Abstract
Fabry disease (FD) is a rare genetic disorder caused by mutations in the GLA gene, affecting multiple organs. Over 60% of patients experience heart-related issues, primarily arrhythmias. Unlike typical cases, these arrhythmias are complex and often do not respond well to standard antiarrhythmic drugs, sometimes worsening symptoms. This paper presents a case study of a FD patient with paroxysmal atrial fibrillation successfully treated using bilateral pulmonary vein cryoablation, resulting in positive outcomes. Additionally, we conducted molecular docking studies for the first time to assess the enzyme-substrate binding of E358del, confirming findings similar to laboratory experiments. Our findings underscore the potential role of artificial intelligence in better understanding FD, and aim to provide insights into managing arrhythmias associated with FD.
1 Introduction
Fabry disease (FD) is a rare genetic disorder due to mutations in the galactosidase alpha (GLA) gene, leading to partial or complete loss of activity of the encoded α-galactosidase A (α-Gal A). This deficiency inhibits the breakdown of globotriaosylceramide (Gb3), resulting in its accumulation in lysosomes and causing a multiple-system disease (1). Clinically, the presentations of FD patients vary significantly, from asymptotic to classic form, but generally, female patients tend to have less typical presentation compared with male patients (2).
Among the affected systems, more than 60% of FD patients display symptoms of cardiac involvement, which is also the primary cause of death for such patients. FD patients often present with left ventricular hypertrophy, arrhythmia, and heart valvular disease (3–5). Notably, arrhythmias induced by FD exhibit diversity, with commonly prescribed antiarrhythmic medications showing limited effectiveness and even worsening their symptoms (5, 6).
Presently, more than 900 types of GLA gene mutations have been identified as pathogenic or likely pathogenic. Different gene mutation sites determine distinct residual enzyme activities, thereby impacting the age of onset and the progression in individuals with FD (7, 8). Here we report a female FD patient with delayed onset, primarily presented with atrial fibrillation (AF), and rhythm control was used successfully with cryo-ablation. Through SWISS-MODEL analysis, the enzyme protein structure in this patient was compared with wild-type control. This case contributes to the clinical understanding of the c.1072_1074delGAG (p.E358del) mutation, offering insights for treating FD patients presenting with arrhythmia complications.
2 Case presentation
2.1 Clinical presentation and physical examination
A 50-year-old female patient was admitted due to recurrent chest pain. During the past six years, the patient experienced recurrent episodes of unexplained dull pain in the precordial region, exacerbated by physical activity. Throughout the clinical course, the patient exhibited no signs of lower extremity edema, paroxysmal nocturnal dyspnea, or systemic manifestations such as hypohidrosis, oliguria, acral pain, gastrointestinal disturbances, or visual impairment. Furthermore, there was no evidence of stroke-related symptoms, including transient ischemic attacks, focal neurological deficits, or other cerebrovascular events. Comprehensive clinical evaluations revealed no abnormalities in renal function or proteinuria. The day before admission, the patient felt palpitation and meanwhile, the symptom of chest pain worsened with sweating. An emergency electrocardiogram (ECG) indicated atrial flutter (Figure 1A), prompting her admission to the cardiovascular department for further assessment and treatment of the arrhythmia.
Figure 1
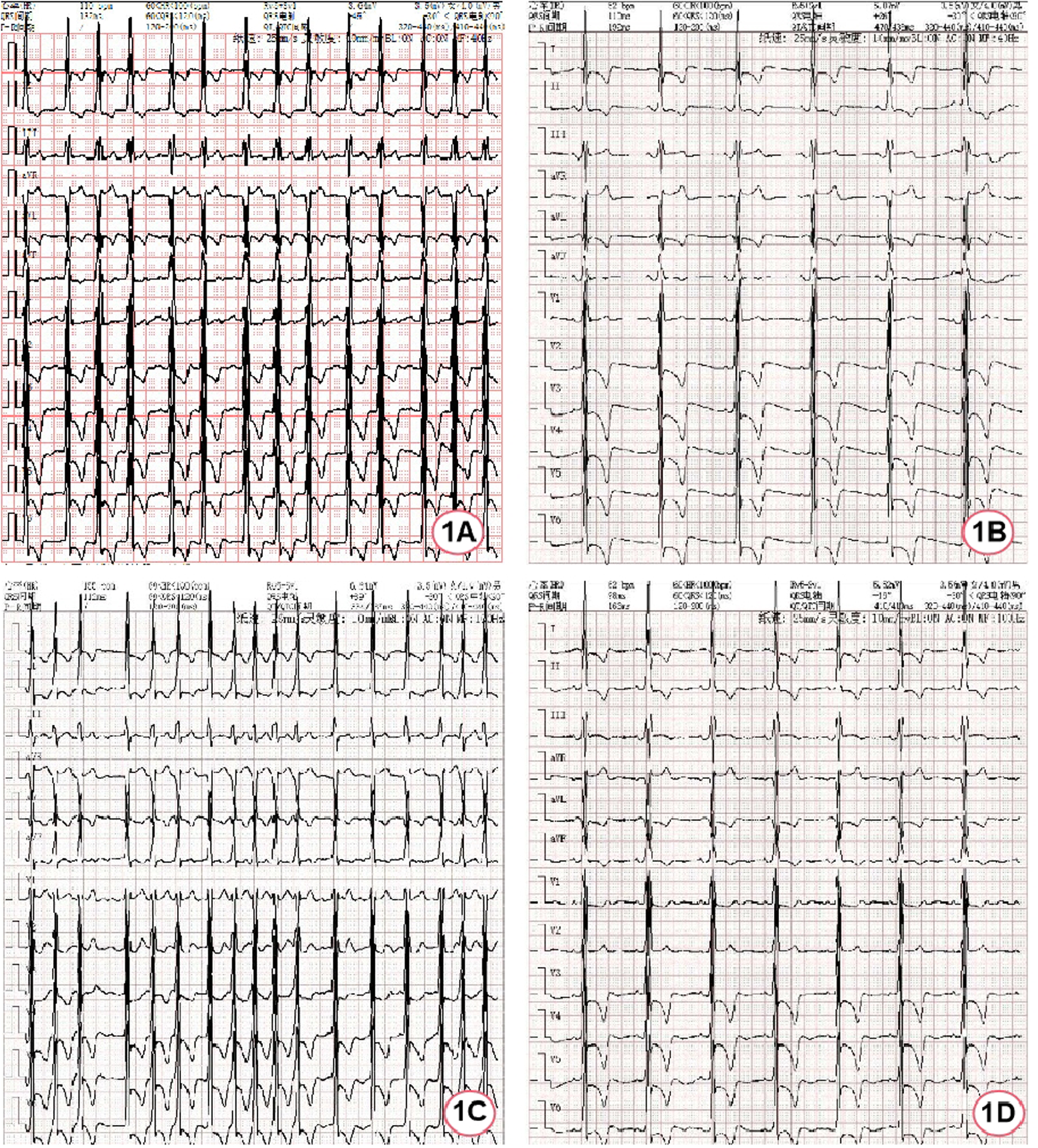
ECG of the patient during hospitalization. (A) ECG at admission; (B) ECG after spontaneous restoration of sinus rhythm; (C) ECG during atrial fibrillation episodes; (D) ECG after bilateral pulmonary vein cryoablation.
Four years ago, the patient was diagnosed with hypertrophic cardiomyopathy and echocardiography revealed symmetric left ventricular hypertrophy (14 mm). Electrocardiography showed sinus bradycardia, frequent atrial premature beats, and paroxysmal atrial tachycardia. She denied the history of hypertension, diabetes mellitus, or coronary atherosclerotic heart disease. Her mother passed away 20 years ago due to “heart disease”. She also denied the presence of cardiovascular diseases in other family members.
Upon admission, her vital signs were stable with blood pressure of 113/76 mmHg (1 mmHg = 0.133 kPa). Physical examination showed no rash or other skin lesions were observed on the patient's body. Hearing and vision were normal, with no abnormalities detected on eye examination. Pulmonary auscultation revealed no rales. The cardiac borders were not enlarged; heart rate was 87 beats per minute, with a regular rhythm and no murmurs detected. No edema was noted in the lower extremities. Neurological examination revealed no positive signs.
2.2 Assistant examination
After hospitalization, myocardial enzymes revealed elevated levels of troponin I at 0.27 ng/ml (normal range 0–0.023 ng/ml) and N-terminal pro-B-type natriuretic peptide were 3,910 ng/L (normal range 0–300 ng/L). The first electrocardiogram after hospitalization was completed 40 min after the emergency electrocardiogram and it had restored to sinus rhythm without using antiarrhythmic drugs, but it was remarkable sinus bradycardia (47 bpm) (Figure 1B). Afterwards, recurrent episodes of palpitation occurred and ECG indicated rapid ventricular response AF (Figure 1C). Due to the obvious symptoms, rhythm control strategy was used with intravenous use of amiodarone. Meanwhile, anticoagulation with dabigatran etexilate was administered. Nineteen hours later, it successfully restored to sinus rhythm, showing sinus bradycardia (52 bpm).
During the hospitalization, Holter displayed an average ventricular rate of 53 bpm and echocardiography showed a left ventricular ejection fraction of 65%, interventricular septal end-diastolic thickness of 15 mm, left ventricular posterior wall end-diastolic thickness of 15 mm, maximum thickness at apex 18 mm without left ventricular outflow obstruction. Cardiac magnetic resonance (CMR) imaging also revealed diffuse hypertrophy in the apical and mid-ventricular walls of the left ventricle, approximately 21 mm in the apical (Figure 2A) and the T1 mapping value was 1,210 ± 23.5 ms (Figure 2B). Ambulatory blood pressure test showed the average 24-hour blood pressure was 120/72 mmHg. Coronary angiography did not show obvious stenosis or slow flow of coronary artery. Considering her recurrent episodes of paroxysmal AF/atrial flutter with obvious symptoms, the patient received bilateral pulmonary vein cryo-ablation, after which it reverted to sinus rhythm (Figure 1D).
Figure 2
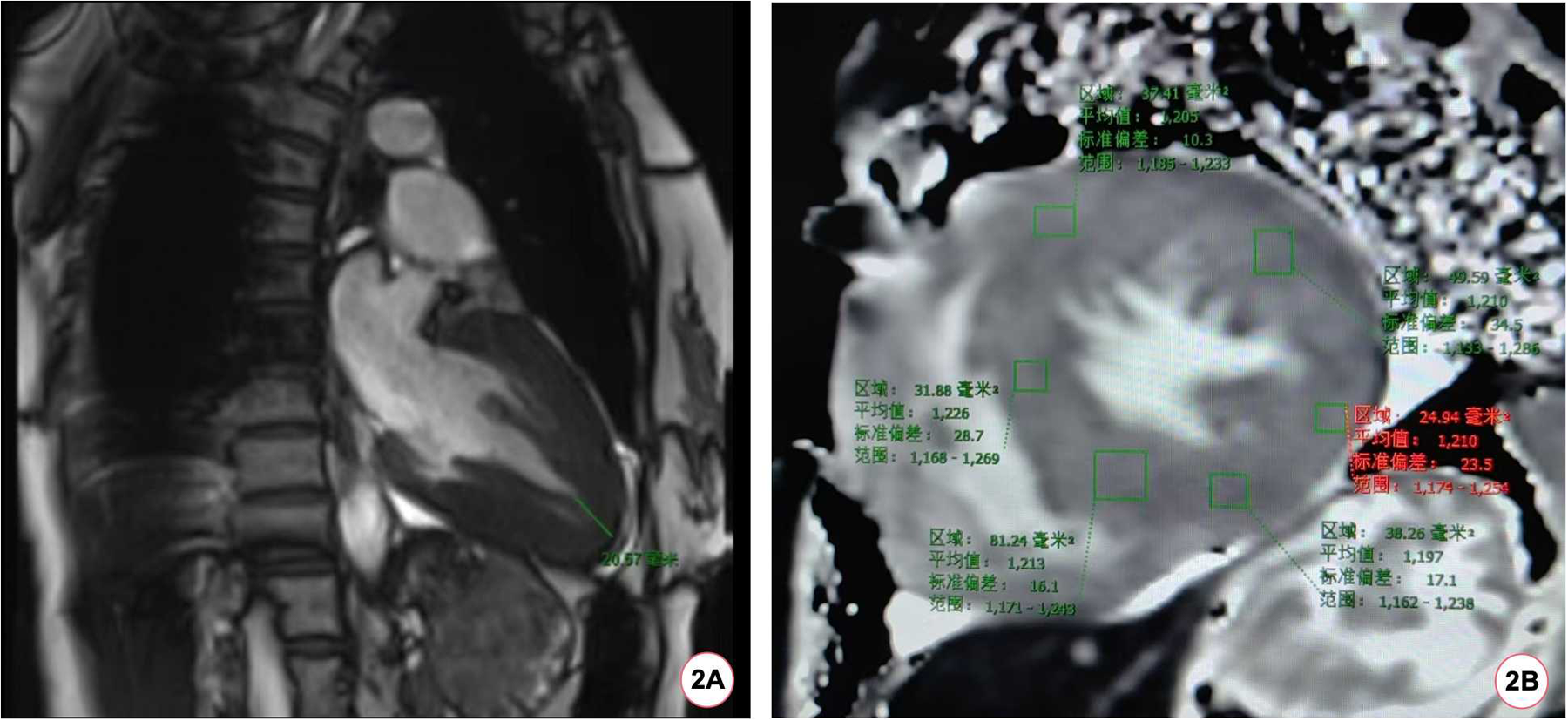
The patient's CMR scan. (A) The patient's CMR imaging revealed a ventricular wall thickness exceeding 20 mm at the apex of the heart. (B) The baseline myocardial T1 mapping value of the patient's CMR was 1,210 (±23.5) ms.
2.3 Etiological results
Due to the patient's unexplained left ventricular hypertrophy, along with arrhythmias, and suspicious cardiac family history, disease associated with gene mutation was suspected. Subsequent examination showed her α-Gal A activity was decreased and globotriaosylsphingosine (Lyso-GL-3) concentration was increased (Table 1). Finally, whole-genome sequencing identified a heterozygous mutation at c.1072_1074delGAG (p.E358del) in the patient's GLA gene (Table 2).
Table 1
| Methodology: MSMS | ||||
|---|---|---|---|---|
| Test item | Abbr. | Test result | Reference interval | Unit |
| Alpha-galactosidase A | α-Gal A | 1.09 ↓ | 2.40–17.65 | umol/L/h |
| Biomarkers(Lyso-GL-3) | Lyso-GL-3 | 8.61 ↑ | <1.11 | ng/ml |
Patient's peripheral blood α-Gal A enzyme activity and globotriaosylsphingosine (Lyso-GL-3) concentration.
Table 2
| Genes and transcripts | Related genes and genetic patterns | Chromosome position | Variable site |
|---|---|---|---|
| GLA NM_000169.2 | Fabry disease (XL) | chrX:10065301 3-100653015 | c.1072_1074 delGAG p.E358del |
| Exon/Intron | Heterozygosity | gnomAD maximum frequency | Mutation degree |
| exon7 | heterozygote | - | likely pathogenic |
Whole gene sequencing results of the patient.
The MutationTaster tool demonstrated this mutation was pathogenic (prob: 0.999) (9). The American College of Medical Genetics and Genomics also classified this mutation as likely pathogenic. Finally, we used SWISS-MODEL to visually simulate the structural site (Figures 3A,B) of E358del mutation (10–14). As α-Gal A is sheared and assembled into dimers in vivo, we utilize the dimeric form of α-Gal A for subsequent experiments. Then we performed a consistency analysis using wild-type and mutant (E358del) dimer structures. We observed significant disparities not only in amino acid 358 but also in the vicinity of amino acids 236, 278, 335, and 405 (Figure 3D). To assess the docking capability of α-Gal A with substrates, we employed the JAMDA Protein-ligand docking function in ProteinsPlus for docking (15–19). Initially, we retrieved the molecular coordinate file of globotriaosylceramide (Gb3, Compound CID: 66616222) from the PubChem website and identified pockets formed by W47, D92, D93, Y134, C142, K168, D170, C172, E203, L206, Y207, R227, D231, D266, and M267 amino acids for docking (20) (Figure 3E). The wild-type yielded 29 poses and JAMDA scores, whereas the E358del variant produced 32 poses and JAMDA scores (19). Finally, we validated the significant difference between wild-type and E358del using non-parametric tests using the SPSS statistical software, version 26.0 (IBM, USA) (Figure 3F), which suggested that the E358del mutant exhibits significantly diminished substrate binding ability compared to that of the wild-type.
Figure 3
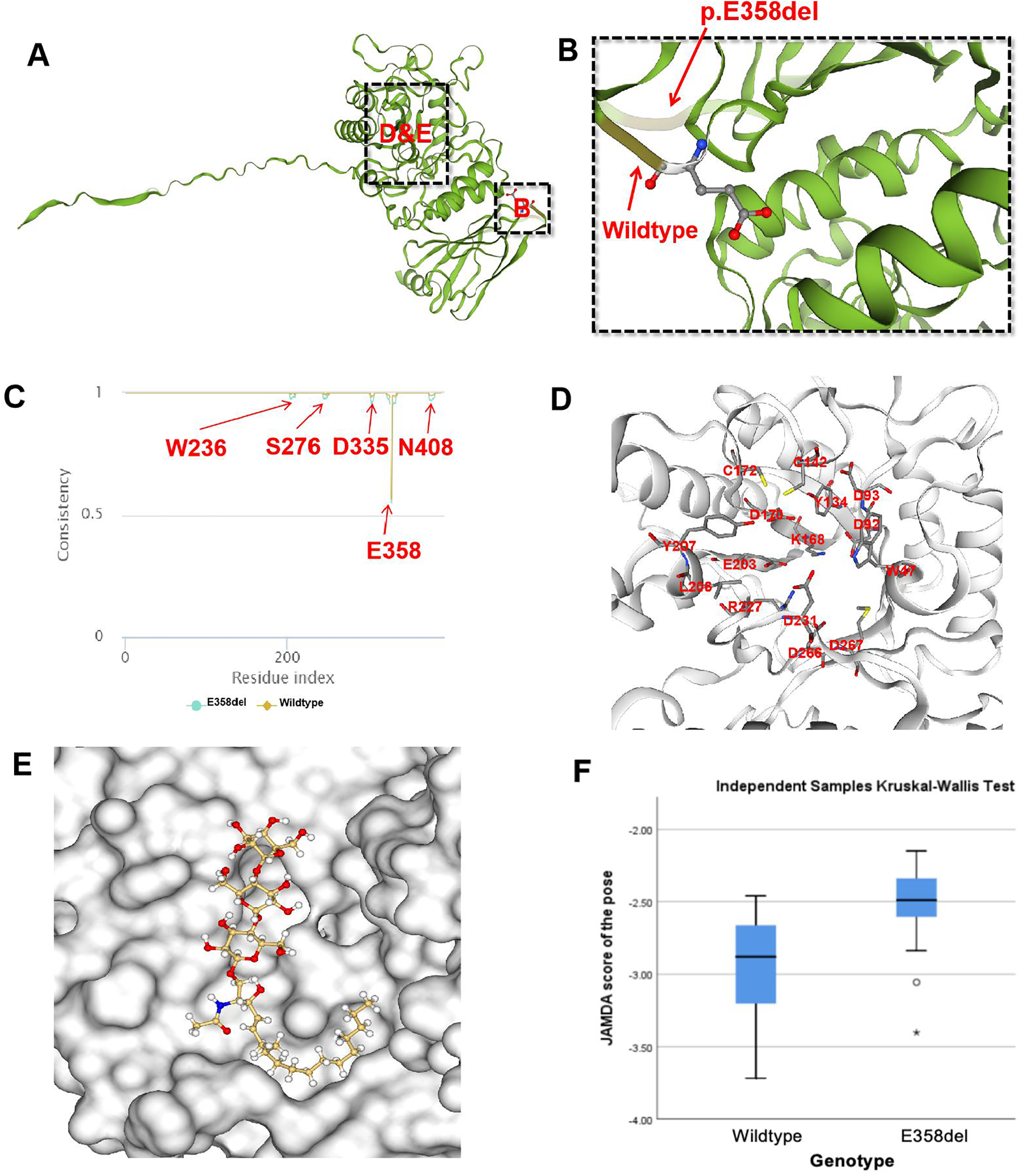
Molecular docking of E358del mutation. (A,B) SWISS-MODEL simulation comparing wild-type and p.E358del; (C) consistency analysis of α-Gal A encoded by wild-type and E358del mutations; (D) One of the molecular pockets in α-Gal A dimer; (E) schematic diagram of Gb3 binding to α-Gal A in E358del mutant (JAMDA score = −3.4018); (F) non-parametric test results of JAMDA scores for wild-type and E358del mutant docking with Gb3.
2.4 Follow-up
After cryo-ablation for AF, sinus rhythm was maintained. The patient was advised to receive enzyme replacement therapy (ERT), but she refused. Over a 2-year follow-up, the patient had no symptoms of discomfort and ECG confirmed the maintenance of sinus rhythm (Figure 1D).
3 Discussion
FD is a rare lysosomal storage disorder caused by mutations in the GLA gene on the X chromosome. Epidemiological study shows that more than 60% of patients with FD have cardiac involvement, among which left ventricular hypertrophy is common (3). Therefore, the phenotype of left ventricular hypertrophy is usually an important clue to proceed the subsequent examination to exclude FD.
In clinical practice, the common cause of left ventricular hypertrophy includes hypertension, aortic valve stenosis, hypertrophic cardiomyopathy, amyloidosis, FD, glycogen storage syndrome, etc (21). In this patient, hypertension and aortic valve stenosis were excluded based on the medical history, ambulatory blood pressure monitoring, and echocardiography. In such a case, hypertrophic cardiomyopathy is usually considered. However, hypertrophic cardiomyopathy mainly characterized by asymmetric hypertrophy of the interventricular septum mostly caused by mutations in the sarcomere gene (22), whereas this patient's left ventricular hypertrophy was symmetrical. Another characteristic of FD is the involvement of the cardiac conduction system, leading to sinus bradycardia or bradycardia-tachycardia syndrome caused by sinus node involvement in the early stages of the disease (23, 24).
AF is the most common arrhythmia with an incidence of nearly 20% in FD (4, 5). Notably, arrhythmias in patients with FD have multiplicity and complexity. In this case, the patient mainly presented with paroxysmal atrial flutter and AF, but the heart rate was slow during sinus rhythm, making the use of antiarrhythmic drugs cautious (25). In addition, literature reports suggest that amiodarone can inhibit lysosomal degradation and induce phospholipid deposition in organs, which may exacerbate the symptoms of FD (6). Therefore, rhythm control with antiarrhythmic drugs for such patients were challenging and ablation is an alternative treatment. Up to now, there have been very scarce reports on the use of ablation for AF in patients with FD. Qian et al (25). reported a case with FD complicated with AF and successfully treated with ablation, but in this study, a traditional radiofrequency ablation was adopted and underwent repeated ablation. In contrast, we used cryo-ablation to treat AF in our case. Although previous studies did not demonstrated the superiority of cryo-ablation to traditional radiofrequency ablation (26), whether cryo-ablation is superior to radiofrequency ablation in patients with FD deserves further study.
Currently, ERT is the main treatment for FD via intravenous administration of exogenous agalsidase alpha/beta every two weeks to replace the function of deficient endogenous α-Gal A (27). ERT has demonstrated efficacy in enhancing the quality of life and mitigating adverse events among FD patients (28). However, ERT treatment has several limitations. For instance, most exogenous enzymes are primarily taken up by the liver, resulting in limited amounts reaching the heart and kidneys (27). Furthermore, postmortem analyses have demonstrated some Gb3 accumulation in the brains in FD patients; however, no exogenous enzyme therapy currently available has been shown to traverse the blood-brain barrier although the clinical significance of Gb3 accumulation in the brain remains unclear (27). Moreover, the autopsy results indicated that FD patients had extensive accumulation of Gb3 in their brains, but currently, there is no exogenous enzyme capable of crossing the blood-brain barrier (27). In addition, approximately 40% of male patients undergoing ERT may develop drug antibodies, which may neutralize the drug's efficacy (28). In this context, novel treatment strategies are being explored. Missense mutations in the GLA gene have been shown to have normal or slightly decreased enzyme activity, but α-Gal A cannot enter lysosome and is degraded by endoplasmic reticulum due to structural abnormalities (29). These mutations are now considered to be more effective through the addition of an oral chaperone therapy regimen (30). In comparison to ERT, oral chaperone therapy does not stimulate antibody production and maintains enzyme levels more closely to physiological states over an extended period (28, 31). In our molecular docking experiments, we demonstrated a notable decrease in docking efficacy with substrates following the c.1072_1074 delGAG mutation. The consistency analysis between the wild-type and E358del mutant revealed variances in the protein structure surrounding W236, S276, D335, E358, and N408 (Figure 3E). This observation aligns with the findings of Seiji et al. (32), who illustrated the significant role of hydrogen bonding between E358 and W236 in maintaining the α-Gal A conformation. However, the roles other amino acids played in E358del mutations require further experimental verification.
Changes in critical sites of enzyme structure significantly influence molecular docking capability (Figure 3H), which aligns with previous reports indicating the limited effectiveness of chaperone therapy for E358del mutation (33). Moreover, some GLA gene mutations are related to specific clinical phenotypes, for instance, mutation at N215S or R112H is commonly referred as “cardiac mutations” (34). Multiple studies targeting heterozygous female FD patients have shown that up to 60%–100% of patients have subjective and/or objective evidence of FD, with approximately 10% exhibiting severe symptoms, suggesting that simply X-linked single-gene inheritance patterns cannot explain the complex phenotype in FD (35).
We searched for c.1072_1074delGAG mutations in the GLA gene in the ClinVar database, and found a total of 9 patients with clinical symptoms and phenotypes reported in 6 articles (36–41). The characteristics of all patients associated with this gene mutation, including the one reported in our study, are summarized in Table 3. Among the affected organs, the heart, kidneys, and nervous system were each involved in 5 cases, followed by the cornea, which was involved in 4 cases.
Table 3
| Authors | Lianne C (36) | Wu (37) | Markus (38) | Lorenzo (39) | Takaaki (40) | Kenichi (41) | Our present case | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Clinical phenotype | Classic | Classic | Classic | Non-classic | Classic | Classic | Classic | Non-classic | ||
| Age (years) | In thirties | 25 | 22 | 6 | 50 | 4 | 8 | 50 | ||
| Gender | Male | Male | Male | Female | Male | Male | Female | Male | Male | Female |
| Race | Australian | Chinese | Chinese | Chinese | Caucasian | Japanese | Japanese | Chinese | ||
| Skin lesions | + | + | + | – | – | – | ||||
| Neurosensory abnormalities | + | + | – | + | + | + | – | |||
| Cardiovascular involvements | + | – | – | – | + | + | + | + | ||
| Renal insufficiency | + | + | + | – | – | + | + | – | ||
| Hypohidrosis | – | + | – | – | + | + | – | |||
| Enteropathy | – | – | – | + | – | |||||
| Cornea verticillata | + | + | + | + | ||||||
| Enzyme activity | 0.4 nmol/ml/h | 0.9 nmol/h/ml | 1.0 nmol/h/ml | 2.6 nmol/h/ml | 1.4% of WT | 12% of WT | 4.7 AgalU | 1.09 umol/L/h | ||
Summary of clinical manifestations of all NM:000169.3 (GLA): c.1072_1074del mutations in the ClinVar database.
Despite the patient did not receive ERT treatment, her sinus rhythm remained for more than 2 years following bilateral pulmonary vein cryo-ablation, which indicated, for one hand, cryo-ablation is effective for such patients; for the other hand, cryo-ablation can be served as a temporary symptom-alleviated method even if ERT is not administered. Actually, previous studies have demonstrated the benefit of early rhythm control vs. rate control in patients with AF (42). Although the efficacy of ablation in FD patients is still needed to be confirmed, ablation remains a therapeutic option for FD with symptomatic AF.
4 Conclusion
For patients with unexplained cardiac hypertrophy, especially middle-aged women, even if there is a lack of multi-system involvement, genetic test should be administered to exclude FD. Cryo-ablation of bilateral pulmonary veins may be an effective way to treat AF secondary to FD.
Statements
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving humans were approved by Institutional Review Board of the First Affiliated Hospital of Chongqing Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
YL: Data curation, Formal analysis, Methodology, Software, Writing – original draft. BH: Funding acquisition, Resources, Supervision, Validation, Writing – review & editing. SL: Funding acquisition, Resources, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was supported by Project of Chongqing Talent plan (cstc2022ycjh-bgzxm0231) and CQMU Program for Youth Innovation in Future Medicine (W0184).
Acknowledgments
We would like to thank the patients for her participation in this study. Generative AI MutationTaster2021 (Charité - Universitätsmedizin Berlin & Berliner Institut für Gesundheitsforschung), SWISS-MODEL (Computational Structural Biology Group) and ProteinsPlus (de.NBI - German Network for Bioinformatics Infrastructure) were used for molecular docking.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Averbuch T White JA Fine NM . Anderson-Fabry disease cardiomyopathy: an update on epidemiology, diagnostic approach, management and monitoring strategies. Front Cardiovasc Med. (2023) 10:1152568. 10.3389/fcvm.2023.1152568
2.
Arends M Wanner C Hughes D Mehta A Oder D Watkinson OT et al Characterization of classical and nonclassical Fabry disease: a multicenter study. J Am Soc Nephrol. (2017) 28(5):1631–41. 10.1681/ASN.2016090964
3.
Umer M Kalra DK . Cardiac MRI in Fabry disease. Front Cardiovasc Med. (2022) 9:1075639. 10.3389/fcvm.2022.1075639
4.
Shah JS Hughes DA Sachdev B Tome M Ward D Lee P et al Prevalence and clinical significance of cardiac arrhythmia in Anderson-Fabry disease. Am J Cardiol. (2005) 96(6):842–6. 10.1016/j.amjcard.2005.05.033
5.
Linhart A Palecek T . Narrative review on Morbus Fabry: diagnosis and management of cardiac manifestations. Cardiovasc Diagn Ther. (2021) 11(2):650–60. 10.21037/cdt-20-593
6.
Fine NM Wang Y Khan A . Acute decompensated heart failure after initiation of amiodarone in a patient with Anderson-Fabry disease. Can J Cardiol. (2019) 35(1):104–5. 10.1016/j.cjca.2018.10.004
7.
Spada M Pagliardini S Yasuda M Tukel T Thiagarajan G Sakuraba H et al High incidence of later-onset Fabry disease revealed by newborn screening. Am J Hum Genet. (2006) 79(1):31–40. 10.1086/504601
8.
Izhar R Borriello M La Russa A Di Paola R De A Capasso G et al Fabry disease in women: genetic basis, available biomarkers, and clinical manifestations. Genes (Basel). (2024) 15(1):37. 10.3390/genes15010037
9.
Schwarz JM Cooper DN Schuelke M Seelow D . Mutationtaster2: mutation prediction for the deep-sequencing age. Nat Methods. (2014) 11(4):361–2. 10.1038/nmeth.2890
10.
Waterhouse A Bertoni M Bienert S Studer G Tauriello G Gumienny R et al SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. (2018) 46(W1):W296–303. 10.1093/nar/gky427
11.
Bienert S Waterhouse A de Beer TA Tauriello G Studer G Bordoli L et al The SWISS-MODEL repository-new features and functionality. Nucleic Acids Res. (2017) 45(D1):D313–9. 10.1093/nar/gkw1132
12.
Guex N Peitsch MC Schwede T . Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis. (2009) 30(Suppl 1):S162–73. 10.1002/elps.200900140
13.
Studer G Rempfer C Waterhouse AM Gumienny R Haas J Schwede T . QMEANDisCo-distance constraints applied on model quality estimation. Bioinformatics. (2020) 36(6):1765–71. 10.1093/bioinformatics/btz828
14.
Bertoni M Kiefer F Biasini M Bordoli L Schwede T . Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci Rep. (2017) 7(1):10480. 10.1038/s41598-017-09654-8
15.
Flachsenberg F Ehrt C Gutermuth T Rarey M . Redocking the PDB. J Chem Inf Model. (2024) 64(1):219–37. 10.1021/acs.jcim.3c01573
16.
Schellhammer I Rarey M . Trixx: structure-based molecule indexing for large-scale virtual screening in sublinear time. J Comput Aided Mol Des. (2007) 21(5):223–38. 10.1007/s10822-007-9103-5
17.
Henzler AM Urbaczek S Hilbig M Rarey M . An integrated approach to knowledge-driven structure-based virtual screening. J Comput Aided Mol Des. (2014) 28(9):927–39. 10.1007/s10822-014-9769-4
18.
Volkamer A Griewel A Grombacher T Rarey M . Analyzing the topology of active sites: on the prediction of pockets and subpockets. J Chem Inf Model. (2010) 50(11):2041–52. 10.1021/ci100241y
19.
Flachsenberg F Meyder A Sommer K Penner P Rarey M . A consistent scheme for gradient-based optimization of protein-ligand poses. J Chem Inf Model. (2020) 60(12):6502–22. 10.1021/acs.jcim.0c01095
20.
Garman SC Garboczi DN . The molecular defect leading to Fabry disease: structure of human alpha-galactosidase. J Mol Biol. (2004) 337(2):319–35. 10.1016/j.jmb.2004.01.035
21.
Grajewski KG Stojanovska J Ibrahim EH Sayyouh M Attili A . Left ventricular hypertrophy: evaluation with cardiac MRI. Curr Probl Diagn Radiol. (2020) 49(6):460–75. 10.1067/j.cpradiol.2019.09.005
22.
Mattos BP Torres MA Freitas VC . Diagnostic evaluation of hypertrophic cardiomyopathy in its clinical and preclinical phases. Arq Bras Cardiol. (2008) 91(1):51–62. 10.1590/s0066-782x2008001300009
23.
Hiestand R Nowak A Sokolska JM Chan R Ruschitzka F Manka R et al Clinical and CMR characteristics associated with cardiac events in patients with Fabry disease. Int J Cardiol. (2023) 382:46–51. 10.1016/j.ijcard.2023.04.016
24.
El Sayed M Postema PG Datema M van Dussen L Kors JA Ter Haar CC et al ECG changes during adult life in Fabry disease: results from a large longitudinal cohort study. Diagnostics (Basel). (2023) 13(3):354. 10.3390/diagnostics13030354
25.
Qian P Ross D Tchan M Sadick N . A patient with recurrent disabling atrial fibrillation and Fabry cardiomyopathy successfully treated with single ring pulmonary vein isolation. Int J Cardiol. (2015) 182:375–6. 10.1016/j.ijcard.2015.01.001
26.
Kuck KH Brugada J Furnkranz A Metzner A Ouyang F Chun KR et al Cryoballoon or radiofrequency ablation for paroxysmal atrial fibrillation. N Engl J Med. (2016) 374(23):2235–45. 10.1056/NEJMoa1602014
27.
van der Veen SJ Hollak C van Kuilenburg A Langeveld M . Developments in the treatment of Fabry disease. J Inherit Metab Dis. (2020) 43(5):908–21. 10.1002/jimd.12228
28.
Lenders M Brand E . Fabry disease: the current treatment landscape. Drugs. (2021) 81(6):635–45. 10.1007/s40265-021-01486-1
29.
Ishii S . Pharmacological chaperone therapy for Fabry disease. Proc Jpn Acad Ser B Phys Biol Sci. (2012) 88(1):18–30. 10.2183/pjab.88.18
30.
Nowicki M Bazan-Socha S Blazejewska-Hyzorek B Klopotowski MM Komar M Kusztal MA et al A review and recommendations for oral chaperone therapy in adult patients with Fabry disease. Orphanet J Rare Dis. (2024) 19(1):16. 10.1186/s13023-024-03028-w
31.
Weidemann F Jovanovic A Herrmann K Vardarli I . Chaperone therapy in Fabry disease. Int J Mol Sci. (2022) 23(3):1887. 10.3390/ijms23031887
32.
Saito S Ohno K Sakuraba H . Comparative study of structural changes caused by different substitutions at the same residue on alpha-galactosidase A. PLoS One. (2013) 8(12):e84267. 10.1371/journal.pone.0084267
33.
Shin SH Kluepfel-Stahl S Cooney AM Kaneski CR Quirk JM Schiffmann R et al Prediction of response of mutated alpha-galactosidase A to a pharmacological chaperone. Pharmacogenet Genomics. (2008) 18(9):773–80. 10.1097/FPC.0b013e32830500f4
34.
Akhtar MM Elliott PM . Anderson-Fabry disease in heart failure. Biophys Rev. (2018) 10(4):1107–19. 10.1007/s12551-018-0432-5
35.
Laney DA Fernhoff PM . Diagnosis of Fabry disease via analysis of family history. J Genet Couns. (2008) 17(1):79–83. 10.1007/s10897-007-9128-x
36.
Blanch LC Meaney C Morris CP . A sensitive mutation screening strategy for Fabry disease: detection of nine mutations in the alpha-galactosidase A gene. Hum Mutat. (1996) 8(1):38–43. 10.1002/(SICI)1098-1004(1996)8:1%3C38::AID-HUMU5%3E3.0.CO;2-L
37.
Wu KH Tzung TY Ro LS Hsiao KJ . A novel mutation (c. 1072_1074delGAG) in the alpha-galactosidase gene of a Taiwanese family with Fabry disease. Acta Derm Venereol. (2004) 84(4):310–1. 10.1080/00015550410024643
38.
Ries M Gupta S Moore DF Sachdev V Quirk JM Murray GJ et al Pediatric Fabry disease. Pediatrics. (2005) 115(3):e344–55. 10.1542/peds.2004-1678
39.
Monserrat L Gimeno-Blanes JR Marín F Hermida-Prieto M García-Honrubia A Pérez I et al Prevalence of Fabry disease in a cohort of 508 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. (2007) 50(25):2399–403. 10.1016/j.jacc.2007.06.062
40.
Sawada T Kido J Yoshida S Sugawara K Momosaki K Inoue T et al Newborn screening for Fabry disease in the western region of Japan. Mol Genet Metab Rep. (2020) 22:100562. 10.1016/j.ymgmr.2019.100562
41.
Hongo K Harada T Fukuro E Kobayashi M Ohashi T Eto Y . Massive accumulation of globotriaosylceramide in various tissues from a Fabry patient with a high antibody titer against alpha-galactosidase A after 6 years of enzyme replacement therapy. Mol Genet Metab Rep. (2020) 24:100623. 10.1016/j.ymgmr.2020.100623
42.
Kirchhof P Camm AJ Goette A Brandes A Eckardt L Elvan A et al Early rhythm-control therapy in patients with atrial fibrillation. N Engl J Med. (2020) 383(14):1305–16. 10.1056/NEJMoa2019422
Summary
Keywords
Fabry disease (FD), paroxysmal atrial fibrillation, cryo-ablation, ventricular hypertropy, molecular docking
Citation
Li Y, Huang B and Luo S (2025) Cryo-ablation management of atrial fibrillation in Fabry disease without agalsidase alpha: a case report. Front. Cardiovasc. Med. 12:1483283. doi: 10.3389/fcvm.2025.1483283
Received
19 August 2024
Accepted
15 May 2025
Published
30 May 2025
Volume
12 - 2025
Edited by
Neil Morgan, University of Birmingham, United Kingdom
Reviewed by
Qi Guo, Capital Medical University, China
Sandro Feriozzi, Azienda Sanitaria Locale di Viterbo, Italy
Updates
Copyright
© 2025 Li, Huang and Luo.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
* Correspondence: Bi Huang huangbi120@163.com Suxin Luo luosuxin0204@163.com
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.