/physrev.00052.2009
55.
Kleniewska P Piechota A Skibska B Goraca A . The NADPH oxidase family and its inhibitors. Arch Immunol Ther Exp. (2012) 60:277–94. 10.1007/s00005-012-0176-z
56.
Roos D Van Leeuwen K Hsu AP Priel DL Begtrup A Brandon R et al . Hematologically important mutations: the autosomal forms of chronic granulomatous disease (third update). Blood Cells Mol Dis. (2021) 92:102596. 10.1016/j.bcmd.2021.102596
57.
Megías-Vericat JE Montesinos P Herrero MJ Moscardó F Bosó V Rojas L et al . Impact of NADPH oxidase functional polymorphisms in acute myeloid leukemia induction chemotherapy. Pharmacogenom J. (2018) 18:301–7. 10.1038/tpj.2017.19
58.
Kopeva KV Grakova EV Shilov SN Berezikova EN Popova AA Neupokoeva MN et al . Anthracycline-induced cardiotoxicity in women without cardiovascular diseases: molecular and genetic predictors. Acta Cardiol. (2021) 16:1–10. 10.1080/00015385.2021.2003061
59.
Leong SL Chaiyakunapruk N Lee SW . Candidate gene association studies of anthracycline-induced cardiotoxicity: a systematic review and meta-analysis. Sci Rep. (2017) 7:39. 10.1038/s41598-017-00075-1
60.
Wang X Liu W Sun CL Armenian SH Hakonarson H Hageman L et al . Hyaluronan synthase 3 variant and anthracycline-related cardiomyopathy: a report from the children's oncology group. J Clin Oncol. (2014) 32:647–53. 10.1200/JCO.2013.50.3557
61.
Vaitiekus D Muckiene G Vaitiekiene A Sereikaite L Inciuraite R Insodaite R et al . HFE gene variants' impact on anthracycline-based chemotherapy-induced subclinical cardiotoxicity. Cardiovasc Toxicol. (2021) 21:59–66. 10.1007/s12012-020-09595-1
62.
Hannuksela J Leppilampi M Peuhkurinen K Kärkkäinen S Saastamoinen E Heliö T et al . Hereditary hemochromatosis gene (HFE) mutations C282Y, H63D and S65C in patients with idiopathic dilated cardiomyopathy. Eur J Heart Fail. (2005) 7:103–8. 10.1016/j.ejheart.2004.03.007
63.
Gomes AV Venkatraman G Davis JP Tikunova SB Engel P Solaro RJ et al . Cardiac troponin T isoforms affect the Ca(2+) sensitivity of force development in the presence of slow skeletal troponin I: insights into the role of troponin T isoforms in the fetal heart. J Biol Chem. (2004) 279:49579–87. 10.1074/jbc.M407340200
64.
Anderson PA Greig A Mark TM Malouf NN Oakeley AE Ungerleider RM et al . Molecular basis of human cardiac troponin T isoforms expressed in the developing, adult, and failing heart. Circ Res. (1995) 76:681–6. 10.1161/01.RES.76.4.681
65.
Cejas RB Tamaño-Blanco M Fontecha JE Blanco JG . Impact of DYRK1A expression on TNNT2 splicing and daunorubicin toxicity in human iPSC-derived cardiomyocytes. Cardiovasc Toxicol. (2022) 22:701–12. 10.1007/s12012-022-09746-6
66.
Wang X Sun CL Quiñones-Lombraña A Singh P Landier W Hageman L et al . CELF4 variant and anthracycline-related cardiomyopathy: A Children's Oncology Group Genome-Wide Association Study. J Clin Oncol. (2016) 34:863–70. 10.1200/JCO.2015.63.4550
67.
Aminkeng F Bhavsar AP Visscher H Rassekh SR Li Y Lee JWAminkeng F et al . A coding variant in RARG confers susceptibility to anthracycline-induced cardiotoxicity in childhood cancer. Nat Genet. (2015) 47:1079–84. 10.1038/ng.3374
68.
Christidi E Huang H Shafaattalab S Maillet A Lin E Huang K et al . Variation in RARG increases susceptibility to doxorubicin-induced cardiotoxicity in patient specific induced pluripotent stem cell-derived cardiomyocytes. Sci Rep. (2020) 10:10363. 10.1038/s41598-020-65979-x
69.
Huang H Christidi E Shafaattalab S Davis MK Tibbits GF Brunham LR . RARG S427L attenuates the DNA repair response to doxorubicin in induced pluripotent stem cell-derived cardiomyocytes. Stem Cell Rep. (2022) 17:756–65. 10.1016/j.stemcr.2022.03.002
70.
Liu B An T Li M Yi Z Li C Sun X et al . The association between early-onset cardiac events caused by neoadjuvant or adjuvant chemotherapy in triple-negative breast cancer patients and some novel autophagy-related polymorphisms in their genomic DNA: a real-world study. Cancer Commun. (2018) 38:71. 10.1186/s40880-018-0343-7
71.
Colhoun HM McKeigue PM Davey Smith G . Problems of reporting genetic associations with complex outcomes. Lancet. (2003) 361:865–72. 10.1016/S0140-6736(03)12715-8
72.
Hewitt JK . Editorial policy on candidate gene association and candidate gene-by-environment interaction studies of complex traits. Behav Genet. (2012) 42:1–2. 10.1007/s10519-011-9504-z
73.
Thudi M Li Y Jackson SA May GD Varshney RK . Current state-of-art of sequencing technologies for plant genomics research. Brief Funct Genom. (2012) 11:3–11. 10.1093/bfgp/elr045
74.
Genomes Project Consortium Abecasis GR Auton A Brooks LD DePristo MA Durbin RM et al . An integrated map of genetic variation from 1,092 human genomes. Nature. (2012) 491:56–65. 10.1038/nature11632
75.
Yanhu L Lu W Li Y . The principle and application of the single-molecule real-time sequencing technology. Yi Chuan. (2015) 37:259–68. 10.1186/s41021-015-0017-5
76.
Roberts RJ Carneiro MO Schatz MC . The advantages of SMRT sequencing. Genome Biol. (2013) 14:405. 10.1186/gb-2013-14-6-405
77.
Wang Y Tian K Shi R Gu A Pennell M Alberts L et al . Nanolock-nanopore facilitated digital diagnostics of cancer driver mutation in tumor tissue. ACS Sens. (2017) 2:975–81. 10.1021/acssensors.7b00235
78.
Magdy T Kuo HH Burridge PW . Precise and cost-Effective nanopore sequencing for post-GWAS Fine-Mapping and causal variant identification. iScience. (2020) 23:100971. 10.1016/j.isci.2020.100971
79.
Sloan SA Darmanis S Huber N Khan TA Birey F Caneda C et al . Human astrocyte maturation captured in 3D cerebral cortical spheroids derived from pluripotent stem cells. Neuron. (2017) 95:779–90.e6. 10.1016/j.neuron.2017.07.035
80.
Zheng GX Terry JM Belgrader P Ryvkin P Bent ZW Wilson R et al . Massively parallel digital transcriptional profiling of single cells. Nat Commun. (2017) 8:14049. 10.1038/ncomms14049
81.
Simão D Gomes CM Alves PM Brito C . Capturing the third dimension in drug discovery: spatially-resolved tools for interrogation of complex 3D cell models. Biotechnol Adv. (2022) 55:107883. 10.1016/j.biotechadv.2021.107883
82.
Ståhl PL Salmén F Vickovic S Lundmark A Navarro JF Magnusson J et al . Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. (2016) 353:78–82. 10.1126/science.aaf2403
83.
Asnani A Shi X Farrell L Lall R Sebag IA Plana JC et al . Changes in citric acid cycle and nucleoside metabolism are associated with anthracycline cardiotoxicity in patients with breast cancer. J Cardiovasc Transl Res. (2020) 13:349–56. 10.1007/s12265-019-09897-y
84.
Aslam B Basit M Nisar MA Khurshid M Rasool MH . Proteomics: technologies and their applications. J Chromatogr Sci. (2017) 55:182–96. 10.1093/chromsci/bmw167
85.
Beer LA Kossenkov AV Liu Q Luning Prak E Domchek S Speicher DW et al . Baseline immunoglobulin E levels as a marker of doxorubicin- and trastuzumab-associated cardiac dysfunction. Circ Res. (2016) 119:1135–44. 10.1161/CIRCRESAHA.116.309004
86.
Yuan Y Fan S Shu L Huang W Xie L Bi C et al . Exploration the mechanism of doxorubicin-induced heart failure in rats by integration of proteomics and metabolomics data. Front Pharmacol. (2020) 11:600561. 10.3389/fphar.2020.600561
87.
Selevsek N Caiment F Nudischer R Gmuender H Agarkova I Atkinson FL et al . Network integration and modelling of dynamic drug responses at multi-omics levels. Commun Biol. (2020) 3:573. 10.1038/s42003-020-01302-8
88.
Liu X Liu Y Chen X Wang C Chen X Liu W et al . Multi-walled carbon nanotubes exacerbate doxorubicin-induced cardiotoxicity by altering gut microbiota and pulmonary and colonic macrophage phenotype in mice. Toxicology. (2020) 435:152410. 10.1016/j.tox.2020.152410
89.
Russo M Guida F Paparo L Trinchese G Aitoro R Avagliano C et al . The novel butyrate derivative phenylalanine-butyramide protects from doxorubicin-induced cardiotoxicity. Eur J Heart Fail. (2019) 21:519–28. 10.1002/ejhf.1439
90.
Pinheiro EA Fetterman KA Burridge PW . hiPSCs in cardio-oncology: deciphering the genomics. Cardiovasc Res. (2019) 115:935–48. 10.1093/cvr/cvz018
91.
Babiarz JE Ravon M Sridhar S Ravindran P Swanson B Bitter H et al . Determination of the human cardiomyocyte mRNA and miRNA differentiation network by fine-scale profiling. Stem Cells Dev. (2012) 21:1956–65. 10.1089/scd.2011.0357
92.
Ma J Guo L Fiene SJ Anson BD Thomson JA Kamp TJ et al . High purity human-induced pluripotent stem cell-derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents. Am J Physiol Heart Circ Physiol. (2011) 301:H2006–17. 10.1152/ajpheart.00694.2011
93.
Burridge PW Li YF Matsa E Wu H Ong SG Sharma A et al . Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat Med. (2016) 22:547–56. 10.1038/nm.4087
94.
Kitani T Ong SG Lam CK Rhee JW Zhang JZ Oikonomopoulos A et al . Human-induced pluripotent stem cell model of trastuzumab-induced cardiac dysfunction in patients with breast cancer. Circulation. (2019) 139:2451–65. 10.1161/CIRCULATIONAHA.118.037357
95.
Sharma A Burridge PW McKeithan WL Serrano R Shukla P Sayed N et al . High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci Transl Med. (2017) 9:eaaf2584. 10.1126/scitranslmed.aaf2584
96.
Nemade H Chaudhari U Acharya A Hescheler J Hengstler JG Papadopoulos S et al . Cell death mechanisms of the anti-cancer drug etoposide on human cardiomyocytes isolated from pluripotent stem cells. Arch Toxicol. (2018) 92:1507–24. 10.1007/s00204-018-2170-7
97.
Yan M Feng L Shi Y Wang J Liu Y Li F et al . Mechanism of As2O3-induced action potential prolongation and using hiPS-CMs to evaluate the rescue efficacy of drugs with different rescue mechanism. Toxicol Sci. (2017) 158:379–90. 10.1093/toxsci/kfx098
98.
Goineau S Castagne V . Proarrhythmic risk assessment using conventional and new in vitro assays. Regul Toxicol Pharmacol. (2017) 88:1–11. 10.1016/j.yrtph.2017.05.012
99.
Hsu WT Huang CY Yen CYT Cheng AL Hsieh PCH . The HER2 inhibitor lapatinib potentiates doxorubicin-induced cardiotoxicity through iNOS signaling. Theranostics. (2018) 8:3176–88. 10.7150/thno.23207
100.
Kopljar I Gallacher DJ De Bondt A Cougnaud L Vlaminckx E Van den Wyngaert I et al . Functional and transcriptional characterization of histone deacetylase inhibitor-mediated cardiac adverse effects in human induced pluripotent stem cell-derived cardiomyocytes. Stem Cells Transl Med. (2016) 5:602–12. 10.5966/sctm.2015-0279
101.
Xu Q Patel D Zhang X Veenstra RD . Changes in cardiac Nav1.5 expression, function, and acetylation by pan-histone deacetylase inhibitors. Am J Physiol Heart Circ Physiol. (2016) 311:H1139–49. 10.1152/ajpheart.00156.2016
102.
Richards DJ Li Y Kerr CM Yao J Beeson GC Coyle RC et al . Human cardiac organoids for the modelling of myocardial infarction and drug cardiotoxicity. Nat Biomed Eng. (2020) 4:446–62. 10.1038/s41551-020-0539-4
103.
Truitt R Mu A Corbin EA Vite A Brandimarto J Ky B et al . Increased afterload augments sunitinib-induced cardiotoxicity in an engineered cardiac microtissue model. JACC Basic Transl Sci. (2018) 3:265–76. 10.1016/j.jacbts.2017.12.007
104.
Zhang YS Aleman J Shin SR Kilic T Kim D Mousavi Shaegh SA et al . Multisensor-integrated organs-on-chips platform for automated and continual in situ monitoring of organoid behaviors. Proc Natl Acad Sci USA. (2017) 114:E2293–302. 10.1073/pnas.1612906114
105.
Higo S Hikoso S Miyagawa S Sakata Y . Genome editing in human induced pluripotent stem cells (hiPSCs). Methods Mol Biol. (2021) 2320:235–45. 10.1007/978-1-0716-1484-6_21
106.
Maillet A Tan K Chai X Sadananda SN Mehta A Ooi J et al . Modeling doxorubicin-Induced cardiotoxicity in human pluripotent stem cell derived-cardiomyocytes. Sci Rep. (2016) 6:25333. 10.1038/srep25333
Summary
Keywords
drug-induced cardiotoxicity, pharmacogenomics, single nucleotide polymorphisms (SNPs), biomarker, new technologies in pharmacogenomics
Citation
Li M-Y, Peng L-M and Chen X-P (2022) Pharmacogenomics in drug-induced cardiotoxicity: Current status and the future. Front. Cardiovasc. Med. 9:966261. doi: 10.3389/fcvm.2022.966261
Received
10 June 2022
Accepted
05 September 2022
Published
13 October 2022
Volume
9 - 2022
Edited by
Shusen Sun, Western New England University, United States
Reviewed by
Tarek Magdy, Northwestern University, United States; Jiali Li, School of Pharmaceutical Sciences, Sun Yat-sen University, China
Updates
Copyright
© 2022 Li, Peng and Chen.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li-Ming Peng limingpeng@csu.edu.cnXiao-Ping Chen chenxiaoping@csu.edu.cn
This article was submitted to Cardiovascular Therapeutics, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.