 Le-Kun Gui
Le-Kun Gui Huang-Jun Liu
Huang-Jun Liu Li-Jun Jin1*
Li-Jun Jin1* Xiao-Chun Peng
Xiao-Chun Peng- 1Department of Cardiology, The First Affiliated Hospital of Yangtze University, Jingzhou, Hubei, China
- 2School of Medicine, Yangtze University, Jingzhou, Hubei, China
- 3Department of Pathophysiology, School of Basic Medicine, Health Science Center, Yangtze University, Jingzhou, Hubei, China
- 4Laboratory of Oncology, School of Basic Medicine, Center for Molecular Medicine, Health Science Center, Yangtze University, Jingzhou, Hubei, China
Cardiomyopathy, a heterogeneous pathological condition characterized by changes in cardiac structure or function, represents a significant risk factor for the prevalence and mortality of cardiovascular disease (CVD). Research conducted over the years has led to the modification of definition and classification of cardiomyopathy. Herein, we reviewed seven of the most common types of cardiomyopathies, including Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC), diabetic cardiomyopathy, Dilated Cardiomyopathy (DCM), desmin-associated cardiomyopathy, Hypertrophic Cardiomyopathy (HCM), Ischemic Cardiomyopathy (ICM), and obesity cardiomyopathy, focusing on their definitions, epidemiology, and influencing factors. Cardiomyopathies manifest in various ways ranging from microscopic alterations in cardiomyocytes, to tissue hypoperfusion, cardiac failure, and arrhythmias caused by electrical conduction abnormalities. As pleiotropic Transcription Factors (TFs), the Krüppel-Like Factors (KLFs), a family of zinc finger proteins, are involved in regulating the setting and development of cardiomyopathies, and play critical roles in associated biological processes, including Oxidative Stress (OS), inflammatory reactions, myocardial hypertrophy and fibrosis, and cellular autophagy and apoptosis, particularly in diabetic cardiomyopathy. However, research into KLFs in cardiomyopathy is still in its early stages, and the pathophysiologic mechanisms of some KLF members in various types of cardiomyopathies remain unclear. This article reviews the roles and recent research advances in KLFs, specifically those targeting and regulating several cardiomyopathy-associated processes.
1 Introduction
Cardiomyopathies are a group of heterogeneous pathological disorders characterized by alterations in cardiac structure and function (1). Their conception can be traced to Fiedler's discovery of a series of fatal cases of cardiac hypertrophy and Heart Failure (HF) in young people in 1899 (2). With advancements in medicine and an enhanced understanding of diseases, cardiomyopathies have been updated and categorized into several groups (1, 3–5), including Dilated Cardiomyopathies (DCM), Restrictive Cardiomyopathies (RCM), Hypertrophic Cardiomyopathies (HCM) and Arrhythmogenic Right Ventricular Cardiomyopathies (ARVC), among others (3, 6). Furthermore, research is expanding into the disease(cardiomyopathy)-causing genes (7, 8). Cardiomyopathy etiology is multifactorial and has not been fully elucidated hitherto. The molecular mechanisms underlying cardiomyopathy-associated myocardial remodeling and cardiac dysfunction are highly complex and warrant further research. In recent years, the Krüppel-Like Transcription Factors (KLFs) family has gained renewed attention as research advances have revealed the involvement of KLFs in various processes, including cardiomyopathy progression. Krüppel-Like Factors (KLFs) are a group of DNA-binding proteins first discovered in the early 1990s as erythroid cell-specific Transcription Factors (TFs) (9). As important gene transcription regulators, KLFs are involved in multiple processes regulating the occurrence and development of myocardial diseases, including Oxidative Stress (OS), inflammatory responses, and myocardial hypertrophic fibrosis, as well as cell proliferation, differentiation, apoptosis, and regeneration (10–14), via amino acid terminal region regulation of protein-DNA and Protein-Protein Interactions (PPIs). Although research has revealed key insights on KLFs and their involvement in cardiomyopathies, much is unknown about the KLF-induced pathophysiologic alterations in cardiomyopathies and the potential therapeutic targets for treating these diseases. Therefore, this article aims to summarize the current roles and molecular mechanisms of KLF family members in different types of cardiomyopathies and to outline the key roles KLFs play in Cardiovascular Diseases (CVDs).
2 Krüppel-Like Factors
Although KLFs are found in multiple organ systems, including the cardiovascular, respiratory, gastrointestinal, urinary, neurological, and hematopoietic systems (15), their tissue expression varies (Figure 1). In other words, some family members are universally expressed, while others are specifically expressed (16–18). Studies have shown that KLFs possess three highly conserved C2H2 zinc-finger domains in their carboxy-terminal region. These domains facilitate interactions with common GC-rich sites during transcriptional regulation, enabling them to activate or inhibit cellular development (17, 19). It has been reported that KLF proteins show homology in their gene sequences, with the structural similarity allowing for overlapping transcriptional targets. For example, KLF2 is expressed in the cardiovascular, respiratory, urinary, and nervous systems, while KLF5 is found in the cardiovascular, gastrointestinal, urinary, nervous, and hematopoietic systems. On the other hand, KLF6 can be found in all of the above-mentioned systems. However, KLF proteins possess unique amino-terminal sequences that provide specific regions for interaction with distinct binding partners. For instance, KLF1 features a minimal transactivation domain (TAD) within its first 100 amino acids. Research has classified KLF1 TAD into two functional subdomains, TAD1 and TAD2, with the latter conserved in four additional KLF proteins (KLF2, 4, 5, and 15) (20). KLF1 is predominantly expressed in mast and erythroid cells and is associated with β-thalassemia (21), whereas KLF2 is highly expressed in the lungs and is an essential regulator involved in lung development (22, 23). On the other hand, KLF15 is abundantly expressed in the cardiovascular system and is a negative regulator of cardiac hypertrophy, ensuring appropriate cardiac responses to physiological stress signals (24). Additionally, KLF3, 8, and 12 are characterized by an N-terminal repression domain containing a CtBP recognition motif (25–27), while the N-terminal regions of KLF9, 10, 11, 13, 14, and 16 contain a Cabut domain with a Sin3 interaction domain (SID), serving as a transcriptional regulatory repression domain (28–32).
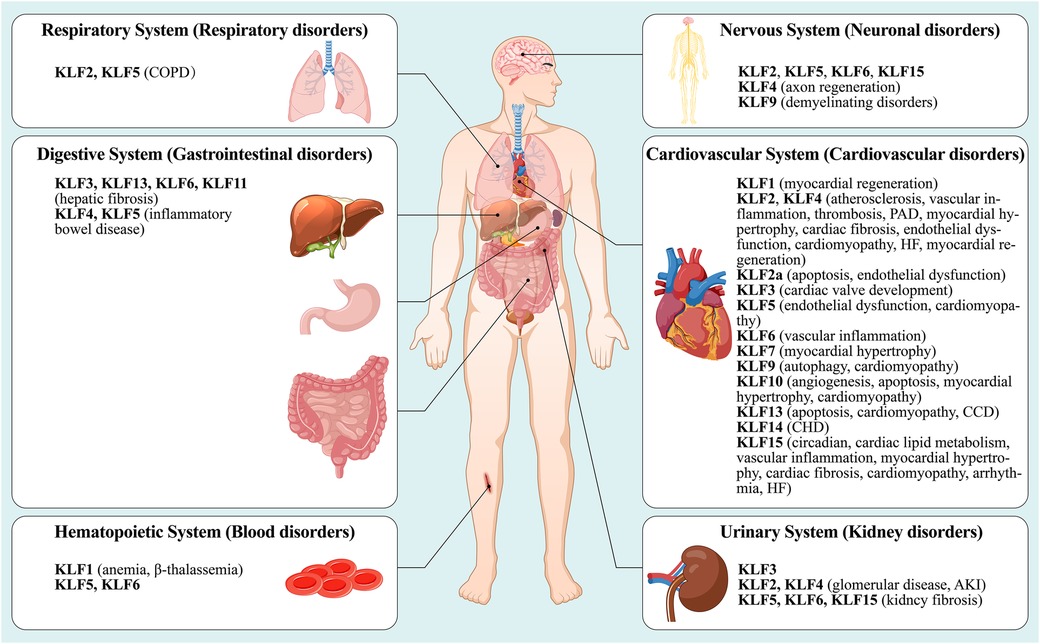
Figure 1. A summary nof the type distribution of KLFs in humans and their roles in diseases. The figure describes the physiological distribution of KLFs in various body systems (cardiovascular system, respiratory system, nervous system, urinary system, digestive system and hematopoietic system), and its role in the initiation and development of the corresponding pathologic diseases or disorders. See text for details. PAD, peripheral arterial disease; HF, heart failure; CCD, congenital cardiovascular diseases; CHD, coronary heart disease; COPD, chronic obstructive pulmonary disease; AKI, acute kidney injury.
Krüppel-Like Factors (KLFs) represent a conserved family of TFs, with the initial discovery of KLF1 (EKLF) in erythrocytes in 1993 (9). Since then, researchers have identified 18 members (KLF1-KLF18) in mammals. However, some debate surrounds whether there are only 17 KLF members, excluding KLF18, which is considered a duplicate of KLF17 (33). These KLF proteins can be classified into three groups based on their functional characteristics (Table 1). Group 1 includes KLFs 3, 8, and 12, acting as transcriptional repressors that interact with CtBP (C-terminal binding protein) (17, 26, 34, 35). Group 2 consists of KLFs 1, 2, 4, 5, 6, and 7, primarily functioning as transcriptional activators (9, 36–40). In contrast, KLFs 15, 17, and 18 form the non-consensus group, exhibiting more distant relationships and lacking clear protein interaction motifs (17, 24, 33, 46). The remaining KLFs, categorized as group 3 members, exert repressive effects similar to those in group 1 but their effects are dependent on its interaction with the transcriptional co-repressor Sin3A (15, 41–45).
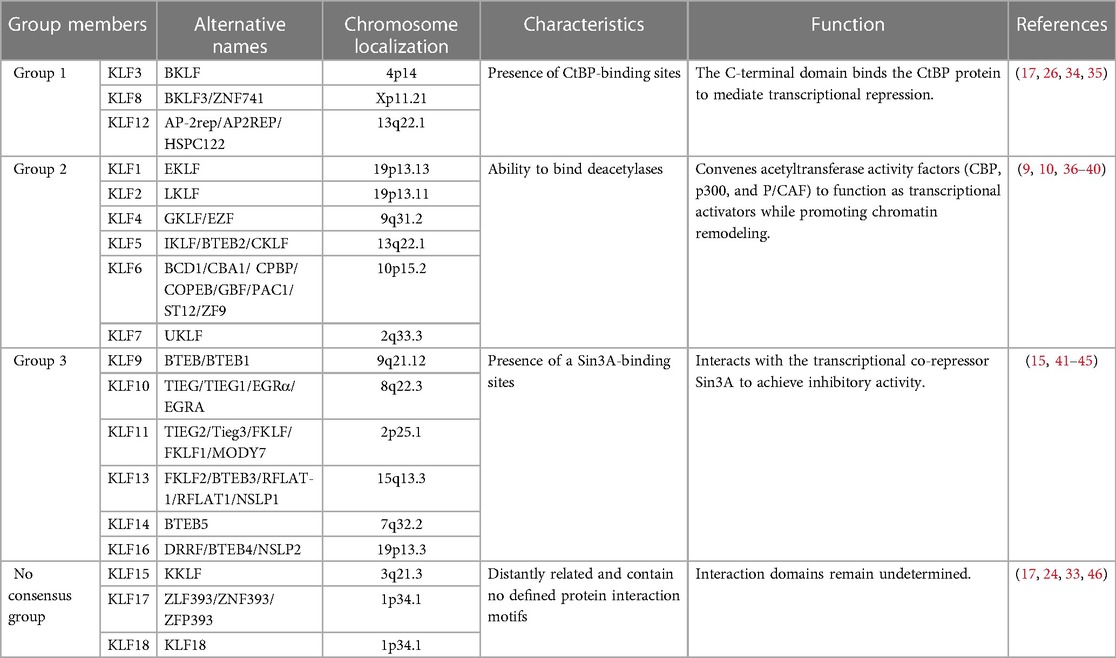
Table 1. Phylogenetic classification of Krüppel-Like Factors.
3 Cardiomyopathy
Cardiomyopathy manifests in various ways, ranging from microscopic changes in myocardial cells, to fulminant HF with inadequate tissue perfusion and arrhythmias caused by electrical conduction abnormalities (47). Cardiomyopathies, which means myocardium diseases, were traditionally categorized as hypertrophic, dilated, and restrictive. However, advances in genomics have demonstrated the diversity in their phenotypic expression (1).
In 1980, the World Health Organization (WHO) published its first report on cardiomyopathies (3), defining them as muscle diseases of unknown cause and categorizing them as DCM, RCM, and HCM. This diagnostic criterion was updated by subsequent classification iterations, leading to the WHO amending the definition of cardiomyopathy in 1995 (4), redefining it as myocardial illnesses associated with cardiac dysfunction, including ARVC in the previous classification, and further elucidating and highlighting the term “specific cardiomyopathy”. Based on previously published clinical practice guidelines and recent advances in the characterization of myocardial diseases (48–50), the American Heart Association (AHA) Committee proposed a new, more rigorous classification in 2006 that reflects the evolving molecular genetics of cardiology and standardizes nomenclature inconsistencies (1). The panel proposed a new definition of cardiomyopathies, describing them as a heterogeneous group of myocardial diseases with etiologies related to mechanical and electrical dysfunction, often exhibiting inappropriate ventricular hypertrophy and/or dilatation and arising from multiple causes, often genetic. Regarding classification, the expert consensus panel recommended the categorization of cardiomyopathies as primary or secondary. Primary cardiomyopathies referred to diseases occurring exclusively or predominantly in the myocardium, while secondary cardiomyopathies described pathologic myocardial illnesses associated with a multisystem disease. Primary cardiomyopathies were further grouped into the genetic, mixed, and acquired classes, with genetic cardiomyopathies including HCM and ARVC; mixed cardiomyopathies including genetic and non-genetic DCM; and acquired cardiomyopathies (also known as inflammatory cardiomyopathies) including myocarditis. Notably, ion channelopathies were also listed as primary cardiomyopathies in the scientific statement. Adding to the ongoing updates, the European Society of Cardiology (ESC) introduced a new cardiomyopathy classification criteria in 2008 (5). This classification criteria were oriented towards clinical utility, was based on ventricular structure and function, and defined cardiomyopathies as structural and/or functional abnormalities of the myocardium not caused by Coronary Artery Diseases (CADs), Hypertension (HTN), valvular diseases, and congenital heart defects. Based on morphologic and functional characteristics, cardiomyopathies were categorized into five groups (DCM, RCM, HCM, ARVC, and unclassified), each of which was further divided into several subtypes, including familial (genetic), non-familial (non-genetic), and unidentified gene defect classes, as well as disease sub-types, and idiopathic subgroups. Notably, this version does not distinguish between primary and secondary cardiomyopathies. In 2013, Arbustini and other cardiovascular experts proposed a novel set of phenotypic-genotypic MOGE(S) classification criteria for cardiomyopathies (51), which was supported by the World Heart Federation (WHF). This classification criteria described cardiomyopathies as diseases characterized by morphologically and/or functionally abnormal myocardium, devoid of disruptions caused by the clinical manifestations of other diseases. The criteria classify cardiomyopathic disorders based on five characteristics: M (Morphofunctional features), O (Organ involvement), G (Genetic or familial inheritance patterns), E (Clear etiologic annotations), and optionally, S (Functional status information). The most current version is the 2023 ESC Guidelines for the Management of Cardiomyopathies (52). Here, the ESC defines cardiomyopathy as a disease of the myocardium with structural and functional abnormalities. It should be noted that the presence of such a disease does not preclude the occurrence of other diseases, such as CADs, HTN, and valvular and congenital heart diseases. The guideline task force updated the description of the phenotype of non-dilated left ventricular cardiomyopathy (NDLVC) and did not recommend both left ventricular non-compaction (LVNC) as well as Takotsubo syndrome (stress cardiomyopathy) as separate subtypes and no longer uses arrhythmogenic cardiomyopathy (ACM, from the original terminology of ARVC) as a distinct cardiomyopathic subtype. The guidelines further highlight the diagnostic value of multimodality imaging and the prognostic importance of genetic testing, recommend a multidisciplinary team approach to the management of cardiomyopathy that focuses on the patient and his or her family, and recommend clinical evaluation and genetic cascade screening for relatives of patients with cardiomyopathy (Figure 2). Although there are various types of cardiomyopathies, our understanding of these diseases and their current typing methods remains limited. However, with progress in cardiomyopathy research, future cardiomyopathy definitions and typing methods will be more refined and clinically applicable.
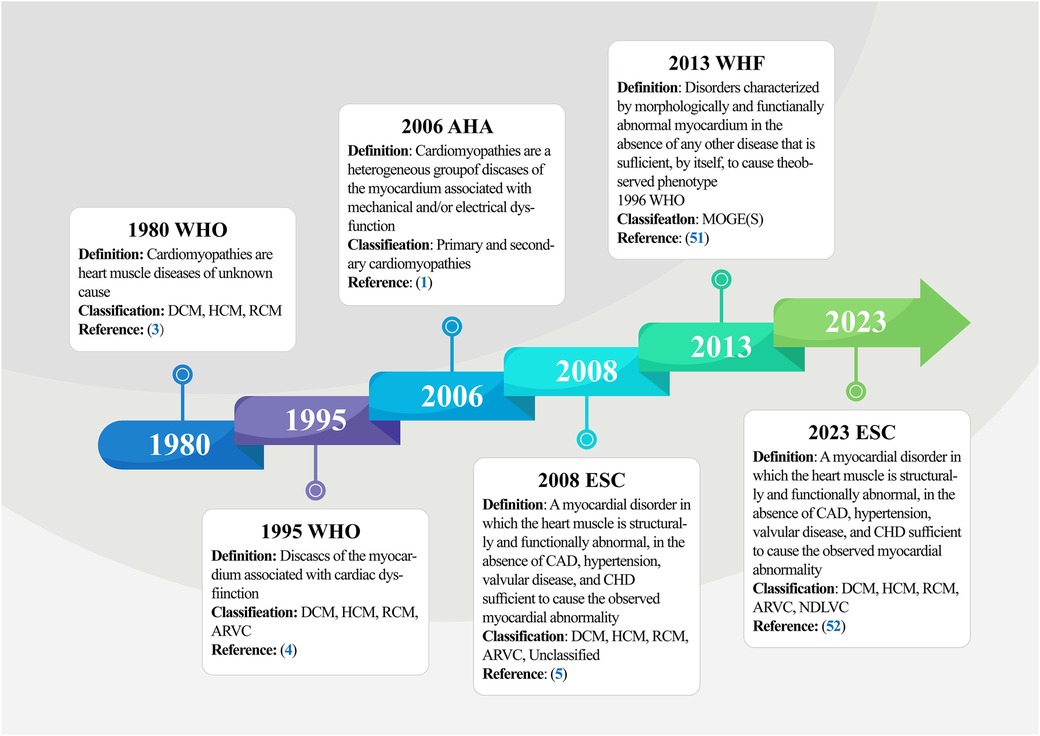
Figure 2. Timeline of definitions and classifications of cardiomyopathy. The figure summarizes the five main stages involved in the initiation and progression of cardiomyopathy, from the formal naming of cardiomyopathies and primary classification by the WHO in 1980 to the current more comprehensive definition and classification of cardiomyopathy by the ESC in 2023. WHO, world health organization; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; RCM, restrictive cardiomyopathy; AVRC, arrhythmogenic right ventricular cardiomyopathy; AHA, American heart association; WHF, world heart federation; ESC, European society of cardiology; CAD, coronary artery disease; CHD, congenital heart disease.
4 KLFs and cardiomyopathy
Many KLFs are involved in cardiovascular system regulation and in diversely controlling cell, tissue, and system metabolism. For example, KLFs 2 and 4 act as nodal regulators of endothelial function, promoting anti-inflammatory and anti-thrombotic gene expression, which collectively keep blood vessels healthy (53, 54). However, in pathological states, the heart undergoes cardiac remodeling due to disease and stress-induced long-term metabolic changes, triggering structural and functional abnormalities of the myocardium that eventually lead to cardiomyopathy. Cardiomyopathies are a heterogeneous group of pathological conditions. As some of the TFs affecting various pathophysiological processes in myocardial diseases, KLFs play vital roles in different cardiomyopathies (Table 2, Figure 3).

Table 2. Role of KLFs family members in various cardiomyopathies.
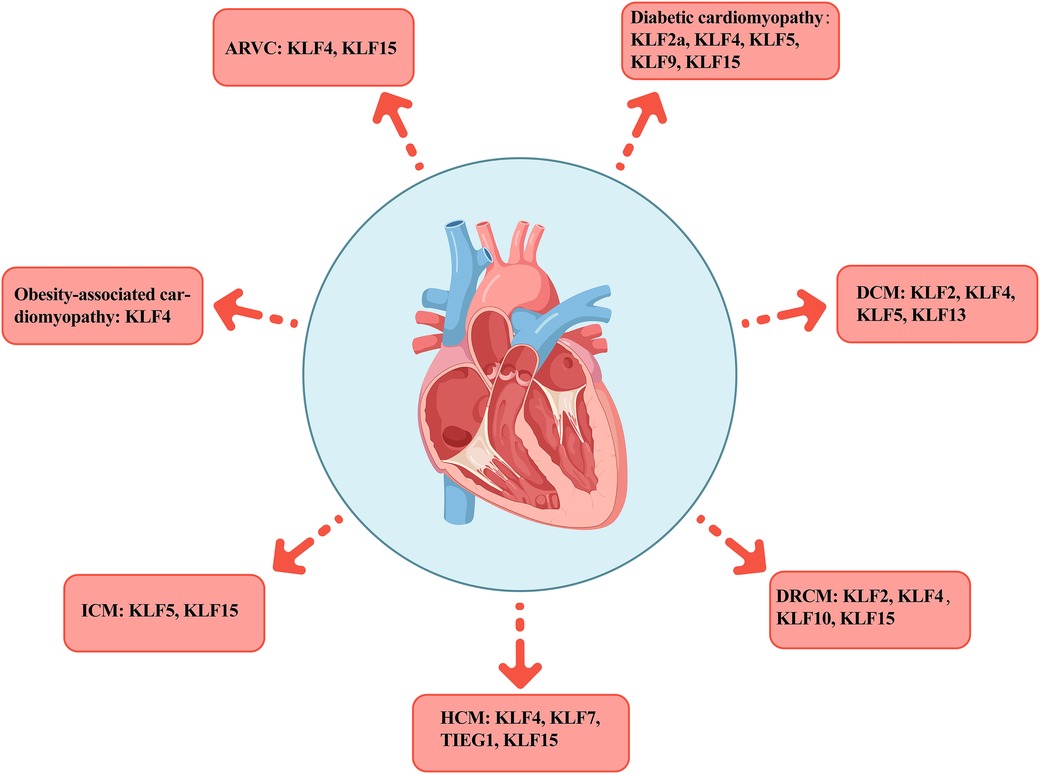
Figure 3. Distribution of KLFs across various cardiomyopathies. The figure presents seven typical cardiomyopathies including AVRC, diabetic cardiomyopathy, DCM, DRCM, HCM, ICM, and obesity cardiomyopathy, as well as the distribution of the KLFs family members in each subtype. AVRC, arrhythmogenic right ventricular cardiomyopathy; DCM, dilated cardiomyopathy; DRCM, desmin-related cardiomyopathy; HCM, hypertrophic cardiomyopathy; ICM, ischemic cardiomyopathy.
4.1 Arrhythmogenic right ventricular cardiomyopathy
It has been reported that Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an autosomal dominant cardiomyopathy (100) characterized by cardiomyocyte replacement with fibro-adipose tissues, resulting in abnormal Excitation-Contraction (EC) coupling and a series of malignant events such as Ventricular Arrhythmias (VA), HF, and Sudden Cardiac Death (SCD) (101, 102). The disease is considered a major cause of sudden death in young adults, especially athletes (6, 103), and its prevalence is estimated to be between 1/2,500 and 1/5,000, with a male predominance (female to male ratio of 1:2.7), which may be related to the disease genes and androgens (104, 105).
Intercalated disks (connective structures between cardiomyocytes) are the functional unit that provides the mechanical and/or electrical coupling that enables the coordinated and synchronized contraction of cardiomyocytes. According to research, variants affecting the gene encoding the desmosomal protein are responsible for approximately 50%–60% of ARVC cases (106). Five of the eight genes that account for pathogenic or potentially pathogenic variants are desmosomal genes (107). The primary function of “glue” desmosomes is transmitting mechanical strength between myocardial cells. When the desmosomes are mutated, the myocardium undergoes cell detachment and death, later replaced by fibro-adipose tissues, thereby resulting in scarring, wall thinning, and aneurysms. This adhesion defect is exacerbated by exercise, with a greater impact on the thinner right ventricle (55, 108). Li et al. (58) identified a novel heterozygous mutation in the KLF15 gene in a family with atrial fibrillation (AF), ventricular arrhythmia, and hypertrophic cardiomyopathy. Subsequent investigations revealed that the loss-of-function mutation in KLF15 could potentially trigger AF by disrupting myocardial energy metabolism (59). Moreover, this mutation could exacerbate AF by prolonging repolarization and extending the effective refractory period (60, 109).
Plakophilin-2 (PKP2) mutations are the most widespread genetic association in ARVC. Khudiakov et al. (56) obtained an iPSC line carrying 2 mutations in the PKP2 gene from a 14-year-old female with severe ARVC, and detected high OCT4, NANOG, and SOX2 mRNA levels after reprogramming and transducing them with Sendai virus vectors, confirming the pluripotency of the iPSC line. Yang et al. (75) used a similar approach to transduce PBMCs carrying three reprogramming factors (KOS, KLF4, and cMYC) obtained from a 41-year-old female ARVC patient. The iPSCs obtained exhibited pluripotency marker expression, intact karyotype, and the potential to differentiate into multiple germ layers. Similarly, KLF4 produces iPSCs via reprogramming in DCM (110, 111). Drawing on KLF4's pivotal role in regulating cardiac potassium (K+) channels, Chowdhury et al. (57) established a connection between mitogen-activated kinase kinase-7 (MKK7) deficiency and heightened susceptibility to arrhythmia. MKK7 deficiency prevents the phosphorylation of histone deacetylase-2, leading to the accumulation of filamentin A in the nucleus. This filamentin A then forms a complex with KLF4, causing KLF4 to dissociate from the promoter regions of several potassium channel genes. Consequently, this disrupts transcription levels, delays repolarization, and ultimately precipitates ventricular arrhythmia.
4.2 Diabetic cardiomyopathy
Diabetic Cardiovascular Diseases (DCVDs) account for over 50% of diabetes-related mortalities (112), including diabetic cardiomyopathy (a specific type of heart disease) cases (113, 114). The existence of a specific diabetic heart muscle disease that does not involve CAD or HTN was first proposed by Lundbaek in 1954 (115, 116). In 2008, the European Society of Cardiology (ESC) defined the disease as an abnormality of the heart muscle in terms of its structure and function, along with the absence of CADs, HTN, and valvular and congenital heart diseases sufficient to cause the observed myocardial abnormality (5). Nevertheless, the ESC stated again in 2018 that there was no definition of diabetic cardiomyopathy (117). Given the lack of consensus on its definition, it remains difficult to accurately assess epidemiologic data on diabetic cardiomyopathy-related morbidity and mortality. However, clinical trials in Type 2 Diabetes Mellitus (T2DM) patients revealed an HF prevalence of 10%–30% (117).
Diabetic cardiomyopathy is a major risk factor for diabetes-related morbidity and mortality. It is characterized by hypoinsulinemia and hyperglycemia in Type 1 Diabetes Mellitus (T1DM) patients and hyperinsulinemia or insulin resistance in T2DM patients (118). Although the underlying mechanism of some KLFs in diabetes remains unclear (44), studies have acknowledged the essential role KLFs play in many types of diabetes (71, 89, 119–124) (Table 3). For example, it was reported that KLF7 can regulate insulin sensitivity and susceptibility to type 2 diabetes by lowering adiponectin and leptin levels (126, 127), and can negatively regulate miR-132-3p to aggravate the transition of Human Umbilical Vein Endothelial Cells (HUVECs) to a mesenchymal state after high glucose exposure (128). Similarly, KLF14 has been implicated in increasing susceptibility to type 2 diabetes by regulating key genes associated with insulin resistance (122, 123). For instance, the risk alleles G of rs972283 and rs4731702 have been linked to this effect. Additionally, KLF11 plays a negative regulatory role in NDM, Maturity Onset Diabetes in Young (MODY) (129, 132), as well as T2DM (130) and Type 1B Diabetes (131). Mutations in KLF11 may hinder insulin secretion in pancreatic β-cells by inhibiting insulin promoter regulatory activity, consequently impairing insulin gene transcription in NDM and MODY. Moreover, KLF11 is involved in regulating hepatic glucose metabolism. Zhang et al. (130) found that overexpression of KLF11 in mouse hepatocytes inhibited the expression of gluconeogenic genes, such as peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) and phosphoenolpyruvate carboxykinase (PEPCK-C), thereby reducing cellular glucose output. As a negative regulator of adipogenesis, KLF2 is highly expressed in preadipocytes and regulates glucolipid metabolism and insulin sensitivity by directly inhibiting the PPARγ2 promoter activity (119). Furthermore, although KLFs 2, 4, and 9 have been strongly associated with Gestational Diabetes Mellitus (GDM) development, the specific regulatory roles of KLFs 2 and 4 remain unclear, necessitating additional research in a larger patient population (121, 125). Research indicates that KLF9 plays a crucial role in regulating hepatic glucose metabolism. Hepatic KLF9 overexpression induced by dexamethasone (Dex) and fasting directly binds to its promoter, stimulating the expression of the PGC-1α gene and activating the gluconeogenesis program. A mutation in KLF9 eliminates the stimulatory effect of Dex on cellular glucose output and effectively attenuates Dex-induced hyperglycemia (120), a critical finding for patients requiring long-term glucocorticoid therapy.
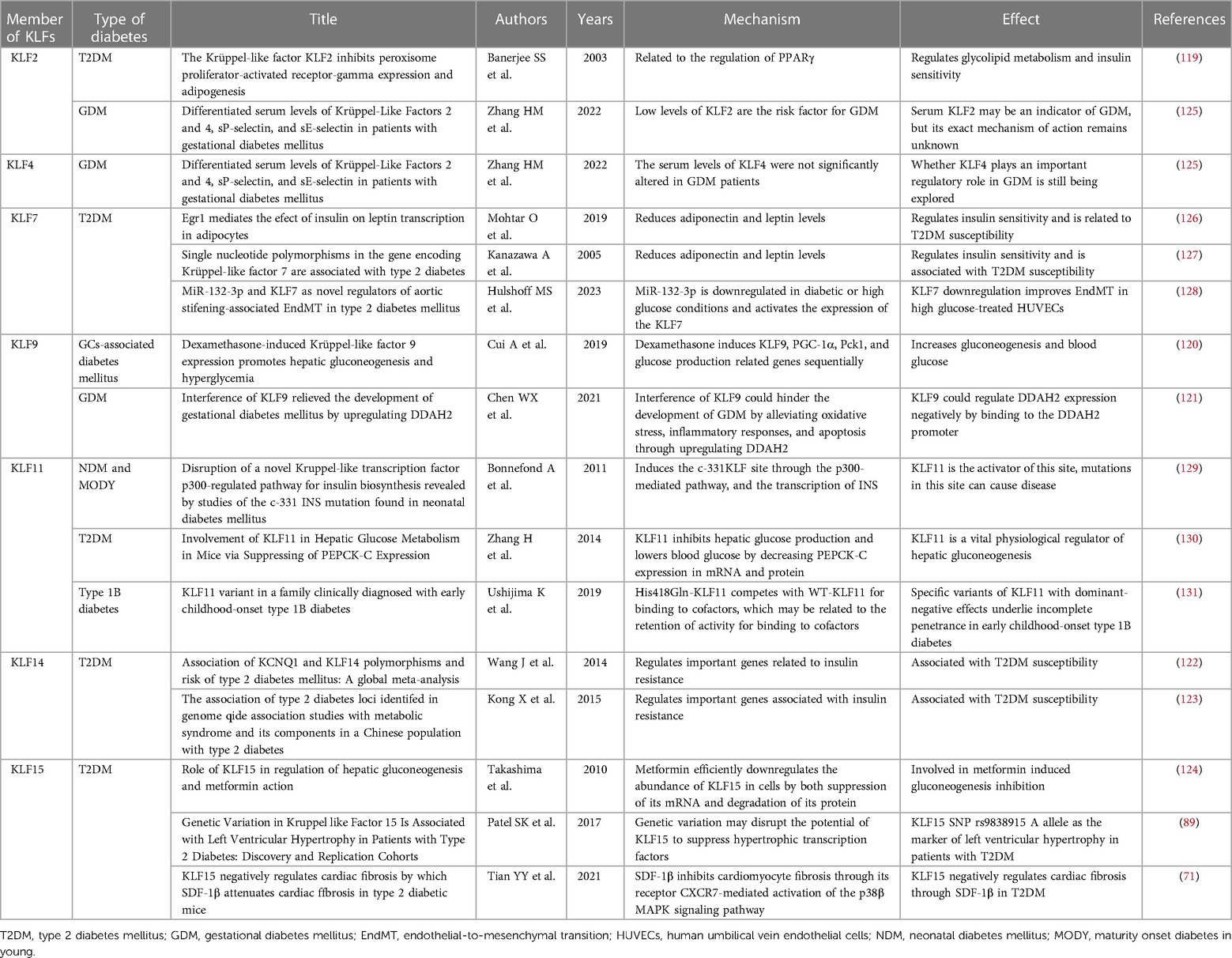
Table 3. Effect and mechanism of KLFs in diabetes mellitus.
While genetic susceptibility plays a significant role in the pathogenesis of T2DM, the identified genes influencing susceptibility to T2DM are still limited (133, 134). Kanazawa et al. (127) found that KLFs not only play vital roles in cellular differentiation and tissue development but are also implicated in the pathogenesis of T2DM. In their study, they genotyped 33 single nucleotide polymorphisms (SNPs) in 12 KLF genes in T2DM patients and found a direct association between an allele of the SNP site in the second intron of KLF7 and T2DM, suggesting KLF7 as a novel candidate gene for genetic susceptibility to T2DM and its role in promoting the development of diabetic cardiomyopathy. Diabetes mellitus is an independent predictor of left ventricular hypertrophy (LVH), but not all diabetic patients develop LVH, indicating genetic components are involved. A clinical study investigated the association between the KLF15 gene and LVH in T2DM patients. Patel et al. (89) prospectively recruited 318 T2DM patients without known cardiac disease for transthoracic echocardiographic evaluation and genotyping for two KLF15 SNPs (rs9838915 and rs6796325). They found that the A allele of the rs9838915 SNP in the KLF15 gene was associated with increased left ventricular mass in patients, providing more accurate risk stratification for developing HF. Diabetic cardiomyopathy pathophysiology is multifaceted and involves complex metabolic pathways, with the main pathological features being myocardial hypertrophy and fibrosis, inflammation, cellular autophagy and apoptosis, and elevated OS markers (135–137). Myocardial hypertrophy makes the myocardium less compliant, causing diastolic dysfunction, which ultimately results in arrhythmia, HF, and even SCD (138, 139). In these processes, KLFs regulates the upstream and downstream pathways (Figure 4).
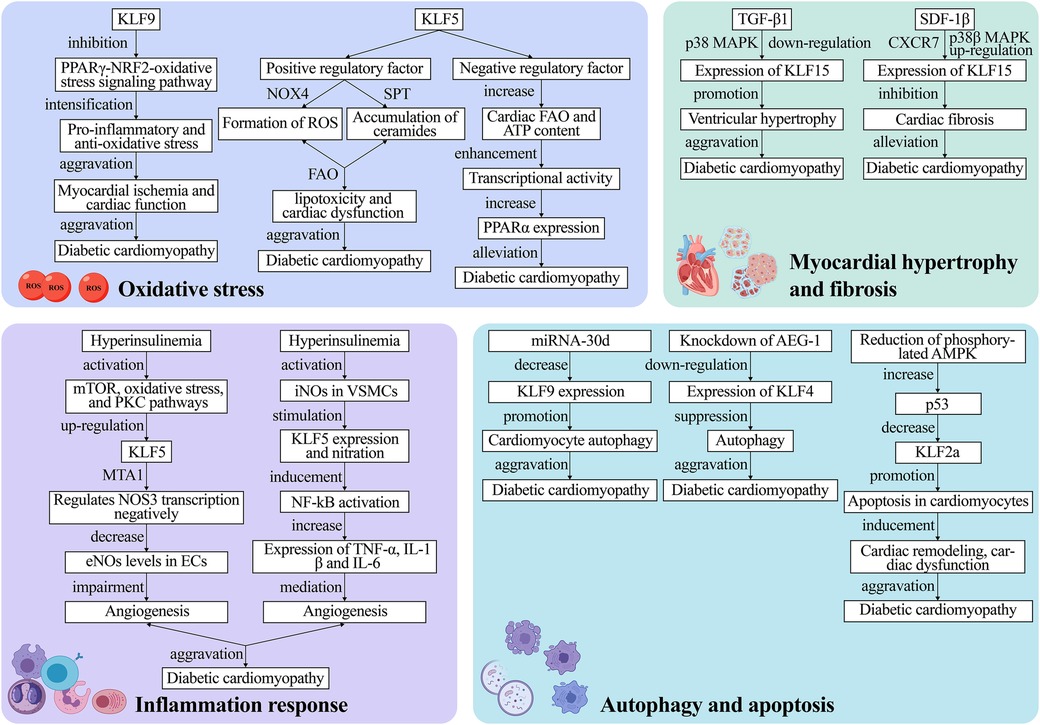
Figure 4. The mechanism of KLFs in regulation of diabetic cardiomyopathy. The pathophysiological process of diabetic cardiomyopathy mainly comprises four major pathways: oxidative stress, inflammation, myocardial hypertrophy and fibrosis as well as cell autophagy and apoptosis. These processes lead to irreversible heart failure. KLFs, as upstream and downstream regulators, can exacerbate or alleviate diabetic cardiomyopathy by targeting various metabolic pathways or signaling pathways, and the specific mechanisms of their action are presented in the figure. NOX4, NADPH-oxidase 4; ROS, reactive oxygen species; FAO, fatty acid oxidation; PPARα, peroxisome proliferator-activated receptor; mTOR, mammalian target of rapamycin; PKC, protein kinase C; eNOS, endothelial nitric oxide synthase; ECs, endothelial cells; iNOS, inducible nitric oxide synthase; VSMCs, vascular smooth muscle cells; p38 MAPK, p38 mitogen-activated protein kinase; SDF-1β, stromal cell-derived factor-1β; TGF-β1, transforming growth factor-β1; AEG-1, astrocyte elevated gene-1.
4.2.1 Oxidative stress
Multiple studies have discovered that OS mediated by excessive Reactive Oxygen Species (ROS) levels is the pathogenic mechanism underlying type 1 and type 2 diabetes-related cardiomyopathy (140–142). The PPARγ-NRF2-OS signaling pathway stimulates cell survival signals, regulates autophagy, and exerts cardio-protective effects in cardiomyocytes (143). On the other hand, KLF9 aggravates ischemic injury in cardiomyocytes by exerting pro-inflammatory and anti-oxidative stress effects and deteriorates cardiac function by inhibiting PPARγ expression and transcriptionally lowering NRF2 expression and nuclear translocation (68, 69).
Diabetic cardiomyopathy is also associated with altered Fatty Acid Oxidation (FAO). The cardiomyocyte KLF5 can regulate cardiac FAO and induce de novo synthesis of ceramides through various pathways such as NADPH-Oxidase 4 (NOX4)-mediated ROS formation or SPT-mediated ceramide accumulation, leading to lipotoxicity and cardiac dysfunction (11). Correction of hyperglycemia with the SGLT2 inhibitor dapagliflozin reverses KLF5 expression in early diabetes (63). While KLF5 appears detrimental to cardiac function, another study proposed that transient KLF5 over-expression could be beneficial (63). Specifically, they discovered that cardiomyocyte-specific KLF5-deficient aMHC-KLF5-/- mice progressively developed cardiac dysfunction with signs of DCM. This outcome may be linked to the inhibition of cardiac Peroxisome Proliferator-Activated Receptor-α (PPARα) expression and reduced KLF5 transcriptional activity (64, 65). In the heart, KLF5 positively regulates FAO through PPAR transcription (66). Furthermore, KLF5 transcriptionally activates PPARα and decreases with PPARα gene expression (67). Additionally, cardiomyocyte-specific KLF5 ablation decreases cardiac FAO and ATP content, as well as transcriptional activity, ultimately triggering cardiac dysfunction. The above-mentioned findings imply that KLF5 has a dual function that could be exploited to treat or even reverse cardiomyopathy. However, this hypothesis requires further verification in future clinical trials.
4.2.2 Inflammation response
Hyperinsulinemia and hyperglycemia could negatively affect angiogenesis by impairing the Endothelial Cell (EC) and Vascular Smooth Muscle Cell (VSMC) proliferation and transport. It has been reported that KLF5 induces vascular inflammation in diabetic VSMCs (144). Specifically, this process is divided into two parts: Negative regulation of endothelial Nitric Oxide Synthase (eNOS) expression and activation of inducible Nitric Oxide Synthase (iNOS) production. Inhibiting insulin signaling activates the mammalian Target of Rapamycin (mTOR), Protein Kinase C (PKC) pathways, and OS, up-regulates KLF5 along with MTA1, and negatively regulates NOS3 transcription, thereby leading to reduced eNOS levels in ECs, which impairs in vitro and in vivo angiogenesis (144). On the other hand, hyperglycemia activates iNOS in VSMCs, which, in turn, stimulates KLF5 expression and nitration. Stimulated KLF5 binds to NF-κB, inducing NF-κB activation, which increases TNFα, IL1β, and IL-6 expression and mediates vascular inflammation (145).
4.2.3 Myocardial hypertrophy and fibrosis
Myocardial hypertrophy and cardiac fibrosis are prominent pathological remodeling features in hemodynamic or neurohormonal stress induced diabetic cardiomyopathy. Myocardial hypertrophy causes cardiac systolic and diastolic dysfunction, and KLF15 is a crucial negative regulator of diabetic cardiomyopathy-induced cardiac dysfunction and myocardial fibrosis (24, 72). Leenders et al. (73) discovered that TGF-β1 downregulates KLF15 in a p38 Mitogen-Activated Protein Kinase (p38 MAPK)-dependent manner and promotes ventricular hypertrophy. It was reported that KLF15 negatively regulated cardiac fibrosis in mice with type 2 diabetes, and the cardio-protective effects of Stromal Cell-Derived Factor-1β (SDF-1β) are mediated by binding to the CXCR7 receptor and the p38β MAPK-mediated upregulation of KLF15 (71). Furthermore, Takashima et al. (124) demonstrated that KLF15 plays an emerging role in regulating energy production and amino acid degradation. Gu et al. (146) recently found that the amide alkaloid Piperlongumine (PLG) extracted from piper longum exerted anti-hypertrophic and anti-fibrotic effects after angiotensin II (Ang II) treatment. This mechanism entails reduced levels of phosphorylated Akt and transcriptional regulation of fibrotic gene expression. Overall, this study enhances our understanding of the use of natural compounds for the targeted treatment of clinical diseases, implying that the co-mediation of the negative regulator KLF15 with natural compounds may be an entirely new therapeutic strategy for cardiomyopathy treatment in the future.
4.2.4 Autophagy and apoptosis
Autophagy, a dynamic intracellular degradation mechanism, is a highly conserved eukaryotic cellular process that plays an important role in maintaining intracellular homeostasis as well as the synthesis, degradation, and recycling of cellular products. Growing evidence suggests that autophagy exerts a beneficial effect on diabetic hearts (147–150). Zhang and Wang recently found that decreased autophagic activity in cardiomyocytes is closely associated with diabetes-related cardiomyopathy and that restoring autophagy levels hinders diabetic cardiomyopathy development (70). According to research, KLF9 is a target gene of miR-30d that correlates negatively with miR-30d expression. Therefore, reduced KLF9 expression regulates autophagy in cardiomyocytes and exacerbates diabetic cardiomyopathy. Furthermore, the SGLT-2 inhibitor promoted cardiomyocyte autophagy by blocking miRNA-30d expression, and miRNA-30d negatively regulated KLF9, thereby improving cardiac function in diabetic cardiomyopathy mice (70). Additionally, KLF4 upregulation exacerbates cardiomyocyte autophagy; hence, aggravating diabetic cardiomyopathy. For the first time, Zhao et al. (62) found that the Astrocyte Elevated Gene-1 (AEG-1) can modulate autophagy in diabetic cardiomyopathy by regulating KLF4 expression, which is expected to be a new therapeutic target for diabetic cardiomyopathy treatment.
Increased cardiomyocyte apoptosis was found to be a major cause of systolic and diastolic dysfunction in diabetic models (151). Therefore, inhibiting cardiomyocyte apoptosis may effectively prevent myocardial remodeling in diabetic cardiomyopathy (152–154). It has been reported that KLF2a exerts cardio-protective effects through the AMPK-p53-KLF2a pathway. In a Streptozocin (STZ)-induced hyperglycemic zebrafish model, Wang et al. (61) found that decreasing phosphorylated AMPK elevated p53, leading to KLF2a downregulation, which in turn, promoted cardiomyocyte apoptosis and induced cardiac remodeling and dysfunction. KLF10 participates in the regulation of various aspects of tissue homeostasis and cellular functions, including proliferation, differentiation, and apoptosis (155, 156). In a study by Kong et al. (157), loss-of-function analyses of the zebrafish homologs of human KLF10 were conducted using antisense morpholino (MO). The results revealed that embryos injected with KLF11b-MO exhibited developmental retardation and cell death, whereas those injected with KLF11a-MO did not display significant abnormalities in development. Moreover, embryos co-injected with KLF11b-MO and p53-MO showed reduced apoptosis. These findings suggest that KLF10 serves as a critical negative regulator of p53-dependent transcription, and the KLF10/p53 complex contributes to apoptosis and hence the maintenance of tissue homeostasis.
4.3 Dilated cardiomyopathy
Dilated Cardiomyopathy (DCM), a multifactorial disorder associated with genetics, immunity, infection, and the environment, is primarily characterized by heart enlargement and systolic dysfunction (51, 158, 159). The prevalence of DCM is at 1:2,500 in 2013 (160) and the diseases is always progressive, eventually leading to irreversible HF and sudden death. The annual SCD incidence in DCM is 2%–4% (161, 162). Given that the disease is multifactorial, effectively targeting treatment to its etiology has become a major research focus (158, 163, 164). In this regard, multiple studies recently demonstrated the critical functions of KLFs in DCM, including regulating molecular mechanisms contributing to DCM progression.
Multiple mechanisms such as inflammation, Endoplasmic Reticulum Stress (ERS), and mitochondrial dysfunction play vital roles in the DCM-associated ventricular dysfunction progression and HF development. Specifically, it has been reported that myocardial injury triggers an inflammatory response. Zhang et al. (74) found that the KLF2 overexpression-derived Extracellular Vesicles (EVs) reduced cardiac inflammation and improved left ventricular dysfunction in DCM mice by inhibiting Ly6Chigh monocyte mobilization via targeting the CCR2 protein. High KLF2 expression in ECs exerts an anti-inflammatory effect, implying that KLF2 may be a potential therapeutic target for DCM treatment.
Furthermore, growing evidence highlights the crucial role of genetic defects in the pathogenic mechanism of DCM (165). As one of the mutation carriers of disease-causing genes, KLF5 mutations co-segregating with DCM have shown complete penetrance in this family, and genetically compromised KLF5 is highly susceptible to DCM (76). Notably, KLF13 is also co-segregated from DCM. Also known as FKLF2/BTEB3/RFLAT-1/RFLAT1/NSLP1, KLF13 is a new DCM susceptibility gene localized on chromosome 15q13.3. Guo et al. (77) conducted sequencing analysis focusing on specific genes within the localization region of human chromosome 15q13.1-q13.3 and identified three mutations that co-segregated with the dilated cardiomyopathy (DCM) phenotype with complete penetrance. These mutations, namely c.430G>T (p.E144X), c.580G>T (p.E194X), and c.595T>C (p.C199R), exhibited impaired transactivation of the target genes ACTC1 and MYH7, both individually and in combination with GATA4, a gene known to induce DCM. Furthermore, these mutations displayed reduced binding capacity to the promoters of ACTC1 and MYH7. Furthermore, the intracellular distribution of E144X mutant KLF13 was disrupted. In summary, detecting the aforementioned new susceptibility genes and loci offers new insights into the DCM molecular pathogenesis and may provide guidance for precision medicine for DCM.
4.4 Desmin-related cardiomyopathy
Desmin, a major intermediate filament protein in cardiomyocytes, is a fundamental component of the cellular structure and the Purkinje fibers of the conduction system (166, 167). Desmin is encoded by the DES gene (OMIM *125660), and mutations in this gene are hypothesized to disrupt mechanical stress in the muscle, leading to rhabdomyolysis (168–170) Additionally, desmin is crucial in balancing ROS generation with antioxidant defense. According to research, MnSOD overexpression and/or catalase are the major antioxidant defense systems that can reduce ROS production and improve cardiac function in desmin-deficient mice hearts (171).
Desmin-Related Cardiomyopathy (DRCM) presents early with lower extremity muscle weakness and gait disturbances before progressing to the proximal, respiratory, fascial, and cardiac muscles, eventually leading to arrhythmias and congestive heart failure. Notably, heart defects occasionally precede skeletal muscle defects (167, 172, 173). A meta-analysis of 159 patients with 40 different DES gene mutations revealed that more than 70% of carriers exhibited myopathy or muscular weakness and that about 50% of carriers had cardiomyopathies. In another study, up to 60% of patients had a cardiac conduction disorder or arrhythmias, such as an atrioventricular block (174). Except for symptomatic treatments for alleviating the symptoms, especially in the cardiac area focusing on cardiovascular complications (175, 176), there are currently no targeted therapies for DRCM. Khudiakov et al. (79) delivered four reprogramming factors (OCT4, KLF4, SOX2, and CMYC) with Sendai virus from patient-specific fatty tissue-derived pluripotent Mesenchymal Stromal Cells (MSCs) carrying heterozygous splice site mutations in the DES gene using a non-integrative reprogramming approach, ultimately yielded a human iPSC line. Establishing an iPSC line holds promise for addressing DRCM and for the future development of innovative drugs. Several KLFs may share similar regulatory mechanisms across various muscle tissues. Moreover, within the same muscle tissue, there could be synergistic or antagonistic interactions among KLFs, crucial for muscle tissue development and functional regulation. For instance, KLF2, KLF4, KLF10, and KLF15 have been implicated in these processes. In mouse skeletal muscle cells, the extracellular signal-regulated kinase 5 (ERK5) signaling pathway can induce the upregulation of KLF2 and KLF4 genes via the Sp1 transcription factor. This activation subsequently promotes the expression of nephronectin (Npnt) genes, facilitating skeletal muscle cell fusion through enhanced cell-matrix adhesion (78). KLF15, on the other hand, can stimulate the expression of the slow-twitch fiber gene Myh7 (MHC-β/slow) by targeting the nuclear factor of activated T-cells, cytoplasmic 1 (NFATc1) gene, thereby positively regulating skeletal muscle differentiation (82). Conversely, KLF10 inhibits myoblast proliferation by suppressing the function of pro-proliferative signaling molecules (80) and the expression of cyclin (81). Most mutations associated with DRCM, such as those affecting CRYAB and BAG3, are missense or small deletion mutations. The formation of aggregates appears to be a significant trigger for DRCM. However, the downstream effects of these mutations are diverse and heterogeneous (177). While a direct link between DRCM and KLFs has yet to be established, ongoing advancements in molecular and cellular biology hold promise for the development of molecular therapies for DRCM. Targeting the regulation of DES gene expression by KLFs may emerge as a promising strategy for reducing the expression of mutant DES alleles and mitigating DRCM pathology.
4.5 Hypertrophic cardiomyopathy
Hypertrophic Cardiomyopathy (HCM), a relatively common monogenic heart disease with autosomal dominant inheritance, is characterized by ventricular hypertrophy on echocardiography, mostly in the left ventricle (51, 178, 179). The disease was once assumed to be a sudden exercise death disease that primarily affected young men, but is no longer restricted to young males. According to the 2018 global hypertrophic cardiomyopathy burden statistics, HCM affected 6.3 billion people or 88% of the world's population (178). Auspiciously, HCM treatment has progressed from limited palliative pharmacotherapy and occasional high-risk surgery to more definitive screening tools and highly efficient interventional therapies, resulting in a 0.5% annual reduction in morbidity and mortality (180–183).
Although HCM is usually a non-progressive disease, HF symptoms may occur or worsen at any age, most often in middle-aged individuals (184, 185). Efficient management of myocardial hypertrophy is crucial given its potential progression to HF. One significant hallmark of this transition is the shift in myocardial substrate preference from FAO to glycolysis, resembling an embryonic metabolic profile. KLFs have emerged as key transcriptional regulators in pathological cardiac hypertrophy and metabolism. For instance, Wang et al. (86) demonstrated that both cardiac-specific knockdown and overexpression of KLF7 disrupt the balance between glycolysis and fatty acid metabolism, leading to cardiac hypertrophy and myocardial fibrosis. Additionally, KLF4 serves as a central transcriptional regulator of cardiac metabolic function and mitochondrial dynamics, reactivating fetal cardiac genes during hypertrophic development (83, 84). In vivo studies have shown that KLF4 can modulate isoproterenol-induced cardiac hypertrophy by regulating myocardin (MYOCD) expression and activity, and cardiomyocyte-specific knockdown of KLF4 exacerbates hypertrophy (85). Oxytocin (OT), a hormone involved in cardiovascular homeostasis, mitigates cardiac hypertrophy by targeting the lncRNA GAS5/miR-375-3p/KLF4 axis to inhibit the PI3K/AKT pathway (186). Moreover, TIEG1 has been identified as a critical player in cardiac hypertrophy (87). The TGFb Inducible Early Gene-1 (TIEG1) is classified as a member of the KLF10 family and is involved in gene transcription regulation (42). Subramaniam et al. (88) studied male mice aged 4–16 months and found that TIEG1-/- mice exhibited cardiac hypertrophy compared to wild-type animals. This finding demonstrates that TIEG1, a gene transcription inducer or repressor, can inhibit cell proliferation and induce apoptosis by activating the TGF-β/Smad pathway. KLF15 is a pivotal regulator of cardiac metabolic homeostasis, primarily governing myocardial lipid flux. It plays a significant role in controlling the transcriptional network associated with cardiac metabolism (187). Notably, KLF15 exerts its regulatory effects by negatively modulating pro-hypertrophic and pro-fibrotic markers such as brain natriuretic peptide (BNP) and connective tissue growth factor (CTGF) through the p38-MAPK signaling pathway. This pathway has been identified as a crucial negative regulator of cardiac hypertrophy (90). Deletion or inhibition of KLF15 reduces the suppression of pro-hypertrophic factors and the stimulation of fibrotic signaling pathways, resulting in Left Ventricular Hypertrophy (LVH) development (58, 89).
4.6 Ischemic cardiomyopathy
Ischemic Cardiomyopathy (ICM), generally referred to as systolic left ventricular dysfunction in the context of obstructive CAD, is the leading cause of HF worldwide (188, 189). Ventricular dysfunction in CAD patients is usually caused by the irreversible loss of viable myocardium after Acute Myocardial Infarction (AMI) (189, 190). According to research, KLF5 regulates ceramide accumulation and lipid metabolism after Myocardial Infarction (MI) (91). Furthermore, KLF5 is involved in the aggravation of Ischemic Heart Failure (IHF), and induced KLF5 increases the expression of some regulatory factors [including Serine-Palmitoyl-Transferase-Long-Chain-Base-Subunit (SPTLC)1 and SPTLC2], and ceramide biosynthesis, leading to systolic dysfunction and eccentric remodeling, thereby decreasing ejection fraction and exacerbating IHF (91).
Following infarction, the remaining surviving segmental myocardium adapts to chronic perfusion insufficiency by reducing its energy requirements blocking myofibrillar protein expression, with the heart preferentially relying on glucose absorption rather than fatty acids for energy supply. This gene expression reprogramming via fluctuations in the cellular microenvironment is known as epigenetic reprogramming (191–193). It has been reported that KLF15 is an upstream regulator of metabolic gene expression, and that it is negatively regulated by EZH2 (an epigenetic regulator) in a SET structural domain-dependent manner for cardiac metabolism and structural reprogramming (92). Additionally, KLF15 suppresses gene expression and negatively regulates adverse cardiac remodeling and hypertrophy in ICM progression (72, 92, 96). Specifically, KLF15 antagonizes the expression of various genes actively involved in cardiac remodeling. Rogers and Otis studied the mechanism of resveratrol (an antioxidant) in ischemic heart treatment. They found that chronic myocardial ischemia significantly reduced KLF15 expression, which then increased the transcription of TGF-β1 and Nox4 mRNA, and that sustained TGF-β1/Nox4 signaling led to the production of large amounts of oxidants, which aggravated OS in diseased hearts, as well as myocardial fibrosis and hypertrophy (194). Haldar et al. (93) reported that the KLF15-p53-p300 pathway may also be a therapeutic target in CVD treatment. Specifically, they found that KLF15 activation inhibits the p300-mediated p53 acetylation. On the other hand, KLF15 deficiency causes p53 hyperacetylation in the aorta and heart (93–95). Mice lacking KLF15 develop aortopathy and cardiomyopathy in p53-dependent and p300 acetyltransferase-dependent manners.
4.7 Obesity-associated cardiomyopathy
Globally, the morbidity and mortality levels due to HF have been on the rise, making HF a major threat to human health. The etiology of HF is multifactorial, and studies have shown that uncorrected overweight and obesity may be independent risk factors for development of CVDs (195). Evidence from previous experimental, clinical, and epidemiological studies has pointed to the existence of a distinct disease entity known as “obesity cardiomyopathy”, which occurs independently of other CVD risk factors such as hyperlipidemia, hypertension, and diabetes mellitus (196). “Obesity cardiomyopathy” refers to metabolic and functional abnormalities of the heart caused by obesity alone (197, 198). In general, chronic obesity is strongly associated with myocardial remodeling, and its clinical features may range from LVH and myocardial fibrosis which eventually evolve to HF (199–201). Obesity also affects myocardial electrophysiology, which increases the prevalence of atrial fibrillation (202, 203).
Multiple studies have shown that berberine prevents heart disease, controls cardiac remodeling (204–207), and ameliorates KLF4-dependent obesity-related cardiac damage (97, 98). Ding et al. (99) treated high fat diet-induced obese mice with berberine and found that this drug ameliorated myocardial mitochondrial biogenesis and activity. However, KLF4 silencing reduced the mitochondrial mass, ATP production, oxygen consumption, and lipid metabolism, which were upregulated by berberine treatment. It also reversed berberine's structural benefits on cardiac anti-hypertrophic and fibrotic, anti-inflammatory, and antioxidant properties. In summary, these findings suggest that KLF4 indirectly mediates obesity-associated cardiac injury, controls myocardial remodeling, and confers protection on the heart.
5 Summary and outlook
Cardiovascular diseases (CVDs), including cardiomyopathy, contribute to high morbidity and mortality worldwide. This calls for investigations to identify targeted strategies for preventing and treating patients with cardiomyopathy. Unfortunately, the etiology of cardiomyopathies has not been fully clarified given its multifactorial nature. Furthermore, the pathophysiological mechanisms underlying cardiac dysfunction and myocardial remodeling in various types of cardiomyopathies are highly intricate and necessitate additional research. As such, the exploration of novel targets or regulators, both upstream and downstream, that exert direct or indirect influence on the progression of cardiomyopathy has become of paramount importance. In recent years, there has been renewed research interest on the role of Krüppel-Like Factors (KLFs) in various biological processes and conditions such as myocardial diseases, such as oxidative stress, inflammatory responses, myocardial hypertrophy and fibrosis and apoptosis. KLFs are a group of DNA-binding proteins associated that can activate or suppress genes, stimulate cell growth, differentiation, apoptosis, and biological processes related to tissue development and maintenance. Here, we review the mechanisms underlying the development of 7 cardiomyopathies focusing on the involvement of 18 members of the KLFs family in terms of their classification. For example, KLF13, localized on chromosome 15q13.3, is a novel gene that increases the risk of cardiomyopathy (DCM), whereas KLF15 can prevent diabetic cardiomyopathy-induced cardiac dysfunction and myocardial fibrosis as well as hypertrophic cardiomyopathy (HCM) by repressing pro-hypertrophic transcription factors to attenuate LVH.
While numerous studies, including those conducted in animal models, cell trials, and clinical settings, have highlighted the importance of KLFs as crucial regulators or markers of cardiomyopathies, the precise pathophysiological roles of certain KLF members in different types of cardiomyopathy remain poorly understood. For instance, it is unclear whether KLF2 and KLF4 participate in gestational diabetes mellitus-associated cardiomyopathy. Therefore, there is a need for more comprehensive investigations to elucidate the signaling pathways modulated by KLFs in various types of cardiomyopathies. These pathways may include, but are not limited to, the Hippo/Yap signaling pathway, Wnt signaling, Hedgehog (Hh) signaling, Notch pathway, and Mitogen-activated protein kinases (MAPKs) signaling. In the article, we describe the importance of KLFs in cardiomyopathies and hope to provide new ideas regarding the cellular and molecular biological mechanisms involved in the regulation of KLFs and reveal new insights into development of precision medicine for patients with cardiomyopathies. In future, we predict that pharmacology and invasive procedures will no longer be the only means of treating cardiomyopathy, preventing sudden death, or heart transplantation. Therefore, a better understanding of the molecular genetics of cardiomyopathy wil boost the development of potential therapeutic targets for cardiomyopathy.
Author contributions
L-KG: Writing – original draft. H-JL: Writing – original draft. L-JJ: Writing – review & editing. X-CP: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article.
This work was supported by National innovation and entrepreneurship training program for College Students (202010489017 to X-CP), Jingzhou Science and Technology Bureau Project (2022HC78 to X-CP), the College Students Innovative Entrepreneurial Training Program in Yangtze University (Yz2022297 to X-CP), the Scientific Research Project of Education Department of Yangtze university (JY2020134 to X-CP), Hubei Province Natural Science Foundation of China (2017CFB786 to X-CP).
Acknowledgments
The authors would like to thank all the reviewers who participated in the review and MJEditor (www.mjeditor.com) for its linguistic assistance during the preparation of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Contemporary definitions and classification of the cardiomyopathies: an American heart association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation. (2006) 113:1807–16. doi: 10.1161/CIRCULATIONAHA.106.174287
2. Fiedler A. Ueber akute interstitielle mvokarditis. In: zu Dresden R, editors. Festschrift zur feier des funfzigjahrigen bestehens des stadtkrankenhauses zu Dresden-friedrichstadt. Part 2. Dresden, Germany: Wilhelm Baensch (1899). p. 3–24.
3. Brandenburg RO, Cherian G, False AO, Grosgogeat Y, Kawai C, Loogen F, et al. Report of the WHO/ISFC task force on the definition and classification of cardiomyopathies. Br Heart J. (1980) 44:672–3. doi: 10.1136/hrt.44.6.672
4. Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O'Connell J, et al. Report of the 1995 world health organization/international society and federation of cardiology task force on the definition and classification of cardiomyopathies. Circulation (1996) 93:841–2. doi: 10.1161/01.cir.93.5.841
5. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: a position statement from the European society of cardiology working group on myocardial and pericardial diseases. Eur Heart J. (2008) 29:270–6. doi: 10.1093/eurheartj/ehm342
6. Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. (1988) 318:129–33. doi: 10.1056/NEJM198801213180301
7. Olson TM, Michels VV, Thibodeau SN, Tai YS, Keating KM. Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science. (1998) 280:750–2. doi: 10.1126/science.280.5364.750
8. Kubo T, Kitaoka H. Genetic testing for cardiomyopathy in Japan 2022: current Status and issues of precision medicine. J Card Fail. (2023) 29:805–14. doi: 10.1016/j.cardfail.2022.11.017
9. Miller IJ, Bieker JJ. A novel, erythroid cell-specific murine transcription factor that binds to the CACCC element and is related to the krüppel family of nuclear proteins. Mol Cell Biol. (1993) 13:2776–86. doi: 10.1128/mcb.13.5.2776-2786.1993
10. Santoyo-Suarez MG, Mares-Montemayor JD, Padilla-Rivas GR, Delgado-Gallegos JL, Quiroz-Reyes AG, Roacho-Perez JA, et al. The involvement of Krüppel-Like Factors in cardiovascular diseases. Life (Basel). (2023) 13:420. doi: 10.3390/life13020420
11. Kyriazis ID, Hoffman M, Gaignebet L, Lucchese AM, Markopoulou E, Palioura D, et al. KLF5 Is induced by FOXO1 and causes oxidative stress and diabetic cardiomyopathy. Circ Res. (2021) 128:335–57. doi: 10.1161/CIRCRESAHA.120.316738
12. Sweet DR, Fan L, Hsieh PN, Jain MK. Krüppel-Like factors in vascular inflammation: mechanistic insights and therapeutic potential. Front Cardiovasc Med. (2018) 5:6. doi: 10.3389/fcvm.2018.00006
13. Zhao Y, Song W, Wang L, Rane MJ, Han F, Cai L. Multiple roles of KLF15 in the heart: underlying mechanisms and therapeutic implications. J Mol Cell Cardiol. (2019) 129:193–6. doi: 10.1016/j.yjmcc.2019.01.024
14. Tetreault MP, Yang Y, Katz JP. Krüppel-Like Factors in cancer. Nat Rev Cancer. (2013) 13:701–13. doi: 10.1038/nrc3582
15. Pearson R, Fleetwood J, Eaton S, Crossley M, Bao S. Krüppel-like transcription factors: a functional family. Int J Biochem Cell Biol. (2008) 40:1996–2001. doi: 10.1016/j.biocel.2007.07.018
16. Chang E, Nayak L, Jain MK. Krüppel-Like Factors in endothelial cell biology. Curr Opin Hematol. (2017) 24:224–9. doi: 10.1097/MOH.0000000000000337
17. McConnell BB, Yang VW. Mammalian Krüppel-Like Factors in health and diseases. Physiol Rev. (2010) 90:1337–81. doi: 10.1152/physrev.00058.2009
18. Kotlyarov S, Kotlyarova A. Participation of Krüppel-Like Factors in atherogenesis. Metabolites. (2023) 13:448. doi: 10.3390/metabo13030448
19. Bialkowska AB, Yang VW, Mallipattu SK. Krüppel-Like Factors in mammalian stem cells and development. Development. (2017) 144:737–54. doi: 10.1242/dev.145441
20. Mas C, Lussier-Price M, Soni S, Morse T, Arseneault G, Di Lello P, et al. Structural and functional characterization of an atypical activation domain in erythroid Kruppel-like factor (EKLF). Proc Natl Acad Sci U S A. (2011) 108:10484–9. doi: 10.1073/pnas.1017029108
21. Perkins AC, Sharpe AH. Orkin SH: lethal beta-thalassaemia in mice lacking the erythroid CACCC-transcription factor EKLF. Nature. (1995) 375:318–22. doi: 10.1038/375318a0
22. Pei L, Leblanc M, Barish G, Atkins A, Nofsinger R, Whyte J, et al. Thyroid hormone receptor repression is linked to type I pneumocyte-associated respiratory distress syndrome. Nat Med. (2011) 17:1466–72. doi: 10.1038/nm.2450
23. Jha P, Das H. KLF2 in regulation of NF-κB-mediated immune cell function and inflammation. Int J Mol Sci. (2017) 18:2383. doi: 10.3390/ijms18112383
24. Fisch S, Gray S, Heymans S, Haldar SM, Wang B, Pfister O, et al. Kruppel-like factor 15 is a regulator of cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. (2007) 104:7074–9. doi: 10.1073/pnas.0701981104
25. Turner J, Crossley M. Cloning and characterization of mCtBP2, a co-repressor that associates with basic Krüppel-like factor and other mammalian transcriptional regulators. Embo j. (1998) 17:5129–40. doi: 10.1093/emboj/17.17.5129
26. van Vliet J, Turner J, Crossley M. Human Krüppel-like factor 8: a CACCC-box binding protein that associates with CtBP and represses transcription. Nucleic Acids Res. (2000) 28:1955–62. doi: 10.1093/nar/28.9.1955
27. Schuierer M, Hilger-Eversheim K, Dobner T, Bosserhoff AK, Moser M, Turner J, et al. Induction of AP-2alpha expression by adenoviral infection involves inactivation of the AP-2rep transcriptional corepressor CtBP1. J Biol Chem. (2001) 276:27944–9. doi: 10.1074/jbc.M100070200
28. Simmen FA, Alhallak I, Simmen RCM. Krüppel-like factor-9 and Krüppel-like factor-13: highly related, multi-functional, transcriptional repressors and activators of oncogenesis. Cancers (Basel). (2023) 15:5667. doi: 10.3390/cancers15235667
29. Cook T, Gebelein B, Belal M, Mesa K, Urrutia R. Three conserved transcriptional repressor domains are a defining feature of the TIEG subfamily of Sp1-like zinc finger proteins. J Biol Chem. (1999) 274:29500–4. doi: 10.1074/jbc.274.41.29500
30. Kaczynski J, Zhang JS, Ellenrieder V, Conley A, Duenes T, Kester H, et al. The Sp1-like protein BTEB3 inhibits transcription via the basic transcription element box by interacting with mSin3A and HDAC-1 co-repressors and competing with Sp1. J Biol Chem. (2001) 276:36749–56. doi: 10.1074/jbc.M105831200
31. Truty MJ, Lomberk G, Fernandez-Zapico ME, Urrutia R. Silencing of the transforming growth factor-beta (TGFbeta) receptor II by Kruppel-like factor 14 underscores the importance of a negative feedback mechanism in TGFbeta signaling. J Biol Chem. (2009) 284:6291–300. doi: 10.1074/jbc.M807791200
32. Daftary GS, Lomberk GA, Buttar NS, Allen TW, Grzenda A, Zhang J, et al. Detailed structural-functional analysis of the Krüppel-like factor 16 (KLF16) transcription factor reveals novel mechanisms for silencing Sp/KLF sites involved in metabolism and endocrinology. J Biol Chem. (2012) 287:7010–25. doi: 10.1074/jbc.M111.266007
33. Pei J, Grishin NV. A new family of predicted Krüppel-like factor genes and pseudogenes in placental mammals. PLoS One. (2013) 8(11):e81109. doi: 10.1371/journal.pone.0081109
34. Imhof A, Schuierer M, Werner O, Moser M, Roth C, Bauer R, et al. Transcriptional regulation of the AP-2alpha promoter by BTEB-1 and AP-2rep, a novel wt-1/egr-related zinc finger repressor. Mol Cell Biol. (1999) 19:194–204. doi: 10.1128/MCB.19.1.194
35. Shao M, Ge GZ, Liu WJ, Xiao J, Xia HJ, Fan Y, et al. Characterization and phylogenetic analysis of Krüppel-like transcription factor (KLF) gene family in tree shrews (tupaia belangeri chinensis). Oncotarget. (2017) 8:16325–39. doi: 10.18632/oncotarget.13883
36. Anderson KP, Kern CB, Crable SC, Lingrel JB. Isolation of a gene encoding a functional zinc finger protein homologous to erythroid Krüppel-like factor: identification of a new multigene family. Mol Cell Biol. (1995) 15:5957–65. doi: 10.1128/MCB.15.11.5957
37. Shields JM, Christy RJ, Yang VW. Identification and characterization of a gene encoding a gut-enriched Krüppel-like factor expressed during growth arrest. J Biol Chem. (1996) 271:20009–17. doi: 10.1074/jbc.271.33.20009
38. Conkright MD, Wani MA, Anderson KP, Lingrel JB. A gene encoding an intestinal-enriched member of the Krüppel-like factor family expressed in intestinal epithelial cells. Nucleic Acids Res. (1999) 27:1263–70. doi: 10.1093/nar/27.5.1263
39. Suzuki T, Yamamoto T, Kurabayashi M, Nagai R, Yazaki Y, Horikoshi M. Isolation and initial characterization of GBF, a novel DNA-binding zinc finger protein that binds to the GC-rich binding sites of the HIV-1 promoter. J Biochem. (1998) 124:389–95. doi: 10.1093/oxfordjournals.jbchem.a022124
40. Matsumoto N, Laub F, Aldabe R, Zhang W, Ramirez F, Yoshida T, et al. Cloning the cDNA for a new human zinc finger protein defines a group of closely related Krüppel-like transcription factors. J Biol Chem. (1998) 273:28229–37. doi: 10.1074/jbc.273.43.28229
41. Imataka H, Sogawa K, Yasumoto K, Kikuchi Y, Sasano K, Kobayashi A, et al. Two regulatory proteins that bind to the basic transcription element (BTE), a GC box sequence in the promoter region of the rat P-4501A1 gene. Embo J. (1992) 11:3663–71. doi: 10.1002/j.1460-2075.1992.tb05451.x
42. Cook T, Urrutia R. TIEG Proteins join the smads as TGF-beta-regulated transcription factors that control pancreatic cell growth. Am J Physiol Gastrointest Liver Physiol. (2000) 278:G513–21. doi: 10.1152/ajpgi.2000.278.4.G513
43. Cook T, Gebelein B, Mesa K, Mladek A, Urrutia R. Molecular cloning and characterization of TIEG2 reveals a new subfamily of transforming growth factor-beta-inducible Sp1-like zinc finger-encoding genes involved in the regulation of cell growth. J Biol Chem. (1998) 273:25929–36. doi: 10.1074/jbc.273.40.25929
44. Chen X, Shi W, Zhang H. The role of KLF14 in multiple disease processes. Biofactors. (2020) 46:276–82. doi: 10.1002/biof.1612
45. Xin Y, Su P, Liu Y, Gu S, An X, Zhang X, et al. Knock out hepatic Krüppel-like factor 16 (KLF16) improve myocardial damage and promoted myocardial protection of myocardial ischemia-reperfusion via anti-oxidative and anti-inflammation effects by TFAM/PPARβ signal passage. Bioengineered. (2021) 12:10219–31. doi: 10.1080/21655979.2021.1982302
46. van Vliet J, Crofts LA, Quinlan KG, Czolij R, Perkins AC, Crossley M. Human KLF17 is a new member of the Sp/KLF family of transcription factors. Genomics. (2006) 87:474–82. doi: 10.1016/j.ygeno.2005.12.011
47. Ciarambino T, Menna G, Sansone G, Giordano M. Cardiomyopathies: an overview. Int J Mol Sci. (2021) 22:7722. doi: 10.3390/ijms22147722
48. Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, et al. ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines (writing committee to update the 2001 guidelines for the evaluation and management of heart failure): developed in collaboration with the American college of chest physicians and the international society for heart and lung transplantation: endorsed by the heart rhythm society. Circulation. (2005) 112:e154–235. doi: 10.1161/CIRCULATIONAHA.105.167586
49. Strickberger SA, Conti J, Daoud EG, Havranek E, Mehra MR, Piña IL, et al. Council on clinical cardiology subcommittee on electrocardiography and arrhythmias and the quality of care and outcomes research interdisciplinary working group; heart rhythm society. Patient selection for cardiac resynchronization therapy: from the council on clinical cardiology subcommittee on electrocardiography and arrhythmias and the quality of care and outcomes research interdisciplinary working group, in collaboration with the heart rhythm society. Circulation. (2005) 111:2146–50. doi: 10.1161/01.CIR.0000161276.09685.4A
50. Gregoratos G, Abrams J, Epstein AE, Freedman RA, Hayes DL, Hlatky MA, et al. ACC/AHA/NASPE 2002 guideline update for implantation of cardiac pacemakers and antiarrhythmia devices: summary article: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines (ACC/AHA/NASPE committee to update the 1998 pacemaker guidelines). Circulation. (2002) 106:2145–61. doi: 10.1161/01.cir.0000035996.46455.09
51. Arbustini E, Narula N, Dec GW, Reddy KS, Greenberg B, Kushwaha S, et al. The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: endorsed by the world heart federation. Glob Heart. (2013) 8:355–82. doi: 10.1016/j.jacc.2013.08.1644
52. Arbelo E, Protonotarios A, Gimeno JR, Arbustini E, Barriales-Villa R, Basso C, et al. 2023 ESC guidelines for the management of cardiomyopathies. Eur Heart J. (2023) 44:3503–626. doi: 10.1093/eurheartj/ehad194
53. Sweet DR, Padmanabhan R, Liao X, Dashora HR, Tang X, Nayak L, et al. Krüppel-Like factors orchestrate endothelial gene expression through redundant and non-redundant enhancer networks. J Am Heart Assoc. (2023) 12:e024303. doi: 10.1161/JAHA.121.024303
54. Sweet DR, Lam C, Jain MK. Evolutionary protection of Krüppel-Like Factors 2 and 4 in the development of the mature hemovascular system. Front Cardiovasc Med. (2021) 8:645719. doi: 10.3389/fcvm.2021.645719
55. Alblaihed L, Kositz C, Brady WJ, Al-Salamah T, Mattu A. Diagnosis and management of arrhythmogenic right ventricular cardiomyopathy. Am J Emerg Med. (2023) 65:146–53. doi: 10.1016/j.ajem.2022.12.010
56. Khudiakov A, Kostina D, Zlotina A, Yany N, Sergushichev A, Pervunina T, et al. Generation of iPSC line from patient with arrhythmogenic right ventricular cardiomyopathy carrying mutations in PKP2 gene. Stem Cell Res. (2017) 24:85–8. doi: 10.1016/j.scr.2017.08.014
57. Chowdhury SK, Liu W, Zi M, Li Y, Wang S, Tsui H, et al. Stress-activated kinase mitogen-activated kinase kinase-7 governs epigenetics of cardiac repolarization for arrhythmia prevention. Circulation. (2017) 135:683–99. doi: 10.1161/CIRCULATIONAHA.116.022941
58. Li N, Xu YJ, Shi HY, Yang CX, Guo YH, Li RG, et al. KLF15 loss-of-function mutation underlying atrial fibrillation as well as ventricular arrhythmias and cardiomyopathy. Genes (Basel). (2021) 12:408. doi: 10.3390/genes12030408
59. Yu LM, Dong X, Zhao JK, Xu YL, Xu DY, Xue XD, et al. Activation of PKG-CREB-KLF15 by melatonin attenuates angiotensin II-induced vulnerability to atrial fibrillation via enhancing branched-chain amino acids catabolism. Free Radic Biol Med. (2022) 178:202–14. doi: 10.1016/j.freeradbiomed
60. Jeyaraj D, Haldar SM, Wan X, McCauley MD, Ripperger JA, Hu K, et al. Circadian rhythms govern cardiac repolarization and arrhythmogenesis. Nature. (2012) 483:96–9. doi: 10.1038/nature10852
61. Wang Q, Luo C, Lu G, Chen Z. Effect of adenosine monophosphate-activated protein kinase-p53-krüppel-like factor 2a pathway in hyperglycemia-induced cardiac remodeling in adult zebrafish. J Diabetes Investig. (2021) 12:320–33. doi: 10.1111/jdi.13393
62. Zhao L, Zhang Q, Liang J, Li J, Tan X, Tang N. Astrocyte elevated gene-1 induces autophagy in diabetic cardiomyopathy through upregulation of KLF4. J Cell Biochem. (2019) 120:9709–15. doi: 10.1002/jcb.28249
63. Palioura D, Lazou A, Drosatos K. Krüppel-like factor (KLF)5: an emerging foe of cardiovascular health. J Mol Cell Cardiol. (2022) 163:56–66. doi: 10.1016/j.yjmcc.2021.10.002
64. Timmers L, van Keulen JK, Hoefer IE, Meijs MF, van Middelaar B, den Ouden K, et al. Targeted deletion of nuclear factor kappaB p50 enhances cardiac remodeling and dysfunction following myocardial infarction. Circ Res. (2009) 104:699–706. doi: 10.1161/CIRCRESAHA.108.189746
65. Duncan JG, Finck BN. The PPARalpha-PGC-1alpha axis controls cardiac energy metabolism in healthy and diseased myocardium. PPAR Res. (2008) 2008:253817. doi: 10.1155/2008/253817
66. Roe ND, Standage SW, Tian R. The relationship between KLF5 and PPARα in the heart: it’s complicated. Circ Res. (2016) 118:193–5. doi: 10.1161/CIRCRESAHA.115.308069
67. Drosatos K, Pollak NM, Pol CJ, Ntziachristos P, Willecke F, Valenti MC, et al. Cardiac myocyte KLF5 regulates ppara expression and cardiac function. Circ Res. (2016) 118:241–53. doi: 10.1161/CIRCRESAHA.115.306383
68. Li F, Peng J, Feng H, Yang Y, Gao J, Liu C, et al. KLF9 Aggravates streptozotocin-induced diabetic cardiomyopathy by inhibiting PPARγ/NRF2 signalling. Cells. (2022) 11:3393. doi: 10.3390/cells11213393
69. Yan Q, He B, Hao G, Liu Z, Tang J, Fu Q, et al. KLF9 aggravates ischemic injury in cardiomyocytes through augmenting oxidative stress. Life Sci. (2019) 233:116641. doi: 10.1016/j.lfs.2019.116641
70. Zhang WY, Wang J, Li AZ. A study of the effects of SGLT-2 inhibitors on diabetic cardiomyopathy through miR-30d/KLF9/VEGFA pathway. Eur Rev Med Pharmacol Sci. (2020) 24:6346–59. doi: 10.26355/eurrev_202006_21533
71. Tian Y, Wang Z, Zheng X, Song W, Cai L, Rane M, et al. KLF15 negatively regulates cardiac fibrosis by which SDF-1β attenuates cardiac fibrosis in type 2 diabetic mice. Toxicol Appl Pharmacol. (2021) 427:115654. doi: 10.1016/j.taap.2021.115654
72. Wang B, Haldar SM, Lu Y, Ibrahim OA, Fisch S, Gray S, et al. The Kruppel-like factor KLF15 inhibits connective tissue growth factor (CTGF) expression in cardiac fibroblasts. J Mol Cell Cardiol. (2008) 45:193–7. doi: 10.1016/j.yjmcc.2008.05.005
73. Leenders JJ, Wijnen WJ, Hiller M, van der Made I, Lentink V, van Leeuwen REW, et al. Regulation of cardiac gene expression by KLF15, a repressor of myocardin activity. J Biol Chem. (2010) 285:27449–56. doi: 10.1074/jbc.M110.107292
74. Zhang W, Chen Z, Qiao S, Chen S, Zheng H, Wei X, et al. The effects of extracellular vesicles derived from Krüppel-like factor 2 overexpressing endothelial cells on the regulation of cardiac inflammation in the dilated cardiomyopathy. J Nanobiotechnology. (2022) 20:76. doi: 10.1186/s12951-022-01284-1
75. Yang J, Samal E, Burgos Angulo M, Bertalovitz A, McDonald TV. Establishment of an arrhythmogenic right ventricular cardiomyopathy derived iPSC cell line (USFi004-A) carrying a heterozygous mutation in PKP2 (c.1799delA). Stem Cell Res. (2021) 54:102398. doi: 10.1016/j.scr.2021.102398
76. Di RM, Yang CX, Zhao CM, Yuan F, Qiao Q, Gu JN, et al. Identification and functional characterization of KLF5 as a novel disease gene responsible for familial dilated cardiomyopathy. Eur J Med Genet. (2020) 63:103827. doi: 10.1016/j.ejmg.2019.103827
77. Guo YH, Wang J, Guo XJ, Gao RF, Yang CX, Li L, et al. KLF13 Loss-of-Function mutations underlying familial dilated cardiomyopathy. J Am Heart Assoc. (2022) 11:e027578. doi: 10.1161/JAHA.122.027578
78. Sunadome K, Yamamoto T, Ebisuya M, Kondoh K, Sehara-Fujisawa A, Nishida E. ERK5 Regulates muscle cell fusion through klf transcription factors. Dev Cell. (2011) 20:192–205. doi: 10.1016/j.devcel.2010.12.005
79. Khudiakov A, Kostina D, Zlotina A, Nikulina T, Sergushichev A, Gudkova A, et al. Generation of iPSC line from desmin-related cardiomyopathy patient carrying splice site mutation of DES gene. Stem Cell Res. (2017) 24:77–80. doi: 10.1016/j.scr.2017.08.015
80. Parakati R, DiMario JX. Repression of myoblast proliferation and fibroblast growth factor receptor 1 promoter activity by KLF10 protein. J Biol Chem. (2013) 288:13876–84. doi: 10.1074/jbc.M113.457648
81. Miyake M, Hayashi S, Iwasaki S, Uchida T, Watanabe K, Ohwada S, et al. TIEG1 Negatively controls the myoblast pool indispensable for fusion during myogenic differentiation of C2C12 cells. J Cell Physiol. (2011) 226:1128–36. doi: 10.1002/jcp.22434
82. Wang J, Chen T, Feng F, Wei H, Pang W, Yang G, et al. KLF15 Regulates slow myosin heavy chain expression through NFATc1 in C2C12 myotubes. Biochem Biophys Res Commun. (2014) 446:1231–6. doi: 10.1016/j.bbrc.2014.03.091
83. Liao X, Zhang R, Lu Y, Prosdocimo DA, Sangwung P, Zhang L, et al. Kruppel-like factor 4 is critical for transcriptional control of cardiac mitochondrial homeostasis. J Clin Invest. (2015) 125:3461–76. doi: 10.1172/JCI79964
84. Liao X, Haldar SM, Lu Y, Jeyaraj D, Paruchuri K, Nahori M, et al. Krüppel-like factor 4 regulates pressure-induced cardiac hypertrophy. J Mol Cell Cardiol. (2010) 49:334–8. doi: 10.1016/j.yjmcc.2010.04.008
85. Yoshida T, Yamashita M, Horimai C, Hayashi M. Kruppel-like factor 4 protein regulates isoproterenol-induced cardiac hypertrophy by modulating myocardin expression and activity. J Biol Chem. (2014) 289:26107–18. doi: 10.1074/jbc.M114.582809
86. Wang C, Qiao S, Zhao Y, Tian H, Yan W, Hou X, et al. The KLF7/PFKL/ACADL axis modulates cardiac metabolic remodelling during cardiac hypertrophy in male mice. Nat Commun. (2023) 14:959. doi: 10.1038/s41467-023-36712-9
87. Rajamannan NM, Subramaniam M, Abraham TP, Vasile VC, Ackerman MJ, Monroe DG, et al. TGFbeta inducible early gene-1 (TIEG1) and cardiac hypertrophy: discovery and characterization of a novel signaling pathway. J Cell Biochem. (2007) 100:315–25. doi: 10.1002/jcb.21049.s
88. Subramaniam M, Hawse JR, Johnsen SA, Spelsberg TC. Role of TIEG1 in biological processes and disease states. J Cell Biochem. (2007) 102:539–48. doi: 10.1002/jcb.21492
89. Patel SK, Wai B, Lang CC, Levin D, Palmer CNA, Parry HM, et al. Genetic variation in Kruppel like factor 15 is associated with left ventricular hypertrophy in patients with type 2 diabetes: discovery and replication cohorts. EBioMedicine. (2017) 18:171–8. doi: 10.1016/j.ebiom.2017.03.036
90. Patel SK, Ramchand J, Crocitti V, Burrell LM. Kruppel-like factor 15 is critical for the development of left ventricular hypertrophy. Int J Mol Sci. (2018) 19:1303. doi: 10.3390/ijms19051303
91. Hoffman M, Palioura D, Kyriazis ID, Cimini M, Badolia R, Rajan S, et al. Cardiomyocyte krüppel-like factor 5 promotes de novo ceramide biosynthesis and contributes to eccentric remodeling in ischemic cardiomyopathy. Circulation. (2021) 143:1139–56. doi: 10.1161/CIRCULATIONAHA.120.047420
92. Pepin ME, Ha CM, Crossman DK, Litovsky SH, Varambally S, Barchue JP, et al. Genome-wide DNA methylation encodes cardiac transcriptional reprogramming in human ischemic heart failure. Lab Invest. (2019) 99:371–86. doi: 10.1038/s41374-018-0104-x
93. Haldar SM, Lu Y, Jeyaraj D, Kawanami D, Cui Y, Eapen SJ, et al. Klf15 deficiency is a molecular link between heart failure and aortic aneurysm formation. Sci Transl Med. (2010) 2:26ra26. doi: 10.1126/scitranslmed.3000502
94. Brooks CL, Gu W. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell. (2011) 2:456–62. doi: 10.1007/s13238-011-1063-9
95. Yao TP, Oh SP, Fuchs M, Zhou ND, Ch'ng LE, Newsome D, et al. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell. (1998) 93:361–72. doi: 10.1016/s0092-8674(00)81165-4
96. Lu X, Yang B, Qi R, Xie Q, Li T, Yang J, et al. Targeting WWP1 ameliorates cardiac ischemic injury by suppressing KLF15-ubiquitination mediated myocardial inflammation. Theranostics. (2023) 13:417–37. doi: 10.7150/thno.77694
97. Kee HJ, Kook H. Krüppel-like factor 4 mediates histone deacetylase inhibitor-induced prevention of cardiac hypertrophy. J Mol Cell Cardiol. (2009) 47:770–80. doi: 10.1016/j.yjmcc.2009.08.022
98. Lee C, Cho S, Jeong D. Inhibition of miR-25 ameliorates cardiac dysfunction and fibrosis by restoring krüppel-like factor 4 expression. Int J Mol Sci. (2023) 24:12434. doi: 10.3390/ijms241512434
99. Ding L, Li S, Wang F, Xu J, Li S, Wang B, et al. Berberine improves dietary-induced cardiac remodeling by upregulating Kruppel-like factor 4-dependent mitochondrial function. Biol Chem. (2021) 402:795–803. doi: 10.1515/hsz-2020-0267
100. Nava A, Bauce B, Basso C, Muriago M, Rampazzo A, Villanova C, et al. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. (2000) 36:2226–33. doi: 10.1016/s0735-1097(00)00997-9
101. Krahn AD, Wilde AAM, Calkins H, La Gerche A, Cadrin-Tourigny J, Roberts JD, et al. Arrhythmogenic right ventricular cardiomyopathy. JACC Clin Electrophysiol. (2022) 8:533–53. doi: 10.1016/j.jacep.2021.12.002
102. Bosman LP, Te Riele A. Arrhythmogenic right ventricular cardiomyopathy: a focused update on diagnosis and risk stratification. Heart. (2022) 108:90–7. doi: 10.1136/heartjnl-2021-319113
103. Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. (2009) 373:1289–300. doi: 10.1016/S0140-6736(09)60256-7
104. Shah SN, Umapathi KK, Oliver TI. Arrhythmogenic Right Ventricular Cardiomyopath. Treasure Island, FL: StatPearls Publishing (2023).
105. Te Riele A, James CA, Sawant AC, Bhonsale A, Groeneweg JA, Mast TP, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy in the pediatric population: clinical characterization and comparison with adult-onset disease. JACC Clin Electrophysiol. (2015) 1:551–60. doi: 10.1016/j.jacep.2015.08.004
106. Xu Z, Zhu W, Wang C, Huang L, Zhou Q, Hu J, et al. Genotype-phenotype relationship in patients with arrhythmogenic right ventricular cardiomyopathy caused by desmosomal gene mutations: a systematic review and meta-analysis. Sci Rep. (2017) 7:41387. doi: 10.1038/srep41387
107. James CA, Jongbloed JDH, Hershberger RE, Morales A, Judge DP, Syrris P, et al. International evidence based reappraisal of genes associated with arrhythmogenic right ventricular cardiomyopathy using the clinical genome resource framework. Circ Genom Precis Med. (2021) 14:e003273. doi: 10.1161/CIRCGEN.120.003273
108. McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (naxos disease). Lancet. (2000) 355:2119–24. doi: 10.1016/S0140-6736(00)02379-5
109. Young ME. The cardiac circadian clock: implications for cardiovascular disease and its treatment. JACC Basic Transl Sci. (2023) 8:1613–28. doi: 10.1016/j.jacbts.2023.03.024
110. Lin YH, Chen XM, Zhang JW, He XQ, Dai WJ, Chen MS. Preclinical study on induction of pluripotent stem cells from urine of dilated cardiomyopathy patients. Eur Rev Med Pharmacol Sci. (2016) 20:1450–7. PMID: 27160114
111. Zhu S, Law AHY, Deng R, Poon ENY, Lo CW, Kwong AKY, et al. Generation of genomic-integration-free human induced pluripotent stem cells and the derived cardiomyocytes of X-linked dilated cardiomyopathy from DMD gene mutation. Stem Cell Res. (2020) 49:102040. doi: 10.1016/j.scr.2020.102040
112. Einarson TR, Acs A, Ludwig C, Panton UH. Prevalence of cardiovascular disease in type 2 diabetes: a systematic literature review of scientific evidence from across the world in 2007–2017. Cardiovasc Diabetol. (2018) 17:83. doi: 10.1186/s12933-018-0728-6
113. Ritchie RH, Abel ED. Basic mechanisms of diabetic heart disease. Circ Res. (2020) 126:1501–25. doi: 10.1161/CIRCRESAHA.120.315913
114. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF Diabetes atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. (2022) 183:109119. doi: 10.1016/j.diabres.2021.109119
115. Lundbaek K. Diabetic angiopathy: a specific vascular disease. Lancet. (1954) 266:377–9. doi: 10.1016/s0140-6736(54)90924-1
116. Lundbaek K. Is there a diabetic cardiopathy? Pathogenetische Fakt des Myokardinfarkts. (1969):63–71.
117. Seferović PM, Petrie MC, Filippatos GS, Anker SD, Rosano G, Bauersachs J, et al. Type 2 diabetes mellitus and heart failure: a position statement from the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. (2018) 20:853–72. doi: 10.1002/ejhf.1170
118. Lorenzo O, Ramírez E, Picatoste B, Egido J, Tuñón J. Alteration of energy substrates and ROS production in diabetic cardiomyopathy. Mediators Inflamm. (2013) 2013:461967. doi: 10.1155/2013/461967
119. Banerjee SS, Feinberg MW, Watanabe M, Gray S, Haspel RL, Denkinger DJ, et al. The Krüppel-like factor KLF2 inhibits peroxisome proliferator-activated receptor-gamma expression and adipogenesis. J Biol Chem. (2003) 278:2581–4. doi: 10.1074/jbc.M210859200
120. Cui A, Fan H, Zhang Y, Zhang Y, Niu D, Liu S, et al. Dexamethasone-induced Krüppel-like factor 9 expression promotes hepatic gluconeogenesis and hyperglycemia. J Clin Invest. (2019) 129:2266–78. doi: 10.1172/JCI66062
121. Chen W, Wang H, Liu J, Li K. Interference of KLF9 relieved the development of gestational diabetes mellitus by upregulating DDAH2. Bioengineered. (2022) 13:395–406. doi: 10.1080/21655979.2021.2005929
122. Wang J, Zhang J, Shen J, Hu D, Yan G, Liu X, et al. Association of KCNQ1 and KLF14 polymorphisms and risk of type 2 diabetes mellitus: a global meta-analysis. Hum Immunol. (2014) 75:342–7. doi: 10.1016/j.humimm.2014.01.008
123. Kong X, Zhang X, Xing X, Zhang B, Hong J, Yang W. The association of type 2 diabetes loci identified in genome-wide association studies with metabolic syndrome and its components in a Chinese population with type 2 diabetes. PLoS One. (2015) 10:e0143607. doi: 10.1371/journal.pone.0143607
124. Takashima M, Ogawa W, Hayashi K, Inoue H, Kinoshita S, Okamoto Y, et al. Role of KLF15 in regulation of hepatic gluconeogenesis and metformin action. Diabetes. (2010) 59:1608–15. doi: 10.2337/db09-1679
125. Zhang H, Chen Z, Wang X. Differentiated serum levels of Krüppel-Like Factors 2 and 4, sP-selectin, and sE-selectin in patients with gestational diabetes mellitus. Gynecol Endocrinol. (2022) 38:1121–4. doi: 10.1080/09513590.2022.2164762
126. Mohtar O, Ozdemir C, Roy D, Shantaram D, Emili A, Kandror KV. Egr1 mediates the effect of insulin on leptin transcription in adipocytes. J Biol Chem. (2019) 294:5784–9. doi: 10.1074/jbc.AC119.007855
127. Kanazawa A, Kawamura Y, Sekine A, Iida A, Tsunoda T, Kashiwagi A, et al. Single nucleotide polymorphisms in the gene encoding Krüppel-like factor 7 are associated with type 2 diabetes. Diabetologia. (2005) 48:1315–22. doi: 10.1007/s00125-005-1797-0
128. Hulshoff MS, Schellinger IN, Xu X, Fledderus J, Rath SK, Wong FC, et al. miR-132-3p and KLF7 as novel regulators of aortic stiffening-associated EndMT in type 2 diabetes mellitus. Diabetol Metab Syndr. (2023) 15:11. doi: 10.1186/s13098-022-00966-y
129. Bonnefond A, Lomberk G, Buttar N, Busiah K, Vaillant E, Lobbens S, et al. Disruption of a novel Kruppel-like transcription factor p300-regulated pathway for insulin biosynthesis revealed by studies of the c.-331 INS mutation found in neonatal diabetes mellitus. J Biol Chem. (2011) 286:28414–24. doi: 10.1074/jbc.M110.215822
130. Zhang H, Chen Q, Jiao T, Cui A, Sun X, Fang W, et al. Involvement of KLF11 in hepatic glucose metabolism in mice via suppressing of PEPCK-C expression. PLoS One. (2014) 9:e89552. doi: 10.1371/journal.pone.0089552
131. Ushijima K, Narumi S, Ogata T, Yokota I, Sugihara S, Kaname T, et al. KLF11 variant in a family clinically diagnosed with early childhood-onset type 1B diabetes. Pediatr Diabetes. (2019) 20:712–9. doi: 10.1111/pedi.12868
132. Mancera-Rincón P, Luna-España MC, Rincon O, Guzmán I, Alvarez M. Maturity-onset diabetes of the young type 7 (MODY7) and the krüppellike factor 11 mutation (KLF11). A review. Curr Diabetes Rev. (2024) 20:e210323214817. doi: 10.2174/1573399819666230321114456
133. Reza-López SA, González-Gurrola S, Morales-Morales OO, Moreno-González JG, Rivas-Gómez AM, González-Rodríguez E, et al. Metabolic biomarkers in adults with type 2 diabetes: the role of PPAR-γ2 and PPAR-β/δ polymorphisms. Biomolecules. (2023) 13:1791. doi: 10.3390/biom13121791
134. Miyazaki T. Calpain and cardiometabolic diseases. Int J Mol Sci. (2023) 24:16782. doi: 10.3390/ijms242316782
135. De Geest B, Mishra M. Role of oxidative stress in diabetic cardiomyopathy. Antioxidants (Basel). (2022) 11:784. doi: 10.3390/antiox11040784
136. Avagimyan A, Popov S, Shalnova S. The pathophysiological basis of diabetic cardiomyopathy development. Curr Probl Cardiol. (2022) 47:01156. doi: 10.1016/j.cpcardiol.2022.101156
137. Nakamura K, Miyoshi T, Yoshida M, Akagi S, Saito Y, Ejiri K, et al. Pathophysiology and treatment of diabetic cardiomyopathy and heart failure in patients with diabetes Mellitus. Int J Mol Sci. (2022) 23:3587. doi: 10.3390/ijms23073587
138. Tang Z, Wang P, Dong C, Zhang J, Wang X, Pei H. Oxidative stress signaling mediated pathogenesis of diabetic cardiomyopathy. Oxid Med Cell Longev. (2022) 2022:5913374. doi: 10.1155/2022/5913374
139. Lorenzo-Almorós A, Cepeda-Rodrigo JM, Lorenzo Ó. Diabetic cardiomyopathy. Rev Clin Esp (Barc). (2022) 222:100–11. doi: 10.1016/j.rce.2019.10.013
140. Zhang H, Chen X, Zong B, Yuan H, Wang Z, Wei Y, et al. Gypenosides improve diabetic cardiomyopathy by inhibiting ROS-mediated NLRP3 inflammasome activation. J Cell Mol Med. (2018) 22:4437–48. doi: 10.1111/jcmm.13743
141. You S, Qian J, Sun C, Zhang H, Ye S, Chen T, et al. An aza resveratrol-chalcone derivative 6b protects mice against diabetic cardiomyopathy by alleviating inflammation and oxidative stress. J Cell Mol Med. (2018) 22:1931–43. doi: 10.1111/jcmm.13477
142. Jiang Z, Fu L, Xu Y, Hu X, Yang H, Zhang Y, et al. Cyclovirobuxine D protects against diabetic cardiomyopathy by activating Nrf2-mediated antioxidant responses. Sci Rep. (2020) 10:6427. doi: 10.1038/s41598-020-63498-3
143. Gao L, Liu Y, Guo S, Xiao L, Wu L, Wang Z, et al. LAZ3 Protects cardiac remodeling in diabetic cardiomyopathy via regulating miR-21/PPARa signaling. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:3322–38. doi: 10.1016/j.bbadis.2018.07.019
144. Wang XH, Yan CY, Liu JR. Hyperinsulinemia-induced KLF5 mediates endothelial angiogenic dysfunction in diabetic endothelial cells. J Mol Histol. (2019) 50:239–51. doi: 10.1007/s10735-019-09821-3
145. Zhang ML, Zheng B, Tong F, Yang Z, Wang ZB, Yang BM, et al. iNOS-derived peroxynitrite mediates high glucose-induced inflammatory gene expression in vascular smooth muscle cells through promoting KLF5 expression and nitration. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:2821–34. doi: 10.1016/j.bbadis.2017.07.004
146. Gu J, Qiu M, Lu Y, Ji Y, Qian Z, Sun W. Piperlongumine attenuates angiotensin-II-induced cardiac hypertrophy and fibrosis by inhibiting akt-FoxO1 signalling. Phytomedicine. (2021) 82:153461. doi: 10.1016/j.phymed.2021.153461
147. Zhang J, Cheng Y, Gu J, Wang S, Zhou S, Wang Y, et al. Fenofibrate increases cardiac autophagy via FGF21/SIRT1 and prevents fibrosis and inflammation in the hearts of type 1 diabetic mice. Clin Sci (Lond). (2016) 130:625–41. doi: 10.1042/CS20150623
148. He C, Zhu H, Li H, Zou MH, Xie Z. Dissociation of bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes. (2013) 62:1270–81. doi: 10.2337/db12-0533
149. Kanamori H, Takemura G, Goto K, Tsujimoto A, Mikami A, Ogino A, et al. Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy. (2015) 11:1146–60. doi: 10.1080/15548627.2015.1051295
150. Yarmohammadi F, Barangi S, Aghaee-Bakhtiari SH, Hosseinzadeh H, Moosavi Z, Reiter RJ, et al. Melatonin ameliorates arsenic-induced cardiotoxicity through the regulation of the Sirt1/Nrf2 pathway in rats. Biofactors. (2023) 49:620–35. doi: 10.1002/biof.1934
151. Cai L, Wang Y, Zhou G, Chen T, Song Y, Li X, et al. Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in a prevention of diabetic cardiomyopathy. J Am Coll Cardiol. (2006) 48:1688–97. doi: 10.1016/j.jacc.2006.07.022
152. Yu H, Zhen J, Yang Y, Gu J, Wu S, Liu Q. Ginsenoside Rg1 ameliorates diabetic cardiomyopathy by inhibiting endoplasmic reticulum stress-induced apoptosis in a streptozotocin-induced diabetes rat model. J Cell Mol Med. (2016) 20:623–31. doi: 10.1111/jcmm.12739
153. Gao P, Cao M, Jiang X, Wang X, Zhang G, Tang X, et al. Cannabinoid receptor 2-centric molecular feedback loop drives necroptosis in diabetic heart injuries. Circulation. (2023) 147:158–74. doi: 10.1161/CIRCULATIONAHA.122.059304
154. Gong DF, Sun SC, Wang RR, Dawuti A, Kong DW, Liu RQ, et al. Salvianolic acid A improve mitochondrial respiration and cardiac function via inhibiting apoptosis pathway through CRYAB in diabetic cardiomyopathy. Biomed Pharmacother. (2023) 160:114382. doi: 10.1016/j.biopha.2023.114382
155. Baroukh N, Canteleux N, Lefèvre A, Dupuy C, Martias C, Presset A, et al. Serum and soleus metabolomics signature of Klf10 knockout mice to identify potential biomarkers. Metabolites. (2022) 12:556. doi: 10.3390/metabo12060556
156. Luo HY, Zhu JY, Chen M, Mu WJ, Guo L. Krüppel-like factor 10 (KLF10) as a critical signaling mediator: versatile functions in physiological and pathophysiological processes. Genes Dis. (2022) 10:915–30. doi: 10.1016/j.gendis.2022.06.005
157. Kong HJ, Lee JJ, Kim JW, Kim J, Kim YO, Yeo SY. Zebrafish Klf11b is required to maintain cell viability by inhibiting p53-mediated apoptosis. Dev Reprod. (2022) 26:79–90. doi: 10.12717/DR.2022.26.2.79
158. Orphanou N, Papatheodorou E, Anastasakis A. Dilated cardiomyopathy in the era of precision medicine: latest concepts and developments. Heart Fail Rev. (2022) 27:1173–91. doi: 10.1007/s10741-021-10139-0
159. Furquim SR, Linnenkamp B, Olivetti NQS, Giugni FR, Lipari L, Andrade FA, et al. Challenges and applications of genetic testing in dilated cardiomyopathy: genotype, phenotype and clinical implications. Arq Bras Cardiol. (2023) 120:e20230174. doi: 10.36660/abc.20230174
160. Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. (2013) 10:531–47. doi: 10.1038/nrcardio.2013.105
161. Goldberger JJ, Subačius H, Patel T, Cunnane R, Kadish AH. Sudden cardiac death risk stratification in patients with nonischemic dilated cardiomyopathy. J Am Coll Cardiol. (2014) 63:1879–89. doi: 10.1016/j.jacc.2013.12.021
162. D'Ascenzi F, Valentini F, Pistoresi S, Frascaro F, Piu P, Cavigli L, et al. Causes of sudden cardiac death in young athletes and non-athletes: systematic review and meta-analysis: sudden cardiac death in the young. Trends Cardiovasc Med. (2022) 32:299–308. doi: 10.1016/j.tcm.2021.06.001
163. Alonso-Villa E, Bonet F, Hernandez-Torres F, Campuzano Ó, Sarquella-Brugada G, Quezada-Feijoo M, et al. The role of MicroRNAs in dilated cardiomyopathy: new insights for an old entity. Int J Mol Sci. (2022) 23:13573. doi: 10.3390/ijms232113573
164. El Hadi H, Freund A, Desch S, Thiele H, Majunke N. Hypertrophic, dilated, and arrhythmogenic cardiomyopathy: where are we? Biomedicines. (2023) 11:524. doi: 10.3390/biomedicines11020524
165. McNally EM, Mestroni L. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ Res. (2017) 121:731–48. doi: 10.1161/CIRCRESAHA.116.309396
166. Carlsson L, Thornell LE. Desmin-related myopathies in mice and man. Acta Physiol Scand. (2001) 171:341–8. doi: 10.1046/j.1365-201x.2001.00837.x
167. McLendon PM, Robbins J. Desmin-related cardiomyopathy: an unfolding story. Am J Physiol Heart Circ Physiol. (2011) 301:H1220–8. doi: 10.1152/ajpheart.00601.2011
168. Tsikitis M, Galata Z, Mavroidis M, Psarras S, Capetanaki Y. Intermediate filaments in cardiomyopathy. Biophys Rev. (2018) 10:1007–31. doi: 10.1007/s12551-018-0443-2
169. Maggi L, Mavroidis M, Psarras S, Capetanaki Y, Lattanzi G. Skeletal and cardiac muscle disorders caused by mutations in genes encoding intermediate filament proteins. Int J Mol Sci. (2021) 22:4256. doi: 10.3390/ijms22084256
170. Papadopoulos C, Malfatti E, Métay C, Keren B, Lejeune E, Buratti J, et al. Deep characterization of a Greek patient with desmin-related myofibrillar myopathy and cardiomyopathy. Int J Mol Sci. (2023) 24:11181. doi: 10.3390/ijms241311181
171. Rapti K, Diokmetzidou A, Kloukina I, Milner DJ, Varela A, Davos CH, et al. Opposite effects of catalase and MnSOD ectopic expression on stress induced defects and mortality in the desmin deficient cardiomyopathy model. Free Radic Biol Med. (2017) 110:206–18. doi: 10.1016/j.freeradbiomed.2017.06.010
172. Goldfarb LG, Park KY, Cervenáková L, Gorokhova S, Lee HS, Vasconcelos O, et al. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet. (1998) 19:402–3. doi: 10.1038/1300
173. Dalakas MC, Park KY, Semino-Mora C, Lee HS, Sivakumar K, Goldfarb LG. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med. (2000) 342:770–80. doi: 10.1056/NEJM200003163421104
174. van Spaendonck-Zwarts KY, van Hessem L, Jongbloed JD, de Walle HE, Capetanaki Y, van der Kooi AJ, et al. Desmin-related myopathy. Clin Genet. (2011) 80:354–66. doi: 10.1111/j.1399-0004.2010.01512.x
175. Peretto G, Di Resta C, Perversi J, Forleo C, Maggi L, Politano L, et al. Cardiac and neuromuscular features of patients with LMNA-related cardiomyopathy. Ann Intern Med. (2019) 171:458–63. doi: 10.7326/M18-2768
176. Boriani G, Biagini E, Ziacchi M, Malavasi VL, Vitolo M, Talarico M, et al. Cardiolaminopathies from bench to bedside: challenges in clinical decision-making with focus on arrhythmia-related outcomes. Nucleus. (2018) 9:442–59. doi: 10.1080/19491034.2018.1506680
177. Brodehl A, Gaertner-Rommel A, Milting H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys Rev. (2018) 10:983–1006. doi: 10.1007/s12551-018-0429-0
178. Maron BJ, Rowin EJ, Maron MS. Global burden of hypertrophic cardiomyopathy. JACC Heart Fail. (2018) 6:376–8. doi: 10.1016/j.jchf.2018.03.004
179. The Joint Committee of Cardiomyopathy Specialty Alliance; National Center for Cardiovascular Diseases/Cardiovascular Precision Medicine Branch of China International Exchange and Promotive Association for Medical and Health Care. 2023 Guideline for diagnosis and treatment of patients with hypertrophic cardiomyopathy. Mol Cardiol China. (2023) 23:5115–49. doi: 10.16563/j.cnki.1671-6272.2023.02.002
180. Maron BJ, Rowin EJ, Maron MS. Hypertrophic cardiomyopathy: new concepts and therapies. Annu Rev Med. (2022) 73:363–75. doi: 10.1146/annurev-med-042220-021539
181. Matthia EL, Setteducato ML, Elzeneini M, Vernace N, Salerno M, Kramer CM, et al. Circulating biomarkers in hypertrophic cardiomyopathy. J Am Heart Assoc. (2022) 11:e027618. doi: 10.1161/JAHA.122.027618
182. Sebastian SA, Panthangi V, Singh K, Rayaroth S, Gupta A, Shantharam D, et al. Hypertrophic cardiomyopathy: current treatment and future options. Curr Probl Cardiol. (2023) 48:101552. doi: 10.1016/j.cpcardiol.2022.101552
183. Sawan MA, Prabakaran S, D’Souza M, Behbahani-Nejad O, Gold ME, Williams BR, et al. A systematic review of present and future pharmaco-structural therapies for hypertrophic cardiomyopathy. Clin Cardiol. (2024) 47:e24207. doi: 10.1002/clc.24207
184. Maron BJ. Clinical course and management of hypertrophic cardiomyopathy. N Engl J Med. (2018) 379:1977. doi: 10.1056/NEJMra1710575
185. Maron MS, Olivotto I, Zenovich AG, Link MS, Pandian NG, Kuvin JT, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation. (2006) 114:2232–9. doi: 10.1161/CIRCULATIONAHA.106.644682
186. Yang Y, Wang Z, Yao M, Xiong W, Wang J, Fang Y, et al. Oxytocin protects against isoproterenol-induced cardiac hypertrophy by inhibiting PI3K/AKT pathway via a lncRNA GAS5/miR-375-3p/KLF4-dependent mechanism. Front Pharmacol. (2021) 12:766024. doi: 10.3389/fphar.2021.766024
187. Prosdocimo DA, Anand P, Liao X, Zhu H, Shelkay S, Artero-Calderon P, et al. Kruppel-like factor 15 is a critical regulator of cardiac lipid metabolism. J Biol Chem. (2014) 289:5914–24. doi: 10.1074/jbc.M113.531384
188. Del Buono MG, Moroni F, Montone RA, Azzalini L, Sanna T, Abbate A. Ischemic cardiomyopathy and heart failure after acute myocardial infarction. Curr Cardiol Rep. (2022) 24:1505–15. doi: 10.1007/s11886-022-01766-6
189. Felker GM, Shaw LK, O'Connor CM. A standardized definition of ischemic cardiomyopathy for use in clinical research. J Am Coll Cardiol. (2002) 39:210–8. doi: 10.1016/s0735-1097(01)01738-7
190. Almeida AG, Carpenter JP, Cameli M, Donal E, Dweck MR, Flachskampf FA, et al. Multimodality imaging of myocardial viability: an expert consensus document from the European Association of Cardiovascular Imaging (EACVI). Eur Heart J Cardiovasc Imaging. (2021) 22:e97–e125. doi: 10.1093/ehjci/jeab053
191. Marwick TH. The viable myocardium: epidemiology, detection, and clinical implications. Lancet. (1998) 351:815–9. doi: 10.1016/S0140-6736(97)08080-X
192. Taegtmeyer H, Sen S, Vela D. Return to the fetal gene program: a suggested metabolic link to gene expression in the heart. Ann N Y Acad Sci. (2010) 1188:191–8. doi: 10.1111/j.1749-6632.2009.05100.x
193. Da Dalt L, Cabodevilla AG, Goldberg IJ, Norata GD. Cardiac lipid metabolism, mitochondrial function, and heart failure. Cardiovasc Res. (2023) 119:1905–14. doi: 10.1093/cvr/cvad100
194. Rogers RG, Otis JS. Resveratrol-mediated expression of KLF15 in the ischemic myocardium is associated with an improved cardiac phenotype. Cardiovasc Drugs Ther. (2017) 31:29–38. doi: 10.1007/s10557-016-6707-9
195. Castañeda D, Gabani M, Choi SK, Nguyen QM, Chen C, Mapara A, et al. Targeting autophagy in obesity-associated heart disease. Obesity (Silver Spring). (2019) 27:1050–8. doi: 10.1002/oby.22455
196. Ren J, Wu NN, Wang S, Sowers JR, Zhang Y. Obesity cardiomyopathy: evidence, mechanisms, and therapeutic implications. Physiol Rev. (2021) 101:1745–807. doi: 10.1152/physrev.00030.2020
197. Aurigemma GP, de Simone G, Fitzgibbons TP. Cardiac remodeling in obesity. Circ Cardiovasc Imaging. (2013) 6:142–52. doi: 10.1161/CIRCIMAGING.111.964627
198. Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiol Rev. (2008) 88:389–419. doi: 10.1152/physrev.00017.2007
199. Alpert MA. Obesity cardiomyopathy: pathophysiology and evolution of the clinical syndrome. Am J Med Sci. (2001) 321:225–36. doi: 10.1097/00000441-200104000-00003
200. Powell-Wiley TM, Poirier P, Burke LE, Després JP, Gordon-Larsen P, Lavie CJ, et al. Obesity and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. (2021) 143:e984–e1010. doi: 10.1161/CIR.0000000000000973
201. Aryee EK, Ozkan B, Ndumele CE. Heart failure and obesity: the latest pandemic. Prog Cardiovasc Dis. (2023) 78:43–8. doi: 10.1016/j.pcad.2023.05.003
202. Middeldorp ME, Kamsani SH, Sanders P. Obesity and atrial fibrillation: prevalence, pathogenesis, and prognosis. Prog Cardiovasc Dis. (2023) 78:34–42. doi: 10.1016/j.pcad.2023.04.010
203. Shu H, Cheng J, Li N, Zhang Z, Nie J, Peng Y, et al. Obesity and atrial fibrillation: a narrative review from arrhythmogenic mechanisms to clinical significance. Cardiovasc Diabetol. (2023) 22:192. doi: 10.1186/s12933-023-01913-5
204. Chang W, Zhang M, Meng Z, Yu Y, Yao F, Hatch GM, et al. Berberine treatment prevents cardiac dysfunction and remodeling through activation of 5'-adenosine monophosphate-activated protein kinase in type 2 diabetic rats and in palmitate-induced hypertrophic H9c2 cells. Eur J Pharmacol. (2015) 769:55–63. doi: 10.1016/j.ejphar.2015.10.043
205. Chen K, Li G, Geng F, Zhang Z, Li J, Yang M, et al. Berberine reduces ischemia/reperfusion-induced myocardial apoptosis via activating AMPK and PI3K-akt signaling in diabetic rats. Apoptosis. (2014) 19:946–57. doi: 10.1007/s10495-014-0977-0
206. Tian CX, Li MY, Shuai XX, Jiang F, Dong YL, Gui Y, et al. Berberine plays a cardioprotective role by inhibiting macrophage Wnt5a/β-catenin pathway in the myocardium of mice after myocardial infarction. Phytother Res. (2023) 37:50–61. doi: 10.1002/ptr.7592
Keywords: Krüppel-Like Factors, cardiomyopathy, genetic screen, diabetic cardiomyopathy, heart failure
Citation: Gui L-K, Liu H-J, Jin L-J and Peng X-C (2024) Krüpple-like factors in cardiomyopathy: emerging player and therapeutic opportunities. Front. Cardiovasc. Med. 11:1342173. doi: 10.3389/fcvm.2024.1342173
Received: 22 November 2023; Accepted: 23 February 2024;
Published: 7 March 2024.
Edited by:
Yi Cao, University of South China, ChinaReviewed by:
Jose Francisco Islas, Autonomous University of Nuevo León, MexicoFuyang Zhang, Air Force Medical University, China
© 2024 Gui, Liu, Jin and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li-Jun Jin amxqc2lyQHNpbmEuY29t Xiao-Chun Peng cHhjd2Q3ODlAc2luYS5jb20=
†These authors have contributed equally to this work and share first authorship